

Abstract
Understanding the dynamics of protein–ligand interactions, which lie at the heart of host–pathogen recognition, represents a crucial step to clarify the molecular determinants implicated in binding events, as well as to optimize the design of new molecules with therapeutic aims. Over the last decade, advances in complementary biophysical and spectroscopic methods permitted us to deeply dissect the fine structural details of biologically relevant molecular recognition processes with high resolution. This Review focuses on the development and use of modern nuclear magnetic resonance (NMR) techniques to dissect binding events. These spectroscopic methods, complementing X‐ray crystallography and molecular modeling methodologies, will be taken into account as indispensable tools to provide a complete picture of protein–glycoconjugate binding mechanisms related to biomedicine applications against infectious diseases.
Keywords: glycoconjugates, molecular modeling, molecular recognition, NMR spectroscopy, proteins
1. Introduction and Overview
1.1. Biological Relevance of Protein–Glycoconjugate Interactions
In nature, carbohydrates constitute an important class of biomolecules; in the form of oligosaccharides, polysaccharides, glycoconjugates like glycosaminoglycans, glycoproteins, glycopeptides, glycolipids, proteoglycans, they have long been known to participate in many biological processes. The remarkable degree of complexity, typical of the three‐dimensional structure of glycans compared to other classes of biomolecules, originates from the many ways they can be assembled from simple sugar building blocks. Although, in mammals, only around ten monosaccharides are used to build longer glycans, they can be connected, in turn, at different positions of the sugar unit, differently substituted, and adopt various spatial orientations, thus creating both linear and branched polymers with a large range of shapes.1 The structural diversity can be further increased through other variables such as the anomeric configuration, the sugar ring size, the introduction of non‐carbohydrate substituents, such as “chemical decoration” with phosphate, sulfate,2 or/and acetyl3 substituents, often in a nonstoichiometric fashion. Bacterial glycans are even more numerous and complex than their eukaryotic counterparts, owing to the further presence of peculiar sugars, including pentoses, heptoses, and nonuloses. Given their extreme structural variability, glycans potentially hold a high information content and are able to trigger specific biochemical cascades upon the tight regulated binding with different molecular receptors including lectins, antibodies, and enzymes.4 Thus, glycan biomolecules set the molecular basis of cell–cell interactions, signal transduction, inflammation, viral entry, and host–bacteria recognition, thereby participating in disease, defense, and symbiosis.5
Recognition of microbial glycans by host proteins, microbial recognition, as well as molecular mimicry of host glycans by microbes can lead to either beneficial or detrimental outcomes; on the one hand, microbes establishing symbiotic or pathogenic interactions use host glycans for adherence or invasion, whereas, on the other hand, peculiar microbial glycosylated molecules, mostly found on the cell surface, are recognized by the innate immune system during early stages of infection, activating inflammation and host defense pathways.6 The molecular comprehension of the fundamental roles played by glycans and glycoconjugates in the dynamic interplay between host and microbes is not well understood, thereby precluding us from the ability to modulate them in beneficial ways.
Given the above premises, improving the knowledge on the molecular features at the basis of glycoconjugates perception, at the maximum possible resolution, is pivotal for the comprehension and modulation of several biological processes closely related to health and disease.
The understanding that a large part of the biological information is encoded in the glycan structures (glycocode) has led to one of the central concepts in glycobiology, that is, the ability of complex carbohydrates to transfer molecular interactions into biological signaling.5 Deconvoluting the roles played by glycans in biological events is a major challenge, owing to various factors. These include their structural complexity, the multivalent nature of their interactions with proteins, as well as their complex biosynthesis and the subtly different phenotypes of glycans that often manifest throughout multicellular environments. Determination of the three‐dimensional structural and dynamic features of complex carbohydrates, along with the molecular basis of their interactions and associations with proteins, constitutes the main challenge of modern structural glycoscience. Nowadays, it is recognized that, to unveil these phenomena at atomic level, the nuclear magnetic resonance (NMR) approach represents a key angle of observation. In this Review article, we survey the significant contributions and the current status of the applications of NMR spectroscopy, when aided by molecular modeling and other biophysical methods, to the characterization of protein–glycoconjugate interactions.
1.2. Biophysical Methods to Unveil Interaction Processes
A wide range of methods is being used to characterize protein–carbohydrate interactions, offering access to various types of quantitative information such as thermodynamic data (stoichiometry of binding, binding constants, enthalpy and entropy components of the binding) as well as kinetics and mechanistic information. Certainly X‐ray crystallography, but also surface plasmon resonance (SPR), isothermal titration microcalorimetry (ITC), electron microscopy, and electron paramagnetic resonance (EPR) spectroscopy, are complementary techniques and would be worth of description; however, in this review, we will focus on the role of NMR spectroscopy and molecular modeling approaches.
1.2.1. NMR Spectroscopy
NMR is an extremely powerful and versatile technique for the detection and characterization of binding events, and provides key structural and dynamic information over a wide range of systems. NMR usually applies a reductionist approach, in which either one or both of the players can be handled by the currently available NMR techniques.
In general, carbohydrate binding to different molecular sensors is governed by relative weak forces including van der Waals interactions, hydrogen bonding, and hydrophobic CH–π associations.7 Salt bonds, coordination with divalent cations, and reorganization of water molecules upon binding are additional factors invoked in glycoconjugate–protein interactions. The result is that cognate ligand–receptor interactions usually occur with weak equilibrium dissociation constants, ranging from micromolar to millimolar, appropriate for analysis with modern NMR techniques. Although several types of classification could be considered, the most intuitive way to gather the numerous NMR techniques used to monitor molecular recognition events is to split them into two broad categories: the “ligand‐based” and the “receptor‐based” approaches. When a ligand binds to its receptor, indeed, the mutual binding affinity drives an exchange process that modulates the NMR parameters of both players of the interaction. Thus, the screening may proceed by ligand and/or receptor point of view, describing the binding event from the different nature of the interacting partners, where the receptor is a large biomolecule (slow tumbling rate and fast relaxation) and the interacting ligand can be simplified as a small molecule (fast tumbling and slow relaxation), whose NMR signals can be better observed, offering information from the ligand perspective (Section 2.1). The observation of large biomolecules by using NMR usually requires isotopic labeling (13C/15N/2H) or the use of specific chemical tags (paramagnetic) (Section 2.2).
One of the main hindrances of NMR in terms of in‐depth structural studies is its relatively intrinsic low sensitivity. Nevertheless, much effort has been made to improve this drawback,8 developing different protocols to unravel the interactions at high resolution. NMR has indeed become one of the most powerful and versatile methods for the investigation of transiently forming complexes, well suited to provide a detailed description of receptor–ligand interactions.
Although in this Review we will basically focus on solution‐state NMR, seminal advanced studies have also been carried out by using solid‐state NMR spectroscopy. In particular, the high‐resolution magic angle spinning (HR‐MAS) NMR approach has been applied to study the structures of bacterial capsular polysaccharides9 and cell‐wall components10 in intact cells. An interesting issue is the ability to study in vivo changes of the composition in the constituent glycoconjugates, depending on factors like bacterial phase growth, gene mutation, or drug administration, which also allow the use of on‐cell HR‐MAS methods to study glycan–protein interactions.11
1.2.2. Molecular Modeling
1.2.2.1. Molecular Mechanics and Dynamics
Characterization of the structural and dynamic features of carbohydrates constitutes a challenge, both from the theoretical and experimental point of view. The conformations of complex carbohydrates depend on 1) the sequence and nature of the monosaccharides in the complex glycan (i.e. glucose vs. mannose), 2) the anomeric centers (i.e. α vs. β), 3) the linkage positions (i.e. 1–3 vs. 1–4), and 4) the chemical modification of the core structure (i.e. sulfation, phosphorylation, methylation, acetylation). These molecules have highly polar functionality and the consequences of the electronic arrangements, such as the anomeric, exo‐anomeric and gauche effects, have to be taken into consideration during conformational and configurational changes.12
As carbohydrates and their derivatives possess many hydroxyl groups, their structures are characterized by a large number of rotatable bonds, which, on top of the torsional movements occurring at the glycosidic linkages (so called Φ and Ψ torsion angles), are sources of conformational flexibility. Besides, the orientation of such hydroxyl groups relative to the sugar ring is at the origin of the existence of hydrophilic patches (formed by polar hydrogens) and hydrophobic patches (formed by nonpolar aliphatic protons). This results in an anisotropic solvent density around carbohydrate molecules. To address these issues, molecular modeling methods have been developed for molecular mechanics and dynamics calculations. Appropriate energy functions and/or parameter sets are available in the literature. Some of them have the capability of treating carbohydrates in interactions with proteins taking solvation into consideration.13
Molecular dynamics offers a way to explore the conformational hyperspace of complex carbohydrates, and at the same time to take into account the subtle interplay between carbohydrate and water molecules. In molecular dynamics simulations, an ensemble of configurations is generated by applying the laws of motion to the atoms of the molecule. The concept behind molecular dynamics simulation involves calculating the displacement co‐ordinates in time (trajectory) of a molecular system at a given temperature. Finding positions and velocities of a set of particles as a function of time is done classically by integrating Newtons's equation of motion in time. Several algorithms have been developed for molecular dynamics simulations. Such simulations follow a system for a limited time. Physically observed properties are computed as the appropriate time averages through the collective behavior of individual molecules. For the results to be meaningful, the simulations must be sufficiently long, so that the important motions are statistically well sampled. Experimentally accessible spectroscopic and thermodynamic quantities can be computed, compared, and related to microscopic interactions.
It should be noted that molecular dynamics is severely limited by the available computer power. Very recently, it became feasible to perform a simulation with several thousand explicit atoms for a total time of up to the microsecond scale, but most of the published simulations have a duration of less than a microsecond. To explore the conformational space adequately, it is necessary to perform many such simulations. In addition, it may be possible that carbohydrate molecules undergo dynamic events on longer time scales. These motions cannot be investigated with standard molecular dynamics techniques, and such a limitation makes it difficult to compare situations that occur on a much larger timescale that normally occur throughout NMR experiments. At present, the best approach is the inclusion of the environment in the simulation, that is, a molecular dynamics simulation with explicit water molecules or other surrounding molecules. Carbohydrates have a very high affinity towards water, with the majority of hydrogen bonding between water and carbohydrates occurring throughout their hydroxyl groups. The carbohydrates affect the surrounding water structure, and, in return, the water affects the structure of the dissolved carbohydrate molecules. Molecular dynamics provides a most promising way to investigate the hydration features of carbohydrates and set up a firm basis for docking simulations.
1.2.2.2. Docking Simulations
When used in conformational studies of carbohydrates, computational molecular modeling methods offer alternatives for the study of protein–carbohydrate interactions (Figure 1). Significant steps have been made, among which are the developments and implementations of force fields capable of accounting for the specificity of carbohydrates and their compatibility with those developed for proteins. The conformational flexibility of carbohydrates needs to be characterized and taken into account at each step of the investigation. Protein–carbohydrate docking has come of age; reliable and insightful results have started to be produced.14 The question of choosing the appropriate software with regard to the problem to be investigated still stands, and remains critical with respect to the proposed solution. This is particularly true for cases of small ligands in large and poorly defined binding sites.
Figure 1.
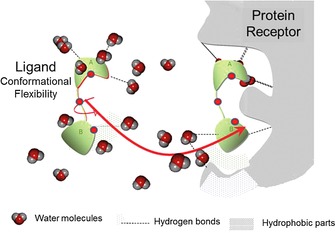
Representation of protein–ligand interactions. Theoretical and computational methods are used to the predict ligand orientation in the binding pocket.
Docking is a computational method that places a small molecule (ligand) in the combining site of its macromolecular target (receptor), and provides an estimate of the binding affinity. Molecular docking requires (at least some) structural knowledge of the ligand and the receptor of interest. The carbohydrate ligands are typically built by using molecular mechanics methods or directly sourced from structural databases. Energy parameters suitable for energy minimization and/or molecular dynamics of protein–carbohydrate complexes are available for different force fields.15 Receptors structures are currently obtained from X‐ray crystallography and NMR spectroscopy; those that are unavailable can be generated by homology modeling, threading, and de novo methods. Despite the fact that several docking programs that operate in slightly different ways are available, they all involve two main features, that is sampling and scoring.
Sampling entails the conformational and orientational location of the ligand in the receptor binding site.
To predict the carbohydrate orientation in binding sites, flexible docking methods are used to account for possible orientations of pendent groups (i.e. hydrogen bond network directed by the orientation of hydroxyl and hydroxymethyl groups) and the conformational flexibility occurring at each glycosidic linkage. In most docking programs, the ligand is treated as flexible, whereas the protein conformation is often kept rigid. Programs exist that have the capability to carry such “soft docking”. Proper accounting for receptor flexibility is computationally much more expensive, and it is not yet a common practice.
The docking algorithms can be grouped into deterministic approaches that provide reproducibility and stochastic approaches in which the algorithm includes random factors that do not allow for full reproducibility. Incremental construction algorithms consist of the division of a ligand in rigid fragments, as implemented in program DOCK.16 One of the fragments is selected and placed in the protein binding site. The reconstruction of the ligand is performed in situ, adding the remaining fragments. Among the stochastic searching approaches, the genetic algorithm (inspired by evolutionary biology) is implemented in AutoDock.17 A variety of other sampling methods has been implemented in docking programs. Some of them include simulated annealing protocols and Monte Carlo simulations. The algorithm used in Glide18 can be defined as a hierarchical algorithm.
Scoring functions are used to evaluate the best conformation, orientation, and translation (referred to as poses), which classify the ligands in rank order. Energy scoring functions evaluate the free energy of binding between proteins and ligands, using the Gibbs–Helmhotz equation that describes ligand–receptor affinity. Empirical scoring functions use a set of parametrized terms describing properties known to be decisive in protein–ligand binding to formulate an equation for predicting affinities. These terms generally describe polar–apolar interactions, loss of ligand flexibility, and desolvation effects. A distinct feature of protein–carbohydrate recognition is the interaction between aromatic side chains of the proteins and C−H bonds of the carbohydrate's hydrophobic faces, which results in the formation of crucial CH–π contacts.7 Widely used docking programs, which account differently for these types of interactions, may not perform as well for protein–carbohydrate complexes. It has been recognized that various docking programs and scoring functions perform differently for different targets, and that varying performance has been observed for different ligand types. It should be recognized that accurate determination of carbohydrate–protein complexes remains a non‐trivial matter. In the case of protein–lectin complexes, the difficulties are attributed to the shallow and multichambered binding sites of many lectins. Comprehensive studies on the validation of carbohydrate–lectin docking have been performed and compared to experimentally crystallographically determined complexes. In comparison with the large number of docking studies performed on carbohydrate–lectin complexes, there are relatively few published docking studies on carbohydrate–antibody recognition, reflecting the limited number of suitable validation tests (i.e. high‐resolution carbohydrate–antibody crystal structure complexes) and the inherent difficulty in modeling such systems.
Despite inherent difficulties arising from the challenges of protein–carbohydrate complexes, molecular docking has started producing reliable and insightful results. However, many challenges remain, and it is still a non‐trivial exercise to perform and far from being a turn‐key tool. In particular, the ability of docking programs to correctly score docking poses (especially in the cases of small ligands in large and poorly defined binding sites) calls for a critical inspection of the results.
1.2.3. Interplay of Molecular Modeling and NMR
Nowadays, the increasing interplay of NMR with other biophysical methods supports structural biology, especially in the determination of carbohydrate and protein 3D structures, as well as the comprehension, at high resolution, of protein–glycoconjugate system interactions (Figure 2).
Figure 2.
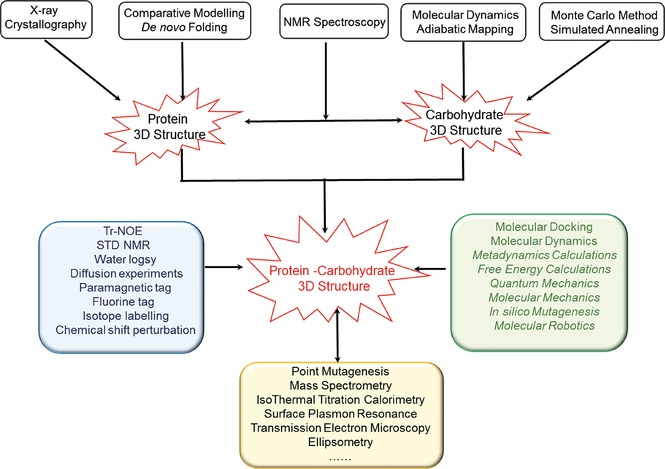
Interplay of NMR with other biophysical methods in the 3D structure determination of carbohydrates, proteins, and protein–glycoconjugate complexes.
Above all, the use of computational methods to complement data gathered from NMR experiments is becoming an accepted protocol to elucidate the structural features of protein–carbohydrate interactions. Most of the recently published articles make use, in one way or another, of molecular modeling tools. Given the recognized high conformational flexibility of oligo‐ and polysaccharides, a preliminary level of investigation requires the characterization of their starting conformation(s) from molecular dynamics calculations. A large number of the protein–carbohydrate systems investigated so far (see Table 2) deal with carbohydrates of low to moderate molecular weight. It may be anticipated that future investigations, aiming at exploring the binding of larger oligosaccharides in their interactions with antibodies, will require a more complete considerations of both conformational flexibility and influence of hydration.
Table 2.
Summary of the most relevant studies of protein–glycoconjugate interactions, showing the NMR methods discussed in Section 2. Some of these are described in detail in this Review article.
| System of interaction | M W protein | NMR approach | Ref. |
|---|---|---|---|
| Lectins | |||
| Dectin‐1+glucans | ca. 30 kDa | STD NMR | 64, 65 |
| ZG16P+mycobacterial phosphatidilinositol mannosides | ca. 14 kDa | STD NMR; tr‐NOE; CSP | 66 |
| DC‐SIGN+virus associated ligands | ca. 44 kDa; CRD ca. 18 kDa | STD NMR; tr‐NOE; CSP | 67, 68, 69, 70 |
| DC‐SIGNR+mannosides | ca. 17 kDa | CSP | 71 |
| langerin+GAGs | ca. 35 kDa | STD NMR; tr‐NOE | 72 |
| MBL+carbohydrates of viral surface | ca. 70 kDa | STD NMR; tr‐NOE | 73 |
| E‐selectin+sialyl lewis x | ca. 115 kDa | STD NMR; tr‐NOE | 74 |
| galectins+digalactosides | ca. 14 kDa | CSP | 75 |
| BTL+N‐glycans | ca. 9 kDa | STD NMR | 76 |
| siglec+oligosaccharides | ca. 100 kDa | STD NMR | 77 |
| HA+oligosaccharides | ca. 60 kDa | STD NMR | 78, 79 |
| cyanovirin‐N+mannosides | ca. 11 kDa | STD NMR | 80 |
| MVN+mannosides | ca. 12 kDa | CSP | 81 |
| FimH+mannosides | ca. 30 kDa | CSP | 82 |
| CCL2+oligosaccharide | ca. 15 kDa | CSP | 83 |
| hevein‐+chitoligosaccharides | ca. 4.7 kDa | CSP | 84 |
| Viruses | |||
| RHDV+HBGA | VLPs ca. 10 MDa | STD NMR | 85 |
| NV+HBGA | VLPs ca. 10 MDa | STD NMR; tr‐NOE | 86 |
| VP8+oligosaccharides | ca. 25 kDa | STD NMR | 87 |
| Antibodies | |||
| ScFv+TF antigen | ca. 30 kDa | STD NMR | 88 |
| mAb+LPS from Yersinia pestis | ca. 50 kDa | STD NMR | 89 |
| TT conjugate antibodies+LPS from Bordetella pertussis | – | STD NMR | 90 |
| mAb 5D8+LPS from Burkholderia anthina | >150 kDa | STD NMR; tr‐NOE | 91 |
| mAb CS35+oligosaccharide from B.anthracis | >150 kDa | STD NMR | 92 |
| mAb 4C4+CPS from B. pseudomallei | >150 kDa | STD NMR; tr‐NOE; DOSY | 41 |
| mAb+O‐polysaccharide from Shigella flexnery | – | STD NMR; tr‐NOE; water LOGSY | 36, 93 |
| mAb 2G12+mannosylated glycans | – | STD NMR; tr‐NOE; CORCEMA‐ST | 23 |
| mAb+N‐glycans (HIV) | ca. 50 kDa | STD NMR | 94, 95 |
| mAb+oligosaccharides from Mycobacterium | – | STD NMR; tr‐NOE | 96, 97 |
| Eukaryotic immune receptors | |||
| LysM+chitooligosaccharides | ca. 20 kDa | STD NMR; tr‐NOE | 98 |
| El tor Cholera+oligosaccharides | ca. 20 kDa | STD NMR | 99 |
| FH+sialylated oligosaccharides | ca. 50 kDa | STD NMR | 100 |
With regard to the protein partner, the starting 3D structures can be generated either by considering available X‐ray structures or by using homology modeling methods. The use of atomic coordinates derived from the high‐resolution X‐ray structure offers the most reliable source prior to any docking calculations. When starting from an X‐ray elucidation, the protein structure has to be fully ‘re‐optimized’ prior to any docking calculation, by 1) giving the proper ionization states to the amino‐acid residues, 2) removing or completing the “crystalline” water molecules with addition of explicit solvation, and 3) running a full molecular dynamics simulation of the so‐reconstructed protein in its hydrated environment. It is likely that such a computational protocol will become standard practice in the future in order to generate a set of starting structures for docking experiments.
As stated previously, the evaluation of several computational programs in terms of their performance for docking carbohydrate molecules into lectins and antibodies differ widely. Special attention needs to be given to the ranking of docking poses, a difficulty mainly caused by scoring problems that are not systematically performed. The most commonly used programs are Glide, GOLD, AutoDock, and Flex; they are used under different conditions, with regard to the flexibility of the ligands and the proteins. Cases can also be found where positioning the carbohydrate in the protein binding site was achieved by performing energy minimization throughout a molecular dynamics simulation over 10–30 ns.
Besides providing a detailed picture of the key moieties of the molecules involved in the interactions, the results derived from computational methods offer ways to correct some difficult experimental bias. Computational simulations can also help to establish and ascertain the occurrence of single or multiple binding sites for the carbohydrate to the protein. This is particularly important in deciphering the eventual contributions arising from two or more sites, and takes into account their respective contributions to the observed NMR data. Practically, it should be acknowledged that existing software, energy functions, and scoring functions need to be evaluated further for protein–carbohydrate systems before they can be used routinely and reliably. It should be also understood that the timescales of NMR experiments are far from those covered by molecular dynamics simulations (rarely in the range of few microseconds). Another feature to be elucidated is the influence of multivalency can on the experimental and computational levels.
2. NMR for Protein–Carbohydrate Interactions
2.1. Ligand‐Based NMR Methods
The ligand‐based methods rely on the differential behavior of the ligand in the free and bound state. They are particularly useful in the medium–low affinity range, characterized by dissociation binding constant K D≥100 μm (see Table 1). Consequently, they have been adopted to detect a wide range of different systems of interaction in “fast‐exchange” regime, with the slowest exchange rate k off values lying in the range 1000<k off<100 000 s−1. Such an approach exhibits several advantages: 1) it does not require labeled protein, as only the ligand signals are detected; 2) only a small amount of receptor is necessary (typically in the micromolar range); 3) there are no limitations about the upper size of the protein, that is, using a high‐molecular‐weight receptor results in readily detectable ligand signals. However, as labeling with stable isotopes is not used in these experiments and the structure of the receptor is not known, it is not possible to directly extract information about the location of the protein binding site or about the specificity of the interaction; competition experiments could be used to partially avoid this last issue by measuring signals of ligands with different affinities. In addition, it is worth noting that most of these techniques require an excess of the substrate with respect to the receptor (typically in the millimolar range); therefore, a low solubility of the ligand represents a further limitation.
Table 1.
Typical range of applicability and delivered information from the main NMR methods for the study of protein–ligand interactions.
| NMR methods | Range of applicability | Delivered information | |||||
|---|---|---|---|---|---|---|---|
| K D [M] | Target M W [kDa] | Typical protein/ligand ratio | Labeled target required | Target binding site | Ligand epitope mapping | Ligand selectivity in a mixture | |
| TR‐NOE | 10 −6–10 −3 | no limit | 1:5/1:50 | no | ✓ | ||
| STD NMR | 10 −6–10 −3 | >15 | 1:50/1:200 | no | ✓ | ✓ | |
| waterLogsy | 10 −6–10 −3 | no limit | 1:5/1:50 | no | ✓ | ✓ | |
| diffusion experiments | 10 −6–10 −3 | no limit | 1:1/1:20 | no | ✓ | ✓ | |
| CSP | 10 −9–10 −3 | <100 | 1:1/1:10 | yes | ✓ | ||
2.1.1. Exchange‐Transferred Nuclear Overhauser Effect
The nuclear Overhauser enhancement or effect (NOE), namely the dipolar interaction between two spins, is the most important phenomenon in NMR spectroscopy. The NOE decreases very fast with distance (1/r 6) and occurs between nuclei close in space with a distance of <5–6 Å (500–600 pm). The magnitude and the sign of NOE are intimately related to the Brownian motion of the molecular structure.19 Molecules with a short correlation time τ c, which corresponds to a fast random rotation in solution, exhibit positive NOE contacts, whereas molecules with a long τ c, which means a rather sluggish rotation, undergo negative NOE. The correlation time is influenced by different factors including temperature (the higher the temperature, the shorter τ c), solvent viscosity (the more viscous, the longer τ c), aggregation in solution, but, overall, depends on the molecular weight (the larger a molecule, the slower its re‐orientation; therefore, the longer its τ c). The exchange‐transferred NOE (tr‐NOE) is evaluated whenever the pulse sequence of a NOESY experiment is applied to an interacting protein–ligand system in dynamic exchange. Such methods provide key information on the ligands’ binding mode in the natural environment by measuring intra ligand and inter ligand–protein distances. It allows: 1) monitoring possible changes in the conformational equilibrium of ligand upon binding; 2) deducing the recognized conformation in the bound state, the so‐called “bioactive conformation”; 3) determining the orientation of the ligand in the receptor binding pocket. The observation of tr‐NOEs relies on the different effective correlation times τ c of free and bound molecules. When a small ligand binds to a high‐molecular‐weight protein, it behaves as a part of a large molecule and adopts the corresponding NOE behavior. Therefore, the NOE signs undergo a drastic changes and lead to the appearance of transferred NOEs (Figure 3).
Figure 3.
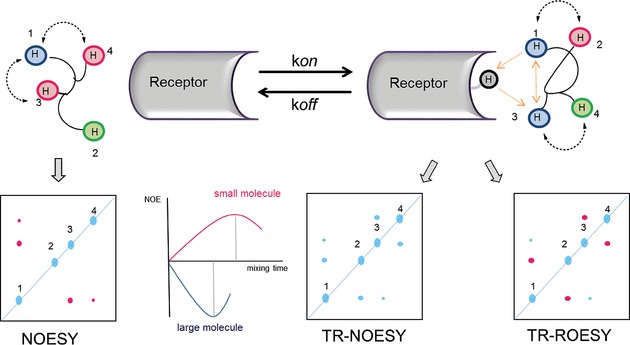
Schematic representation of a NOESY spectrum of a small ligand in the free state, which reaches the maximum of NOE intensity at longer mixing times; cross peaks and diagonal peaks have different signs (left). Schematic representation of tr‐NOESY and tr‐ROESY spectra recorded on the ligand in the bound state, characterized by faster build up rate (right). In the tr‐NOESY spectrum, cross peaks and diagonal peaks show the same signs as expected for a large molecule, thus indicating binding to the protein. The relative sizes of the peaks and the appearance/disappearance of NOE contacts may be used to detect conformational variations. The tr‐ROESY spin‐diffusion cross peaks (H1/H3) and diagonal peaks display the same signs, whereas direct cross peaks (H1/H2; H3/H4) have a different sign to the diagonal peaks.20
The conditions for the applicability of this method are established considering the equilibrium between free and protein‐bound ligands and their molar fractions. To perform tr‐NOESY experiments, it is important that the dissociation of the ligand occurs sufficiently quickly; otherwise, for the slow off‐rate, the ligand relaxes before dissociation from the protein target takes place and no tr‐NOE will be detected. Under ideal conditions, the existence of binding of a given ligand to a receptor protein can easily be distinguished by merely monitoring the sign and size of the observed NOEs. However, if a larger ligand is studied (M W>2000 Da), negative NOE contacts will also be observed for the free state. In such a case, a quantitative analysis based on the construction of NOE build‐up curves may help in defining protein‐induced conformational variations. Indeed, the time required to achieve maximum NOE intensity (build‐up rate) is in the range of 50–100 ms for tr‐NOEs originating from the bound state, whereas it is four‐ to ten‐times longer for nonbinding molecules (Figure 3). The accurate evaluation of the pattern of intermolecular NOE contacts provides key information that allows us to define whether a specific conformer has been selected upon protein binding from an ensemble of conformers present in solution.
Usually, experiments with different ligand target concentration ratios are acquired, ranging from 1:10 to 1:50, depending on the size of the receptor and the kinetic constant involved.
One of the major drawbacks of tr‐NOE experiments is related to spin diffusion effects, common for large molecules. Indeed, they lead to the appearance of negative NOE contacts between ligand protons that are actually far apart in the bound state, but that arise because of the exchange of magnetization mediated by other spins, often including those of the receptor. Thus, receptor‐mediated indirect tr‐NOE effects may produce interpretation errors in the analysis of the ligand bioactive conformation. However, by using tr‐NOEs in the rotating frame (tr‐ROESY) experiments,20 it is possible to distinguish between cross peaks that are dominated by spin diffusion effects and direct NOE enhancements. In a tr‐ROESY spectrum, indeed, all the molecules are in the fast‐tumbling limit and all direct ROEs effects appear as positive cross‐peaks; on the contrary, spin‐diffusion enhancements result in negative ROE signals (Figure 3). Hence, complementing the NOE data with ROE spectra represents a good approach to avoid errors in the calculated inter‐nuclear distances and, therefore, in the predicted bioactive conformation.
2.1.2. Saturation Transfer Difference NMR
The intermolecular magnetization transfer occurring through the 1H−1H cross‐relaxation pathways between protein and ligand spins in transient complexes stands at the basis of the saturation transfer difference (STD) NMR method.21 Originally proposed as a technique for the rapid screening of compound libraries, it is now one of the most powerful and widespread NMR techniques for the detection and characterization of receptor–ligand interactions in solution. STD NMR, indeed, allows not only to deduce the existence of interactions in solution, but it permits also to determine the binding epitope of ligands, detecting the regions in closer contact with the receptor.
An STD NMR spectrum is produced by the difference between on‐resonance and off‐resonance experiments (Figure 4). In the on‐resonance experiment, resonances of the protein are selectively irradiated by applying a selective RF(radio frequency)‐pulse train for a given time (saturation time) to a region of the spectrum only containing protein signals, and where ligand resonances are absent. As a rule of thumb, if the ligand is a sugar, the on‐resonance irradiation frequency is set to around 0/−1 ppm, or it could be placed in the aromatic proton spectral region, as no saccharide resonances are found in these ppm ranges. The off‐resonance experiment, recorded with no protein saturation, represents the reference spectrum. The STD spectrum is obtained by subtracting the off‐ and on‐resonance experiments, which are recorded in an interleaved fashion, and yields only signals of the ligand(s) that received saturation transfer from the protein.
Figure 4.
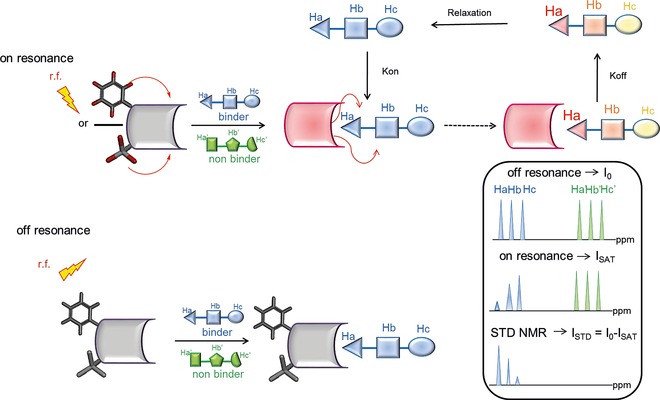
Schematic representation of the STD NMR method. During on‐resonance (upper panel), frequency‐selective irradiation (r.f.), applied for a sustained period (saturation time), causes saturation of the receptor protein. Then, the saturation is transferred by spin diffusion through intermolecular NOEs to the ligand–protein interface, that is, to the bound compounds during their residence time in the protein binding site. Next, by chemical exchange, the ligand molecules go into solution, where the saturated state persists, owing to their small R 1 (enthalpic relaxation) values, and can be detected. In the off‐resonance experiment (lower panel), r.f. saturation is applied in an off‐resonance region from both receptor and ligand, producing a reference spectrum. As a rule of thumb, an irradiation time of 2 s and a 100‐fold excess of ligand give good results. Given the high ligand/protein ratios, only a relatively small amount of protein is required for the measurements (10−9–10−6 m). It is worth noting that receptor resonances are not usually visible, as the protein concentration in solution is very low, and they easily can be deleted by R 2 relaxation filtering prior to detection.
STD involves two experiments: the on‐ and the off‐ resonance.
It is worth noting that, for a given ligand, not all protons will receive the same amount of saturation, as the transfer of magnetization from the receptor will depend on the inverse sixth power of the intermolecular 1H−1H distance in the bound state. Thus, in the STD NMR spectrum, resonance intensities of binding compounds will be dependent on their proximity to the binding pocket. Ligand protons in closer contact to the protein binding site will receive a higher degree of saturation and, therefore, will give rise to stronger STD signals.
The degree of ligand saturation naturally depends on its residence time in the protein‐binding pocket. The STD sensitivity also depends on the number of ligands receiving the saturation from the receptor and can be described in terms of the average number of saturated ligands produced per receptor molecule. Indeed, the saturation of the protein and the bound ligand is fast (about 100 ms) and, assuming fast ligand exchange, as in the case of protein–carbohydrate interactions, the information about saturation is transferred very quickly into solution. If a large excess of the ligand is present, one binding site can be used to saturate many ligand molecules in a few seconds. Ligands in solution lose their information by normal T 1/T 2 relaxation, which is on the order of one second for carbohydrates; thus, the proportion of saturated ligands in solution increases during the sustained RF‐pulse train (saturation time), and the information about the bound state resulting from the saturated protein is amplified. This means that the more ligand that is used, the longer the irradiation time and the stronger the STD signal is. As a rule of thumb, an irradiation time of 2 s and a 50‐ to100‐fold excess of ligand give good results.
Ideally, epitope information would not depend on the chosen saturation time; however, significantly different R 1 relaxation rates of the ligand protons can produce artifacts in the epitope definition. Indeed, protons with slower R 1 relaxations efficiently accumulate saturation in solution, giving rise to higher STD relative intensity, which means that their proximity to the binding pocket may be overestimated. Thus, to quantitatively and correctly estimate the binding epitope, the use of STD build‐up curves has been proposed.22, 23 The experimental STD build‐up curves can be further compared with STD intensities predicted for a model of the protein–ligand complex that could be obtained by using different techniques, including X‐ray, docking, and molecular modeling techniques. This quantitative structural interpretation of experimental STD data, known as CORCEMA (complete relaxation and conformational exchange matrix)‐ST and developed by Jayalakshmi and Krishna, allows us to define how well the molecular model reproduces the experimental NMR data.24
As the STD intensity reflects the concentration of ligand–receptor complex present in solution, data obtained from STD NMR experiments are fundamental not only in the definition of the epitope mapping, but they can even be used in the determination of K D values.23, 25 Furthermore, competition STD NMR experiments have been proposed to derive an approximate K D value of strong binding ligands with slow dissociation rates.26 In this case, a reference compound with intermediate affinity for the receptor and a known K D is used. The observation of a substantial decrease in the reference compound resonances upon the addition of the ligand gives an indication of its K D. As extensively shown by Pinto and co‐workers, this technique is also useful to determine if different ligands in a compound mixture bind at or near to the same binding site.27
It is worth noting that the STD effects depend largely on the off rate; the K D range of the STD method has been estimated to be 10−8–10−3 m. For weak binders, the probability of the ligand to be in the receptor pocket is very low. As the K D value further increases, the population of the complex decreases, leading to a reduction and ultimately the disappearance of the STD signals. Conversely, if binding is very tight, and off rates are in the range of 0.1–0.01 Hz, decreasing K D implies increasing of the receptor–ligand lifetime, and the STD spectra are poorly detectable. Fortunately, the STD NMR method is well suited for the study of protein–carbohydrate complexes, as their typical K D values are in the 10−3–10−6 m range. Therefore, STD NMR is a solid and versatile technique that gives essential information, at a molecular level, on receptor–glycoconjugate ligand interactions. Starting from the simplest version of the methodology, elaborate alternatives have been developed to enhance the resolution and to allow the study of complex systems of interactions, such as the interactions of small molecules in live cells. In this context, saturation transfer double difference (STDD) NMR has been reported for the analysis of ligands binding directly on the surface of living cells.28 As the name suggests, this method differs from the standard procedure in that the final spectrum is obtained from a double difference between the STD spectrum of the cell/ligand mixture and those acquired for the cell only. More recently, a modified STD protocol, called “clean‐STD”, has been proposed to avoid accidental saturation of the ligand.29 Finally, with the aim to overcome problems related to the huge proton overlapping typical of carbohydrate molecules, STD information can be extended into a second dimension by running 2D experiments like STD‐TOCSY, STD‐HSQC, STD‐NOESY.30
2.1.3. WaterLOGSY
Water–ligand observed via gradient spectroscopy (waterLOGSY) is a powerful method for the identification of compounds interacting with macromolecules.31 It may be considered a peculiar variant of the STD NMR method, which relies on the transfer of magnetization from the bulk water molecules to the free ligand at the protein–ligand interface.32 It allows us to easily discriminate between binders and non‐binders, with higher sensitivity with respect to STD NMR experiments.
In this experiment, the water molecules surrounding the receptor protein are excited through a selective irradiation or by using pulsed field gradients.33 Once inverted, the water magnetization is transferred, during the mixing time, to the bound ligand through different pathways (Figure 5):
Figure 5.
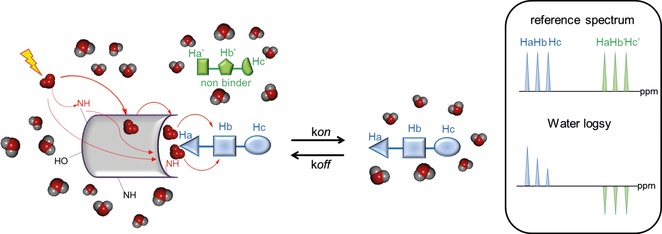
Schematic representation of the waterLOGSY experiment. The different cross‐relaxation pathways are shown with red lines. High ligand/protein ratios should be avoided; otherwise, negative peaks would be observed even if the ligand is bound to the receptor, as the contribution from the free state would be predominant. Typically, protein/ligand ratios of 1:10 or 1:50 are chosen. Recording waterLOGSY spectra at low temperature, typically 5 °C, has been reported to give an improved signal‐to‐noise ratio.34
By direct transfer from the water molecules immobilized on the binding site of the protein, the so called “bound waters”, as their receptor residence time is longer than 1 ns.
A second pathway involves a chemical exchange between excited water and labile receptor NH and OH protons within the binding site, with a subsequent spread of the inversion to the bound ligand through intermolecular dipole–dipole cross relaxation.
Another mechanism of magnetization transfer relies on indirect cross relaxation with exchangeable receptor protons distant from the binding pocket and the propagation of the inverted magnetization by spin diffusion through a protein–ligand complex.
All these pathways act constructively, enhancing the sensitivity of waterLOGSY experiments.
In all three pathways described above, the bound ligands interact, directly or indirectly, with inverted water with rotational correlation time, yielding negative cross‐relaxation rates and exhibiting negative NOEs with water. On the contrary, small non‐binder molecules interacting with bulk water with much shorter correlation times will experience much faster tumbling, which translates into positive NOEs. Therefore, in a WaterLOGSY spectrum, free versus protein‐bound ligands will display peaks of opposite signs, which enables one to easily discriminate between binders and non‐binders.
As this experiment is less dependent on the spin diffusion, it is a widely applied 1D ligand‐observation technique, especially for the study of low‐proton‐density receptors, such as nucleic acids. However, in contrast to most other ligand‐observed NMR methods, it takes fully into account the influence of protein and ligand solvation during complex formation.35 Therefore, its results are particularly useful for a deep understanding of protein–carbohydrate system interactions that require knowledge of the role of water molecules in the binding pocket. As elegantly shown by Pinto and co‐workers,36 combination of the waterLOGSY experiment with STD NMR data may indeed allow the identification of bound water molecules at the interface between the protein and the ligand upon binding. In addition, a novel application of waterLOGSY, known as SALMON (solvent accessibility and protein–ligand binding studied by NMR spectroscopy),37 has also been shown to provide valuable information about the ligand orientation in the protein binding pocket, deducing the portions of the ligand that are more exposed to the solvent upon the interaction.
The major drawback of the waterLOGSY method is related to its inability to directly detect strong binding ligands with slow dissociation rates. However, as for the STD, competition water LOGSY experiments have been proposed38 to overcome this issue.
2.1.4. Diffusion Experiments
The comparison of the ligand diffusion coefficient in the presence and in the absence of a target protein offers a fast method to characterize molecular interactions between small ligands and intermediate size receptors.39 The diffusion coefficient reflects the translational mobility of a molecule and it is closely related to its molecular size, as shown in the Stokes–Einstein equation.
A ligand and its receptor exhibit, in the free state, their own diffusion coefficients, which are dependent on their size and shape. When a ligand forms a complex with its receptor protein, it transiently adopts the properties of the large molecule, its hydrodynamic radius apparently increases, and, as consequence, a change in the translational diffusion is observed. In detail, if the host–guest association is strong, both the ligand and the protein exhibit the same diffusion coefficient that reflects the tight complex. In the case of fast exchange on the NMR timescale, the measured diffusion coefficient is the weighted average of that of the free and bound ligand.
Given the actual improvement of gradient technology for high‐resolution NMR spectroscopy, several pulse sequences may be used for diffusion ordered spectroscopy (DOSY) experiments,40 with the pulsed‐field gradient stimulated echo (PFG STE) and the bipolar longitudinal eddy current (BPLED) being more versatile for the analysis of ligand–protein binding. An example of the use of the PFG NMR method for the study of receptor–carbohydrate molecular interactions has been recently published.41
One of the most intuitive ways to present diffusion data is a pseudo‐2D DOSY spectrum, in which the vertical dimension represents the log of the diffusion coefficient, whereas the horizontal axis displays the chemical shift. A change in the second dimension upon the addition of the receptor protein is a clear indication of binding; by contrast, the diffusion coefficient of non‐interacting molecules in the mixture will not be affected (Figure 6).
Figure 6.
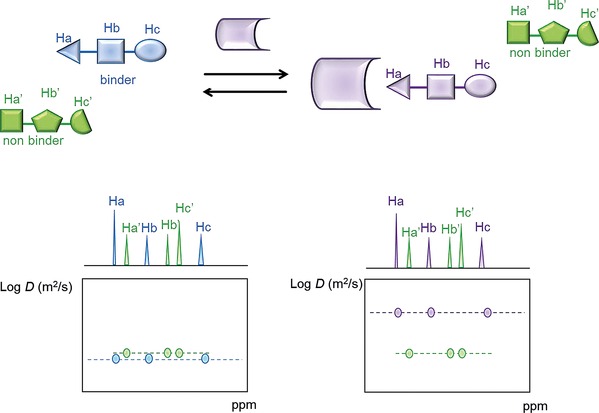
Schematic representation of a pseudo‐2D DOSY experiment. Upon the addition of the receptor in solution, a change in the diffusion coefficient is only observed for binder molecules.
This technique can also help determine the epitope mapping, avoiding eventual bias from different relaxation T 1 between protons within the same ligand, as described by Yan et al.42 Furthermore, diffusion NMR can be used to directly calculate the ligand dissociation constants (in the range 10–105 m −1) for systems in fast exchange.
The major limitation of NMR diffusion experiments is the relatively large amount of protein that is necessary for this technique; the best results are indeed obtained at a protein concentration in the millimolar range. This is attributed to the difficulty in identifying ligand resonances in the presence of the receptor because of the line broadening. To overcome this problem, a large excess of the ligand should be used. However, it may decrease the expected difference in the diffusion coefficient; therefore, high‐concentration protein and low ligand/target ratios (1:1 to 1:10) are usually preferred for diffusion measurements.
2.1.5. Paramagnetic Tagging
Oligosaccharides are characterized by numerous degrees of motional freedom, possessing significant conformational flexibility. NMR spectroscopy has been used to monitor the dynamic conformations of oligosaccharides in solution, mainly through the analysis of NOEs and scalar couplings. However, these classical NMR parameters are often influenced by strong signal overlap and provide only local information. Therefore, to define the global shape of flexible molecules, lanthanide‐assisted NMR spectroscopy, combined with molecular dynamics, can be used as a powerful tool for an accurate conformational analysis. This approach consists of the introduction, onto sugar moieties, of a paramagnetic ion with an anisotropic susceptibility tensor (Δχ), which induces molecular alignment in the presence of magnetic fields (RDCs),43 paramagnetic relaxation enhancement (PRE),44 and pseudo‐contact shifts (PCSs).45 All of these paramagnetic effects provide long‐distance information that is pivotal for the characterization of the overall conformation of non‐globular molecules in solution. Particular attention is given to pseudo‐contact shifts.
The pseudo‐contact shifts originate from the dipolar interaction occurring between the unpaired electrons of a paramagnetic ion and the nuclei in its vicinity. They are measured as the differences in the chemical shift (Δδ PCS) between paramagnetic and diamagnetic samples. For carbohydrates, they can readily be carried out through standard 1H−13C HSQC experiments. As the PCSs decrease with the distance between the nuclear spin and the metal ion with a 1/r 3 dependence, their magnitude can be tuned by varying the length of the linker used to attach the paramagnetic tag. On the other side, given the relative differences in magnetic susceptibility anisotropy of lanthanoids, it is possible to modulate the magnitude of PCSs by using different metal ions. It allows increased accuracy in cases where only few PCSs are observed or assigned, as the complexation of different lanthanoids provides several sets of δ PCS data for the same system of interaction. Originally used to derive binding site information on protein–protein46 or protein–ligand47 interactions by tagging the protein with a paramagnetic ion, the study of PCSs with the opposite approach, by tagging the ligand, has been carried out in recent years. In particular, this methodology has been used in the carbohydrate field with the aim to establish the conformation of oligosaccharides, from disaccharides up to nonasaccharide structures.48 The standard protocol foresees a conformational study, through molecular dynamic simulations, followed by the calculation of the expected pseudo‐contact shifts for each conformation. Finally, a comparison between the experimental and calculated PCS values permits us to define the overall ligand conformation. The use of this approach is particularly useful in the analysis of complex N‐glycans possessing different branches with identical terminal arms, with a consequent strong overlapping between several NMR signals (Figure 7).48b By using lanthanide binding tags, it is possible to break the pseudo‐symmetry in multi‐antennary glycans, providing a global perspective of their conformation. Recently, it has also been shown that the interaction features of complex molecules in solution can be derived through the use of a lanthanoid‐tagged sugar moiety.49 By following the protein resonance changes upon binding to a tagged ligand, it is possible to study protein–ligand binding, identifying the protein binding site with improved accuracy with respect to standard chemical‐shift mapping for low‐affinity interactions (Section 2.2.3).
Figure 7.
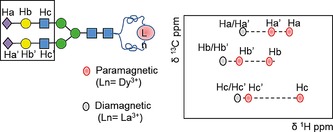
Schematic representation of PCSs. When the ligand (i.e. a complex oligosaccharide) is chelated to a diamagnetic ion (i.e. La3+), some proton resonances are isochronous; when the ligand bears a paramagnetic lanthanide binding tag, significant shifts are observed.
One of the major limitations of this technique is the compatibility between the ligand functional groups and the condition of metal‐ion complexation (60 °C, pH 5–6). Furthermore, it must be taken into account that the synthetic modification could affect the binding, as a pivotal group for the recognition could be modified. To ensure that the tag does not interfere in the binding process, control experiments using the unmodified ligand should be performed. Otherwise, the introduction of a paramagnetic label at different ligand functionalities may be necessary.
2.1.6. Fluorine Tagging
In recent years, a number of robust and versatile experiments, relying on 19F observation, have been developed to characterize protein–ligand binding.
Given its ubiquity in organic molecules, 1H is so far the most used nucleus in ligand‐based NMR methods. Nevertheless, the 19F nucleus exhibits peculiar favorable NMR properties that render it a valuable alternative. It occurs at 100 % natural abundance and its theoretical sensitivity is 0.83 times that of the 1H. Furthermore, the large chemical shift range (ca. 900 ppm) and the absence of endogenous 19F in biomolecules, buffers, and solvents allow clean observation of ligand spectra, avoiding issues of signal overlap and baseline distortion. In addition, owing to the large chemical shift anisotropy of 19F, a large broadening effect is observed for ligand fluorine signals upon binding. Thus, 19F detection represents a sensitive method to monitor interactions between small ligands and large receptors.
19F‐detected STD experiments have been developed and successfully applied to detect the interaction of fluorinated carbohydrates with lectins. Two different NMR schemes have been presented. In the simplest version, the protons of the protein are saturated and the transfer of saturation is detected on 19F. In an alternative scheme, conventional homonuclear 1H−1H STD is first allowed, and subsequent 1H polarization is transferred to 19F through scalar J HF, where it is detected.50 19F‐based methods are clearly limited by the necessity to have 19F atoms incorporated in the chemical structure under study or alternatively use a labeled spy molecule. The use of other NMR‐active nuclei, including 13C and 15N, has been proposed by several research groups.51 However, these methods are less sensitive than 19F approaches, and there is a lack of commercially available fragments containing such nuclei.
2.2. Receptor‐Based NMR Methods
The receptor‐based methods rely on the observation of protein signals in the absence and in the presence of one or more ligands. These techniques can be applied to systems with a large range of affinity, without the requirement of fast exchange, and provide with a wide set of information on the ligand binding mode (see Table 1). However, contrary to ligand‐based methods, a large amount of soluble, non‐aggregated, and stable isotope‐labeled protein is required. Moreover, a previous assignment of the receptor NMR resonances has to be performed to clearly identify the target regions that become more perturbed upon ligand binding. Until recently, the application of receptor‐based techniques was limited to “small” proteins, typically under 40 KDa. As structural information about the protein binding site is pivotal, especially in the drug discovery field, several advances have been made with the aim to extend the molecular size amenable to NMR studies; current progress permits the observation of receptors beyond 100 KDa.
2.2.1. Isotope Labeling
As mentioned above, a detailed analysis of protein–ligand interactions from the receptor point of view requires the assignment of the target resonances. To this aim, it is necessary to label the protein under study with stable isotopes. This is achieved by expressing the protein in bacteria cultured in enriched minimum media. The three most commonly used isotopes are 15N, 13C, and 2H.
Uniform isotope labeling of the proteins allows the use of heteronuclear multidimensional NMR experiments for the sequential assignment of receptor resonances.52 In general, one of the most widespread methods for the analysis of proteins is 1H−15N HSQC. In this experiment, one cross peak for each amide is observed; therefore, the HSQC spectrum displays a number of signals corresponding, to some extent, to the number of amino‐acid residues in the protein. As the size of the protein increases, the spectra are more crowded and the signal overlap complicates the full interpretation of the NMR data. With the aim to expand the applicability of receptor‐based approaches, several selective isotopic labeling strategies, focused on the simplification of the assignment process, have been developed.53
In any case, the relaxation mechanisms of high‐molecular‐weight proteins remain an issue, as they cause severe line broadening and the disappearance of some signals, impeding the investigation of large receptors. The introduction of alternative methods, including transverse relaxation optimized spectroscopy (TROSY),54 has allowed the limit of the protein molecular weight to be pushed to 100 KDa. Several modified versions of this experiment allow the problems of resonances overlap to be further reduced.54c, 55
2.2.2. Chemical Shift Perturbation Mapping
Provided that the assignment of protein resonances is known, monitoring its chemical shift changes in the 1H−15N HSQC spectrum upon the addition of a ligand in solution constitutes a powerful tool for binding studies.56 The comparison between a reference spectrum of the protein in the free state and those acquired in the presence of a potential ligand allows us to identify the amide whose environment is perturbed by ligand binding (Figure 8). In the same way, 1H−13C HSQC spectra may be used to detect perturbations in aromatic and aliphatic chemical shifts of the side chains. The magnitude of the chemical shift change depends on the vicinity to the binding site as well as on the type of protein–ligand interactions.57
Figure 8.
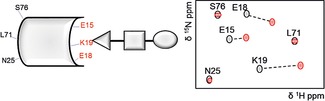
Schematic representation of 1H−15N HSQC spectra. A zoom of 1H−15N HSQC spectrum of the protein alone in solution is depicted in grey. Upon the addition of the ligand, significant shifts are observed for the amide protons more involved in the interaction (red cross peaks).
Usually, titration NMR experiments are performed by gradually increasing the concentrations of the ligand in a solution, in which the protein concentration is held constant. This approach provides an adequate means to accurately measure the equilibrium association constant by correlating the chemical shift change, δ, with the ligand concentration.58
Although, in some cases, the protein skeleton is pre‐organized to accommodate the ligand and the “lock and key” interaction produces no effects on receptor residues not directly involved,59 often ligand binding induces conformational changes on the protein and the majority of protein resonances are affected.60 Some of the chemical shift perturbations are produced, in latter cases, by long‐range effects, owing to the reorganization of the whole protein, resulting in an ambiguous interpretation of NMR data. The comparison of chemical shift changes between two slightly different ligands may help in overcoming this issue.58
Important applications of chemical shift perturbation mapping can be found in drug discovery research.61 For example, structure–activity relationships (SARs) through NMR is one of most widespread receptor‐based strategies for the screening of low‐affinity ligands and the development of strong inhibitors. This technique relies on the chemical shift perturbation mapping produced by small molecules that weakly bind to two proximal sites of the target protein. Knowledge about the orientation of the bound ligands is used to guide the synthesis of new potential drugs, in which the two small molecules are linked, and thus the binding affinity is greatly increased. The main limitation of this method remains the ability to isotopically enrich the protein, as well as its size, solubility, and stability, in concentrations of 0.1–1 mm, during the experiments time.
2.2.3. Paramagnetic Tag
The use of a tagged protein with a paramagnetic ion constitutes a powerful tool for the characterization of ligand binding. Covalently attaching spin labels to a selected site of the protein leads to a significantly enhanced T 2 relaxation of neighboring protons (within a distance of 15–20 Å); therefore, any ligand that interacts with the receptor experiences a paramagnetic relaxation enhancement effect and exhibits weakened and broadened resonances.62, 63 The major limitation of this technique is related to the necessity of a covalent modification on the protein, close to the binding site, which could affect the binding activity. Furthermore, given that bleaching of resonances occurs around the tag, no information will be available from sites very close to it.
3. Applications
As stated above, the present Review paper aims to especially disclose a precise and undercovered arena, the protein–glycoconjugate recognition field, as glyco‐molecules play a pivotal role in cell–cell interactions in all kingdoms of life. In fact, all of the NMR approaches described above have been extensively used to investigate a wide range of protein–glyco‐molecule systems and many outstanding papers have been published on the structural events that mediate the molecular recognition processes. Table 2 gathers a collection of relevant studies of protein–glycoconjugate interactions published in recent years involving lectins, antibodies, viruses, and eukaryotic immune receptors; in the interest of space, only some of them are further described in the following paragraphs. In particular, the reviewed examples emphasize the strength of the aforementioned NMR‐based methods in shedding light on the role that bacterial/viral cell‐wall components play in the interaction with the eukaryotic host, and on the structure‐to‐function relationships in these systems.
3.1. Lectin–Carbohydrate Interactions
Lectins are proteins of non‐immune origin, found in all living organisms ranging from viruses and bacteria to plants and animals, which bind to specific carbohydrates without modifying them. They are able to interact reversibly and specifically with oligosaccharides or glycoconjugates.101 According to present knowledge, lectins act as molecular readers to decipher sugar‐encoded information, playing important biological roles in recognition processes involved in fertilization, embryogenesis, inflammation, metastasis, and parasite‐symbiont recognition in microbes and invertebrates to plants vertebrates.
To date, the NMR approach has exhaustively been used to monitor and unravel structural features of carbohydrate–lectin interactions (Table 2). An “iconic” example in the field is the DC‐SIGN recognition, one of the pathogen recognition lectin that has received much attention.102 This lectin is highly expressed on immature dendritic cells (DC) of mucosal tissues, and it is known to bind and uptake a wide variety of viruses (HIV, Ebola virus, hepatitis C), bacteria (Mycobacterium tuberculosis, Helicobacter pylori), or yeast (Candida albicans). The first step of the infection takes place through the specific recognition of high‐mannose‐ and fucose‐containing glycans of the pathogens by the lectin at the surface of the DC. The outcome of this interaction differs widely depending on the pathogen. Whereas in some cases the recognition event triggers the proper immune response, in others the pathogens are able to subvert the DC function, escaping the immune response and enhancing infectivity, as in the case of HIV.103 From the NMR perspective, the recognition of natural oligomannosides and Lewisx fragments by DC‐SIGN has been thoroughly explored. In the case of mannosides, the use of STD NMR22, 104 methods has led to the characterization of the binding epitope for the interaction in solution. Further, in conjunction with CORCEMA‐ST,24 the nature of the multimodal recognition was unambiguously established in agreement with the X‐ray crystallographic findings.105 As for the conformation, the bound geometry showed the same torsion angles around the glycosidic linkages as those found in solution. In contrast, for the Lewisx trisaccharide, the application of an analogous protocol (STD NMR in combination with CORCEMA‐ST and HADDOCK106) shed light on a bound conformation that significantly differs from the one shown in the previous studies and determined by using X‐ray analysis (Figure 9).107 It has been shown to also be different from the recently published solution conformation.108 Therefore, the ligands and the receptor exhibit certain degrees of flexibility that could have important implications for target recognition in the “natural” context of the tetrameric DC‐SIGN when it interacts with glycans at the membrane surface.
Figure 9.
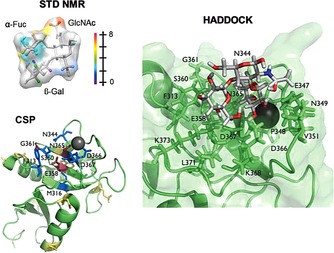
The application of both ligand‐ and receptor‐based techniques, combined with molecular modeling, allowed the ligand binding epitope, the protein residues more involved in the interaction, and the bioactive conformation of the Lewisx to be found when bound to the DC‐SIGN binding pocket. Adapted with permission from Ref. 106. Copyright (2014) American Chemical Society.
With the aim of disrupting DC‐SIGN hijacking by pathogens (like HIV) and getting insights into the intriguing roles of DC‐SIGN in immunity, there has been much interest in the design of glycomimetics as DC‐SIGN inhibitors.70, 109 Similar strategies have been applied to characterize the binding of Lewisx[110] and Manα1–2 Man68 mimetics to the DC‐SIGN carbohydrate recognition domain (CRD). In the former case, the occurrence of two binding modes needs to be assumed to fit the experimental STD data with the theoretical computational/CORCEMA‐ST model. In the latter case, only one binding pose of the mimic in the binding site was found, strongly contrasting to the situation found for the corresponding natural ligand.
For DC‐SIGN, it is known that langerin (Lg), a C‐type lectin expressed almost exclusively on Langherans cells, binds both endogenous and pathogenic cell surface carbohydrates, acting as a PRR. Endogenous binders are the blood group B antigen Galα1–3(Fuca1‐2)βGal, 6‐sulfated galactosides, and high‐mannose N‐linked oligosaccharides, whereas pathogenic ones are high‐mannose, mannan, and β‐glucan entities. NMR has been used72 to gain structural insights into the intriguing capacity of Lg to recognize glycosaminoglycans (GAG) in a non‐Ca2+‐dependant manner, and only in the trimeric form of the extracellular Lg domains.111 For the sake of simplicity and to comply with the requirement of the NMR ligand‐based method, small heparin‐like trisaccharides were used. Comparison of the STD results for different GAG fragments with distinct sulfation patterns led the authors to postulate that the interaction is independent of the presence and the position of the sulfate groups of the glucosamine residues (Figure 10 a). STD experiments, when performed in the presence and absence of Ca2+, showed that the interaction is Ca2+ dependent. This finding was corroborated throughout competition STD experiments with 6‐O‐sulfate‐galactose, which is a ligand known to bind at the Ca2+ binding site. Interestingly, opposite results were obtained for a longer GAG fragment (hexasaccharide). In this particular case, STD responses were observed both in the presence and absence of Ca2+ ions. The tr‐NOESY analysis for the interaction with the trisaccharides concluded that Lg CRD is able to recognize the two co‐existing 1C4 and 2SO solution conformations of the central iduronate ring (Figure 10 b).
Figure 10.
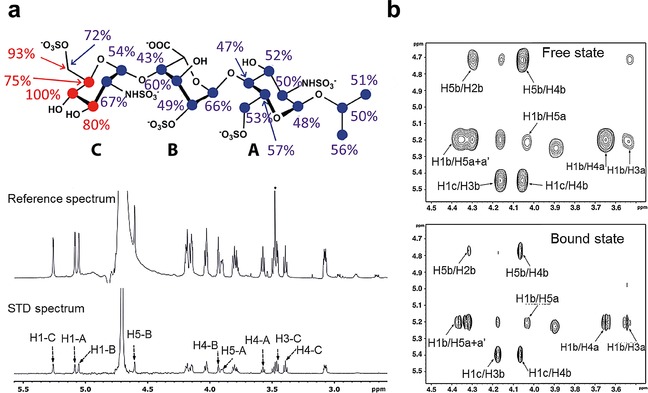
a) Binding epitope derived from STD NMR experiments on the trisaccharide fragment in the presence of Lg. b) Zoom of tr‐NOESY spectra of the trisaccharide in the free and bound state. Adapted with permission from Ref. 72 Copyright (2015) American Chemical Society.
Dectin‐1 is another C‐type lectin PRR that recognizes β‐1,3‐linked and β‐1,6‐linked glucans from fungi and plants and induces innate immune responses. As β‐glucans are composed of repetitive units of the same monosaccharide, the drastic overlap of NMR signals makes it difficult to extract non‐ambiguous information about the ligand epitope. As such, the NMR investigation of these interactions remains an important technical and methodological challenge. Different STD NMR studies64, 65 confirmed that the binding affinity of β‐glucans to Dectin‐1 depends on the number of repeating units; this is also confirmed by other biophysical techniques. Laminarihexose, for instance, showed very weak or no STD response, whereas Laminarin (18 to 31 units) provided clear STD signals. Furthermore, based on the measured STD intensities, the authors came to the conclusion that the binding mode is such that the α‐face of the β‐glucose moieties (with H1, H3, and H5 pointing in the same spatial direction) interacts with hydrophobic residues of the protein. This increased affinity with the degree of polymerization, together with studies that showed that 1–3‐linked β‐glucans have a tendency to form helical structures in solution,112 permits us to hypothesize that this organization is important for the interaction with this PRR. To further explore this possibility,113 the propensity of the hydroxyl protons to establish hydrogen bonding as a stabilizing factor of a helical structure was studied,114 also employing NMR methods. The use of deuterium‐induced isotope shifts (DISs)115 suggested that the H/D exchange rate at the C4 hydroxyl is significantly slower for laminarin when compared to that for shorter oligomers. Nevertheless, DOSY NMR experiments showed that, under the employed experimental conditions, all of the tested β‐glucans were monomers, and did not show any signatures of the existence of a supramolecular triple helix structure. Therefore, the helical‐like interaction model for long β‐glucans has not yet been fully demonstrated.
Pathogens also exploit lectins for the recognition of glycans at the cell surface to infect host cells. An interesting example in the field is the role of influenza hemagglutinin (HA). As well as other analogous envelope proteins from Ebola and HIV, HA mediates virus entry through binding to specific glycan epitopes. This process is followed by dramatic conformational changes that result in fusion of the viral and target cell membranes. Many examples in this field made use of NMR ligand‐based approaches, especially STD.78, 79 In a seminal work, the receptor binding properties of influenza A HA from different subtypes (H1, H5, and H9) were studied by using NMR spectroscopy.78 Strikingly, the STD experiments showed that all HAs were able to interact with both 2,3‐sialyllactose (most abundant in the digestive tract in avian species) and 2,6‐sialyllactose (most abundant in the upper respiratory tract in humans); only subtle differences in the binding mode were identified. As such, STD experiments were proposed as a fast method to quickly assess the binding abilities of newly found HA.
The majority of examples reported above underline the potential of ligand‐based methods in the study of protein–carbohydrate system interactions. However, as already mentioned, receptor‐based NMR approaches have also been widely used in the glycan recognition arena, including the interaction of galectins with different natural and synthetic ligands.75 With regard to infection, one illustrative example is the study of the glycan recognition properties of human RegIV. This protein is highly expressed in mucosa cells of the gastrointestinal tract during pathogen infection. RegIV contains a sequence motif homologous to Ca2+‐dependent C‐type lectin‐like domains,116 despite the fact that glycan binding has been shown to be Ca2+ independent.
Recently, receptor‐based NMR methods have also been used to study the recognition properties of a counterpart of DC‐SIGN (see above), DC‐SIGNR,71 toward large glycan structures such as Man9GlcNAc. Interestingly, the authors found that the chemical shift perturbations produced by different Man‐containing N‐glycan fragments differ significantly. The larger glycan, with the highest binding affinity, produced the most localized chemical shift perturbations. Relaxation NMR data showed that the protein is very dynamic in the apo state, and that this flexibility persists upon glycan binding, although at a slightly increased rate. This interesting observation allowed the authors to speculate on the role of the flexibility of C‐type lectin in its capacity of transmitting information to the intracellular regions throughout the whole polypeptide chain, as previously suggested from X‐ray results.117
In the context of viral infections, a number of lectins have been described that inhibit HIV entry with high potencies. Actually, several of them, which have been shown to be effective in primate models, are being investigated to be used as topical microbicides to prevent sexual transmission of HIV. Among them, the molecular recognition features of cyanovirin‐N (CVN) have been extensively studied by using both the ligand‐ and receptor‐based approaches. Despite the relatively small size of CVN (11 kDa), STD NMR allowed the determination of the epitope of different di‐ and trimannoside substructures of Man‐9, which is the predominant oligosaccharide on gp120, the key HIV viral surface glycoprotein.80 The use of fluorinated glycomimetic analogues of the mannose oligosaccharides82 was instrumental in the identification of major structural and kinetic features underlying the binding process. This type of study illustrates the wide range of possibilities offered by 19F NMR spectroscopy for ligand screening and fast detection of glycan binding processes, as stated above.50
The examples described in this section clearly give an idea of the wide range of applicability of NMR methodologies in the full understanding of protein–glycoconjugate mechanisms of interaction, providing different levels of information. It can range from the mere confirmation of the binding event to the definition of the ligand key structural elements for the interaction, as well as the achievement of the complex 3D structure.
3.2. Antibody–Carbohydrate Interactions
Antibodies are a specific class of immunoglobulins that exist in a membrane‐bound form attached to the surface of a B cell or in a soluble form freely in the bloodstream as part of the humoral immune system. Circulating antibodies are produced by clonal B cells that specifically respond to only one antigen (i.e. a virus capsid or a glycoprotein fragment). They contribute to immunity in three ways: 1) they prevent pathogens from entering or damaging cells by binding to them; 2) stimulate removal of pathogens by macrophages and other cells by coating the pathogen; 3) trigger destruction of pathogens by stimulating other immune responses such as the complement pathway. In all cases, they bind to non‐self‐chemical structures belonging to microbes, which are a typical hallmark of microbial structure/metabolism.5
On this ground, understanding how complex carbohydrates chemically interact with antibodies is an important first step towards establishing rules for designing carbohydrate antigens to be used in a vaccination strategy. In this particular field of structural biology, the NMR approach has been and remains useful to elucidate the structural basis of the interaction of antibodies with their respective ligands (Table 2).
One very significant example is the dissection of carbohydrate antibody interactions by using a synthesized tetrasaccharide from the Bacillus anthracis (BclA) cell wall.92 Understanding which are the essential structural features of the BclA cell‐wall oligosaccharide essential for the antibody recognition is a mandatory step in the design of efficient carbohydrate‐based anthrax vaccines. Based on the tetrasaccharide exposed by BclA cell wall, a plethora of oligosaccharides has been synthesized and selected by microarray screening. Different antibodies were produced by using B‐cell hybridoma technology with spleen cells of mice immunized with the BclA‐related disaccharide. A proper oligosaccharide (tetra‐) and antibody (antibody MTA1) were selected and the quantification of their interactions by using SPR analysis was achieved. Afterward, a complete epitope mapping of the BclA tetrasaccharide/MTA1 interaction was established by using STD NMR spectroscopy. As a result, a precise cartography of the molecular elements of the BclA tetrasaccharide that participate in tight antibody binding was drawn. This study is a classic example of the efficient combination of different biophysical and biochemical approaches to shed light on the binding requirements of the ligand versus the antibody. The approach has triggered the discovery of mAbs that are currently under development as a highly sensitive spore‐detection system. Such multivariate functional approach will serve in the near future to study antigen–antibody interactions and will be of utmost importance to design vaccines or to obtain functional antibodies against any carbohydrate antigens.
Another striking example of the potential of NMR spectroscopy in assessing the binding modes of glycans to antibodies is given by the work of Pancera et al.95 One of the ways that HIV can escape antibody recognition is to make use of the eukaryotic machinery to synthesize an array of N‐glycans that decorate the virus envelope glycoprotein gp120. Because of the prominent coverage of its surface by host N‐linked glycans, antibodies generated from HIV‐1‐infected individuals generally result in a very low glycan‐dependent reactivity. Such a low frequency of glycan‐reactive antibodies has been attributed to cross‐reactivity in antibody recognition of N‐linked glycans on both HIV‐1 and of N‐linked glycan on host or self‐proteins. The antigenic structure of HIV‐1 gp120 displays a dense cluster of N‐glycans, which is infrequently recognized by the host immune system. Nevertheless, an antibody able to selectively recognize a cluster of high‐mannose N‐glycan was produced and, from this, a new class of antibodies with such characteristics were obtained.95 Deciphering such a complex series of molecular interactions, that is, the binding mode of the N‐glycans from HIV virus to PG9 and PG16 antibodies, could only be attained through the use of a range of complementary methods of investigation: crystallography, molecular biology, and NMR spectroscopy. In particular, the STD NMR approach provided a full epitope mapping of the key atoms involved in the interaction with the antibodies and, particularly, shed light on the key role played by the sialic acid residue. From the protein side, information was attained by mutagenesis of the amino acids involved in the binding and subsequent STD NMR experiments. These results are among the first evidences of an HIV‐1‐neutralizing antibody whose epitope comprises a terminal sialic acid containing glycan. They will help to decipher the complex interplay between viral glycan diversity and antibody‐mediated recognition and neutralization.
Another case of the use of NMR in the study at the atomic level of antibody interactions with cell surface glycans is illustrated by the characterization of a glycoprotein, MUC1, that carries a tandem repeating domain with five possible O‐glycosylation sites of conserved 20 amino acids.118 In normal tissues, the protein backbone carries complex oligosaccharides, with the α‐O‐GalNAc unit directly linked to the hydroxyl group of serine (Ser) or threonine (Thr). In tumor cells, the expression of MUC1 is usually increased with aberrant glycosylation, as the carbohydrate side chains are incomplete. As a result, different carbohydrate epitopes, such as the Tn antigen (α‐GalNAc‐1‐O‐Ser/Thr), are exposed to the immune system and can be used to design synthetic MUC1‐based antitumor vaccines. The application of experimental approaches (synthetic chemistry methods, mAb generation, microarray methods, NMR) and computational methods has been instrumental in the identification of the molecular elements of the recognition region of the antigens for two different families of cancer‐related monoclonal antibodies that have been produced on an ad hoc basis (Figure 11).119 The combination of microarrays and STD NMR shed light on the functional significance of glycosylated peptides as antigens, providing detailed information for the design of tailored Tn‐based vaccines such as MAG‐Tn3. In particular, it has been demonstrated that the amino‐acid sequence, to which Tn antigen is attached, modulates the affinity of the mAb, whereas the sugar residue modulates the affinity and strength of binding.
Figure 11.
Comparison of STD NMR spectra of two different families of cancer‐related monoclonal antibodies in the presence of different peptides. As shown by the NMR data, anti‐MUC1 recognizes MUC1‐derived peptides, whereas anti‐Tn mAb preferentially binds glycopeptides‐containing Tn–Ser antigen. Adapted with permission from Ref. 119a. Copyright (2015) American Chemical Society.
The examples described above illustrate the importance of the ligand‐based NMR approach in assessing glycan binding to antibodies from the ligand point of view. Throughout such an approach, it is possible to draw the epitope mapping of any given glycan with the antibody and establish a tight structure–activity relationship as a first step towards drug design studies. It is clear, however, that this ligand‐based NMR approach has to be conjugated to other biophysical and biomolecular approaches such as molecular biology, selected amino‐acid mutagenesis, SPR, ITC, or/and glycan ligands microarrays to get a clear idea of the interaction either in terms of thermodynamics or kinetics. Such a multivariate functional approach will serve in the near future to study antigen–antibody interactions and will be of utmost importance in the design of vaccines with functional antibodies against any carbohydrate antigens.
3.3. Virus–Ligand and Virus‐Like‐Particle–Ligand Interactions
Ligand‐based NMR methods have emerged as well‐suited techniques to investigate the molecular basis of viral entry into host cells. Starting from the year 2003,120a the potential of NMR in the study of the interactions between low‐molecular‐weight glycan ligands and receptors on the surface of native viruses or VLPs (virus‐like particles) has been extensively demonstrated85, 87, 120 (Table 2). This approach has been elegantly used, for example, to identify molecules able to interfere with the attachment of Norovirus (NV) to histo‐blood group antigens.86 STD NMR experiments with NV VLPs allowed the binding epitopes of several synthetic compounds to be determined from a large library. Further ligand‐based approaches, including NOE‐based methods, have been employed to investigate the bioactive conformation and to identify adjacent ligand binding sites. The combination of the NMR results with X‐ray, SPR, and docking analyses was the basis of the development of potent entry inhibitors to fight human NV infections, against which there is no effective vaccine so far.
A similar strategy has been applied to investigate the binding specificity of the rabbit hemorrhagic disease virus (RHDV), an animal pathogen that binds to the carbohydrate structures of blood‐group determinants.85 The analysis of STD NMR spectra performed on VLPs in the presence of different HBGAs revealed the minimal structural requirements for their specific recognition by RHDV (Figure 12), opening an avenue for the design of antiviral entry inhibitors.
Figure 12.
STD NMR spectra of H trisaccharide I/II in the presence of RHDV VLPs, showing a preference for the binding to the H‐antigen type II structure. Binding epitope for H antigen II. Adapted with permission from Ref. 85. Copyright (2008) American Chemical Society.
Interesting experiments have also been performed on enveloped viruses such as the influenza virus. Haselhorst et al.79 have shown that competitive STD experiments may be safely used to monitor ligand binding, employing virus‐like particles decorated with subtype H5 of influenza hemagglutinin. An STD NMR competition assay allowed the preferential binding of this particular HA to 2,3‐sialyllactose to be demonstrated, complementing previous studies on the isolated HA.78, 79
All NMR studies on virus–ligand interactions published so far underline the need for modifications to the standard STD NMR approach, with a complex setup of experimental conditions. However, the possibility to investigate binding processes in a near‐physiological environments, the requirement of a small amount of the viruses and their corresponding ligands, as well as the large size of the viruses and VLPs have rendered STD NMR a suitable tool for the rational design of potential antiviral entry inhibitors.
4. Summary and Outlook
4.1. Improvement of NMR Methodologies (Instrumentation/Sequences/Processing, Protein and Carbohydrate Labeling)
Generally speaking, the main limit of the NMR approach is its low sensitivity. Solutions to this problem can come from different directions, for example using higher magnetic fields or setting particular sequences and filters to improve the overall signal‐to‐noise ratio. An extensive and very interesting Review has been recently published on this issue.121
Protein and carbohydrate labeling methods will certainly profit from further developments in expression systems (mammalian, insects, yeast) that are able to provide intact mammalian glycoproteins in high yields and even decorated with 15N/13C‐isotope‐labeled glycans. Most probably, the development of expression approaches combined with enzymatic glycosylation122 will provide avenues to tackle this difficult and interesting field. This type of approach has already generated seminal examples in the antibody/glycan interactions field.123
Moreover, the availability of labeled glycan ligands will also be instrumental in developing ligand‐based and receptor‐based NMR techniques by selecting those NMR resonances with a heteronuclear label or by filtering them out. An elegant example in the innate immunity field has been presented to assess key structural properties of the fatty acyl chains of lipooligosaccharides present in TLR4‐agonistic and TLR4‐antagonistic binary and ternary complexes,124 and to determine the conformation of a rough‐type lipopolysaccharide from Capnocytophaga canimorsus.125
4.2. Improvement of Modeling Methodologies
The full characterization of sugar recognition by using proteins requires the quantitative measurements of the strength and specificity of the interaction, which can be assessed through biophysical methods and computer simulations. The present Review article has provided ample examples of how NMR methods in conjunction with docking simulation have dealt with several classes of physical interactions at the recognition sites. But, challenges still remain, owing to the well‐identified features present in carbohydrates, which display numerous functional groups and enhanced conformational flexibility. Among some novel features that may need to be considered is the implementation of models of water clusters in the molecular dynamics computation. It has been shown that some carbohydrate molecules can fit into a network structure of an icosahedral water cluster. Such types of interaction can modulate the structure and dynamics of carbohydrates prior and during the course of their interactions with proteins, which opens a field of investigation relevant to the scope of the present chapter. There is also the need for analytical methods aimed at revealing the magnitude of the free energy that determines the strength and the mechanism underlying the interaction between carbohydrates and proteins. There are a large number of computational methods that derive free‐energy changes. One group of methods, including molecular mechanics Poisson–Boltzman surface‐area calculations can provide the relative free energy. This has been shown to be suitable for the calculation of the binding free energy of relatively large carbohydrate systems. Another group of methods uses free‐energy perturbation/thermodynamics integration methods. By applying rigorous statistical mechanics, the absolute binding free energy or accurate free‐energy changes between different states can be obtained. It should, nevertheless, be understood that accurate determination of the absolute free energies remains challenging, owing to the considerable variations of translational and rotational as well as conformational entropies underlying the binding process. Some of these variations cannot be fully captured in routine, finite‐length statistical simulations. For the time being, this group of methods has been shown to be suitable for mono‐ and disaccharide systems, and to predict the enthalpy‐/entropic‐dominant nature of the interaction between chitobiose and hevein.126 The accumulation of other successful predictions is indicative of the adequacy of the method that still needs to be calibrated and automated before its full integration with NMR and other experimental methods.
4.3. Integration of NMR with Other Approaches
In the field of molecular recognition, in which glycans play a part, the joint approach of NMR and molecular modeling is still a pivotal method of choice. Nevertheless, a complete structural biology study must be accompanied by several other techniques, such as ITC, SPR, and X‐ray, when possible. Also, most of the time, the organic synthesis of the carbohydrate “binder” is required, as well as its variations, for SAR studies. If the glycan binder is totally unknown, the production of a plethora of glycans through synthetic methods is required, usually to be placed on a microarray platform for a first screening. As for the protein ligand, molecular biology expertise is also necessary for the recombinant synthesis of the protein and/or a single domain of it.
All of this represents the (almost) full set of knowledge and expertise required to study the intriguing field of cell–cell interactions by means of glycans or glycoconjugates molecules.
Biographical Information
Alba Silipo obtained her degree in Chemistry at the University of Naples Federico II in 2001 with summa cum laude. She is currently Professor of Organic Chemistry at the Department of Chemical Sciences of University of Naples Federico II (Italy). Her research experience started with the isolation, purification, and structural characterization of glycoconjugates of bacterial origin by means of state‐of‐the‐art techniques for the characterization of glycoconjugates. During her PhD and Postdoctoral experiences, she improved her knowledge of NMR techniques and learnt the most advanced NMR experiments to study the NMR‐based conformational behavior of the carbohydrate ligand alone and when interacting with a protein. She also learnt in‐depth molecular mechanics and dynamics applied to the field of carbohydrates. She is currently studying the structure–function relationships of glycoconjugates of microbial origin and their molecular mechanisms at the basis of protein–carbohydrate interactions at host–pathogen interfaces.

Biographical Information
Antonio (Tony) Molinaro is Professor of Organic Chemistry and Carbohydrate Chemistry at the Department of Chemical Sciences of University of Naples Federico II (Italy). He is also special appointed Professor of Organic Chemistry at the Graduate School of Science of Osaka University (Japan). He has been President of the European Carbohydrate Organization and is currently Coordinator of the Interdivisional Group of Carbohydrate Chemistry with the Italian Society of Chemistry. He is deeply interested in any aspect of glycosciences with particular focus on eukaryotic–microbe interactions mediated by glycoconjugates. He is member of the editorial board of ChemBioChem, Carbohydrate Research, Glycoconjugate journal, Marine Drugs, and Innate Immunity Journal. Tony likes sport and rock music and is a devoted fan of D. A. Maradona and Led Zeppelin.

Acknowledgements
A.S. and A.M. acknowledge PRIN‐MIUR 2010‐2011, COST actions BM1003, and H2020‐MSCA‐ITN‐2014‐ETN TOLLERANT.
R. Marchetti, S. Perez, A. Arda, A. Imberty, J. Jimenez-Barbero, A. Silipo, A. Molinaro, ChemistryOpen 2016, 5, 274.
Contributor Information
Prof. Alba Silipo, Email: silipo@unina.it
Prof. Antonio Molinaro, Email: molinaro@unina.it
References
- 1.
- 1a. Gabius H., The Sugar Code: Fundamentals of Glycosciences, Wiley-VCH, Weinheim, 2009, pp. 11–28; [Google Scholar]
- 1b. Silipo A., Larsbrink J., Marchetti R., Lanzetta R., Brumer H., Molinaro A., Chemistry 2012, 18, 13395; [DOI] [PubMed] [Google Scholar]
- 1c. Silipo A., Sturiale L., De Castro C., Lanzetta R., Parrilli M., Garozzo D., Molinaro A., Carbohydr. Res. 2012, 357, 75–82. [DOI] [PubMed] [Google Scholar]
- 2. Nieto L., Canales A., Gimenez-Gallego G., Nieto P. M., Jimenez-Barbero J., Chemistry 2011, 17, 11204–11209. [DOI] [PubMed] [Google Scholar]
- 3. Perepelov A. V., Shekht M. E., Liu B., Shevelev S. D., Ledov V. A., Senchenkova S. N., L. L′Vov V , Shashkov A. S., Feng L., Aparin P. G., Wang L., Knirel Y. A., FEMS Immunol. Med. Microbiol. 2012, 66, 201–210. [DOI] [PubMed] [Google Scholar]
- 4. Smith A. E., Helenius A., Science 2004, 304, 237–42. [DOI] [PubMed] [Google Scholar]
- 5.S. Flitsch, S. Perez, et al. “A Roadmap for Glycoscience in Europe”, http://www.egsf.org/2014/a-roadmap-for-glycoscience-in-europe-now-available/.
- 6. De Castro C., Lanzetta R., Holst O., Parrilli M., Molinaro A., Prot. Pept. Lett. 2012, 19, 1040–1044. [DOI] [PubMed] [Google Scholar]
- 7. Hudson K. L., Bartlett G. J., Diehl R. C., Agirre J., Gallagher T., Kiessling L. L., Woolfson D. N., J. Am. Chem. Soc. 2015, 137, 15152–15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schubert M., Walczak M. J., Aebi M., Wider G., Angew. Chem. Int. Ed. 2015, 54, 7096–7100; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7202–7206. [Google Scholar]
- 9.
- 9a. Gudlavalleti S. K., Szymanski C. M., Jarrell H. C., Stephens D. S., Carbohydr. Res. 2006, 341, 557–562; [DOI] [PubMed] [Google Scholar]
- 9b. Szymanski C. M., Michael F. St., Jarrell H. C., Li J., Gilbert M., Larocque S., Vinogradov E., Brisson J.-R., J. Biol. Chem. 2003, 278, 24509–24520. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Lee R. E. B., Li W., Chatterjee D., Lee R. E., Glycobiology 2005, 15, 139–151; [DOI] [PubMed] [Google Scholar]
- 10b. Maes E., Mille C., Trivelli X., Janbon G., Poulain D., Guerardel Y., J. Biochem. 2009, 145, 413–419. [DOI] [PubMed] [Google Scholar]
- 11. Airoldi C., Giovannardi S., La Ferla B., Jiménez-Barbero J., Nicotra F., Chem. Eur. J. 2011, 17, 13395–13399. [DOI] [PubMed] [Google Scholar]
- 12. Pérez S., Tvaroška I., Adv. Carbohydr. Chem. Biochem. 2014, 71, 9–136. [DOI] [PubMed] [Google Scholar]
- 13. Grant O. C., Woods R. J., Curr. Opin. Struct. Biol. 2014, 28, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Agostino M., Ramsland P. A., Yuriev E., Structural Glycobiology 2012, 111. [Google Scholar]
- 15. Sarkar A., Perez S., Structural Glycobiology 2012, 71, 110. [Google Scholar]
- 16. Ewing T. J. A., Makino S., Skillman A. G., Kinz I. D., J. Comput. Aided Mol. Des. 2001, 15, 411–428. [DOI] [PubMed] [Google Scholar]
- 17. Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J., J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- 18. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 19. Neuhaus D., Willianson M., The Nuclear Overhauser Effect in Structural and Conformational Analysis, 2nd Edition, Wiley-VCH, New York, 2000. [Google Scholar]
- 20. Asensio J. L., Canada F. J., Bruix M., Rodriguez-Romero A., Jimenez-Barbero J., Eur. J. Biochem. 1995, 230, 621–633. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Mayer M., Meyer C., J. Am. Chem. Soc. 2001, 123, 6108–6117; [DOI] [PubMed] [Google Scholar]
- 21b. Lepre C. A., Moore J. M., Peng J. W., Chem. Rev. 2004, 104, 3641–3676. [DOI] [PubMed] [Google Scholar]
- 22. Angulo J., Diaz I., Reina J. J., Tabarani G., Fieschi F., Rojo J., Nieto P. M., ChemBioChem 2008, 9, 2225–2227. [DOI] [PubMed] [Google Scholar]
- 23. Enríquez-Navas P. M., Marradi M., Padro D., Angulo J., Penadés S., Chem. Eur. J. 2011, 17, 1547–1560. [DOI] [PubMed] [Google Scholar]
- 24. Jayalakshmi V., Krishna N. R., J. Magn. Reson. 2002, 155, 106–118. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Angulo J., Enriquez-Navas P. M., Nieto P. M., Chem. Eur. J. 2010, 16, 7803–7812; [DOI] [PubMed] [Google Scholar]
- 25b. Angulo J., Nieto P. M., Eur. Biophys. J. 2011, 40, 1357–1369. [DOI] [PubMed] [Google Scholar]
- 26. Wang Y. S., Liu D., Wyss D. F., Magn. Reson. Chem. 2004, 42, 485–489. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Yuan Y., Wen X., Sanders D. A. R., Pinto B. M., Biochemistry 2005, 44, 14080–14084; [DOI] [PubMed] [Google Scholar]
- 27b. Yuan Y., Bleile D. W., Wen X., Sanders D. A. R., Itoh K i., Liu H.-w., Pinto B. M., J. Am. Chem. Soc. 2008, 130, 3157–3168; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27c. Szczepina M. G., Zheng R. B., Completo G. C., Lowary T. L., Pinto B. M., ChemBioChem 2009, 10, 2052–2059. [DOI] [PubMed] [Google Scholar]
- 28. Claasen B., Axmann M., Meinecke R., Meyer B., J. Am. Chem. Soc. 2005, 127, 916–919. [DOI] [PubMed] [Google Scholar]
- 29. Xia Y., Zhu Q., Jun K. Y., Wang J., Gao X., Magn. Reson. Chem. 2010, 48, 918–924. [DOI] [PubMed] [Google Scholar]
- 30. Vogtherr M., Peters T. J., J. Am. Chem. Soc. 2000, 122, 6093–6099. [Google Scholar]
- 31. Dalvit C., Pevarello P., Tato M., Veronesi M., Vulpetti A., Sundstrom M., J. Biomol. NMR 2000, 18, 65–68. [DOI] [PubMed] [Google Scholar]
- 32. Dalvit C., Fogliatto G., Stewart A., Veronesi M., Stockman B., J. Biomol. NMR 2001, 21, 349–359. [DOI] [PubMed] [Google Scholar]
- 33. Bertini I., Dalvit C., Huber J. G., Luchinat C., Piccioli M., FEBS Lett. 1997, 415, 45–48. [DOI] [PubMed] [Google Scholar]
- 34. Murray C. W., Carr M. G., Callaghan O., Chessari G., Congreve M., Cowan S., Coyle J. E., Downham R., Figueroa E., Frederickson M., Graham B., McMenamin R., O'Brien M. A., Patel S., Phillips T. R., Williams G., Woodhead A. J., Woolford A. J., J. Med. Chem. 2010, 53, 5942–5955. [DOI] [PubMed] [Google Scholar]
- 35. Qi P. X., Urbauer J. L., Fuentes E. J., Leopold M. F., Wand A. J., Nat. Struct. Biol. 1994, 1, 378–382. [DOI] [PubMed] [Google Scholar]
- 36. Szczepina M. G., Bleile D. W., Müllegger J., Lewis A. R., Pinto B. M., Chem. Eur. J. 2011, 17, 11438–11445. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Ludwig C., Guenther U. L., Front. Biosci. 2009, 14, 4565–4574; [DOI] [PubMed] [Google Scholar]
- 37b. Ludwig C., Michiels P. J. A., Wu X., Kavanagh K. L., Pilka E., Jansson A., Oppermann U., Gunther U. L., J. Med. Chem. 2008, 51, 1–3. [DOI] [PubMed] [Google Scholar]
- 38. Dalvit C., Fasolini M., Flocco M., Knapp S., Pevarello P., Veronesi M., J. Med. Chem. 2002, 45, 2610–2614. [DOI] [PubMed] [Google Scholar]
- 39. Lucas L. H., Larive C. K., Concepts Magn. Reson. 2004, 20, 24–41. [Google Scholar]
- 40.
- 40a. Stejskal E. O., Tanner J. E., J. Chem. Phys. 1965, 42, 288–292; [Google Scholar]
- 40b. Tanner J. E., J. Chem. Phys. 1970, 52, 2523–2527; [Google Scholar]
- 40c. Gibbs S. J., C. S. Johnson Jr. , J. Magn. Reson. 1991, 93, 395–402; [Google Scholar]
- 40d. Wu D., Chen A., C. S. Johnson Jr. , J. Magn. Reson. Ser. A 1995, 115, 260–264; [Google Scholar]
- 40e. Otto W. H., Larive C. K., J. Magn. Reson. 2001, 153, 273–276. [DOI] [PubMed] [Google Scholar]
- 41. Marchetti R., Dillon M. J., Burtnick M. N., Hubbard M. A., Tamigney Kenfack M., Bleriot Y., Gauthier C., Brett P. J., AuCoin D. P., Lanzetta R., Silipo A., Molinaro A., ACS Chem. Biol. 2015, 10, 2295–2302. [DOI] [PubMed] [Google Scholar]
- 42. Yan J., Kline A. D., Mo H., Zartler E. R., Shapiro M. J., J. Am. Chem. Soc. 2002, 124, 9984–9985. [DOI] [PubMed] [Google Scholar]
- 43. Lipsitz R. S., Tjandra N., Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 387–413. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. DeMarco M. L., Woods R. J., Prestegard J. H., Tian F., J. Am. Chem. Soc. 2010, 132, 1334–1338; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44b. Yamaguchi T., Kamiya Y., Choo Y.-M., Yamamoto S., Kato K., Chem. Lett. 2013, 42, 544–546. [Google Scholar]
- 45.
- 45a. Yamamoto S., Yamaguchi T., Erdélyi M., Griesinger C., Kato K., Chem. Eur. J. 2011, 17, 9280–9282; [DOI] [PubMed] [Google Scholar]
- 45b. Erdélyi M., d′Auvergne E., Navarro-Vázquez A., Leonov A., Griesinger C., Chem. Eur. J. 2011, 17, 9368–9376; [DOI] [PubMed] [Google Scholar]
- 45c. Yamamoto S., Zhang Y., Yamaguchi T., Kameda T., Kato K., Chem. Commun. 2012, 48, 4752–4754; [DOI] [PubMed] [Google Scholar]
- 45d. Zhang Y., Yamamoto S., Yamaguchi T., Kato K., Molecules 2012, 17, 6658–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.
- 46a. Hass M. A. S., Ubbink M., Curr. Opin. Struct. Biol. 2014, 24, 45–53; [DOI] [PubMed] [Google Scholar]
- 46b. Pintacuda G., Park A. Y., Keniry M. A., Dixon N. E., Otting G., J. Am. Chem. Soc. 2006, 128, 3696–3702; [DOI] [PubMed] [Google Scholar]
- 46c. Saio T., Yokochi M., Kumeta H., Inagaki F., J. Biomol. NMR 2010, 46, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.
- 47a. Guan J.-Y., Keizers P. H. J., Liu W.-M., Loehr F., Skinner S. P., Heeneman E. A., Schwalbe H., Ubbink M., Siegal G., J. Am. Chem. Soc. 2013, 135, 5859–5868; [DOI] [PubMed] [Google Scholar]
- 47b. Zhuang T., Lee H.-S., Imperiali B., Prestegard J. H., Protein Sci. 2008, 17, 1220–1231; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47c. Camacho-Zarco A. R., Munari F., Wegstroth M., Liu W.-M., Ubbink M., Becker S., Zweckstetter M., Angew. Chem. Int. Ed. 2015, 54, 336–339; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 343–346. [Google Scholar]
- 48.
- 48a. Mallagaray A., Canales A., Dominguez G., Jimenez-Barbero J., Perez-Castells J., Chem. Commun. 2011, 47, 7179–7181; [DOI] [PubMed] [Google Scholar]
- 48b. Canales A., Mallagaray A., Perez-Castells J., Boos I., Unverzagt C., Andre S., Gabius H. J., Canada F. J., Jimenez-Barbero J., Angew. Chem. Int. Ed. 2013, 52, 13789–13793; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14034–14038. [Google Scholar]
- 49.
- 49a. Canales A., Mallagaray A., Berbís M. A., Navarro-Vazquez A., Dominguez G., Canada F. J., Andre S., Gabius H.-J., Perez-Castells J., Jimenez-Barbero J., J. Am. Chem. Soc. 2014, 136, 8011–8017; [DOI] [PubMed] [Google Scholar]
- 49b. Brath l., Swamy S. I., Veiga A. X., Tung C.-C., Petegem F. V., Erdelyi M., J. Am. Chem. Soc. 2015, 137, 11391–11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.
- 50a. Diercks T., Ribeiro J. P., Canada F. J., Andre S., Jimenez-Barbero J., Gabius H. J., Chem. Eur. J. 2009, 15, 5666–5668; [DOI] [PubMed] [Google Scholar]
- 50b. Ribeiro J. P., Diercks T., Jiménez-Barbero J., Andre S., Gabius H. J., Cañada F. J., Biomolecules 2015, 5, 3177–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.
- 51a. Fehér K., Groves P., Batta G., Jiménez-Barbero J., Muhle-Goll C., Kövér K. E., J. Am. Chem. Soc. 2008, 130, 17148–17153; [DOI] [PubMed] [Google Scholar]
- 51b. Wagstaff J. L., Taylor S. L., Howard M. J., Mol. BioSyst. 2013, 9, 571–577. [DOI] [PubMed] [Google Scholar]
- 52. Kay L. E., Ikura M., Tschudin R., Bax A., J. Magn. Reson. 1990, 89, 496–514. [DOI] [PubMed] [Google Scholar]
- 53.
- 53a. Sibille N., Hanoulle X., Bonachera F., Verdegem D., Landrieu I., Wieruszeski J. M., Lippens G., J. Biomol. NMR 2009, 43, 219–227; [DOI] [PubMed] [Google Scholar]
- 53b. Yamazaki T., Otomo T., Oda N., Kyogoku Y., Uegaki K., Ito N., Ishino Y., Nakamura H., J. Am. Chem. Soc. 1998, 120, 5591–5592; [Google Scholar]
- 53c. Otomo T., Teruya K., Uegaki K., Yamazaki T., Kyogoku Y., J. Biomol. NMR 1999, 14, 105–114; [DOI] [PubMed] [Google Scholar]
- 53d. Xu R., Ayers B., Cowburn D., Muir T. W., Proc. Natl. Acad. Sci. USA 1999, 96, 388–393; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53e. Weigelt J., van Dongen M., Uppenberg J., Schultz J., Wikstrom M., J. Am. Chem. Soc. 2002, 124, 2446–2447; [DOI] [PubMed] [Google Scholar]
- 53f. Sondej Pochapsky S., Pochapsky T. C., Curr. Top Med. Chem. 2001, 1, 427–441. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Hajduk P. J., Gerfin T., Boehlen J. M., Haberli M., Marek D., Fesik S. W., J. Med. Chem. 1999, 42, 2315–2317; [DOI] [PubMed] [Google Scholar]
- 54b. Pervushin K., Riek R., Wider G., Wuthrich K., Proc. Natl. Acad. Sci. USA 1997, 94, 12366–12371; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54c. Pellecchia M., Meininger D., Shen A. L., Jack R., Kasper C. B., Sem D. S., J. Am. Chem. Soc. 2001, 123, 4633–4634. [DOI] [PubMed] [Google Scholar]
- 55.
- 55a. Frieden C., Hoeltzli S. D., Bann J. G., Methods Enzymol. 2004, 380, 400–415; [DOI] [PubMed] [Google Scholar]
- 55b. Wiesner S., Sprangers R., Curr. Opin. Struct. Biol. 2015, 35, 60–67. [DOI] [PubMed] [Google Scholar]
- 56. Canales A., Lozano R., Lopez-Mendez B., Angulo J., Ojeda R., Nieto P. M., Martın-Lomas M., Gimenez-Gallego G., Jimenez-Barbero J., FEBS J. 2006, 273, 4716–4727. [DOI] [PubMed] [Google Scholar]
- 57. Medek A., Hajduk P., Mack J., Fesik S. W., J. Am. Chem. Soc. 2000, 122, 1241–1242. [Google Scholar]
- 58.
- 58a. Fielding L., Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 219–242; [Google Scholar]
- 58b. Poveda A., Jimenez-Barbero J., Chem. Soc. Rev. 1998, 27, 133–144. [Google Scholar]
- 59. Qin H., Shi J., Noberini R., Pasquale E. B., Song J., J. Biol. Chem. 2008, 283, 29473–29484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kahmann J. D., Sass H.-J., Allan M. G., Seto H., Thompson C. J., Grzesiek S., EMBO J. 2003, 22, 1824–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pellecchia M., Sem D. S., Wuthrich K., Nat. Rev. Drug Discovery 2002, 1, 211–219. [DOI] [PubMed] [Google Scholar]
- 62. Jahnke W., Ruedisser S., Zurini M., J. Am. Chem. Soc. 2001, 123, 3149–3150. [DOI] [PubMed] [Google Scholar]
- 63. Jain N. U., Venot A., Umemoto K., Leffler H., Prestegard J. H., Protein Sci. 2001, 10, 2393–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sylla B., Guegan J. P., Wieruszeski J. M., Nugier-Chauvin C., Legentil L., Daniellou R., Ferrieres V., Carbohydr. Res. 2011, 346, 1490–1494. [DOI] [PubMed] [Google Scholar]
- 65. Tanaka H., Kawai T., Adachi Y., Hanashima S., Yamaguchi Y., Ohno N., Takahashi T., Bioorg. Med. Chem. 2012, 20, 3898–3914. [DOI] [PubMed] [Google Scholar]
- 66. Hanashima S., Gotze S., Liu Y., Ikeda A., Kojima-Aikawa K., Taniguchi N., Varon Silva D., Feizi T., Seeberger P. H., Yamaguchi Y., ChemBioChem 2015, 16, 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Reina J. J., Sattin S., Invernizzi D., Mari S., Martínez-Prats L., Tabarani G., Fieschi F., Delgado R., Nieto P. M., Rojo J., Bernardi A., ChemMedChem 2007, 2, 1030–1036. [DOI] [PubMed] [Google Scholar]
- 68. Thépaut M., Guzzi C., Sutkeviciute I., Sattin S., Ribeiro-Viana R., Varga N., Chabrol E., Rojo J., Bernardi A., Angulo J., Nieto P. M., Fieschi F., J. Am. Chem. Soc. 2013, 135, 2518–2529. [DOI] [PubMed] [Google Scholar]
- 69. Mari S., Serrano-Gomez D., Canada J. F., Corbo A. L., Jimenez-Barbero J., Angew. Chem. Int. Ed. 2005, 44, 296–298; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 300–302. [Google Scholar]
- 70. Prost L. R., Grim J. C., Tonelli M., Kiessling L. L., ACS Chem. Biol. 2012, 7, 1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Probert F., Whittaker S. B., Crispin M., Mitchell D. A., Dixon A. M., J. Biol. Chem. 2013, 288, 22745–22757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Muñoz-García J. C., Chabrol E., Vivès R. R., Thomas A., de Paz J. L., Rojo J., Imberty A., Fieschi F., Nieto P. M., Angulo J., J. Am. Chem. Soc. 2015, 137, 4100–4110. [DOI] [PubMed] [Google Scholar]
- 73. Marchetti R., Lanzetta R., Michelow C. J., Molinaro A., Silipo A., Eur. J. Org. Chem. 2012, 5275–5281. [Google Scholar]
- 74. Rinnbauer M., Ernst B., Wagner B., Magnani J., Benie A. J., Peters T., Glycobiology 2003, 13, 435–443. [DOI] [PubMed] [Google Scholar]
- 75. Miller M. C., Ribeiro J. P., Roldos V., Martin-Santamaria S., Canada F. J., Nesmelova I. A., Andre S., Pang M., Klyosov A. A., Baum L. G., Jimenez-Barbero J., Gabius H. J., Mayo K. H., Glycobiology 2011, 21, 1627–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. do Nascimento A. S. F., Serna S., Beloqui A., Arda A., Sampaio A. H., Walcher J., Ott D., Unverzagt C., Reichardt N.-C., Jimenez-Barbero J., Nascimento K. S., Imberty A., Cavada B. S., Varrot A., Glycobiology 2015, 25, 607–616. [DOI] [PubMed] [Google Scholar]
- 77. Hanashima S., Sato K.-i., Naito Y., Takematsu H., Kozutsumi Y., Ito Y., Yamaguchi Y., Bioorg. Med. Chem. 2010, 18, 3720–3727. [DOI] [PubMed] [Google Scholar]
- 78. McCullough C., Wang M., Rong L., Caffrey M., PLoS One 2012, 7, e33958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Haselhorst T., Garcia J. M., Islam T., Lai J. C., Rose F. J., Nicholls J. M., Peiris J. S., von Itzstein M., Angew. Chem. Int. Ed. 2008, 47, 1910–1912; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1936–1938. [Google Scholar]
- 80. Sandström C., Berteau O., Gemma E., Oscarson S., Kenne L., Gronenborn A. M., Biochemistry 2004, 43, 13926–13931. [DOI] [PubMed] [Google Scholar]
- 81. Matei E., Andre S., Glinschert A., Infantino A. S., Oscarson S., Gabius H.-J., Gronenborn A. M., Chem. Eur. J. 2013, 19, 5364–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vanwetswinkel S., Volkov A. N., Sterckx Y. G. J., Garcia-Pino A., Buts L., Vranken W. F., Bouckaert J., Roy R., Wyns L., van Nuland N. A. J., J. Med. Chem. 2014, 57, 1416–1427. [DOI] [PubMed] [Google Scholar]
- 83. Schubert M., Bleuler-Martinez S., Butschi A., Wälti M. A., Egloff P., Stutz K., Yan S., Collot M., Mallet J. M., Wilson I. B., Hengartner M. O., Aebi M., Allain F. H., Künzler M., PLoS Pathog. 2012, 8, e1002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.
- 84a. Jiménez-Barbero J., Javier Cañada F., Luis Asensio J., Aboitiz N., Vidal P., Canales A., Groves P., Gabius H.-J., Siebert H.-C., Adv. Carbohydr. Chem. Biochem. 2006, 60, 303–354; [DOI] [PubMed] [Google Scholar]
- 84b. Asensio J. L., Canada F. J., Siebert H. C., Laynez J., Poveda A., Nieto P. M., Soedjanaamadja U. M., Gabius H. J., Jimenez-Barbero J., Chem. Biol. 2000, 7, 529–543; [DOI] [PubMed] [Google Scholar]
- 84c. Aboitiz N., Vila-Perello M., Groves P., Asensio J. L., Andreu D., Canada F. J., Jimenez-Barbero J., ChemBioChem 2004, 5, 1245–1255. [DOI] [PubMed] [Google Scholar]
- 85. Rademacher C., Krishna N. R., Palcic M., Parra F., Peters T., J. Am. Chem. Soc. 2008, 130, 3669–3675. [DOI] [PubMed] [Google Scholar]
- 86.
- 86a. Fiege B., Rademacher C., Cartmell J., Kitov P. I., Parra F., Peters T., Angew. Chem. Int. Ed. 2012, 51, 928–932; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 952–956; [Google Scholar]
- 86b. Rademacher C., Guiard J., Kitov P. I., Fiege B., Dalton K. P., Parra F., Bundle D. R., Peters T., Chem. Eur. J. 2011, 17, 7442–7453. [DOI] [PubMed] [Google Scholar]
- 87. Haselhorst T., Blanchard H., Frank M., Kraschnefski M. J., Kiefel M. J., Szyczew A. J., Dyason J. C., Fleming F., Holloway G., Coulson B. S., von Itzstein M., Glycobiology 2006, 17, 68–81. [DOI] [PubMed] [Google Scholar]
- 88. Yuasa N., Koyama T., Subedi G. P., Yamaguchi Y., Matsushita M., Yamaguchi Y. F., J. Biochem. 2013, 154, 521–529. [DOI] [PubMed] [Google Scholar]
- 89. Broecker F., Aretz J., Yang Y., Hanske J., Guo X., Reinhardt A., Wahlbrink A., Rademacher C., Anish C., Seeberger P. H., ACS Chem. Biol. 2014, 9, 867–873. [DOI] [PubMed] [Google Scholar]
- 90. Niedziela T., Letowska I., Lukasiewicz J., Kaszowska M., Czarnecka A., Kenne L., Lugowski C., Infect. Immun. 2005, 73, 7381–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Marchetti R., Canales A., Lanzetta R., Nilsson I., Vogel C., Reed D. E., Aucoin D. P., Jimenez-Barbero J., Molinaro A., Silipo A., ChemBioChem 2013, 14, 1485–1493. [DOI] [PubMed] [Google Scholar]
- 92. Oberli M. A., Tamborrini M., Tsai Y.-H., Werz D. B., Horlacher T., Adibekian A., Gauss D., Moller H. M., Pluschke G., Seeberger P. H., J. Am. Chem. Soc. 2010, 132, 10239–10241. [DOI] [PubMed] [Google Scholar]
- 93. Szczepina M. G., Bleile D. W., Pinto B. M., Chem. Eur. J. 2011, 17, 11446–11455. [DOI] [PubMed] [Google Scholar]
- 94. McLellan J. S., Pancera M., Carrico C., Gorman J., Julien J. P., Khayat R., Louder R., Pejchal R., Sastry M., Dai K., O'Dell S., Patel N., Shahzad-ul-Hussan S., Yang Y., Zhang B., Zhou T., Zhu J., Boyington J. C., Chuang G. Y., Diwanji D., Georgiev I., Kwon Y. D., Lee D., Louder M. K., Moquin S., Schmidt S. D., Yang Z. Y., Bonsignori M., Crump J. A., Kapiga S. H., Sam N. E., Haynes B. F., Burton D. R., Koff W. C., Walker L. M., Phogat S., Wyatt R., Orwenyo J., Wang L. X., Arthos J., Bewley C. A., Mascola J. R., Nabel G. J., Schief W. R., Ward A. B., Wilson I. A., Kwong P. D., Nature 2011, 480, 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pancera M., Shahzad-Ul-Hussan S., Doria-Rose N. A., McLellan J. S., Bailer R. T., Dai K., Loesgen S., Louder M. K., Staupe R. P., Yang Y., Zhang B., Parks R., Eudailey J., Lloyd K. E., Blinn J., Alam S. M., Haynes B. F., Amin M. N., Wang L. X., Burton D. R., Koff W. C., Nabel G. J., Mascola J. R., Bewley C. A., Kwon P. D., Nat. Struct. Mol. Biol. 2013, 20, 804–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rademacher C., Shoemaker G. K., Kim H.-S., Zheng R. B., Taha H., Liu C., Nacario R. C., Schriemer D. C., Klassen J. S., Peters T., Lowary T. L., J. Am. Chem. Soc. 2007, 129, 10489–10502. [DOI] [PubMed] [Google Scholar]
- 97. van de Weerd R., Berbis M. A., Sparrius M., Maaskant J. J., Boot M., Paauw N. J., de Vries N., Boon L., Baba O., Canada F. J., Geurtsen J., Jimenez-Barbero J., Appelmelk B. J., ChemBioChem 2015, 16, 977–989. [DOI] [PubMed] [Google Scholar]
- 98. Hayafune M., Berisio R., Marchetti R., Silipo A., Kayama M., Desaki Y., Arima S., Squeglia F., Ruggiero A., Tokuyasu K., Molinaro A., Kaku H., Shibuya N., Proc. Natl. Acad. Sci. USA 2014, 111, E404–E413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mandal P. K., Branson T. R., Hayes E. D., Ross J. F., Gavın J. A., Daranas A. H., Turnbull W. B., Angew. Chem. Int. Ed. 2012, 51, 5143–5146; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5233–5236. [Google Scholar]
- 100. Blaum B. S., Hannan J. P., Herbert A. P., Kavanagh D., Uhrin D., Stehle T., Nat. Chem. Biol. 2015, 11, 77–82. [DOI] [PubMed] [Google Scholar]
- 101.
- 101a. Dambuza I. M., Brown G. D., Curr. Opin. Immunol. 2015, 32, 21–27; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101b. Lis H., Sharon N., Chem. Rev. 1998, 98, 637–674; [DOI] [PubMed] [Google Scholar]
- 101c. Imberty A., Varrot A., Curr. Opin. Struct. Biol. 2008, 18, 567–576. [DOI] [PubMed] [Google Scholar]
- 102. Geijtenbeek T. B. H., Torensma R., van Vliet S. J., van Duijnhoven G. C. F., Adema G. J., van Kooyk Y., Figdo C. G., Cell 2000, 100, 575–585. [DOI] [PubMed] [Google Scholar]
- 103. van Kooyk Y., Geijtenbeek T. B. H., Nat. Rev. Immunol. 2003, 3, 697–709. [DOI] [PubMed] [Google Scholar]
- 104. Reina J. J., Diaz I., Nieto P. M., Campillo N. E., Paez J. A., Tabarani G., Fieschi F., Rojo J., Org. Biomol. Chem. 2008, 6, 2743–2754. [DOI] [PubMed] [Google Scholar]
- 105. Feinberg H., Castelli R., Drickamer K., Seeberger P. H., Weis W. I., J. Biol. Chem. 2007, 282, 4202–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pederson K., Mitchell D. A., Prestegard J. H., Biochemistry 2014, 53, 5700–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Guo Y., Feinberg H., Conroy E., Mitchell D. A., Alvarez R., Blixt O., Taylor M. E., Weis W. I., Drickamer K., Nat. Struct. Mol. Biol. 2004, 11, 591–598. [DOI] [PubMed] [Google Scholar]
- 108. Zierke M., Smiesko M., Rabbani S., Aeschbacher T., Cutting B., Allain F. H.-T., Schubert M., Ernst B., J. Am. Chem. Soc. 2013, 135, 13464–13472. [DOI] [PubMed] [Google Scholar]
- 109.
- 109a. Ciobanu M., Huang K.-T., Daguer J.-P., Barluenga S., Chaloin O., Schaeffer E., Mueller C. G., Mitchellc D. A., Winssinger N., Chem. Commun. 2011, 47, 9321–9323; [DOI] [PubMed] [Google Scholar]
- 109b. Wang S.-K., Liang P.-H., Astronomo R. D., Hsu T.-L., Hsieh S.-L., Burton D. R., Wong C.-H., Proc. Natl. Acad. Sci. USA 2008, 105, 3690–3695; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109c. Tabarani G., Reina J. J., Ebel C., Vives C., Lortat-Jacob H., Rojo J., Fieschi F., FEBS Lett. 2006, 580, 2402–2408. [DOI] [PubMed] [Google Scholar]
- 110. Guzzi C., Angulo J., Doro F., Reina J. J., Thepaut M., Fieschi F., Bernardi A., Rojo J., Nieto P. M., Org. Biomol. Chem. 2011, 9, 7705–7712. [DOI] [PubMed] [Google Scholar]
- 111. Chabrol E., Nurisso A., Daina A., Vassal-Stermann E., Thepaut M., Girard E., Vivès R. R., Fieschi F., PLoS ONE 2012, 7, e50722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Young S. H., Dong W. J., Jacobs R. R., J. Biol. Chem. 2000, 275, 11874–11879. [DOI] [PubMed] [Google Scholar]
- 113. Hanashima S., Ikeda A., Tanaka H., Adachi Y., Ohno N., Takahashi T., Yamaguchi Y., Glycoconjugate J. 2014, 31, 199–207. [DOI] [PubMed] [Google Scholar]
- 114. Noguchi K., Okuyama K., Kitamura S., Takeo K., Carbohydr. Res. 1992, 237, 33–43. [DOI] [PubMed] [Google Scholar]
- 115. Bates H. A., Rapoport H., J. Am. Chem. Soc. 1979, 101, 1259–1265. [Google Scholar]
- 116. Ho M. R., Lou Y. C., Wei S. Y., Luo S. C., Lin W. C., Lyu P. C., Chen C., J. Mol. Biol. 2010, 402, 682–695. [DOI] [PubMed] [Google Scholar]
- 117. Watson A. A., Lebedev A. A., Hall B. A., Fenton-May A. E., Vagin A. A., Dejnirattisai W., Felce J., Mongkolsapaya J., Palma A. S., Liu Y., Feizi T., Screaton G. R., Murshudov G. N., O'Callaghan C. A., J. Biol. Chem. 2011, 286, 24208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hattrup C. L., Gendler S., J. Annu. Rev. Physiol. 2008, 70, 431–457. [DOI] [PubMed] [Google Scholar]
- 119.
- 119a. Coelho H., Matsushita T., Artigas G., Hinou H., Canada F. J., Lo-Man R., Leclerc C., Cabrita E. J., Jimenez-Barbero J., Nishimura S., Garcia-Martin F., Marcelo F., J. Am. Chem. Soc. 2015, 137, 12438–12441; [DOI] [PubMed] [Google Scholar]
- 119b. Martínez-Sáez N., Castro-López J., Valero-González J., Madariaga D., Compañón I., Somovilla V. J., Salvadó M., Asensio J. L., Jimenez-Barbero J., Avenoza A., Busto J. H., Bernardes G. J., Peregrina J. M., Hurtado-Guerrero R., Corzana F., Angew. Chem. Int. Ed. 2015, 54, 9830–9834; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9968–9972. [Google Scholar]
- 120.
- 120a. Benie A. J., Moser R., Bauml E., Blaas D., Peters T., J. Am. Chem. Soc. 2003, 125, 14–15; [DOI] [PubMed] [Google Scholar]
- 120b. Rademacher C., Peters T., Top. Curr. Chem. 2008, 273, 183–202. [DOI] [PubMed] [Google Scholar]
- 121. Ardenkjaer-Larsen J. H., Boebinger G. S., Comment A., Duckett S., Edison A. S., Engelke F., Griesinger C., Griffin R. G., Hilty C., Maeda H., Parigi G., Prisner T., Ravera E., van Bentum J., Vega S., Webb A., Luchinat C., Schwalbe H., Frydman L., Angew. Chem. Int. Ed. 2015, 54, 9162–9185; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9292–9317. [Google Scholar]
- 122. Barb A. W., Meng L., Gao Z., Johnson R. W., Moremen K. W., Prestegard J. H., Biochemistry 2012, 51, 4618–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.
- 123a. Subedi G. P., Barb A. W., Structure 2015, 23, 1573–1583; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123b. Barb A. W., Biochemistry 2015, 54, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Yu L., Phillips R. L., Zhang D. S., Teghanemt A., Weiss J. P., Gioannini T. L., J. Biol. Chem. 2012, 287, 16346–16355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zahringer U., Ittig S., Lindner B., Moll H., Schombel U., Gisch N., Cornelis G. R., J. Biol. Chem. 2014, 289, 23963–23976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Koppisetty C., Frank M., Lyubartsev A., Nyholm P., J. Comput. Aided Mol. Des. 2015, 29, 13–21. [DOI] [PubMed] [Google Scholar]