

SUMMARY
The c-Jun N-terminal kinases (JNKs), as members of the mitogen-activated protein kinase (MAPK) family, mediate eukaryotic cell responses to a wide range of abiotic and biotic stress insults. JNKs also regulate important physiological processes, including neuronal functions, immunological actions, and embryonic development, via their impact on gene expression, cytoskeletal protein dynamics, and cell death/survival pathways. Although the JNK pathway has been under study for >20 years, its complexity is still perplexing, with multiple protein partners of JNKs underlying the diversity of actions. Here we review the current knowledge of JNK structure and isoforms as well as the partnerships of JNKs with a range of intracellular proteins. Many of these proteins are direct substrates of the JNKs. We analyzed almost 100 of these target proteins in detail within a framework of their classification based on their regulation by JNKs. Examples of these JNK substrates include a diverse assortment of nuclear transcription factors (Jun, ATF2, Myc, Elk1), cytoplasmic proteins involved in cytoskeleton regulation (DCX, Tau, WDR62) or vesicular transport (JIP1, JIP3), cell membrane receptors (BMPR2), and mitochondrial proteins (Mcl1, Bim). In addition, because upstream signaling components impact JNK activity, we critically assessed the involvement of signaling scaffolds and the roles of feedback mechanisms in the JNK pathway. Despite a clarification of many regulatory events in JNK-dependent signaling during the past decade, many other structural and mechanistic insights are just beginning to be revealed. These advances open new opportunities to understand the role of JNK signaling in diverse physiological and pathophysiological states.
INTRODUCTION
Protein kinases are intracellular signaling enzymes that catalyze the phosphorylation of specific residues in their target substrate proteins. Despite a basic appreciation of the regulatory roles played by protein phosphorylation across a broad range of aspects of biology, many questions remain outstanding. Little is known about how phosphorylation directly modifies protein function. In many cases, it is not known how these molecular changes then influence the activity of signaling intermediates to impact ultimately on cellular behavior or how these mechanistic insights into phospho-protein function could be integrated with cellular-level observations to improve our understanding of both health and disease.
In this review, we survey the current understanding of the c-Jun N-terminal kinase (JNK) subfamily of Ser/Thr protein kinases. Signaling by the JNKs has been intensely studied for more than 2 decades, with several previous reviews covering general aspects (1) or some covering more specific aspects, such as JNK signaling in the brain or the opportunities for inhibition of JNK signaling as a therapeutic strategy in cancer (2, 3). Indeed, JNKs have attracted attention as potential pharmaceutical targets through their implication via biochemical, cellular, and systems-level approaches in disease development (4, 5). Although this review is broad in scope, its foundations lie in an exploration of the current molecular and mechanistic understanding of JNK-mediated signaling pathways, including a critical appraisal of how core JNK signaling modules assemble, the diversity of the JNK proteins themselves, and how JNKs connect with partner proteins.
We then assess the functional consequences of JNK-mediated phosphorylation on known substrate proteins. Indeed, the number of known and well-validated JNK substrates is now close to 100. This has prompted our mechanistic classification of the role of JNK-mediated phosphorylation among these functionally diverse substrate proteins; the intense research in the field before and after our former review, published in 2006 in Microbiology and Molecular Biology Reviews (1), provided our framework. Importantly, the functional diversity of JNK substrates readily explains why JNK signaling is so pervasive and how it controls such diverse processes. In our final section, we discuss how the critical roles for JNK signaling in mammals help to explain why microbes often “tinker” with JNK signaling pathways to use them to their own advantage. Although knowledge remains rudimentary for many of these aspects, a molecular-level understanding of JNK enzyme-substrate partnerships holds the promise, in combination with the results of emerging systems-level studies, to ultimately lead to a more complete understanding of JNK signaling.
CONTROL OF ACTIVITY AND LOCALIZATION OF JNK PATHWAYS
The Molecular Architecture of Core JNK Pathways
Protein kinases, such as JNKs of the mitogen-activated protein kinase (MAPK) family, relay, amplify, and integrate signals from a diverse range of intra- and extracellular stimuli. All MAPKs are Ser/Thr kinases that belong to the so-called CMGC kinase group (named after its best-known members: cyclin-dependent kinases [CDKs], MAPKs, glycogen synthase kinase 3 [GSK3], and CDK-like kinases [CLKs]). The CMGC kinases share many similarities within their kinase domains, especially in the vicinity of their catalytic site; as a result, they recognize identical or very similar consensus sequences in their targeted substrate proteins. Apart from some constitutively active members, most CMGC kinases (and all MAPKs) require phosphorylation of their activation loop for full catalytic activity. In the case of classical MAPKs, such as the JNKs, extracellular signal-regulated kinases 1/2 (ERK1/2), p38, or ERK5, two phosphorylation events within a typical Thr-x-Tyr motif (TxY in general, TPY in the case of JNKs) within the activation loop are required. The Ste7 family of kinases, better known as MAPK kinases or MAP2Ks, catalyze these phosphorylation events, whereas several phosphatases catalyze the removal of either or both phosphate groups to inactivate these kinases. Thus, there is direct control of the activities of the MAPKs, such as the JNKs, by the coordinated actions of positive and negative regulators.
More broadly, JNKs are components of canonical signal transduction cascades/pathways described generically as the “three-tiered” MAPK pathways (Fig. 1). Within these pathways, a top tier of kinases (MAP3Ks) receives a variety of inputs, a middle tier of kinases (MAP2Ks) is strictly dependent on the upper tier kinases for activation, and a lower tier of kinases (MAPKs) phosphorylates a large number of substrates to elicit regulatory responses; thus, these MAPKs are considered the effectors of the pathway. Such multitiered kinase pathways are common in regulatory systems; other prominent examples include the Hippo/LATS and the AMP-activated protein kinase pathways (6, 7). Furthermore, the sharing of multiple MAP3Ks and MAP2K between MAPK pathways can facilitate pathway cross talk and signal integration, thus providing a coordinated response to each activating signal.
FIG 1.

Overall organization of JNK signaling pathways. JNK pathways are activated by a variety of extracellular stimuli (e.g., cytokines, pathogens, morphogenic factors, hormones) as well as intracellular stimuli (e.g., oxidative stress, DNA damage), converging on the three JNKs. These phosphorylate a variety of cytoplasmic as well as nuclear substrates and engage in direct (e.g., phosphorylation of MAP3Ks) as well as indirect (e.g., expression of the dual-specificity phosphatases MKP1 and MKP5) feedback circuits. The protein kinase members of the core MAPK pathway are displayed in red, while critical proteins directly controlling MAP3K activation are shown in green. Proteins further upstream of the pathway are colored turquoise, MKPs are blue, and substrates are yellow. Note that, for the purposes of clarity, not all the known proteins or possible pathways are shown here. Continuous arrows imply direct binding or direct enzymatic reactions, while dotted arrows show either indirect, multistep reactions or connections where the exact mechanism(s) is uncertain. Abbreviations (other than the protein names defined in the main text): GPCR, G-protein-coupled receptor; Ubi, ubiquitin (usually nondegradative, with Lys63 linkage).
JNK pathways are activated in response to a wide range of stimuli but most notably following cell exposure to a variety of biotic or abiotic stress events, such as infection, inflammation, oxidative stress, DNA damage, osmotic stress, or cytoskeletal changes, with the best-characterized pathways being activated downstream of receptors, including G-protein coupled receptors (GPCRs), Wnt receptors, transforming growth factor-β (TGF-β) receptors, tumor necrosis factor (TNF) receptors, and the Toll receptor complex (Fig. 1). In addition, JNK activation has been reported in the response to endoplasmic reticulum stress (ER stress), downstream of activation of the ER-resident stress sensor kinase inositol-requiring enzyme 1 (IRE1) (8; see also reference 9 for a review of this topic). However, there have been subsequent studies indicating that the autocrine release of the inflammatory cytokine TNF may mediate these ER stress-activated events (10, 11), akin to initial suggestions that clustering of cytokine receptors could underlie JNK activation in response to UV light or osmotic stress (12).
Consistent with the wide range of inputs, the MAP3Ks of the top MAPK pathway tier are highly variable and do not even form a single kinase family when their kinase domains are considered: the MAP3Ks include both Ste20/Ste11/Ste7 (STE) family kinases (MEKKs, ASK1/2, TAOs) and members of the tyrosine kinase-like (TKL) family (MLKs, DLK, LZK, MTK, TAK1). The additional diversity of domains outside their kinase domains also likely reflects different modes of MAP3K activation. The MAP3Ks of the JNK pathway include TAK1 (in complex with Tab1 and Tab2/3, capable of sensing Lys63-linked polyubiquitinylation events at the TNF receptor and TAK1 itself being regulated by ubiquitination) (13–18), MEKK1 (through a mechanism involving Lys63-linked polyubiquitinylation of the Tab1 adaptor protein or other pathway regulators, including the T-cell receptor [TCR]-associated scaffold Carma 1) (19–22), MEKK4 (sensing GADD45 proteins, induced by DNA damage, or acting downstream of TRAF4) (23, 24), ASK1 (regulated by thioredoxin binding, and thus redox sensitive) (25, 26), and MLKs (regulated by GTP-bound small G-proteins of the Rho family, involved in cytoskeletal rearrangement) (27). Other MAP3Ks have also been described (e.g., MEKK2, MEKK3, TAO1, and TAO2) that primarily regulate the p38 and ERK5 MAPK pathways as well as the Hippo/LATS pathway but which may also have some role in JNK activation (28, 29). This shared use of kinases again emphasizes the possibilities for pathway cross talk and signal integration.
The MAP3Ks have been considered the “gatekeepers” of the MAPK pathways, but despite their importance, most MAP3Ks remain poorly characterized at a molecular level and no full-length structures of MAP3Ks have been determined to date. What is clear is that the MAP3Ks are tightly controlled by multiple mechanisms so that they normally cannot be activated in a single step. Indeed, the activation cycle of most MAP3Ks shows some similarity to that of receptor tyrosine kinases: many MAP3Ks are subject to autoinhibition (e.g., MEKK1, MEKK4, MLK3) by various regulatory domains associating with their kinase domains (23, 30, 31). Following the relief of autoinhibition (e.g., by protein ligands), MAP3Ks such as MLK3 or MEKK1 are allosterically activated due to kinase domain dimerization (30, 31). For MEKK2, dimerization of its kinase domain is required for full JNK activation in cells (32). However, it is still unclear how, at a molecular level, the catalytically competent kinase domain dimers of these MAP3Ks form. In the only well-explored example, observations for B-Raf from the ERK MAPK cascade suggest its symmetric dimerization leaves its kinase domains free to accept substrates (33–35). This is consistent with the dimerization mode observed for the isolated kinase domains of the MAP3K ASK1 (36). However, the existence of other modes cannot be excluded, e.g., MLKs primarily dimerize through coiled-coil interactions (37). Other noncatalytic proteins can also aid in MAP3K dimerization; one notable example is the Tab1-Tab2/3 complex, which is indispensable for TAK1 activity (15). In some cases, as exemplified by ASK2, the MAP3Ks themselves may lose their catalytic activity and become obligate dimerization partners that enhance the activation of other related MAP3Ks (38, 39). MAP3K full activation also requires activation loop autophosphorylation, presumably in trans, through a transient tetrameric complex (similar to the IκB kinases [IKKs]) (40, 41). This phosphorylation of MAP3Ks increases and stabilizes their activity, removing the requirement of a dimeric state for substrate phosphorylation. Other auxiliary kinases, generically classified as MAP4Ks, may mediate these and other modifications of MAP3Ks. The inactivation of MAP3Ks can also involve feedback phosphorylation events (42–44), the association with and phosphorylation by other kinases as reported for MEKK4 (45), or ubiquitin-directed degradation as reported for DLK1 (46, 47). Ultimately, following stimulation, phosphatases can remove the activating phosphate groups, as observed for the negative regulation of ASK1 by phosphatases, including PP5, PPM1L (also known previously as PP2C), Cdc25A and Cdc25C (48–51), or the actions of PP2A or PP6 on TAK1 (52, 53), to allow the kinases to return to a monomeric, autoinhibited basal state.
The most important MAP3K substrates within the JNK pathway are the MAP2Ks known as MKK4 and MKK7 (Fig. 1). MAP2Ks are highly specialized proteins of the STE kinase family, and MAP3K-mediated phosphorylation is the only reported activation mechanism for MKK4 or MKK7 under physiological conditions. Most MAP3Ks involved in the JNK pathway can phosphorylate multiple MAP2Ks, including those belonging to the p38 pathway (MKK3 and/or MKK6). Despite suggestions that a “DVD motif” (which was subsequently shown to be a structured segment of the MAP2K kinase domain critical for domain integrity) (54–56) would direct MAP3K-MAP2K specificity (57), the issue of MAP3K substrate selectivity is still largely unresolved. However, for the DLK-MKK7 complex, the MAP2K-MAP3K interaction has been mapped: the coiled-coil region of DLK binds to the N-terminal disordered segment of MKK7 preceding the kinase domain (58). Depending on the set of MAP3Ks activated, the degree of MKK4/MKK7 phosphorylation also varies with the nature of the pathway-initiating stimulus. For example, interleukin-1 or TNF-α exposure preferentially activated MKK7, whereas MKK4 and MKK7 were both activated following stress; these differences are in agreement with the failure of cytokines to activate JNKs in cells isolated from MKK7−/− mice, whereas JNK activation following exposure to stress was prevented only in MKK4−/− MKK7−/− cells (59).
MAP2Ks display very little activity on generic substrates such as synthetic peptide arrays; their activity appears restricted to the activation loops of intact kinase domains of their targeted MAPKs. MKK4 and MKK7 are capable of phosphorylating and activating JNKs in vitro (60). Under physiological conditions, the two kinases are synergistic in generating double-phosphorylated JNKs (61). Unlike MKK7, which targets JNKs only, MKK4 can also phosphorylate p38α both in vitro and in vivo (reviewed in reference 62). Hence, the JNK and p38 pathways are not truly separate, even at this middle tier of kinases.
Phosphatases and Feedback Mechanisms in Control of JNK Activity
By their dephosphorylation of kinases acting within the different MAPK pathway tiers, protein phosphatases can exert control over the magnitude and timing of MAPK activation. For the JNK pathway, Ser/Thr phosphatases may act as negative regulators, but little is known about those phosphatases specifically targeting the upper tiers of MAP3K or MAP2K enzymes. The PP5 and PP2A-type phosphatases, known as ERK MAPK pathway regulators, are potential candidates for this role (48, 53, 63–68). For JNKs themselves, dual Thr and Tyr phosphorylation within the JNK activation loop TPY motif is required for full JNK activity; thus, the removal of either phosphate can decrease JNK activity toward all substrates. Indeed, Ser/Thr phosphatases can directly regulate JNK actions. For example, the protein Ser/Thr metallophosphatase PPM1J (PP2Cζ) was found to harbor a JNK-binding motif (69). Since this recruitment or docking motif was also found in the related PPM1H phosphatase and is broadly conserved across the animal kingdom, these phosphatases may have important roles in the regulation of JNK pathways. Conversely, MAPK-mediated phosphorylation of regulatory phosphatases is also known (70, 71), and this phosphorylation has the potential to provide important additional control of pathway activation. Thus, specific Ser/Thr phosphatases have the potential to regulate JNK signaling by their actions at multiple tiers of the pathway.
Greater attention has been directed toward the contributions of the dual-specificity phosphatases (DUSPs) that can dephosphorylate both phosphotyrosine and phosphoserine/threonine residues within their substrate proteins. DUSPs are a large and diverse family, but a subset specific for MAPK dephosphorylation are known as MAPK phosphatase (MKPs). The two subfamilies of MKPs targeting JNK can be distinguished as either inducible, primarily nuclear DUSPs (MKP1/DUSP1 and MKP2/DUSP4) or predominantly cytoplasmic DUSPs (MKP5/DUSP10, MKP7/DUSP16, and M3/6 or DUSP8). Notably, MKP1−/− or MKP5−/− mice display JNK hyperactivation in diverse tissues, supporting these phosphatases as regulators of low basal JNK enzymatic activities (72–75). However, most DUSPs may dephosphorylate more than a single substrate, e.g., MKP1 and MKP2 target ERK1/2, p38 kinases, and JNKs (72), whereas MKP5 and MKP7 act on p38 and JNKs (76, 77). In addition to the dual-specificity phosphatase domain, these phosphatases carry a special noncatalytic domain termed the rhodanese domain, due to its structural similarity to bacterial rhodanese enzymes (78). The role of the rhodanese domains in MKP1 and MKP5 in recruiting ERK2 and p38α is well-known: by mimicking a so-called docking motif, this surface enables strong interactions with several MAPKs (79). However, the docking site of JNK cannot bind rhodanese domains, so phosphatases targeting JNK bind in alternative ways (via their catalytic domain or by docking motifs, as observed in MKP5 and MKP7 [80, 81]). In vivo studies suggest that MKP1 is not required for growth factor signaling; however, it is essential for immune cell activation, as MKP1−/− mice show immune defects (82). Similar phenotypes were observed for MKP5−/− mice (75). In addition, MKP1 controls JNK activity that is critical for appropriate axon branching in developing cortical neurons (83) as well as apoptosis of sympathetic neurons due to nerve growth factor withdrawal (84). Such findings emphasize the importance of these MAPK-directed DUSPs.
As the MKPs that dephosphorylate JNKs can also dephosphorylate p38, the regulatory effects in vivo cannot be clearly assigned to either specific MAPK family. However, the results of comparison of MKP1 with MKP5 (by evaluating the levels of individual MAPK activities in MKP1−/− versus MKP5−/− mice) suggest that MKP5 is more important for JNK1 dephosphorylation (75) while MKP1 is likely more critical for p38α inactivation, at least in macrophages (85). Other members of the dual-specificity phosphatase family may also regulate JNK pathways by acting on upstream signaling proteins, such as the Tab1/TAK1 complex (86) or the focal adhesion kinase (87). Depending on their targets, the latter phosphatases can either inhibit (MKP6/DUSP14) or activate (MKP-x/DUSP22) JNK signaling (87–89). Hence, MKPs can also act as either negative or positive regulators of JNK signaling.
Importantly, many MAPK phosphatases are under transcriptional control by the same pathways that they inactivate. For example, mammalian MKP1 is inducible by growth factor pathways (90). Genetic experiments in Drosophila melanogaster suggest that its MKP5 ortholog (Puckered) is also transcriptionally upregulated by the activity of its JNK ortholog (Basket) (91), exerting feedback control over a pathway that is essential for proper antibacterial and antiparasitic responses in many insects (92). In mammals, MKP5 expression levels may be inducible by inflammation (93). Although primarily induced by p38α as part of its feedback mechanism (94), MKP1 expression may also be at least partially controlled by JNK actions on its transcription factor substrates ATF2 and c-Jun (85, 95). However, transcriptional-level feedback is not the only option for MAPK pathways: the JNK pathway is also subject to feedback phosphorylation. It is known that MLK3 and DLK can be directly phosphorylated by activated JNK to exert positive feedback, although the exact sites and mechanisms differ greatly between these two proteins (44, 96). Such positive feedback loops can result in bistable switches (97) that can ensure immediate and robust responses by rapid, local, and maximal kinase activation.
Scaffold Proteins in JNK Signaling
In addition to the essential enzymatic roles played by kinases and phosphatases within signal transduction pathways, scaffold proteins that may lack intrinsic enzyme activities can act as protein-protein interaction hubs for multiple different enzymes to facilitate pathway activation in the response to cell stimulation (98–101). Before the JNK pathway scaffolds are addressed in more detail, it is important to appreciate that the identification and validation of scaffold proteins can be an experimentally difficult task. In contrast to the MAPKs themselves, which can be effectively purified for further analyses, the upper-tier MAP2K and MAP3K enzymes are frequently unstable in a purified form and prone to spurious interactions (102, 103; A. Zeke and A. Reményi, unpublished observations). These undesirable features of the MAP2K and MAP3K enzymes thus necessitate rigorous testing with positive and negative controls to assess their binding partners. Furthermore, several of the proposed scaffold proteins are also large, structurally poorly characterized proteins, so that fragments may be unstable and display artificially “sticky” binding. Therefore, the identification of JNK scaffold proteins from in vitro studies that did not test the structural integrity of all protein partners should be treated with caution. Similarly, in vitro kinase reactions with scaffolds can only yield biologically relevant results if the assay conditions were carefully set, particularly with the inclusion of agents (such as detergents, bovine serum albumin, or cell protein extracts) that mitigate spurious binding and vessel wall effects. To date, most cell-based interaction studies have used only coimmunoprecipitation assays with overexpressed putative interactors that may bias toward the detection of interactions. It is also important to consider that the phosphorylation enhancement effects seen in cell-based studies upon the knockdown or overexpression of a pathway regulator may also arise from complex network behavior and feedback loops rather than a traditional scaffolding action to bring together multiple pathway components. All of these critically important issues must be addressed before the true importance of JNK pathway scaffolds can be clarified; with these caveats in mind, the following paragraphs consider the proteins currently classified as potential JNK pathway scaffolds but focus on JIP1 as the best-described example.
Several proteins have been described to bind JNKs and one or more of its upstream activators. These include JIP1 (104), JIP3 (105), arrestins (106, 107), filamins A and B (108, 109), RACK1 (110), Crk (111), POSH (112), WDR62 (113), DUSP19 (114), and GRASP1 (115). Despite this interest in how scaffold proteins may work, the structural and mechanistic details for the actions of many of these suggested scaffolds remain unresolved (see Table S1 in the supplemental material). Interestingly, studies with mammalian MAPK pathway scaffolds, such as KSR1/2, hint that the original, simple scaffolding model based on a ternary or higher-order catalytic complex needs critical reevaluation (116). Specifically, KSR1 and KSR2 can form a complex with c-Raf or B-Raf, enhancing their activity through dimerization-induced allosteric activation. Furthermore, KSRs coordinate MEK1/2 as if these kinases were their own substrates (i.e., in the same way as B-Raf or c-Raf recruits MEK1/2) (117). ERK1 or ERK2 can also bind and phosphorylate KSR proteins directly to provide negative feedback (118). Thus, certain proteins can regulate MAPK signaling without directly impacting the assembly of MAP3K-MAP2K or MAP2K-MAPK complexes. It is important to consider whether the reported JNK scaffolds assemble ternary kinase complexes; if not, these proteins may be acting as complex network regulators that enhance JNK signaling by other means. The example of the possible actions of JIP1 (Fig. 2A), the protein first described as a JNK pathway scaffold, is evaluated in greater detail below.
FIG 2.

Structure and function of JIP1 acting within the JNK pathway. (A) The domain architecture of the JIP1 protein. The N-terminal regulatory “tail” is largely disordered, while the C-terminal half of JIP1 contains three folded domains as well as a kinesin light chain (KLC)-binding linear motif. The precise function of the intrinsically disordered N terminus (with its conserved acidic motifs) is unknown, yet it is highly phosphorylated by JNK in a D-motif-dependent manner. Currently, only two target sites (T103 and S421) are known to have a role in JNK-dependent physiological regulation of JIP1. This model was built by combining domain signature searches (PFAM), folding tendency predictors (IUPRED), and conservation analyses (multiple alignments among vertebrate proteins) as well as curated data from the literature. The lower line shows the results of conservation analyses (red, highly conserved sequence; blue, nonconserved sequence), when sequences of vertebrate JIP1 and the closely related JIP2 proteins are aligned with each other. Structural domains and key motifs are preserved in both proteins (including the JNK-binding D-motif), while most regulatory phosphorylation sites differ between JIP1 and JIP2. (B) A model of JIP1 actions on the microtubule-dependent transport processes in neurons. The JIP1/2 dimers (turquoise) are capable of transporting a diverse set of membrane-associated proteins (e.g., β-APP, APoE2-R) as well as certain MAP2Ks (MKK7 [red]) and inactive MAP3Ks (MLK3, DLK [magenta]). These complexes are moved along the microtubule filaments with the help of kinesin 1-kinesin light chain 1 motors (blue). At the end of their journey, the transport complexes are uncoupled by a JNK-dependent phosphorylation of JIP1. Since this step also results in the release of upstream components and activators belonging to the JNK pathway, it leads to a positive feedback loop and helps to maintain subcellular compartments with high local JNK activity. The JIP1/2 proteins uncoupled from their cargo are also transported in a reverse direction (likely through a dynein-driven process), although the structural details of the latter complex are poorly known.
One clue in the understanding of JIP1 functions in JNK signaling comes from the observation that JIP1 can bind several MAP3Ks (MLKs and DLK) as well as MKK7 (119, 120). Furthermore, JNK-dependent phosphorylation of JIP1 can regulate not only the release of its own upstream activators but also multiple proteins destined for secretion (121). This influence on pathway proteins rapidly leads to the establishment of subcellular compartments where local JNK pathway activation drives a strong positive feedback loop via JNK recruitment of its own activators through JIP1 (Fig. 2B). Notably, in accordance with this model, a high local JNK activity was detected at the distal end of developing axons (122). This sophisticated positive feedback loop between JNK1 and DLK could explain most of the enhancement of JNK activation following JIP1 transfection, particularly as JIP1 phospho-site mutants could not increase pathway activation (104). Thus, the role of JIP1 in the JNK pathway may be mechanistically very different from prototypical signaling scaffolds, such as the Saccharomyces cerevisiae Ste5 protein that increases overall signaling specificity by binding different pathway components simultaneously and so stimulates pathway throughput via allosteric regulatory mechanisms (123). In addition, the mammalian MKK7 shows high specificity for the JNKs (124–126), and so the mammalian MKK7-JNK pathway would not need to rely on additional scaffold proteins to provide pathway specificity. However, in this context it should be appreciated that scaffold proteins may admit a number of negative and/or positive feedback circuits to set the appropriate level of JNK activity for different subcellular compartments and physiological states.
In addition to these roles in JNK pathway regulation, the involvement of JIP1 and JIP2 as kinesin-dependent transport adaptors has been supported by numerous studies (121, 127, 128). The expression pattern of JIP1 (and the closely related JIP2) entails the highest levels in neurons or neuroendocrine cells that have the greatest need for large-volume axonal transport of vesicles (129). Structural predictions and analyses of sequence conservation suggest that JIP1 can approximately be divided into two halves: an unstructured N-terminal regulatory tail and a mostly folded C-terminal region (Fig. 2A). The last dozen amino acids of the C terminus also appear to be intrinsically disordered and form a kinesin light chain (KLC)-binding motif, allowing JIP1 to directly couple to complexes of kinesin light chain 1 (KLC1) and kinesin 1 (130). Sequence motif analysis suggests that the structured C-terminal half of JIP1 consists of at least three different domains. One of them is a Dab/Numb-type PTB (phosphotyrosine-binding, or protein tail-binding) domain, required for cargo binding. Like most other PTB domains, this domain can associate with the canonical NPxY motifs located in the cytoplasmic tails of transmembrane or perimembrane proteins as well as with other motifs, but importantly, this interaction does not require phosphorylation within the NPxY motifs (131, 132). The immediately preceding structured region is an Src homology 3 (SH3) domain. However, the SH3 domain of JIP1 is unique: it is incapable of binding to Pro-rich motifs and its purpose is to provide a specific dimerization interface for JIP1/2 proteins. Considering that the KLC1-kinesin 1 complexes are also dimeric, this is consistent with the requirement for JIP1 dimerization (133). An extensive region preceding the SH3 domain also appears to be conserved and is likely folded. While this segment shows no clear homology to other known protein domains, it apparently serves as an auxiliary cargo-binding module required to reinforce ligand recruitment by the PTB domain (including canonical NPxY motif-containing ligands, such as the β-amyloid precursor protein [β-APP]) (134). The N-terminal regulatory tail of JIP1 appears to be intrinsically disordered. It is highly phosphorylated by JNK, and phosphorylation at Thr103 can regulate the trafficking of JIP1-cargo complexes (104). As the cargo-binding domain(s) of JIP1 can also accept transmembrane proteins (β-APP, apolipoprotein E receptor 2) (135) or peripheral membrane proteins (ARHGEF28) (136) as ligands, it is clear that JIP1 may play significant, broader roles.
JNK Nucleo-Cytoplasmic Trafficking
The subcellular localization of MAPKs will dictate their access to their substrate proteins. JNKs can directly phosphorylate many cytoplasmic, cytoskeletal, mitochondrial, and cell membrane-associated substrates, but like many other MAPKs they were originally considered to act primarily in the nucleus through their modulation of transcription factor actions to alter gene expression programs. All classical MAPKs, including ERK1/2, JNKs, and the p38 kinases, can change their subcellular localization upon pathway activation via either preferential nuclear localization or enhanced nuclear retention. Although this phenomenon of increased nuclear MAPK populations has been appreciated for more than 2 decades, the mechanism(s) underlying MAPK nuclear entry and retention remains relatively poorly understood.
Most classical MAPKs (apart from ERK5) have no classical nuclear localization sequence (NLS) motifs for their nuclear import. Although a phosphorylation-based NLS has been proposed for ERK2 (137), the implicated site is not functional in JNK or the p38 kinases (138). The ERK2 NLS is also problematic due to structural reasons: the proposed site is rigidly folded and does not appear to be available for intramolecular autophosphorylation. ERK1/2 more likely translocates into the nucleus following activation loop phosphorylation-dependent formation of its FxFP pocket and subsequent interactions with FxFG motif-containing nucleoporin proteins of the nuclear pore complex, a mechanism not possible for JNKs (139–141). Regardless, nuclear translocation of both active and inactive MAPKs may also be mediated by several β-importins. For example, importin-3, importin-7, and importin-9 have all been implicated in mediating JNK nuclear entry (138). The use of these different importin systems could thus provide multiple nuclear entry modes for other MAPKs (142) as well as the JNKs.
However, the movement of proteins into the nucleus may also be mediated by complex formation with other proteins that themselves have bona fide NLS motifs. For example, following coexpression with partners like c-Jun, JNK2 can become predominantly nuclear (143). Such piggyback transport on partner proteins can be independent of activation state and would allow them to form a dynamic equilibrium between the nucleus and the cytoplasm even in the absence of JNK pathway activation. The recent results of a predominantly α-importin-dependent nuclear import of JNK1, but in the absence of a classical NLS motif or direct α-importin binding, are consistent with this mechanism (144). Live-cell imaging, including fluorescence recovery after photobleaching protocols, has also revealed the constitutive nucleo-cytoplasmic shuttling of green fluorescent protein (GFP)-labeled JNK1 (144), implying that JNKs can access substrates in all intracellular compartments rather than being restricted to a single intracellular location. Importantly, consistent with the JNKs interacting with various protein partners, these studies also revealed a decrease in the intranuclear and intracytoplasmic JNK1 mobilities following cell exposure to hyperosmotic stress (144). These observations should prompt further studies evaluating the mechanisms of JNK nuclear entry and export, together with the contributions played by JNK-interacting partners in modulating these nuclear trafficking events under both normal and stress conditions.
STRUCTURE AND ROLES OF DIFFERENT JNK ISOFORMS
Structural Overview of the JNKs
The human genome contains three closely related JNK genes: JNK1, JNK2, and JNK3 (145). All three genes encode ∼400-amino-acid proteins encompassing little more than a canonical Ser/Thr protein kinase domain (146). Within the core kinase fold, JNKs contain several additional structural features well-conserved among MAPKs: the CMGC insert protruding from the C-terminal kinase lobe, the short common docking (CD) helix, and the C-terminal helix binding back to the N-terminal lobe (147, 148). Most nonvertebrate animals possess only a single JNK gene (e.g., Basket in Drosophila) (149); the three vertebrate JNK genes (conserved from mammals to fish) appear to have arisen from a twin whole-genome duplication event at the dawn of vertebrate evolution (150). Thus, the sequences and structures of all three vertebrate paralogs are similar, with JNK2 being the earliest-branching member as judged by the amino acid differences in its kinase domain (151) and JNK3 closely resembling JNK1 but with a JNK3-specific N-terminal extension added through the use of an upstream translational initiation site (145).
Alternative Isoforms Encoded by Each JNK Gene
All three JNK genes encode multiple isoforms generated by transcript alternative splicing (Fig. 3). One such alternative splicing site lies within the sequence specifying the C-terminal lobe of the kinase domain; use of a mutually exclusive exon pair (the sixth exon in most transcripts) results in two similar but not identical kinases, termed the α- and β-isoforms. Unfortunately, the nomenclature for these isoforms is not consistent, with the isoforms incorporating exon 6a being denoted JNK1α and JNK3α but JNK2β, whereas JNK1β, JNK3β, and JNK2α all contain exon 6b (see Table S2 in the supplemental material). Exons 6a and 6b are likely the result of an ancient exon duplication predating JNK gene duplications (152). Since the upstream intron is removed during transcript maturation by a U2 splicing apparatus, whereas the downstream intron is removed by a U12-type splicing apparatus, the hybrid intron between the two exons cannot be excised without eliminating one of the two exons (153). The incorporation of alternative exons is mostly random, but it can also be controlled by specific splicing factors. For example, the polypyrimidine tract preceding exon 6b contains multiple binding sites for the Nova family of neuronal splicing regulator proteins (154, 155). Thus, the neuron-specific generation of the JNK2β isoform depends on the protein Nova2, which masks the polypyrimidine tract of the 6b exon, allowing the preferred incorporation of exon 6a instead of exon 6b (155). Importantly, all JNK genes carry Nova-type splicing regulator binding sites, but the number of these differ: JNK2, JNK3, and JNK1 carry 5, 3, and 1 of these sites, respectively, providing a molecular rationale for tissue-specific JNK gene splicing or the absence thereof. For all three JNK genes, the 5-nucleotide shift that occurs with the use of a 3′-splicing site of the final intron results in different reading frames and so produces JNK proteins with C-terminal extensions of differing lengths. As these C termini are not part of the kinase domain and are predicted to be structurally disordered, their influence on the function or stability of the individual isoforms is still unclear. However, the two splicing variations do freely combine, giving rise to four isoforms for both JNK1 and JNK2. Kinases with a longer C terminus are regarded as isoform 2 (i.e., the so-called “p54” JNKs, which can include α- or β-isoforms), while the shorter JNKs are regarded as isoform 1 (i.e., the so-called “p46” JNKs, which again can include α- or β-isoforms).
FIG 3.

Splice isoforms of the JNK1, JNK2, and JNK3 proteins. (A) The structure of JNKs. The generic structure of the JNK proteins is displayed in beige (represented by the crystal structure of JNK1β1; PDB ID 2XRW), and the variable regions (alternative splice isoforms) are highlighted in green, red, and magenta. The catalytic site of the kinase domain, ATP, is indicated in yellow. Regions that are unstructured or flexible are drawn with dotted lines. (B) All human JNK genes encode multiple splice isoforms. Apart from transcripts lacking a complete kinase domain (and therefore likely not yielding a functional protein), there are two variable regions for JNK1/2 and three for JNK3. All these alternative splicing products (as well as those resulting from alternative initiation with JNK3) combine freely and yield four isoforms for JNK1 and JNK2. For JNK3, there are 8 possible isoforms (including the longer [L] and shorter [S] N-terminal extensions), but only 3 isoforms have been characterized to date. However, mRNA sequences from databases (such as ENSEMBL) suggest that, like JNK1/2, JNK3 also contains the same alternative exons in its kinase domain. This hints at the existence of many more uncharacterized JNK3 isoforms (in blue). In the figure, the alternative segments structurally and evolutionarily corresponding to each other are labeled with the same colors: red, within the kinase domain; magenta, C-terminal flexible extension; green, N-terminal flexible extension. (C) Mechanisms of splice isoform generation in vertebrate JNK genes. The JNK3 gene has an upstream ATG codon, resulting in N-terminally extended proteins (green). However, this upstream initiation site has no Kozak consensus sequence, and so this is expected to result in “leaky scanning” by ribosomes, allowing the translational start to stochastically shift downstream to the site shared with all other JNK proteins. All vertebrate JNK genes have a duplicated exon (exon 6a [beige] and exon 6b [red]), where nonregulated splicing 6b is the preferred (major) exon. Their inclusion in the final transcript is mutually exclusive with each other because of the incompatibility of their U2- and U12-recognized splicing sites. Inclusion of the 6a exon depends on the suppression of exon 6b splicing, which can happen when the Nova2 protein binds to its polypyrimidine tract (Py) in JNK2. The ultimate splicing site is also variable, allowing for a 5-nucleotide shift. This results in a frameshift and an early stop codon in the short (p46) isoforms, while allowing the translation of the last exon in full in the case of the long (p54) isoforms. The sequences of the p46 (blue) and p54 (magenta) isoforms in the figure refer to JNK1. Note that the generic intron-exon pattern (colored to match the alternative protein sequences) shown at the top is not proportional to actual intron-exon sizes. The untranslated regions are displayed in light blue.
In the case of JNK3, an extra ATG codon upstream of the original initiation site can also result in an N-terminal-extended protein. Notably, this upstream initiation site lacks a Kozak sequence, unlike the strong Kozak consensus sequence of the second ATG codon in JNK3. This situation is a hallmark of several genes with alternative translational initiation, hinting that JNK3 could also be produced in short as well as long forms, even from the same mRNA (156). The resulting JNK3 N-terminal extension has a high hydrophobic amino acid content, making it an ideal interaction mediator. Indeed, evidence is mounting that the JNK3 N-terminal extension could direct a number of unique interactions with JNK3 (157, 158). JNK3 also differs from the other two JNK paralogs in its expression patterns: JNK1 and JNK2 are expressed in almost all tissues, but JNK3 expression is largely restricted to the central nervous system. Although less studied than the other two JNKs, JNK3 has 3 distinct isoforms confirmed at a protein level, and at least 5 more (including the conserved, but uncharacterized, JNK3β isoforms using exon 6b) only described at an mRNA level (159). Further work is needed to define the full JNK3 repertoire.
Lastly, in addition to mRNAs encoding full-length JNK proteins, extra transcripts from all three JNK genes have also been recorded in the ENSEMBL database (160). Even if not targeted by nonsense-mediated decay, these shorter mRNA species would only translate to proteins with severely truncated, structurally unstable, nonfunctional kinase domains. Therefore, these transcripts may be involved in RNA-level regulation, but this awaits further exploration.
Activities and Modifications of Individual JNK Isoforms
The enzymatic activities of JNK proteins encoded by separate genes, or as different splice isoforms from each gene, can differ considerably. While the addition of an overtly disordered C-terminal extension would not be expected to cause major differences in the activities of JNKs in vitro, splicing events that swap a segment within the kinase domain, thus giving rise to the α- and β-isoforms, could clearly cause a change in enzyme activity. Thus, each splice variant can display different kinetic parameters. For example, proteins with the 6b exon have similar Michaelis constants (KM) for their common substrate ATF2, but these constants are consistently higher than those containing the 6a exon (161). The catalytic efficacy of phosphatases toward JNK can also be different for the different splice isoforms: DUSP8 preferentially inactivates exon 6a-containing JNKs, at least in vitro (162). Taken together, these observations suggest that the individual JNK isoforms will likely show different enzyme activities and activation/deactivation kinetics in vivo.
The catalytic activity of the individual JNK isoforms might also vary on different substrates, making their roles even more complex. The products of the JNK1 or JNK2 genes are markedly different in their ability to phosphorylate c-Jun. Notably, JNK1 appears to be far more active as a kinase acting on c-Jun: fibroblasts and hematopoietic cells isolated from JNK1−/− mice show lower c-Jun phosphorylation and ensuing c-Jun autoinduction after TNF-α stimulation, while JNK2−/− mice display high c-Jun phosphorylation, even in unstimulated cells, due to compensatory JNK1 hyperactivation (163, 164). The evidence from these in vivo experiments is thus in conflict with earlier in vitro results that indicated that JNK2α2 was more active than JNK1α1 toward c-Jun (165). However, isoforms of both JNK1 and JNK2 actively contribute to c-Jun phosphorylation and are at least partly redundant toward most substrates (166). In neuronal tissues with measurable JNK3 expression levels, JNK1, JNK2, and JNK3 all contribute to c-Jun phosphorylation (167). It is also likely that enzymes encoded by the JNK2 gene have a lower overall in vivo activity toward other substrates (e.g., ATF2) (163). So, the picture of different JNK enzymes fulfilling different roles is more subtle than initially anticipated.
Additional regulatory mechanisms for the different JNK isoforms have also been explored. The α-isoforms of JNK2 (i.e., JNK2α1 and JNK2α2, with exon 6b) appear to be unique in their ability to autophosphorylate and autoactivate efficiently in the absence of activator kinases, at least when grossly overexpressed in cells or assayed in vitro (168–170). Other JNK proteins also retain some residual autophosphorylation activity, but the physiological significance of such observations is not well understood. Furthermore, the long (p54) isoforms of JNK1 and JNK2 harbor a caspase cleavage site and their C termini can be processed during the onset of apoptosis, with unclear functional consequences (171). Lastly, the N- and C-terminal-lengthened JNK3α2(L) isoform of JNK3 was palmitoylated on its C terminus by the DHHC family palmitoyltransferase ZDHHC15, which regulates JNK3α2(L) subcellular localization and activity; in contrast, the almost-identical C terminus of JNK1α2 was not palmitoylated (172). Taken together, these studies reinforce the differences in the potential regulatory mechanisms for the different JNK isoforms.
Expression Patterns of JNK Genes and Splice Isoforms
The results reviewed in the preceding sections suggest an intriguing dichotomy for the α- versus β-isoforms of the JNKs. The α-isoforms of JNK2 (with exon 6b) are preferentially expressed in nonneuronal tissues (155), whereas both α- and β-isoforms of JNK1 are readily detectable in immune cells (173). This is consistent with the sequence of their primary mRNA transcripts, with JNK2 harboring five Nova2-binding sites in the polypyrimidine tract before its 6b exon (allowing tissue-selective splicing), whereas JNK1 has only one such site (152). The available evidence, including the transcripts preferentially detected and the multiple splicing regulator-binding sequences, suggest that the JNK3 α-isoforms are dominant over the more poorly characterized JNK3 β-isoforms (see Table S2 in the supplemental material for the predicted sequence of the JNK3 β-isoforms using exon 6b).
The selection of the last 3′ splicing site also differs considerably for the JNK genes. For JNK1, there is a clear preference toward producing mRNA encoding the shorter (p46) isoforms in both immune cells and fibroblasts (173, 174). Conversely, JNK2 tends to preferentially express the long (p54) isoform in the same tissues. Similar patterns are seen in many other cell lineages, like the rat adrenal medulla pheochromocytoma cell line PC12 (175). In experiments where JNK1 and JNK2 were coexpressed, a preference toward phosphorylation of the shorter p46 isoform was noted across several different tissues (174, 176). However, this preferential phosphorylation may not reflect a difference between JNKs with different C-terminal extensions (i.e., the long and short isoforms) but may be related to other differences between JNK1 and JNK2 (177).
Comparison of JNK1 and JNK2 Functions In Vivo
The creation and phenotyping of different JNK knockout mice have revealed the actions of the individual JNKs. JNK1 and JNK2 (but not JNK3) are likely to fulfill essential but largely overlapping roles, as JNK1/JNK2 double knockouts are embryonic lethal owing to a defect in neural tube closure, but single JNK gene knockout, JNK1/JNK3 double knockout, or JNK2/JNK3 double knockout mice were viable (178, 179). However, important differences between JNK1 and JNK2 in terms of their contributions to cellular regulation could be inferred from the differences in the phenotypes of the JNK1−/− and JNK2−/− mice. Specifically, the phenotype of JNK1−/− mice was more marked, with abnormalities in brain development (abnormal cortical neuronal migration and anterior commissure degeneration) as well as disturbed metabolic regulation (including resistance to obesity and obesity-induced metabolic changes) (180, 181). Conversely, JNK2−/− mice showed a less remarkable phenotype, with epidermal hyperplasia and mild immune abnormalities (182). Furthermore, different JNK gene knockout mice showed that JNK1 and JNK2 regulate fibroblasts, macrophages, and T cells differently (163, 183, 184) and also influence skin wound repair differently (185). Neurogenesis in vitro also primarily requires JNK1, but not JNK2 or JNK3 (186). Maintenance of metabolic homeostasis required both JNK1 and JNK2, but JNK1 played more important roles (187). These studies reinforce the different roles played by the different JNKs.
The compensatory JNK1 hyperactivation and thus paradoxically higher c-Jun phosphorylation observed in JNK2−/− mice may help to explain some of the different observations for JNK1−/− and JNK2−/− mice, such as altered cell cycle regulation (164). That the extent of JNK2 phosphorylation is consistently lower than JNK1 phosphorylation under normal conditions may be due to differences in their activation loops (177). Nevertheless, there are several tissues and experimental models where the actions of JNK1 and JNK2 are cooperative or synergistic. Examples include the development of skin keratinocytes (188), spiral ganglion neurons (189), or the differentiation of pluripotent embryonic cells, where both JNK1 and JNK2 were required for establishing mesodermal and epithelial lineages (190). UV- or arsenite-induced apoptosis of fibroblasts required both JNK1 and JNK2 (191, 192). In addition, lipolysis in adipocytes was also regulated by JNK1 and JNK2 (193). The results of these studies suggest that the different JNKs can act in a concerted manner in some cell and stimulus contexts.
Greater attention has also been directed toward the study of JNK1−/− mice in the area of tumor biology. JNK1−/− mice spontaneously developed intestinal tumors, consistent with the roles of JNKs as negative regulators of cell proliferation and their proapoptotic actions in many cell types (194). In several carcinogenesis models, such as UV- or phorbol ester-induced skin tumorigenesis, JNK1−/− mice were also more prone to develop malignant tumors (195). However, JNK1 is not a clear tumor suppressor: in cases such as nitrosamine-induced gastric and hepatic tumorigenesis, JNK1−/− mice were less responsive, suggesting that JNK1 may be required for tumor development (196). Also, the combined actions of JNK1 and JNK2 have been implicated in the proliferation of glioma cells (197). The underlying causes for these different outcomes remain unresolved, but some researchers have proposed a regeneration-based model as the basis of enhanced tumorigenesis, especially in the liver (198). As JNK1−/− mice show less apoptosis leading to the same cytotoxic insult, they consequently require lower hepatocyte regeneration and thus fewer cell divisions. This reduced proliferative requirement, relative to the higher numbers of proliferating cells exposed to genetic damage in wild-type animals, may help to protect against genotoxic stress. However, it also remains possible that JNK1 and/or JNK2 enhances the survival and metastatic ability of certain malignant cell types in a tissue-specific and/or tumor-specific manner.
JNK RECOGNITION OF ITS PARTNER REGULATORS AND SUBSTRATES
Docking Motifs (D-Motifs) Are a Dominant Molecular Solution in JNK Recruitment
The MAPKs, including the JNKs, are classified as Pro-directed Ser/Thr kinases: the Ser/Thr residues targeted for phosphorylation by these kinases usually are followed immediately by a Pro residue. Although such Ser/Thr-Pro (i.e., Ser-Pro/Thr-Pro [SP/TP]) sites are extremely common in all proteins, only a fraction will likely be bona fide MAPK substrates. This specificity is attributed to docking motifs within targeted substrate proteins that bind to dedicated sites on the kinase domains of MAPKs (199). The most commonly used kinase domain docking site consists of the negatively charged common docking (CD) region and the hydrophobic groove, commonly referred to as the docking groove, which binds to the so-called substrate D-motifs (a name derived from the D region of Elk1 and the δ segment of c-Jun) (Fig. 4). MAPK substrate D motifs are short linear motifs of ∼9 to 18 amino acids found in the disordered segments of proteins, often (but not always) N-terminal from the targeted phosphorylation sites (200). As the catalytic site and the D-motif docking groove are spatially separated on the kinase domain, the phospho-target motif must also be separated by a minimal number of amino acids (>9) from the D-motif for efficient coupling (201). However, other MAPK partners, not only substrates, utilize D-motifs as a molecular solution to recruit MAPKs. Thus, the same binding site is also used by MAP2Ks to access their MAPK partners, many phosphatases responsible for the inactivation of MAPKs, and a wide variety of pathway regulators/scaffolds. These interacting partners thus all compete for the same interaction “hot spot” on the MAPKs. Although the D-motifs are structurally variable, the structural basis of MAPK-partner protein specificity is well understood. It is now becoming increasingly clear that the motifs directing association with JNKs are often specific and distinct from docking motifs targeting other MAPKs. For example, the set of p38 substrates and partners substantially overlaps with those of the mitogen-activated ERK1/2, but not with JNKs (200). This phenomenon is also likely to have network-level implications for the stress-activated MAPKs, because JNKs and p38 kinases could control different sets of stress-activated proteins by phosphorylation.
FIG 4.

Structural features and substrate recognition by JNKs. (A) JNK proteins are comprised of a single protein kinase domain (structure on the left). The docking site, consisting of the negatively charged CD region (blue) and the hydrophobic docking groove (beige), plays an important role in partner binding and recruitment of substrates (red). The phosphotransfer reaction from ATP (yellow) takes place at the opposite side of the kinase, where the catalytic residues (pink) are located. Apart from the docking site, the CMGC insert (orange) is also unique to the MAPKs and a few related protein kinases. This also harbors a docking surface called the FxFP site. Although known to be functional in other MAPKs, no FxFP site-dependent substrates have been identified for JNK. The figure is based on the complex of human JNK1 with a docking motif from NFAT4 (PDB ID 2XRW). The peptide chain modeled at the catalytic site is based on the DYRK1A-substrate complex (PDB ID 2WO6; DYRKs are closely related to MAPKs in structure as well as in substrate preference). The rest of the substrate, which is not associated with JNK, is indicated with a dotted red line. Together, the CD region and the docking groove form the major docking site (D-site) of JNK proteins and play a key role in substrate recruitment (shown on the right). The best-characterized substrate proteins either contain a linear motif capable of interacting with the D-site directly (direct substrates [top]) or interact with a third protein having such a motif through heterologous interactions (indirect substrates [bottom]). (B) JNKs bind most of their known partners by engaging a dedicated recruitment site (D-site) that is distinct from their catalytic site. The same docking site is used to interact with activator kinases (MAP2Ks) responsible for the phosphorylation of the JNK activation loop, with phosphatases that dephosphorylate the same residues, as well as with other proteins involved in the regulation of pathway through intracellular compartmentalization and multiprotein complex formation (i.e., scaffolds). Many substrates also utilize the same docking site to provide access to the kinase. Therefore, most partners of JNKs directly compete with each other for binding and access to the catalytic site. Abbreviations: D, docking motif; K, kinesin-binding motif of JIP1.
In recent years, many novel JNK partners (mostly substrates) have been identified based on the presence of D-motifs, and their number is expected to grow (69). JNK-associating D-motifs can be separated into at least two, structurally different varieties: either resembling the D-motif found in the JNK pathway regulator JIP1 or resembling the motif described for the NFAT4 transcription factor (80, 200) (Fig. 5; see also Table 1 for a summary of D-motif types in known JNK substrates). These two motifs interact with the same region of JNKs but are not equivalent structurally due to the differences in the relative positioning of their hydrophobic and charged residues. While most examples of these JIP1- or NFAT4-type docking motifs have been described in JNK1-interacting proteins, the docking surfaces of JNK1, JNK2, and JNK3 are near identical. That the interaction interfaces for the different JNKs will be very similar is supported by the structures of JNK3-JIP1 and JNK3-Sab complexes, which are nearly identical to the JNK1-JIP1 complex (202). The same D-motifs for JNK1 and JNK3 (at least for the short splice isoforms) also are likely to function equally well (158). Although the docking groove lies next to the “hinge” connecting the N and C lobes of the kinase domain and D-motif binding may exert some allosteric changes on the kinase domain (203), it is likely that the JNK docking groove is available for D-motif binding in all activation states of the kinase domain. As a consequence, it is expected that many of the same binding partners interact with JNKs in both active and inactive conformations.
FIG 5.

The two main classes of D-motifs that interact with JNKs. Most of the known JNK-interacting D-motifs (located in diverse partners) belong to one of two distinct structural types, corresponding either to the JIP1 or to the NFAT4 consensus motifs (top). These two structural classes can be described with related, though different, consensus motifs (middle). Despite the differences, all these motifs bind to the same docking site. A large number of known JNK interactors, together with their evolutionarily closely related paralogs, harbor docking motifs showing sequence similarity to either the docking motif of JIP1 or to the docking motif of NFAT4 (bottom panels). Many of these docking motifs were characterized in in vitro experiments only, a few motifs do not satisfy the complete consensus, and some proteins (e.g., BMPR2, ATF2, ATF7, MKK7) contain more than one motif of the same or different type. (Structural panels were made by using crystal structures of JNK-peptide complexes: PDB IDs 4H39, 4H3B, and 2XRW for JNK3-pepJip1, JNK3-pepSab, and JNK1-pepNFAT4, respectively.)
TABLE 1.
Summary of JNK substratesa
| Protein functional group and nameb | Function | No. of JNK target phospho-sitesc | D-motif typec | Suppl. table(s) | Reference(s) |
|---|---|---|---|---|---|
| 1. Transcription, DNA and chromatin regulation | |||||
| ATF2# | Transcription factor | 2 | B | S3 | 164, 230 |
| Beta-catenin | Transcriptional coactivator | 2 (+2) | D | S5, S6 | 270, 272, 471 |
| c-Jun# | Transcription factor | 4 | B | S3 | 224, 229 |
| Cdt1 | DNA replication factor | 3 | D | S5 | 252 |
| Elk1# | Transcription factor | 2 | A | S3 | 472, 473 |
| Elk3 | Transcription factor | 4 | C | S4 | 343 |
| Elk4 | Transcription factor | 6 | C | S4 | 474 |
| FOXO3 | Transcription factor | 1 | D | S5 | 236 |
| FOXO4 | Transcription factor | 2 | D | S5 | 235 |
| Gli1 | Transcription factor | 1 | C | S4 | 69 |
| Gli3 | Transcription factor | 1 | C | S4 | 69 |
| Hes1† | Transcription factor | 1 | D | S5 | 232 |
| HSF1‡ | Transcription factor | 1 | B | S3 | 316 |
| JDP2# | Transcription factor | 1 | D | S5 | 475 |
| JunD# | Transcription factor | 3 | B | S3 | 476 |
| Myc# | Transcription factor | 2 | D | S5 | 231 |
| NFAT1 (NFATc2) | Transcription factor | 2 | D | S5 | 309 |
| NFAT2 (NFATc1)‡ | Transcription factor | 1 | D | S5 | 308 |
| NFAT4† | Transcription factor | 2 | B | S3 | 307 |
| NF-E2 | Transcription factor | 1 (+1) | D | S5, S6 | 471 |
| Nrl# | Transcription factor | 1 | D | S5 | 347 |
| p53 | Transcription factor | 1 | D | S5 | 238 |
| p73† | Transcription factor | 6 | D | S5 | 239 |
| RRN3 (TIF1A) | Transcription factor | 1 | D | S5 | 477 |
| Sirtuin | Histone deacetylase | 1 (+1) | D | S5, S6 | 253, 254 |
| Smad2# | Transcription factor | 2 | D | S5 | 478, 479 |
| Smad3# | Transcription factor | 2 | D | S5 | 478, 479 |
| Sp1† | Transcription factor | 2 | D | S5 | 234 |
| STAT1 | Transcription factor | 1 | D | S5 | 480 |
| STAT3 | Transcription factor | 1 | D | S5 | 481 |
| Twist1† | Transcription factor | 1 | D | S5 | 233 |
| YAP1† | Transcriptional coactivator | 4 | D | S5 | 246 |
| POU6F1 (TCF-β1) | Transcription factor | (2) | D | S6 | 482 |
| Histone H3.1 | Histone (DNA packaging) | (1) | D | S6 | 483 |
| Runx2 | Transcription factor | (1) | D | S6 | 484 |
| 2. mRNA splicing and translation | |||||
| DCP1a | mRNA decapping enzyme | 1 | D | S5 | 255 |
| eEF1α2 | Elongation factor | 1 (+1) | D | S5, S6 | 259 |
| hnRNPK† | RNA-binding protein | 3 | C | S4 | 256 |
| SP45 | RNA-splicing factor | 1 | D | S5 | 258 |
| AIMP1 | tRNA synthase regulator | (1) | D | S6 | 485 |
| 3. Receptors and sensors | |||||
| Androgen receptor† | Nuclear hormone receptor | 1 | D | S5 | 240 |
| Glucocorticoid receptor† | Nuclear hormone receptor | 1 | D | S5 | 241 |
| GluR2† | Receptor ion channel | 1 | D | S5 | 288 |
| GluR4† | Receptor ion channel | 1 | D | S5 | 288 |
| LRP6 | Wnt coreceptor | 1 | D | S5 | 486 |
| LSR (angulin) | Lipoprotein receptor | 1 | D | S5 | 487 |
| Nur77# | Nuclear hormone receptor | 1 | D | S5 | 488 |
| PPAR㇠| Nuclear hormone receptor | 1 | D | S5 | 242 |
| RARα# | Nuclear hormone receptor | 3 | D | S5 | 243 |
| RXRαd* | Nuclear hormone receptor | 4 | D | S5 | 244, 245 |
| SREBP1 | Lipid sensor | 2 | D | S5 | 489 |
| 4. Protein phosphorylation and dephosphorylation | |||||
| Akt1 | Protein kinase | 1 | D | S5 | 490 |
| DLK | Protein kinase | 4 (+1) | D | S5, S6 | 44 |
| DUSP8 (M3/6) | Protein phosphatase | 3 | D | S5 | 491 |
| S6K (p70RSK)# | Protein kinase | 2 | D | S5 | 333 |
| PPM1J# | Protein phosphatase | 3 | A | S3 | 69, 71 |
| Cdc25B | Protein phosphatase | 2 | D | S5 | 492, 493 |
| Cdc25C | Protein phosphatase | 1 | D | S5 | 494, 495 |
| Raptor | Protein kinase regulator | 3 | D | S5 | 496 |
| 14-3-3-ζ* | Phospho-protein adaptor | 1 | D | S5 | 285, 359 |
| 14-3-3-σ* | Phospho-protein adaptor | 1 | D | S5 | 285 |
| MST1 | Protein kinase | (1) | D | S6 | 497 |
| RSK | Protein kinase | (1) | D | S6 | 498 |
| 5. Diverse scaffolds and adaptors | |||||
| DLG4 (PSD-95)† | Synaptic scaffold protein | 1 | D | S5 | 287 |
| eIF4ET | Nucleopore shuttling protein | 6 | D | S5 | 499 |
| IRS1† | Receptor-associated scaffold | 1 | A | S3 | 273 |
| IRS2† | Receptor-associated scaffold | 1 | A | S3 | 274 |
| Paxillin# | Cell adhesion receptor and cytoskeletal protein | 1 | D | S5 | 268, 269 |
| Shc1 | Receptor-associated scaffold | 1 | D | S5 | 500 |
| LIMD1 | Multipurpose adaptor | (2) | D | S6 | 501 |
| 6. Other signaling systems | |||||
| cPLA2 | Phospholipase | 1 | D | S5 | 502 |
| DAT1 (SLC6A3) | Dopamine transporter | 1 | D | S5 | 503 |
| eNOS* | Nitric oxide synthase | 1 | D | S5 | 358 |
| ITCH‡ | E3 ubiquitin ligase | 2 | D | S5 | 319, 504 |
| Rad18 | E3 ubiquitin ligase | 1 | D | S5 | 250 |
| Aquaporin-2 | Water channel | (1) | D | S6 | 505 |
| CDKN1B (p27Kip1) | Cyclin/Cdk inhibitor | (1) | D | S6 | 506 |
| (SMPD2) | Sphingomyelin phosphodiesterase | (1) | D | S6 | 507 |
| 7. Cytoskeletal proteins | |||||
| Cytokeratin-8‡ | Cytoskeletal protein | 1 | D | S5 | 310 |
| DCX† | Microtubule-associated protein | 3 | B | S3 | 260 – 262 |
| MAP2† | Microtubule-associated protein | 3 | D | S5 | 264 |
| MARCKSL1† | Actin-binding protein | 3 | D | S5 | 266 |
| SMTL2# | Actin-binding protein | 4 | A | S3 | 267 |
| Stathmin 1 | Microtubule-associated protein | 2 | C | S4 | 508 |
| Stathmin 2 (SCG10)# | Microtubule-associated protein | 2 | C | S4 | 509 |
| Taue# | Microtubule-associated protein | 3 | D | S5 | 265 |
| WDR62† | Microtubule-associated protein | 3 | B | S3 | 213 |
| KIF5C (kinesin) | Motor protein | 1 | D | S6 | 510 |
| Moesin | Cytoskeletal anchor protein | (1) | D | S6 | 511 |
| Stathmin 3 (SCLIP) | Microtubule-associated protein | (1) | D | S6 | 508 |
| 8. Vesicular transport | |||||
| APLP2 | Vesicular transport receptor | 1 | D | S5 | 217, 512 |
| β-APP# | Vesicular transport receptor | 1 | D | S5 | 217 |
| JIP1† | Vesicular transport adaptor | 7 | A | S3 | 104 |
| JIP3† | Vesicular transport adaptor | 3 | A | S3 | 275 |
| Synaptotagmin 4 | Vesicle fusion protein | 1 | D | S5 | 513 |
| 9. Mitochondrial control of apoptosis | |||||
| Bad‡ | BH3 only protein | 1 | D | S5 | 303 |
| Bax | Mitochondrial pore regulator | 1 | D | S5 | 514 |
| Bcl2‡ | Mitochondrial pore regulator | 3 | D | S5 | 305 |
| Bim‡ | BH3-only protein | 3 | D | S5 | 302 |
| Mcl1 | Mitochondrial pore regulator | 2 | A | S3 | 281 |
| Noxa (PMAIP) | BH3-only protein | 1 | D | S5 | 515 |
| Bid | BH3 motif protein | (1) | D | S6 | 516 |
| SMAC (Diablo) | Ubiquitin ligase inhibitor | (1) | D | S6 | 517 |
| 10. Unknown | |||||
| Sab (SH3BP5) | Mitochondrial, possible scaffold | 1 | A | S3 | 378, 518 |
The table presents JNK substrate proteins (104 in total; 89 well-validated JNK substrates [indicated in regular font] and 15 less-well-validated JNK substrates [indicated in italics]), listed alphabetically within each of the 10 major functional groupings (see Fig. S1 in the supplemental material for representations of these groupings for the 89 comprehensively and well-validated proteins). In addition to protein function, the number of characterized JNK target phosphorylation sites (again with well-validated information indicated in regular font and less-well-validated values indicated in italics) and the D-motif type are presented (this information represents a summary of the more-detailed information presented in Tables S3 to S6 in the supplemental material). Of these substrates, features of the interaction site with JNK have been defined for 23 protein substrates: 9 with a JIP1-like site (class A), 7 with an NFAT-like site (class B), and 7 with a weak, incomplete, or atypical binding sequence (class C). Class D sites are uncharacterized. The ratios of known JNK target sites per protein are 1.98 for all well-validated JNK substrates (i.e., 176 sites in 89 proteins), 2.70 for well-validated protein substrates possessing a known JNK-binding D-motif (i.e., 62 sites in 23 proteins), and only 1.73 for the well-validated protein substrates devoid of known D-motifs (i.e., 114 sites in 66 proteins).
Commonly used protein names and/or their commonly used acronyms are listed; when a protein is well known by >1 name, the alternative is presented in parentheses. Within a framework of understanding the immediate molecular consequences of JNK-mediated phosphorylation of these substrates, the following symbols indicate the impact of the indicated protein: ‡, negative phospho-switch (i.e., phosphorylation to negatively regulate protein-protein interactions or intramolecular interactions); #, positive phospho-switch (i.e., phosphorylation to positively regulate protein-protein interactions or intramolecular interactions); *, allosteric phospho-switch; †, a less-understood impact, including complex changes such as impacts on intracellular localization, as discussed in the text.
The total number of validated JNK target sites is indicated (additional possible sites are indicated in italics and parentheses). See the indicated table(s) in the supplemental material for further information (and also on D-motif classification, where indicated).
In addition, RXRα can be subjected to more complex regulation (245).
In addition to D-motifs located in flexible protein regions of partner proteins, other domains or linear sequences may direct MAPK-partner interactions. Although folded domains have also been described to interact with the MAPK docking groove, for example, in the p38α-MKP5 complex (204), it is still an open question whether JNK has similar structured domain partners. The elucidation of such complexes will require substantial structural efforts directed toward defining modes of interactions of the JNKs with partners that are apparently devoid of the conventional D-motif consensus sequences. In addition, substrates can also be recruited by most MAPKs via their FxFP site that is located in between the phosphorylated activation loop and the CMGC insert. This site was named based on the optimal consensus of linear motifs targeting it in the case of ERK2; the FxFP-site of JNK1 likely prefers substantially different motifs, as peptide arrays testing the original Phe-x-Phe-Pro motif failed to identify any strong JNK binders (203). However, other assays using artificial substrates designed to include docking domain peptides suggested that the FxFP site of JNKs is still functional (201); recent structural studies indeed suggest that this site mediates JNK1 interaction with the dual-specificity phosphatase MKP7 (81). Despite these alternative modes of interactions with the MAPKs, most JNK interactors use typical D-motifs to partner with JNKs (80).
Multiprotein Complexes and the Prevalence of JNK Substrates in Trans
Many of the reported JNK substrates lack the typical JNK-binding motifs or domains described in the preceding section (Table 1), but many of these substrates do form complexes either with each other or with proteins that do possess JNK-binding motifs. For example, the cooperation between different AP-1 proteins in JNK recruitment is well-known (205); AP-1 transcription factors and certain nuclear receptors commonly occupy adjacent positions in promoters (206, 207). Many leucine zipper transcription factors, including Jun and ATF2, can heterodimerize with each other in a combinatorial manner (208), whereas other JNK substrates, such as YAP1 and p73, are also direct binding partners (209). In these examples, the recruitment of JNK into larger transcription factor complexes enables JNK-mediated phosphorylation of multiple transcription factor protein substrates.
Inside the nucleus, the restriction of JNK activity to certain chromosomal sites opens up the possibility for the same protein to be regulated by one MAPK on one promoter but not on another. Such JNK-regulated complexes may also have dedicated protein components: the histone-like pioneer transcription factor NF-Y is surprisingly a better predictor of JNK presence on the chromatin than canonical AP-1 complexes themselves (210). Again, this may not require NF-Y (which lacks any obvious JNK-binding motifs) to recruit JNK directly: it may be an obligatory protein component of JNK-regulated, phospho-AP-1-containing promoter complexes (211). That the histones themselves can also be phosphorylated by JNK around this assembly may allow further enhancement of gene expression (210). This nano-environment of a chromatin-bound multiprotein complex, with its own combination of JNK-regulated proteins, remains an intense focus of research (reviewed in reference 212).
Compartmentalization effects may also apply to phosphorylation events outside the nucleus. Cytoskeleton-associated JNK partner proteins or microtubule-binding proteins targeted by JNK contain diverse structural features, such as double WD40 domains (in WDR62 and MABP1), tandem doublecortin domains (in DCX and DCLK), or MAP2 repeats (in Tau and MAP2), which may allow them to be recruited onto the surface of the same microtubule, possibly even engaging adjacent sites (213–216). However, only a fraction of these proteins carry dedicated JNK-binding motifs (e.g., WDR62, MABP1), so that the rest (e.g., MAP2, Tau) may rely on their spatial proximity to these partners to be phosphorylated by JNK. Similarly, the MINT2 (APBA2) protein, distantly similar to JIP1 and JIP2, enables JNK-mediated phosphorylation of β-APP with a possible impact on the regulation of amyloid β production (217).
In summary, multiprotein complexes only need a single member to recruit JNK via a typical JNK docking D-motif to catalyze the phosphorylation of accessible SP/TP sites on other members of the complex. Thus, docking motifs are instrumental to recruitment of JNKs into diverse subcellular compartments and macromolecular complexes. As a consequence, many substrates can be indirect (i.e., lacking the kinase recruitment motif characteristic of direct substrates), using heterologous interactions in encounters with activated JNKs. The prevalence of indirectly recruited substrates may also conveniently explain certain peculiar protein architectures. The transmembrane receptor BMPR2 presents an array of two JIP1-type docking motifs but contains relatively few conserved JNK target sites, suggesting that other BMPR2-interacting proteins may be the targets of JNK-mediated phosphorylation (218).
THE MANY SUBSTRATES OF JNK: 20 YEARS AND STILL COUNTING
Substrate Specificity of JNKs
Since the description of the JNKs as the kinases responsible for c-Jun phosphorylation, many additional JNK substrates have been described (Table 1). The consensus constructed from the well-validated JNK substrate sites curated from the available published literature, which deals mostly with JNK1-mediated phosphorylation (176 sites in 89 proteins) (Table 1; see also Tables S3 to S5 in the supplemental material for additional detailed information), reinforces the preference by JNKs to target Ser/Thr residues followed by a Pro, i.e., according to the standard nomenclature in which the phosphorylated site is the P0 position, JNK has a requirement for Pro at the P + 1 position and so prefers SP/TP motifs (see Fig. S1A in the supplemental material). The overwhelming similarity of the JNK catalytic sites suggests almost identical substrate preferences for all 3 JNKs, and this is reinforced by an analysis of the experimentally determined phosphorylation sites (see Fig. S1A in the supplemental material). The substrate site specificity of JNK2 as directly measured with oriented peptide libraries again indicates that there is practically no constraint other than a Pro following the Ser/Thr targeted for phosphorylation (203). Furthermore, as JNK2 does not display any preference for Pro in the P-2 position (i.e., a Pro positioned 2 residues immediately N terminal to the target Ser/Thr residue), its selectivity is lower than that of ERK2 or p38α, which prefer a P-X-[S/T]-P motif (203). Furthermore, like other MAPKs, JNKs may phosphorylate target sites of SA, TA, SS, TS, ST, or TT (where the first residue, underlined, is the target of phosphorylation), yet at a much lower rate than the SP/TP sites; SG or TG sites are still allowed, but amino acids with large side chains are strongly disfavored or completely forbidden at the P + 1 position (203).
Challenges for JNK Substrate Identification and Validation
More than 100 proteins have now been reported as JNK substrates (Table 1; see also Tables S3 to S6 in the supplemental material for more detailed summaries). Although JNKs have been studied intensively for more than 2 decades, the identification of their substrates is still not a simple task. Most difficulties stem from the low stringency of JNKs toward their phosphorylation target sites. Because SP/TP consensus sites are common in all proteins, in vitro kinase assays performed with purified proteins are usually not informative: with supraphysiological amounts of activated JNKs, many such sites can be efficiently phosphorylated in vitro, regardless of the physiological situation within cells where MAPK-dependent phosphorylation is further constrained by compartmentalization and/or dedicated recruitment effects. If used in overtly high quantities, JNKs may phosphorylate several suboptimal (non-SP/TP) sites.
In addition, many SP/TP sites can be targeted by MAPKs or by a plethora of other related kinases, including GSK3 and CDKs, as well as DYRKs, HIPKs, IKKs, or even mTOR. Thus, a single site may be potentially modified by multiple different kinases, activated by different signaling events, adding a further layer of complexity to physiological regulation. Selective inhibitors can be helpful in this case to differentiate between kinases with similar substrate site preferences. However, even inhibitors described as “specific” for a given kinase should be used with caution because slightly higher concentrations may still have the potential to inhibit all structurally related kinases. In addition, “specific” inhibitors may act against other less-studied protein kinases equally well. For example, the commonly used “JNK1/2-specific” SP600125 (219) showed comparable inhibitory activities against SGK1, PDK1, Aurora B and C, CK1δ, MELK, DYRK2, and DYRK3 (Kinase Inhibitor Database; http://www.kinase-screen.mrc.ac.uk/kinase-inhibitors) (220). Peptide or peptido-mimetic inhibitors based on the initial domain-mapping experiments for JIP1 (221), such as the cell-penetrating TI-JIP (interfering with JNK activation by MAP2Ks as well as its targeting to substrates [222]) or its retro-inverso analogs (223), have less favorable pharmacokinetic properties but are expected to be more specific toward JNKs. Thus, only a combination of different approaches will achieve a desired selectivity in targeting JNKs to facilitate a better understanding of the relationships between JNKs and possible physiologically relevant substrates.
A striking observation for many of JNK substrates is that they possess more than one phosphorylation site that can be targeted for phosphorylation by JNKs (Table 1 provides a summary of the data regarding an average of 1.98 sites/substrate protein, representing 176 sites in 89 well-validated protein substrates). These multiple phosphorylation sites may reflect several possible regulatory mechanisms under the control of JNK activity. It may be that only 1 or 2 of these sites will be critical for the regulation of a given physiological function, but the same protein can also have multiple regulatory sites, each performing different, and sometimes even opposing, roles (224). Furthermore, the sites may be phosphorylated in a specific order; this processive phosphorylation (in which phosphorylation events at distinct sites occur without the dissociation of the enzyme from its substrate) is a common situation for enzymes such as the Src-family tyrosine kinases, which also rely on additional substrate recruitment sites, such as SH2 domains (225–227).
Studies using engineered mutants in the phospho-sites of the substrates under investigation (i.e., [S/T] → A or [S/T] → E mutants) can be invaluable in the validation of phosphorylation or give hints on their physiological effects. However, the mapping of critical regulatory sites can be complicated, because the phenotypes of multisite [S/T] → A or [S/T] → E mutants are typically not informative enough to decipher molecular-level regulatory switches, necessitating single-site scanning. If the site in question is subject to complex, multistep regulation (such as phosphorylation-dependent ubiquitinylation or SUMOylation), then the phenotype will also depend on the experimental conditions used. For example, paradoxical stabilization effects were described when several phosphodegron systems were manipulated, potentially due to substrate-specific competition between enzymes (228).
Finally, a bona fide phosphorylation site cannot be confirmed only by obtaining a phenotype after mutating an SP/TP site; the region could participate in phosphorylation-independent interactions or may be a structurally important feature in an otherwise-kinase-inaccessible folded domain. Thus, the array of potential JNK substrates defined in the current published literature should be treated with some caution, especially in the absence of multiple independent lines of evidence for true enzyme-substrate relationships. With this caveat in mind, we present here an overview of the major classes of proteins targeted by JNKs before we discuss in greater detail the established roles for JNKs in mediating a range of physiological and pathological events.
Major Classes of Proteins Targeted by JNKs
The archetypical substrates of JNK are a diverse assortment of transcription factors (summarized in Table 1; see also Tables S3 to S6 in the supplemental material for details regarding the JNK-mediated phosphorylation and docking site sequences). These JNK substrates encompass members of many different families, including those of the bZIP (Jun, ATF2, Myc [164, 224, 229–231]), bHLH (Hes1, Twist1 [232, 233]), Zinc finger (Sp1 [234]), Forkhead (FOXO4, FOXO3 [235–237]), and RUNT (p53, p73 [238, 239]) families, as well as proteins of the nuclear receptor family (androgen receptor, glucocorticoid receptor, peroxisome proliferator-activated receptors, RARα, and RXRα [240–245]). Coactivators and corepressors lacking a direct DNA-binding capacity (such as YAP1 [246]) can also be targeted by JNK.
By acting on these nuclear transcription factor substrates (Table 1 provides a summary of the complete list, including 30 well-described transcription factor or transcriptional activator protein substrates and 2 with less-strong supporting evidence, in addition to 5 nuclear hormone receptors), JNKs can effectively regulate transcription of many target genes. These are required not only for adaptation to stress stimuli or apoptosis, but also for embryonic development. However, at a molecular level, most transcription factors (even those that are closely related) can be regulated by different mechanisms. Notably, a number of their target genes encode proteins that are also JNK substrates themselves (e.g., c-Jun, p73, YAP1), allowing multilevel regulation of the same protein by JNK via both transcriptional and posttranslational mechanisms (247, 248). With >1,000 annotated transcription factors in the human genome (249), this list of JNK targets is likely still far from complete. Indeed, JNK may also phosphorylate many more transcription-associated targets, including the PPARγ receptor coactivator PGC1α, the transcription factors TSHZ3 and HSF4, and the nuclear receptor signaling regulator KANK2 (80). Taken together, these observations reinforce a prominent role for nuclear JNKs as transcriptional regulators (see Fig. S1B and C in the supplemental material).
Despite the importance of transcription factors as JNK substrates, not all DNA-binding JNK substrates are transcription factors (Table 1). Indeed, JNK can also phosphorylate proteins that participate in DNA repair (RAD18), DNA replication (Cdt1), or DNA packaging (via regulation of the histone deacetylase sirtuin) (250–254). Furthermore, JNK can also influence gene expression through phosphorylating proteins involved in mRNA splicing or other components of the translational machinery that impact on the use of the gene transcripts and their ultimate production of products (including DCP1a, heterogeneous nuclear ribonucleoprotein-K [hnRNP-K], SP45, and the α-subunit of eukaryotic elongation factor 1A2 [eEF1α2]) (255–259).
Although most research groups have followed a “MAPK-dependent gene expression” paradigm by focusing on JNK nuclear substrates, further evaluation of nonnuclear proteins may reveal additional JNK substrates, a notion supported by the prevalence of D-motifs in diverse cytoskeleton-regulating proteins (80). Indeed, numerous JNK substrates are found outside the nucleus (see Fig. S1B in the supplemental material for an overall summary of intracellular localizations of the 89 well-validated JNK substrates). For example, cytoskeletal and intracellular transport proteins are also important JNK substrates (Table 1; see also Fig. S1C). These cytoskeletal JNK substrate proteins include those associating with microtubules (DCX [260–262], MAP1B [180, 263], MAP2 [264], Tau [265], and WDR62 [213]), the actin cytoskeleton (MARCKSL1 [266] and SMTL2 [267]), or focal adhesions (paxillin [268, 269] and β-catenin [270–272]). In addition, JNK-mediated phosphorylation of additional proteins may also direct vesicular transport (through phosphorylation of JIP1 and β-APP [104, 217]), as well as the exocytosis of specialized, Glut4-containing vesicles (through insulin-stimulated phosphorylation of IRS1 and IRS2 [273, 274]). Also, it can potentially control Rho-family small G-proteins involved in this process (through JIP3 [275]). Since JNK is implicated in the control of cell migration, especially in the developing central nervous system (276), the purpose of JNK-dependent regulation of diverse cytoskeletal proteins can be understood in this context. Additionally, JNK is an important regulator of cell-cell adhesion. For example, JNK activation can lead to changes in epithelial morphology, adherens, and tight junctions (277, 278). Recent studies have implicated many more JNK target proteins in these processes, including Rho-family small G-protein activators (DOCK5, DOCK7), Rab-family G-protein partners (MADD, RUSC2), vesicular transport adaptors (APBA2/MINT2), actin filament assembly proteins (Formin1, FHOD3), microtubule-associated proteins (CCSER1), and molecular motors (Myosin-9B) (80, 158). These additional potential targets therefore await further testing and validation.
JNKs are also important for the promotion of apoptosis in specific cellular contexts through its phosphorylation of apoptosis-regulating proteins of the Bcl2 family of mitochondrial pore-forming proteins (279–283) (Table 1). Antiapoptotic proteins like Mcl1 and Bcl2 are targeted by JNK similarly to their proapoptotic partners (such as Bad or Bim), as it controls them by diverse molecular mechanisms (284). JNK activation can also influence other signaling pathways (Table 1). Insulin receptor signaling (where JNK-mediated phosphorylation of IRS1/2 inhibits the action of insulin) is one prominent example (273), whereas JNK may also more generally antagonize Akt/PKB-dependent signaling (285). These cross-regulation mechanisms are likely most critical during embryonic development due to JNK-dependent control of developmental signaling pathways. JNKs, activated as part of the noncanonical Wnt and TGF-β/BMP pathways, can provide feedback within these pathways; for example, the bone morphogenic protein receptor BMPR2 is a partner (and likely also a substrate) of JNK (218). Similarly, components of the canonical Wnt pathways, including β-catenin itself, can also become substrates of JNK (270). JNK can also cross-regulate the Hippo/LATS pathway at certain points, such as its phosphorylation of the coactivator YAP1 (246, 286). Furthermore, JNK also regulates the synaptic functions and learning processes of adult neurons through its substrates PSD-95 and GluR2/GluR4, respectively (287, 288). Recent research into the interactome of JNK has uncovered many further potential targets of JNK pathways, including components of cyclic AMP (cAMP)-dependent signaling (e.g., PDE4B, AKAP6), more members of the canonical Wnt pathway (APC2), and Akt/PKB signaling (INPP5F/SAC2), as well as the TNF-α receptor complex (RIPK2) and the JNK pathway itself (MEKK1, MLK2) (80). Together, these studies highlight the possibilities for complex signaling cross talk and feedback mediated by JNK-dependent phosphorylation of its substrates.
Molecular-Level Regulation of JNK Substrates: Looking into the Black Box
The majority of JNK target sites, like the sites targeted by other Ser/Thr kinases (and to a lesser extent, Tyr kinases), are located in intrinsically disordered protein segments (289). The remaining phospho-sites are mostly presumed to be within flexible loops protruding from otherwise-rigidly folded domains. As a consequence, JNK-mediated phosphorylation would rarely be expected to have an effect on the conformation of its targeted proteins. Instead, phosphorylation typically affects target protein function by either creating or disrupting protein-protein interactions. In this regard, the “loose” target site consensus for JNK-mediated phosphorylation is significant; it enables the control of a diverse set of protein-protein interactions, because the simple target site requirement is compatible with a large pool of different linear motifs. These kinase-controlled linear motifs can be considered either positive or negative phospho-switches (290, 291); these switches are considered in relation to JNK-dependent changes in substrate proteins in the later subsections of this review.
Although some JNK-targeted linear motifs require only a single site to be phosphorylated, it is common to find additional phosphorylation sites nearby that synergistically promote the same protein-protein interaction (potentially because the binding energies for the individual phosphate groups are relatively modest). Multisite phosphorylation also tends to result in a more robust and switch-like behavior (292), and several kinase families effectively drive efficient multisite phosphorylation events. Thus, “slave” kinases typically require priming by a “master” kinase, but they may also be primed by sites that they themselves phosphorylate, resulting in a chain reaction of phosphorylation events for the same substrate.
Two major families of “slave” kinases have been identified for the JNKs: the GSK3 and the CK1 families. The GSK3 family (GSK3α and GSK3β) represents the single most important partner and modulator of JNK systems (293). GSK3s can recognize substrates primed by the phosphorylation of Ser/Thr site 4 amino acids downstream of their own targeted phospho-sites (294). Although GSK3 does not strictly require their targeted amino acid to be followed by a Pro, its optimal targeted phosphorylation sequence is still [S/T]-P, allowing it to replace JNK-mediated phosphorylation of appropriate sites. GSK3 is commonly needed for the efficient phosphorylation of most phosphodegrons (Fig. 6A). The CK1 kinase family (casein kinase 1 enzymes CK1α1, CK1α2, CK1δ, and CK1ε) contains additional well-established accomplices of JNK. CK1 kinases recognize Ser/Thr (with Ser preferred) if they are preceded by an already-phosphorylated Ser/Thr amino acid 3 amino acids upstream of the CK1 target site. The best-known example for JNK-driven multisite CK1 phosphorylation is the transcription factor NFAT4 (Fig. 6B). Since CK1 requires an upstream priming site, it can processively phosphorylate substrates in an N-to-C direction with 3-amino-acid spacing, unlike GSK3, which senses downstream priming events and proceeds in an opposing C-to-N direction, with 4-amino-acid spacing. In summary, these examples highlight that single-site phosphorylation by JNKs needs to be examined in the context of the activity to prime the actions of its “slave” kinases.
FIG 6.
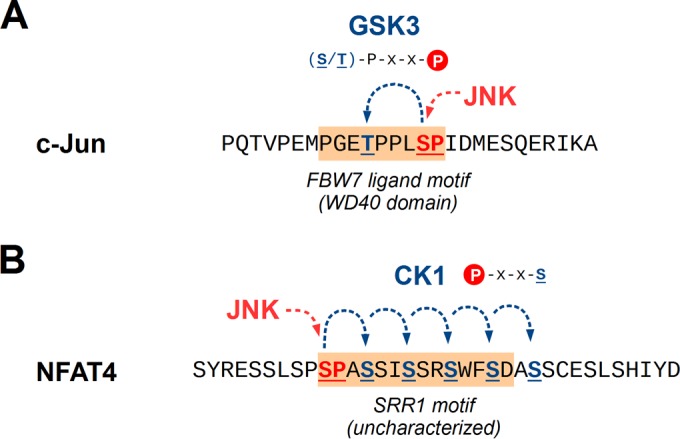
Cooperation between JNK and other kinases in substrate phosphorylation. (A) Phosphorylation by JNK can serve as a priming site for GSK3 enzymes in several substrates. The latter targets the site 4 amino acids upstream of the priming site, with a preference for proteins where the upstream Ser/Thr is also followed by a small amino acid, such as Pro. The double-phosphorylated region can often act as a phosphodegron, as in the case of c-Jun, where the motif is subsequently recognized by the cullin/F-box ubiquitin ligase FBW7. (B) Sites phosphorylated by JNK can also be recognized by casein kinase 1 (CK1). These enzymes phosphorylate Ser/Thr residues 3 amino acids downstream of the original phosphorylation site, with few sequence constraints. Like most other kinases reliant on substrate priming, CK1 can also recognize sites phosphorylated by itself. In the case of NFAT4, this leads to a chain of phosphorylation events initiated by JNK. Multisite phosphorylation of this so-called SRR1 (serine-rich region 1) motif then leads to cytoplasmic anchoring of NFAT4, although its precise binding partners are unknown.
The same protein may also contain multiple, JNK phosphorylation site-regulated motifs, each pertaining to a different regulatory mechanism. A well-known example is c-Jun itself (Fig. 7A). Detailed analysis of phosphorylation sites suggests that they form part of at least three separate linear motifs in the intrinsically disordered Jun proteins. The two N-terminal motifs (each containing two conserved phosphorylation sites), located near the NFAT4-type JNK docking motif, are implicated in transactivation by recruiting yet-unidentified coactivators, histone acetyltransferases, or generic transcription factors (295–298). The third motif, positioned several hundreds of amino acids away from the docking motif, also contains two JNK target sites, where the latter is a phospho-degron controlling ubiquitinylation-dependent degradation of Jun proteins (299). The latter motif also cooperates with GSK3 to provide double-phosphorylated motifs for the FBW7 ubiquitin ligase. Additionally, the latter motif can also be primed by other protein kinases (such as CK2) and provide a basal level of protein turnover in the absence of JNK stimulation (300).
FIG 7.

JNK-dependent phosphorylation of c-Jun elicits diverse effects. (A) The three Jun proteins (c-Jun, JunB, and JunD) were the first JNK substrates to be described and are still perhaps the best-known targets of the JNK pathways. The c-Jun protein can be phosphorylated at several sites by JNK; the most important regulatory sites are located in three different linear motifs. Phosphorylation of the two transactivator motifs located directly next to the docking motif can elicit transcriptional activation, probably due to phosphorylation-dependent recruitment of unknown effector proteins. This results in transcriptional activation of genes containing a Jun (AP-1)-binding element on their promoters, including c-Jun itself. On the other hand, phosphorylation of a much more distant phosphodegron motif in c-Jun, either by JNK alone or by cooperating with GSK3, represents an opposing regulatory mechanism. The latter provides a way for negative regulation of c-Jun levels by JNK, the mechanism for which is interestingly absent in the oncogenic v-Jun. (B) Under low or transient JNK activity, the c-Jun phospho-sites directly adjacent to the D-motif are the first to be phosphorylated, due to their favorable stereochemistry and strong coupling with the docking site. This results in a sharp rise of c-Jun mRNA and protein levels, due to autoinduction of the c-Jun gene. (C) If JNK activity is high and/or sustained over several hours, efficient phosphorylation of the distant C-terminal sites may also occur. The result will be the well-known attenuation of c-Jun levels due to ubiquitinylation and proteasome-dependent degradation of phospho–AP-1 complexes.
Due to the lack of strict spatial constraints, it is possible that the phosphorylation of different target sites is directed by a single JNK recruitment motif. However, the positioning of target sites relative to the docking site likely has a profound impact on the extent of JNK control: sites that are in a near-optimal range from the docking point (10 to 50 amino acids downstream) are expected to be phosphorylated to a much higher extent and at a lower threshold than sites that are located further away or in an otherwise-sterically disfavored position, such as upstream of the D-motif (201). These ideas regarding the spatial constraints for phosphorylation thus explain why JNK pathway activation can increase c-Jun levels (via Jun gene autoinduction due to proximal, N-terminal Jun protein phosphorylation) but not attenuate c-Jun levels, observed only following sustained JNK activity (partly due to the phosphorylation of the distant C-terminal sites in Jun) (301) (Fig. 7B and C).
JNK Phospho-Switches Negatively Regulating Protein-Protein or Intramolecular Interactions
Protein phosphorylation events may disable a particular inter- or intramolecular interaction, thus acting as negative phospho-switches. The addition of a phosphate to a Ser or Thr side chain may make the motif incompatible with partner domain binding due to either direct steric/size effects, electrostatic conflicts if the unmodified amino acid is coordinated by negatively charged side chains, or intrachain competition with the disruption/formation of salt bridges. Several notable examples of JNK substrates have been described for JNK-mediated phosphorylation negatively controlling various protein-protein interactions.
A simple example of a JNK-driven negative phospho-switch is the phosphorylation of the dynein light chain (DLC)-binding motifs of the BH3-only apoptosis regulator protein Bim. Both Bim and its close relative Bmf harbor a canonical TQT DLC-binding motif (302), in which the second Thr (underlined) is followed by a Pro, so that its phosphorylation by JNKs or other Pro-directed Ser/Thr kinases prevents its interaction with DLC (Fig. 8A). The ensuing release of Bim from its transport adaptor-bound complexes likely sensitizes cells to proapoptotic stimuli (302). Another important proapoptotic protein, BAD, can also be modified by JNK (303), leading to release of BAD from 14-3-3 protein-bound complexes. The mechanism of this release probably differs, because within the targeted sequence (SPFRGRSRSA), the JNK-targeted phosphorylation site (Ser [underlined in the sequence]) is located near two Arg residues (in bold), constituting an Akt1-dependent phosphorylation site (the final Ser is shown in bold and italics). Akt1-dependent phosphorylation events are absolutely required for 14-3-3 binding and sequestration of BAD; interference with either event would allow the release of BAD and trigger apoptosis (304). Several members of the Bcl2 family may also be JNK substrates that are subjected to negative regulation by JNKs, but their phosphorylation sites are often poorly conserved and evolutionarily do not correspond to each other. In the case of Bcl2, multisite phosphorylation (at least on Ser70 and Ser87) inhibits its binding to the apoptosis and autophagy regulator Beclin, thus leading to autophagy activation (305, 306).
FIG 8.

Negative phospho-switches and autoinhibitory switches driven by JNK. (A) A simple negative switch, illustrated by Bim. Phosphorylation of a dynein light chain (DLC)-binding motif in the BH3-only apoptosis regulator protein Bim impedes its binding to DLC-DIC (dynein intermediate chain) complexes. This shifts the equilibrium toward free Bim molecules, eventually completely releasing them into the cytoplasm. (B) A complex negative switch occurring on PPARγ. The disordered N terminus of the peroxisome proliferator-activated receptor γ contains a WW domain ligand and an overlapping PDSM (phosphorylation-dependent SUMOylation motif). Phosphorylation by JNK can therefore elicit SUMOylation of PPARγ, making its association with WW domain-containing coactivators (such as YAP1) sterically impossible. Ubc9 is a SUMO-conjugating enzyme. (C) The autoinhibitory switch of the ITCH ubiquitin ligase. As typical for the NEDD4 family of E3 ubiquitin ligases, ITCH is subject to autoinhibition. According to studies, the autoinhibitory interface of ITCH is complex, and the C2 domain, linkers, and its WW domains all contribute to maintenance of the “closed” conformation. JNK-dependent phosphorylation of certain residues located in the linker region interfere with the autoinhibitory interactions. This “opens up” the catalytic HECT domain of the ITCH enzyme, allowing the E3 ligase to recruit and ubiquitinylate its substrates.
Many NFAT proteins are also substrates of JNKs, counteracting calcineurin and Ca2+ signaling (307–309). However, different NFATs may be regulated at different sites and by different mechanisms. NFAT2 (NFATc1), for example, was found to be phosphorylated directly at its PxIxIT motif, which is responsible for calcineurin recruitment (308). Although phosphorylation of this site (APALESPRIEITSCL, in which the PxIxIT motif is indicated in bold) does not result in an obvious steric clash with the surface of calcineurin, it markedly decreases its affinity (possibly due to intrachain effects). The resulting weakening of the association between calcineurin and NFAT2, thus acting as a negative phospho-switch, would in turn lead to NFAT2 hyperphosphorylation by other kinases and its relocalization to the cytoplasm (308).
Within the class of cytoskeletal proteins, the intermediate filament-forming protein cytokeratin-8 was among the earliest-described JNK substrates (310); its Ser74 phosphorylation site lies in the conserved, but disordered, head “domain” of this protein and is critical for the assembly of higher-order filaments, in cooperation with its partner, cytokeratin-18. Although the structural details are unknown, cytokeratin-8 Ser74 phosphorylation can also be achieved downstream of ERK or p38 activation (311), but it is implicated in the disassembly of filaments, possibly through a negative phospho-switch (312). Thus, simple negative phospho-switches have the capacity to mediate the actions of JNKs across multiple cellular compartments.
Phosphorylation can also elicit disruptive effects by more complex, multistep mechanisms. JNK-mediated phosphorylation of the nuclear receptor PPAR-γ severely inhibits its transactivation potency via a complex negative switch (242). Specifically, the immediate surroundings of the target phosphorylation site (IKVEPASPPYY) form a WW domain ligand (SPPxY motif corresponding to the C terminus of this site), while also conforming to a phosphorylation-induced SUMOylation motif (IKxExxSP, corresponding to the N terminus of this site). SUMOylation of the motif by the Ubc9 SUMO-conjugating enzyme following phosphorylation renders this sequence sterically inaccessible for WW domain-containing transcriptional regulators (including TAZ and YAP) (313) (Fig. 8B). The importance of this switch region was highlighted by the observations of mutations at the Pro adjacent to the phosphorylated Ser in a severe, hereditary obesity syndrome (314). These observations reinforce the critical role of this complex as a negative switch in regulating PPAR-γ activity (315). Similarly, the phosphorylation of heat shock factor HSF1 at Ser303 (part of an IKxExxSP motif) by several MAPKs, including JNK, leads to SUMOylation (316–318). This subsequently inhibits their transactivation potency, although the exact interaction partner disabled by steric effects in this case remains unknown.
Not all JNK-dependent phosphorylation events control protein-protein binding events; the disruption of intramolecular interactions following phosphorylation can also be an important regulatory event for several proteins. In the case of the E3 ubiquitin ligase ITCH, JNK-dependent phosphorylation can relieve the autoinhibitory interactions between the array of WW domains (responsible for substrate recruitment) and the catalytic HECT domain (319). Indeed, ITCH autoinhibition is probably even more complex, since one of the key phosphorylation sites controlling autoinhibition is part of a Pro-rich, basic, and highly conserved linear motif (RPPRPSRPPPPTPRRP) near the N terminus that belongs to neither the WW nor the HECT domain. The latter motif could serve as an “autoligand” for the enzyme or its WW domains only, perhaps “sandwiching” the rest of the protein in its autoinhibited, self-associating state (Fig. 8C). Interestingly, neither the phosphorylation sites nor any other features of the motif are found in related NEDD4-like E3 ubiquitin ligases, suggesting that this autoinhibitory mechanism is unique to ITCH.
JNK Phospho-Switches Potentiating New Protein-Protein Binding Events
For many substrates, JNK-mediated phosphorylation may directly establish new protein-protein interactions, i.e., acting as positive phospho-switches. A large number of protein domains have been described that bind to linear motifs containing phospho-Ser or phospho-Thr residues, either in an obligatory fashion or in a facultative fashion in which Asp or Glu can replace some of the phosphorylated residues (320, 321). Unfortunately, the dedicated interactors for relatively few JNK substrates have been identified. However, for several unrelated effector proteins involved in protein degradation, transcriptional activity, cytoskeletal dynamics, or other modifications, intriguing details of their selective recruitment by special JNK target phospho-motifs have been revealed.
Some of the best-explored effectors of JNK-dependent phosphorylation events, at least from a structural perspective, are the E3 ubiquitin ligases, by which the ensuing interactions can drive the proteolytic destruction of the JNK target. The E3 ligase subunit FBW7 (also known as Archipelago or Cdc4) recognizes linear motifs with the sequence TPPxSP (or similar) in its substrates, where both Ser/Thr amino acids are phosphorylated (322). This dual phosphorylation, following actions of JNKs and the auxiliary kinase GSK3, results in K48-linked polyubiquitinylation and subsequent degradation of substrates by their recruitment into the proteasome. Since FBW7-containing cullin/F-box enzymes are dimeric, their optimal substrates either carry multiple phosphodegrons or are also oligomeric (323). This helps explain earlier observations that the levels of the Jun family transcription factors are regulated by JNK in both a positive and a negative manner, the latter mediated by a C-terminal phosphodegron distinct from the transactivator region. Interestingly, this phosphodegron is absent from the oncogenic v-Jun protein (299), explaining v-Jun's prolonged activation downstream of JNK activation. Within their N termini, the proto-oncogene Myc-family proteins also carry an FBW7 phosphodegron that can be phosphorylated by JNK, resulting in ubiquitinylation and degradation (324) (Fig. 9B). More recently, a similar phosphodegron was identified for the Bcl2-family member Mcl1 (282, 325). This degradation of the antiapoptotic Mcl1 provides another mechanism by which JNK can sensitize cells to proapoptotic stimuli (281). Phosphorylation of the PPM1J phosphatase at Ser92 (part of a potential FBW7-type phosphodegron, AVQSPPDTGR) can also profoundly reduce its activity in cells (71), albeit that the impact of JNK-mediated phosphorylation on PPM1J ubiquitinylation and its half-life have not been specifically studied. This putative phosphodegron of PPM1J is not evident in the closely related PPM1H phosphatase, suggesting differential regulation of these phosphatases by JNKs which may, in turn, dephosphorylate and deactivate JNKs. Two phosphorylation sites of the recently identified JNK substrate SMTL2 (a close relative of the actin-binding protein smoothelin) also lie in a conserved motif resembling a near-optimal FBW7 phosphodegron (PLVTPPQSPVS; phosphorylation sites are underlined). Although the functions of SMTL2 require further exploration (69), these examples provide interesting insights into proteins regulated through the interplay of JNK-mediated phosphorylation and their ensuing degradation.
FIG 9.

Positive phospho-switches and allosteric switches controlled by JNK. (A) A simple positive switch acting on Elk1. Phosphorylation of the ETS transcription factor Elk1 by JNK1 (or ERK1/2) on multiple sites at its transactivation region creates a new linear motif. This protein-protein interaction motif binds to the histone acetyltransferase CBP/P300, likely through its second TAZ zinc finger domain. Thus, Elk1 can recruit chromatin-modifying enzymes that enhance transcription of its target genes. (B) A complex positive switch exemplified by phosphodegron systems. Several substrates of JNK, such as c-Myc, contain phosphodegron motifs. Multistep phosphorylation of such a motif (using GSK3) results in the recruitment of an E3 ubiquitin ligase complex containing the recognition subunit FBW7. Subsequent ubiquitinylation will then generate another new protein-protein interaction, this time with the lid of the proteasome. The result is usually the degradation of the ubiquitinylated protein, which in this sense is an inhibitory outcome (despite all protein-protein interactions being positive). (C) An allosteric switch on RXRα elicited by JNK-dependent phosphorylation of a regulatory loop. The retinoid X receptor α is an allosterically sensitive protein which only recruits the coactivator NCoA2 in its ligand (11-cis-retinoic acid [11-CRA])-bound state. The JNK-phosphorylated loop is directly adjacent to the ligand-binding pocket, and all available evidence suggests that it will bind back to the pocket when modified. The consequential distortion of the ligand-binding site would not only elicit release of 11-CRA, but also its coactivators, thus shutting down RXRα-dependent transcription.
Other ubiquitin ligases may also recognize JNK-phosphorylated linear motifs. Smurf1 and its relationships with the SMAD transcription factors and Nur77 provide interesting examples. [S/T]-P phosphorylation of SMADs by multiple kinases, including the JNKs (326, 327), switches their interactions with YAP/TAZ transcription factor proteins toward binding to the NEDD4-like E3 ubiquitin ligase Smurf1 (328). In this example, one of the multiple WW domains of Smurf1 mediating this interaction requires the canonical PPxY motif, but the other recognizes phosphorylated motifs instead and it binds in a reverse orientation, thus promoting interaction and the subsequent degradation of SMAD1 (328). Similarly, the orphan nuclear receptor Nur77, after its multisite phosphorylation by JNKs, can be recognized as a substrate of Smurf1, albeit the structural details are not yet clear (329).
While numerous phosphodegrons have been identified with links to specific enzymes driving these changes, the exact identities of many of the cytosolic or nuclear enzymes involved are still not clear in many cases. Among nuclear substrates, phosphorylation-dependent degradation has been described for the retinoid receptor RARα and the Jun partner protein JDP2. The RARα site is not conserved in the related RARβ or RXR proteins (243), emphasizing differences in JNK-mediated control of these proteins. However, the lack of similarity with other known phosphodegron motifs suggests the involvement of hitherto-unidentified enzymes for RARα degradation. Similarly, the Jun partner protein JDP2 is targeted for proteasomal degradation by unspecified ubiquitin ligases after its phosphorylation at Thr148 (330). A phosphorylation site homologous to Thr148 of JDP2 is also found on the related ATF3 protein, but ATF3 levels appear to be controlled by JNK in a much weaker manner than is JDP2 (330).
In the cytoplasm, JNK-dependent phosphorylation of the microtubule regulatory protein Stathmin-2 (also known as SCG10) at Ser62 and Ser73 can also induce proteasome-dependent degradation of the protein, although the involvement of ubiquitin ligases has not been studied (331). The phosphorylation of Ser69 within the alternative N terminus of the BH3-only protein Bim (distinct from the Thr116 DLC-binding modulatory site discussed above) can trigger ubiquitinylation, but the molecular details of the degradation events require further evaluation (332). Despite the numerous well-characterized examples, it is important to note that not all phosphorylation sites that affect ubiquitinylation are degrons. The phosphorylation of the regulatory C terminus of S6K1 (p70-RSK) also promotes its ubiquitinylation and degradation (333). However, this unusual target region (SPRRFIGSPRTPVSPVKFSP; phosphorylation sites are underlined) possibly elicits its effects in an indirect way, as C-terminally truncated proteins were more unstable. Thus, some JNK-mediated phosphorylation events may also control protein turnover in a more indirect manner, not by recruiting ubiquitin ligases directly but by disrupting complexes that would mask the true degrons.
Ubiquitin ligases are not the only major effectors of JNK phosphorylation. Numerous substrates have been described (e.g., β-APP, Tau, c-Jun) in which phosphorylation triggers an interaction with the peptidyl-prolyl cis-trans isomerase Pin1 (132, 334–336). The importance of Pin1-mediated Pro-peptide bond isomerization lies in its roles in protein folding and because certain linear motifs require particular Pro configurations (i.e., Pro positioned with a turn will likely be of a cis conformation, but a Pro in other contexts will be of a trans conformation) (337). Pin1 contains a substrate-recruiting WW domain that requires barely more than a Ser-Pro or Thr-Pro (SP or TP) site in which the serine/threonine amino acid is phosphorylated (338) and a conserved catalytic domain that shows a preference toward isomerizing phospho-Ser-Pro or phospho-Thr-Pro (pSP/pTP) bonds (339). One should note, however, that the catalytic domain of Pin1 is incapable of accessing a pSP/pTP motif that is bound to the WW domain (in a trans conformation); only other, adjacent pSP/pTP motifs can be targeted for cis-trans isomerization. The generation of cis- pSP/pTP stereoisomers is significant: protein kinases primarily phosphorylate trans-SP/TP sites only, and many known effectors (including the previously mentioned FBW7 ubiquitin ligase or the PP2A phosphatase) can only access trans-pSP/pTP sites. Dedicated binding partners requiring cis-pSP/pTP sites also do exist (340) but still await identification for most JNK substrates.
JNK-mediated phosphorylation can directly induce protein-protein interactions, as exemplified by their regulation of transcription factor proteins. Despite some of the best-known JNK substrates being well-established transcription factors, our knowledge of their specific interacting partners and how these interactions are influenced by phosphorylation is still remarkably poor. One of the structurally clearest cases is provided by the ETS transcription factor family member Elk1, which can be phosphorylated at the same sites by either ERK2 or JNKs. In Elk1, the phosphorylation of a long (∼50 amino acids) and presumably fully disordered transactivator region is required for binding to p300/CBP histone acetyltransferases that are essential for enhanced transcriptional activity. Elk1 lacks the classical transactivator motifs (such as the 9-amino-acid TAD motif) that could enable a strong, constitutive interaction with the TAZ, KIX, or IBID domains of p300/CBPs (341). Instead, it appears to rely on phosphorylatable linear motifs (e.g., the highly conserved SIHFWSSLSP) segment, incorporating the Ser383 site ([underlined], could theoretically be a 9-amino-acid TAD-like helical motif) that bind to the same domains in a regulated manner (342) (Fig. 9A). The same sites and motifs are also seen in the related Elk3 (originally known as Net) and Elk4 proteins, but their corresponding D-motifs may preferentially bind ERK2 and p38α rather than JNK (343). More recently, members of the NuA4 histone acetyltransferase complex, including TRRAP and TIP60, were also suggested to be preferred partners and effectors of phosphorylated Elk1 (344), so p300/CBPs might not be the only effectors and might not even be the main partners of phospho-Elk1.
c-Jun/JunD and ATF2 transcription factor proteins are also well-known JNK substrates. In the case of the ATF2 family proteins, phosphorylation of an intrinsically disordered transactivator motif on two adjacent positions (Thr69 and Thr71 in human ATF2) is a prerequisite for the activation of promoters to which they bind (345). Although these phosphorylation events would directly trigger some protein-protein interactions, their binding partners are still unclear, as experimental data on the role of p300/CBP and others remain controversial (346). The case of Jun family transcription factors is slightly more complex: these proteins possess an N-terminal transactivator motif with two well-conserved phosphorylation sites (Ser63 and Ser73 of c-Jun), in addition to the motif also found in the ATF2 family (Thr91 and Thr93 in human c-Jun). Therefore, it is possible that these two motifs recruit different partners simultaneously to phosphorylated Jun proteins. However, none of the proposed effectors (p300/CBP, TFIID, TCF4, MBD3) have been well characterized as partners (295–298). The same can be said for the AP-1 partner, the Leu zipper transcription factor Nrl, which is required specifically for retinal development. The STPYSSVPPSPTFS motif surrounding Ser50 (underlined; the main JNK-mediated phosphorylation site in Nrl) is highly conserved among the Maf family transcription factors and required for efficient transcription of Nrl target genes; it is still unclear if this effect is mediated by TIP60 histone acetyltransferase or not, since JNK activity and TIP60 binding to Nrl-containing promoters did not correlate in vivo (347). Thus, these proposed partnerships require further evaluation.
JNK also targets diverse cytoskeletal proteins involved in adhesion, cell migration, and the cytoplasmic trafficking of proteins, organelles, and vesicles. Unfortunately, the mechanistic insights into these important processes remain largely limited. One of the better-characterized substrates is the focal adhesion protein paxillin. Although paxillin has been implicated as an ERK2-dependent target, it can also be phosphorylated (at least on Ser 178) by JNK (268, 269). Paxillin displays an intriguing interplay between Ser/Thr and Tyr phosphorylation. Across all proteins, most of the known Tyr phosphorylation sites are flanked N-terminally by one or more negatively charged amino acids (Glu/Asp) because these are needed for their recognition by most Tyr kinases (348, 349). Intriguingly, two major tyrosine phosphorylation sites (shown in bold) in paxillin (Tyr88, SSPVYGSS, and Tyr181, LSPLYGVPE) lack these charged positions. Instead, they contain SP sites (underlined), allowing MAPK-dependent phosphorylation events to provide the necessary negative charges. Thus, ERK2 or JNKs can promote tyrosine phosphorylation and subsequent Crk and/or Src binding of paxillin, which leads to stronger focal adhesion kinase binding, Src kinase activation, and greater tyrosine phosphorylation (350). Thus, the example of paxillin demonstrates how JNK-dependent phosphorylation can potentially expand the repertoire of Tyr kinase protein targets.
Lesser-Understood JNK-Dependent Phospho-Switches
Although the preceding examples illustrate how JNK-mediated phosphorylation can exert actions as either positive or negative phospho-switches, there are numerous examples in which this delineation of effects is less understood. For example, transcription factor complexes such as the YAP/TAZ proteins together with p73 and p63 proteins (relatives of the better-known p53 tumor suppressor protein) constitute a critical transcriptional complex in which both protein partners can be regulated by JNKs. Specifically, the coactivator protein YAP1 is also subject to multisite phosphorylation that can be considered a complex phospho-switch. At least four sites (of which two are highly conserved) were identified as JNK targets in response to genotoxic stress (351), and all play a role in transactivation by YAP1, albeit by different mechanisms. Ser128 (underlined in the following sequence; it is also conserved in TAZ) lies near the TEAD-binding region of YAP1, directly inside one of the two LATS-phosphorylated 14-3-3 binding sites (HVRAHSSP). This endows JNK with the potential to counteract Hippo/LATS kinases, setting YAP1 free to interact with TEAD transcription factors (351). On the other hand, Thr412 (underlined in the following sequence) is part of a conserved, long sequence motif (YSVPRTPDDFLNSVDEMDTGD) lying in a disordered C-terminal region important for transactivation (352). It shows some sequence similarity to the transactivator motifs of p53 and other 9-amino-acid TAD motifs (recruiting TAF9 or p300/CBP); however, its precise binding partners have not been established.
The transcription factor p73 has at least seven potential JNK phosphorylation sites, of which several are highly conserved and also found in its close paralog, p63. Except for the C-terminal Thr482 (TPPPPY motif), which potentially modulates the binding of WW domain-containing partners (e.g., YAP1, WWOX, ITCH, HECW2), the exact functions of the other sites are unknown. Experiments suggest that these motifs (such as the N-terminal SPY motifs) are important for transactivation of p73-dependent genes as well as stabilization of the p73 protein by protecting it from ubiquitinylation (239).
JNK can also regulate translation of specific mRNA transcripts through targeting the RNA-binding protein hnRNP-K (256). Of the three phosphorylation sites identified in human hnRNP-K, two (Ser216 and Ser284) are conserved in most other vertebrate species. Ser216 lies immediately after the second Ku homology (KH) domain, as part of its conserved extension. It is probably required for its interaction with the ribosomes, essential for the translation of associated mRNA in developing neurons, in order to support axon outgrowth (257).
Several cytoskeletal proteins show conserved motifs surrounding their JNK phosphorylation sites. In most cases, it is unclear whether the modification induces a new interaction or abolishes existing ones. Some of the most intriguing examples encompass important microtubule-associated proteins. The microtubule-associated, intrinsically disordered MAP2 and Tau proteins are among the earliest discovered MAPK substrates and are extensively modified in vivo by JNK at several conserved sites (180). One key region is the segment immediately preceding the microtubule-binding repeats in these proteins. For MAP2, the phosphorylation of a conserved “perimicrotubular” motif (TPGTPGTPS, containing Thr1619, Thr1622, and Thr1625 [underlined] of MAP2) increased microtubule binding, although the molecular details of this regulation were not fully explored (353). The phosphorylation of the neighboring, well-conserved KKVAIIRTPPKSPA motif has been better documented in Tau than in MAP2. This MAP2 region is similar to an FBW7 phosphodegron of Tau (Thr231 and Ser235 [underlined in the above sequence]), which is modified in cooperation with GSK3. Although phosphorylation was originally reported to inhibit the microtubule-binding ability of Tau, more recent research suggests that modification of these sites may not be sufficient to induce Tau dissociation from microtubules (354, 355). The phosphorylation of the latter motif may still contribute to neurotoxicity and filament forming when Tau is released from microtubules in Alzheimer's disease (355). These mechanistic links thus can help inform possible approaches to intervene in neurodegenerative diseases.
Many other microtubule-binding proteins are also JNK substrates, including the X-linked lissencephaly protein DCX (doublecortin-X). DCX is a critical regulator of neurogenesis and postmitotic neuroblast migration, capable of binding to JNK directly and also containing numerous JNK phosphorylation sites (261, 262). Interestingly, most of the sites located N-terminally and C-terminally from the microtubule-binding tandem doublecortin domains are also conserved in the closely related doublecortin-like kinases 1 and 2 (DCLK1 and DCLK2), but not in the more-divergent DCLK3. Currently, the most plausible explanation is that these motifs have a role in regulating microtubule-actin cross-linking through binding to unidentified effectors (especially by Ser297 phosphorylation, which is part of the highly conserved AKSPGPMRRSKSPA motif in the disordered C-terminal tail of DCX) (356). Furthermore, the JNK-mediated phosphorylation of WDR62, a microtubule-binding protein and established JNK interactor, regulates its subcellular localization (213, 357). The critical Thr1053 phospho-site, together with its surroundings (PQTPEQEKFLRHHFELTLT), is highly conserved in vertebrates, in comparison to adjacent regions, thus hinting at a true linear motif. The sequence conservation for WDR62 with the related protein MABP1 (MAPK-binding protein 1) extends to the JNK-docking motif (80), but whether the phospho-motif directly associates with the microtubules, or binds to some other protein, is not yet known.
For the synaptic scaffolding protein PSD-95 (also known as Disks large homolog 4 [DLG4]), its phosphorylation by JNK on Ser295, located in the flexible linker between the second and third PDZ domains, enhanced its synaptic localization (287). Again, this site forms part of a larger motif (TPTSPRRYSPVAKDLLGEEDI) that is very conserved, unlike other parts of the linker, and is also encountered in several paralogs (DLG1, DLG2, and DLG3). Despite these observations, the molecular details of the functions of this motif remain completely unknown (287). The highly conserved cytoplasmic tail of the AMPA-type glutamate receptor GluR4 and the long isoform of GluR2 are also JNK substrates (at Thr875 and Thr874 of the human proteins) (288). These receptor ion channels are implicated in learning processes, and the pharmacological blockade of JNK pathways impaired the ability of these channels to be reincorporated into the membrane after internalization. The trafficking complexes mediating this process remain unidentified, but JIP1 remains a candidate (288). Lastly, the fully disordered actin-binding protein MARCSKL1 is another important cytoskeletal JNK substrate. In this case, concomitant phosphorylation of three sites (corresponding to Ser120, Thr148, and Thr178 in human MARCKSL1) enhanced the F-actin-binding capacity (266). Two of the three sites are poorly conserved, but Ser120 lies in a region (SSPTEEEQ) also evident in its fish orthologs. As none of these phosphorylation sites is situated in the central actin-binding motif, the underlying mechanism of regulation by phosphorylation cannot be easily inferred. However, the interaction of the central motif toward actin may be strengthened by the presence of downstream phosphorylated motifs, because F-actin-binding linear motifs may contact multiple surfaces of actin molecules. This role further emphasizes the critical importance of JNK-mediated phospho-switches in the cytosol, beyond stress-induced signaling events.
Phosphorylation-Induced Conformational Changes in Folded Domains
In contrast to the numerous reported JNK-directed phospho-switches acting on linear motifs, relatively few JNK-mediated phosphorylation events are known to elicit their effect through classical allostery. In this case, phosphorylation of a relatively flexible and accessible SP/TP site would direct its binding to a directly adjacent, folded domain, leading to conformational changes in the domain structure. Such effects were described for the retinoid X receptor RXRα (245). Phosphorylation of a mobile loop protruding from the ligand-binding domain of RXRα leads to its direct inhibition by JNK. Since the phosphorylatable amino acid (Ser260) is located in close proximity to the ligand-binding pocket (but far from the coactivator/corepressor recruitment site), the phosphorylation event probably directly modulates ligand (11-cis retinoic acid) binding (Fig. 9C). The endothelial nitric oxide synthase (eNOS) may provide another case of JNK-mediated allosteric regulation. The phosphorylation of eNOS at Ser114 (lying on a loop on the heme domain, directly facing its catalytic site) potentiated its activity (358). Multiple 14-3-3 proteins (such as 14-3-3ζ or 14-3-3σ) were also described as being phosphorylated by JNK (277, 278, 359, 360). Their case is quite unusual, as the phosphorylation site lies in a structural domain, on a short loop connecting two α-helices. Surprisingly, this Ser-Pro amino acid pair (S184-P185 in the human 14-3-3ζ protein) could theoretically fit into the catalytic site of JNK without a clash, allowing phosphorylation of a relatively fixed side chain protruding from a folded domain. The modification of this site has important consequences, as it can potentially distort the relative positioning of the underlying helices, thus dismantling the ligand-binding groove of 14-3-3 proteins. This allows an allosteric, negative regulation of many 14-3-3 ligands controlled by kinases like PKB/Akt (270), emphasizing that these changes likely have broader impact.
Complex Effects of JNK on Its Substrates
JNK phosphorylation can also influence nucleo-cytoplasmic shuttling events, but the exact molecular mechanisms for many JNK substrates are not well understood at a molecular level. The well-known JNK substrate NFAT4 is primarily regulated by its phosphorylation-regulated localization (307). In this case, JNK “primes” the so-called SRR1 (Ser-rich region 1) motif of NFAT4 for sequential phosphorylation by casein kinase 1 (CK1) (307, 361). The distance (>100 amino acids) between the SRR1 and nuclear localization sequence (NLS) motif within the disordered N terminus of NFAT makes a direct interaction between the two unlikely (362). Therefore, it is probable that the phosphorylated SRR1 motif binds to cytoplasmic proteins that anchor NFAT4 in the cytoplasm (thus inhibiting its transcriptional activity) until its subsequent dephosphorylation by calcineurin. JNK-dependent phosphorylation of the androgen receptor likewise inhibited its nuclear localization (240). But in this case, the phosphorylation site within the linker between the DNA binding and ligand binding domains is located close to the NLS motif, making direct intramolecular effects more likely (363). The glucocorticoid receptor is also phosphorylated by JNK on Ser226; this inhibitory phosphorylation was proposed to act through an enhanced nuclear export of receptors (241). Since the amino acids surrounding this site do not correspond to the better-known nuclear export signals (NES), it is possible that the export effect is secondary to other changes. Thus, the impact on protein nuclear localization and/or shuttling can result from different mechanisms impacting cytoplasmic retention, nuclear translocation or nuclear export.
The protection from ubiquitinylation and/or proteolytic degradation (protein stabilization) frequently can also be a result of multistep regulation. Currently, very few degron motifs are known that would be directly inhibited by phosphorylation; however, the protection from degradation can be achieved by the inclusion in vivo of highly unstable, degron-containing proteins into multiprotein complexes, in which their degron motif is masked. Although the basic helix-loop-helix (bHLH) transcription factor Twist1 was described as being stabilized by JNK phosphorylation (233), the targeted site (Ser68, which is also conserved in Twist2) is located within a classic, potentially bipartite NLS motif, only 4 amino acids N-terminal of the major α-importin-binding site (364). Phosphorylation within this region is known to enhance nuclear import of affected proteins (365). Therefore, Ser68 phosphorylation may control the nuclear translocation of Twist1 (which is known to be potentiated by epidermal growth factor stimulation), to which Twist1 stabilization might be a secondary effect (366). In the case of the bHLH transcription factor Hes1, JNK-dependent phosphorylation of Ser263 within its flexible C terminus resulted in its stabilization (232). While the phosphorylation site itself is not strictly conserved in all vertebrates, the directly adjacent (extended WRPW-type) Groucho corepressor-binding motif is always preserved (367). Due to their close proximity (with only 3 amino acids lying between the two motifs), it may be that phosphorylation modulates Groucho binding, possibly providing stabilization as a secondary phenomenon. JNK could also phosphorylate the Zn finger transcription factor Sp1 on at least two distinct sites (Ser278 and Ser739 of human Sp1) in its extensive N- and C-terminal disordered regions. The modification of both, but especially the C-terminal site, provided a profound stabilization effect, thus increasing the cellular Sp1 concentration by prolonging its half-life (234). Examination of the Sp1 sequence reveals that both sites lie in close proximity to potential β-TRCP E3 ubiquitin ligase-recruiting phosphodegron motifs (SSGSQES and DSGAGSEG, with the C-terminal motif being a better match) (368). Phosphorylation of these β-TRCP phosphodegrons may potentially rely on autonomous GSK3 activity (due to Glu residues positioned downstream). However, nearby JNK-mediated phosphorylation events may inhibit the function of these phosphodegrons if they generate new protein-protein interactions that compete with GSK3 and/or β-TRCP recruitment.
JNK-Modulated Adaptors, Scaffolds, and Other Proteins
JNK-mediated phosphorylation can also regulate critically important adaptor proteins. The insulin receptor substrate (IRS) family of docking proteins also carry a conserved JIP1-type JNK docking motif. The JNK-dependent phosphorylation of IRS1 and IRS2 proteins has been examined in a number of studies (273, 274, 369). The best-documented target sites (human IRS1 Ser312 and human IRS2 Thr350) lie close to a conserved 14-3-3 protein-binding motif (RSRTESITATSPAS and RSRTDSLAATPPAA; boldface residues conform to the 14-3-3 binding consensus). Although these JNK target sites are not identical, IRS2 Thr350 could assist GSK3-directed phosphorylation of the 14-3-3-binding motif. However, the extensive disordered C termini of both IRS1 and IRS2 contain a large number of better conserved SP/TP sites, and it is likely that JNK can phosphorylate more than just one of them. For example, in IRS2, another site was identified more recently (Ser491 in human IRS2, within the conserved motif SASASGSPSDPGFMS, also found in IRS1), where JNK also provides a priming site for GSK3 (274). However, the function of the latter motif is also obscure. That JNK-dependent phosphorylation of IRS1 and IRS2 leads to a strong inhibition of its tyrosine phosphorylation during insulin stimulation (273) suggests a critical role for JNKs in the development of insulin resistance (reviewed in reference 370).
The role of JNK-dependent phosphorylation is even less clear for microtubule-associated transport adaptors, which are not only critical for trafficking of vesicles but also involved in JNK pathway regulation. Despite extensive research, little is known of the functional consequences of JIP1 phosphorylation. JNK can phosphorylate JIP1 at over a dozen different sites, most of them lying within its disordered N terminus or possible flexible loops (371). With the exception of a single site (Thr448), none of these is conserved in JIP2, although the JNK-docking motif itself is almost identical; that JIP2 also harbors the same, JNK-specific motif as JIP1 casts doubts on JIP2's possible links with p38 rather than JNK (372). Known regulatory sites of JIP1 (Thr103, located in the second acidic motif, and Ser421, in the auxiliary cargo-binding domain) are not found in JIP2. Thus, it is possible that despite their redundant function in kinesin-dependent transport, JIP1 and JIP2 are regulated differently by JNK. It can be assumed that one role of the phosphorylation is to regulate JIP1/2-cargo or JIP1/2-kinesin linkage or both in an indirect manner. Although nonvertebrate animals do not contain JNK-docking motifs in their JIP1/2 proteins, these proteins are essential for vesicular transport in Drosophila, hinting at a JIP1/2 trafficking mechanism which is less strictly coupled to JNK but also not completely independent from it (373).
JIP3 and JIP4 (previously known as JLP or SPAG9) constitute a separate protein family, although they share a name with the unrelated JIP1/2 family (372). The sequence similarity of the C-terminal halves of JIP3 and JIP4 to the Rho-GAP domains of several ARHGAP proteins suggests that they might possess a GTPase activator ability toward Rho family small G-proteins (such as Rac1 or Cdc42). In addition, they are also implicated to be critical regulators of kinesin-dependent vesicular transport, binding the small G-protein Arf and being members of the same transport complex that incorporates JIP1/2 (128, 374–376). JIP4 has been linked more strongly with p38 pathway activation (377). While reports of JIP3 interaction with the MAP3K ASK1 require further evaluation, JIP3 appears to be a genuine JNK substrate (105). Several phosphorylation sites have been described on JIP3, with Thr276 (within the motif GTKSNTPTSS) also being conserved in JIP4. Interestingly, many of the SP/TP sites on JIP3 are incorporated into conserved “islands” of an otherwise-largely unstructured N terminus of JIP3/4, suggestive of multiple, JNK-regulated linear motifs. Since the JNK pathway is activated by several GTP-bound small G-proteins (through MLK/DLK kinases), feedback mechanisms through GTPase regulator proteins are plausible. However, the lack of detailed structural and functional studies continues to hamper our understanding of JIP3 and JIP4.
Lastly, JNK can bind and phosphorylate the mitochondrial outer membrane coiled-coil protein Sab (or SH3BP5) (378). We know very little of the physiological role of Sab Ser330 phosphorylation, but the extensive conservation of sequence over a long stretch (PRSECSGASSPECEVERGDRAEGAE) surrounding the phosphorylation site across all vertebrates suggests a likely regulatory role, which is supported by recent studies implicating the Sab/JNK relationship in initiating regulatory control over intramitochondrial Src actions and reactive oxygen species production (379).
BIOLOGICAL AND PATHOLOGICAL ROLES OF JNK PATHWAYS
In the last 2 decades there has been enormous research interest in discovering the specific functional roles of JNKs. These studies established the involvement of JNK signaling pathways in embryonic development, neuronal functions, immunity, or in different pathological states, such as insulin resistance, oncogenic transformation, and excitotoxicity. In the following section, we briefly review this large body of research to highlight the breadth of biological and pathological actions of JNKs.
JNK Involvement in Embryonic Development
The JNK pathway has attracted considerable interest as a target for gene knockout studies in mice to probe the developmental roles played by the JNKs. While single-JNK knockout mice, JNK1−/−, JNK2−/−, or JNK3−/−, are viable, as are double-knockout JNK2−/− JNK3−/− mice; JNK1−/− JNK2−/− mice die early in embryogenesis (embryonic day 11.5 [E11.5]) (for a detailed summary of cellular and behavioral phenotypes of JNK knockout animal models, see reference 2). These multiple knockout mice do not undergo neural tube closure, and they also display exencephaly (179). The latter severe developmental defect, in which the brain extrudes outside the skull, is suggested to be a consequence of deregulated apoptosis (178). However, JNKs do not only contribute to apoptosis regulation, and neural tube closure is a morphogenetic process that also involves the coordinated regulation of cell migration and cell proliferation. JNKs play a crucial role in cell motility and in the developmental morphogenesis of epithelial organs (380–384). In the developing central nervous system, JNKs are activated by various guidance cues, including netrins and semaphorins, and JNK activity is required for correct axon trajectories (180, 385, 386). In line with this, the genetic deletion of JIP3, an important target and regulator of JNK and its upstream kinases, causes axon guidance defects (387).
Studies on MKK4−/− and MKK7−/− mice have highlighted the nonredundant roles of these JNK-activating MAP2Ks (reviewed in references 62 and 388). Indeed, in vivo RNA interference (RNAi) screens have identified MKK4 as a key regulator of liver regeneration (389), whereas studies in MKK7−/− mice have revealed the critical need for MKK7 in axon elongation in the developing cerebral cortex (390). The compound disruption of both the MKK4 and MKK7 genes results in severe growth retardation and embryonic lethality (E9.5). It is striking that genetic deletion of single JNK substrates may also phenocopy JNK1−/− JNK2−/− mice, as observed for mice lacking the actin regulatory protein MARCKS-like protein 1 (266). While MARCKS-like protein 1 gene deletion causes neural tube defects (391), knockout mice for c-Jun or ATF2 live longer but die of heart failure or respiratory failure, respectively (392, 393).
JNK also plays a prominent role in Wnt signaling, with implications for the regulation of cell polarity (394). One of the three main branches of the Wnt pathway, the so-called planar cell polarity pathway (the PCP pathway), involves recruitment of Rho-GEF proteins (e.g., Daam1) and activation of Rho family small GTPases (most notably, Rac1). In turn, the GTP-bound Rac1 activates the JNK pathway to generate polarity and drive morphogenesis of several organs (395). The extent of PCP pathway activation depends on the exact nature of the receptor complex: several Wnt ligands are known that preferentially activate the PCP pathway and increase JNK activity. Notable examples include Wnt-11, which is indispensable in heart formation and in renal epithelial cells, and Wnt-7b, which drives maturation and dendritic arborization of hippocampal neurons (395–397). Cross talk of JNK with two other developmentally important signaling pathways (Hedgehog and Hippo) has also been suggested. Gli is an important terminal regulator of Hedgehog signaling, and its JNK-mediated phosphorylation suggests that a direct connection between MAPK and Hedgehog signaling may occur at the level of these key regulators (69). Recently, JNK-mediated inhibition of Hippo signaling was described as a mechanism allowing mechanical strain to influence cell proliferation (286). Taken together, these studies highlight the multiple actions of JNK signaling in important cellular events underlying embryonic development.
Neuronal Functions of JNKs
In addition to their roles in embryonic development in brain morphogenesis and neuronal pathfinding, JNKs also affect many neuronal functions in adults. In adults, the brain is the site of highest JNK expression, and all single-JNK knockout mice have CNS defects to a variable degree (reviewed in reference 2). Since normal JNK activity is implicated in neuronal development and regeneration but overactivation of JNK can induce apoptosis of neurons, inhibitors of JNKs or their upstream regulators have been proposed as desirable neuroprotective agents (398–400). These observations highlight JNKs as targets for pharmaceutical intervention to ameliorate stroke and epilepsy, Alzheimer's disease, Parkinson's disease, and Huntington's disease (reviewed in reference 2). JNKs may contribute to the earliest phases of Alzheimer's disease (AD); the Tau protein is a well-established substrate of JNK, and the phosphorylation of Tau might occur at the earliest stages of synaptic dysfunction and microtubule disassembly, thus preceding cognitive deficits in AD models (265). The inhibition of JNK activity (causing microtubule disassembly, Tau release, and/or aggregation) may thus represent one option to prevent the formation of neurofibrillary tangles and so should be further tested for its efficacy as a treatment modality.
JNK has also emerged as a central mediator of excitotoxic damage in the adult nervous system. Excitotoxic insults can induce JNK activation, which leads to neuronal death and contributes to many neurological conditions, such as cerebral ischemia and neurodegenerative disorders (401, 402). JNK3−/− mice are resistant to kainite-induced seizures (403). Conversely, the pharmacological inhibition of JNK protects neurons from N-nitrosyldimethylamine (NMDA)-induced excitotoxicity in vitro and from cerebral ischemia in animal models (402, 404). Most studies have highlighted the involvement of JNK3 in triggering neuronal death, but other JNK isoforms likely also contribute. For example, presynaptic JNK2 has been implicated in the regulation of glutamate release downstream of NMDA receptor activation (405). However, only a simultaneous knockdown of JNK1, -2, and -3 was sufficient to confer neuroprotection, indicating that a single form of JNK is sufficient to produce a dominant apoptotic signal (406). Furthermore, the inhibition of JNK activity in the nucleus, rather than blocking specific JNK isoforms globally, protected cells from undergoing apoptosis (406). These results further emphasize the critical nature of JNK localization in exerting its downstream biological effects. More recent studies have demonstrated the neuroprotective efficacy of a peptide targeting MKK7 (407), again highlighting that blocking JNK activation can provide an important strategy in the protection of neurons.
Although JNKs are best known as mediators of neuronal degeneration after stress and injury, they may be important also for neural development and survival. JNKs are implicated in axonal regeneration of adult neurons, as their specific inhibition in vitro reduces neuritogenesis (408). Similarly, JNK inhibition slowed nerve regeneration in a sciatic nerve transection-regeneration model (409). Moreover, distinct roles for different JNK isoforms were suggested in neurite initiation and elongation during axonal regeneration (410). Mutations disrupting the JNK3 gene have recently been identified in patients with hereditary intellectual disability (411), and so these studies support the roles for JNKs in maintenance of neuronal populations for normal development and in the response to injury. The mechanisms underlying these developmental roles require further evaluation, but the interaction of JNK1 with neuronal RNA transport granule proteins (412) will provide exciting new avenues for exploration.
JNK in Apoptosis and Cancer
JNKs are involved in the extrinsic apoptotic pathway initiated by death receptors as well as in the intrinsic pathway initiated at the mitochondrial level. In response to both extrinsic and intrinsic apoptotic stimuli, JNKs play an essential role through their interactions that modulate the activities of diverse pro- and antiapoptotic proteins (reviewed in reference 279). JNK activation is critical for TNF-α-stimulated AP-1-dependent gene expression (413) and has a dual role in the TNF-α-stimulated death of fibroblasts by not only suppressing TNF-α-stimulated apoptosis but also potentiating TNF-α-stimulated necrosis (414). Moreover, JNKs negatively regulate the autocrine expression of TGF-β1 in fibroblasts (414). This allows for complex, indirect effects on survival and cell growth through the TGF-β pathway (reviewed in reference 415).
Apoptotic cell death is an important process for tumor suppression, and JNK activation could be expected to be important as a negative regulator of cancer. However, depending on the cell type and lineage studied, JNK signal transduction pathways have been implicated not only in apoptosis but also in cell survival. Accordingly, JNKs have also been implicated in the malignant transformation and tumorigenesis of cells (reviewed in reference 416). In transformed B lymphoblasts, JNK1 signals cell survival, as its genetic disruption in mice causes defective transformation of pre-B cells by BCR-ABL (417). Moreover, JNKs play a key role in Ras-induced tumorigenesis, for example, in lung tumor development caused by mutational activation of the endogenous KRas gene (278). Thus, these studies of the fundamental biological processes highlight that JNKs can play a balancing role in the regulation of apoptosis and tumorigenesis.
The kinase domain-encoding portions for MKK4 (MAP2K4) and to a lesser extent the MKK7 (MAP2K7) genes appear to harbor a high number of missense and nonsense mutations (418). The upstream-acting MEKK1 (MAP3K1) gene (419) was found to be often mutated in breast cancer, and 12% of luminal A tumors contained inactivating mutations in MAP3K1 and MAP2K4 (420). Interestingly, MKK4 mutants harboring cancer-associated somatic mutations had lower enzymatic activity and also caused anchorage-independent cell growth (421). In summary, large-scale tumor genome sequencing studies further reinforce the tumor-suppressive role of JNK pathways in certain tissues (419).
JNK Actions in Insulin Resistance and Diabetes
Maintenance of insulin signaling is critical for metabolic homeostasis, with its failure in the form of insulin resistance being a hallmark of type 2 diabetes. Feeding mice a high-fat diet causes activation of JNK1 in insulin-responsive peripheral tissues, including fat, muscle, and liver, and ultimately leads to insulin resistance in these tissues (181). JNKs are among the key kinases activated by cytokines TNF-α, interleukin-6, and resistin, which are produced by the adipocytes of obese individuals (337). In these pathways, JNK1 and JNK2 act in close concert with members of the IKK family to inhibit insulin signaling through their effectors, including the insulin receptor substrates IRS1 and IRS2 (422).
JNK1 knockout prevents both diet-induced obesity and insulin resistance. JNK1−/− mice, but not wild-type or JNK2−/− mice, are resistant to obesity-induced insulin resistance (336). Nonetheless, JNK2 is also involved in metabolic regulation, but its contributions are not obvious in the presence of functional JNK1 signaling (187). Genetic or pharmacological inhibition studies have demonstrated that the chronic activation of JNK1 in obesity may be a direct cause of insulin resistance (reviewed in reference 423). The numerous studies using germ line or tissue-specific genetic ablation of JNKs established JNK1 as a potential pharmacological target for the development of drugs that could be used to counteract insulin resistance. These observations spurred an intense, but short-lived, interest in developing JNK inhibitors for therapeutic agents in type II diabetes (181), but a major portion of the protective effect seen in JNK1−/− mice can stem from dysregulation of the central nervous system. In the last 10 years, the picture has become more detailed, and JNK1 (and even JNK2) may also play a role in peripheral insulin resistance: JNK-dependent insulin resistance involves immune cells (424) and skeletal muscle cells to some extent (423). The presence of JNK in macrophages appears to be required for the establishment of obesity-induced insulin resistance and inflammation (424). JNK1 in adipose tissue or in the muscle regulates insulin resistance, and JNK1 deficiency in these tissues promotes insulin sensitivity (423, 425). In contrast, hepatocyte-specific JNK1 deficiency promotes insulin resistance (423, 426). Interestingly, the phenotype of these latter mice is similar to that of IRS1 Ser307Ala substitution in vivo (427). JNK1-mediated insulin resistance was initially considered mainly mediated by the insulin receptor adapter protein IRS1, phosphorylated on Ser312 (Ser307 in mice), which disrupts the interaction of the IRS1 phosphotyrosine-binding (PTB) domain with the tyrosine-phosphorylated receptor (428). However, analysis of mutant, knock-in mice demonstrated that the Ser312 site of IRS1 is not essential for the development of insulin resistance (427). While the influence of other phosphorylation sites in IRS1 and IRS2 cannot be excluded, most deleterious defects and the origin of insulin resistance are suggested to occur independently of IRS1 (429). IRS proteins are not the only targets of the JNK pathway: the insulin secretion-regulating protein MADD and the insulin sensitivity-influencing PPARγ coactivator PGC1α have also been implicated to be novel JNK targets (80). The role of MADD polymorphisms in type II diabetes susceptibility is strongly supported by genome-wide association studies and mouse models, while PPARγ agonist “glitazones” are among the most widely used antidiabetic agents (422, 430).
Additionally, since cell death in type I diabetes also involves JNK hyperactivation in pancreatic β-cells (causing their death), various JNK inhibitors have also been studied for the latter indication (431). However, β-cells also express JNK3, and this may have protective roles, unlike those prodeath roles described for JNK1 or JNK2 (432). Thus, approaches targeting the JNKs as possible therapeutic strategies will need to consider both tissue- and isoform-specific differences.
JNK in Immunity
JNKs play evolutionarily conserved roles in immunity: antibacterial, antiviral, or antiparasitic responses depend on JNK, even in insects (92, 433). JNKs can be strongly activated in multiple cell types by lipopolysaccharides (LPS) or inflammatory cytokines such as TNF-α and interleukin-1. Although JNKs, particularly JNK2, influence T cell functions and Th1/Th2 polarization, they are nonessential for T cell proliferation and for many aspects of adaptive immunity (434). JNK1−/− or JNK2−/− mice have normal lymphocytes, and JNK activity is not required for all forms of thymocyte apoptosis (435). Nonetheless, both JNK1 and JNK2 respond to T cell activators (436) and are required for the induction of T cell cytokines (182, 437). In addition, JNKs have a stage-specific role in T cell proliferation and apoptosis; while c-Jun is a target of both JNKs in thymocytes, it does not seem to be an important target in peripheral T cells, where both JNK isozymes contribute to the induction of NF-AT rather than AP-1 activity (435, 438).
It is well-established that viral infection can result in stimulation of c-Jun–ATF2 via JNK activation; JNK2-deficient cells exhibit a defect in induction of type I interferons in response to viral infection or double-stranded RNA that allows more robust viral infection (439). More recent studies have indicated that the JNK pathways mainly control processes of innate immunity (reviewed in reference 440). The innate immune response provides the first line of defense following infection, using a limited number of germ line-encoded pattern recognition receptors, such as the Toll or NOD receptors, which recognize invariant microbial components, termed pathogen-associated molecular patterns. All of these receptors activate MAPK and nuclear factor-κB (NF-κB) pathways in innate immune cells such as macrophages and dendritic cells. During an inflammatory response, NF-κB activation antagonizes apoptosis induced by TNF-α. The prosurvival NF-κB activity suppresses proapoptotic JNK signaling, and this is crucial for numerous physiological processes, such as the response of the liver to injury and the survival of cells during an inflammatory reaction, as well as for chronic inflammatory diseases (reviewed in reference 441). For example, JNK was essential for hepatitis by being critically required for TNF-α expression in hematopoietic cells, including resident inflammatory cells in the liver (e.g., Kupffer cells, which are specialized macrophages located in the liver, and natural killer T cells) (442), rather than being required for TNF-α-stimulated cell death during the development of the disease. These observations support the notion that JNK can act as a genuine immune modulator, not merely an apoptosis-inducing effector.
MICROBIAL PATHOGENS AFFECTING JNK SIGNALING
In addition to inducing an inflammatory response, pathogens themselves can also modify JNK signaling events. Since JNK is a key component of innate immunity pathways of many animals, this is not surprising: many bacteria, viruses, and eukaryotic parasites directly “tamper with” the JNK pathway in order to evade, suppress, or modify immune responses to their advantage (440, 443, 444). Enzymes produced by pathogenic bacteria and injected into the host cells, in most cases by a type III secretion system, are among the best examples (reviewed in references 445 and 446). These can impact different levels in the JNK signaling pathway, and various catalytic mechanisms have been described.
The YopJ enzyme of Yersinia pestis was one of the first pathway-subverting enzymes identified; its use of acetyl-coenzyme A (CoA) as a substrate in an acetyltransferase-mediated modification of critical residues in MAP2K activation loops thereby blocked their phosphorylation and thus activation by MAP3Ks (447). While YopJ is an acetyltransferase and its recognition of MAP2Ks has been mapped (448), it is also structurally related to eukaryotic cysteine peptidases involved in deubiquitinylation and deSUMOylation. Some experiments have suggested these activities for the YopJ family of enzymes (449). Unlike YopJ, which also targets the NF-κB pathway, the AvrA enzyme from Salmonella enterica serovar Typhimurium is more selective for JNK pathway inhibition. AvrA was found to directly acetylate the activation loop of MKK7, rendering it permanently inactive, unable to be phosphorylated (450). Other bacterial acetyltransferases work by different mechanisms: the VopA acetyltransferase of Vibrio parahaemolyticus targets a catalytic site lysine of different MAP2K enzymes (including MKK6, but others as well), disrupting ATP coordination (451). VopA thus efficiently shuts down both the JNK and p38 pathways and suppresses host cytokine production. Instead of targeting kinases, the Eis acetyltransferase from Mycobacterium tuberculosis modifies the MAPK phosphatase MKP7 (also known as DUSP16) to produce a hyperactive phosphatase that suppresses both JNK and p38 activity (452). Thus, these acetyltransferases act to suppress JNK signaling via targeting different pathway components, not just the JNKs themselves.
Bacterial proteases can elicit similarly insidious effects. The NleD protease produced by enteropathogenic Escherichia coli (EPEC) strains cleaves JNK at its activation loop, resulting in JNK degradation and immune suppression (453). The lethal factor (LF) of Bacillus anthracis exerts its effects by a more complex way: the catalytic site of this protease recognizes substrates with a consensus similar to many MAPK docking motifs found in the host and is known to cleave the N-terminal docking motifs from MAP2Ks (454). The truncated MAP2Ks are unable to propagate ERK1/2, JNK, and p38 signaling, and they may even act as dominant-negative inhibitors at the MAP3K level of these MAPK pathways. The G63 protease produced by the eukaryotic intracellular parasite Leishmania major cleaves multiple substrates within host cells, including the adaptor protein Tab1 (455). Without this critical scaffolding component acting at the MAP3K level, the TNF-α, Toll, and NOD receptors are unable to recruit the TAK1 kinase, which is necessary for both JNK and p38 pathway activation.
Some pathogen-derived enzymes act through different, perhaps more exotic mechanisms. The OspF enzyme of Shigella flexneri, originally believed to be a phosphatase, is a MAPK-phosphothreonine-lyase (456). After removal of a phosphate from the activation loop of MAPKs, the lyase leaves no hydroxyl group behind, making rephosphorylation and hence MAPK reactivation impossible (457). Although OspF displays a much higher activity on ERK2 or p38 than on JNKs, the related Spvc enzyme from Salmonella Typhimurium targets phospho-JNK and phospho-ERK2 (458). Interestingly, even plant pathogens use similar enzymes (such as HopAI1 from Pseudomonas syringae) to interfere with MAPK-driven innate immunity responses of the host (459). Members of the Theileria genus of intracellular parasites (responsible for human as well as animal diseases) are unique among eukaryotes in that they can induce oncogenic transformation of infected cells. In the case of Theileria annulata, oncogenic transformation is elicited via secretion of the protein TaPIN1 into the host cells. This enzyme is related to the mammalian Pin1 peptidyl-prolyl cis-trans isomerase and acts by disabling the host FBW7 ubiquitin ligase system, thereby allowing c-Jun overproduction (460).
While suppression of JNK activity is advantageous to many pathogens, there are some pathogens that intentionally activate the JNK pathway. The latent membrane protein 1 (LMP1) encoded by Epstein-Barr virus is absolutely necessary to promote survival of infected B lymphocytes. LMP1 is a multipass membrane protein with an N-terminal oligomerization domain and a C-terminal cytoplasmic segment. The latter contains two critical linear motifs, one for the recruitment of TRAF6 and another for the interaction with TRADD that is necessary to recruit TRAF2 (461). Thus, LMP1 can mimic activated TNF-α-like receptors; it interferes with the B cell proliferation response by turning on TRAFs and increasing JNK and NF-κB activation. In infected B cells, JNK pathway activation can promote survival and stop apoptosis when coactivated with NF-κB (462). Furthermore, several other viruses also intentionally activate the JNK pathway as a mechanism in the induction of cell cycle arrest or even apoptosis. This can help human immunodeficiency virus (HIV) to modulate T cell functions, enhance varicella-zoster virus (VZV) replication more efficiently, and promote virion release for coxsackievirus group B3 (463–465). However, the molecular-level details of these latter interactions await characterization.
CONCLUSIONS
JNK functions in specialized tissues can greatly differ. However, one overarching problem remains unresolved even after decades of research: how do JNK pathways “decide” between responses that promote survival and those ultimately leading to cell death?
Experimental findings suggest multiple possible answers to this conundrum. That overexpression or overactivation of JNK1 almost invariably results in cell death in most cell types suggests that the level of JNK pathway activation must be important. In this “threshold model,” low levels of JNK activity drive essentially prosurvival responses, while overactivation causes switching to cell cycle arrest and sensitization to apoptotic stimuli. The observation that some JNK pathways contain positive feedback loops is significant in this regard. Other experiments have suggested the importance of spatiotemporal separation of JNK activity. In this “localization model,” cytoplasmic JNK activity is part of normal developmental procedures, while the actions of activated JNKs in the nucleus trigger events culminating apoptosis. These two models do not necessarily contradict each other. In the context of neuroblasts, highly localized JNK activity is detected at the axonal growth cone tips (122) and is required to guide the axon to its destination as part of normal physiology. On the other hand, an increase of JNK activity around the nucleus would prevent growth and, if sustained, drive the outcome toward cell death.
The subcellular localizations of JNKs and their upstream regulators will continue to add further complexities to our understanding of JNK signal transduction pathways. Thus, apart from the well-accepted roles for JNKs in their phosphorylation of nuclear substrates, their roles in regulating cytoplasmic targets and mitochondrial proteins are emerging. In addition, studies on upstream kinases have begun to reveal the importance of their localization in influencing JNK pathway actions (466, 467). More in-depth analyses of compartmentalization of JNK signaling will require greater development and use of biosensors capable of reporting JNK activities in different subcellular locations. This has been shown with a Jun-based fluorescence resonance energy transfer (FRET) biosensor, which reported JNK activity and its subsequent targeting to different intracellular locations (468). Furthermore, such cell-based assays have and will allow the examination of system-level behavior of the JNK cascade and have been instrumental in revealing the all-or-none behavior of stress-activated JNK signaling (468). Clearly, a more thorough consideration of the time course of JNK activation will also be critical in this context (469).
Lastly, greater efforts must be directed toward understanding the complexities of the JNK signaling events at a theoretical level if we are to integrate the information on upstream regulatory events, scaffolds, and substrates into a working model of JNK actions. Of interest are recently developed models of JNK activation that indicate that the double phosphorylation events observed at numerous upstream points in the MAPK pathways, although not as efficient as monophosphorylation events, may have evolved together with scaffold proteins to drive both tighter control and higher specificity in signaling events (470). After >20 years of intense interest and research on JNKs, we propose that the baffling complexity of JNK signaling may be best tackled by the exploration of the complex protein-protein interactions of the JNKs with their regulators and substrates. These studies will provide new mechanistic insights into how JNKs can integrate their protein kinase activity into higher-order protein network activities at the cellular or organism level. We anticipate that the next decade will bring a more quantitative, system-level understanding of JNK signaling.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an Australian Research Council Discovery Grant (DP160100374, awarded to M.A.B. and A.R.) and an OTKA grant (NN 114309 awarded to A.R.). A.R. is the recipient of the Lendület grant from the Hungarian Academy of Sciences (LP2013-57).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MMBR.00043-14.
REFERENCES
- 1.Bogoyevitch MA, Kobe B. 2006. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev 70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coffey ET. 2014. Nuclear and cytosolic JNK signalling in neurons. Nat Rev Neurosci 15:285–299. doi: 10.1038/nrn3729. [DOI] [PubMed] [Google Scholar]
- 3.Bubici C, Papa S. 2014. JNK signalling in cancer: in need of new, smarter therapeutic targets. Br J Pharmacol 171:24–37. doi: 10.1111/bph.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogoyevitch MA, Arthur PG. 2008. Inhibitors of c-Jun N-terminal kinases: JuNK no more? Biochim Biophys Acta 1784:76–93. doi: 10.1016/j.bbapap.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabapathy K. 2012. Role of the JNK pathway in human diseases. Prog Mol Biol Transl Sci 106:145–169. doi: 10.1016/B978-0-12-396456-4.00013-4. [DOI] [PubMed] [Google Scholar]
- 6.Zhao B, Tumaneng K, Guan KL. 2011. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol 13:877–883. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shackelford DB, Shaw RJ. 2009. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. 2000. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 9.Sano R, Reed JC. 2013. ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. 2006. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol 26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Q, Kim YS, Lin Y, Lewis J, Neckers L, Liu ZG. 2006. Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress-induced activation of the MAP kinase JNK. EMBO Rep 7:622–627. doi: 10.1038/sj.embor.7400687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosette C, Karin M. 1996. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 13.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 14.Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. 2004. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell 15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Brown K, Vial SC, Dedi N, Long JM, Dunster NJ, Cheetham GM. 2005. Structural basis for the interaction of TAK1 kinase with its activating protein TAB1. J Mol Biol 354:1013–1020. doi: 10.1016/j.jmb.2005.09.098. [DOI] [PubMed] [Google Scholar]
- 16.Fan Y, Yu Y, Shi Y, Sun W, Xie M, Ge N, Mao R, Chang A, Xu G, Schneider MD, Zhang H, Fu S, Qin J, Yang J. 2010. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J Biol Chem 285:5347–5360. doi: 10.1074/jbc.M109.076976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan Y, Yu Y, Mao R, Zhang H, Yang J. 2011. TAK1 Lys-158 but not Lys-209 is required for IL-1beta-induced Lys63-linked TAK1 polyubiquitination and IKK/NF-kappaB activation. Cell Signal 23:660–665. doi: 10.1016/j.cellsig.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen IT, Hsu PH, Hsu WC, Chen NJ, Tseng PH. 2015. Polyubiquitination of transforming growth factor beta-activated kinase 1 (TAK1) at lysine 562 residue regulates TLR4-mediated JNK and p38 MAPK activation. Sci Rep 5:12300. doi: 10.1038/srep12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallagher ED, Gutowski S, Sternweis PC, Cobb MH. 2004. RhoA binds to the amino terminus of MEKK1 and regulates its kinase activity. J Biol Chem 279:1872–1877. doi: 10.1074/jbc.M309525200. [DOI] [PubMed] [Google Scholar]
- 20.Charlaftis N, Suddason T, Wu X, Anwar S, Karin M, Gallagher E. 2014. The MEKK1 PHD ubiquitinates TAB1 to activate MAPKs in response to cytokines. EMBO J 33:2581–2596. doi: 10.15252/embj.201488351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suddason T, Gallagher E. 2015. A RING to rule them all? Insights into the Map3k1 PHD motif provide a new mechanistic understanding into the diverse roles of Map3k1. Cell Death Differ 22:540–548. doi: 10.1038/cdd.2014.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suddason T, Anwar S, Charlaftis N, Gallagher E. 2016. T-cell-specific deletion of Map3k1 reveals the critical role for Mekk1 and Jnks in Cdkn1b-dependent proliferative expansion. Cell Rep 14:449–457. doi: 10.1016/j.celrep.2015.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mita H, Tsutsui J, Takekawa M, Witten EA, Saito H. 2002. Regulation of MTK1/MEKK4 kinase activity by its N-terminal autoinhibitory domain and GADD45 binding. Mol Cell Biol 22:4544–4555. doi: 10.1128/MCB.22.13.4544-4555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abell AN, Johnson GL. 2005. MEKK4 is an effector of the embryonic TRAF4 for JNK activation. J Biol Chem 280:35793–35796. doi: 10.1074/jbc.C500260200. [DOI] [PubMed] [Google Scholar]
- 25.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. 1998. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katagiri K, Matsuzawa A, Ichijo H. 2010. Regulation of apoptosis signal-regulating kinase 1 in redox signaling. Methods Enzymol 474:277–288. doi: 10.1016/S0076-6879(10)74016-7. [DOI] [PubMed] [Google Scholar]
- 27.Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkind JS. 1996. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J Biol Chem 271:27225–27228. [DOI] [PubMed] [Google Scholar]
- 28.Matitau AE, Gabor TV, Gill RM, Scheid MP. 2013. MEKK2 kinase association with 14-3-3 protein regulates activation of c-Jun N-terminal kinase. J Biol Chem 288:28293–28302. doi: 10.1074/jbc.M113.511352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeda AN, Oberoi-Khanuja TK, Glatz G, Schulenburg K, Scholz RP, Carpy A, Macek B, Remenyi A, Rajalingam K. 2014. Ubiquitin-dependent regulation of MEKK2/3-MEK5-ERK5 signaling module by XIAP and cIAP1. EMBO J 33:1784–1801. doi: 10.15252/embj.201487808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du Y, Bock BC, Schachter KA, Chao M, Gallo KA. 2005. Cdc42 induces activation loop phosphorylation and membrane targeting of mixed lineage kinase 3. J Biol Chem 280:42984–42993. doi: 10.1074/jbc.M502671200. [DOI] [PubMed] [Google Scholar]
- 31.Chadee DN, Yuasa T, Kyriakis JM. 2002. Direct activation of mitogen-activated protein kinase kinase kinase MEKK1 by the Ste20p homologue GCK and the adapter protein TRAF2. Mol Cell Biol 22:737–749. doi: 10.1128/MCB.22.3.737-749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng J, Yu L, Zhang D, Huang Q, Spencer D, Su B. 2005. Dimerization through the catalytic domain is essential for MEKK2 activation. J Biol Chem 280:13477–13482. doi: 10.1074/jbc.M414258200. [DOI] [PubMed] [Google Scholar]
- 33.Luo Z, Tzivion G, Belshaw PJ, Vavvas D, Marshall M, Avruch J. 1996. Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature 383:181–185. doi: 10.1038/383181a0. [DOI] [PubMed] [Google Scholar]
- 34.Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, Bravo BJ, Giannetti AM, Peck A, Masselot A, Morales T, Smith D, Brandhuber BJ, Hymowitz SG, Malek S. 2014. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 26:402–413. doi: 10.1016/j.ccr.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, Therrien M. 2015. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol 22:37–43. doi: 10.1038/nsmb.2924. [DOI] [PubMed] [Google Scholar]
- 36.Bunkoczi G, Salah E, Filippakopoulos P, Fedorov O, Muller S, Sobott F, Parker SA, Zhang H, Min W, Turk BE, Knapp S. 2007. Structural and functional characterization of the human protein kinase ASK1. Structure 15:1215–1226. doi: 10.1016/j.str.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leung IW, Lassam N. 1998. Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J Biol Chem 273:32408–32415. doi: 10.1074/jbc.273.49.32408. [DOI] [PubMed] [Google Scholar]
- 38.Ortner E, Moelling K. 2007. Heteromeric complex formation of ASK2 and ASK1 regulates stress-induced signaling. Biochem Biophys Res Commun 362:454–459. doi: 10.1016/j.bbrc.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 39.Takeda K, Shimozono R, Noguchi T, Umeda T, Morimoto Y, Naguro I, Tobiume K, Saitoh M, Matsuzawa A, Ichijo H. 2007. Apoptosis signal-regulating kinase (ASK) 2 functions as a mitogen-activated protein kinase kinase kinase in a heteromeric complex with ASK1. J Biol Chem 282:7522–7531. doi: 10.1074/jbc.M607177200. [DOI] [PubMed] [Google Scholar]
- 40.Tegethoff S, Behlke J, Scheidereit C. 2003. Tetrameric oligomerization of IkappaB kinase gamma (IKKgamma) is obligatory for IKK complex activity and NF-kappaB activation. Mol Cell Biol 23:2029–2041. doi: 10.1128/MCB.23.6.2029-2041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polley S, Huang DB, Hauenstein AV, Fusco AJ, Zhong X, Vu D, Schrofelbauer B, Kim Y, Hoffmann A, Verma IM, Ghosh G, Huxford T. 2013. A structural basis for IkappaB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS Biol 11:e1001581. doi: 10.1371/journal.pbio.1001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brummer T, Naegele H, Reth M, Misawa Y. 2003. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of B-Raf. Oncogene 22:8823–8834. doi: 10.1038/sj.onc.1207185. [DOI] [PubMed] [Google Scholar]
- 43.Ritt DA, Monson DM, Specht SI, Morrison DK. 2010. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol 30:806–819. doi: 10.1128/MCB.00569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huntwork-Rodriguez S, Wang B, Watkins T, Ghosh AS, Pozniak CD, Bustos D, Newton K, Kirkpatrick DS, Lewcock JW. 2013. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J Cell Biol 202:747–763. doi: 10.1083/jcb.201303066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abell AN, Granger DA, Johnson GL. 2007. MEKK4 stimulation of p38 and JNK activity is negatively regulated by GSK3beta. J Biol Chem 282:30476–30484. doi: 10.1074/jbc.M705783200. [DOI] [PubMed] [Google Scholar]
- 46.Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin Y. 2005. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell 120:407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 47.Baker ST, Opperman KJ, Tulgren ED, Turgeon SM, Bienvenut W, Grill B. 2014. RPM-1 uses both ubiquitin ligase and phosphatase-based mechanisms to regulate DLK-1 during neuronal development. PLoS Genet 10:e1004297. doi: 10.1371/journal.pgen.1004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H. 2001. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J 20:6028–6036. doi: 10.1093/emboj/20.21.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho YC, Park JE, Park BC, Kim JH, Jeong DG, Park SG, Cho S. 2015. Cell cycle-dependent Cdc25C phosphatase determines cell survival by regulating apoptosis signal-regulating kinase 1. Cell Death Differ 22:1605–1617. doi: 10.1038/cdd.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zou X, Tsutsui T, Ray D, Blomquist JF, Ichijo H, Ucker DS, Kiyokawa H. 2001. The cell cycle-regulatory CDC25A phosphatase inhibits apoptosis signal-regulating kinase 1. Mol Cell Biol 21:4818–4828. doi: 10.1128/MCB.21.14.4818-4828.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saito J, Toriumi S, Awano K, Ichijo H, Sasaki K, Kobayashi T, Tamura S. 2007. Regulation of apoptosis signal-regulating kinase 1 by protein phosphatase 2Cepsilon. Biochem J 405:591–596. doi: 10.1042/BJ20070231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim SI, Kwak JH, Wang L, Choi ME. 2008. Protein phosphatase 2A is a negative regulator of transforming growth factor-beta1-induced TAK1 activation in mesangial cells. J Biol Chem 283:10753–10763. doi: 10.1074/jbc.M801263200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kajino T, Ren H, Iemura S, Natsume T, Stefansson B, Brautigan DL, Matsumoto K, Ninomiya-Tsuji J. 2006. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J Biol Chem 281:39891–39896. doi: 10.1074/jbc.M608155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsumoto T, Kinoshita T, Kirii Y, Yokota K, Hamada K, Tada T. 2010. Crystal structures of MKK4 kinase domain reveal that substrate peptide binds to an allosteric site and induces an auto-inhibition state. Biochem Biophys Res Commun 400:369–373. doi: 10.1016/j.bbrc.2010.08.071. [DOI] [PubMed] [Google Scholar]
- 55.Matsumoto T, Kinoshita T, Matsuzaka H, Nakai R, Kirii Y, Yokota K, Tada T. 2012. Crystal structure of non-phosphorylated MAP2K6 in a putative auto-inhibition state. J Biochem 151:541–549. doi: 10.1093/jb/mvs023. [DOI] [PubMed] [Google Scholar]
- 56.Sogabe Y, Matsumoto T, Hashimoto T, Kirii Y, Sawa M, Kinoshita T. 2015. 5Z-7-Oxozeaenol covalently binds to MAP2K7 at Cys218 in an unprecedented manner. Bioorg Med Chem Lett 25:593–596. doi: 10.1016/j.bmcl.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 57.Takekawa M, Tatebayashi K, Saito H. 2005. Conserved docking site is essential for activation of mammalian MAP kinase kinases by specific MAP kinase kinase kinases. Mol Cell 18:295–306. doi: 10.1016/j.molcel.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 58.Mooney LM, Whitmarsh AJ. 2004. Docking interactions in the c-Jun N-terminal kinase pathway. J Biol Chem 279:11843–11852. doi: 10.1074/jbc.M311841200. [DOI] [PubMed] [Google Scholar]
- 59.Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. 2001. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev 15:1419–1426. doi: 10.1101/gad.888501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lisnock J, Griffin P, Calaycay J, Frantz B, Parsons J, O'Keefe SJ, LoGrasso P. 2000. Activation of JNK3 alpha 1 requires both MKK4 and MKK7: kinetic characterization of in vitro phosphorylated JNK3 alpha 1. Biochemistry 39:3141–3148. doi: 10.1021/bi992410+. [DOI] [PubMed] [Google Scholar]
- 61.Fleming Y, Armstrong CG, Morrice N, Paterson A, Goedert M, Cohen P. 2000. Synergistic activation of stress-activated protein kinase 1/c-Jun N-terminal kinase (SAPK1/JNK) isoforms by mitogen-activated protein kinase kinase 4 (MKK4) and MKK7. Biochem J 352:145–154. doi: 10.1042/bj3520145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang X, Destrument A, Tournier C. 2007. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta 1773:1349–1357. doi: 10.1016/j.bbamcr.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 63.von Kriegsheim A, Pitt A, Grindlay GJ, Kolch W, Dhillon AS. 2006. Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat Cell Biol 8:1011–1016. doi: 10.1038/ncb1465. [DOI] [PubMed] [Google Scholar]
- 64.Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. 2003. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol 13:1356–1364. doi: 10.1016/S0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- 65.Zhou G, Golden T, Aragon IV, Honkanen RE. 2004. Ser/Thr protein phosphatase 5 inactivates hypoxia-induced activation of an apoptosis signal-regulating kinase 1/MKK-4/JNK signaling cascade. J Biol Chem 279:46595–46605. doi: 10.1074/jbc.M408320200. [DOI] [PubMed] [Google Scholar]
- 66.Chen L, Liu L, Huang S. 2008. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med 45:1035–1044. doi: 10.1016/j.freeradbiomed.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 67.Sekine Y, Hatanaka R, Watanabe T, Sono N, Iemura S, Natsume T, Kuranaga E, Miura M, Takeda K, Ichijo H. 2012. The Kelch repeat protein KLHDC10 regulates oxidative stress-induced ASK1 activation by suppressing PP5. Mol Cell 48:692–704. doi: 10.1016/j.molcel.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 68.Vazquez-Carballo A, Ceperuelo-Mallafre V, Chacon MR, Maymo-Masip E, Lorenzo M, Porras A, Vendrell J, Fernandez-Veledo S. 2013. TWEAK prevents TNF-alpha-induced insulin resistance through PP2A activation in human adipocytes. Am J Physiol Endocrinol Metab 305:E101–E112. doi: 10.1152/ajpendo.00589.2012. [DOI] [PubMed] [Google Scholar]
- 69.Whisenant TC, Ho DT, Benz RW, Rogers JS, Kaake RM, Gordon EA, Huang L, Baldi P, Bardwell L. 2010. Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput Biol 6:e1000908. doi: 10.1371/journal.pcbi.1000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mustelin T, Tautz L, Page R. 2005. Structure of the hematopoietic tyrosine phosphatase (HePTP) catalytic domain: structure of a KIM phosphatase with phosphate bound at the active site. J Mol Biol 354:150–163. doi: 10.1016/j.jmb.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 71.Awano K, Amano K, Nagaura Y, Kanno S, Echigo S, Tamura S, Kobayashi T. 2008. Phosphorylation of protein phosphatase 2Czeta by c-Jun NH2-terminal kinase at Ser92 attenuates its phosphatase activity. Biochemistry 47:7248–7255. doi: 10.1021/bi800067p. [DOI] [PubMed] [Google Scholar]
- 72.Wu JJ, Bennett AM. 2005. Essential role for mitogen-activated protein (MAP) kinase phosphatase-1 in stress-responsive MAP kinase and cell survival signaling. J Biol Chem 280:16461–16466. doi: 10.1074/jbc.M501762200. [DOI] [PubMed] [Google Scholar]
- 73.Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, Chang CH, Liu Y. 2006. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med 203:131–140. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi H, Verma M, Zhang L, Dong C, Flavell RA, Bennett AM. 2013. Improved regenerative myogenesis and muscular dystrophy in mice lacking Mkp5. J Clin Invest 123:2064–2077. doi: 10.1172/JCI64375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Blattman JN, Kennedy NJ, Duong J, Nguyen T, Wang Y, Davis RJ, Greenberg PD, Flavell RA, Dong C. 2004. Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature 430:793–797. doi: 10.1038/nature02764. [DOI] [PubMed] [Google Scholar]
- 76.Tanoue T, Moriguchi T, Nishida E. 1999. Molecular cloning and characterization of a novel dual specificity phosphatase, MKP-5. J Biol Chem 274:19949–19956. doi: 10.1074/jbc.274.28.19949. [DOI] [PubMed] [Google Scholar]
- 77.Tanoue T, Yamamoto T, Maeda R, Nishida E. 2001. A novel MAPK phosphatase MKP-7 acts preferentially on JNK/SAPK and p38 alpha and beta MAPKs. J Biol Chem 276:26629–26639. doi: 10.1074/jbc.M101981200. [DOI] [PubMed] [Google Scholar]
- 78.Bordo D, Bork P. 2002. The rhodanese/Cdc25 phosphatase superfamily. Sequence-structure-function relations. EMBO Rep 3:741–746. doi: 10.1093/embo-reports/kvf150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang YY, Wu JW, Wang ZX. 2011. A distinct interaction mode revealed by the crystal structure of the kinase p38alpha with the MAPK binding domain of the phosphatase MKP5. Sci Signal 4:ra88. doi: 10.1126/scisignal.2002241. [DOI] [PubMed] [Google Scholar]
- 80.Zeke A, Bastys T, Alexa A, Garai A, Meszaros B, Kirsch K, Dosztanyi Z, Kalinina OV, Remenyi A. 2015. Systematic discovery of linear binding motifs targeting an ancient protein interaction surface on MAP kinases. Mol Syst Biol 11:837. doi: 10.15252/msb.20156269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu X, Zhang CS, Lu C, Lin SC, Wu JW, Wang ZX. 2016. A conserved motif in JNK/p38-specific MAPK phosphatases as a determinant for JNK1 recognition and inactivation. Nat Commun 7:10879. doi: 10.1038/ncomms10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang Y, Reynolds JM, Chang SH, Martin-Orozco N, Chung Y, Nurieva RI, Dong C. 2009. MKP-1 is necessary for T cell activation and function. J Biol Chem 284:30815–30824. doi: 10.1074/jbc.M109.052472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeanneteau F, Deinhardt K, Miyoshi G, Bennett AM, Chao MV. 2010. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat Neurosci 13:1373–1379. doi: 10.1038/nn.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kristiansen M, Hughes R, Patel P, Jacques TS, Clark AR, Ham J. 2010. Mkp1 is a c-Jun target gene that antagonizes JNK-dependent apoptosis in sympathetic neurons. J Neurosci 30:10820–10832. doi: 10.1523/JNEUROSCI.2824-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Comalada M, Lloberas J, Celada A. 2012. MKP-1: a critical phosphatase in the biology of macrophages controlling the switch between proliferation and activation. Eur J Immunol 42:1938–1948. doi: 10.1002/eji.201242441. [DOI] [PubMed] [Google Scholar]
- 86.Yang CY, Li JP, Chiu LL, Lan JL, Chen DY, Chuang HC, Huang CY, Tan TH. 2014. Dual-specificity phosphatase 14 (DUSP14/MKP6) negatively regulates TCR signaling by inhibiting TAB1 activation. J Immunol 192:1547–1557. doi: 10.4049/jimmunol.1300989. [DOI] [PubMed] [Google Scholar]
- 87.Li JP, Fu YN, Chen YR, Tan TH. 2010. JNK pathway-associated phosphatase dephosphorylates focal adhesion kinase and suppresses cell migration. J Biol Chem 285:5472–5478. doi: 10.1074/jbc.M109.060186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zheng H, Li Q, Chen R, Zhang J, Ran Y, He X, Li S, Shu HB. 2013. The dual-specificity phosphatase DUSP14 negatively regulates tumor necrosis factor- and interleukin-1-induced nuclear factor-kappaB activation by dephosphorylating the protein kinase TAK1. J Biol Chem 288:819–825. doi: 10.1074/jbc.M112.412643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen AJ, Zhou G, Juan T, Colicos SM, Cannon JP, Cabriera-Hansen M, Meyer CF, Jurecic R, Copeland NG, Gilbert DJ, Jenkins NA, Fletcher F, Tan TH, Belmont JW. 2002. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J Biol Chem 277:36592–36601. doi: 10.1074/jbc.M200453200. [DOI] [PubMed] [Google Scholar]
- 90.Shapiro PS, Ahn NG. 1998. Feedback regulation of Raf-1 and mitogen-activated protein kinase (MAP) kinase kinases 1 and 2 by MAP kinase phosphatase-1 (MKP-1). J Biol Chem 273:1788–1793. doi: 10.1074/jbc.273.3.1788. [DOI] [PubMed] [Google Scholar]
- 91.Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A. 1998. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 12:557–570. doi: 10.1101/gad.12.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garver LS, de Almeida Oliveira G, Barillas-Mury C. 2013. The JNK pathway is a key mediator of Anopheles gambiae antiplasmodial immunity. PLoS Pathog 9:e1003622. doi: 10.1371/journal.ppat.1003622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hofken T, Keller N, Fleischer F, Goke B, Wagner AC. 2000. Map kinase phosphatases (MKP's) are early responsive genes during induction of cerulein hyperstimulation pancreatitis. Biochem Biophys Res Commun 276:680–685. doi: 10.1006/bbrc.2000.3530. [DOI] [PubMed] [Google Scholar]
- 94.Staples CJ, Owens DM, Maier JV, Cato AC, Keyse SM. 2010. Cross-talk between the p38alpha and JNK MAPK pathways mediated by MAP kinase phosphatase-1 determines cellular sensitivity to UV radiation. J Biol Chem 285:25928–25940. doi: 10.1074/jbc.M110.117911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sanchez-Tillo E, Comalada M, Xaus J, Farrera C, Valledor AF, Caelles C, Lloberas J, Celada A. 2007. JNK1 Is required for the induction of Mkp1 expression in macrophages during proliferation and lipopolysaccharide-dependent activation. J Biol Chem 282:12566–12573. doi: 10.1074/jbc.M609662200. [DOI] [PubMed] [Google Scholar]
- 96.Schachter KA, Du Y, Lin A, Gallo KA. 2006. Dynamic positive feedback phosphorylation of mixed lineage kinase 3 by JNK reversibly regulates its distribution to Triton-soluble domains. J Biol Chem 281:19134–19144. doi: 10.1074/jbc.M603324200. [DOI] [PubMed] [Google Scholar]
- 97.Ferrell JE, Xiong W. 2001. Bistability in cell signaling: how to make continuous processes discontinuous, and reversible processes irreversible. Chaos 11:227–236. doi: 10.1063/1.1349894. [DOI] [PubMed] [Google Scholar]
- 98.Morrison DK, Davis RJ. 2003. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol 19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 99.Shaw AS, Filbert EL. 2009. Scaffold proteins and immune-cell signalling. Nat Rev Immunol 9:47–56. doi: 10.1038/nri2473. [DOI] [PubMed] [Google Scholar]
- 100.Zeke A, Lukacs M, Lim WA, Remenyi A. 2009. Scaffolds: interaction platforms for cellular signalling circuits. Trends Cell Biol 19:364–374. doi: 10.1016/j.tcb.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Good MC, Zalatan JG, Lim WA. 2011. Scaffold proteins: hubs for controlling the flow of cellular information. Science 332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R, Cancer Genome Project. 2004. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867. doi: 10.1016/S0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 103.Park S, Rath O, Beach S, Xiang X, Kelly SM, Luo Z, Kolch W, Yeung KC. 2006. Regulation of RKIP binding to the N-region of the Raf-1 kinase. FEBS Lett 580:6405–6412. doi: 10.1016/j.febslet.2006.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nihalani D, Wong HN, Holzman LB. 2003. Recruitment of JNK to JIP1 and JNK-dependent JIP1 phosphorylation regulates JNK module dynamics and activation. J Biol Chem 278:28694–28702. doi: 10.1074/jbc.M304212200. [DOI] [PubMed] [Google Scholar]
- 105.Matsuura H, Nishitoh H, Takeda K, Matsuzawa A, Amagasa T, Ito M, Yoshioka K, Ichijo H. 2002. Phosphorylation-dependent scaffolding role of JSAP1/JIP3 in the ASK1-JNK signaling pathway. A new mode of regulation of the MAP kinase cascade. J Biol Chem 277:40703–40709. doi: 10.1074/jbc.M202004200. [DOI] [PubMed] [Google Scholar]
- 106.Song X, Coffa S, Fu H, Gurevich VV. 2009. How does arrestin assemble MAPKs into a signaling complex? J Biol Chem 284:685–695. doi: 10.1074/jbc.M806124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guo C, Whitmarsh AJ. 2008. The beta-arrestin-2 scaffold protein promotes c-Jun N-terminal kinase-3 activation by binding to its nonconserved N terminus. J Biol Chem 283:15903–15911. doi: 10.1074/jbc.M710006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Marti A, Luo Z, Cunningham C, Ohta Y, Hartwig J, Stossel TP, Kyriakis JM, Avruch J. 1997. Actin-binding protein-280 binds the stress-activated protein kinase (SAPK) activator SEK-1 and is required for tumor necrosis factor-alpha activation of SAPK in melanoma cells. J Biol Chem 272:2620–2628. doi: 10.1074/jbc.272.5.2620. [DOI] [PubMed] [Google Scholar]
- 109.Jeon YJ, Choi JS, Lee JY, Yu KR, Ka SH, Cho Y, Choi EJ, Baek SH, Seol JH, Park D, Bang OS, Chung CH. 2008. Filamin B serves as a molecular scaffold for type I interferon-induced c-Jun NH2-terminal kinase signaling pathway. Mol Biol Cell 19:5116–5130. doi: 10.1091/mbc.E08-06-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lopez-Bergami P, Habelhah H, Bhoumik A, Zhang W, Wang LH, Ronai Z. 2005. RACK1 mediates activation of JNK by protein kinase C. Mol Cell 19:309–320. doi: 10.1016/j.molcel.2005.06.025 (Erratum, 19:578-.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Girardin SE, Yaniv M. 2001. A direct interaction between JNK1 and CrkII is critical for Rac1-induced JNK activation. EMBO J 20:3437–3446. doi: 10.1093/emboj/20.13.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu Z, Kukekov NV, Greene LA. 2003. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J 22:252–261. doi: 10.1093/emboj/cdg021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cohen-Katsenelson K, Wasserman T, Khateb S, Whitmarsh AJ, Aronheim A. 2011. Docking interactions of the JNK scaffold protein WDR62. Biochem J 439:381–390. doi: 10.1042/BJ20110284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zama T, Aoki R, Kamimoto T, Inoue K, Ikeda Y, Hagiwara M. 2002. Scaffold role of a mitogen-activated protein kinase phosphatase, SKRP1, for the JNK signaling pathway. J Biol Chem 277:23919–23926. doi: 10.1074/jbc.M200838200. [DOI] [PubMed] [Google Scholar]
- 115.Ye B, Yu WP, Thomas GM, Huganir RL. 2007. GRASP-1 is a neuronal scaffold protein for the JNK signaling pathway. FEBS Lett 581:4403–4410. doi: 10.1016/j.febslet.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shi F, Lemmon MA. 2011. Biochemistry KSR plays CRAF-ty. Science 332:1043–1044. doi: 10.1126/science.1208063. [DOI] [PubMed] [Google Scholar]
- 117.Brennan DF, Dar AC, Hertz NT, Chao WC, Burlingame AL, Shokat KM, Barford D. 2011. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472:366–369. doi: 10.1038/nature09860. [DOI] [PubMed] [Google Scholar]
- 118.McKay MM, Ritt DA, Morrison DK. 2009. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci U S A 106:11022–11027. doi: 10.1073/pnas.0901590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. 1999. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol 19:7245–7254. doi: 10.1128/MCB.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. 1998. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science 281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 121.Fu MM, Holzbaur EL. 2013. JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J Cell Biol 202:495–508. doi: 10.1083/jcb.201302078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Oliva AA Jr, Atkins CM, Copenagle L, Banker GA. 2006. Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci 26:9462–9470. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Good M, Tang G, Singleton J, Remenyi A, Lim WA. 2009. The Ste5 scaffold directs mating signaling by catalytically unlocking the Fus3 MAP kinase for activation. Cell 136:1085–1097. doi: 10.1016/j.cell.2009.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Moriguchi T, Toyoshima F, Masuyama N, Hanafusa H, Gotoh Y, Nishida E. 1997. A novel SAPK/JNK kinase, MKK7, stimulated by TNFalpha and cellular stresses. EMBO J 16:7045–7053. doi: 10.1093/emboj/16.23.7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ho DT, Bardwell AJ, Grewal S, Iverson C, Bardwell L. 2006. Interacting JNK-docking sites in MKK7 promote binding and activation of JNK mitogen-activated protein kinases. J Biol Chem 281:13169–13179. doi: 10.1074/jbc.M601010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kragelj J, Palencia A, Nanao MH, Maurin D, Bouvignies G, Blackledge M, Jensen MR. 2015. Structure and dynamics of the MKK7-JNK signaling complex. Proc Natl Acad Sci U S A 112:3409–3414. doi: 10.1073/pnas.1419528112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Koushika SP. 2008. “JIP”ing along the axon: the complex roles of JIPs in axonal transport. Bioessays 30:10–14. doi: 10.1002/bies.20695. [DOI] [PubMed] [Google Scholar]
- 128.Satake T, Otsuki K, Banba Y, Suenaga J, Hirano H, Yamanaka Y, Ohno S, Hirai S. 2013. The interaction of Kinesin-1 with its adaptor protein JIP1 can be regulated via proteins binding to the JIP1-PTB domain. BMC Cell Biol 14:12. doi: 10.1186/1471-2121-14-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.GTEx Consortium. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Blasius TL, Cai D, Jih GT, Toret CP, Verhey KJ. 2007. Two binding partners cooperate to activate the molecular motor Kinesin-1. J Cell Biol 176:11–17. doi: 10.1083/jcb.200605099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Smith MJ, Hardy WR, Murphy JM, Jones N, Pawson T. 2006. Screening for PTB domain binding partners and ligand specificity using proteome-derived NPXY peptide arrays. Mol Cell Biol 26:8461–8474. doi: 10.1128/MCB.01491-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tamayev R, Zhou D, D'Adamio L. 2009. The interactome of the amyloid beta precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol Neurodegener 4:28. doi: 10.1186/1750-1326-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kristensen O, Guenat S, Dar I, Allaman-Pillet N, Abderrahmani A, Ferdaoussi M, Roduit R, Maurer F, Beckmann JS, Kastrup JS, Gajhede M, Bonny C. 2006. A unique set of SH3-SH3 interactions controls IB1 homodimerization. EMBO J 25:785–797. doi: 10.1038/sj.emboj.7600982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Scheinfeld MH, Roncarati R, Vito P, Lopez PA, Abdallah M, D'Adamio L. 2002. Jun NH2-terminal kinase (JNK) interacting protein 1 (JIP1) binds the cytoplasmic domain of the Alzheimer's beta-amyloid precursor protein (APP). J Biol Chem 277:3767–3775. doi: 10.1074/jbc.M108357200. [DOI] [PubMed] [Google Scholar]
- 135.Stockinger W, Brandes C, Fasching D, Hermann M, Gotthardt M, Herz J, Schneider WJ, Nimpf J. 2000. The reelin receptor ApoER2 recruits JNK-interacting proteins-1 and -2. J Biol Chem 275:25625–25632. doi: 10.1074/jbc.M004119200. [DOI] [PubMed] [Google Scholar]
- 136.Meyer D, Liu A, Margolis B. 1999. Interaction of c-Jun amino-terminal kinase interacting protein-1 with p190 RhoGEF and its localization in differentiated neurons. J Biol Chem 274:35113–35118. doi: 10.1074/jbc.274.49.35113. [DOI] [PubMed] [Google Scholar]
- 137.Chuderland D, Konson A, Seger R. 2008. Identification and characterization of a general nuclear translocation signal in signaling proteins. Mol Cell 31:850–861. doi: 10.1016/j.molcel.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 138.Zehorai E, Seger R. 2014. Beta-like importins mediate the nuclear translocation of mitogen-activated protein kinases. Mol Cell Biol 34:259–270. doi: 10.1128/MCB.00799-13. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 139.Khokhlatchev AV, Canagarajah B, Wilsbacher J, Robinson M, Atkinson M, Goldsmith E, Cobb MH. 1998. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 93:605–615. doi: 10.1016/S0092-8674(00)81189-7. [DOI] [PubMed] [Google Scholar]
- 140.Whitehurst AW, Wilsbacher JL, You Y, Luby-Phelps K, Moore MS, Cobb MH. 2002. ERK2 enters the nucleus by a carrier-independent mechanism. Proc Natl Acad Sci U S A 99:7496–7501. doi: 10.1073/pnas.112495999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lidke DS, Huang F, Post JN, Rieger B, Wilsbacher J, Thomas JL, Pouyssegur J, Jovin TM, Lenormand P. 2010. ERK nuclear translocation is dimerization-independent but controlled by the rate of phosphorylation. J Biol Chem 285:3092–3102. doi: 10.1074/jbc.M109.064972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ranganathan A, Yazicioglu MN, Cobb MH. 2006. The nuclear localization of ERK2 occurs by mechanisms both independent of and dependent on energy. J Biol Chem 281:15645–15652. doi: 10.1074/jbc.M513866200. [DOI] [PubMed] [Google Scholar]
- 143.Schreck I, Al-Rawi M, Mingot JM, Scholl C, Diefenbacher ME, O'Donnell P, Bohmann D, Weiss C. 2011. c-Jun localizes to the nucleus independent of its phosphorylation by and interaction with JNK and vice versa promotes nuclear accumulation of JNK. Biochem Biophys Res Commun 407:735–740. doi: 10.1016/j.bbrc.2011.03.092. [DOI] [PubMed] [Google Scholar]
- 144.Misheva M, Kaur G, Ngoei KR, Yeap YY, Ng IH, Wagstaff KM, Ng DC, Jans DA, Bogoyevitch MA. 2014. Intracellular mobility and nuclear trafficking of the stress-activated kinase JNK1 are impeded by hyperosmotic stress. Biochim Biophys Acta 1843:253–264. doi: 10.1016/j.bbamcr.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 145.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. 1996. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J 15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 146.Hanks SK, Hunter T. 1995. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J 9:576–596. [PubMed] [Google Scholar]
- 147.Kannan N, Neuwald AF. 2004. Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2alpha. Protein Sci 13:2059–2077. doi: 10.1110/ps.04637904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nguyen T, Ruan Z, Oruganty K, Kannan N. 2015. Co-conserved MAPK features couple D-domain docking groove to distal allosteric sites via the C-terminal flanking tail. PLoS One 10:e0119636. doi: 10.1371/journal.pone.0119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sluss HK, Han Z, Barrett T, Goberdhan DC, Wilson C, Davis RJ, Ip YT. 1996. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev 10:2745–2758. doi: 10.1101/gad.10.21.2745. [DOI] [PubMed] [Google Scholar]
- 150.Wolfe KH. 2001. Yesterday's polyploids and the mystery of diploidization. Nat Rev Genet 2:333–341. doi: 10.1038/35072009. [DOI] [PubMed] [Google Scholar]
- 151.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. 2002. The protein kinase complement of the human genome. Science 298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 152.Letunic I, Copley RR, Bork P. 2002. Common exon duplication in animals and its role in alternative splicing. Hum Mol Genet 11:1561–1567. doi: 10.1093/hmg/11.13.1561. [DOI] [PubMed] [Google Scholar]
- 153.Chang WC, Chen YC, Lee KM, Tarn WY. 2007. Alternative splicing and bioinformatic analysis of human U12-type introns. Nucleic Acids Res 35:1833–1841. doi: 10.1093/nar/gkm026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jensen KB, Dredge BK, Stefani G, Zhong R, Buckanovich RJ, Okano HJ, Yang YY, Darnell RB. 2000. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 25:359–371. doi: 10.1016/S0896-6273(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 155.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. 2003. CLIP identifies Nova-regulated RNA networks in the brain. Science 302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 156.Smith E, Meyerrose TE, Kohler T, Namdar-Attar M, Bab N, Lahat O, Noh T, Li J, Karaman MW, Hacia JG, Chen TT, Nolta JA, Muller R, Bab I, Frenkel B. 2005. Leaky ribosomal scanning in mammalian genomes: significance of histone H4 alternative translation in vivo. Nucleic Acids Res 33:1298–1308. doi: 10.1093/nar/gki248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li X, MacLeod R, Dunlop AJ, Edwards HV, Advant N, Gibson LC, Devine NM, Brown KM, Adams DR, Houslay MD, Baillie GS. 2009. A scanning peptide array approach uncovers association sites within the JNK/beta arrestin signalling complex. FEBS Lett 583:3310–3316. doi: 10.1016/j.febslet.2009.09.035. [DOI] [PubMed] [Google Scholar]
- 158.Chen WK, Yeap YY, Bogoyevitch MA. 2014. The JNK1/JNK3 interactome; contributions by the JNK3 unique N-terminus and JNK common docking site residues. Biochem Biophys Res Commun 453:576–581. doi: 10.1016/j.bbrc.2014.09.122. [DOI] [PubMed] [Google Scholar]
- 159.Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Keenan S, Lavidas I, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Nuhn M, Parker A, Patricio M, Pignatelli M, Rahtz M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Birney E, Harrow J, Muffato M, Perry E, Ruffier M, Spudich G, Trevanion SJ, Cunningham F, Aken BL, Zerbino DR, Flicek P. 2016. Ensembl 2016. Nucleic Acids Res 44:D710–D716. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Kahari AK, Keenan S, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Overduin B, Parker A, Patricio M, Perry E, Pignatelli M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Aken BL, Birney E, Harrow J, Kinsella R, Muffato M, Ruffier M, Searle SM, Spudich G, Trevanion SJ, Yates A, Zerbino DR, Flicek P. 2015. Ensembl 2015. Nucleic Acids Res 43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Figuera-Losada M, LoGrasso PV. 2012. Enzyme kinetics and interaction studies for human JNK1beta1 and substrates activating transcription factor 2 (ATF2) and c-Jun N-terminal kinase (c-Jun). J Biol Chem 287:13291–13302. doi: 10.1074/jbc.M111.323766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Oehrl W, Cotsiki M, Panayotou G. 2013. Differential regulation of M3/6 (DUSP8) signaling complexes in response to arsenite-induced oxidative stress. Cell Signal 25:429–438. doi: 10.1016/j.cellsig.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 163.Liu J, Minemoto Y, Lin A. 2004. c-Jun N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for tumor necrosis factor alpha-induced c-Jun kinase activation and apoptosis. Mol Cell Biol 24:10844–10856. doi: 10.1128/MCB.24.24.10844-10856.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. 2004. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell 15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 165.Kallunki T, Su B, Tsigelny I, Sluss HK, Derijard B, Moore G, Davis R, Karin M. 1994. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev 8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- 166.Jaeschke A, Karasarides M, Ventura JJ, Ehrhardt A, Zhang C, Flavell RA, Shokat KM, Davis RJ. 2006. JNK2 is a positive regulator of the cJun transcription factor. Mol Cell 23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 167.Waetzig V, Herdegen T. 2003. A single c-Jun N-terminal kinase isoform (JNK3-p54) is an effector in both neuronal differentiation and cell death. J Biol Chem 278:567–572. doi: 10.1074/jbc.M207391200. [DOI] [PubMed] [Google Scholar]
- 168.Tsuiki H, Tnani M, Okamoto I, Kenyon LC, Emlet DR, Holgado-Madruga M, Lanham IS, Joynes CJ, Vo KT, Wong AJ. 2003. Constitutively active forms of c-Jun NH2-terminal kinase are expressed in primary glial tumors. Cancer Res 63:250–255. [PubMed] [Google Scholar]
- 169.Cui J, Holgado-Madruga M, Su W, Tsuiki H, Wedegaertner P, Wong AJ. 2005. Identification of a specific domain responsible for JNK2alpha2 autophosphorylation. J Biol Chem 280:9913–9920. doi: 10.1074/jbc.M412165200. [DOI] [PubMed] [Google Scholar]
- 170.Pimienta G, Ficarro SB, Gutierrez GJ, Bhoumik A, Peters EC, Ronai Z, Pascual J. 2007. Autophosphorylation properties of inactive and active JNK2. Cell Cycle 6:1762–1771. doi: 10.4161/cc.6.14.4434. [DOI] [PubMed] [Google Scholar]
- 171.Enomoto A, Suzuki N, Morita A, Ito M, Liu CQ, Matsumoto Y, Yoshioka K, Shiba T, Hosoi Y. 2003. Caspase-mediated cleavage of JNK during stress-induced apoptosis. Biochem Biophys Res Commun 306:837–842. doi: 10.1016/S0006-291X(03)01050-7. [DOI] [PubMed] [Google Scholar]
- 172.Yang G, Liu Y, Yang K, Liu R, Zhu S, Coquinco A, Wen W, Kojic L, Jia W, Cynader M. 2012. Isoform-specific palmitoylation of JNK regulates axonal development. Cell Death Differ 19:553–561. doi: 10.1038/cdd.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dreskin SC, Thomas GW, Dale SN, Heasley LE. 2001. Isoforms of Jun kinase are differentially expressed and activated in human monocyte/macrophage (THP-1) cells. J Immunol 166:5646–5653. doi: 10.4049/jimmunol.166.9.5646. [DOI] [PubMed] [Google Scholar]
- 174.Chan ED, Winston BW, Jarpe MB, Wynes MW, Riches DW. 1997. Preferential activation of the p46 isoform of JNK/SAPK in mouse macrophages by TNF alpha. Proc Natl Acad Sci U S A 94:13169–13174. doi: 10.1073/pnas.94.24.13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yao R, Yoshihara M, Osada H. 1997. Specific activation of a c-Jun NH2-terminal kinase isoform and induction of neurite outgrowth in PC-12 cells by staurosporine. J Biol Chem 272:18261–18266. doi: 10.1074/jbc.272.29.18261. [DOI] [PubMed] [Google Scholar]
- 176.Zhang N, Gao G, Bu X, Han S, Fang L, Li J. 2007. Neuron-specific phosphorylation of c-Jun N-terminal kinase increased in the brain of hypoxic preconditioned mice. Neurosci Lett 423:219–224. doi: 10.1016/j.neulet.2007.07.028. [DOI] [PubMed] [Google Scholar]
- 177.Takatori A, Geh E, Chen L, Zhang L, Meller J, Xia Y. 2008. Differential transmission of MEKK1 morphogenetic signals by JNK1 and JNK2. Development 135:23–32. doi: 10.1242/dev.007120. [DOI] [PubMed] [Google Scholar]
- 178.Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF. 1999. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech Dev 89:115–124. doi: 10.1016/S0925-4773(99)00213-0. [DOI] [PubMed] [Google Scholar]
- 179.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. 1999. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22:667–676. doi: 10.1016/S0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 180.Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M. 2003. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell 4:521–533. doi: 10.1016/S1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- 181.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. 2002. A central role for JNK in obesity and insulin resistance. Nature 420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 182.Yang DD, Conze D, Whitmarsh AJ, Barrett T, Davis RJ, Rincon M, Flavell RA. 1998. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity 9:575–585. doi: 10.1016/S1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- 183.Denninger K, Rasmussen S, Larsen JM, Orskov C, Seier Poulsen S, Sorensen P, Christensen JP, Illges H, Odum N, Labuda T. 2011. JNK1, but not JNK2, is required in two mechanistically distinct models of inflammatory arthritis. Am J Pathol 179:1884–1893. doi: 10.1016/j.ajpath.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Arbour N, Naniche D, Homann D, Davis RJ, Flavell RA, Oldstone MB. 2002. c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8(+) T cell-mediated antiviral immunity. J Exp Med 195:801–810. doi: 10.1084/jem.20011481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Koehler K, Mielke K, Schunck M, Neumann C, Herdegen T, Proksch E. 2011. Distinct roles of JNK-1 and ERK-2 isoforms in permeability barrier repair and wound healing. Eur J Cell Biol 90:565–571. doi: 10.1016/j.ejcb.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 186.Amura CR, Marek L, Winn RA, Heasley LE. 2005. Inhibited neurogenesis in JNK1-deficient embryonic stem cells. Mol Cell Biol 25:10791–10802. doi: 10.1128/MCB.25.24.10791-10802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. 2006. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci U S A 103:10741–10746. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Schumacher M, Schuster C, Rogon ZM, Bauer T, Caushaj N, Baars S, Szabowski S, Bauer C, Schorpp-Kistner M, Hess J, Holland-Cunz S, Wagner EF, Eils R, Angel P, Hartenstein B. 2014. Efficient keratinocyte differentiation strictly depends on JNK-induced soluble factors in fibroblasts. J Invest Dermatol 134:1332–1341. doi: 10.1038/jid.2013.535. [DOI] [PubMed] [Google Scholar]
- 189.Atkinson PJ, Cho CH, Hansen MR, Green SH. 2011. Activity of all JNK isoforms contributes to neurite growth in spiral ganglion neurons. Hear Res 278:77–85. doi: 10.1016/j.heares.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Xu P, Davis RJ. 2010. c-Jun NH2-terminal kinase is required for lineage-specific differentiation but not stem cell self-renewal. Mol Cell Biol 30:1329–1340. doi: 10.1128/MCB.00795-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Choi HS, Bode AM, Shim JH, Lee SY, Dong Z. 2009. c-Jun N-terminal kinase 1 phosphorylates Myt1 to prevent UVA-induced skin cancer. Mol Cell Biol 29:2168–2180. doi: 10.1128/MCB.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang D, Song L, Li J, Wu K, Huang C. 2006. Coordination of JNK1 and JNK2 is critical for GADD45alpha induction and its mediated cell apoptosis in arsenite responses. J Biol Chem 281:34113–34123. doi: 10.1074/jbc.M602821200. [DOI] [PubMed] [Google Scholar]
- 193.Rozo AV, Vijayvargia R, Weiss HR, Ruan H. 2008. Silencing Jnk1 and Jnk2 accelerates basal lipolysis and promotes fatty acid re-esterification in mouse adipocytes. Diabetologia 51:1493–1504. doi: 10.1007/s00125-008-1036-6. [DOI] [PubMed] [Google Scholar]
- 194.Tong C, Yin Z, Song Z, Dockendorff A, Huang C, Mariadason J, Flavell RA, Davis RJ, Augenlicht LH, Yang W. 2007. c-Jun NH2-terminal kinase 1 plays a critical role in intestinal homeostasis and tumor suppression. Am J Pathol 171:297–303. doi: 10.2353/ajpath.2007.061036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chen N, She QB, Bode AM, Dong Z. 2002. Differential gene expression profiles of Jnk1- and Jnk2-deficient murine fibroblast cells. Cancer Res 62:1300–1304. [PubMed] [Google Scholar]
- 196.Sakurai T, Maeda S, Chang L, Karin M. 2006. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A 103:10544–10551. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yoon CH, Kim MJ, Kim RK, Lim EJ, Choi KS, An S, Hwang SG, Kang SG, Suh Y, Park MJ, Lee SJ. 2012. c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene 31:4655–4666. doi: 10.1038/onc.2011.634. [DOI] [PubMed] [Google Scholar]
- 198.Das M, Garlick DS, Greiner DL, Davis RJ. 2011. The role of JNK in the development of hepatocellular carcinoma. Genes Dev 25:634–645. doi: 10.1101/gad.1989311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Biondi RM, Nebreda AR. 2003. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J 372:1–13. doi: 10.1042/bj20021641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Garai A, Zeke A, Gogl G, Toro I, Fordos F, Blankenburg H, Barkai T, Varga J, Alexa A, Emig D, Albrecht M, Remenyi A. 2012. Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci Signal 5:ra74. doi: 10.1126/scisignal.2003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Fernandes N, Bailey DE, Vanvranken DL, Allbritton NL. 2007. Use of docking peptides to design modular substrates with high efficiency for mitogen-activated protein kinase extracellular signal-regulated kinase. ACS Chem Biol 2:665–673. doi: 10.1021/cb700158q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Laughlin JD, Nwachukwu JC, Figuera-Losada M, Cherry L, Nettles KW, LoGrasso PV. 2012. Structural mechanisms of allostery and autoinhibition in JNK family kinases. Structure 20:2174–2184. doi: 10.1016/j.str.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sheridan DL, Kong Y, Parker SA, Dalby KN, Turk BE. 2008. Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J Biol Chem 283:19511–19520. doi: 10.1074/jbc.M801074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Goldsmith EJ. 2011. Three-dimensional docking in the MAPK p38alpha. Sci Signal 4:pe47. doi: 10.1126/scisignal.2002697. [DOI] [PubMed] [Google Scholar]
- 205.Kallunki T, Deng T, Hibi M, Karin M. 1996. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 87:929–939. doi: 10.1016/S0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- 206.Herrlich P. 2001. Cross-talk between glucocorticoid receptor and AP-1. Oncogene 20:2465–2475. doi: 10.1038/sj.onc.1204388. [DOI] [PubMed] [Google Scholar]
- 207.Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, Vinson C, Stamatoyannopoulos JA, Hager GL. 2011. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43:145–155. doi: 10.1016/j.molcel.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lopez-Bergami P, Lau E, Ronai Z. 2010. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer 10:65–76. doi: 10.1038/nrc2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, Oren M, Sudol M, Cesareni G, Blandino G. 2001. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem 276:15164–15173. doi: 10.1074/jbc.M010484200. [DOI] [PubMed] [Google Scholar]
- 210.Tiwari VK, Stadler MB, Wirbelauer C, Paro R, Schubeler D, Beisel C. 2012. A chromatin-modifying function of JNK during stem cell differentiation. Nat Genet 44:94–100. doi: 10.1038/ng.1036. [DOI] [PubMed] [Google Scholar]
- 211.Lindaman LL, Yeh DM, Xie C, Breen KM, Coss D. 2013. Phosphorylation of ATF2 and interaction with NFY induces c-Jun in the gonadotrope. Mol Cell Endocrinol 365:316–326. doi: 10.1016/j.mce.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Klein AM, Zaganjor E, Cobb MH. 2013. Chromatin-tethered MAPKs. Curr Opin Cell Biol 25:272–277. doi: 10.1016/j.ceb.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lim NR, Yeap YY, Zhao TT, Yip YY, Wong SC, Xu D, Ang CS, Williamson NA, Xu Z, Bogoyevitch MA, Ng DC. 2015. Opposing roles for JNK and Aurora A in regulating the association of WDR62 with spindle microtubules. J Cell Sci 128:527–540. doi: 10.1242/jcs.157537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Richards MW, Law EW, Rennalls LP, Busacca S, O'Regan L, Fry AM, Fennell DA, Bayliss R. 2014. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical beta-propeller domain. Proc Natl Acad Sci U S A 111:5195–5200. doi: 10.1073/pnas.1322892111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fourniol F, Perderiset M, Houdusse A, Moores C. 2013. Structural studies of the doublecortin family of MAPs. Methods Cell Biol 115:27–48. doi: 10.1016/B978-0-12-407757-7.00003-7. [DOI] [PubMed] [Google Scholar]
- 216.Al-Bassam J, Ozer RS, Safer D, Halpain S, Milligan RA. 2002. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol 157:1187–1196. doi: 10.1083/jcb.200201048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Taru H, Suzuki T. 2004. Facilitation of stress-induced phosphorylation of beta-amyloid precursor protein family members by X11-like/Mint2 protein. J Biol Chem 279:21628–21636. doi: 10.1074/jbc.M312007200. [DOI] [PubMed] [Google Scholar]
- 218.Podkowa M, Zhao X, Chow CW, Coffey ET, Davis RJ, Attisano L. 2010. Microtubule stabilization by bone morphogenetic protein receptor-mediated scaffolding of c-Jun N-terminal kinase promotes dendrite formation. Mol Cell Biol 30:2241–2250. doi: 10.1128/MCB.01166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A 98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. 2007. The selectivity of protein kinase inhibitors: a further update. Biochem J 408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. 1997. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 222.Barr RK, Kendrick TS, Bogoyevitch MA. 2002. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem 277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 223.Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. 2001. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- 224.Reddy CE, Albanito L, De Marco P, Aiello D, Maggiolini M, Napoli A, Musti AM. 2013. Multisite phosphorylation of c-Jun at threonine 91/93/95 triggers the onset of c-Jun pro-apoptotic activity in cerebellar granule neurons. Cell Death Dis 4:e852. doi: 10.1038/cddis.2013.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Scott MP, Miller WT. 2000. A peptide model system for processive phosphorylation by Src family kinases. Biochemistry 39:14531–14537. doi: 10.1021/bi001850u. [DOI] [PubMed] [Google Scholar]
- 226.Patwardhan P, Shen Y, Goldberg GS, Miller WT. 2006. Individual Cas phosphorylation sites are dispensable for processive phosphorylation by Src and anchorage-independent cell growth. J Biol Chem 281:20689–20697. doi: 10.1074/jbc.M602311200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Patwardhan P, Miller WT. 2007. Processive phosphorylation: mechanism and biological importance. Cell Signal 19:2218–2226. doi: 10.1016/j.cellsig.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hirata Y, Sugie A, Matsuda A, Matsuda S, Koyasu S. 2013. TAK1-JNK axis mediates survival signal through Mcl1 stabilization in activated T cells. J Immunol 190:4621–4626. doi: 10.1049/jimmunol.1202809. [DOI] [PubMed] [Google Scholar]
- 229.Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. 1994. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 230.Gupta S, Campbell D, Derijard B, Davis RJ. 1995. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 231.Noguchi K, Kitanaka C, Yamana H, Kokubu A, Mochizuki T, Kuchino Y. 1999. Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J Biol Chem 274:32580–32587. doi: 10.1074/jbc.274.46.32580. [DOI] [PubMed] [Google Scholar]
- 232.Lin CH, Lee EH. 2012. JNK1 inhibits GluR1 expression and GluR1-mediated calcium influx through phosphorylation and stabilization of Hes-1. J Neurosci 32:1826–1846. doi: 10.1523/JNEUROSCI.3380-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hong J, Zhou J, Fu J, He T, Qin J, Wang L, Liao L, Xu J. 2011. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res 71:3980–3990. doi: 10.1158/0008-5472.CAN-10-2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Chuang JY, Wang YT, Yeh SH, Liu YW, Chang WC, Hung JJ. 2008. Phosphorylation by c-Jun NH2-terminal kinase 1 regulates the stability of transcription factor Sp1 during mitosis. Mol Biol Cell 19:1139–1151. doi: 10.1091/mbc.E07-09-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, Burgering BM. 2004. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J 23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Tikhanovich I, Kuravi S, Campbell RV, Kharbanda KK, Artigues A, Villar MT, Weinman SA. 2014. Regulation of FOXO3 by phosphorylation and methylation in hepatitis C virus infection and alcohol exposure. Hepatology 59:58–70. doi: 10.1002/hep.26618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Li Z, Zhao J, Tikhanovich I, Kuravi S, Helzberg J, Dorko K, Roberts B, Kumer S, Weinman SA. 2016. Serine 574 phosphorylation alters transcriptional programming of FOXO3 by selectively enhancing apoptotic gene expression. Cell Death Differ 23:583–595. doi: 10.1038/cdd.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Buschmann T, Potapova O, Bar-Shira A, Ivanov VN, Fuchs SY, Henderson S, Fried VA, Minamoto T, Alarcon-Vargas D, Pincus MR, Gaarde WA, Holbrook NJ, Shiloh Y, Ronai Z. 2001. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol Cell Biol 21:2743–2754. doi: 10.1128/MCB.21.8.2743-2754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Jones EV, Dickman MJ, Whitmarsh AJ. 2007. Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochem J 405:617–623. doi: 10.1042/BJ20061778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, Paschal BM, Weber MJ. 2006. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol Endocrinol 20:503–515. doi: 10.1210/me.2005-0351. [DOI] [PubMed] [Google Scholar]
- 241.Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. 2002. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol 16:2382–2392. doi: 10.1210/me.2002-0144. [DOI] [PubMed] [Google Scholar]
- 242.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. 1997. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem 272:5128–5132. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 243.Srinivas H, Juroske DM, Kalyankrishna S, Cody DD, Price RE, Xu XC, Narayanan R, Weigel NL, Kurie JM. 2005. c-Jun N-terminal kinase contributes to aberrant retinoid signaling in lung cancer cells by phosphorylating and inducing proteasomal degradation of retinoic acid receptor alpha. Mol Cell Biol 25:1054–1069. doi: 10.1128/MCB.25.3.1054-1069.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Adam-Stitah S, Penna L, Chambon P, Rochette-Egly C. 1999. Hyperphosphorylation of the retinoid X receptor alpha by activated c-Jun NH2-terminal kinases. J Biol Chem 274:18932–18941. doi: 10.1074/jbc.274.27.18932. [DOI] [PubMed] [Google Scholar]
- 245.Bruck N, Bastien J, Bour G, Tarrade A, Plassat JL, Bauer A, Adam-Stitah S, Rochette-Egly C. 2005. Phosphorylation of the retinoid x receptor at the omega loop, modulates the expression of retinoic-acid-target genes with a promoter context specificity. Cell Signal 17:1229–1239. doi: 10.1016/j.cellsig.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 246.Tomlinson V, Gudmundsdottir K, Luong P, Leung KY, Knebel A, Basu S. 2010. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis 1:e29. doi: 10.1038/cddis.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Toh WH, Siddique MM, Boominathan L, Lin KW, Sabapathy K. 2004. c-Jun regulates the stability and activity of the p53 homologue, p73. J Biol Chem 279:44713–44722. doi: 10.1074/jbc.M407672200. [DOI] [PubMed] [Google Scholar]
- 248.Danovi SA, Rossi M, Gudmundsdottir K, Yuan M, Melino G, Basu S. 2008. Yes-associated protein (YAP) is a critical mediator of c-Jun-dependent apoptosis. Cell Death Differ 15:217–219. doi: 10.1038/sj.cdd.4402226. [DOI] [PubMed] [Google Scholar]
- 249.Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. 2009. A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- 250.Barkley LR, Palle K, Durando M, Day TA, Gurkar A, Kakusho N, Li J, Masai H, Vaziri C. 2012. c-Jun N-terminal kinase-mediated Rad18 phosphorylation facilitates Poleta recruitment to stalled replication forks. Mol Biol Cell 23:1943–1954. doi: 10.1091/mbc.E11-10-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Chandrasekaran S, Tan TX, Hall JR, Cook JG. 2011. Stress-stimulated mitogen-activated protein kinases control the stability and activity of the Cdt1 DNA replication licensing factor. Mol Cell Biol 31:4405–4416. doi: 10.1128/MCB.06163-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Miotto B, Struhl K. 2011. JNK1 phosphorylation of Cdt1 inhibits recruitment of HBO1 histone acetylase and blocks replication licensing in response to stress. Mol Cell 44:62–71. doi: 10.1016/j.molcel.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ford J, Ahmed S, Allison S, Jiang M, Milner J. 2008. JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle 7:3091–3097. doi: 10.4161/cc.7.19.6799. [DOI] [PubMed] [Google Scholar]
- 254.Gao Z, Zhang J, Kheterpal I, Kennedy N, Davis RJ, Ye J. 2011. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J Biol Chem 286:22227–22234. doi: 10.1074/jbc.M111.228874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Rzeczkowski K, Beuerlein K, Muller H, Dittrich-Breiholz O, Schneider H, Kettner-Buhrow D, Holtmann H, Kracht M. 2011. c-Jun N-terminal kinase phosphorylates DCP1a to control formation of P bodies. J Cell Biol 194:581–596. doi: 10.1083/jcb.201006089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Habelhah H, Shah K, Huang L, Burlingame AL, Shokat KM, Ronai Z. 2001. Identification of new JNK substrate using ATP pocket mutant JNK and a corresponding ATP analogue. J Biol Chem 276:18090–18095. doi: 10.1074/jbc.M011396200. [DOI] [PubMed] [Google Scholar]
- 257.Hutchins EJ, Szaro BG. 2013. c-Jun N-terminal kinase phosphorylation of heterogeneous nuclear ribonucleoprotein K regulates vertebrate axon outgrowth via a posttranscriptional mechanism. J Neurosci 33:14666–14680. doi: 10.1523/JNEUROSCI.4821-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Al-Ayoubi AM, Zheng H, Liu Y, Bai T, Eblen ST. 2012. Mitogen-activated protein kinase phosphorylation of splicing factor 45 (SPF45) regulates SPF45 alternative splicing site utilization, proliferation, and cell adhesion. Mol Cell Biol 32:2880–2893. doi: 10.1128/MCB.06327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Gandin V, Gutierrez GJ, Brill LM, Varsano T, Feng Y, Aza-Blanc P, Au Q, McLaughlan S, Ferreira TA, Alain T, Sonenberg N, Topisirovic I, Ronai ZA. 2013. Degradation of newly synthesized polypeptides by ribosome-associated RACK1/c-Jun N-terminal kinase/eukaryotic elongation factor 1A2 complex. Mol Cell Biol 33:2510–2526. doi: 10.1128/MCB.01362-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Gdalyahu A, Ghosh I, Levy T, Sapir T, Sapoznik S, Fishler Y, Azoulai D, Reiner O. 2004. DCX, a new mediator of the JNK pathway. EMBO J 23:823–832. doi: 10.1038/sj.emboj.7600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Singh SA, Winter D, Bilimoria PM, Bonni A, Steen H, Steen JA. 2012. FLEXIQinase, a mass spectrometry-based assay, to unveil multikinase mechanisms. Nat Methods 9:504–508. doi: 10.1038/nmeth.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Jin J, Suzuki H, Hirai S, Mikoshiba K, Ohshima T. 2010. JNK phosphorylates Ser332 of doublecortin and regulates its function in neurite extension and neuronal migration. Dev Neurobiol 70:929–942. doi: 10.1002/dneu.20833. [DOI] [PubMed] [Google Scholar]
- 263.Kawauchi T, Chihama K, Nishimura YV, Nabeshima Y, Hoshino M. 2005. MAP1B phosphorylation is differentially regulated by Cdk5/p35, Cdk5/p25, and JNK. Biochem Biophys Res Commun 331:50–55. doi: 10.1016/j.bbrc.2005.03.132. [DOI] [PubMed] [Google Scholar]
- 264.Komulainen E, Zdrojewska J, Freemantle E, Mohammad H, Kulesskaya N, Deshpande P, Marchisella F, Mysore R, Hollos P, Michelsen KA, Magard M, Rauvala H, James P, Coffey ET. 2014. JNK1 controls dendritic field size in L2/3 and L5 of the motor cortex, constrains soma size, and influences fine motor coordination. Front Cell Neurosci 8:272. doi: 10.3389/fncel.2014.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Ploia C, Antoniou X, Sclip A, Grande V, Cardinetti D, Colombo A, Canu N, Benussi L, Ghidoni R, Forloni G, Borsello T. 2011. JNK plays a key role in tau hyperphosphorylation in Alzheimer's disease models. J Alzheimers Dis 26:315–329. doi: 10.3323/JAD-2011-110320. [DOI] [PubMed] [Google Scholar]
- 266.Bjorkblom B, Padzik A, Mohammad H, Westerlund N, Komulainen E, Hollos P, Parviainen L, Papageorgiou AC, Iljin K, Kallioniemi O, Kallajoki M, Courtney MJ, Magard M, James P, Coffey ET. 2012. c-Jun N-terminal kinase phosphorylation of MARCKSL1 determines actin stability and migration in neurons and in cancer cells. Mol Cell Biol 32:3513–3526. doi: 10.1128/MCB.00713-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Gordon EA, Whisenant TC, Zeller M, Kaake RM, Gordon WM, Krotee P, Patel V, Huang L, Baldi P, Bardwell L. 2013. Combining docking site and phosphosite predictions to find new substrates: identification of smoothelin-like-2 (SMTNL2) as a c-Jun N-terminal kinase (JNK) substrate. Cell Signal 25:2518–2529. doi: 10.1016/j.cellsig.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. 2003. JNK phosphorylates paxillin and regulates cell migration. Nature 424:219–223. doi: 10.1038/nature01745. [DOI] [PubMed] [Google Scholar]
- 269.Lee YC, Chang AY, Lin-Feng MH, Tsou WI, Chiang IH, Lai MZ. 2012. Paxillin phosphorylation by JNK and p38 is required for NFAT activation. Eur J Immunol 42:2165–2175. doi: 10.1002/eji.201142192. [DOI] [PubMed] [Google Scholar]
- 270.Lee MH, Koria P, Qu J, Andreadis ST. 2009. JNK phosphorylates beta-catenin and regulates adherens junctions. FASEB J 23:3874–3883. doi: 10.1096/fj.08-117804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Lee TL, Shyu YC, Hsu PH, Chang CW, Wen SC, Hsiao WY, Tsai MD, Shen CK. 2010. JNK-mediated turnover and stabilization of the transcription factor p45/NF-E2 during differentiation of murine erythroleukemia cells. Proc Natl Acad Sci U S A 107:52–57. doi: 10.1073/pnas.0909153107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. 2008. Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell 133:340–353. doi: 10.1016/j.cell.2008.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Lee YH, Giraud J, Davis RJ, White MF. 2003. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem 278:2896–2902. doi: 10.1074/jbc.M208359200. [DOI] [PubMed] [Google Scholar]
- 274.Sharfi H, Eldar-Finkelman H. 2008. Sequential phosphorylation of insulin receptor substrate-2 by glycogen synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in hepatic insulin signaling. Am J Physiol Endocrinol Metab 294:E307–E315. doi: 10.1152/ajpendo.00534.2007. [DOI] [PubMed] [Google Scholar]
- 275.Kelkar N, Gupta S, Dickens M, Davis RJ. 2000. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol Cell Biol 20:1030–1043. doi: 10.1128/MCB.20.3.1030-1043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Zdrojewska J, Coffey ET. 2014. The impact of JNK on neuronal migration. Adv Exp Med Biol 800:37–57. doi: 10.1007/978-94-007-7687-6_3. [DOI] [PubMed] [Google Scholar]
- 277.Samak G, Suzuki T, Bhargava A, Rao RK. 2010. c-Jun NH2-terminal kinase-2 mediates osmotic stress-induced tight junction disruption in the intestinal epithelium. Am J Physiol Gastrointest Liver Physiol 299:G572–G584. doi: 10.1152/ajpgi.00265.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Cellurale C, Sabio G, Kennedy NJ, Das M, Barlow M, Sandy P, Jacks T, Davis RJ. 2011. Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol Cell Biol 31:1565–1576. doi: 10.1128/MCB.01122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Dhanasekaran DN, Reddy EP. 2008. JNK signaling in apoptosis. Oncogene 27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Deng X, Xiao L, Lang W, Gao F, Ruvolo P, May WS Jr. 2001. Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem 276:23681–23688. doi: 10.1074/jbc.M100279200. [DOI] [PubMed] [Google Scholar]
- 281.Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J, Umezawa A, Ichijo H. 2002. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem 277:43730–43734. doi: 10.1074/jbc.M207951200. [DOI] [PubMed] [Google Scholar]
- 282.Morel C, Carlson SM, White FM, Davis RJ. 2009. Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol Cell Biol 29:3845–3852. doi: 10.1128/MCB.00279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Hubner A, Barrett T, Flavell RA, Davis RJ. 2008. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell 30:415–425. doi: 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Liu J, Lin A. 2005. Role of JNK activation in apoptosis: a double-edged sword. Cell Res 15:36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 285.Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. 2005. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J Cell Biol 170:295–304. doi: 10.1083/jcb.200409117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Codelia VA, Sun G, Irvine KD. 2014. Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr Biol 24:2012–2017. doi: 10.1016/j.cub.2014.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. 2007. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron 56:488–502. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 288.Thomas GM, Lin DT, Nuriya M, Huganir RL. 2008. Rapid and bi-directional regulation of AMPA receptor phosphorylation and trafficking by JNK. EMBO J 27:361–372. doi: 10.1038/sj.emboj.7601969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK. 2004. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res 32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Van Roey K, Gibson TJ, Davey NE. 2012. Motif switches: decision-making in cell regulation. Curr Opin Struct Biol 22:378–385. doi: 10.1016/j.sbi.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 291.Akiva E, Friedlander G, Itzhaki Z, Margalit H. 2012. A dynamic view of domain-motif interactions. PLoS Comput Biol 8:e1002341. doi: 10.1371/journal.pcbi.1002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Liu X, Bardwell L, Nie Q. 2010. A combination of multisite phosphorylation and substrate sequestration produces switchlike responses. Biophys J 98:1396–1407. doi: 10.1016/j.bpj.2009.12.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Sutherland C. 2011. What are the bona fide GSK3 substrates? Int J Alzheimers Dis 2011:505607. doi: 10.4061/2011/505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.ter Haar E, Coll JT, Austen DA, Hsiao HM, Swenson L, Jain J. 2001. Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat Struct Biol 8:593–596. doi: 10.1038/89624. [DOI] [PubMed] [Google Scholar]
- 295.Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M, Feramisco J, Montminy M. 1994. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature 370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- 296.Liu WL, Coleman RA, Ma E, Grob P, Yang JL, Zhang Y, Dailey G, Nogales E, Tjian R. 2009. Structures of three distinct activator-TFIID complexes. Genes Dev 23:1510–1521. doi: 10.1101/gad.1790709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Nateri AS, Spencer-Dene B, Behrens A. 2005. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 437:281–285. doi: 10.1038/nature03914. [DOI] [PubMed] [Google Scholar]
- 298.Aguilera C, Nakagawa K, Sancho R, Chakraborty A, Hendrich B, Behrens A. 2011. c-Jun N-terminal phosphorylation antagonises recruitment of the Mbd3/NuRD repressor complex. Nature 469:231–235. doi: 10.1038/nature09607. [DOI] [PubMed] [Google Scholar]
- 299.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG Jr. 2005. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 300.Lin A, Frost J, Deng T, Smeal T, al-Alawi N, Kikkawa U, Hunter T, Brenner D, Karin M. 1992. Casein kinase II is a negative regulator of c-Jun DNA binding and AP-1 activity. Cell 70:777–789. doi: 10.1016/0092-8674(92)90311-Y. [DOI] [PubMed] [Google Scholar]
- 301.Angel PE, Herrlich P. 1994. The FOS and JUN families of transcription factors. CRC Press, Boca Raton, FL. [Google Scholar]
- 302.Lei K, Davis RJ. 2003. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A 100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Donovan N, Becker EB, Konishi Y, Bonni A. 2002. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem 277:40944–40949. doi: 10.1074/jbc.M206113200. [DOI] [PubMed] [Google Scholar]
- 304.Wang XT, Pei DS, Xu J, Guan QH, Sun YF, Liu XM, Zhang GY. 2007. Opposing effects of Bad phosphorylation at two distinct sites by Akt1 and JNK1/2 on ischemic brain injury. Cell Signal 19:1844–1856. doi: 10.1016/j.cellsig.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 305.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. 2008. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. 2009. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem 284:2719–2728. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Chow CW, Rincon M, Cavanagh J, Dickens M, Davis RJ. 1997. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science 278:1638–1641. doi: 10.1126/science.278.5343.1638. [DOI] [PubMed] [Google Scholar]
- 308.Chow CW, Dong C, Flavell RA, Davis RJ. 2000. c-Jun NH(2)-terminal kinase inhibits targeting of the protein phosphatase calcineurin to NFATc1. Mol Cell Biol 20:5227–5234. doi: 10.1128/MCB.20.14.5227-5234.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Ortega-Perez I, Cano E, Were F, Villar M, Vazquez J, Redondo JM. 2005. c-Jun N-terminal kinase (JNK) positively regulates NFATc2 transactivation through phosphorylation within the N-terminal regulatory domain. J Biol Chem 280:20867–20878. doi: 10.1074/jbc.M501898200. [DOI] [PubMed] [Google Scholar]
- 310.He T, Stepulak A, Holmstrom TH, Omary MB, Eriksson JE. 2002. The intermediate filament protein keratin 8 is a novel cytoplasmic substrate for c-Jun N-terminal kinase. J Biol Chem 277:10767–10774. doi: 10.1074/jbc.M111436200. [DOI] [PubMed] [Google Scholar]
- 311.Tanaka T, Iino M. 2015. Sec8 regulates cytokeratin 8 phosphorylation and cell migration by controlling the ERK and p38 MAPK signalling pathways. Cell Signal 27:1110–1119. doi: 10.1016/j.cellsig.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 312.Ku NO, Azhar S, Omary MB. 2002. Keratin 8 phosphorylation by p38 kinase regulates cellular keratin filament reorganization: modulation by a keratin 1-like disease causing mutation. J Biol Chem 277:10775–10782. doi: 10.1074/jbc.M107623200. [DOI] [PubMed] [Google Scholar]
- 313.Yang XJ, Gregoire S. 2006. A recurrent phospho-sumoyl switch in transcriptional repression and beyond. Mol Cell 23:779–786. doi: 10.1016/j.molcel.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 314.Ristow M, Muller-Wieland D, Pfeiffer A, Krone W, Kahn CR. 1998. Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. N Engl J Med 339:953–959. doi: 10.1056/NEJM199810013391403. [DOI] [PubMed] [Google Scholar]
- 315.Rangwala SM, Rhoades B, Shapiro JS, Rich AS, Kim JK, Shulman GI, Kaestner KH, Lazar MA. 2003. Genetic modulation of PPARgamma phosphorylation regulates insulin sensitivity. Dev Cell 5:657–663. doi: 10.1016/S1534-5807(03)00274-0. [DOI] [PubMed] [Google Scholar]
- 316.Dai R, Frejtag W, He B, Zhang Y, Mivechi NF. 2000. c-Jun NH2-terminal kinase targeting and phosphorylation of heat shock factor-1 suppress its transcriptional activity. J Biol Chem 275:18210–18218. doi: 10.1074/jbc.M000958200. [DOI] [PubMed] [Google Scholar]
- 317.Park J, Liu AY. 2001. JNK phosphorylates the HSF1 transcriptional activation domain: role of JNK in the regulation of the heat shock response. J Cell Biochem 82:326–338. doi: 10.1002/jcb.1163. [DOI] [PubMed] [Google Scholar]
- 318.Hietakangas V, Ahlskog JK, Jakobsson AM, Hellesuo M, Sahlberg NM, Holmberg CI, Mikhailov A, Palvimo JJ, Pirkkala L, Sistonen L. 2003. Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol Cell Biol 23:2953–2968. doi: 10.1128/MCB.23.8.2953-2968.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, Karin M. 2004. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 306:271–275. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- 320.Seet BT, Dikic I, Zhou MM, Pawson T. 2006. Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol 7:473–483. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- 321.Reinhardt HC, Yaffe MB. 2013. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol 14:563–580. doi: 10.1038/nrm3640. [DOI] [PubMed] [Google Scholar]
- 322.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. 2007. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 323.Welcker M, Larimore EA, Swanger J, Bengoechea-Alonso MT, Grim JE, Ericsson J, Zheng N, Clurman BE. 2013. Fbw7 dimerization determines the specificity and robustness of substrate degradation. Genes Dev 27:2531–2536. doi: 10.1101/gad.229195.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. 2004. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, Xiao Y, Christie AL, Aster J, Settleman J, Gygi SP, Kung AL, Look T, Nakayama KI, DePinho RA, Wei W. 2011. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471:104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Engel ME, McDonnell MA, Law BK, Moses HL. 1999. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem 274:37413–37420. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- 327.Kamato D, Rostam MA, Piva TJ, Babaahmadi Rezaei H, Getachew R, Thach L, Bernard R, Zheng W, Little PJ, Osman N. 2014. Transforming growth factor beta-mediated site-specific Smad linker region phosphorylation in vascular endothelial cells. J Pharm Pharmacol 66:1722–1733. doi: 10.1111/jphp.12298. [DOI] [PubMed] [Google Scholar]
- 328.Aragon E, Goerner N, Zaromytidou AI, Xi Q, Escobedo A, Massague J, Macias MJ. 2011. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev 25:1275–1288. doi: 10.1101/gad.2060811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Lin H, Lin Q, Liu M, Lin Y, Wang X, Chen H, Xia Z, Lu B, Ding F, Wu Q, Wang HR. 2014. PKA/Smurf1 signaling-mediated stabilization of Nur77 is required for anticancer drug cisplatin-induced apoptosis. Oncogene 33:1629–1639. doi: 10.1038/onc.2013.116. [DOI] [PubMed] [Google Scholar]
- 330.Weidenfeld-Baranboim K, Koren L, Aronheim A. 2011. Phosphorylation of JDP2 on threonine-148 by the c-Jun N-terminal kinase targets it for proteosomal degradation. Biochem J 436:661–669. doi: 10.1042/BJ20101031. [DOI] [PubMed] [Google Scholar]
- 331.Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, DiAntonio A. 2012. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci U S A 109:E3696–E3705. doi: 10.1073/pnas.1216204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Leung KT, Li KK, Sun SS, Chan PK, Ooi VE, Chiu LC. 2008. Activation of the JNK pathway promotes phosphorylation and degradation of BimEL: a novel mechanism of chemoresistance in T-cell acute lymphoblastic leukemia. Carcinogenesis 29:544–551. doi: 10.1093/carcin/bgm294. [DOI] [PubMed] [Google Scholar]
- 333.Zhang J, Gao Z, Ye J. 2013. Phosphorylation and degradation of S6K1 (p70S6K1) in response to persistent JNK1 activation. Biochim Biophys Acta 1832:1980–1988. doi: 10.1016/j.bbadis.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. 2001. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J 20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Rudrabhatla P, Zheng YL, Amin ND, Kesavapany S, Albers W, Pant HC. 2008. Pin1-dependent prolyl isomerization modulates the stress-induced phosphorylation of high molecular weight neurofilament protein. J Biol Chem 283:26737–26747. doi: 10.1074/jbc.M801633200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Kang C, Bharatham N, Chia J, Mu Y, Baek K, Yoon HS. 2012. The natively disordered loop of Bcl-2 undergoes phosphorylation-dependent conformational change and interacts with Pin1. PLoS One 7:e52047. doi: 10.1371/journal.pone.0052047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Pahlke D, Freund C, Leitner D, Labudde D. 2005. Statistically significant dependence of the Xaa-Pro peptide bond conformation on secondary structure and amino acid sequence. BMC Struct Biol 5:8. doi: 10.1186/1472-6807-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. 2000. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol 7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 339.Lu KP, Zhou XZ. 2007. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol 8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 340.Werner-Allen JW, Lee CJ, Liu P, Nicely NI, Wang S, Greenleaf AL, Zhou P. 2011. cis-Proline-mediated Ser(P)5 dephosphorylation by the RNA polymerase II C-terminal domain phosphatase Ssu72. J Biol Chem 286:5717–5726. doi: 10.1074/jbc.M110.197129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Piskacek S, Gregor M, Nemethova M, Grabner M, Kovarik P, Piskacek M. 2007. Nine-amino-acid transactivation domain: establishment and prediction utilities. Genomics 89:756–768. doi: 10.1016/j.ygeno.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 342.Li QJ, Yang SH, Maeda Y, Sladek FM, Sharrocks AD, Martins-Green M. 2003. MAP kinase phosphorylation-dependent activation of Elk-1 leads to activation of the co-activator p300. EMBO J 22:281–291. doi: 10.1093/emboj/cdg028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Ducret C, Maira SM, Lutz Y, Wasylyk B. 2000. The ternary complex factor Net contains two distinct elements that mediate different responses to MAP kinase signalling cascades. Oncogene 19:5063–5072. doi: 10.1038/sj.onc.1203892. [DOI] [PubMed] [Google Scholar]
- 344.Mahajan MA, Stanley FM. 2014. Insulin-activated Elk-1 recruits the TIP60/NuA4 complex to increase prolactin gene transcription. Mol Cell Endocrinol 382:159–169. doi: 10.1016/j.mce.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 345.Nagadoi A, Nakazawa K, Uda H, Okuno K, Maekawa T, Ishii S, Nishimura Y. 1999. Solution structure of the transactivation domain of ATF-2 comprising a zinc finger-like subdomain and a flexible subdomain. J Mol Biol 287:593–607. doi: 10.1006/jmbi.1999.2620. [DOI] [PubMed] [Google Scholar]
- 346.Duyndam MC, van Dam H, Smits PH, Verlaan M, van der Eb AJ, Zantema A. 1999. The N-terminal transactivation domain of ATF2 is a target for the co-operative activation of the c-jun promoter by p300 and 12S E1A. Oncogene 18:2311–2321. doi: 10.1038/sj.onc.1202584. [DOI] [PubMed] [Google Scholar]
- 347.Kim JW, Jang SM, Kim CH, An JH, Choi KH. 2012. Transcriptional activity of neural retina leucine zipper (Nrl) is regulated by c-Jun N-terminal kinase and Tip60 during retina development. Mol Cell Biol 32:1720–1732. doi: 10.1128/MCB.06440-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Songyang Z, Cantley LC. 1995. Recognition and specificity in protein tyrosine kinase-mediated signalling. Trends Biochem Sci 20:470–475. doi: 10.1016/S0968-0004(00)89103-3. [DOI] [PubMed] [Google Scholar]
- 349.Amanchy R, Periaswamy B, Mathivanan S, Reddy R, Tattikota SG, Pandey A. 2007. A curated compendium of phosphorylation motifs. Nat Biotechnol 25:285–286. doi: 10.1038/nbt0307-285. [DOI] [PubMed] [Google Scholar]
- 350.Ishibe S, Joly D, Liu ZX, Cantley LG. 2004. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol Cell 16:257–267. doi: 10.1016/j.molcel.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 351.Lee KK, Yonehara S. 2012. Identification of mechanism that couples multisite phosphorylation of Yes-associated protein (YAP) with transcriptional coactivation and regulation of apoptosis. J Biol Chem 287:9568–9578. doi: 10.1074/jbc.M111.296954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. 1999. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J 18:2551–2562. doi: 10.1093/emboj/18.9.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Komulainen E. 2015. JNK regulates dendrite and spine architecture in the central nervous system influencing spatial learning and motor tasks. Ph.D. dissertaion. Åbo Akademi University, Turku, Finland. [Google Scholar]
- 354.Cho JH, Johnson GV. 2004. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau's ability to bind and stabilize microtubules. J Neurochem 88:349–358. doi: 10.1111/j.1471-4159.2004.02155.x. [DOI] [PubMed] [Google Scholar]
- 355.Alonso AD, Di Clerico J, Li B, Corbo CP, Alaniz ME, Grundke-Iqbal I, Iqbal K. 2010. Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. J Biol Chem 285:30851–30860. doi: 10.1074/jbc.M110.110957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Bielas SL, Serneo FF, Chechlacz M, Deerinck TJ, Perkins GA, Allen PB, Ellisman MH, Gleeson JG. 2007. Spinophilin facilitates dephosphorylation of doublecortin by PP1 to mediate microtubule bundling at the axonal wrist. Cell 129:579–591. doi: 10.1016/j.cell.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Bogoyevitch MA, Yeap YY, Qu Z, Ngoei KR, Yip YY, Zhao TT, Heng JI, Ng DC. 2012. WD40-repeat protein 62 is a JNK-phosphorylated spindle pole protein required for spindle maintenance and timely mitotic progression. J Cell Sci 125:5096–5109. doi: 10.1242/jcs.107326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Park JH, Park M, Byun CJ, Jo I. 2012. c-Jun N-terminal kinase 2 phosphorylates endothelial nitric oxide synthase at serine 116 and regulates nitric oxide production. Biochem Biophys Res Commun 417:340–345. doi: 10.1016/j.bbrc.2011.11.112. [DOI] [PubMed] [Google Scholar]
- 359.Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. 2005. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat Cell Biol 7:278–285. doi: 10.1038/ncb1228. [DOI] [PubMed] [Google Scholar]
- 360.Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, Yoshioka K, Masuyama N, Gotoh Y. 2004. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J 23:1889–1899. doi: 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Okamura H, Garcia-Rodriguez C, Martinson H, Qin J, Virshup DM, Rao A. 2004. A conserved docking motif for CK1 binding controls the nuclear localization of NFAT1. Mol Cell Biol 24:4184–4195. doi: 10.1128/MCB.24.10.4184-4195.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Park S, Uesugi M, Verdine GL. 2000. A second calcineurin binding site on the NFAT regulatory domain. Proc Natl Acad Sci U S A 97:7130–7135. doi: 10.1073/pnas.97.13.7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Gao Y, Chen S. 2013. Proline-directed androgen receptor phosphorylation. J Mol Genet Med 7:75. doi: 10.4172/1747-0862.1000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Funato N, Twigg SR, Higashihori N, Ohyama K, Wall SA, Wilkie AO, Nakamura M. 2005. Functional analysis of natural mutations in two TWIST protein motifs. Hum Mutat 25:550–556. doi: 10.1002/humu.20176. [DOI] [PubMed] [Google Scholar]
- 365.Rona G, Borsos M, Ellis JJ, Mehdi AM, Christie M, Kornyei Z, Neubrandt M, Toth J, Bozoky Z, Buday L, Madarasz E, Boden M, Kobe B, Vertessy BG. 2014. Dynamics of re-constitution of the human nuclear proteome after cell division is regulated by NLS-adjacent phosphorylation. Cell Cycle 13:3551–3564. doi: 10.4161/15384101.2014.960740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Pallier K, Cessot A, Cote JF, Just PA, Cazes A, Fabre E, Danel C, Riquet M, Devouassoux-Shisheboran M, Ansieau S, Puisieux A, Laurent-Puig P, Blons H. 2012. TWIST1 a new determinant of epithelial to mesenchymal transition in EGFR mutated lung adenocarcinoma. PLoS One 7:e29954. doi: 10.1371/journal.pone.0029954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Jennings BH, Pickles LM, Wainwright SM, Roe SM, Pearl LH, Ish-Horowicz D. 2006. Molecular recognition of transcriptional repressor motifs by the WD domain of the Groucho/TLE corepressor. Mol Cell 22:645–655. doi: 10.1016/j.molcel.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 368.Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. 2003. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell 11:1445–1456. doi: 10.1016/S1097-2765(03)00234-X. [DOI] [PubMed] [Google Scholar]
- 369.Solinas G, Naugler W, Galimi F, Lee MS, Karin M. 2006. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci U S A 103:16454–16459. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Li H, Yu X. 2013. Emerging role of JNK in insulin resistance. Curr Diabetes Rev 9:422–428. doi: 10.2174/15733998113099990074. [DOI] [PubMed] [Google Scholar]
- 371.D'Ambrosio C, Arena S, Fulcoli G, Scheinfeld MH, Zhou D, D'Adamio L, Scaloni A. 2006. Hyperphosphorylation of JNK-interacting protein 1, a protein associated with Alzheimer disease. Mol Cell Proteomics 5:97–113. doi: 10.1074/mcp.M500226-MCP200. [DOI] [PubMed] [Google Scholar]
- 372.Whitmarsh AJ. 2006. The JIP family of MAPK scaffold proteins. Biochem Soc Trans 34:828–832. doi: 10.1042/BST0340828. [DOI] [PubMed] [Google Scholar]
- 373.Horiuchi D, Collins CA, Bhat P, Barkus RV, Diantonio A, Saxton WM. 2007. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr Biol 17:1313–1317. doi: 10.1016/j.cub.2007.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Hammond JW, Griffin K, Jih GT, Stuckey J, Verhey KJ. 2008. Co-operative versus independent transport of different cargoes by Kinesin-1. Traffic 9:725–741. doi: 10.1111/j.1600-0854.2008.00722.x. [DOI] [PubMed] [Google Scholar]
- 375.Isabet T, Montagnac G, Regazzoni K, Raynal B, El Khadali F, England P, Franco M, Chavrier P, Houdusse A, Menetrey J. 2009. The structural basis of Arf effector specificity: the crystal structure of ARF6 in a complex with JIP4. EMBO J 28:2835–2845. doi: 10.1038/emboj.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Montagnac G, Sibarita JB, Loubery S, Daviet L, Romao M, Raposo G, Chavrier P. 2009. ARF6 Interacts with JIP4 to control a motor switch mechanism regulating endosome traffic in cytokinesis. Curr Biol 19:184–195. doi: 10.1016/j.cub.2008.12.043. [DOI] [PubMed] [Google Scholar]
- 377.Kelkar N, Standen CL, Davis RJ. 2005. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol Cell Biol 25:2733–2743. doi: 10.1128/MCB.25.7.2733-2743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Wiltshire C, Matsushita M, Tsukada S, Gillespie DA, May GH. 2002. A new c-Jun N-terminal kinase (JNK)-interacting protein, Sab (SH3BP5), associates with mitochondria. Biochem J 367:577–585. doi: 10.1042/bj20020553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Win S, Than TA, Min RW, Aghajan M, Kaplowitz N. 2016. JNK mediates mouse liver injury through a novel Sab (SH3BP5) dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 63:1987–2003. doi: 10.1002/hep.28486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Xia Y, Karin M. 2004. The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol 14:94–101. doi: 10.1016/j.tcb.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 381.Weston CR, Wong A, Hall JP, Goad ME, Flavell RA, Davis RJ. 2003. JNK initiates a cytokine cascade that causes Pax2 expression and closure of the optic fissure. Genes Dev 17:1271–1280. doi: 10.1101/gad.1087303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Weston CR, Wong A, Hall JP, Goad ME, Flavell RA, Davis RJ. 2004. The c-Jun NH2-terminal kinase is essential for epidermal growth factor expression during epidermal morphogenesis. Proc Natl Acad Sci U S A 101:14114–14119. doi: 10.1073/pnas.0406061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Murtagh J, McArdle E, Gilligan E, Thornton L, Furlong F, Martin F. 2004. Organization of mammary epithelial cells into 3D acinar structures requires glucocorticoid and JNK signaling. J Cell Biol 166:133–143. doi: 10.1083/jcb.200403020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Cellurale C, Girnius N, Jiang F, Cavanagh-Kyros J, Lu S, Garlick DS, Mercurio AM, Davis RJ. 2012. Role of JNK in mammary gland development and breast cancer. Cancer Res 72:472–481. doi: 10.1158/0008-5472.CAN-11-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Qu C, Li W, Shao Q, Dwyer T, Huang H, Yang T, Liu G. 2013. c-Jun N-terminal kinase 1 (JNK1) is required for coordination of netrin signaling in axon guidance. J Biol Chem 288:1883–1895. doi: 10.1074/jbc.M112.417881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Ben-Zvi A, Yagil Z, Hagalili Y, Klein H, Lerman O, Behar O. 2006. Semaphorin 3A and neurotrophins: a balance between apoptosis and survival signaling in embryonic DRG neurons. J Neurochem 96:585–597. doi: 10.1111/j.1471-4159.2005.03580.x. [DOI] [PubMed] [Google Scholar]
- 387.Ha HY, Cho IH, Lee KW, Lee KW, Song JY, Kim KS, Yu YM, Lee JK, Song JS, Yang SD, Shin HS, Han PL. 2005. The axon guidance defect of the telencephalic commissures of the JSAP1-deficient brain was partially rescued by the transgenic expression of JIP1. Dev Biol 277:184–199. doi: 10.1016/j.ydbio.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 388.Asaoka Y, Nishina H. 2010. Diverse physiological functions of MKK4 and MKK7 during early embryogenesis. J Biochem 148:393–401. 148:393–401. doi: 10.1093/jb/mvq098. [DOI] [PubMed] [Google Scholar]
- 389.Wuestefeld T, Pesic M, Rudalska R, Dauch D, Longerich T, Kang TW, Yevsa T, Heinzmann F, Hoenicke L, Hohmeyer A, Potapova A, Rittelmeier I, Jarek M, Geffers R, Scharfe M, Klawonn F, Schirmacher P, Malek NP, Ott M, Nordheim A, Vogel A, Manns MP, Zender L. 2013. A direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell 153:389–401. doi: 10.1016/j.cell.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 390.Yamasaki T, Kawasaki H, Arakawa S, Shimizu K, Shimizu S, Reiner O, Okano H, Nishina S, Azuma N, Penninger JM, Katada T, Nishina H. 2011. Stress-activated protein kinase MKK7 regulates axon elongation in the developing cerebral cortex. J Neurosci 31:16872–16883. doi: 10.1523/JNEUROSCI.1111-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Chen J, Chang S, Duncan SA, Okano HJ, Fishell G, Aderem A. 1996. Disruption of the MacMARCKS gene prevents cranial neural tube closure and results in anencephaly. Proc Natl Acad Sci U S A 93:6275–6279. doi: 10.1073/pnas.93.13.6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Eferl R, Sibilia M, Hilberg F, Fuchsbichler A, Kufferath I, Guertl B, Zenz R, Wagner EF, Zatloukal K. 1999. Functions of c-Jun in liver and heart development. J Cell Biol 145:1049–1061. doi: 10.1083/jcb.145.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Maekawa T, Bernier F, Sato M, Nomura S, Singh M, Inoue Y, Tokunaga T, Imai H, Yokoyama M, Reimold A, Glimcher LH, Ishii S. 1999. Mouse ATF-2 null mutants display features of a severe type of meconium aspiration syndrome. J Biol Chem 274:17813–17819. doi: 10.1074/jbc.274.25.17813. [DOI] [PubMed] [Google Scholar]
- 394.Yamanaka H, Moriguchi T, Masuyama N, Kusakabe M, Hanafusa H, Takada R, Takada S, Nishida E. 2002. JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep 3:69–75. doi: 10.1093/embo-reports/kvf008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Rosso SB, Sussman D, Wynshaw-Boris A, Salinas PC. 2005. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci 8:34–42. doi: 10.1038/nn1374. [DOI] [PubMed] [Google Scholar]
- 396.Zhang P, Cai Y, Soofi A, Dressler GR. 2012. Activation of Wnt11 by transforming growth factor-beta drives mesenchymal gene expression through non-canonical Wnt protein signaling in renal epithelial cells. J Biol Chem 287:21290–21302. doi: 10.1074/jbc.M112.357202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Pandur P, Lasche M, Eisenberg LM, Kuhl M. 2002. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature 418:636–641. doi: 10.1038/nature00921. [DOI] [PubMed] [Google Scholar]
- 398.Ferraris D, Yang Z, Welsbie D. 2013. Dual leucine zipper kinase as a therapeutic target for neurodegenerative conditions. Future Med Chem 5:1923–1934. doi: 10.4155/fmc.13.150. [DOI] [PubMed] [Google Scholar]
- 399.Antoniou X, Falconi M, Di Marino D, Borsello T. 2011. JNK3 as a therapeutic target for neurodegenerative diseases. J Alzheimers Dis 24:633–642. doi: 10.3323/JAD-2011-091567. [DOI] [PubMed] [Google Scholar]
- 400.Borsello T, Forloni G. 2007. JNK signalling: a possible target to prevent neurodegeneration. Curr Pharm Des 13:1875–1886. doi: 10.2174/138161207780858384. [DOI] [PubMed] [Google Scholar]
- 401.Spigolon G, Veronesi C, Bonny C, Vercelli A. 2010. c-Jun N-terminal kinase signaling pathway in excitotoxic cell death following kainic acid-induced status epilepticus. Eur J Neurosci 31:1261–1272. doi: 10.1111/j.1460-9568.2010.07158.x. [DOI] [PubMed] [Google Scholar]
- 402.Centeno C, Repici M, Chatton JY, Riederer BM, Bonny C, Nicod P, Price M, Clarke PG, Papa S, Franzoso G, Borsello T. 2007. Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ 14:240–253. doi: 10.1038/sj.cdd.4401988. [DOI] [PubMed] [Google Scholar]
- 403.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. 1997. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 404.Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. 2003. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med 9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 405.Nistico R, Florenzano F, Mango D, Ferraina C, Grilli M, Di Prisco S, Nobili A, Saccucci S, D'Amelio M, Morbin M, Marchi M, Mercuri NB, Davis RJ, Pittaluga A, Feligioni M. 2015. Presynaptic c-Jun N-terminal kinase 2 regulates NMDA receptor-dependent glutamate release. Sci Rep 5:9035. doi: 10.1038/srep09035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Bjorkblom B, Vainio JC, Hongisto V, Herdegen T, Courtney MJ, Coffey ET. 2008. All JNKs can kill, but nuclear localization is critical for neuronal death. J Biol Chem 283:19704–19713. doi: 10.1074/jbc.M707744200. [DOI] [PubMed] [Google Scholar]
- 407.Vercelli A, Biggi S, Sclip A, Repetto IE, Cimini S, Falleroni F, Tomasi S, Monti R, Tonna N, Morelli F, Grande V, Stravalaci M, Biasini E, Marin O, Bianco F, di Marino D, Borsello T. 2015. Exploring the role of MKK7 in excitotoxicity and cerebral ischemia: a novel pharmacological strategy against brain injury. Cell Death Dis 6:e1854. doi: 10.1038/cddis.2015.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Kenney AM, Kocsis JD. 1998. Peripheral axotomy induces long-term c-Jun amino-terminal kinase-1 activation and activator protein-1 binding activity by c-Jun and junD in adult rat dorsal root ganglia in vivo. J Neurosci 18:1318–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Tu NH, Katano T, Matsumura S, Pham VM, Muratani T, Minami T, Ito S. 2016. Role of c-Jun N-terminal kinase in late nerve regeneration monitored by in vivo imaging of thy1-yellow fluorescent protein transgenic mice. Eur J Neurosci 43:548–560. doi: 10.1111/ejn.13139. [DOI] [PubMed] [Google Scholar]
- 410.Barnat M, Enslen H, Propst F, Davis RJ, Soares S, Nothias F. 2010. Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J Neurosci 30:7804–7816. doi: 10.1523/JNEUROSCI.0372-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Kunde SA, Rademacher N, Tzschach A, Wiedersberg E, Ullmann R, Kalscheuer VM, Shoichet SA. 2013. Characterisation of de novo MAPK10/JNK3 truncation mutations associated with cognitive disorders in two unrelated patients. Hum Genet 132:461–471. doi: 10.1007/s00439-012-1260-5. [DOI] [PubMed] [Google Scholar]
- 412.Sury MD, McShane E, Hernandez-Miranda LR, Birchmeier C, Selbach M. 2015. Quantitative proteomics reveals dynamic interaction of c-Jun N-terminal kinase (JNK) with RNA transport granule proteins splicing factor proline- and glutamine-rich (Sfpq) and non-POU domain-containing octamer-binding protein (Nono) during neuronal differentiation. Mol Cell Proteomics 14:50–65. doi: 10.1074/mcp.M114.039370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Ventura JJ, Kennedy NJ, Lamb JA, Flavell RA, Davis RJ. 2003. c-Jun NH(2)-terminal kinase is essential for the regulation of AP-1 by tumor necrosis factor. Mol Cell Biol 23:2871–2882. doi: 10.1128/MCB.23.8.2871-2882.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Ventura JJ, Kennedy NJ, Flavell RA, Davis RJ. 2004. JNK regulates autocrine expression of TGF-beta1. Mol Cell 15:269–278. doi: 10.1016/j.molcel.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 415.Zarzynska JM. 2014. Two faces of TGF-beta1 in breast cancer. Mediators Inflamm 2014:141747. doi: 10.1155/2014/141747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Chen F. 2012. JNK-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res 72:379–386. doi: 10.1158/0008-5472.CAN-11-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Hess P, Pihan G, Sawyers CL, Flavell RA, Davis RJ. 2002. Survival signaling mediated by c-Jun NH(2)-terminal kinase in transformed B lymphoblasts. Nat Genet 32:201–205. doi: 10.1038/ng946. [DOI] [PubMed] [Google Scholar]
- 418.Su GH, Hilgers W, Shekher MC, Tang DJ, Yeo CJ, Hruban RH, Kern SE. 1998. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Res 58:2339–2342. [PubMed] [Google Scholar]
- 419.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ. 2015. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43:D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Cancer Genome Atlas Network. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP, Peters BA, Pujara K, Cordes S, Davis DP, Carlton VE, Yuan W, Li L, Wang W, Eigenbrot C, Kaminker JS, Eberhard DA, Waring P, Schuster SC, Modrusan Z, Zhang Z, Stokoe D, de Sauvage FJ, Faham M, Seshagiri S. 2010. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 422.Goldstein BJ, Müller-Wieland D. 2007. Type 2 diabetes: principles and practice, 2nd ed. CRC Press, Boca Raton, FL. [Google Scholar]
- 423.Sabio G, Kennedy NJ, Cavanagh-Kyros J, Jung DY, Ko HJ, Ong H, Barrett T, Kim JK, Davis RJ. 2010. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol 30:106–115. doi: 10.1128/MCB.01162-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ. 2013. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339:218–222. doi: 10.1126/science.1227568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. 2008. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322:1539–1543. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Sabio G, Cavanagh-Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, Barrett T, Mora A, Kim JK, Davis RJ. 2009. Prevention of steatosis by hepatic JNK1. Cell Metab 10:491–498. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. 2010. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab 11:84–92. doi: 10.1016/j.cmet.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. 2002. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem 277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 429.Hoehn KL, Hohnen-Behrens C, Cederberg A, Wu LE, Turner N, Yuasa T, Ebina Y, James DE. 2008. IRS1-independent defects define major nodes of insulin resistance. Cell Metab 7:421–433. doi: 10.1016/j.cmet.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Li LC, Wang Y, Carr R, Haddad CS, Li Z, Qian L, Oberholzer J, Maker AV, Wang Q, Prabhakar BS. 2014. IG20/MADD plays a critical role in glucose-induced insulin secretion. Diabetes 63:1612–1623. doi: 10.2337/db13-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Mokhtari D, Myers JW, Welsh N. 2008. The MAPK kinase kinase-1 is essential for stress-induced pancreatic islet cell death. Endocrinology 149:3046–3053. doi: 10.1210/en.2007-0438. [DOI] [PubMed] [Google Scholar]
- 432.Abdelli S, Puyal J, Bielmann C, Buchillier V, Abderrahmani A, Clarke PG, Beckmann JS, Bonny C. 2009. JNK3 is abundant in insulin-secreting cells and protects against cytokine-induced apoptosis. Diabetologia 52:1871–1880. doi: 10.1007/s00125-009-1431-7. [DOI] [PubMed] [Google Scholar]
- 433.Delaney JR, Stoven S, Uvell H, Anderson KV, Engstrom Y, Mlodzik M. 2006. Cooperative control of Drosophila immune responses by the JNK and NF-kappaB signaling pathways. EMBO J 25:3068–3077. doi: 10.1038/sj.emboj.7601182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Sabapathy K, Hu Y, Kallunki T, Schreiber M, David JP, Jochum W, Wagner EF, Karin M. 1999. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr Biol 9:116–125. doi: 10.1016/S0960-9822(99)80065-7. [DOI] [PubMed] [Google Scholar]
- 435.Sabapathy K, Kallunki T, David JP, Graef I, Karin M, Wagner EF. 2001. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have similar and stage-dependent roles in regulating T cell apoptosis and proliferation. J Exp Med 193:317–328. doi: 10.1084/jem.193.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben-Neriah Y. 1994. JNK is involved in signal integration during costimulation of T lymphocytes. Cell 77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 437.Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. 1998. Defective T cell differentiation in the absence of Jnk1. Science 282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 438.Behrens A, Sabapathy K, Graef I, Cleary M, Crabtree GR, Wagner EF. 2001. Jun N-terminal kinase 2 modulates thymocyte apoptosis and T cell activation through c-Jun and nuclear factor of activated T cell (NF-AT). Proc Natl Acad Sci U S A 98:1769–1774. doi: 10.1073/pnas.98.4.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Chu WM, Ostertag D, Li ZW, Chang L, Chen Y, Hu Y, Williams B, Perrault J, Karin M. 1999. JNK2 and IKKbeta are required for activating the innate response to viral infection. Immunity 11:721–731. doi: 10.1016/S1074-7613(00)80146-6. [DOI] [PubMed] [Google Scholar]
- 440.Arthur JS, Ley SC. 2013. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol 13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 441.Papa S, Zazzeroni F, Pham CG, Bubici C, Franzoso G. 2004. Linking JNK signaling to NF-kappaB: a key to survival. J Cell Sci 117:5197–5208. doi: 10.1242/jcs.01483. [DOI] [PubMed] [Google Scholar]
- 442.Das M, Sabio G, Jiang F, Rincon M, Flavell RA, Davis RJ. 2009. Induction of hepatitis by JNK-mediated expression of TNF-alpha. Cell 136:249–260. doi: 10.1016/j.cell.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Filardy AA, Costa-da-Silva AC, Koeller CM, Guimaraes-Pinto K, Ribeiro-Gomes FL, Lopes MF, Heise N, Freire-de-Lima CG, Nunes MP, DosReis GA. 2014. Infection with Leishmania major induces a cellular stress response in macrophages. PLoS One 9:e85715. doi: 10.1371/journal.pone.0085715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Ramphul UN, Garver LS, Molina-Cruz A, Canepa GE, Barillas-Mury C. 2015. Plasmodium falciparum evades mosquito immunity by disrupting JNK-mediated apoptosis of invaded midgut cells. Proc Natl Acad Sci U S A 112:1273–1280. doi: 10.1073/pnas.1423586112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Krachler AM, Woolery AR, Orth K. 2011. Manipulation of kinase signaling by bacterial pathogens. J Cell Biol 195:1083–1092. doi: 10.1083/jcb.201107132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Shan L, He P, Sheen J. 2007. Intercepting host MAPK signaling cascades by bacterial type III effectors. Cell Host Microbe 1:167–174. doi: 10.1016/j.chom.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 447.Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, Orth K. 2006. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312:1211–1214. doi: 10.1126/science.1126867. [DOI] [PubMed] [Google Scholar]
- 448.Hao YH, Wang Y, Burdette D, Mukherjee S, Keitany G, Goldsmith E, Orth K. 2008. Structural requirements for Yersinia YopJ inhibition of MAP kinase pathways. PLoS One 3:e1375. doi: 10.1371/journal.pone.0001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Sweet CR, Conlon J, Golenbock DT, Goguen J, Silverman N. 2007. YopJ targets TRAF proteins to inhibit TLR-mediated NF-kappaB, MAPK and IRF3 signal transduction. Cell Microbiol 9:2700–2715. doi: 10.1111/j.1462-5822.2007.00990.x. [DOI] [PubMed] [Google Scholar]
- 450.Jones RM, Wu H, Wentworth C, Luo L, Collier-Hyams L, Neish AS. 2008. Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe 3:233–244. doi: 10.1016/j.chom.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 451.Trosky JE, Li Y, Mukherjee S, Keitany G, Ball H, Orth K. 2007. VopA inhibits ATP binding by acetylating the catalytic loop of MAPK kinases. J Biol Chem 282:34299–34305. doi: 10.1074/jbc.M706970200. [DOI] [PubMed] [Google Scholar]
- 452.Kim KH, An DR, Song J, Yoon JY, Kim HS, Yoon HJ, Im HN, Kim J, Kim do, Lee JSJ, Kim KH, Lee HM, Kim HJ, Jo EK, Lee JY, Suh SW. 2012. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci U S A 109:7729–7734. doi: 10.1073/pnas.1120251109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Baruch K, Gur-Arie L, Nadler C, Koby S, Yerushalmi G, Ben-Neriah Y, Yogev O, Shaulian E, Guttman C, Zarivach R, Rosenshine I. 2011. Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. EMBO J 30:221–231. doi: 10.1038/emboj.2010.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. 2000. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J 352:739–745. doi: 10.1042/bj3520739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Halle M, Gomez MA, Stuible M, Shimizu H, McMaster WR, Olivier M, Tremblay ML. 2009. The Leishmania surface protease GP63 cleaves multiple intracellular proteins and actively participates in p38 mitogen-activated protein kinase inactivation. J Biol Chem 284:6893–6908. doi: 10.1074/jbc.M805861200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, Chen S, Zhou JM, Shao F. 2007. The phosphothreonine lyase activity of a bacterial type III effector family. Science 315:1000–1003. doi: 10.1126/science.1138960. [DOI] [PubMed] [Google Scholar]
- 457.Brennan DF, Barford D. 2009. Eliminylation: a post-translational modification catalyzed by phosphothreonine lyases. Trends Biochem Sci 34:108–114. doi: 10.1016/j.tibs.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 458.Mazurkiewicz P, Thomas J, Thompson JA, Liu M, Arbibe L, Sansonetti P, Holden DW. 2008. SpvC is a Salmonella effector with phosphothreonine lyase activity on host mitogen-activated protein kinases. Mol Microbiol 67:1371–1383. doi: 10.1111/j.1365-2958.2008.06134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Zhang J, Shao F, Li Y, Cui H, Chen L, Li H, Zou Y, Long C, Lan L, Chai J, Chen S, Tang X, Zhou JM. 2007. A Pseudomonas syringae effector inactivates MAPKs to suppress PAMP-induced immunity in plants. Cell Host Microbe 1:175–185. doi: 10.1016/j.chom.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 460.Marsolier J, Perichon M, DeBarry JD, Villoutreix BO, Chluba J, Lopez T, Garrido C, Zhou XZ, Lu KP, Fritsch L, Ait-Si-Ali S, Mhadhbi M, Medjkane S, Weitzman JB. 2015. Theileria parasites secrete a prolyl isomerase to maintain host leukocyte transformation. Nature 520:378–382. doi: 10.1038/nature14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Busch LK, Bishop GA. 2001. Multiple carboxyl-terminal regions of the EBV oncoprotein, latent membrane protein 1, cooperatively regulate signaling to B lymphocytes via TNF receptor-associated factor (TRAF)-dependent and TRAF-independent mechanisms. J Immunol 167:5805–5813. doi: 10.4049/jimmunol.167.10.5805. [DOI] [PubMed] [Google Scholar]
- 462.Kutz H, Reisbach G, Schultheiss U, Kieser A. 2008. The c-Jun N-terminal kinase pathway is critical for cell transformation by the latent membrane protein 1 of Epstein-Barr virus. Virology 371:246–256. doi: 10.1016/j.virol.2007.09.044. [DOI] [PubMed] [Google Scholar]
- 463.Manganaro L, Lusic M, Gutierrez MI, Cereseto A, Del Sal G, Giacca M. 2010. Concerted action of cellular JNK and Pin1 restricts HIV-1 genome integration to activated CD4+ T lymphocytes. Nat Med 16:329–333. doi: 10.1038/nm.2102. [DOI] [PubMed] [Google Scholar]
- 464.Rahaus M, Desloges N, Wolff MH. 2004. Replication of varicella-zoster virus is influenced by the levels of JNK/SAPK and p38/MAPK activation. J Gen Virol 85:3529–3540. doi: 10.1099/vir.0.80347-0. [DOI] [PubMed] [Google Scholar]
- 465.Kim SM, Park JH, Chung SK, Kim JY, Hwang HY, Chung KC, Jo I, Park SI, Nam JH. 2004. Coxsackievirus B3 infection induces cyr61 activation via JNK to mediate cell death. J Virol 78:13479–13488. doi: 10.1128/JVI.78.24.13479-13488.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Stronach B, Lennox AL, Garlena RA. 2014. Domain specificity of MAP3K family members, MLK and Tak1, for JNK signaling in Drosophila. Genetics 197:497–513. doi: 10.1534/genetics.113.160937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Wallbach M, Duque Escobar J, Babaeikelishomi R, Stahnke MJ, Blume R, Schroder S, Kruegel J, Maedler K, Kluth O, Kehlenbach RH, Miosge N, Oetjen E. 2016. Distinct functions of the dual leucine zipper kinase depending on its subcellular localization. Cell Signal 28:272–283. doi: 10.1016/j.cellsig.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 468.Fosbrink M, Aye-Han NN, Cheong R, Levchenko A, Zhang J. 2010. Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc Natl Acad Sci U S A 107:5459–5464. doi: 10.1073/pnas.0909671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. 2006. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell 21:701–710. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 470.Kocieniewski P, Faeder JR, Lipniacki T. 2012. The interplay of double phosphorylation and scaffolding in MAPK pathways. J Theor Biol 295:116–124. doi: 10.1016/j.jtbi.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Lee MH, Padmashali R, Koria P, Andreadis ST. 2011. JNK regulates binding of alpha-catenin to adherens junctions and cell-cell adhesion. FASEB J 25:613–623. doi: 10.1096/fj.10-161380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. 1995. Integration of MAP kinase signal transduction pathways at the serum response element. Science 269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 473.Cavigelli M, Dolfi F, Claret FX, Karin M. 1995. Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J 14:5957–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Janknecht R, Hunter T. 1997. Activation of the Sap-1a transcription factor by the c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase. J Biol Chem 272:4219–4224. doi: 10.1074/jbc.272.7.4219. [DOI] [PubMed] [Google Scholar]
- 475.Katz S, Heinrich R, Aronheim A. 2001. The AP-1 repressor, JDP2, is a bona fide substrate for the c-Jun N-terminal kinase. FEBS Lett 506:196–200. doi: 10.1016/S0014-5793(01)02907-6. [DOI] [PubMed] [Google Scholar]
- 476.Yazgan O, Pfarr CM. 2002. Regulation of two JunD isoforms by Jun N-terminal kinases. J Biol Chem 277:29710–29718. doi: 10.1074/jbc.M204552200. [DOI] [PubMed] [Google Scholar]
- 477.Mayer C, Bierhoff H, Grummt I. 2005. The nucleolus as a stress sensor: JNK2 inactivates the transcription factor TIF-IA and down-regulates rRNA synthesis. Genes Dev 19:933–941. doi: 10.1101/gad.333205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa M, Fujisawa J, Okazaki K. 2004. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene 23:7416–7429. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- 479.Yoshida K, Matsuzaki K, Mori S, Tahashi Y, Yamagata H, Furukawa F, Seki T, Nishizawa M, Fujisawa J, Okazaki K. 2005. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol 166:1029–1039. doi: 10.1016/S0002-9440(10)62324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Zhang Y, Cho YY, Petersen BL, Zhu F, Dong Z. 2004. Evidence of STAT1 phosphorylation modulated by MAPKs, MEK1 and MSK1 Carcinogenesis 25:1165–1175. doi: 10.1093/carcin/bgh115. [DOI] [PubMed] [Google Scholar]
- 481.Liu J, Chen B, Lu Y, Guan Y, Chen F. 2012. JNK-dependent Stat3 phosphorylation contributes to Akt activation in response to arsenic exposure. Toxicol Sci 129:363–371. doi: 10.1093/toxsci/kfs199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Kasibhatla S, Tailor P, Bonefoy-Berard N, Mustelin T, Altman A, Fotedar A. 1999. Jun kinase phosphorylates and regulates the DNA binding activity of an octamer binding protein, T-cell factor beta1. Mol Cell Biol 19:2021–2031. doi: 10.1128/MCB.19.3.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Yoshida I, Ibuki Y. 2014. Formaldehyde-induced histone H3 phosphorylation via JNK and the expression of proto-oncogenes. Mutat Res 770:9–18. doi: 10.1016/j.mrfmmm.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 484.Huang YF, Lin JJ, Lin CH, Su Y, Hung SC. 2012. c-Jun N-terminal kinase 1 negatively regulates osteoblastic differentiation induced by BMP2 via phosphorylation of Runx2 at Ser104. J Bone Miner Res 27:1093–1105. doi: 10.1002/jbmr.1548. [DOI] [PubMed] [Google Scholar]
- 485.Kim G, Han JM, Kim S. 2010. Toll-like receptor 4-mediated c-Jun N-terminal kinase activation induces gp96 cell surface expression via AIMP1 phosphorylation. Biochem Biophys Res Commun 397:100–105. doi: 10.1016/j.bbrc.2010.05.075. [DOI] [PubMed] [Google Scholar]
- 486.Cervenka I, Wolf J, Masek J, Krejci P, Wilcox WR, Kozubik A, Schulte G, Gutkind JS, Bryja V. 2011. Mitogen-activated protein kinases promote WNT/beta-catenin signaling via phosphorylation of LRP6. Mol Cell Biol 31:179–189. doi: 10.1128/MCB.00550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Nakatsu D, Kano F, Taguchi Y, Sugawara T, Nishizono T, Nishikawa K, Oda Y, Furuse M, Murata M. 2014. JNK1/2-dependent phosphorylation of angulin-1/LSR is required for the exclusive localization of angulin-1/LSR and tricellulin at tricellular contacts in EpH4 epithelial sheet. Genes Cells 19:565–581. doi: 10.1111/gtc.12158. [DOI] [PubMed] [Google Scholar]
- 488.Liu B, Wu JF, Zhan YY, Chen HZ, Zhang XY, Wu Q. 2007. Regulation of the orphan receptor TR3 nuclear functions by c-Jun N terminal kinase phosphorylation. Endocrinology 148:34–44. doi: 10.1210/en.2006-0800. [DOI] [PubMed] [Google Scholar]
- 489.Knebel B, Lehr S, Hartwig S, Haas J, Kaber G, Dicken HD, Susanto F, Bohne L, Jacob S, Nitzgen U, Passlack W, Muller-Wieland D, Kotzka J. 2014. Phosphorylation of sterol regulatory element-binding protein (SREBP)-1c by p38 kinases, ERK and JNK influences lipid metabolism and the secretome of human liver cell line HepG2. Arch Physiol Biochem 120:216–227. doi: 10.3109/13813455.2014.973418. [DOI] [PubMed] [Google Scholar]
- 490.Shao Z, Bhattacharya K, Hsich E, Park L, Walters B, Germann U, Wang YM, Kyriakis J, Mohanlal R, Kuida K, Namchuk M, Salituro F, Yao YM, Hou WM, Chen X, Aronovitz M, Tsichlis PN, Bhattacharya S, Force T, Kilter H. 2006. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ Res 98:111–118. doi: 10.1161/01.RES.0000197781.20524.b9. [DOI] [PubMed] [Google Scholar]
- 491.Cotsiki M, Oehrl W, Samiotaki M, Theodosiou A, Panayotou G. 2012. Phosphorylation of the M3/6 dual-specificity phosphatase enhances the activation of JNK by arsenite. Cell Signal 24:664–676. doi: 10.1016/j.cellsig.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 492.Uchida S, Yoshioka K, Kizu R, Nakagama H, Matsunaga T, Ishizaka Y, Poon RY, Yamashita K. 2009. Stress-activated mitogen-activated protein kinases c-Jun NH2-terminal kinase and p38 target Cdc25B for degradation. Cancer Res 69:6438–6444. doi: 10.1158/0008-5472.CAN-09-0869. [DOI] [PubMed] [Google Scholar]
- 493.Uchida S, Watanabe N, Kudo Y, Yoshioka K, Matsunaga T, Ishizaka Y, Nakagama H, Poon RY, Yamashita K. 2011. SCFbeta(TrCP) mediates stress-activated MAPK-induced Cdc25B degradation. J Cell Sci 124:2816–2825. doi: 10.1242/jcs.083931. [DOI] [PubMed] [Google Scholar]
- 494.Goss VL, Cross JV, Ma K, Qian Y, Mola PW, Templeton DJ. 2003. SAPK/JNK regulates cdc2/cyclin B kinase through phosphorylation and inhibition of cdc25c. Cell Signal 15:709–718. doi: 10.1016/S0898-6568(03)00009-3. [DOI] [PubMed] [Google Scholar]
- 495.Gutierrez GJ, Tsuji T, Cross JV, Davis RJ, Templeton DJ, Jiang W, Ronai ZA. 2010. JNK-mediated phosphorylation of Cdc25C regulates cell cycle entry and G(2)/M DNA damage checkpoint. J Biol Chem 285:14217–14228. doi: 10.1074/jbc.M110.121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Kwak D, Choi S, Jeong H, Jang JH, Lee Y, Jeon H, Lee MN, Noh J, Cho K, Yoo JS, Hwang D, Suh PG, Ryu SH. 2012. Osmotic stress regulates mammalian target of rapamycin (mTOR) complex 1 via c-Jun N-terminal kinase (JNK)-mediated Raptor protein phosphorylation. J Biol Chem 287:18398–18407. doi: 10.1074/jbc.M111.326538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Bi W, Xiao L, Jia Y, Wu J, Xie Q, Ren J, Ji G, Yuan Z. 2010. c-Jun N-terminal kinase enhances MST1-mediated pro-apoptotic signaling through phosphorylation at serine 82. J Biol Chem 285:6259–6264. doi: 10.1074/jbc.M109.038570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Zhang Y, Zhong S, Dong Z, Chen N, Bode AM, Ma W, Dong Z. 2001. UVA induces Ser381 phosphorylation of p90RSK/MAPKAP-K1 via ERK and JNK pathways. J Biol Chem 276:14572–14580. doi: 10.1074/jbc.M004615200. [DOI] [PubMed] [Google Scholar]
- 499.Cargnello M, Tcherkezian J, Dorn JF, Huttlin EL, Maddox PS, Gygi SP, Roux PP. 2012. Phosphorylation of the eukaryotic translation initiation factor 4E-transporter (4E-T) by c-Jun N-terminal kinase promotes stress-dependent P-body assembly. Mol Cell Biol 32:4572–4584. doi: 10.1128/MCB.00544-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Le S, Connors TJ, Maroney AC. 2001. c-Jun N-terminal kinase specifically phosphorylates p66ShcA at serine 36 in response to ultraviolet irradiation. J Biol Chem 276:48332–48336. doi: 10.1074/jbc.M106612200. [DOI] [PubMed] [Google Scholar]
- 501.Sun G, Irvine KD. 2013. Ajuba family proteins link JNK to Hippo signaling. Sci Signal 6:ra81. doi: 10.1126/scisignal.2004324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Casas J, Meana C, Esquinas E, Valdearcos M, Pindado J, Balsinde J, Balboa MA. 2009. Requirement of JNK-mediated phosphorylation for translocation of group IVA phospholipase A2 to phagosomes in human macrophages. J Immunol 183:2767–2774. doi: 10.4049/jimmunol.0901530. [DOI] [PubMed] [Google Scholar]
- 503.Gorentla BK, Moritz AE, Foster JD, Vaughan RA. 2009. Proline-directed phosphorylation of the dopamine transporter N-terminal domain. Biochemistry 48:1067–1076. doi: 10.1021/bi801696n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Gallagher E, Gao M, Liu YC, Karin M. 2006. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc Natl Acad Sci U S A 103:1717–1722. doi: 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Rinschen MM, Yu MJ, Wang G, Boja ES, Hoffert JD, Pisitkun T, Knepper MA. 2010. Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-dependent signaling pathways in renal collecting duct cells. Proc Natl Acad Sci U S A 107:3882–3887. doi: 10.1073/pnas.0910646107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Kim H, Jung O, Kang M, Lee MS, Jeong D, Ryu J, Ko Y, Choi YJ, Lee JW. 2012. JNK signaling activity regulates cell-cell adhesions via TM4SF5-mediated p27(Kip1) phosphorylation. Cancer Lett 314:198–205. doi: 10.1016/j.canlet.2011.09.030. [DOI] [PubMed] [Google Scholar]
- 507.Yabu T, Shiba H, Shibasaki Y, Nakanishi T, Imamura S, Touhata K, Yamashita M. 2015. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Differ 22:258–273. doi: 10.1038/cdd.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Yip YY, Yeap YY, Bogoyevitch MA, Ng DC. 2014. Differences in c-Jun N-terminal kinase recognition and phosphorylation of closely related stathmin-family members. Biochem Biophys Res Commun 446:248–254. doi: 10.1016/j.bbrc.2014.02.101. [DOI] [PubMed] [Google Scholar]
- 509.Tararuk T, Ostman N, Li W, Bjorkblom B, Padzik A, Zdrojewska J, Hongisto V, Herdegen T, Konopka W, Courtney MJ, Coffey ET. 2006. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J Cell Biol 173:265–277. doi: 10.1083/jcb.200511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Morfini GA, You YM, Pollema SL, Kaminska A, Liu K, Yoshioka K, Bjorkblom B, Coffey ET, Bagnato C, Han D, Huang CF, Banker G, Pigino G, Brady ST. 2009. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat Neurosci 12:864–871. doi: 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Pan YR, Tseng WS, Chang PW, Chen HC. 2013. Phosphorylation of moesin by Jun N-terminal kinase is important for podosome rosette formation in Src-transformed fibroblasts. J Cell Sci 126:5670–5680. doi: 10.1242/jcs.134361. [DOI] [PubMed] [Google Scholar]
- 512.Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N. 2003. A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem 278:22946–22955. doi: 10.1074/jbc.M212160200. [DOI] [PubMed] [Google Scholar]
- 513.Mori Y, Higuchi M, Hirabayashi Y, Fukuda M, Gotoh Y. 2008. JNK phosphorylates synaptotagmin-4 and enhances Ca2+-evoked release. EMBO J 27:76–87. doi: 10.1038/sj.emboj.7601935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Kim BJ, Ryu SW, Song BJ. 2006. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem 281:21256–21265. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 515.Rath S, Das L, Kokate SB, Pratheek BM, Chattopadhyay S, Goswami C, Chattopadhyay R, Crowe SE, Bhattacharyya A. 2015. Regulation of Noxa-mediated apoptosis in Helicobacter pylori-infected gastric epithelial cells. FASEB J 29:796–806. doi: 10.1096/fj.14-257501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Prakasam A, Ghose S, Oleinik NV, Bethard JR, Peterson YK, Krupenko NI, Krupenko SA. 2014. JNK1/2 regulate Bid by direct phosphorylation at Thr59 in response to ALDH1L1. Cell Death Dis 5:e1358. doi: 10.1038/cddis.2014.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Park B. 2014. JNK1mediated phosphorylation of Smac/DIABLO at the serine 6 residue is functionally linked to its mitochondrial release during TNFalpha-induced apoptosis of HeLa cells. Mol Med Rep 10:3205–3210. doi: 10.3892/mmr.2014.2625. [DOI] [PubMed] [Google Scholar]
- 518.Court NW, Kuo I, Quigley O, Bogoyevitch MA. 2004. Phosphorylation of the mitochondrial protein Sab by stress-activated protein kinase 3. Biochem Biophys Res Commun 319:130–137. doi: 10.1016/j.bbrc.2004.04.148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.