

Abstract
The cellular membrane constitutes the first element that encounters a wide variety of molecular species to which a cell might be exposed. Hosting a large number of structurally and functionally diverse proteins associated with this key metabolic compartment, the membrane not only directly controls the traffic of various molecules in and out of the cell, it also participates in such diverse and important processes as signal transduction and chemical processing of incoming molecular species. In this article, we present a number of cases where details of interaction of small molecular species such as drugs with the membrane, which are often experimentally inaccessible, have been studied using advanced molecular simulation techniques. We have selected systems in which partitioning of the small molecule with the membrane constitutes a key step for its final biological function, often binding to and interacting with a protein associated with the membrane. These examples demonstrate that membrane partitioning is not only important for the overall distribution of drugs and other small molecules into different compartments of the body, it may also play a key role in determining the efficiency and the mode of interaction of the drug with its target protein.
Keywords: Membrane, Membrane Proteins, Molecular Dynamics, Small Molecules
Graphical Abstract
Membrane Activity is a Critical Factor in the Biology of Small Molecules
Membranes are abundant in biology, acting as the effective barrier that compartmentalizes life into cells and organelles that can accumulate resources and reproduce [1, 2]. This compartmentalization of life allows for concentration gradients to exist between cellular structures, which can be established or exploited by membrane-embedded proteins, such as the creation and use of a proton gradient by the oxidative phosphorylation pathway in mitochondria [3]. Membrane-embedded or membrane-associated proteins also act as the intermediaries for cellular signaling [4], allowing biological systems to respond to changing environmental conditions over a range of timescales, for instance by triggering muscle contraction [5, 6] or altering gene expression [7, 8]. Thus while membrane proteins account for only ~ 30% of the human proteome [9, 10], they play an outsized role in controlling biological processes by regulating the transit of matter and information across the membrane. Indeed, dysregulation of membrane crossing events can have profound effects leading to disease [11, 12] or even death [13, 14].
Some molecules impact biological processes without crossing the bilayer, and instead partition into the membrane, penetrating below the lipid headgroups, and diffuse within a single bilayer leaflet. This partitioning is fundamentally a result of the bilayer structure (Fig. 1) and the detailed interactions of the small molecule with individual lipids. Biological membranes are composed of two leaflets of amphipathic phospholipid molecules that can be roughly described as three chemically distinct regions: the hydrophillic headgroup, the hydrophobic fatty acid acyl tails, and a connector between the two, typically derived from glycerol (Fig. 1A). Each individual lipid can accommodate a large number of conformational states, particularly as it relates to its two acyl tails (Fig. 1B), bestowing a flexibility that helps to eliminate gaps in the membrane (Fig. 1C) while permitting a hospitable environment for specific types of molecules, as required for partitioning. Molecules that partition strongly into the hydrophobic core of the membrane, as measured by their large membrane partition coefficient logP, tend to be only weakly soluble in aqueous solution[15]. At the molecular level, weak direct interactions between solvent and solute cause solute to be excluded from the solvent, which seeks to maximize its own entropy [16]. As a result, the membrane accumulates these species, yielding up to a 10-million-fold enrichment of these molecules within the membrane relative to their concentration in solution [17, 18].
Figure 1.

Membrane structure is defined by the chemical structure of lipids. A) The chemical structure of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), a prototypical lipid present in eukaryotic membranes, is comprised of a zwitterionic head group (blue), a polar glycerol group (red), and non-polar carbon tails (black/grey). B) A three-dimensional structure of an isolated phospholipid highlights the flexibility of the carbon tails which engenders the fluid and highly dynamic nature of membranes. C) By color coding the different chemical groups within each lipid, three distinct regions within a fully equilibrated POPC membrane become clear. The chemical details defined by small molecule interactions with the different lipid functional groups are major determinants of how molecules partition into the membrane.
The increased concentration of molecular species within membranes is heavily utilized in biological processes. Common gases such as O2, NO and CO2 congregate in lipid membranes [19–24], and reach their target sites within membrane-embedded proteins in rapid fashion, presumably by the existence of protein tunnels leading from the membrane core [25–28]. Additionally, contemporary pharmaceuticals and drug-like signaling molecules often depend on partitioning into the membrane to find their binding target, approximately ~ 60% of which are membrane-associated [29]. Membrane-associated proteins themselves may also insert structural elements into the membrane to better carry out their function [30–33]. This includes antimicrobial peptides, which are thought embed into the membrane and generate a pore-like structure that kills the target [34], as well as other signaling or metabolic proteins that depend on access to membrane-embedded substrates, and may anchor themselves to the membrane via electrostatic or hydrophobic interactions [35–37].
Designing experiments to directly observe membrane partitioning or insertion phenomena is hindered by the exacting requirements the membrane places upon the experimenter. The membrane is a fluid environment [38], undergoing macroscopic phase changes within minutes [39] and nanoscopic lipid rearrangements on the microsecond timescale [40], precluding static diffraction techniques from being an effective tool at elucidating the details of molecule-membrane interaction. Membranes are also too thin (~ 4 nm thick) to measure membrane penetration depth using super-resolution fluorescence techniques, although other methods such as AFM can determine the fine structure of membranes [41]. The greatest experimental successes have come with ensemble spectroscopic techniques such as NMR [42–44], EPR [45, 46], and fluorescence quenching [47, 48] which have the sensitivity needed for reliable measurements, but are still more complex than their equivalent solution experiments.
As a result of the complexity and technical difficulties associated with experimental methods, modeling membrane processes through molecular dynamics (MD) simulations has a long history [49–51] in advancing our understanding of membrane biology by simultaneously exploring length and timescales unobtainable by other means [52, 53]. Membrane modeling itself has advanced significantly over the intervening 40 years; the computing power available for the simulations has grown exponentially through Moore’s Law, and the forcefields used to describe the internal dynamics and intermolecular interactions have become increasingly accurate over successive generations [54–57]. Recently developed small molecule forcefields (eg. GAFF [58], CGenFF [59], OPLS [60]) and tools for expanding parameter coverage (Antechamber [61], CGenFF Program [62], MATCH [63], flTK [64]) have greatly expanded the chemical library that molecular dynamics can access. Leveraging these advances, it is now possible to simulate real membrane systems for microseconds [65] to potentially milliseconds [66], for the first time allowing direct observations of the interactions between molecules and the membrane on the single molecule level.
For processes operating on timescales that exceed what can be achieved using simple equilibrium simulations to collect adequate statistics, working with in silico models allows a great deal of freedom and creativity to apply computational techniques that are not bound to constraints of the physical world. For example, the use of alternative membrane representations can greatly extend the timescale accessible through simulations. Coarse grained and united atom membrane representations map multiple atoms into a single simulated particle, which reduces the simulation particle count and allows for longer simulation timesteps [67, 68]. The particle count can be reduced even further by removing the explicit membrane atoms altogether, replacing them with an implicit membrane model in which the effect of the membrane is applied as a continuum with varying dielectric [69], again enabling longer simulations. Membrane representations that increase the diffusion constant of the lipids [70] by making them shorter and adding an organic solvent to fill the void have also been developed [71], which accelerates the kinetics of insertion processes [72] while preserving the energetics and specificity of a conventional membrane representation at the membrane periphery [73]. While each of these methods has their advantages, they make sacrifices in the accuracy of the molecular interactions; hence, the gold standard for MD simulation remains at the fully atomic scale.
Alternatively, enhanced sampling techniques can extend the reach of equilibrium simulation to studying membrane partitioning and insertion by biasing the sampling in favor of rarely visited states. This sampling can be explicitly defined by the user, such as in umbrella sampling where an external harmonic potential is added to a specific point on the reaction coordinate [74, 75], or guided by the system, such as in metadynamics where the resulting free energy profile is built up as the system traverses reaction coordinate space [76]. In either case, a complete free energy profile emerges, providing invaluable insight as to the location and height of energetic barriers and troughs along the reaction pathway. Combined with advanced analyses to complement these methods [77–79], computational scientists are well armed to answer fundamental questions of how small molecule interactions have a big impact in biology.
In this review, we present specific examples taken from our own research in which the interaction of small molecules with membrane proteins is mediated by and facilitated through their interaction with the lipid bilayer. This includes entrance of gas molecules into bioenergetic proteins from the membrane, binding of anesthetics to a membrane channel via a membrane-accessible pathway, membrane-mediated conformational change in proteins that bind and metabolize drugs, and lateral binding of lipids into an ABC transporter. In all cases the small molecules initially partition into the membrane, from where they finally reach their protein target. Both of these steps can be effectively described using MD simulations by combining equilibrium simulation with enhanced sampling techniques.
Lipid mediated essential functions of gases in membrane proteins
Membrane partitioning and permeability of gases are vital processes to life. Small dioxygen (O2), nitric oxide (NO), and carbon dioxide (CO2) gases function as substrates or ligands to many proteins, such as myoglobin, guanylyl cyclase and cytochrome c oxidase, triggering and regulating biochemical processes, which include but are not limited to cell signaling and metabolism. Some processes occur at the cell surface, whereas others occur inside the membrane or inside the cell. Due to the hydrophobic nature of lipids, the primary constituents of membranes, hydrophilic, charged, or large molecules require membrane channels or transporter proteins to facilitate translocation across the membrane. O2, NO, and CO2 molecules, on the other hand, can spontaneously diffuse across the membrane without having to overcome large energetic barriers, and represent some of the simplest small molecules that interact with the membrane both as a function of their size and chemical properties.
The process of gas partitioning into proteins or lipids is particularly well-suited using MD simulations. In one of the most effective of simulation-based approaches, which we term explicit ligand sampling (ELS) simulations [80–84], gas molecules are randomly placed initially in the simulation system and allowed to diffuse in an unbiased manner during the run. The ELS approach enables direct monitoring of partitioning events for individual gas molecules and provides dynamic descriptions of proteins or lipids associated with the gas partitioning processes; however, ELS is sensitive to the timescale of simulation relative to protein motion and ligand diffusion rates. In a protein or other complex systems, the target binding sites(s) for gases may be buried deep within the structure and there may be non-specific binding sites. Conformational changes ranging from localized, small breathing motions of side chains to global structural changes may be required for small molecules, even gases [85, 86], to gain access to their target binding site. Hence, with a small copy number of a gas molecule, full binding events may not be captured on the simulation timescale. Moreover, the estimations of thermodynamic and kinetic quantities, such as partitioning and permeability coefficients, require sufficient sampling of multiple events across the entire configuration space. One method within the ELS repertoire to enhance sampling for ligands that interact weakly with their surroundings is so-called “Flooding” simulations, in which ligands are added to the simulation system in high copy numbers that may exceed typical physiological concentrations [81, 87–89]. With such high numbers of ligands in the simulation system, sampling coverage of binding sites and binding pathways of molecules is maximized while the total simulation cost is significantly reduced.
An alternative and complementary approach to ELS for characterizing binding pathways of gas molecules to target binding sites in proteins is termed implicit ligand sampling (ILS) [80, 90–94]. Since individual gas molecules have small volumes and interact weakly with proteins and lipids, their presence at reasonable concentrations negligibly perturbs the overall protein or membrane structure. Following this assumption, snapshots of protein structures taken from crystal structures or trajectory frames generated by MD simulation in the absence of a targeting molecule may be used to quantitatively analyze the binding affinity for the ligand over the entire structure. Performing this affinity analysis over an ensemble of structures allows for massive ligand sampling to search for viable access pathways and binding sites. ILS is performed in a defined three-dimensional grid covering the protein or membrane, in which the grid is divided into several subgrids or positions where the targeting molecule is independently sampled. ILS calculates interaction energies between the targeting molecule and its surroundings (Ei) in any position inside the grid over an ensemble of protein conformations and ligand orientations [90], which estimate a 3D free energy map for inserting a molecule at a particular position (ΔGi),
where, p0 (in vacuum) = 1 and pi is the probability of inserting a molecule at position i. The power of the ILS approach is that it can simultaneously sample a molecule in all positions of the configuration space which cannot be achieved by ELS or flooding simulations.
Partitioning of Small Gases in Membranes
Our group and others have studied the partitioning of O2, NO, and CO2 in phospholipid bilayers using both the ELS and ILS methods [80, 82, 93–97]. As an example, in one of our studies using the ELS technique, independent simulations of O2 and NO membrane partitioning were performed using 125 copies of the gas molecule (300 mM) initially placed in the bulk aqueous solution, followed by 220 ns of equilibrium simulation. The distribution of gas molecules in the membrane vs. the aqueous phase plateaued within the first 20 ns of the simulation (Fig. 2a). Accordingly, the last 200 ns of the simulation trajectory were used to calculate the partitioning free energy profiles with respect to the equilibrated gas concentration in bulk solvent. The partitioning free energy profiles calculated from ELS align with those calculated from ILS simulations (Fig. 2a). Features present in the profile, such as maxima, minima, and inflection points, correspond to three important regions of the membrane (Fig. 1): polar head groups, hydrophobic lipid tails, and the interface between the two membrane leaflets. These profiles demonstrate that O2 and NO spontaneously partition into the membrane from the aqueous solution by first overcoming a small free energy barrier of only 0.5 kcal/mol located in the polar head group region, followed by favorable penetration into the lipid tail region, and finally settle largely near the midpoint of the bilayer corresponding to the global minimum of −2 to −1.5 kcal/mol. These results are consistent with those from obtained from fluorescence, NMR, and ESR experimental studies [22–24] and with those from previous simulations using free energy methods (e.g., ILS and umbrella sampling) [80, 94–98], establishing that small, hydrophobic gases are 5–10 fold more soluble in the membrane than in the aqueous solution. The location of the global minimum allows for O2 and NO molecules to readily migrate between upper and lower membrane leaflets, a characteristic that will be contrasted by larger amphipathic molecules described in later sections.
Figure 2.

Favorable membrane partitioning of gases. A) Illustration of the partitioning of gas molecules into the membrane from the aqueous solution from an ELS trajectory. This illustration is taken from a flooding simulation of O2 molecules (shown in red) which are initially distributed in the aqueous solution. As time progresses, O2 molecules spontaneously enter the membrane and become steadily distributed in both aqueous and lipid phases. More O2 molecules localize in the membrane than in the aqueous solution. B) Partitioning free energy profiles of O2 and NO gases in a POPC bilayer calculated using ELS trajectories and ILS. Shaded blue boxes in each plot represent the aqueous solution. Shaded green boxes represent the polar head group of the membrane. Shaded orange boxes represent hydrophobic lipid tails region. A shaded yellow box represent the interface between two membrane leaflets.
The computational studies of gas molecule partitioning into membrane described above employed a fixed charge model for the gas molecules. The absence of polarizability, in general, is one of the limitations in the majority of classical MD simulations. Small diatomic gases such as O2 are commonly described either as a completely apolar species (q = 0.0 e on both atoms) or by a slightly charged (e.g., q = ±0.02 e) model [28, 80, 90, 93], with the latter model designed to capture part of the polarizability through the molecular rotation of the small molecule. In another study, Hub and de Groot modeled the quadrupole moment of O2 (−0.82 D Å) by introducing a dummy atom with a charge of q = +0.452 e at the midpoint of the two oxygen atoms, neutralized by negative charges of q = −0.226 e on each oxygen atom [95]. As most of the above-described models result in comparable O2 membrane partitioning profiles, lack of polarizability does not seem to introduce a major shortcoming for O2 simulations. We note, however, that the inclusion of polarizability can significantly affect the partitioning of halogenated or hydroxylated compound, which are highly polarizable, into the membrane [99].
Membrane Delivery Pathway for O2 in Cytochrome c Oxidase
Molecular O2 plays a crucial role in bioenergetics as the final electron acceptor of aerobic respiration, which occurs in the inner mitochondria membrane of eukaryotes and in the periplasmic membrane of prokaryotes. In aerobic respiration, O2 is exergonically reduced to water by its terminal oxidase, enzyme cytochrome c oxidase (CcO) [3, 100–107]. Reduction of O2 occurs in the bimetallic site of CcO composed of a heme cofactor bearing a copper ion sequestered inside. The reaction requires protons which enter from the cytoplasmic side of the bacterial membrane via hydrophilic channel(s) [101, 104, 108, 109], electrons which are delivered by cytochrome c on the periplasmic surface, and the entry of O2 from bulk solution. The free energy released from the reaction is coupled to generate the proton motive force for the biosynthesis of ATP, as well as other energy-requiring processes [100, 102, 103, 105, 107, 110, 111].
Our study of a bacterial Thermus thermophilus CcO or cytochrome ba3 (CcO ba3) exemplifies the role of lipids in delivering hydrophobic gaseous substrates to the reaction center [28]. CcO ba3 is one of functionally and structurally well-characterized CcO enzymes [26, 27, 112–116]. The binding of O2 has been experimentally studied using X-ray crystallographic [117, 118] and spectroscopic techniques [26, 119]. From these methods, O2 was determined to be delivered to the bimetallic site of CcO ba3 at 1×109 M−1s−1, which is at the diffusion limit. Hybrid pressurization X-ray crystallography employing xenon gas (Xe) as an electron-dense stand-in for O2 with similar size and chemical properties, resolved the position of several Xe atoms in CcO ba3 [117, 118]. The Xe atoms were all located in a 25 Along hydrophobic tunnel within the protein, which is parallel and exposed to the membrane core [117, 118]. This hydrophobic tunnel is also observed in the structures resolved in the absence of Xe [112, 115, 120], indicating that the tunnel structure is not a result ligand-induced perturbations.
To provide quantitative descriptions of O2 delivery process in CcO ba3 in atomistic detail, we employed both ELS and ILS approaches (Fig. 3a–c). As described for the ILS method, a simulation was performed in the absence of O2 and the resulting trajectory was analyzed for potential O2 binding regions. These calculations reveal highly favorable O2 binding regions within the protein shown as red isosurfaces in Fig. 3b, in which the partitioning free energy (ΔG) of O2 with respect to the aqueous solution is −3.0 kcal/mol. One of these favorable regions forms a Y-shaped pathway in the same position as the experimentally observed Xe-bound tunnel, further supporting this tunnel as potential O2 delivery pathway.
Figure 3.

Protein tunnels facilitate the rapid delivery of O2 in CcO ba3. A) A side-view image of an O2 simulation in CcO ba3 illustrates the entry of substrate O2 to CcO ba3 from the membrane. Small red balls represent O2 molecules distributed during the simulations. Dashed green box highlights the catalytic site of O2, consisting of a heme iron (green ball) and a copper ion (orange ball). Cyan spheres represent phosphorus atoms of membrane lipids. B) Top-view image highlight highly favorable O2 binding regions calculated from ILS. The red surfaces correspond to the partitioning ΔG isosurface of −3.0 kcal/mol. C) Top-view image highlights the collection of O2 molecules that partition in the protein during ELS simulations.
Complementary ELS simulations were performed for tens of nanoseconds and were repeated to ensure the reproducibility of results (Fig. 3a). In each simulation, one hundred copies of O2, corresponding to 210 mM concentration, were added to the system. O2 molecules were initially distributed in the membrane, aqueous solution, or both phases to allow O2 to unequivocally search for its delivery pathways. ELS simulations spontaneously captured the migration of O2 molecules to the catalytic site exclusively via the Xe-bound tunnel, and are consistent with ILS calculations (Fig. 3c). No additional pathways were found to lead O2 into the catalytic site during the course of the simulations. Further, the first delivery event of O2 to the reaction center required only a few nanoseconds of simulation, reproducing the experimentally measured delivery rate of O2 at the diffusion-controlled limit.
General Anesthetics: Utilizing the Membrane as a Conduit to Modulation Sites in Ion Channels
Although the simulation of simple gases also applies to the study of general anesthetics (GAs), such as for xenon [121–125] which is both a gas and a known anesthetic, the simulation of other structuraly complex GAs in the membrane environment introduces additional obstacles and complexity. The need for de novo parametrization, together with the additional degrees of freedom available to GAs, allows for more diverse and complex interactions with membrane lipids and associated proteins. The discovery of general anesthetics over 150 years ago has generated one question that has yet to be answered; namely, what is the exact molecular mechanism(s) by which anesthetics exert their action? Early theories proposed a non-specific biological membrane disruption mechanism [126]. The first studies on the physical properties of anesthetics [127] found a direct correlation between the potency of an anesthetic and its oil:water partition coefficient, lending support to the theory. This phenomenon, deemed the “Meyer-Overton Hypothesis”, dominated clinical and scientific thinking on the molecular mechanism of anesthesia for well over a century [128]. Research on this subject has shifted significantly, however, as physicochemical studies started to show stereoisomers having different anesthetic potency [129–131] and long-chain alcohols having less anesthetic potency than short-chain alcohols [132, 133], both being contradictory to a non-specific membrane disruption mechanism. Moreover, biochemical [134–137], structural [138–140], and computational [87, 141, 142] studies have provided additional evidence that argues against the Meyer-Overton hypothesis and, instead, point to a family of pentameric ligand-gated ion channels (pLGICs), namely the Cys-loop family of receptors, as the main targets of general anesthetics, although other targets have been described [143, 144]. In this section, we describe the our simulations to study how a class of clinically used inhaled anesthetics partition into the membrane and bind to the membrane embedded pLGIC proteins.
Membrane Partitioning of Inhaled Anesthetics
In constrast to the O2 and NO gases discussed above, which are described by Lennard-Jones terms and a single bond parameter, dynamic descriptions of small molecule anesthetics require substantially more parameters. Moreover, due to the intimate interaction between anesthetics and ion channels, the quality of the parameters heavily influences the quality of the simulation data. Early endeavors to perform MD simulations on the interaction of anesthetics and membranes required de novo parametrization. These early studies fit classical molecular mechanics force field parameters by multiple iterations of manual adjustment against quantum mechanical data [141, 145, 146]; beyond this, however, little benchmarking in the form of comparing simulated physical properties of these molecules to experimentally determined physical properties had been reported. These studies predate the release of generalized variants of popular biopolymer force fields, such as the Generalized AMBER Force Filed (GAFF) [58], and the CHARMM General Force Field (CGenFF) [59], which aim to provide coverage for substructures and functional groups that are commonly found in biomedically relevant small molecules, and do so quite well for a limited subset of drug-like molecules [147]. Coverage for inhaled anesthetics, which are often heavily halogenated, has remained poor due to the complexity of reproducing the physical properties of halogens using a classical force field. Alternatively, the OPLS force field is well parametrized to describe small molecules [148, 149]; however, robust support for lipid parameters has only recently been developed [150, 151], relative to the long-standing availability and highly optimized parameters for CHARMM [54, 152–154] and AMBER [55, 155] parameter sets.
In order to conduct studies on the effect of anesthetics on membrane structure, we needed to first parametrize the inhaled anesthetics, desflurane, isoflurane, sevoflurane, and propofol. While there are several resources available to obtain parameters, typically by analogy [62, 63, 156], we used the Force Field Toolkit (ffTK) [64]. ffTK was explicitly designed to develop parameters from first principles in accordance with the best practices for maintaining compatibility with the CHARMM family of force fields. This approach leveraged highly transferable parameters for the Lennard-Jones (LJ) term while optimizing all partial atomic charges and bonded parameters (i.e., bonds, angles, and dihedrals) required to describe each anesthetic molecule in a manner that can be directly combined with existing parameters for the protein, lipid, water, and ion components of the molecular system. Further, ffTK provided the flexibility to deviate from standard protocols. Specifically, using the hybrid B3LYP density-functional theory (DFT) has been shown to yield more appropriate target data for fitting fixed charges to polarizable atoms, such as halogens [146, 157, 158], compared to the commonly prescribed Møller-Plesset (MP2) level of theory. In the special case of isoflurane, the parameter describing LJ well-depth for the chlorine atom required additional tuning, performed by hand, to correctly reproduce intermolecular interactions in the bulk phase. The resulting parameters for all anesthetics were benchmarked against experimentally measured condensed phase properties, and were found to reproduce free energies of solvation in water and dodecane (used to represent an oily environment) to within 1 kcal/mol in all cases, often below a kT, in addition to excellent reproduction of density and enthalpy of vaporization [89] measures. The ability to satisfactorily reproduce both bulk phase and aqueous/organic phase properties speaks to the accuracy and robustness of the developed parameters.
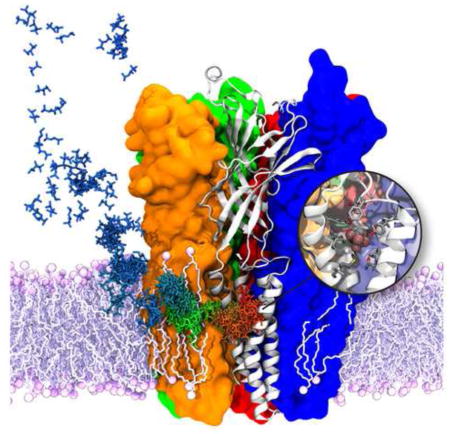
With the parameters for several GAs thoroughly vetted against available experimental data, we began to explore the interaction between GAs and the membrane itself. In order to understand how anesthetics interact with their membrane protein targets, we first had to determine where these molecules partitioned in the membrane. Utilizing two approaches, namely umbrella sampling calculations and flooding simulations, we determined that anesthetics preferentially partition to the amphipathic glycerol backbone region of the membrane due to their own amphipathic nature [89]. Building on previous free energy calculations in simple polar:non-polar systems [146, 159, 160], which showed an energetic minimum at the interface between the two phases, our calculations in a more complex water:POPC system showed a 4–5 kcal/mol energetic minimum in the interfacial glycerol backbone region. This region provides interaction for both the hydrophilic and hydrophobic moieties of each GA [89], in contrast to the entirely hydrophobic gas molecules which partitioned to the center of the membrane. In fact, flooding simulations placing many copies of the drug randomly in solution followed by equilibrium simulation [87, 161–164] to probe unbiased interactions with the membrane (Fig. 5A) showed the same distribution [89] for all simulated GAs. Atomic distributions from these simulations (Fig. 4B) showed that each anesthetic also has a preferential orientation within the membrane, namely with the non-polar moiety oriented inward and the polar moiety oriented outward. These studies, therefore, provide a molecular mechanism for the strong preference of anesthetics for the membrane, namely their amphipathic nature.
Figure 5.

Membrane partitioning of anesthetics facilitates binding to modulation sites of ion channels. A) Simulating high concentrations of desflurane (43 desflurane molecules, 1:5 ratio with lipid) demonstrated partitioning to the amphipathic glycerol region of the membrane within 100 ns. Two different paths of desflurane are shown, colored by simulation time, which exemplify membrane partitioning of inhaled anesthetics. B) Simulations employing high concentrations of desflurane were used to “flood” the GLIC protein embedded in a POPC membrane. Following the rapid partitioning of desflurane molecules into the membrane, several molecules spontaneously bound to the transmembrane domain, revealing an inner and outer binding site. The desflurane molecule forms several non-specific contacts within the inner binding site (top inset). Notably, the binding site near the same location within the membrane where desflurane partitions (bottom inset).
Figure 4.

Partitioning profiles of inhaled anesthetics into a POPC membrane. A) Snapshot of the POPC membrane used in the simulations. Water and ions are omitted for clarity. The color of the atom groups in this image corresponds to the color of the curves in the density profiles. B) Density profile of the simulated systems used to demarcate the regions of the membrane for analysis of anesthetic–membrane interactions. Here, total POPC density is shown as the black dashed line and water density is shown as the light blue line. POPC density was further subdivided into tail (gray), glycerol (red), phosphate (gold), and choline (blue) density. The colors of the curves correspond to the color of the atoms shown in (a). C) PMF for inserting desflurane (blue), isoflurane (green), sevoflurane (orange), and propofol (red) into the membrane. All anesthetics show a distinct energy minimum at the interfacial region, while there is negligible energy difference between the bulk aqueous environment and midpoint of the lipid bilayer. D) Representative snapshot of (starting on left and moving right) desflurane, isoflurane, sevoflurane, and propofol in the umbrella sampling simulations showing the low energy conformation at the amphipathic boundary of the membrane. Figure reprinted from Arcario et al. [89].
As opposed to the Meyer-Overton hypothesis, however, once partitioned, GAs do not appear to perturb the physical, chemical, or electrical properties of the membrane [89], though they have been noted to lower the miscibility phase transition temperature [165]. Assessing the structure of a POPC membrane after anesthetic partitioning by analyzing the atomic distribution profile of the lipid chemical components (e.g., carbon tail, glycerol linker, phosphate and choline portions of the head group) and lipid order parameters did not demonstrate any significant signs of membrane disruption. Moreover, the dipole potential and lateral pressure profile of the POPC membrane remained unchanged. In contrast, MD simulations of ketamine, an injectable general anesthetic, have shown small changes in the lateral pressure of a DOPC membrane [166] with supraclinical concentrations. These studies, however, use the R-(−) enantiomer of ketamine that is less potent than the S-(+) form used clinically, making the generalizability of these studies unclear. Therefore, our simulations have demonstrated that while GAs readily partition to the glycerol interfacial region of the membrane, they do not appear to alter the physicochemical properties of the membrane itself.
Anesthetics Accumulate in the Membrane and Bind to Multiple Modulation Sites of Ion Channels
The discovery that anesthetics bind to and affect ion conduction in pLGICs [167, 168], as well as experimental [169] and our computational [89] studies demonstrating that anesthetics do not appreciably affect membrane properties, we were inspired to explore how anesthetics bind to an interact with pLGICs. While previous cryo-EM structures [170] were of too low resolution to perform MD simulations, discovery of a family of bacterial homologues [171] that are sensitive to clinical concentrations of anesthetics [167, 168] opened the field to higher resolution X-ray crystal structures [138, 140, 172–178]. Simulating anesthetics in an a priori bound state determined by crystallography [138] of the Gloeobacter violaceus ligand-gated ion channel (GLIC), a bacterial homologue of eukaryotic pLGIC, we observed that the anesthetics are loosely bound and spontaneously dissociate from the GLIC binding site within 50 ns of simulation. Moreover, once dissociated from the initial binding pocket, the anesthetics diffuse through an intrasubunit, membrane-accessible lumen, eventually partitioning to the glycerol region of the membrane. Analysis of the binding pocket interactions showed that there is not a complimentary arrangement of residues that tightly binds the anesthetic, but a general amphipathic pocket that is accessible to the membrane. The exceedingly low affinity of the general anesthetic for the binding site provides rationale for why such a large concentration is required for clinical effect [167, 168]
Due to the instability of desflurane in the binding pocket uncovered from crystal structures [138], we performed flooding simulations with GLIC (Fig. 5B) to determine a stable anesthetic binding site. From these simulations, two binding sites were elucidated, the site observed by crystallization studies (TM1 site) and a previously undiscovered site, which occupies a proximal location along the binding path. In addition to crystallographic and simulation studies of GLIC supporting the presence of the TM1 site, flooding simulations on a eukaryotic nicotinic acetylcholine receptor homology model [87] have demonstrated the same binding site. Interestingly, in order to reach both the TM1 and TM2 binding sites, the anesthetics must first partition to the membrane (Fig. 5B). This result helps to shed light on why the Meyer and Overton observed a correlation between hydrophobicity and anesthetic potency; hydrophobicity is necessary for membrane partitioning which is required for anesthetic binding. Once anesthetics are bound to the TM1 and TM2 sites simultaneously, the anesthetic becomes more stably bound, occupying the pocket for over 200 ns in the flooding simulations, compared to a little over 40 ns in the a priori state simulations. At this point, however, further work is needed to clarify whether the second binding site identified in these simulations causes a simple kinetic barrier to prevent the anesthetic from leaving the low affinity binding region or whether occupation of this binding site alters the energy landscape of the TM1 site.
Interactions of Steroid Hormones with Lipid Bilayers and Membrane–Bound Proteins
Steroid hormones regulate many cellular processes, including gene expression and cell signaling [179, 180], by binding to steroid hormone receptors typically located in the cell nucleus [181, 182], but also found in the cytoplasm [180, 183]. Steroid hormones are synthesized from cholesterol [184] which retain the lipophilic core and are often decorated with pendant groups of diverse chemical properties, which facilitates crossing the plasma membrane to bind to their respective steroid receptors inside the cell. Cholesterol is one of the primary components of mammalian cell membranes [185]. The conversion of cholesterol into steroid hormones is mainly mediated by enzymes belonging to two main families, namely hydroxysteroid dehydrogenases (HSD) and the cytochrome P450 (CYP) family [186]. While HSD enzymes are mostly involved in the biotransformation of steroid hormones, CYPs constitute a large family of proteins with diverse functions [187] involved in the metabolism of drugs and a broad spectrum of endogenous compounds, including steroids hormones [188].
Importantly, both clases of enzymes involved in steroid biosynthesis are primarily membrane–bound proteins [189–191], and because of the amphipathic nature of steroid hormones, it is presumed that precursor partitioning into the membrane is a crucial step for steroid synthesis. Additionally, a growing body of evidence suggests that steroid hormones can modulate and inhibit the function of integral membrane proteins, including neuroreceptors and ion channels [192–195]. Hence, it is important to understand the partitioning and localization of steroid hormones in lipid-bilayer environments.
We have employed a combination of different computational approaches to study the interaction steroid hormones with lipid bilayers and membrane–bound proteins. To gain insights of steroid–membrane interactions, we have performed a series of equilibrium simulations to investigate the spontaneous membrane binding of two prototypical steroid molecules, namely estradiol and progesterone. Using this approach allowed the characterization of the insertion depth and orientation of estradiol within the membrane. To characterize the binding pathway of steroids to CYP enzymes, we have performed a series of simulations of progesterone bound to Cytochrome P450 3A4 (CYP3A4). CYP3A4 is the most abundant isoform in the human body [196] and is responsible for the metabolism of more than 50% of drugs [197], including a variety of steroid hormones [188]. This approach not only allowed us to characterize the steroid binding pathway to CYP, but also provided insights into the general drug binding mechanism of CYP3A4 mediated by the membrane.
Steroid Hormone Partitioning Into the Cellular Membrane
Herein, to explore the interactions of steroid molecules with lipid bilayers, we present preliminary simulation results obtained for estradiol in a POPC membrane. Estradiol is a prototypical example of amphipathic steroid hormones (Fig. 6), and it is the most prevalent endogenous estrogen [184]. The general approach employed to study steroid–membrane interactions was to perform simulations where multiple estradiol copies were initially placed at random positions and orientations in the bulk solution, in order to capture the unbiased membrane insertion of the steroid. Primary simulations resulted in significant aggregation of the estradiol copies located in bulk solution, which retarded the penetration of the molecules to the lipid bilayer. To overcome this phenomenon, an additional non–bonded interaction parameter (a short-range force acting only between the oxygen atoms of estradiol) was applied in order to artificially introduce a minimally repulsive force between estradiol molecules, thereby preventing aggregation.
Figure 6.

Partitioning profile of estradiol in a POPC membrane. A) Snapshot after 200 ns illustrating multiple copies of estradiol in their preferred locations and orientations. The membrane consisted of POPC lipids and water molecules have been omitted for clarity. Estradiol is shown in stick (licorice) representation (C, cyan; O, red). B) To describe the orientation of the estradiol within the bilayer, an axis was defined pointing from O-17 to O-3, and an angle was measured with respect to the bilayer normal. C) The angle distribution of estradiol is shown in the lipid bilayer (red) or in bulk solution (blue). The angle distribution computed for an isotropic system (green) is shown for reference. D) Dependence of probability function for orientation of estradiol in the bilayer; the probability is shown in 2D plot, where the color changes from red to blue with increasing probability.
In the extended simulations in the POPC membrane (performed for 200 ns) estradiol molecules were often found in ~90° orientations with respect to the membrane normal (i.e., the z–axis), such that they were parallel to the membrane surface (Fig. 6A–C). The preference for a perpendicular orientation is distinct in comparison to the distribution measured for estradiol in bulk solution (Fig. 6C, red vs. blue). After partitioning into the membrane, estradiol molecules spent most of the time in the glycerol region at the interface of the polar head groups and lipophilic acyl tails (Fig. 6D), similarly to the anesthetic drugs described in previous section. The preferred perpendicular orientation of estradiol in the membrane can be understood in terms of the amphipathic nature of estradiol in which the polar hydroxyl groups, located at opposite ends of the molecule, formed hydrogen bonds with lipid phosphate and carbonyl groups at the lipid-water interface of the membrane, therefore facilitating an almost parallel orientation with respect to the membrane surface. Cholesterol, in contrast, possess only a single hydroxyl group and has been shown to adopt an almost parallel orientation with respect to the membrane normal [198, 199], presumably to facilitate the insertion of its extended hydrophobic acyl chain towards the lipophilic membrane center. These observations therefore suggest that steroid hormones, despite sharing the same scaffold derived from cholesterol, adopt significantly different membrane orientations and interactions which have implications mediating protein–steroid interactions within the membrane.
Membrane-Mediated Drug Recruitment by Cytochrome P450
Cytochrome P450 (CYP) enzymes are large family of heme–containing proteins [187], involved in the metabolism of a wide range of chemical compounds. In the human body, CYPs are involved in the biotransformation of drugs and endogenous compounds [200], such as steroid hormones [186], most of which partition substantially into membranes in the cell. Thus, a key aspect of the function of CYPs is the mechanism by which various molecular species gain access to the active site, a process that is particularly important for membrane-associated CYPs. It has been suggested that the interaction of CYPs with the cell membrane is crucial for the recruitment of lipophilic and amphiphilic substrates, such as steroid hormones and drugs, to the active site of the enzyme [201, 202].
In our studies of CYP3A4, the most abundant CYP isoform in the human body, we have been able to characterize the membrane-bound form by employing a combined simulation/experimental approach [203]. First, to obtain the membrane–bound form of CYP3A4, the spontaneous membrane–association of the globular domain of the enzyme was captured with a membrane mimetic model [71]. Multiple independent simulations were started by placing CYP3A4 away from a PC membrane, each with different initial orientations, achieving the unbiased binding of the protein to the membrane. It is important to note that the initial membrane-binding simulations started with the globular domain of CYP3A4 only, i.e., without including the transmembrane (TM) helix which has not been solved in most crystal structures. This was done since the initial goal of the study was to characterize membrane-interactions of the globular domain alone. The TM helix was added later to CYP3A4 once a membrane-bound model of the globular domain was generated. The resulting membrane–bound model provided details about key protein–lipid interactions, such as the involvement of structural elements (e.g., the F′-G′ loop) in membrane interaction. Moreover, the simulations showed that CYP3A4 takes a specific orientation on the membrane after binding, rather than adhering non-specifically to the lipid bilayer. Furthermore, these interactions were preserved after the membrane mimetic model was converted to a full membrane system by extending the lipid tails, and after adding the missing TM helix, confirming the stability of the model. Importantly, the orientation did not significantly change once the TM helix was added to the simulation system, suggesting that it is the membrane interaction of globular domain, not the TM helix, that determines the orientation of the enzyme on the surface of the membrane. In parallel to the simulations, linear dichroism measurements of nanodisc-bound CYP3A4 were performed to characterize the orientation of the enzyme in the membrane [203]. These measurements further confirmed that CYP3A4 adopts a specific orientation in the membrane, with the heme moiety oriented ~60° with respect to the membrane normal.
One of the observations of our CYP3A4 study is the transient opening of access tunnels that lead to the active site of the enzyme, resulting from protein–membrane interactions. Given their location with respect to the membrane, the observed access tunnels could be involved in the mechanism of drug–binding to CYP3A4. However, the relevance of the access tunnels is not immediately clear given the relative small size (i.e., < 1.5 Å) of their average opening [203], making it difficult for any of the typical substrates of CYP3A4 to bind. We hypothesized that these tunnels could be the initial openings that would require a longer equilibration time to fully open. Thus, to further explore the relevance of these access tunnels in CYP3A4 drug recruitment in the presence of the membrane, we performed a long-timescale (1μs) simulation, starting from our membrane-bound model derived previously [203]. However, although the observed access tunnels in the microsecond simulation are consistent with our shorter simulations, the extended timescale did not reveal any significant opening beyond what was previously achieved.
To take advantage of the conformational sampling of membrane–bound CYP3A4 in the microsecond–long simulation, we adopted an alternative approach to study drug–binding, in which we combined MD simulations with ensemble molecular docking [204, 205] of progesterone, a prototypical steroid substrate of CYP3A4. The proposed approach consisted in performing molecular docking of progesterone on multiple snapshots (e.g., ~ 8500) of the protein obtained from the microsecond simulation, allowing to account for transient conformational changes captured during the simulation that would facilitate accommodating progesterone along a putative access pathway leading to the active site of the enzyme. The analysis of the resulting docked poses revealed a continuous distribution of progesterone docked poses in the membrane-bound CYP3A4, connecting the active site of the enzyme to the membrane, suggesting a putative binding pathway for progesterone (Fig. 7). Interestingly, these results suggest that the access tunnels are not necessarily open in a continuous manner (i.e., connecting the membrane and the active site at once), and instead consist of smaller, contiguous segments, which openings are controlled by transient sidechain fluctuations.
Figure 7.

Membrane-mediated drug binding mechanism of CYP3A4. The binding pathway of progesterone was reconstructed by combining independent equilibrium simulations of progesterone along the putative access tunnel (shown in green-to-blue colorscale, stick representation), and membrane-partition simulations (shown in dark red-to-gold colosrcale). Heme cofactor, located in the active site of the enzyme, is shown in magenta stick representation.
The relevance of the putative progesterone binding pathway identified with molecular docking was further explored with additional MD simulations, starting from a selection of progesterone docked poses, consisting of structures with the best docking score. The simulations started with progesterone located at different positions along its putative binding pathway, in order to increase the sampling of important protein–ligand interactions. Remarkably, we were able to reconstruct an access pathway for progesterone from the protein–ligand conformations generated from multiple independent unbiased simulations, as shown in Fig. 7. Clustering analysis of the resulting simulations revealed significant spatial overlap of progesterone along different simulations in which the molecule preserves its overall orientation and direction through the overlapping regions, indicating that progesterone is able to sample a continuous pathway connecting the membrane to the active site in the ensemble generated by combining independent simulations (Fig. 7). Thus, the combined molecular docking/MD approach revealed the presence of a complete binding pathway that could facilitate the binding of amphipathic molecules partitioning into the membrane into the active site of CYP3A4. Indeed, our simulation of progesterone binding to a POPC membrane revealed that this steroid remains at the phosphate level of the lipid bilayer, where it could be recruited by CYP3A4, as shown in our fully reconstructed binding pathway (Fig. 7). Given the propensity of other molecules discussed in this review (including steroids and anesthetics) to partition into this same region of the lipid bilayer, our findings provide insights into a general mechanism for drug binding to peripheral membrane proteins, including other CYP enzymes, in which the membrane plays a major role in facilitating drug recruitment by serving as an intermediate binding partner for small molecules.
Phospholipid Binding in a Multi-drug Resistant Export Pump
Membrane partitioning studies of anesthetics and steroid hormones discussed in previous sections demonstrated how amphipathic small molecules tend to accumulate at the interfacial region between the head groups and lipid tails of the cellular membrane. Examining their free energy profiles calculated along the membrane normal (Fig. 4) clearly shows that these amphipathic molecules face a high energy barrier at the highly lipophilic membrane core during translocation from one leaflet to another. This aspect is highly relevant to the rate of passive absorption of many drugs through protein-independent pathways (i.e., diffusion through the lipid bilayer). Promiscuous membrane transport proteins with no substrate specificity facilitate the binding and transport of a variety of amphipathic small molecules from one side of the membrane to the other. In this section, we report an interesting aspect of phospholipid binding laterally from the membrane bilayer to a large molecular transport protein with multi-drug resistance, P-glycoprotein (Pgp), observed in MD simulations. Binding characteristics of amphipathic phospholipid molecules can be studied as a surrogate for the binding of any amphipathic substrate, including important drugs, which are believed to be transported through similar mechanisms and pathways.
In both the prokaryotic and eukaryotic kingdoms, many transporters confer multidrug resistance to the cell in which they reside. Some of these transporters, such as ATP-powered ABC transporters [206, 207], undergo a complex and large-scale structural transition while others, such as EmrD [208], perform comparatively simple rocker-switch structural transitions, harnessing the concentration gradients of different ion species to drive the transport. These transporters are mostly known to transport the drugs from the inner leaflet of the lipid bilayer, where the amphipathic drug molecules accumulate [18, 209, 210]. Several ABC transporters, including multidrug-resistant human Pgp and E. coli MsbA, are also lipid flippases [211–213], which move lipids between leaflets in the same manner as drug export occurs. Despite of 40 years of research, the polyspecific transport capability of Pgp and how drug molecules enter the central binding cavity still remain unclear. Existing models claim that the substrate is recruited directly from the inner leaflet based on experimental evidence [210, 211, 214, 215].
Crystal structures of mouse Pgp [206, 207] are important starting points for various structural studies focusing on mechanistic details of its function. Pgp is comprised of two transmembrane (TM) domains (TMDs), each of which contains six TM helices and undergoes a large conformational change when transitioning between the inward(intracellular)-facing (IF) and outward(extracellular)-facing (OF) states during molecular transport. These TMDs fold with respect to each other to form a central drug binding cavity. The regions between these domains are open either to inner or outer leaflets of the lipid bilayer [206]. In addition to the known high membrane partition coefficients of Pgp substrates, MD simulations have shown that many of these drugs accumulate near the head group level of the membrane [216], where the location of several charged residues in Pgp near the membrane interface could provide a “solvation exchange mechanism” to facilitate drug binding from the inner leaflet into the lumen of Pgp [216]. Our equilibrium and nonequilibrium MD simulations of IF and modeled OF states of Pgp have revealed spontaneous binding of lipid molecules from inner and outer leaflets in IF and OF states, respectively.
In the IF state of Pgp, the lipid molecules initially interact with the charged residues of the protein present at the level of lower leaflet and gain entry into the translocation chamber (Fig. 8A left). Independent simulations were performed using two (pre-refined [206] and refined [207]) crystal structures of IF state of Pgp. Simulations starting from the pre-refined structure have shown a few lipid atoms protruding between the transmembrane domains [217]. However, in one of our recent longer (300 ns) simulations of the IF state using the refined crystal structure of Pgp, two full inner leaflet lipid molecules have gained entry into the central chamber through the openings formed between key transmembrane helices (TM4 and TM6; TM10 and TM12). Though our simulations did not include a drug substrate, we expect similar observations because of the similar amphiphilic character of lipids [210]. The observed lipid binding events were either initiated by the head groups entering the cavity followed by lipid tails or vice versa (Fig. 8B). The first molecule entered the cavity interacted with both helices; however, the consequent lipids have only interacted with one of the two helices that form the opening as they moved away from each other after the binding of the first lipid. Extending and repeating these simulations may reveal further information on how these bound lipid molecules would influence the dynamics of IF state of Pgp.
Figure 8.

Lipid binding to Pgp in its IF and OF states. a) Schematic representation of IF and OF conformations of Pgp. Half-inserted and fully inserted lipid molecules are shown in broken and solid yellow lines, respectively. b) Lipid binding through both probable drug entry portals (TM4&6 and TM10&12) in IF conformation. c) Lipid binding through both probable drug entry portals (TM1&3 and TM7&9) in OF conformation. TM helices are in cartoon representation while the initial and final conformations of lipid molecules are in white and yellow sticks, respectively. Hydrogen atoms are not shown for clarity.
We have recently modeled a stable OF state of Pgp using a combination of structure-based sequence alignment and nonequilibrium MD simulations. A 300 ns-long MD simulation of the stable OF model of Pgp has shown that lipids enter between the openings formed between helices (TM1 and TM3; TM7 and TM9) from the outer leaflet of the lipid bilayer (Fig. 8A right). Though we did not observe a full lipid molecule entering into the extracellular side of the translocation chamber, a total ~160 lipid heavy atoms occupy the extracellular opening (Fig. 8C). In terms of how the lipids enter, the same trend has been observed as for the IF state, either head groups or tails initiate the entry. Presence of these lipid molecules at the extracellular opening certainly influence the dynamics and stability of the OF state and may facilitate substrate exit into the upper leaflet supporting the flippase model over the hydrophobic vacuum cleaner model. The discussed results, though preliminary, provide general mechanistic insights on how amphipathic molecules may bind to biomedically important multi-drug transporter, Pgp.
Conclusions and Future Perspectives
Membranes are one of the central compartments of the living cell, providing a critical barrier that defines the inside versus outside of cells [218]. In addition to controlling the traffic of small molecules across this border, membranes are also host to many diverse processes including signaling, energy transduction, and recognition. In order to understand the mechanism of action and cellular effects of both endogenous and xenobiotic small molecules, it is critical to understand how they interact with and distribute within or across the membrane. Although some experimental techniques can be successfully employed to study these processes, the fluid and dynamic nature of the membrane, both at the microscopic and macroscopic levels, introduces a number of technical difficulties. The spatiotemporal resolution of molecular dynamics simulations [52, 53], in contrast, are well-suited to investigations of highly dynamic properties like those that occur near or in the membrane with atomistic resolution.
We have described four examples from our own research which encompass different aspects of membrane processes at both qualitative and quantitative levels. It is clear from these studies that the chemical properties of the small molecule are the primary determinants for partitioning based on how they interact with lipid molecules. Small non-polar species, such as O2 and NO gas molecules, spontaneously partition to the center of the membrane where the lipid tails create a favorable and strongly hydrophobic environment. While gas molecules are able to penetrate the membrane structure by non-specific diffusion, we have found that the rapid activity of oxidase proteins, e.g., cytochrome C oxidase detailed here, requires that gas molecules access the reaction center buried deeply within the protein structure using explicit tunnels. The partitioning profiles of larger, amphipathic molecules, such as anesthetics, hormones, and lipids themselves, in contrast to the hydrophobic gases, indicate that these molecules prefer to reside near the glycerol region of the membrane. Accumulating to establish high concentrations of such molecules within the localized region of the membrane might facilitate binding to their target sites, both for proteins that are embedded and those that are otherwise associated with the membrane. X-ray crystal structures of GLIC (a channel protein) have revealed binding sites for anesthetics in the transmembrane region, which we show to be positioned near the glycerol-tail interfacial region when embedded into a POPC lipid bilayer. Our simulation studies, however, further revealed a second putative binding site just proximal to the first site and that the anesthetic binding to the second site is required to stabilize binding to the first site, a unique feature not previously hypothesized. These observations finally explain why anesthetic potency was correlated with hydrophobicity, yet was in some cases sensitive to stereochemical configurations, and why anesthetics are necessary at high concentrations to exert their action. Even in cases where the ligand binding site exists at a level outside of the membrane, such as in cytochrome P450, small molecules like hormones and xenobiotics reach the reaction center via access tunnels leading into the protein interior from the glycerol region of the membrane where amphipathic molecules tend to accumulate [203]. Amphipathic molecules also differ from the hydrophobic gases, as characterized by free energy techniques, in that they must overcome an energetic barrier to move between the two leaflets of the membrane. This situation suggests that while these molecules can spontaneously partition into one leaflet of the membrane, they are shuttled across the hydrophobic core by membrane proteins to facilitate traversal through the entirety of the membrane. Such shuttling proteins, e.g., the case of Pgp described here, are involved in moving chemicals into and out of the cell, and can serve to export xenobiotics from the cell. In addition to conventional small molecules, Pgp also binds and acts on lipid molecules, moving them across the membrane interface giving rise to its flipase activity, which is necessary to modify the membrane lipid composition during signaling processes. While full details of this process remain to be determined, our initial simulation results begin to elucidate the interactions between individual lipids and Pgp, presumably priming the lipid for a flipping event.
Characterizing the broader properties of small molecules and membranes, such as structure of different regions of the membrane or the accumulation of molecules within the membrane, are driven by the chemical composition of each component. Accurately capturing and characterizing the processes that small molecules undergo within the context of the membrane requires a robust set of force field parameters that are able to accurately describe internal dynamics and intermolecular interactions within a diverse set of environments (e.g., bulk solvent, membrane regions varying from polar to hydrophobic, protein surface, protein core). While quality parameter sets for water, proteins, and lipids have been extensively refined over time, the parametrization of small molecules has only recently been supported in a broader way with the development of force field extensions specifically catering to small molecules, particularly for drug-like compounds. The great expanse of small molecule space, however, precludes the ability of a single parameter set from providing universal coverage. Several toolsets, such as the Force Field Toolkit [64], developed in our lab and employed in our anesthetic studies presented here, and the CGenFF Program [62] used for the hormones, have been developed to facilitate and automate the process of computing and refining parameters for a given molecule, enabling a whole new area of simulation-based studies like those described in this review. As we illustrated for the heavily halogenated anesthetics, molecules bearing complex functional groups often require deviations from standard parametrization protocols; the details surrounding when and how to achieve this currently must be assessed on a case-by-case basis, and better tools are required to make this process more straightforward for non-experts. Further, these parameters apply to the CHARMM force field, which contains a number of limitations, the most prominent of these being a fixed point charge model that cannot account for atomic polarizability. Work is currently progressing on this front using alternative force field models, such as the Drude Force Field [219, 220] and AMOEBA [221], however, their use has yet to become widespread and the need for parametrization tools have not been addressed to the same level as for the non-polarizable CHARMM force field.
All of the studies covered herein utilized membranes consisting of POPC lipids, which are prominent in mammalian membranes [222]. Membranes, however, are rarely homogeneous and the composition of the membrane with respect to lipid species can have significant impact on membrane properties. Many membrane processes are actually dependent on changes in membrane composition, such as cell signaling. Therefore, the actual environments in which drugs and other small molecules exert their activity are much more complex. How specific mixtures of components that can be present in high concentrations in mammalian cells, such as charged lipids (e.g., POPS), sphingomyelin, or cholesterol, affect membrane dynamics and small molecule interactions remains an active area of research. Simulations employing realistic membrane compositions have begun to address these questions [223, 224]. One remaining challenge, however, is insufficient sampling due to the slow mixing of lipids within the membrane relative to the timescale accessible to molecular dynamics. Embedding proteins into such complex membranes further exacerbates the sampling problem due to increased viscosity, yielding protein structures that exhibit dynamics very different from their solution form. These scenarios may be alleviated by alternative methods that enhance sampling, e.g., reduced representations of the membrane as done in the HMMM model [70, 71], coarse-grained membrane models [67], or implicit membrane models for protein complex formation [225–227], and will likely be required in future studies.
In the future, we can only expect more examples in which the importance and specific role of biological membranes in determining the fate of small molecules will be demonstrated. Molecular simulations will continue to play a central role in these studies, since they offer the most detailed microscopic view of such processes. Along with more realistic representations of the cellular membrane, accurate force field parameters for a larger number of small molecules, and our increased ability to access the configuration space by longer simulations and better sampling, we should look forward to discovering more surprises and specific molecular phenomena for drugs and other small molecules such as endogenous hormones and neurotransmitters that may substantiate some of their characteristic chemical behavior within a biological context.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health (Grants R01-GM101048, R01-GM086749, U54-GM087519, and P41-GM104601 to E.T.) and XSEDE compute resources (grant TG-MCA06N060 to E.T.). M.J.A. acknowledges past support from the NSF GRF Program. J.V.V. acknowledges support from the Sandia National Laboratories Campus Executive Program, which is funded by the Laboratory Directed Research and Development (LDRD) Program. Sandia is a multi-program laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the US Department of Energy’s National Nuclear Security Administration under Contract No. DE-AC04-94AL85000.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Christopher G. Mayne, Email: cmayne2@illinois.edu.
Mark J. Arcario, Email: marcar2@illinois.edu.
Paween Mahinthichaichan, Email: mahinth1@illinois.edu.
Javier L. Baylon, Email: baylonc2@illinois.edu.
Josh V. Vermaas, Email: vermaas2@illinois.edu.
Latifeh Navidpour, Email: lnavid@illinois.edu.
Po-Chao Wen, Email: pwen2@illinois.edu.
Sundarapandian Thangapandian, Email: sundar81@illinois.edu.
References
- 1.Kurihara K, Okura Y, Matsuo M, Toyota T, Suzuki K, Sugawara T. A recursive vesicle-based model protocell with a primitive model cell cycle. Nat Commun. 2015;6:8352. doi: 10.1038/ncomms9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ichihashi N, Yomo T. Positive roles of compartmentalization in internal reactions. Curr Opin Cell Biol. 2014;22:12–17. doi: 10.1016/j.cbpa.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 3.Saraste M. Oxidative Phosphorylation at the fin de siécle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 4.Cho W, Stahelin RV. Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 5.Murthy KS. Signaling for Contraction and Relaxation in Smooth Muscle of the Gut. Annu Rev Physiol. 2006;68(1):345–374. doi: 10.1146/annurev.physiol.68.040504.094707. [DOI] [PubMed] [Google Scholar]
- 6.Stiber J, Hawkins A, Zhang ZS, Wang S, Burch J, Graham V, Ward CC, Seth M, Finch E, Malouf N, Williams RS, Eu JP, Rosenberg P. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10(6):688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morozov VA, Dao Thi VL, Denner J. The Transmembrane Protein of the Human Endogenous Retrovirus - K (HERV-K) Modulates Cytokine Release and Gene Expression. PLoS One. 2013;8(8):e70399. doi: 10.1371/journal.pone.0070399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brombacher E, Baratto A, Dorel C, Landini P. Gene Expression Regulation by the Curli Activator CsgD Protein: Modulation of Cellulose Biosynthesis and Control of Negative Determinants for Microbial Adhesion. J Bacteriol. 2006;188(6):2027–2037. doi: 10.1128/JB.188.6.2027-2037.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10(6):1141–1149. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- 10.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480(7376):254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hediger MA, Clémençon B, Burrier RE, Bruford EA. The ABCs of membrane transporters in health and disease (SLC series): Introduction. Mol Aspects Med. 2013;34(2–3):95–107. doi: 10.1016/j.mam.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73(7):1251–1254. doi: 10.1016/0092-8674(93)90353-R. [DOI] [PubMed] [Google Scholar]
- 13.Feng X, Zhu W, Schurig-Briccio LA, Lindert S, Shoen C, Hitchings R, Li J, Wang Y, Baig N, Zhou T, Kim BK, Crick DC, Cynamon M, Mc-Cammon JA, Gennis RB, Oldfield E. Antiinfectives targeting enzymes and the proton motive force. Proc Natl Acad Sci USA. 2015;112(51):E7073–E7082. doi: 10.1073/pnas.1521988112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ajao C, Andersson MA, Teplova VV, Nagy S, Gahmberg CG, Andersson LC, Hautaniemi M, Kakasi B, Roivainen M, Salkinoja-Salonen M. Mitochondrial toxicity of triclosan on mammalian cells. Toxicol Rep. 2015;2:624–637. doi: 10.1016/j.toxrep.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansch C, Quinlan JE, Lawrence GL. Linear free-energy relationship between partition coefficients and the aqueous solubility of organic liquids. J Org Chem. 1968;33(1):347–350. doi: 10.1021/jo01265a071. [DOI] [Google Scholar]
- 16.Fernández-Vidal M, White SH, Ladokhin AS. Membrane Partitioning: “Classical” and “Nonclassical” Hydrophobic Effects. J Mol Biol. 2011;239(1–2):5–14. doi: 10.1007/s00232-010-9321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martel S, Gillerat F, Carosati E, Maiarelli D, Tetko IV, Mannhold R, Carrupt PA. Large, chemically diverse dataset of logP measurements for benchmarking studies. Eur J Pharm Sci. 2013;48(1–2):21–29. doi: 10.1016/j.ejps.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Paloncýová M, DeVane R, Murch B, Berka K, Otyepka M. Amphiphilic Drug-Like Molecules Accumulate in a Membrane below the Head Group Region. J Phys Chem B. 2014;118(4):1030–1039. doi: 10.1021/jp4112052. [DOI] [PubMed] [Google Scholar]
- 19.Fischkoff S, Vanderkooi J. Oxygen diffusion in biological and artificial membranes determined by the fluorochrome pyrene. J Gen Physiol. 1975;65:663–676. doi: 10.1085/jgp.65.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Miller MJ, Joshi MS, Thomas DD, Lancaster JR., Jr Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc Natl Acad Sci USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA. 2001;98:7212–7217. doi: 10.1073/pnas.131128898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Möller M, Botti H, Batthyany C, Rubbo H, Radi R, Denicola A. Direct measurement of nitric oxide and oxygen partitioning into liposomes and low density lipoprotein. J Biol Chem. 2005;280:8850–8854. doi: 10.1074/jbc.M413699200. [DOI] [PubMed] [Google Scholar]
- 23.Marsh D, Dzikovski BG, Livshits VA. Oxygen Profiles in Membranes. Biophys J. 2006;90:L49–L51. doi: 10.1529/biophysj.106.081042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Abdul-Wahid MS, Evanics F, Prosser RS. Dioxygen transmembrane distributions and partitioning thermodynamics in lipid bilayers and micelles. Biochemistry. 2011;50:3975–3983. doi: 10.1021/bi200168n. [DOI] [PubMed] [Google Scholar]
- 25.Salomonsson L, Lee A, Gennis RB, Brzezinski P. A single-amino-acid lid renders a gas-tight compartment within a membrane-bound transporter. Proc Natl Acad Sci USA. 2004;101(32):11617–11621. doi: 10.1073/pnas.0402242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szundi I, Funatogawa C, Fee JA, Soulimane T, Einarsdóttir O. CO impedes superfast O2 binding in ba3 cytochrome oxidase from Thermus thermophilus. Proc Natl Acad Sci USA. 2010;107:21010–21015. doi: 10.1073/pnas.1008603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han H, Hemp J, Pace LA, Ouyang H, Ganesan K, Roh JH, Daldal F, Blanke SR, Gennis RB. Adaptation of aerobic respiration to low O2 environments. Proc Natl Acad Sci USA. 2011;108:14109–14114. doi: 10.1073/pnas.1018958108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahinthichaichan P, Gennis R, Tajkhorshid E. All the O 2 consumed by Thermus thermophilus cytochrome ba3 is delivered to the active site through a long, open hydrophobic tunnel with entrances within the lipid bilayer. Biochemistry. 2016;55(8):1265–1278. doi: 10.1021/acs.biochem.5b01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug–target network. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 30.Adu-Gyamfi E, Digman MA, Gratton E, Stahelin RV. The Ebola virus matrix protein penetrates into the plasma membrane: A key step in viral protein 40 (VP40) oligomerization and viral egress. J Biol Chem. 2013;288:5779–5789. doi: 10.1074/jbc.M112.443960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mim C, Cui H, Gawronski-Salerno JA, Frost A, Lyman E, Voth GA, Unger VM. Structural basis of membrane bending by the N-BAR protein endophilin. Cell. 2012;149(1):137–145. doi: 10.1016/j.cell.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsigelny IF, Sharikov Y, Wrasidlo W, Gonzalez T, Desplats PA, Crews L, Spencer B, Masliah E. Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012;279(6):1000–1013. doi: 10.1111/j.1742-4658.2012.08489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arcario MJ, Tajkhorshid E. Membrane-Induced Structural Rearrangement and Identification of a Novel Membrane Anchor in Talin F2F3. Biophys J. 2014;107(9):2059–2069. doi: 10.1016/j.bpj.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin LM, Edwards MA, Li J, Yip CM, Deber CM. Roles of Hydrophobicity and Charge Distribution of Cationic Antimicrobial Peptides in Peptide-Membrane Interactions. J Biol Chem. 2012;287(10):7738–7745. doi: 10.1074/jbc.M111.303602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miranda-Laferte E, Ewers D, Guzman RE, Jordan N, Schmidt S, Hidalgo P. The N-terminal Domain Tethers the Voltage-gated Calcium Channel β2e-subunit to the Plasma Membrane via Electrostatic and Hydrophobic Interactions. J Biol Chem. 2014;289(15):10387–10398. doi: 10.1074/jbc.M113.507244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janosi L, Li Z, Hancock JF, Gorfe AA. Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc Natl Acad Sci USA. 2012;109(21):8097–8102. doi: 10.1073/pnas.1200773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Jesus AJ, Allen TW. The role of tryptophan side chains in membrane protein anchoring and hydrophobic mismatch. Biochim Biophys Acta Biomembr. 2013;1828(2):864–876. doi: 10.1016/j.bbamem.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 38.Singer S, Nicolson G. The fluid mosaic model of the structure of cell membranes. Science. 1972;173:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 39.Veatch SL, Cicuta P, Sengupta P, Honerkamp-Smith A, Holowka D, Baird B. Critical fluctuations in plasma membrane vesicles. ACS Chem Biol. 2008;3(5):287–293. doi: 10.1021/cb800012x. [DOI] [PubMed] [Google Scholar]
- 40.Hong C, Tieleman DP, Wang Y. Microsecond Molecular Dynamics Simulations of Lipid Mixing. Langmuir. 2014;30(40):11993–12001. doi: 10.1021/la502363b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shan Y, Wang H. The structure and function of cell membranes examined by atomic force microscopy and single-molecule force spectroscopy. Chem Soc Rev. 2015;44(11):3617–3638. doi: 10.1039/C4CS00508B. [DOI] [PubMed] [Google Scholar]
- 42.Prosser RS, Luchette PA, Westerman PW. Using O2 to probe membrane immersion depth by 19F NMR. Proc Natl Acad Sci USA. 2000;97(18):9967–9971. doi: 10.1073/pnas.170295297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazhab-Jafari MT, Marshall CB, Stathopulos PB, Kobashigawa Y, Stambolic V, Kay LE, Inagaki F, Ikura M. Membrane-Dependent Modulation of the mTOR Activator Rheb: NMR Observations of a GTPase Tethered to a Lipid-Bilayer Nanodisc. J Am Chem Soc. 2013;135(9):3367–3370. doi: 10.1021/ja312508w. [DOI] [PubMed] [Google Scholar]
- 44.Maltsev S, Hudson SM, Sahu ID, Liu L, Lorigan GA. Solid-State NMR 31P Paramagnetic Relaxation Enhancement Membrane Protein Immersion Depth Measurements. J Phys Chem B. 2014;118(16):4370–4377. doi: 10.1021/jp500267y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malmberg NJ, Falke JJ. Use of EPR Power Saturation to Analyze the Membrane-Docking Geometries of Peripheral Proteins: Applications to C2 Domains. Annu Rev Biophys Biomol Struct. 2005;34(1):71–90. doi: 10.1146/annurev.biophys.34.040204.144534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osterberg JR, Chon NL, Boo A, Maynard FA, Lin H, Knight JD. Membrane Docking of the Synaptotagmin 7 C2A Domain: Electron Paramagnetic Resonance Measurements Show Contributions from Two Membrane Binding Loops. Biochemistry. 2015;57(37):5684–5695. doi: 10.1021/acs.biochem.5b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfefferkorn CM, Heinrich F, Sodt AJ, Maltsev AS, Pastor RW, Lee JC. Depth of α-Synuclein in a Bilayer Determined by Fluoresence, Neutron Reflectometry, and Computation. Biophys J. 2012;102:613–621. doi: 10.1016/j.bpj.2011.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kyrychenko A, Tobias DJ, Ladokhin AS. Validation of Depth-Dependent Fluorescence Quenching in Membranes by Molecular Dynamics Simulation of Tryptophan Octyl Ester in POPC Bilayer. J Phys Chem B. 2013;117(17):4770–4778. doi: 10.1021/jp310638f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cotterill R. Computer simulation of model lipid membrane dynamics. Biochim Biophys Acta. 1976;433(2):264–270. doi: 10.1016/0005-2736(76)90092-4. [DOI] [Google Scholar]
- 50.van der Ploeg P, Berendsen HJC. Molecular dynamics simulation of a bilayer membrane. J Chem Phys. 1982;76:3271–3276. [Google Scholar]
- 51.Heller H, Schaefer M, Schulten K. Molecular Dynamics Simulation of a Bilayer of 200 Lipids in the Gel and in the Liquid Crystal-Phases. J Phys Chem. 1993;97:8343–8360. [Google Scholar]
- 52.Dror RO, Dirks RM, Grossman J, Xu H, Shaw DE. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu Rev Biophys. 2012;41(1):429–452. doi: 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- 53.Lee EH, Hsin J, Sotomayor M, Comellas G, Schulten K. Discovery Through the Computational Microscope. Structure. 2009;17:1295–1306. doi: 10.1016/j.str.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J Phys Chem B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dickson CJ, Madej BD, Skjevik ÅA, Betz RM, Teigen K, Gould IR, Walker RC. Lipid14: The Amber Lipid Force Field. J Chem Theor Comp. 2014;10(2):865–879. doi: 10.1021/ct4010307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siu SWI, Pluhackova K, Böckmann RA. Optimization of the OPLS-AA Force Field for Long Hydrocarbons. J Chem Theor Comp. 2012;8(4):1459–1470. doi: 10.1021/ct200908r. [DOI] [PubMed] [Google Scholar]
- 57.Botan A, Favela-Rosales F, Fuchs PFJ, Javanainen M, Kanduč M, Kulig W, Lamberg A, Loison C, Lyubartsev A, Miettinen MS, Monticelli L, Määttä J, Ollila OHS, Retegan M, Róg T, Santuz H, Tynkkynen J. Toward Atomistic Resolution Structure of Phosphatidylcholine Headgroup and Glycerol Backbone at Different Ambient Conditions. J Phys Chem B. 2015;119(49):15075–15088. doi: 10.1021/acs.jpcb.5b04878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comp Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 59.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD., Jr CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J Comp Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theor Comp. 2016;12(1):281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 62.Vanommeslaeghe K, MacKerell AD., Jr Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J Chem Inf Model. 2012;52(12):3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yesselman JD, Price DJ, Knight JL, Brooks CL., III MATCH: An atom–typing toolset for molecular mechanics force fields. J Comp Chem. 2012;33:189–202. doi: 10.1002/jcc.21963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mayne CG, Saam J, Schulten K, Tajkhorshid E, Gumbart JC. Rapid parameterization of small molecules using the Force Field Toolkit. J Comp Chem. 2013;34:2757–2770. doi: 10.1002/jcc.23422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ingólfsson HI, Melo MN, van Eerden FJ, Arnarez C, López CA, Wassenaar TA, Periole X, de Vries AH, Tieleman DP, Marrink SJ. Lipid Organization of the Plasma Membrane. J Am Chem Soc. 2014;136(14):14554–14559. doi: 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- 66.Shaw DE, Grossman JP, Bank JA, Batson B, Butts JA, Chao JC, Deneroff MM, Dror RO, Even A, Fenton CH, Forte A, Gagliardo J, Gill G, Greskamp B, Ho CR, Ierardi DJ, Iserovich L, Kuskin JS, Larson RH, Layman T, Lee L-S, Lerer AK, Li C, Killebrew D, Mackenzie KM, Mok SY-H, Moraes MA, Mueller R, Nociolo LJ, Peticolas JL, Quan T, Ramot D, Salmon JK, Scarpazza DP, Ben Schafer U, Siddique N, Snyder CW, Spengler J, Tang PTP, Theobald M, Toma H, Towles B, Vitale B, Wang SC, Young C. Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. IEEE Press; 2014. Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer; pp. 41–53. [Google Scholar]
- 67.Marrink SJ, Tieleman DP. Perspective on the MARTINI model. Chem Soc Rev. 2013;42:6801–6822. doi: 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- 68.Shinoda W, DeVane R, Klein ML. Zwitterionic Lipid Assemblies: Molecular Dynamics Studies of Monolayers, Bilayers, and Vesicles Using a New Coarse Grain Force Field. J Phys Chem B. 2010;114(20):6836–6849. doi: 10.1021/jp9107206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grossfield A. Implicit modeling of membranes. Curr Top Membr. 2008;60:131–157. [Google Scholar]
- 70.Vermaas JV, Baylon JL, Arcario MJ, Muller MP, Wu Z, Pogorelov TV, Tajkhorshid E. Efficient Exploration of Membrane-Associated Phenomena at Atomic Resolution. J Membr Biol. 2015;248(3):563–582. doi: 10.1007/s00232-015-9806-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ohkubo YZ, Pogorelov TV, Arcario MJ, Christensen GA, Tajkhorshid E. Accelerating Membrane Insertion of Peripheral Proteins with a Novel Membrane Mimetic Model. Biophys J. 2012;102:2130–2139. doi: 10.1016/j.bpj.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vermaas JV, Tajkhorshid E. A Microscopic View of Phospholipid Insertion into Biological Membranes. J Phys Chem B. 2014;118:1754–1764. doi: 10.1021/jp409854w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pogorelov TV, Vermaas JV, Arcario MJ, Tajkhorshid E. Partitioning of Amino Acids into a Model Membrane: Capturing the Interface. J Phys Chem B. 2014;118:1481–1492. doi: 10.1021/jp4089113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella Sampling. J Comp Phys. 1977;23:187–199. [Google Scholar]
- 75.Jiang W, Luo Y, Maragliano L, Roux B. Calculation of Free Energy Landscape in Multi-Dimensions with Hamiltonian-Exchange Umbrella Sampling on Petascale Supercomputer. J Chem Theor Comp. 2012;8(11):4672–4680. doi: 10.1021/ct300468g. [DOI] [PubMed] [Google Scholar]
- 76.Barducci A, Bussi G, Parrinello M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys Rev Lett. 2008;100:020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 77.Roux B. The calculation of the potential of mean force using computer simulations. Comput Phys Commun. 1995;91:275–282. [Google Scholar]
- 78.Fiorin G, Klein ML, Hénin J. Using collective variables to drive molecular dynamics simulations. Mol Phys. 2013;111(22–23):3345–3362. doi: 10.1080/00268976.2013.813594. [DOI] [Google Scholar]
- 79.Hénin J, Forin G, Chipot C, Klein ML. Exploring multidimensional free energy landscapes using time-dependent biases on collective variables. J Chem Theor Comp. 2010;6(1):35–47. doi: 10.1021/ct9004432. [DOI] [PubMed] [Google Scholar]
- 80.Wang Y, Cohen J, Boron WF, Schulten K, Tajkhorshid E. Exploring Gas Permeability of Cellular Membranes and Membrane Channels with Molecular Dynamics. J Struct Biol. 2007;157:534–544. doi: 10.1016/j.jsb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 81.Ruscio JZ, Kumar D, Shukla M, Prisant MG, Murali T, Onufreiv AV. Atomic level computational identification of ligand migration pathways between solvent and binding site in myoglobin. Proc Natl Acad Sci USA. 2008;105:9204–9209. doi: 10.1073/pnas.0710825105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hub JS, Winkler FK, Merrick M, de Groot BL. Potentials of Mean Force and Permeabilities for Carbon Dioxide, Ammonia, and Water Flux across a Rhesus Protein Channel and Lipid Membranes. J Am Chem Soc. 2010;132(38):13251–13263. doi: 10.1021/ja102133x. [DOI] [PubMed] [Google Scholar]
- 83.Wang PH, Best RB, Blumberger J. Multiscale Simulation Reveals Multiple Pathways for H2 and O2 transport in a [NiFe]-Hydrogenase. J Am Chem Soc. 2011;133:3548–3556. doi: 10.1021/ja109712q. [DOI] [PubMed] [Google Scholar]
- 84.Wang PH, Bruschi M, Gioia LD, Blumberger J. Uncovering a Dynamically Formed Substrate Access Tunnel in Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase. J Am Chem Soc. 2013;135:9493–9502. doi: 10.1021/ja403110s. [DOI] [PubMed] [Google Scholar]
- 85.Gora A, Brezovsky J, Damborsky J. Gates of enzymes. Chem Rev. 2013;113:5871–5923. doi: 10.1021/cr300384w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shaikh SA, Li J, Enkavi G, Wen PC, Huang Z, Tajkhorshid E. Visualizing functional motions of membrane transporters with molecular dynamics simulations. Biochemistry. 2013;52(4):569–587. doi: 10.1021/bi301086x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brannigan G, LeBard DN, Hénin J, Eckenhoff RG, Klein ML. Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc Natl Acad Sci USA. 2010;107:14122–14127. doi: 10.1073/pnas.1008534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murali S, Wallner B, Trudell JR, Bertaccini E, Lindahl E. Microsecond simulations indicate that ethanol binds between subunits and could stabilize an open-state model of a glycine receptor. Biophys J. 2011;100:1642–1650. doi: 10.1016/j.bpj.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arcario MJ, Mayne CG, Tajkhorshid E. Atomistic Models of General Anesthetics for Use in in silico Biological Studies. J Phys Chem B. 2014;118:12075–12086. doi: 10.1021/jp502716m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cohen J, Arkhipov A, Braun R, Schulten K. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys J. 2006;91:1844–1857. doi: 10.1529/biophysj.106.085746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saam J, Ivanov I, Walther M, Holzhutter H, Kuhn H. Molecular dioxygen enters the active site of 12/15 lipoxygenase via dynamic oxygen access channels. Proc Natl Acad Sci USA. 2007;104:13319–13324. doi: 10.1073/pnas.0702401104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saam J, Rosini E, Molla G, Schulten K, Pollegioni L, Ghisla S. O2-reactivity of flavoproteins: Dynamic access of dioxygen to the active site and role of a H+ relay system in D-amino acid oxidase. J Biol Chem. 2010;285:24439–24446. doi: 10.1074/jbc.M110.131193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y, Tajkhorshid E. Nitric oxide conduction by the brain aquaporin AQP4. Proteins: Struct, Func, Bioinf. 2010;78:661–670. doi: 10.1002/prot.22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y, Shaikh SA, Tajkhorshid E. Exploring transmembrane diffusion pathways with molecular dynamics. Physiology. 2010;25:142–154. doi: 10.1152/physiol.00046.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hub J, de Groot B. Mechanism of selectivity in aquaporins and aquaglyceroporins. Proc Natl Acad Sci USA. 2008;105:1198–203. doi: 10.1073/pnas.0707662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wennberg CL, van der Spoel D, Hub JS. Large Influence of Cholesterol on Solute Partitioning into Lipid Membranes. J Am Chem Soc. 2012;134:5351–5361. doi: 10.1021/ja211929h. [DOI] [PubMed] [Google Scholar]
- 97.Zocher F, van der Spoel D, Pohl P, Hub JS. Local partition coefficients govern solute permeability of cholesterol-containing membranes. Biophys J. 2013;105:2760–2770. doi: 10.1016/j.bpj.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jedlovszky P, Mezei M. Calculations of the Free Energy Profiles of H 2O, O2, CO, NO and CHCl3 in a Lipid Bilayler with a Cavity Insertion Variant of the Widom Method. J Am Chem Soc. 2000;122:5125–5131. [Google Scholar]
- 99.Jämbeck JPM, Lyubartsev AP. Implicit inclusion of atomic polarization in modeling of partitioning between water and lipid bilayer. Phys Chem Chem Phys. 2013;15:4677–4686. doi: 10.1039/c3cp44472d. [DOI] [PubMed] [Google Scholar]
- 100.Ferguson-Miller S, Babcock GT. Heme/Copper Terminal Oxidases. Chem Rev. 1996;96:2889. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 101.Pereira M, Santana M, Teixeira M. A novel scenario for the evolution of haem-copper oxygen reductases. Biochim Biophys Acta – Bioener. 2001;1505:185–208. doi: 10.1016/s0005-2728(01)00169-4. [DOI] [PubMed] [Google Scholar]
- 102.Hosler JP, Ferguson-Miller S, Mills DA. Energy Transduction: Proton Transfer Through the Respiratory Complexes. Annu Rev Biochem. 2006;75:165–187. doi: 10.1146/annurev.biochem.75.062003.101730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brzezinski P, Gennis RB. Cytochrome c oxidase: exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hemp J, Gennis RB. Diversity of the Heme-Copper superfamily in Archaea: Insights from Genomics and Structural Modeling. Results Probl Cell Differ. 2008;45:1–31. doi: 10.1007/400_2007_046. [DOI] [PubMed] [Google Scholar]
- 105.Rich PR, Marechal A. Functions of the hydrophilic channels in protonmotive cytochrome c oxidase. J R Soc Interface. 2013;10:20130183. doi: 10.1098/rsif.2013.0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yoshikawa S, Shimada A. Reaction Mechanism of Cytochrome c Oxidase. Chem Rev. 2015;115:1936–1989. doi: 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- 107.Wikström M, Sharma V, Kaila VR, Hosler JP, Hummer G. New Perspective on Proton Pumping in Cellular Respiration. Chem Rev. 2015;115:2196–2221. doi: 10.1021/cr500448t. [DOI] [PubMed] [Google Scholar]
- 108.Wraight CA. Chance and design – Proton transfer in water, channels and bioenergetic proteins. Biochim Biophys Acta – Bioener. 2006;1757:886–912. doi: 10.1016/j.bbabio.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 109.Lee H, Reimann J, Huang Y, Ådelroth P. Functional proton transfer pathways in the heme-copper oxidase superfamily. Biochim Biophys Acta – Bioener. 2012;1817:537–544. doi: 10.1016/j.bbabio.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 110.Babcock G, Wikström M. Oxygen activation and the conservation of energy in cell respiration. Nature. 1992;256:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 111.Ducluzeau AL, Schoepp-Cothenet B, van Lis R, Baymann F, Russell MJ, Nitschke W. The evolution of respiratory O2/NO reductases: an out-of-the-phylogenetic-box perspective. J R Soc Interface. 2014;11:20140196. doi: 10.1098/rsif.2014.0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soulimane T, Buse G, Bourenkov GP, Bartunik HD, Huber R, Than ME. Structure and mechanism of the abberrant ba3-cytochrome c oxidase from Thermus thermophilus. EMBO J. 2000;19:1766–1776. doi: 10.1093/emboj/19.8.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chang HY, Hemp J, Chen Y, Fee JA, Gennis RB. The cytochrome ba3 oxygen reductase from Thermus thermophilus uses a single input channel for proton delivery to the active site and for proton pumping. Proc Natl Acad Sci USA. 2009;106:16169–16173. doi: 10.1073/pnas.0905264106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.von Ballmoos C, Gennis RB, Ädelroth P, Brzezinski P. Kinetic design of the respiratory oxidases. Proc Natl Acad Sci USA. 2011;108:11057–11062. doi: 10.1073/pnas.1104103108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tiefenbrunn T, Liu W, Chen Y, Katritch V, Stout CD, Fee JA, Cherezov V. High Resolution Structure of the ba3 Cytochrome c Oxidase from Thermus thermophilus in a Lipidic Environment. PLoS One. 2011;6:e22348. doi: 10.1371/journal.pone.0022348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chang HY, Choi SK, Vakkasoglu AS, Chen Y, Hemp J, Fee JA, Gennis RB. Exploring the proton pumps and exit pathway for pumped protons in cytochrome ba3 oxygen reductase from Thermus thermophilus. Proc Natl Acad Sci USA. 2012;109:5259–5264. doi: 10.1073/pnas.1107345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Luna VM, Chen Y, Fee JA, Stout CD. Crystallographic Studies of Xe and Kr Binding within the Large Internal Cavity of Cytochrome ba3 from Thermus thermophilus: Structural Analysis and Role of Oxygen Transport Channels in the Heme-Cu Oxidases. Biochemistry. 2008;47:4657–4665. doi: 10.1021/bi800045y. [DOI] [PubMed] [Google Scholar]
- 118.Luna VM, Fee JA, Deniz AA, Stout CD. Mobility of Xe Atoms within the Oxygen Diffusion Channel of Cytochrome ba3 Oxidase. Biochemistry. 2012;51:4669–4676. doi: 10.1021/bi3003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Einarsdóttir O, Funatogawa C, Soulimane T, Szundi I. Kinetic studies of the reactions of O2 and NO with reduced Thermus thermophilus ba3 and bovine aa3 using photolabile carriers. Biochim Biophys Acta – Bioener. 2012;1817:672–679. doi: 10.1016/j.bbabio.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hunsicker-Wang L, Pacoma R, Chen Y, Fee J, Stout C. A novel cryoprotection scheme for enhancing the diffraction of crystals of recombinants cytochrome ba3 oxidase from Thermus thermophilus. Acta Cryst D. 2005;61:340–343. doi: 10.1107/S0907444904033906. [DOI] [PubMed] [Google Scholar]
- 121.Moskovitz Y, Yang H. Modelling of noble anaesthetic gases and high hydrostatic pressure effects in lipid bilayers. Soft Mat. 2015;11:2125–2138. doi: 10.1039/c4sm02667e. [DOI] [PubMed] [Google Scholar]
- 122.Chen J, Chen L, Wang Y, Wang X, Zeng S. Exploring the Effects of Lipid Bilayer Induced by Noble Gases via Molecular Dynamics Simulations. Sci Rep. 2015;5:17235. doi: 10.1038/srep17235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Booker RD, Sum AK. Biophysical changes induced by xenon on phospholipid bilayers. Biochim Biophys Acta Biomembr. 2013;1828:1347–1356. doi: 10.1016/j.bbamem.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 124.Yamamoto E, Akimoto T, Shimizu H, Hirano Y, Yasui M, Yasuoka K. Diffusive nature of xenon anesthetic changes properties of a lipid bilayer: Molecular dynamics simulations. J Phys Chem B. 2012;116:8989–8995. doi: 10.1021/jp303330c. [DOI] [PubMed] [Google Scholar]
- 125.Stimson LM, Vattulainen I, Róg T, Karttunen M. Exploring the effect of xenon on biomembranes. Cell Mol Biol Letters. 2005;10:563–569. [PubMed] [Google Scholar]
- 126.Harless E, von Bibra E. Erlangen. Die ergebnisse der versuche über die wirkung des schwefeläthers. [Google Scholar]
- 127.Meyer HH. Zur theorie der Alkoholnarkose. Arch Exp Pathol Pharmakol. 1899;42:109–118. [Google Scholar]
- 128.Seeman P. The membrane actions of anesthetics and tranquilizers. Pharmacol Rev. 1972;24:583–655. [PubMed] [Google Scholar]
- 129.Fransson T, Jakobsson S, Johansson P, Kullberg C, Lind J, Vallin A. Magnetic cues trigger extensive refuelling. Nature. 2001;414:35–36. doi: 10.1038/35102115. [DOI] [PubMed] [Google Scholar]
- 130.Harris BD, Moody EJ, Basile AS, Skolnick P. Volatile anesthetics bidirectionally and stereospecifically modulate ligand binding to GABA receptors. Eur J Pharmacol. 1994;267:269–274. doi: 10.1016/0922-4106(94)90150-3. [DOI] [PubMed] [Google Scholar]
- 131.Dickinson R, Franks NP, Lieb WR. Can the stereoselective effects of the anesthetic isoflurane be accounted for by lipid solubility? Biophys J. 1994;66:2019–2023. doi: 10.1016/S0006-3495(94)80994-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pringle MJ, Brown KB, Miller KW. Can the lipid theory of anesthesia account for the cutoff in anesthetic potency in homologous series of alcohols? Mol Pharmacol. 1981;19:49–55. [PubMed] [Google Scholar]
- 133.Franks NP, Lieb WR. Partitioning of long-chain alcohols into lipid bilayers: Implications for mechanisms of general anesthesia. Proc Natl Acad Sci USA. 1986;83:5116–5120. doi: 10.1073/pnas.83.14.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dodson BA, Braswell LM, Miller KW. Barbiturates bind to an allosteric regulatory site on nicotinic acetylcholine receptor-rich membranes. Mol Pharmacol. 1987;32:119–126. [PubMed] [Google Scholar]
- 135.Pratt MB, Husain SS, Miller KW, Cohen JB. Identification of sites of incorporation in the nicotinic acetylcholine receptor of a photoactivatable general anesthetic. J Biol Chem. 2000;275:29441–29451. doi: 10.1074/jbc.M004710200. [DOI] [PubMed] [Google Scholar]
- 136.Husain SS, Ziebell MR, Ruesch D, Hong F, Arevalo E, Kosterlitz JA, Olsen RW, Forman SA, Cohen JB, Miller KW. 2-(3-methyl-3H-diaziren-3-yl)ethyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate: A derivative of the stereoselective general anesthetic etomidate for photolabeling ligand-gated ion channels. J Med Chem. 2003;46:1257–1265. doi: 10.1021/jm020465v. [DOI] [PubMed] [Google Scholar]
- 137.Ghosh B, Satyshur KA, Czajkowski C. Propofol binding to the resting state of the Gloeobacter violaceus ligand–gated ion channel (GLIC) induces structural changes in the inter– and intrasubunit transmembrane domain (TMD) cavities. J Biol Chem. 2013;288:17420–17431. doi: 10.1074/jbc.M113.464040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M, Corringer PJ. X-ray structures of general anesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- 139.Willenbring D, Liu LT, Mowrey D, Xu Y, Tang P. Isoflurane alters the structure and dynamics of GLIC. Biophys J. 2011;101:1905–1912. doi: 10.1016/j.bpj.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pan J, Chen Q, Willenbring D, Yoshida K, Tillman T, Kashlan OB, Cohen A, Kong XP, Xu Y, Tang P. Structure of the pentameric ligand–gated ion channel ELIC cocrystallized with its competitive antagonist acetylcholine. Nat Commun. 2012;3:1–8. doi: 10.1038/ncomms1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.LeBard DN, Hénin J, Eckenhoff RG, Klein ML, Brannigan G. General anesthetics predicted to block the GLIC pore with micromolar affinity. PLoS Comput Biol. 2012;8:e1002532. doi: 10.1371/journal.pcbi.1002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mowrey D, Cheng MH, Liu LT, Willenbring D, Lu X, Wymore T, Xu Y, Tang P. Asymmetric ligand binding facilitates conformational transitions in pentameric ligand–gated ion channels. J Am Chem Soc. 2013;135:2172–2180. doi: 10.1021/ja307275v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hemmings HC, Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 144.Franks NP. General anaesthesia: From molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–386. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- 145.Pohorille A, Wilson MA, Chipot C. Interactions of alcohols and anesthetics with the water-hexane interface: A molecular dynamics study. Prog Colloid Polym Sci. 1997;103:29–40. doi: 10.1021/jp961513o. [DOI] [PubMed] [Google Scholar]
- 146.Hénin J, Brannigan G, Dailey WP, Eckenhoff R, Klein ML. An atomistic model for simulations of the general anesthetic isoflurane. J Phys Chem B. 2010;114:604–612. doi: 10.1021/jp9088035. [DOI] [PubMed] [Google Scholar]
- 147.Paloncýová M, Fabre G, DeVane RH, Trouillas P, Berka K, Otyepka M. Benchmarking of Force Fields for Molecule? Membrane Interactions. J Chem Theor Comp. 2014;10(9):4143–4151. doi: 10.1021/ct500419b. [DOI] [PubMed] [Google Scholar]
- 148.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 149.Robertson MJ, Tirado-Rives J, Jorgensen WL. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J Chem Theor Comp. 2015;11:3499–3509. doi: 10.1021/acs.jctc.5b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Maciejewski A, Pasenkiewicz-Gierula M, Cramariuc O, Vattulainen I, Ròg T. Refined OPLS All-Atom Force Field for Saturated Phosphatidylcholine Bilayers at Full Hydration. J Phys Chem B. 2014;118:4571–4581. doi: 10.1021/jp5016627. [DOI] [PubMed] [Google Scholar]
- 151.Kulig W, Pasenkiewicz-Gierula M, Ròg T. Cis and trans unsaturated phosphatidylcholine bilayers: A molecular dynamics simulation study. Chem Phys of Lipids. 2015;195:12–20. doi: 10.1016/j.chemphyslip.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 152.Feller SE, MacKerell AD., Jr An Improved Empirical Potential Energy Function for Molecular Simulations of Phospholipids. J Phys Chem B. 2000;104(31):7510–7515. [Google Scholar]
- 153.Klauda JB, Brooks BR, MacKerell AD, Jr, Venable RM, Pastor RW. An ab Initio Study on the Torsional Surface of Alkanes and Its Effect on Molecular Simulations of Alkanes and a DPPC Bilayer. J Phys Chem B. 2005;109:5300–5311. doi: 10.1021/jp0468096. [DOI] [PubMed] [Google Scholar]
- 154.Klauda JB, Pastor RW, Brooks BR. Adjacent Gauche Stabilization in Linear Alkanes: Implications for Polymer Models and Conformational Analysis. J Phys Chem B. 2005;109:15684–15686. doi: 10.1021/jp0527608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dickson CJ, Rosso L, Betz RM, Walker RC, Gould IR. GAFFlipid: a General Amber Force Field for the accurate molecular dynamics simulation of phospholipid. Soft Mat. 2012;8(37):9617. doi: 10.1039/c2sm26007g. [DOI] [Google Scholar]
- 156.Vanommeslaeghe K, Raman EP, MacKerell AD., Jr Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J Chem Inf Model. 2012;52(12):3155–3168. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cheng YC, Silbey RJ. Coherence in the B800 Ring of Purple Bacteria LH2. Phys Rev Lett. 2006;96:028103. doi: 10.1103/PhysRevLett.96.028103. [DOI] [PubMed] [Google Scholar]
- 158.Liu Z, Xu Y, Saladino AC, Wymore T, Tang P. Parameterization of 2-bromo-2-chloro-1,1,1-trifluoroethane (halothane) and hexfluoroethane for nonbonded interactions. J Phys Chem A. 2004;108:781–786. [Google Scholar]
- 159.Pohorille A, Cieplak P, Wilson MA. Interactions of anesthetics with the membrane–water interface. Chem Phys. 1996;204:337–345. doi: 10.1016/0301-0104(95)00292-8. [DOI] [PubMed] [Google Scholar]
- 160.Pohorille A, Wilson MA, New MH, Chipot C. Concentrations of anesthetics across the water–membrane interface; the Meyer–Overton hypothesis revisited. Toxicol Lett. 1998;100:421–430. doi: 10.1016/s0378-4274(98)00216-1. [DOI] [PubMed] [Google Scholar]
- 161.Tu K, Tarek M, Klein ML, Scarf D. Effects of anesthetics on the structure of a phospholipid bilayer: Molecular dynamics investigation of halothane in the hydrated liquid crystal phase of dipalmitoylphosphatidylcholine. Biophys J. 1998;75:2123–2134. doi: 10.1016/S0006-3495(98)77655-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Koubi L, Tarek M, Klein ML, Scharf D. Distribution of halothane in a dipalmitoylphosphatidylcholine bilayer from molecular dynamics calculations. Biophys J. 2000;78:800–811. doi: 10.1016/S0006-3495(00)76637-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vemparala S, Saiz L, Eckenhoff RG, Klein ML. Partitioning of Anesthetics into a Lipid Bilayer and their Interaction with Membrane-Bound Peptide Bundles. Biophys J. 2006;91:2815–2825. doi: 10.1529/biophysj.106.085324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Murali S, Howard RJ, Broemstrup T, Bertaccini EJ, Harris RA, Trudell JR, Lindahl E. Molecular Mechanism for the dual alcohol modulation of Cys–loop receptors. PLoS Comput Biol. 2012;8:e1002710. doi: 10.1371/journal.pcbi.1002710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gray E, Karslake J, Machta BB, Veatch SL. Liquid General Anesthetics Lower Critical Temperatures in Plasma Membrane Vesicles. Biophys J. 2013;105(12):2751–2759. doi: 10.1016/j.bpj.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jerabek H, Pabst G, Rappolt M, Stockner T. Membrane–mediated effect on ion channels induced by the anesthetic drug ketamine. J Am Chem Soc. 2010;132:7990–7997. doi: 10.1021/ja910843d. [DOI] [PubMed] [Google Scholar]
- 167.Weng Y, Yang L, Corringer PJ, Sonner JM. Anesthetic sensitivity of the Gloeobacter voilaceus proton-gated ion channel. Anesth Analg. 2010;110:59–63. doi: 10.1213/ANE.0b013e3181c4bc69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Spurny R, Billen B, Howard RJ, Brams M, Debaveye S, Price KL, Weston DA, Strelkov SV, Tygat J, Bertrand S, Bertrand D, Lummis SC, Ulens C. Multisite binding of a general anesthetic to the prokaryotic pentameric Erwinia chrysanthemi ligand–gated ion channel (ELIC) J Biol Chem. 2013;288:8355–8364. doi: 10.1074/jbc.M112.424507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tibbs GR, Rowley TJ, Sanford RL, Herold KF, Proekt A, Hemmings HC, Jr, Andersen OS, Goldstein PA, Flood PD. HCN1 channels as targets for anesthetic and nonanesthetic propofol analogs in the amelioration of mechanical and thermal hyperalgesia in a mouse model of neuropathic pain. J Pharmacol Exp Ther. 2013;345:363–373. doi: 10.1124/jpet.113.203620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resoultion. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 171.Tasneem A, Iyer LM, Jakobsson E, Aravind L. Identification of the prokaryotic ligand-gated ion channels and their implications for the mechanisms and origins of animal Cys–loop ion channels. Gen Biol. 2005;6:R4. doi: 10.1186/gb-2004-6-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 173.Hilf RJC, Bertozzi C, Zimmermann I, Reiter A, Trauner D, Dutzler R. Structural basis of open channel block in a prokaryotic pentameric ligand-gated ion channel. Nat Struct Mol Biol. 2010;17:1330–1336. doi: 10.1038/nsmb.1933. [DOI] [PubMed] [Google Scholar]
- 174.Prevost MS, Sauguet L, Nury H, Van Renterghem C, Huon C, Poitevin F, Baaden M, Delarue M, Corringer PJ. A locally closed conformation of a bacterial pentameric proton–gated ion channel. Nat Struct Mol Biol. 2012;19:642–649. doi: 10.1038/nsmb.2307. [DOI] [PubMed] [Google Scholar]
- 175.Pan J, Chen Q, Willenbring D, Mowrey D, Kong XP, Cohen A, Divito CB, Xu Y, Tang P. Structure of the pentameric ligand–gated ion channel GLIC bound with anesthetic ketamine. Structure. 2012;20:1463–1469. doi: 10.1016/j.str.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hilf RJC, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 177.Gonzalez–Gutierrez G, Lukk T, Agarwal V, Papke D, Nair SK, Grosman C. Mutations that stabilize the open state of the Erwinia chrisanthemi ligand–gated ion channel fail to change the conformation of the pore domain in crystals. Proc Natl Acad Sci USA. 2012;109:6331–6336. doi: 10.1073/pnas.1119268109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hibbs RE, Gouaux E. Principles of activation and permeation in an anion–selective Cys–loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Beato M. Gene regulation by steroid hormones. Cell. 1989;56:335–344. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 180.Levin ER. Minireview: Extranuclear Steroid Receptors: Roles in Modulation of Cell Functions. Mol Endocr. 2011;25:377–384. doi: 10.1210/me.2010-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81(3):1269–1304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 182.Olefsky JM. Nuclear receptor minireview series. J Biol Chem. 2001;276(40):36863–36864. doi: 10.1074/jbc.R100047200. [DOI] [PubMed] [Google Scholar]
- 183.Hammes SR, Levin ER. Minireview: Recent advances in extranuclear steroid receptor actions. Endocrinology. 2011;152:4489–4495. doi: 10.1210/en.2011-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Stanisic V, Lonard DM, O’Malley BW. Modulation of steroid hormone receptor activity. Mol Endocr. 2010;181:153–176. doi: 10.1016/S0079-6123(08)81009-6. [DOI] [PubMed] [Google Scholar]
- 185.Yeagle PL. Cholesterol and the cell membrane. Biochim Biophys Acta Rev Biomembr. 1985;822(3):267–287. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 186.Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine Rev. 2011;32(1):81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, Gunsalus IC, Nebert DW. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6(1):1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 188.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105(6):2253–77. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 189.Sakaguchi M, Mihara K, Sato R. A short amino-terminal segment of microsomal cytochrome P-450 functions both as an insertion signal and as a stop-transfer sequence. EMBO J. 1987;6(8):2425–2431. doi: 10.1002/j.1460-2075.1987.tb02521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Black SD. Membrane topology of the mammalian P450 cytochromes. FASEB J. 1992;6(2):680–685. doi: 10.1096/fasebj.6.2.1537456. [DOI] [PubMed] [Google Scholar]
- 191.Thomas JL, Myers RP, Strickler RC. Human placental 3β-hydroxy-5-ene-steroid dehydrogenase and steroid 5→ 4-ene-isomerase: Purification from mitochondria and kinetic profiles, biophysical characterization of the purified mitochondrial and microsomal enzymes. J Steroid Biochem. 1989;33:209–217. doi: 10.1016/0022-4731(89)90296-3. [DOI] [PubMed] [Google Scholar]
- 192.Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M. Multiple actions of steroid hormones–a focus on rapid, nongenomic effects. Pharmacol Rev. 2000;52:513–556. [PubMed] [Google Scholar]
- 193.Lösel RM, Falkenstein E, Feuring M, Schultz A, Tillmann HC, Rossol-Haseroth K, Wehling M. Nongenomic steroid action: controversies, questions, and answers. Physiol Rev. 2003;83:965–1016. doi: 10.1152/physrev.00003.2003. [DOI] [PubMed] [Google Scholar]
- 194.Nievas GAF, Barrantes FJ, Antollini SS. Conformation–sensitive steroid and fatty acid sites in the transmembrane domain of the nicotinic acetylcholine receptor. Biochemistry. 2007;46:3503–3512. doi: 10.1021/bi061388z. [DOI] [PubMed] [Google Scholar]
- 195.Jones KT, Zhen J, Reith ME. Importance of cholesterol in dopamine transporter function. J Neurochem. 2012;123(5):700–715. doi: 10.1111/jnc.12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Guengerich PF. Cytochrome P450 and Chemical Toxicology. Chem Res Toxicol. 2008;21(1):70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 197.Guengerich PF. Cytochrome P450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 198.Hofsáß C, Lindahl E, Edholm O. Molecular Dynamics Simulations of Phospholipid Bilayers with Cholestrol. Biophys J. 2003;84(4):2192–2206. doi: 10.1016/S0006-3495(03)75025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Khelashvili G, Pabst G, Harries D. Cholesterol Orientation and Tilt Modulus in DMPC Bilayers. J Phys Chem B. 2010;114(22):7524–7534. doi: 10.1021/jp101889k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 2001;58:737–747. doi: 10.1007/PL00000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Williams PA, Cosme J, Ward A, Angove HC, Matak Vinković D, Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424(6947):464–8. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 202.Schleinkofer K, Sudarko P, Winn J, Lüdemann SK, Wade RC. Do mammalian cytochrome P450s show multiple ligand access pathways and ligand channelling? EMBO Reports. 2005;6(6):584–9. doi: 10.1038/sj.embor.7400420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. Characterizing the Membrane-Bound State of Cytochrome P450 3A4: Structure, Depth of Insertion, and Orientation. J Am Chem Soc. 2013;135(23):8542–8551. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lin JH, Perryman AL, Schames JR, McCammon JA. Computational Drug Design Accommodating Receptor Flexibility: The Relaxed Complex Scheme. J Am Chem Soc. 2002;124(20):5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- 205.Amaro RE, Baron R, McCammon JA. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J Comp-Aided Mol Design. 2008;22(9):693–705. doi: 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science. 2009;323(5922):1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Prot Sci. 2014;23(1):34–36. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the Multidrug Transporter EmrD from Escherichia coli. Science. 2006;312(5774):741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yacoub TJ, Reddy AS, Szleifer I. Structural Effects and Translocation of Doxorubicin in a DPPC/Chol Bilayer: The Role of Cholesterol. Biophys J. 2011;101(2):378–385. doi: 10.1016/j.bpj.2011.06.015. http://dx.doi.org/10.1016/j.bpj.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sharom FJ. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front Oncol. 2014;4:1–19. doi: 10.3389/fonc.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Higgins CF, Gottesman MM. Is the multidrug transporter a flippase? Trends Biochem Sci. 1992;17(1):18–21. doi: 10.1016/0968-0004(92)90419-a. [DOI] [PubMed] [Google Scholar]
- 212.Doerrler WT, Gibbons HS, Raetz CRH. MsbA-dependent Translocation of Lipids across the Inner Membrane of Escherichia coli. J Biol Chem. 2004;279(43):45102–45109. doi: 10.1074/jbc.M408106200. [DOI] [PubMed] [Google Scholar]
- 213.Doerrler WT. Lipid trafficking to the outer membrane of Gram-negative bacteria. Mol Microbiol. 2006;60(3):542–552. doi: 10.1111/j.1365-2958.2006.05130.x. [DOI] [PubMed] [Google Scholar]
- 214.Siarheyeva A, Lopez JJ, Glaubitz C. Localization of Multidrug Transporter Substrates within Model Membranes? Biochemistry. 2006;45(19):6203–6211. doi: 10.1021/bi0524870. [DOI] [PubMed] [Google Scholar]
- 215.Homolya L, Holló Z, Germann UA, Pastan I, Gottesman MM, Sarkadi B. Fluorescent cellular indicators are extruded by the multidrug resistance protein. J Biol Chem. 1993;268(29):21493–21496. [PubMed] [Google Scholar]
- 216.Omote H, Al-Shawi MK. Interaction of Transported Drugs with the Lipid Bilayer and P-Glycoprotein through a Solvation Exchange Mechanism. Biophys J. 2006;90(11):4046–4059. doi: 10.1529/biophysj.105.077743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wen PC, Verhalen B, Wilkens S, Mchaourab HS, Tajkhorshid E. On the origin of large flexibility of P-glycoprotein in the inward-facing state. J Biol Chem. 2013;288(26):19211–19220. doi: 10.1074/jbc.M113.450114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct. 2004;33:269–295. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 219.Lopes PEM, Huang J, Shim J, Luo Y, Li H, Roux B, MacKerell AD., Jr Polarizable Force Field for Peptides and Proteins Based on the Classical Drude Oscillator. J Chem Theor Comp. 2013;9(12):5430–5449. doi: 10.1021/ct400781b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chowdhary J, Harder E, Lopes PEM, Huang L, MacKerell AD, Jr, Roux B. A Polarizable Force Field of Dipalmitoylphosphatidylcholine Based on the Classical Drude Model for Molecular Dynamics Simulations of Lipids. J Phys Chem B. 2013;117(31):9142–9160. doi: 10.1021/jp402860e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Shi Y, Xia Z, Zhang J, Best R, Wu C, Ponder JW, Ren P. Polarizable Atomic Multipole-Based AMOEBA Force Field for Proteins. J Chem Theor Comp. 2013;9(9):4046–4063. doi: 10.1021/ct4003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.van Meer G, de Kroon AIPM. Lipid map of the mammalian cell. J Cell Sci. 2011;124(Pt 1):5–8. doi: 10.1242/jcs.071233. [DOI] [PubMed] [Google Scholar]
- 223.Wu E, Cheng X, Jo S, Rui H, Song K, Dávila-Contreras E, Qi Y, Lee J, Monje-Galvan V, Venable R, Klauda J, Im W. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comp Chem. 2014;35(27):1997–2004. doi: 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Khakbaz P, Klauda JB. Probing the importance of lipid diversity in cell membranes via molecular simulation. Chem Phys of Lipids. 2015;192:12–22. doi: 10.1016/j.chemphyslip.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 225.Setzler J, Seith C, Brieg M, Wenzel W. SLIM: An improved generalized Born implicit membrane model. J Comp Chem. 2014;35:2027–2039. doi: 10.1002/jcc.23717. [DOI] [PubMed] [Google Scholar]
- 226.Kim BL, Schafer NP, Wolynes PG. Predictive energy landscapes for folding α-helical transmembrane proteins. Proc Natl Acad Sci USA. 2014;111:11031–11036. doi: 10.1073/pnas.1410529111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Truong HH, Kim BL, Schafer NP, Wolynes PG. Predictive energy landscapes for folding membrane protein assemblies. J Chem Phys. 2014;143:243101. doi: 10.1063/1.4929598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.