

Abstract
Toll-like receptors (TLRs) have been shown to play an important role in the immune system, which warrants their remarkable pharmacological potentials. Activation of TLRs requires participation from specific PAMPs (pathogen associated molecular patterns) and accessory proteins such as MD2 (myeloid differentiation protein 2), LBP (lipopolysaccharide binding protein), and CD-14. Assembly of the TLR4-MD2-LPS complex is essential in TLR4 activation. Recent studies have revealed that TLR4 activation is a significant trigger of signal transmission pathways in the nervous system, which could result in chronic pain as well as opioid tolerance and dependence. Researchers of the molecular structure of TLRs and their accessory proteins have opened a door to syntheses of TLRs agonists and antagonists, such as Eritoran. Small molecule modulators of TLR4, such as MD2-I and tricyclic anti-depressants, offer more promising prospects than peptides for their convenient oral usage and lower cost. We mainly discuss the mechanism and clinical prospect of TLR4 agonists and antagonists in this review.
Keywords: Toll-like receptor, inflammation mechanism, neuropathic pain, analgesia, accessory protein, agonist, antagonist, small molecule modulator, drug discovery
Graphical Abstract
Toll-like receptors (TLRs) are a family of membrane proteins crucial to the cellular innate immune response. Pain is one of the most intractable and widespread conditions that people face today. At the crossroads of the immune system and pain is TLR4. This review aims to provide a brief summary about TLRs and how they their signaling affects pain. Furthermore, this review also takes a look at the molecules that are able to modulate TLR4 signaling as potential drug candidates.
1. Introduction
Since ancient times, the secrets of the body’s self-defense system have been of interest to the earliest doctors. Centuries later, much of the human immune system has been explained, while some details still remain unsolved. Toll-like receptors (TLRs) are one of the greatest discoveries of immune science, and have been widely known to play an important role in the inflammation response [1] Inflammation is one of the most significant immune reactions, triggered by the binding of specific ligands called PAMPs (pathogen associated molecular patterns) to their respective TLRs [2]. As one of the earliest discovered pattern recognition receptors (PRRs) [3], the TLRs were named after the toll receptor of Drosophila, because of their structural similarity and function in host defense [2]. Binding of specific ligands with TLRs (Table 1) will initiate a particular signal transduction pathway, whose activation would lead to the expression of specific effector genes (Figure 1) [4]. Macrophages would also be activated upon recognition of the pathogen signals [5].
Table 1.
List of human and rodent TLRs
| TLR | Exogenous ligands | Origin of ligands | References | Endogenous ligands | References | Localizations |
|---|---|---|---|---|---|---|
| TLR1 | Triacyl lipopeptides | bacteria and mycobacteria | [56] | cell surfaces | ||
| Soluble factors | Neisseria meningitidis | |||||
| TLR2 | Lipoprotein/lipopeptides | various pathogens | [57] | Heat-shock protein 70 | [7d] | cell surfaces |
| Peptidoglycan | gram-positive bacteria | [58] | HGMB1 | [59] | ||
| Lipoteichoic acid | gram-positive bacteria | [58b] | ||||
| Lipoarabinomannan | mycobacteria | [60] | ||||
| Phenol-soluble modulin | Stapylococcus epidermidis | [61] | ||||
| Glycoinositolphospholipids | Trypanosoma cruzi | [62] | ||||
| Glycolipids | Treponema maltophilum | [63] | ||||
| Porins | Neisseria | [64] | ||||
| Atypical lipopolysaccharide | Leptospira interrogans | [65] | ||||
| Atypical lipopolysaccharide | Porphyromanas gingivalis | [66] | ||||
| Zymosan | fungi | [67] | ||||
| TLR3 | Double-stranded RNA | viruses | [68] | Double-stranded DNA | [69] | endosomes |
| TLR4 | Lipopolysaccharide | gram-negative bacteria | [70] | Heat-shock protein 70 | [71] | cell surfaces |
| Fusion protein | respiratory syncytial virus | [72] | Type III repeat extra domain A of fibronectin | [73] | ||
| envelope protein | mouse mammary-tumor virus | [74] | Oligosaccharides of hyaluronic acid | [75] | ||
| Heat-shock protein 60 | Chlamydia pneumoniae | [76] | Polysaccharide fragment of heparan sulphate | [77] | ||
| Fibrinogen | [78] | |||||
| TLR5 | Flagellin | bacteria | [79] | cell surfaces | ||
| TLR6 | Diacyl lipopeptides | Mycoplasma | [80] | cell surfaces | ||
| Lipoteichoic acid | gram-negative bacteria | [58b] | ||||
| Zymosan | fungi | [81] | ||||
| TLR7 | Single-stranded RNA | viruses | [82] | Single-stranded RNA | [83] | endosomes |
| Imiquimod (R837) | synthetic compound (FDA approved drug) | [84] | ||||
| TLR8 | Single-stranded RNA | viruses | [82a] | Single-stranded RNA | [83] | endosomes |
| TLR9 | CpG-containing DNA | bacteria and viruses | [85] | DNA | [86] | endosomes |
| TLR10 | Not Determined | Not Determined | Not Determined | |||
| TLR11 | profilin | Uropathogenic pathogen, toxoplasma | [87] | Not Determined | ||
| TLR13 | 23s rRNA | gram-negative/positive bacteria | [88] | Not Determined |
Figure 1.

Myeloid differentiation primary response gene 88 (MyD88) is the key signaling adaptor for TLR1, TLR2, TLR4, TLR5, TLR6, TLR7, TLR8 and TLR9. Only TLR3 and TLR4 signal via TIR-domain-containing adapter-inducing interferon-β (TRIF). MAL, MyD88 adaptor-like protein; TRAM, TRIF-related adaptor molecule; TBK1, tumor necrosis factor receptor-associated factor (TRAF) family member-associated NF-κB activator (TANK)- binding kinase 1; IKK, inhibitor of NF-κB kinase; IκB, inhibitor of NF-κB; NF-κB, nuclear factor-κB; IRF, interferon regulatory factor; IFN, interferon.
1.1 Ligands of TLRs
TLRs make up one of the biggest family of PRRs. There are at least 13 Toll-like receptors that have been discovered in mammals [6]. Among them, TLRs 1-10 have been found functioning inside the human body [2]. The different members of the TLR family can recognize diverse ligands shown in Table 1 [2, 7].
1.2. TLRs in the development of immune diseases
While TLRs play a protective role as an integral part of the innate immune system, misidentification or excessive activity of TLRs may lead to chronic immune diseases such as osteoarthritis, asthma, neuralgia and sepsis [8]. The inappropriately activated signaling pathway will result in either attack of immune cells, such as macrophage or dendritic cells, or apoptosis of healthy cells [9].
2. TLR4-mediated neuroinflammation
2.1. TLR4 signaling pathway
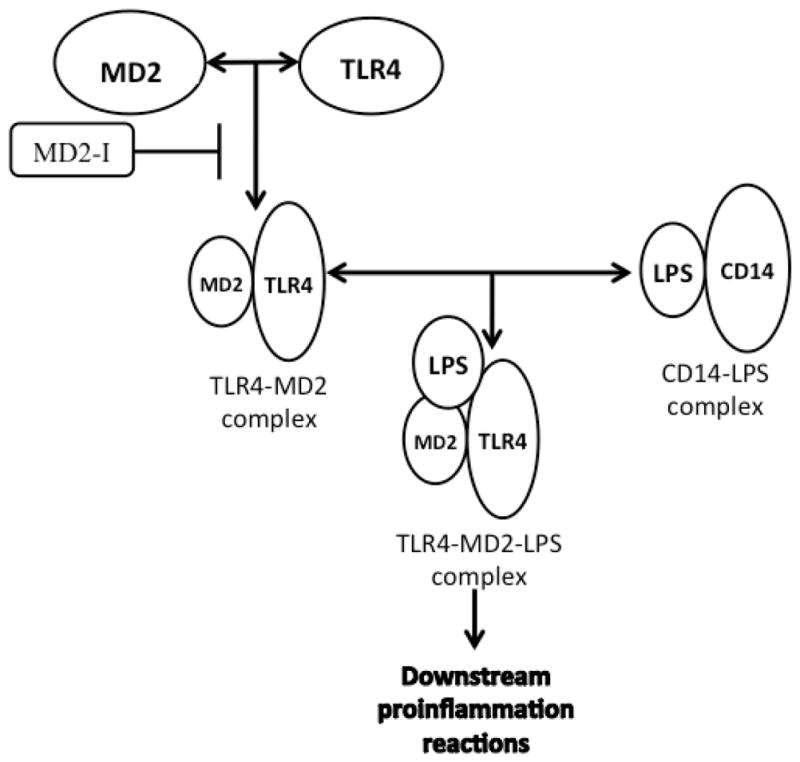
In the study of TLR4 and its signaling pathway, a route diagram of the TLR4 signaling pathway in microglia was clearly developed [9a]. There are several important processes prior to TLR4 activation, which is triggered by a PAMP (usually LPS) or danger associated molecular pattern (DAMP) molecule. Firstly, with the assistance of LPS-binding-protein (LBP), LPS is extracted from the membrane structure of the pathogen or damaged cell [10]. Then, LBP transports an LPS molecule to cluster differentiation antigen 14 (CD14) to form a dimer complex (soluble CD14-LPS)2 or monomer complex (membrane CD14-LPS). Either complex can deliver LPS to the MD2-TLR4 heterodimer complex (MD2, myeloid differentiation protein 2, a TLR4 specific accessory protein) [11]. The interaction between those complexes results in the formation of a new ternary complex (TLR4-MD2-LPS)2 (Figure 2) [12]. The formation of (TLR4-MD2-LPS)2 triggers two intracellular signaling pathways, which results in the production of pro-inflammatory cytokines [13]. Those two signaling pathways begin with the binding of intracellular adapter proteins, MyD88 (myeloid differentiation primary-response gene 88) or TRIF (Toll/IL-1R (TIR)-domain-containing adaptor protein) (Figure 1) [14].
Figure 2.

TLR4 activation requires the participation of some accessory proteins besides TLR4 and LPS. LBP extracts LPS from bacteria and transports it to CD-14 to form a dimer (soluble CD14-LPS)2 complex or monomer (membrane CD14-LPS). Either of the CD14-LPS complexs can bind to the MD2-TLR4 complex to form a ternary TLR2-MD2-LPS complex, which triggers downstream pro-inflammatory signaling.
The specific details about how the previously mentioned cytokines and signaling proteins bind or react with each other are not completely understood [14a]. Studies have shown that one of the final products of those signaling pathways is nitric oxide (NO), which is generally recognized as an important neural signaling molecule. For this reason, the detection of NO concentration has become a popular method to evaluate the activity of TLR4 [4].
2.2. TLR4 activation in pain
Recent studies of chronic immune diseases have indicated that development of chronic pain is closely associated with the activation of TLR4 [9a]. Chronic pain is one of the most intractable diseases with high prevalence. Recently, a systematic review containing 13 studies demonstrated that the prevalence of chronic pain in developed countries may be as much as 55% [15]. Chronic pain is pain without biological value that has persisted beyond the normal tissue healing time (usually considered to be 3 months), according to the International Association of the Study of Pain (IASP) definition [15]. Usually chronic pain is due to the physical damage of specific neural cells [15]. Emerging evidence has demonstrated that TLRs would not only respond to the PAMPs of pathogens, but also could be activated by DAMPs [16]. DAMPs include molecules released by damaged cells during apoptosis, and other endogenous ligands which are involved in cell apoptosis, such as heat shock proteins, extracellular matrix degradation products, protein HMGB-1 etc (Table 1) [16–17]. These findings connect the neural pain to the activation of TLRs.
Expressions of TLRs are found in diverse components of the nervous system such as microglia, astrocytes, oligodendrocytes, Schwann cells and neurons (Table 2) [16, 18]. Unlike TLRs of the immune system, these specific TLRs, especially TLR4, may release different kinds of signaling molecules after activation (Figure 1) [19]. Acting as macrophages in the nervous system, microglia may help to eradicate the damaged neural cells and invasive pathogen in the central nervous system (CNS) [16, 20]. On the other hand, release of signaling molecules at the end of the TLR signaling pathways would promote the inflammatory reaction inside the nervous system, which could result in neuralgia [21]. Although various types of TLRs mRNA are found in microglia, only a part of mRNA is eventually translated into protein, mainly those of TLR4 [16, 22].
Table 2.
The expression of TLR mRNA and TLRs in different human neural cells
2.3. The opioid effect on TLR4
Opioids, such as morphine, are widely used to treat pain. But long-term usage of opioids may lead to opioid tolerance or dependency [23]. The analgesic effect of opioids is thought to be transmitted through the opioid receptors (such as μ, κ, and δ receptor) in a stereoselective manner. However there is another non-stereoselective opioid activation that exists, which is not well understood (Figure 3) [24]. Recent studies suggest that non-stereoselective activation of opioids is related to the activation of glial cells and TLR4 [23].
Figure 3.
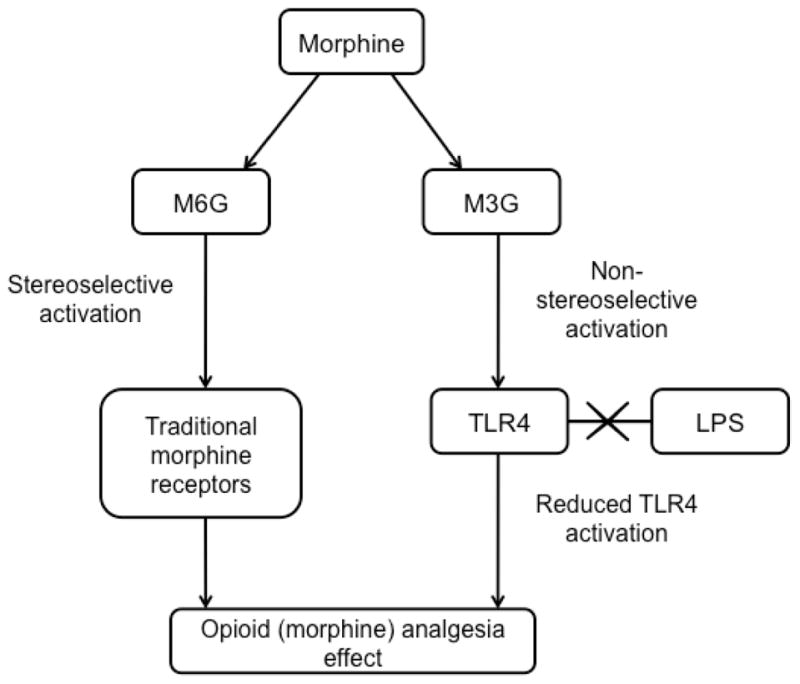
Morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G) are both morphine metabolites. In the stereoselective morphine activation, specific receptors of morphine bind with M6G. However, non-stereoselective morphine activation requires the participation of M3G, for TLR4 on glia cells. M3G competes with LPS for the same binding site on TLR4, which in turn results in the potentiation of the opioid analgesic effect.
TLR4 activation could decrease opioid efficacy, and is linked to the development and maintenance of neuropathic pain [21b]. Both bio-based and in silico-based experiments have shown that opioids could activate TLR4 on glial cells [25]. Their activation could result in the synthesis of nociceptive cytokines, which might exacerbate neuropathic pain and counteract the analgesic effects of opioids [26]. Different from the classical opioid receptors, TLR4 is activated by the morphine metabolite morphine-3-glucuronide (M3G) instead of morphine-6-glucuronide (M6G) [24]. The most recent studies from the Watkins group show that both estradiol-3-glucuronide (E2-3-G) and estradiol-17-glucuronide (E2-17-G) can enhance neuropathic pain [27]. In silico docking was able to predict that E2-3-G and E2-17-G could bind to MD2 in the TLR4-MD2 complex. A cell based, secreted embryonic alkaline phosphatase (SEAP) reporter assay was employed to showcase that this binding resulted in agonistic activity. Finally, an in vivo experiment using Sprague-Dawley rats in a von Frey test showed that both E2-3-G and E2-17-G caused enhanced pain, and that the potent antagonist, LPS-RS (the LPS found in the bacteria Rhodobacter sphaeroides), was able to reverse the effects.
Further studies with the MD2 X-ray crystal structure suggested that the activation effect of opioids on TLR4 might be related to the LPS-mimic structure of opioids and the LPS binding junction part of MD2 [28]. Opioids can mimic LPS and bind to MD2 as an LPS substitute to activate TLR4 directly. Therefore opioids may activate TLR4 without participation of LPS [29]. However Yin and co-workers have taken morphine to study the neuroinflammation activated by opioids. In this previous work, the binding experiment of morphine and MD2, morphine demonstrated a strong binding ability to MD2, and its binding interaction was observed to compete with LPS-MD2 binding [18g]. Further experimental results suggest that not only can morphine bind to MD2 as LPS does, but the morphine-MD2 complex can also induce TLR4 oligomerization, just as the LPS-MD2 complex does [28a, 29]. TLR4 oligomerization induced by the morphine-MD2 complex would lead to the formation of the TLR4-MD2-morphine complex, which would trigger the downstream pro-inflammatory cascade reactions [28a, 29]. These findings lead to the assumptions that TLR4 activation may aggravate pain, TLR4 may play an important role in opioid-induced hyperalgesia, and TLR4 may function against the acute or chronic analgesic effect of opioids [24]. The total of these works suggests that opioids can potentially activate TLR4 signaling, in the presence of MD2 or simply on their own, by binding MD2 and inducing TLR4 dimerization.
The most recent findings, pertaining to opioid analgesia, show that a series of novel (+)-Naltrexone-inspired analogs were able to antagonize TLR4 signaling. The lead opioid in this study was also tested in vivo in a Hargreaves assay, and was able to demonstrate efficacy as compared to a morphine/vehicle[30].
3. Agonists and antagonists of TLR4 with pharmaceutical potential
3.1. Designing TLR4 agonists and antagonists
Both antagonists and agonists of TLR4 were demonstrated to hold high pharmaceutical value, with indications in chronic neuralgia or opioid hyperalgesia with TLR4 activation (Table 3). Designing and screening of potential antagonist or agonist molecules benefits from recent studies on structural biology [10c, 24, 31]. The mechanism of TLR4 binding to its accessory proteins has been revealed via the crystal structure of the (TLR4-MD2-LPS)2 dimer complex and the crystallographic data of MD2 binding to TLR4 antagonists (such as Eritoran) [28b, 32]. The most popular design strategy of antagonists utilizes the native ligand as inspiration for the antagonist molecule, which encourages competitive binding [10c, 33].
Table 3.
TLR4 antagonists and agonists
| Mechanism | Antagonist/agonist | References |
|---|---|---|
| Block binding between MD2 and TLR4 | MD2 I | [12b] |
| YH1/YH3 | [36] | |
| β-amino alcohol derivatives | [37a] | |
| Block binding between MD2 and LPS | Tricyclic anti-depressants | [37b] |
| LPS sequestrants | Polymixin B | [18g, 40, 47] |
| Block binding of CD-14 | Haemin and three sugar-derived small molecules |
[54] [55] |
3.2. Formation of the LPS-MD2-TLR4 and Eritoran-MD2-TLR4 complexes
The natural TLR4 agonist, (LPS) and structurally related antagonist, (Eritoran), both contain an N-acetylglucosamine disaccharide scaffold with two phosphates in positions 1 and 4. One way in which these two molecules differ is that LPS contains six large lipid chains, where as Eritoran contains only four [10c]. The head portion of LPS, known as Lipid A, is able to fill a hydrophobic cavity formed on the MD2 protein with its six lipid chains. When all six lipid chains occupy the MD2 cavity, the disaccharide scaffold is situated just above the cavity, such that the two phosphate groups are able to interact with positively charged residues on both TLR4 molecules to induce dimerization [28a].
Further crystallographic studies have demonstrated that the size of the MD2-binding cavity is relatively unchanged when bound to Eritoran as compared to LPS. Since Eritoran has two fewer lipid chains, the MD2 binding cavity can easily accommodate the smaller ligand [28a]. Because of the smaller volume requirements of Eritoran, the ligand is able to settle further into the MD2 binding cavity as compared to LPS. As a result, the glucosamine backbone of Eritoran is lowered closer to the rim of the cavity by approximately 5Å. This backbone shift allows the two phosphate groups to interact with positively charged residues on MD2, rather than with either of the TLR4 molecules, thus inhibiting TLR4 signaling [28a, 32].
3.3. Targeting MD2-TLR4 binding
3.3.1 Design of MD2-I
Peptide MD2-I was reported in 2009 as an antagonist of TLR4 by reproducing the TLR4-binding region of MD2 [12b]. By rationalizing a 17-residue peptide (CRGSDDDYSFCRALKGE) with a disulfide bridge between Cys95 and Cys105, MD2-I can mimic the critical TLR4-binding region of MD2, which is confirmed by circular dichroism (CD) experiments [34]. In vivo experiments using HEK293 cells suggest that MD2-I behaves similarly to the Cys95Tyr MD2 mutant, which proved the hypothesis that MD2-I might block TLR4 downstream proinflammatory effectors by blocking the TLR4-MD2 interaction [12b] (Figure 4).
Figure 4.
Design of MD2-based TLR4 antagonists utilizes the structural similarity of agonists and MD2, which results in competition between antagonists and MD2 for TLR4 binding. For TLR4 binding with antagonists, such as MD2-I, the formation of the MD2-TLR4 complex may be blocked.
3.3.2. Designs based on the MD2-I structure
Recent studies by Yin’s group on MD2-I have proved that the key structure of MD2 in MD2-TLR4 binding is a short peptide segment with only 10 residues. With Rosetta software, contribution of this 10-residue segment in binding energy of TLR4 and MD2 interaction can be calculated accurately as 53% [35]. It is reasonable to predict that a more ridged version of this sequence could be a template of TLR4 agonists. This was achieved by the synthesis of a disulfide-bridged macrocycle By substituting an alanine (Ala) for a Cysteine (Cys) in YH1 and YH3, which prohibits disulfide bond formation, two linear-structured peptide YH2 and YH4 were synthesized as reference samples (Table 4). In vivo experiments demonstrated that YH1 and YH3 with disulfide-bridged structure show much stronger ability to synergistically activate TLR4 signaling in the presence of LPS than their linear counterparts YH2 and YH4 [36]. Detection of NO production in RAW264.7 cells also suggests that TLR4 activity may be related to the size of macrocycle formed by disulfide bond. The activity of TLR4 stimulated by YH3 is higher than YH1, which may suggest that larger rings might be less effective in binding TLR4 [36].
Table 4.
Peptide sequences and chemical formulas of synthesized TLR4 agonists, antagonists, and analogs
| Peptides | Amino acid sequences |
|---|---|
| MD2-I |
|
| YH1 |
|
| YH2 |

|
| YH3 |
|
| YH4 |
|
Bold and italic styles are used on residues that are supposed to form disulfide bonds.
Underlined font indicates the mutated residues.
3.3.3. β-Amino alcohol derivatives
Besides MD2-based antagonists, other antagonists have been designed to block MD2-TLR4 binding. β-Amino alcohol derivatives have proved to be effective suppressors of TLR4 activation, and further experiments on clinical uses are ongoing [37]. Yin and co-researchers have discovered an innovative, robust method to synthesize β-amino alcohol derivatives, such as T5342126, 8, which is a TLR4 antagonist (Table 5) [37a]. Besides T5342126, Yin’s group also used this new synthetic method to prepare a focused library of analogs, and test their potential inhibitive effects on TLR4 activation in HEK293 cells by SEAP assay [12b, 38]. In this library, compound 10 was confirmed to be the most efficient TLR4 inhibitor. While compound 9 having little inhibition suggests that the functional groups on the aromatic rings (R1 and R2 in Table 5) might have great influence on the activity of these TLR4 antagonists [38]. We can only speculate that the electronic nature of these two rings play an important role in the binding of these molecules to TLR4, and must be fine-tuned to facilitate inhibition. Further results have demonstrated the ability of TLR inhibitors to enhance morphine’s analgesic effect by blocking TLR4 activation [39]. Hargreaves test is a common method to test the intensity of a compound’s analgesic effect, by measuring the time taken to observe radiant heat-induced withdrawal response by hindpaws and tails of unrestrained rats after compound injection [40]. Both compound 8 and compound 10 have proven to be able to enhance morphine’s analgesic effects, while no analgesic effect was detected when these compounds were injected alone [37a, 38].
Table 5.
Results from the SEAP Reporter Gene Activation Assay*
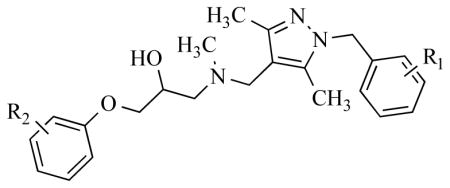
The percent of inhibition was determined by measuring LPS-induced TLR4 activation in HEK293 cells, in the presence of 50 μM drug (compound) as compared to blank control.
Et = Ethyl group.
R1 & R2 = functional groups on the aromatic rings of T5342126-based inhibitors.
Additional efforts based on 8 have recently been published [41]. An in silico screening was performed, favoring molecules with high similarity to 8. These virtual screening results provided 45 potential hit molecules to be further evaluated. The field was further narrowed to fourteen compounds, by elimination of toxic and insoluble compounds. Three of the fourteen compounds (Figure 5) showed favorable IC50 values (ranging from 16.6 μM to 167.5 μM) in a SEAP assay utilizing HEK293 cells. Surface plasmon resonance (SPR) was used to further demonstrate binding of their lead compound to immobilized recombinant human TLR4.
Figure 5.

A T5342126 (8,) inspired similarity search identified three new potential TLR4 signaling inhibitors in silico. The lead compounds (shown in box, with an IC50 of 16.6 μM) ability to bind TLR4 was confirmed in an SPR experiment with chip immobilized TLR4.
3.3.4. Tanshinone IIA
Another small molecule that has been shown, in a recent study, to effectively modulate TLR4 activity is Tanshinone IIA (Tan IIA)[42]. Spinal nerve ligation (SNL), a popular model used to generate neuropathic pain via the ligation of the L5 and L6 spinal nerves [43], was performed on male Sprague-Dawley (SD) SPF rats. The rats were divided into three random groups post-operation: sham group, vehicle treated group, and Tan IIA treated group. Paw mechanical withdraw threshold (PWT) was used to measure mechanical allodynia, and paw thermal withdrawal latency (PWL) was used to measure thermal hyperalgesia. The Tan IIA treated rats displayed a higher threshold for pain in both the PWT and PWL tests as compared to the sham and vehicle groups. Furthermore, HMGB1 and TLR4 genes and protein were downregulated in the spinal cords of the Tan IIA treated group. Finally, the proinflammatory cytokines IL-1β, IL-10, and TNFα were also downregulated in the Tan IIA treated group. This study serves as an example that small molecules can effectively treat pain in mammals.
3.3.5. TAK-242
Perhaps the most advanced small molecule modulator of TLR4 is TAK-242 [44] (Figure 6). TAK-242 has been show to covalently bind to Cys747 of the TIR domain of TLR4, rendering TLR4 completely inactive. However, TAK-242 failed in phase 3 clinical trials in the treatment of sepsis because this inhibition failed to effectively reduce the levels of pro-inflammatory cytokines in humans with severe sepsis. The reason for this is unknown. One theory is that alternative pathways may be able to mediate the inflammation response without TLR4. This is supported by a recent study showing that caspase-11 and caspase-4 are capable of binding intracellular LPS, thus killing the cell [45]. These results, however, should not discourage researchers pursuing TLR4 inhibitors for neuropathic pain. A recent study demonstrated that naturally TLR4 deficient C3H/HeJ mice are less sensitive to stress-induced visceral pain than wild-type C3H/HeN mice [46]. The researchers further supported this finding by administering TAK-242 to mice with functional TLR4, and observing that the mice had acquired a higher threshold for visceral pain. Futhermore, TAK-242 was able to counteract chronic stress-induced visceral hypersensitivity. These results were not reproduced with the TLR4 deficient C3H/HeJ mice. As expected, the levels of pro-inflammatory cytokines IL-6 and TNFα were reduced after TAK-242 treatment. A recent study, however, cautions that TLR4 inhibitors, such as TAK-242, can impair the endogenous opioid-mediated analgesia [47]. The researchers demonstrate this effect in Wistar rats with complete Freund’s adjuvant (CFA)-induced paw inflammation. TAK-242 was shown to block the release of endogenous endorphins, which are normally released to provide natural analgesia.
Figure 6.

Various small molecules which modulate TLR4 activity.
3.4 LPS sequestrants
LPS sequestrants can block TLR4 activation by sequestering the LPS molecule [44]. Structural analysis of the LPS-binding domains of various proteins which have strong affinity for LPS (such as bactericidal/permeability-increasing protein (BPI), and Limulus anti-LPS factor (LALF), and LBP), reveals an LPS-binding motif which is functionally independent of LPS transport or neutralization [10c, 27, 48]. With the help of this structural motif, several peptides were discovered which could specifically bind to LPS on Gram-negative bacteria. These peptides consist of a long β-strand, which has alternating bulky and basic hydrophobic amino acids. Binding of these peptides to LPS suppresses TLR4 activation and TLR4 downstream cytokine production in vitro and in animal models [49]. Polymixin B (PMB) was an LPS sequestrant with this motif structure, whose therapeutic potential has been proved, and PMB has already been approved for clinical use in Japan [10c, 47, 50].
3.5. Blocking LPS-LBP or LPS-CD14 binding
Interfering with LPS-LBP or LPS-CD14 dimer formation could also block TLR4 activation. Structural analysis of the CD14 monomer shows a horseshoe-shaped structure containing a concave surface formed by a large β-sheet, which consists of leucine-rich repeats (LRR). Those LRRs result in the formation of several grooves and pockets, which are essential for LPS binding [51]. Based on this structure, a glycoconjugate obtained from Treponema spirochetes (Tm-Gp) has been demonstrated to be a potential TLR4 antagonist, by blocking LPS binding to CD14 and LBP (Figure 7) [52]. New artificially synthesized molecules, compounds 1-4 (Figure 6), were reported in 2009 as possible antagonists for TLR4 [53]. The inhibition of LPS- and lipid A-promoted cytokine production in macrophages and dendritic cells was observed during relevant experiments [10c, 53].
Figure 7.

A glycoconjugate obtained from Treponema spirochetes (Tm-Gp) has been demonstrated to be a potential TLR4 antagonist by blocking LPS binding to CD14 and LBP. Tm-Gp was designed based on the CD14 monomer structure, which has a horseshoe-shaped structure containing concave surface formed by a large β-sheet.
3.6. Tricyclic anti-depressants work on TLR4 with an opioid-like mechanism
Opioids compete with LPS for binding to MD2 [24]. Many morphine-like compounds are speculated to be potential TLR4 antagonists or agonists by the same mechanism. Hutchinson and co-researchers’ studies have demonstrated the potential of tricyclic anti-depressants to be TLR4 antagonists [37b]. Tricyclic compounds have proved to be effective anti-depressant drugs for the nervous system, and they may share the same mechanism with opioids for activating TLR4 [45–46]. Eight tricyclic compounds were tested as potential antagonists or agonists of TLR4. Compounds were added into HEK293 cell cultures, either alone or with classical TLR4 agonists, to test their influence on TLR4 activation. Six compounds presented varying degrees of inhibition on TLR4 activation, including amitriptyline, imipramine, mianserin, cyclobenzaprine, ketotifen and desipramine (Figure 6). Other two compounds, carbamazepineand and oxcarbazepine, presented a mild agonistic effect on TLR4 [37b]. Those two compounds induced more distinct TLR4 activity without LPS addition. Hutchinson’s group speculated that tricyclic compounds share a similar mechanism with opioids on activating TLR4. They used AutoDock4, the latest version of the popular docking software, to confirm the binding of compounds docked to MD2 in silico [37b]. Most of the compounds were docked with a greater preference to the LPS binding cleft of MD2, instead of the TLR4 binding cleft. These docking results support the idea that tricyclic anti-depressants are competitive with LPS in their binding to MD2, just as the opioids are [37b]. Together with experimental in vivo results, Hutchinson’s group built a new prediction model for both TLR4 activators and inhibitors. The prediction model is mainly based on experimental results of four tricyclic ligands (oxcarbazepine, carbamazepine, amitriptyline and mianserin). Agonism and antagonism of TLR4 by other potential small molecules are predicted under this new model in order to confirm their applicability. Compared to experimental data obtained in vivo, predicted results are very close to actual results, with most errors being less than 1% [37b]. Further experimental results in vivo, such as intracellular TLR4 signaling in RAW264.7 cells and TLR-induced interleukin-1 release in murine microglial BV-2 cells, also support those conclusions. [37b] (Figure 8)
Figure 8.

Different from MD2-I based antagonists, opioid and opioid-like TLR4 antagonists compete with LPS for MD2 binding which reduces TLR4 activity.
3.7. Blocking CD14 binding
Peri’s group mainly focuses on the accessory protein CD14. Haemin (Figure 6) is a common compound that can activate macrophages [53b]. Since haemin activation requires participation of CD14 and TLR4, it is speculated that haemin is a CD14 dependent TLR4 agonist [54]. Besides haemin, Peri’s group also synthesized several sugar-derived small molecules, composed of a glucose unit linked to two C14 chains, and either nitrogen substituent or ether linkage on C-6, as potential CD14-dependent antagonists. While those three compounds showed obvious inhibition of TLR4 in both HEK293 cells and murine cells, another similar sugar-derived compound with a positive charge on C-6 has little inhibition [55]. Thus, the electronic properties of the C-6 nitrogen atom may be key to blocking CD14 binding [53b].
4. Prospective
The mechanistic differences between TLR4 agonists and TLR4 antagonists still remain unsolved. Before TLR4 modulating drugs can be introduced for neuroinflammation, further preclinical and clinical research must be developed. Another focus of TLR4-based drug development is decreasing the molecular weight of new drugs, especially analogs of LPS, which could improve drug absorption, and more effectively inhibit or stimulate TLR4 activity in the human body.
As highlighted in this review, the literature in the field of Toll-like receptors and neuropathic pain is continually growing. Researchers, armed with a growing list of TLR4 and MD2 based crystal structures, are finding more success with in silico docking simulations, which aids them in launching new projects. Additionally, more projects are yielding pre-clinical candidates with promising results from in vivo animal studies. With so much effort and early success in this ever-growing field, there should be applications for TLR4 modulators to treat pain in neurologic or analgesic clinics in near future.
Acknowledgments
We thank the National Institutes of Health (R01GM101279) to H. Y. and the National Natural Science Foundation of China to J. L. (81401333) for financial supports of this work.
Abbreviations
- TLRs
Toll-like receptors
- DAMPs
danger associated molecular patterns
- PAMPs
pathogen associated molecular patterns
- MyD88
myeloid differentiation primary-response gene 88
- TRIF
Toll/IL-1R (TIR)-domain-containing adaptor protein
- TIRAP
TIR domain containing adaptor protein
- MAL
MyD88 adaptor-like protein
- TRAM
TRIF-related adaptor molecule
- TRAF
tumor-necrosis-factor-receptor-associated factor
- TBK1
TRAF-family-member-associated NF-κB activator (TANK)-binding kinase 1
- RIP1
receptor-interacting protein 1
- IRAK
IL-1R-associated kinase
- TAK1
transforming-growth-factor-β-activated kinase
- TAB
TAK1-binding protein
- NF-κB
nuclear factor-κB
- IKK
inhibitor of NF-κB kinase
- IκB
inhibitor of NF-κB
- TNF
tumor-necrosis factor
- IRF
interferon regulatory factor
- IFN
interferon
- MKK
mitogen-activated protein kinase kinase
- LPS
lipopolysaccharide
- LBP
lipopolysaccharide binding protein
- MD-2
myeloid differentiation protein 2
- CNS
central nervous system
- NO
nitric oxide
- PRRs
pattern recognition receptors
- CD14
cluster differentiation antigen 14
- M3G
morphine-3-glucuronide
- M6G
morphine-6-glucuronide
- LRR
leucine-rich repeats
- Tm-Gp
glycoconjugate preparation from Treponema spirochetes
- Et
Ethyl group
- BPI
bactericidal/permeability-increasing protein
- LALF
Limulus anti-LPS factor
- MAPK
mitogen-activated protein kinase
- RelA
p65 subunit of NF-κB
- PI3K
phosphoinositide-3-kinase
- Akt (PKB)
protein kinase B
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
Author Contributions
All authors made substantial contributions to the conception and design of this study. All authors read and approved the final manuscript.
References
- 1.a) Matusik P, Guzik B, Weber C, Guzik TJ. Thrombosis and haemostasis. 2012;108:443–456. doi: 10.1160/TH12-05-0341. [DOI] [PubMed] [Google Scholar]; b) McCusker RH, Kelley KW. The Journal of experimental biology. 2013;216:84–98. doi: 10.1242/jeb.073411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khan KN, Kitajima M, Hiraki K, Fujishita A, Sekine I, Ishimaru T, Masuzaki H. Gynecol Obstet Invest. 2009;68:40–52. doi: 10.1159/000212061. [DOI] [PubMed] [Google Scholar]
- 3.Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Annual review of immunology. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 4.Jia ZJ, Wu FX, Huang QH, Liu JM. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2012;34:168–173. doi: 10.3881/j.issn.1000-503X.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Kielian T, Haney A, Mayes PM, Garg S, Esen N. Infection and immunity. 2005;73:7428–7435. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scanzello CR, Goldring SR. Bone. 2012;51:249–257. doi: 10.1016/j.bone.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Akira S, Takeda K. Nature reviews Immunology. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]; b) Akira S, Uematsu S, Takeuchi O. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]; c) Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H. The Journal of biological chemistry. 2001;276:31332–31339. doi: 10.1074/jbc.M103217200. [DOI] [PubMed] [Google Scholar]; d) Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. The Journal of biological chemistry. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]; e) Li J, Wang X, Zhang F, Yin H. Pharmacology & therapeutics. 2013;138:441–451. doi: 10.1016/j.pharmthera.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Beck C, Morbach H, Richl P, Stenzel M, Girschick HJ. Rheumatology international. 2009;29:229–238. doi: 10.1007/s00296-008-0710-9. [DOI] [PubMed] [Google Scholar]; b) Botturi K, Langelot M, Lair D, Pipet A, Pain M, Chesne J, Hassoun D, Lacoeuille Y, Cavailles A, Magnan A. Pharmacology & therapeutics. 2011;131:114–129. doi: 10.1016/j.pharmthera.2011.03.010. [DOI] [PubMed] [Google Scholar]; c) Khan KN, Kitajima M, Fujishita A, Nakashima M, Masuzaki H. The journal of obstetrics and gynaecology research. 2013;39:1281–1292. doi: 10.1111/jog.12117. [DOI] [PubMed] [Google Scholar]
- 9.a) Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Pharmacological reviews. 2011;63:772–810. doi: 10.1124/pr.110.004135. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Iwasaki A, Medzhitov R. Nature immunology. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 10.a) Dziarski R, Tapping RI, Tobias PS. The Journal of biological chemistry. 1998;273:8680–8690. doi: 10.1074/jbc.273.15.8680. [DOI] [PubMed] [Google Scholar]; b) Weiss J. Biochemical Society transactions. 2003;31:785–790. doi: 10.1042/bst0310785. [DOI] [PubMed] [Google Scholar]; c) Peri F, Piazza M. Biotechnology advances. 2012;30:251–260. doi: 10.1016/j.biotechadv.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 11.a) Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, Tobias PS, Ulevitch RJ. Science. 1990;249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]; b) Gioannini TL, Zhang D, Teghanemt A, Weiss JP. The Journal of biological chemistry. 2002;277:47818–47825. doi: 10.1074/jbc.M206404200. [DOI] [PubMed] [Google Scholar]; c) Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. The Journal of experimental medicine. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR, Yin H. Proceedings of the National Academy of Sciences. 2012;109:6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Slivka PF, Shridhar M, Lee GI, Sammond DW, Hutchinson MR, Martinko AJ, Buchanan MM, Sholar PW, Kearney JJ, Harrison JA, Watkins LR, Yin H. Chembiochem: a European journal of chemical biology. 2009;10:645–649. doi: 10.1002/cbic.200800769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Liu T, Gao YJ, Ji RR. Neuroscience bulletin. 2012;28:131–144. doi: 10.1007/s12264-012-1219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hayashi F, Means TK, Luster AD. Blood. 2003;102:2660–2669. doi: 10.1182/blood-2003-04-1078. [DOI] [PubMed] [Google Scholar]
- 14.a) Buchanan MM, Hutchinson M, Watkins LR, Yin H. Journal of neurochemistry. 2010;114:13–27. doi: 10.1111/j.1471-4159.2010.06736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, Vogel SN. Journal of leukocyte biology. 2009;85:966–977. doi: 10.1189/jlb.1208763. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu S, Kielian T. Journal of immunology. 2009;183:5537–5547. doi: 10.4049/jimmunol.0900083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeLeo JA, Tanga FY, Tawfik VL. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- 16.Lee H, Lee S, Cho IH, Lee SJ. Curr Protein Pept Sci. 2013;14:33–42. doi: 10.2174/1389203711314010006. [DOI] [PubMed] [Google Scholar]
- 17.Miyake K. Seminars in immunology. 2007;19:3–10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 18.a) Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. Journal of immunology. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]; b) Olson JK, Miller SD. Journal of immunology. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]; c) Cassiani-Ingoni R, Cabral ES, Lunemann JD, Garza Z, Magnus T, Gelderblom H, Munson PJ, Marques A, Martin R. Journal of neuropathology and experimental neurology. 2006;65:540–548. doi: 10.1097/00005072-200606000-00002. [DOI] [PubMed] [Google Scholar]; d) Kielian T, Esen N, Bearden ED. Glia. 2005;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Oliveira RB, Ochoa MT, Sieling PA, Rea TH, Rambukkana A, Sarno EN, Modlin RL. Infection and immunity. 2003;71:1427–1433. doi: 10.1128/IAI.71.3.1427-1433.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zhang F, Liu F, Yan M, Ji H, Hu L, Li X, Qian J, He X, Zhang L, Shen A, Cheng C. Journal of neuroimmunology. 2010;218:36–47. doi: 10.1016/j.jneuroim.2009.10.016. [DOI] [PubMed] [Google Scholar]; g) Lee H, Park C, Cho IH, Kim HY, Jo EK, Lee S, Kho HS, Choi SY, Oh SB, Park K, Kim JS, Lee SJ. Glia. 2007;55:712–722. doi: 10.1002/glia.20493. [DOI] [PubMed] [Google Scholar]
- 19.a) Beutler B. Curr Top Microbiol Immunol. 2002;270:109–120. doi: 10.1007/978-3-642-59430-4_7. [DOI] [PubMed] [Google Scholar]; b) Beutler B, Du X, Poltorak A. J Endotoxin Res. 2001;7:277–280. [PubMed] [Google Scholar]
- 20.a) Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Annals of neurology. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]; b) Lee SJ, Lee S. Curr Drug Targets Inflamm Allergy. 2002;1:181–191. doi: 10.2174/1568010023344698. [DOI] [PubMed] [Google Scholar]
- 21.a) Tanga FY, Nutile-McMenemy N, DeLeo JA. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. ScientificWorldJournal. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinsner A, Boveri M, Hareng L, Brown GC, Coecke S, Hartung T, Bal-Price A. Journal of neurochemistry. 2006;99:596–607. doi: 10.1111/j.1471-4159.2006.04085.x. [DOI] [PubMed] [Google Scholar]
- 23.Hameed H, Hameed M, Christo PJ. Current pain and headache reports. 2010;14:96–104. doi: 10.1007/s11916-010-0093-y. [DOI] [PubMed] [Google Scholar]
- 24.Watkins LR, Hutchinson MR, Rice KC, Maier SF. Trends in pharmacological sciences. 2009;30:581–591. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joce C, Stahl JA, Shridhar M, Hutchinson MR, Watkins LR, Fedichev PO, Yin H. Bioorganic & medicinal chemistry letters. 2010;20:5411–5413. doi: 10.1016/j.bmcl.2010.07.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milligan ED, Watkins LR. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis SS, Hutchinson MR, Frick MM, Zhang Y, Maier SF, Sammakia T, Rice KC, Watkins LR. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.a) Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]; b) Ohto U, Fukase K, Miyake K, Satow Y. Science. 2007;316:1632–1634. doi: 10.1126/science.1139111. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR, Yin H. Proc Natl Acad Sci U S A. 2012;109:6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selfridge BR, Wang X, Zhang Y, Yin H, Grace PM, Watkins LR, Jacobson AE, Rice KC. Journal of medicinal chemistry. 2015 doi: 10.1021/acs.jmedchem.5b00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jerala R. International journal of medical microbiology: IJMM. 2007;297:353–363. doi: 10.1016/j.ijmm.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Cheng K, Wang X, Yin H. Journal of the American Chemical Society. 2011;133:3764–3767. doi: 10.1021/ja111312h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L, Ghosh N, Slivka PF, Fiorini Z, Hutchinson MR, Watkins LR, Yin H. Chembiochem: a European journal of chemical biology. 2011;12:1827–1831. doi: 10.1002/cbic.201100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Kuhlman B, Baker D. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Khatib F, Cooper S, Tyka MD, Xu K, Makedon I, Popovic Z, Baker D, Players F. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18949–18953. doi: 10.1073/pnas.1115898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao M, London N, Cheng K, Tamura R, Jin JL, Schueler-Furman O, Yin H. Tetrahedron. 2014;70:7664–7668. doi: 10.1016/j.tet.2014.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.a) Chavez SA, Martinko AJ, Lau C, Pham MN, Cheng K, Bevan DE, Mollnes TE, Yin H. Journal of medicinal chemistry. 2011;54:4659–4669. doi: 10.1021/jm2003365. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hutchinson MR, Loram LC, Zhang Y, Shridhar M, Rezvani N, Berkelhammer D, Phipps S, Foster PS, Landgraf K, Falke JJ, Rice KC, Maier SF, Yin H, Watkins LR. Neuroscience. 2010;168:551–563. doi: 10.1016/j.neuroscience.2010.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bevan DE, Martinko AJ, Loram LC, Stahl JA, Taylor FR, Joshee S, Watkins LR, Yin H. ACS medicinal chemistry letters. 2010;1:194–198. doi: 10.1021/ml100041f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu T, Li G, Mu R, Ye H, Li W, Li Z. Lupus. 2014;23:958–963. doi: 10.1177/0961203314526439. [DOI] [PubMed] [Google Scholar]
- 40.Sloane E, Langer S, Jekich B, Mahoney J, Hughes T, Frank M, Seibert W, Huberty G, Coats B, Harrison J, Klinman D, Poole S, Maier S, Johnson K, Chavez R, Watkins LR, Leinwand L, Milligan E. Gene therapy. 2009;16:1210–1222. doi: 10.1038/gt.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Svajger U, Brus B, Turk S, Sova M, Hodnik V, Anderluh G, Gobec S. European journal of medicinal chemistry. 2013;70:393–399. doi: 10.1016/j.ejmech.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 42.Ma YQ, Chen YR, Leng YF, Wu ZW. Evid Based Complement Alternat Med. 2014;2014:639563. doi: 10.1155/2014/639563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SH, Chung JM. Pain. 1992;50:355–363. [Google Scholar]
- 44.Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J. Crit Care Med. 2010;38:1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 45.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 46.Tramullas M, Finger BC, Moloney RD, Golubeva AV, Moloney G, Dinan TG, Cryan JF. Biol Psychiatry. 2014;76:340–348. doi: 10.1016/j.biopsych.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 47.Sauer RS, Hackel D, Morschel L, Sahlbach H, Wang Y, Mousa SA, Roewer N, Brack A, Rittner HL. Mol Pain. 2014;10:10. doi: 10.1186/1744-8069-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.a) Schumann RR, Lamping N, Hoess A. Journal of immunology. 1997;159:5599–5605. [PubMed] [Google Scholar]; b) Lamping N, Dettmer R, Schroder NW, Pfeil D, Hallatschek W, Burger R, Schumann RR. The Journal of clinical investigation. 1998;101:2065–2071. doi: 10.1172/JCI2338. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Weersink AJ, van Kessel KP, van den Tol ME, van Strijp JA, Torensma R, Verhoef J, Elsbach P, Weiss J. Journal of immunology. 1993;150:253–263. [PubMed] [Google Scholar]
- 49.a) Muhle SA, Tam JP. Biochemistry. 2001;40:5777–5785. doi: 10.1021/bi0100384. [DOI] [PubMed] [Google Scholar]; b) Li J, Wu H, Huang X, Xu D, Zheng W, Zhao Y, Liu W, Zeng X. Medicine (Baltimore) 2014;93:e49. doi: 10.1097/MD.0000000000000049. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Larrick JW, Hirata M, Balint RF, Lee J, Zhong J, Wright SC. Infection and immunity. 1995;63:1291–1297. doi: 10.1128/iai.63.4.1291-1297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Levy O, Ooi CE, Elsbach P, Doerfler ME, Lehrer RI, Weiss J. Journal of immunology. 1995;154:5403–5410. [PubMed] [Google Scholar]; e) Scott MG, Rosenberger CM, Gold MR, Finlay BB, Hancock RE. Journal of immunology. 2000;165:3358–3365. doi: 10.4049/jimmunol.165.6.3358. [DOI] [PubMed] [Google Scholar]; f) Scott MG, Vreugdenhil AC, Buurman WA, Hancock RE, Gold MR. Journal of immunology. 2000;164:549–553. doi: 10.4049/jimmunol.164.2.549. [DOI] [PubMed] [Google Scholar]
- 50.a) Morrison DC, Jacobs DM. Immunochemistry. 1976;13:813–818. doi: 10.1016/0019-2791(76)90181-6. [DOI] [PubMed] [Google Scholar]; b) Vincent JL, Laterre PF, Cohen J, Burchardi H, Bruining H, Lerma FA, Wittebole X, De Backer D, Brett S, Marzo D, Nakamura H, John S. Shock. 2005;23:400–405. doi: 10.1097/01.shk.0000159930.87737.8a. [DOI] [PubMed] [Google Scholar]
- 51.Kim JI, Lee CJ, Jin MS, Lee CH, Paik SG, Lee H, Lee JO. The Journal of biological chemistry. 2005;280:11347–11351. doi: 10.1074/jbc.M414607200. [DOI] [PubMed] [Google Scholar]
- 52.Asai Y, Hashimoto M, Ogawa T. Eur J Immunol. 2003;33:3196–3204. doi: 10.1002/eji.200324219. [DOI] [PubMed] [Google Scholar]
- 53.a) Piazza M, Yu L, Teghanemt A, Gioannini T, Weiss J, Peri F. Biochemistry. 2009;48:12337–12344. doi: 10.1021/bi901601b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Peri F, Piazza M, Calabrese V, Damore G, Cighetti R. Biochemical Society transactions. 2010;38:1390–1395. doi: 10.1042/BST0381390. [DOI] [PubMed] [Google Scholar]
- 54.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT. The Journal of biological chemistry. 2007;282:20221–20229. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 55.a) Bettoni I, Comelli F, Rossini C, Granucci F, Giagnoni G, Peri F, Costa B. Glia. 2008;56:1312–1319. doi: 10.1002/glia.20699. [DOI] [PubMed] [Google Scholar]; b) Piazza M, Rossini C, Della Fiorentina S, Pozzi C, Comelli F, Bettoni I, Fusi P, Costa B, Peri F. Journal of medicinal chemistry. 2009;52:1209–1213. doi: 10.1021/jm801333m. [DOI] [PubMed] [Google Scholar]
- 56.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. Journal of immunology. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 57.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 58.a) Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]; b) Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. The Journal of biological chemistry. 1999;274:17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- 59.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. The Journal of biological chemistry. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 60.Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT, Fenton MJ. Journal of immunology. 1999;163:3920–3927. [PubMed] [Google Scholar]
- 61.Hajjar AM, O’Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, Wilson CB. Journal of immunology. 2001;166:15–19. doi: 10.4049/jimmunol.166.1.15. [DOI] [PubMed] [Google Scholar]
- 62.Coelho PS, Klein A, Talvani A, Coutinho SF, Takeuchi O, Akira S, Silva JS, Canizzaro H, Gazzinelli RT, Teixeira MM. Journal of leukocyte biology. 2002;71:837–844. [PubMed] [Google Scholar]
- 63.Opitz B, Schroder NW, Spreitzer I, Michelsen KS, Kirschning CJ, Hallatschek W, Zahringer U, Hartung T, Gobel UB, Schumann RR. The Journal of biological chemistry. 2001;276:22041–22047. doi: 10.1074/jbc.M010481200. [DOI] [PubMed] [Google Scholar]
- 64.Massari P, Henneke P, Ho Y, Latz E, Golenbock DT, Wetzler LM. Journal of immunology. 2002;168:1533–1537. doi: 10.4049/jimmunol.168.4.1533. [DOI] [PubMed] [Google Scholar]
- 65.Werts C, Tapping RI, Mathison JC, Chuang TH, Kravchenko V, Saint Girons I, Haake DA, Godowski PJ, Hayashi F, Ozinsky A, Underhill DM, Kirschning CJ, Wagner H, Aderem A, Tobias PS, Ulevitch RJ. Nature immunology. 2001;2:346–352. doi: 10.1038/86354. [DOI] [PubMed] [Google Scholar]
- 66.Hirschfeld M, Weis JJ, Toshchakov V, Salkowski CA, Cody MJ, Ward DC, Qureshi N, Michalek SM, Vogel SN. Infection and immunity. 2001;69:1477–1482. doi: 10.1128/IAI.69.3.1477-1482.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Underhill DM, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 68.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 69.Brentano F, Schorr O, Gay RE, Gay S, Kyburz D. Arthritis Rheum. 2005;52:2656–2665. doi: 10.1002/art.21273. [DOI] [PubMed] [Google Scholar]
- 70.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 71.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. The Journal of biological chemistry. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 72.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. Nature immunology. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 73.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., 3rd The Journal of biological chemistry. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 74.Rassa JC, Meyers JL, Zhang Y, Kudaravalli R, Ross SR. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2281–2286. doi: 10.1073/pnas.042355399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. The Journal of experimental medicine. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.a) Bulut Y, Faure E, Thomas L, Karahashi H, Michelsen KS, Equils O, Morrison SG, Morrison RP, Arditi M. Journal of immunology. 2002;168:1435–1440. doi: 10.4049/jimmunol.168.3.1435. [DOI] [PubMed] [Google Scholar]; b) Ohashi K, Burkart V, Flohe S, Kolb H. Journal of immunology. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 77.Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Journal of immunology. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 78.Smiley ST, King JA, Hancock WW. Journal of immunology. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 79.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 80.Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. Int Immunol. 2001;13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 81.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.a) Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]; b) Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 83.Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, Akira S, Kelly KM, Reeves WH, Bauer S, Krieg AM. The Journal of experimental medicine. 2005;202:1575–1585. doi: 10.1084/jem.20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 85.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 86.Yasuda K, Richez C, Uccellini MB, Richards RJ, Bonegio RG, Akira S, Monestier M, Corley RB, Viglianti GA, Marshak-Rothstein A, Rifkin IR. Journal of immunology. 2009;183:3109–3117. doi: 10.4049/jimmunol.0900399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.a) Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]; b) Kucera K, Koblansky AA, Saunders LP, Frederick KB, De La Cruz EM, Ghosh S, Modis Y. J Mol Biol. 2010;403:616–629. doi: 10.1016/j.jmb.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.a) Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, Bathke B, Lauterbach H, Suter M, Dreher S, Koedel U, Akira S, Kawai T, Buer J, Wagner H, Bauer S, Hochrein H, Kirschning CJ. Science. 2012;337:1111–1115. doi: 10.1126/science.1220363. [DOI] [PubMed] [Google Scholar]; b) Li XD, Chen ZJ. elife. 2012;1:e00102. doi: 10.7554/eLife.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bsibsi M, Ravid R, Gveric D, van Noort JM. Journal of neuropathology and experimental neurology. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 90.a) Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]; b) Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Glia. 2006;53:688–695. doi: 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- 91.a) Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yao SY, Soutto M, Sriram S. Journal of neuroimmunology. 2008;200:17–26. doi: 10.1016/j.jneuroim.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 92.a) Qi J, Buzas K, Fan H, Cohen JI, Wang K, Mont E, Klinman D, Oppenheim JJ, Howard OM. Journal of immunology. 2011;186:6417–6426. doi: 10.4049/jimmunol.1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Prehaud C, Megret F, Lafage M, Lafon M. Journal of virology. 2005;79:12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]