

Abstract
Skeletal muscles undergo structural and functional decline with ageing, culminating in sarcopenia. The underlying neuromuscular mechanisms have been the subject of intense investigation, revealing mitochondrial abnormalities as potential culprits within both nerve and muscle cells. Implicated mechanisms involve impaired mitochondrial dynamics, reduced organelle biogenesis and quality control via mitophagy, accumulation of mitochondrial DNA (mtDNA) damage and respiratory chain defect, metabolic disturbance, pro‐apoptotic signalling, and oxidative stress. This article provides an overview of the cellular mechanisms whereby mitochondria may promote maladaptive changes within motor neurons, the neuromuscular junction (NMJ) and muscle fibres. Lifelong physical activity, which promotes mitochondrial health across tissues, is emerging as an effective countermeasure for sarcopenia.
Introduction
Life expectancy in the modern world is rapidly increasing but the quality of life is often compromised towards the end of the mature years. A determinant of functional capacity and autonomy is the integrity of the neuromuscular system, which wires the brain and skeletal muscles via motor neurons and the neuromuscular junction. With age, different components of this system may fail, leading to loss of muscle mass and function, and decreased ability to remain physically active. This review will focus on structural and functional features of muscle ageing and, more specifically, on the role of mitochondria in relation to various maladaptive mechanisms contributing to age‐related neuronal and muscular decline. We also discuss the role of exercise as currently the most successful intervention against age‐related neuromuscular degeneration.
Sarcopenia
It has been known for a few decades that skeletal muscle structure and function deteriorate with age. Muscle mass decreases, reflected in both the reduced cross‐sectional area of individual myofibres and of the total number of fibres (Lexell & Taylor, 1991). This is associated with reduced strength and physical performance, culminating in the frailty syndrome and falls that lead, in extreme cases, to immobility and loss of autonomy (Visser & Schaap, 2011; Landi et al. 2012). Sarcopenia is defined as a measurable level of muscle wasting based on lean mass, grip strength and gate speed (Cruz‐Jentoft et al. 2010). Although sarcopenia eventually affects everyone, the time point at which it begins shows substantial inter‐individual variability – some experience only modest changes in old age, while others become severely disabled as early as in their seventh decade of life.
The causes of sarcopenia are still debated but it has become apparent that involvement of both the muscle itself and the innervating nerve play a role. Voluntary movement is a highly specialized and orchestrated function of the body that requires efficient communication between the nervous and muscular systems. A decision to move triggers excitation of the upper motor neurons residing in the motor cortex of the brain. These cells transmit an action potential to the lower motor neurons within the posterior area of the spinal cord. The electrical impulse then spreads from the cell body of a motor neuron through its axon to the neuromuscular junction (NMJ), a specialized synapse between the neuron and the muscle. There, the incoming neuronal action potential is transmitted by the neurotransmitter acetylcholine to the sarcolemma where it triggers depolarization of the myofibre and initiates muscle contraction.
Because the maintenance of muscle mass requires normal innervation and regular activation, malfunction of any of these elements can lead to the muscle deterioration. The exact causes underlying the age‐related changes in the neuromuscular system are still unknown but there is evidence that mitochondria, a critical cellular organelle involved in energy production and cellular signalling, may be either a primary trigger or at least an important player in this process. Here we will discuss known mitochondrial changes and their potential role in the development of sarcopenic phenotype.
Mitochondrial structure and functions
Mitochondrial DNA and the respiratory chain
Mitochondria contain their own genetic material, the mitochondrial DNA (mtDNA). The mtDNA differs from the nuclear DNA (nDNA) in multiple aspects: it is short (∼16.6 kb) and circular, uniparentally inherited from the mother, contains little (<2%) non‐coding regions, and is unprotected by histones. In addition, multiple mtDNA copies exist within a single mitochondrion, and multiple mitochondria reside in the cytoplasm of each cell, creating the possibility of heteroplasmy where different mtDNAs (e.g. mutant and normal) can coexist in a single cell (Li, Schroder et al. 2015). Despite its small size, mtDNA is essential for cellular function. It contains 37 genes encoding two ribosomal RNA (rRNA) and 22 transfer RNA (tRNA), which are essential to the translation of its 13 messenger RNA (mRNA)‐coding genes (Taylor & Turnbull, 2005). The resulting polypeptides combine with a number of nuclear‐encoded subunits to make up the multiprotein complexes I, III, IV and V of the mitochondrial respiratory chain. Only complex II is entirely encoded by the nuclear genome.
Attesting to the significance of mtDNA gene products, maternally inherited mtDNA mutations result in mitochondrial disease that is often lethal or severely debilitating, affecting primarily high energy demanding tissues such as the central nervous system and the muscle (Taylor & Turnbull, 2005). In addition, the mtDNA can accumulate defects (point mutations, deletions, duplications) with ageing, which are proposed to cause of age‐related functional decline systemically (Wallace, 2013). This includes the neuromuscular system, as discussed later in this article.
Mitochondrial dynamics
Mitochondria are highly dynamic organelles. Not only do they change shape and travel significant distances but they also fuse with or fragment from the neighbouring mitochondria (Westermann, 2010; Archer, 2013). A few proteins are required for mitochondrial dynamics: mitofusin 1 and 2 (Mnf1, Mnf2) and optic atrophy 1 (OPA1) for fusion; and dynamin‐related protein 1 (Drp1), mitochondrial fission factor (Mff) and fission protein 1 (Fis1) for fission (Detmer & Chan, 2007). These processes of fusion and fission allow communication and exchange of matrix content between individual mitochondria, including proteins (Chen et al. 2010) and mtDNA (Ono et al. 2001).
Normal mitochondrial dynamics is crucial for the health of the mitochondrial population as well as for that of the entire cell and organism (Archer, 2013). Alterations of both fusion and fission processes have pervasive effects on several aspects of mitochondrial function including respiratory capacity, coupling, reactive oxygen species production and apoptotic sensitivity (Picard et al. 2013). Overall, excessively fragmented mitochondria tend to exhibit reduced respiratory chain capacity, increased ROS production, and increased susceptibility to the release of mitochondria‐derived activators of caspases. Notably, inhibition of mitochondrial fusion by the removal of Mfn 1/2 in skeletal muscle promotes the accumulation of mtDNA defects and small muscle size (Chen et al. 2010).
Quality control
One of the most efficient methods of recycling of damaged/dysfunctional mitochondria is a specialized type of autophagy called mitophagy. Mitophagy is a process closely linked to mitochondrial dynamics (Youle & van der Bliek, 2012), by which a fragmented (small) mitochondrion becomes encapsulated with a double membrane (isolation membrane, see Fig. 2) to form an autophagosome delivered to a proximal lysosome. Upon fusion with the lysosome, the autophagosome cargo undergoes hydrolytic lysis (Kim et al. 2007). Mitophagy is designed to remove entire organelles whereas selected mitochondrial proteins are degraded via one of three systems: the ubiquitin–proteasome pathway (Hershko & Ciechanover, 1992), Lon protease (Bota & Davies, 2002), and mitochondria‐derived vesicles (MDVs) (McLelland et al. 2014). Removal of dysfunctional mitochondria by mitophagy is essential for maintaining muscle mass (Masiero et al. 2009). Several overlapping pathways thus exist to ensure the maintenance of a healthy pool of mitochondria in different cell types as alterations of these pathways may adversely affect muscle health in ageing.
Figure 2. Proposed mechanism of neuronal mitochondria involvement in sarcopenia .
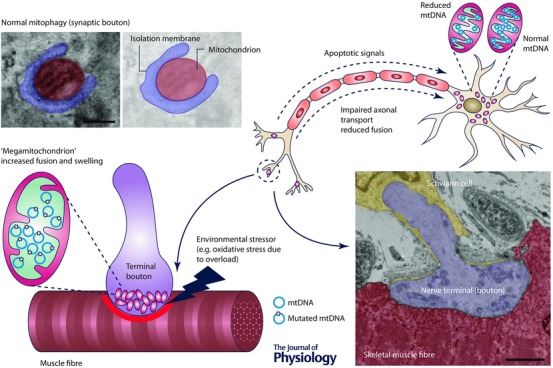
Age‐related increase in oxidative damage or down‐regulation of anti‐oxidant defences leads to mitochondrial damage (swelling, diminished cristae and elevated fusion) in terminal boutons of the neuromuscular junction (NMJ). Reduced fission may prevent mitophagy‐driven clearance of the damaged mitochondria, which accumulate abnormal proteins and mutated mtDNA. These mitochondria become respiratory‐deficient, which leads to inefficient neuromuscular transmission, reduced membrane potential and release of pro‐apoptotic factors (e.g. cytochrome c). Apoptotic signals are transduced via retrograde transport to the neuronal cell body where they induce apoptosis. Impaired mitochondrial dynamics in the terminal bouton affects axonal transport, and hence distal mitochondria do not mix with their counterparts located in the soma. This potentially results in reduced mtDNA content and down‐regulation of mitochondrial respiratory chain proteins with complex I being affected the earliest. Image at bottom right shows an NMJ from a mouse skeletal muscle (diaphragm) fibre visualized by transmission electron microscopy (TEM). Electron micrograph in the top left corner shows a mitochondrion undergoing mitophagy. Scale bar: 2 μm (NMJ, bottom right); 200 nm (mitophagosome, top left). Images kindly provided by the Basil Petrof Laboratory and the Newcastle University EM Research Services.
Role of the lower motor neuron and the neuromuscular junction in sarcopenia
Lower motor neuron
Neuronal stimulation of the muscle via the NMJ is an essential step for muscle contraction and human movement. Without it the muscle atrophies, losing strength and power (Scelsi et al. 1980). It has been reported that the spinal motor neuron population becomes depleted with advancing age (Fig. 1). Stereological analysis of lumbar spinal cords from a rat model of sarcopenia demonstrated 27% reduction in motor neuron pool between young adults and senescent animals (Rowan et al. 2012). A similar rate of motor neuron depletion was noted for human post‐mortem samples where 25% loss of these cells occurred between the second and tenth decade of life (Tomlinson & Irving, 1977).
Figure 1. Lower motor neuron changes with advancing age .
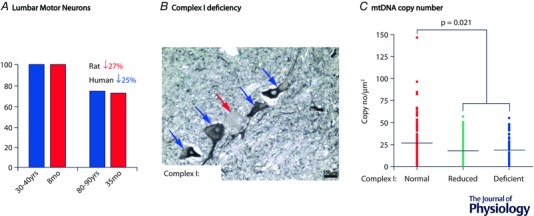
A, spinal motor neuron population undergoes a similar rate of depletion in ageing in both rats and humans. B, some of the remaining motor neurons accumulate somatic mitochondrial damage including severe down‐regulation of mitochondrial respiratory chain complex I, shown here by the absence of labelling for NDUFB8. C, it is likely that complex I deficiency is a result of mtDNA depletion in ageing spinal motor neurons.
Because muscle fibres cannot remain denervated (Carlson, 2014), the loss of motor neurons promotes lateral sprouting of the neighbouring ones, which reinnervate ‘orphelin’ denervated muscle fibres. This increases the size of the individual motor units, thus elevating the metabolic and biosynthetic burden on a single nerve cell, possibly making it prone to overload‐driven degeneration (Stalberg & Fawcett, 1982; Brown et al. 1988). This results in fibre type grouping in the muscle (i.e. the physical clustering of myofibres with the same myosin heavy chain isoform) as multiple muscle fibres are innervated, and fibre‐type dictated by a single motor neuron (Lexell & Downham, 1991; Lexell, 1997).
Neuromuscular junction
The NMJs have also shown age‐related changes. Data obtained from animal models and humans, and across different muscle groups, are sometimes conflicting. The general consensus, however, is that with ageing the complexity and morphology of the pre‐ and postsynaptic regions become altered, the number of neurotransmitter‐containing synaptic vesicles decreases, and the axonal transport is slower (Jang & Van Remmen, 2011). We now discuss the potential role of mitochondrial defects in NMJ maintenance.
Mitochondrial deficiency in the ageing neuron
Mitochondria have been implicated in the age‐related alterations of the motor neuron soma as well as the pre‐ and postsynaptic regions of the NMJ. Our recent study in elderly (68–99 years old) post‐mortem spinal cord samples showed a pronounced mitochondrial dysfunction in the lumbar motor neurons (Rygiel et al. 2014) (Fig. 1). Around 10% of these cells demonstrated a complete loss of mitochondrial respiratory chain complex I and a further 25% had markedly reduced levels of complex I proteins. Mitochondrial DNA analysis carried out on individual complex I‐deficient motor neurons revealed significantly reduced copy numbers (Fig. 1). Interestingly, the respiratory‐deficient cell bodies were smaller than their unaffected counterparts suggesting mitochondrial deficiency‐driven atrophy (Rygiel et al. 2014), not unlike cell atrophy observed in a cytoplasmic hybrid cell of mtDNA heteroplasmy (Picard et al. 2014). This acquired mitochondrial dysfunction could originate within the soma of a motor neuron itself, or in the peripheral axonal/dendritic mitochondria, and then be retrograde‐transported back to the soma where it can impact central cellular functions including gene expression.
Mitochondrial abnormalities in the neuromuscular junction
Mitochondrial abnormalities within the NMJ may also contribute to impaired signal transduction between the motor neuron and muscle. Indeed, mitochondria exist in presynaptic terminals where they modulate neurotransmitter release (Vos et al. 2010). A recent study in aged rats demonstrated substantial changes in the majority of axonal mitochondria residing within the terminal boutons of the tibial nerve (Garcia et al. 2013). Electron microscopy analysis revealed unusual features among presynaptic mitochondria: they were swollen, up to 3‐fold larger than their normal counterparts, with ‘hollow’ matrix, virtually devoid of cristae, and with ruptured membranes. A marked proportion of these mitochondria appeared hyperfused, forming gigantic ‘megamitochondria’ with multiple surrounding membrane layers. None of these morphological alterations were observed in the neuronal cell bodies, suggesting specific alterations of NMJ mitochondria (Fig. 2). Indeed, several differences exist between synaptic and non‐synaptic mitochondria, notably in protein composition, which is skewed towards a pro‐fission phenotype (more Drp1, less OPA1 and Mfn1/2) in the mouse brain (Stauch et al. 2014). Synaptic mitochondria may also be more prone to mtDNA deletions (Stauch et al. 2014). Moreover, calcium‐dense inclusions were reported to be abundant in the aged presynaptic mitochondria, potentially indicating calcium overload (Garcia et al. 2013).
A consequence of mitochondrial calcium overload is the release of mitochondria‐derived apoptotic activators such as cytochrome c (Brookes et al. 2004). Cytochrome c was indeed found in the cytosol in the distal portion of motor neurons, suggesting mitochondrial permeability transition and possibly downstream activation of the apoptosome. Caspase 3 was detected in the soma and nuclei of the aged motor neurons as well as the proximal and distal axons, but the activated caspase 3 was clearly associated with vesicles along the microtubule system and colocalized with dynein, implying retrograde transport (Garcia et al. 2013) (Fig. 2). The underlying causes of the described mitochondrial pathology and activation of the apoptotic machinery in ageing nerve terminals are unclear but they could be at least partially associated with up‐regulation of the oxidative damage and down‐regulation of the natural cellular anti‐oxidant defences. Studies on superoxide dysmutase 1 (Sod 1) knockout mice revealed a sarcopenic phenotype with the accumulation of abnormal giant mitochondria, higher susceptibility to calcium‐induced mitochondrial permeability transition pore opening and apoptosis, linking oxidative damage with mitochondrial dysfunction and neuromuscular junction denervation (Jang et al. 2010).
All of these findings are consistent with the ‘dying back’ phenomenon, where the neuronal damage is inflicted on the axon terminal, causing degeneration of the entire cell. A number of neurodegenerative disorders including motor neuron disease, Alzheimer's disease, Parkinson's disease and glaucoma share this pattern of neuronal degeneration, and therefore it is not surprising that a similar mechanism could be engaged in sarcopenia (Adalbert & Coleman, 2013). Accumulation of abnormal mitochondria primarily in the terminal boutons rather than the neuronal cell bodies can also be explained by the impaired mitochondrial dynamics. A balance between mitochondrial fusion and fission has to be maintained in order to support mitochondrial function and motility. Neurodegenerative diseases such as Charcot–Marie–Tooth 2A (CMT2A) and dominant optic atrophy (DOA) manifest with degeneration of sensory and motor nerves and optic nerve as a direct consequence of mutations in genes encoding mitochondrial fusion proteins Mfn2 and OPA1, respectively (Chan, 2012). It has been proposed that impaired fusion not only leads to reduced function of the fragmented mitochondria but also to accumulation of them in various areas within the neuron resulting in ineffective distribution (Chen & Chan, 2006). Given their potentially skewed protein profile towards fission (Stauch et al. 2014), synaptic mitochondria may be particularly vulnerable to factors that impair normal mitochondrial fusion.
It is possible that due to some environmental insult (e.g. oxidative stress) impaired mitochondrial dynamics in the ageing motor neuron leads to dysmorphic and abnormally large mitochondria in terminal NMJ boutons. Because of physical and possibly other constraints, these mitochondria may be unable to move via axonal transport and communicate with other mitochondria, and would escape mitotophagy, failing to be degraded or removed, and thus accumulate protein and mtDNA damage and become dysfunctional. Because of the mitochondria's dynamic role in regulating synaptic function (Sun et al. 2013), this would promote dysregulation of neuromuscular transmission, NMJ alteration and ultimately denervation of the associated myofibre (Fig. 2).
Skeletal muscle
Neuronal dysfunction undoubtedly contributes to age‐related muscle wasting, but the muscle itself also undergoes complex remodelling that exacerbates the sarcopenic phenotype observed in the elderly. The most commonly reported muscle‐specific changes include lower rate of anabolism, reduced regenerative capacity due to the senescence or depletion of the satellite cell pool and higher rate of cell death. The following sections discuss the role of mitochondria in relation to these aspects of age‐related muscle tissue deterioration.
Skeletal muscle anabolism and catabolism
The major hallmark of the sarcopenic muscle is its reduced size, which can only be explained by a dysregulated protein ‘economy’, namely an imbalance between the rates of protein synthesis (i.e. anabolism) and degradation (i.e. catabolism). These processes are tightly controlled, among other pathways, by insulin signalling via the mammalian target of rapamycin (mTOR) serine/threonine kinase pathway (Bonaldo & Sandri, 2013). Lack of insulin in type I diabetes patients promotes loss of muscle protein content and consequent wasting (Tessari et al. 1990). Furthermore, insulin signalling in conjunction with amino acids not only stimulates protein synthesis but also inhibits proteolysis.
As protein synthesis requires energy input, it is not surprising that mTOR also augments mitochondrial function, mitochondrial respiratory chain protein levels and ATP production (Albert & Hall, 2015). Although experimental muscle insulin infusion in both animals and humans resulted in an increased rate of synthesis of mitochondrial proteins including citrate synthase and cytochrome c oxidase, it had no effect on structural muscle proteins, including contractile myosin heavy chain, (Boirie et al. 2001; Stump et al. 2003), indicating that mitochondrial biogenesis and muscle anabolism may be independently regulated.
Skeletal muscle lipids and metabolism
Insulin resistance is thought to be connected with the intramyocellular lipid accumulation and excess mitochondrial reactive oxygen species in ageing muscle (Anderson et al. 2009). Muscle of older people (∼70 year old) has higher lipid content (individual lipid droplets are larger) than muscle of younger people (∼20 year old), and the lipid stores are rarely associated with mitochondria. Physical association of mitochondria with lipid droplets is increased in myofibres after exercise (Tarnopolsky et al. 2007), which implies an optimal position for oxidation. Mitochondrial mass is reduced in the old, as a result of fewer individual mitochondria rather than their size, which agrees with reduction in the mitochondrial enzymes (Crane et al. 2010). In middle‐aged primates accumulation of larger lipid droplets was also observed together with a shift in fibre type distribution, reduced oxidative phosphorylation capacity and a metabolic shift (increased FAD and NADH levels in situ) (Pugh et al. 2013). This suggests reduced activity of mitochondrial respiratory chain dehydrogenase enzymes, which may become a limiting factor in aged muscle mitochondria.
The difficulty with interpreting the age‐related lipid accumulation in muscle is that physical inactivity also results in a similar phenomenon. In human diaphragm, compared with normally contracting diaphragm muscle, inactivity during mechanical ventilation exhibited reduced respiratory chain complex IV activity, which was associated with significantly higher intramuscular lipid content manifesting as an increased lipid density as well as droplet volume (Picard et al. 2012). Interestingly, lipid accumulation in muscle fibres colocalized with mitochondrial respiratory deficiency measured using histochemical detection of enzymatic activity, indicating a potential causative link between mitochondrial dysfunction and dysregulated lipid metabolism.
Mitochondrial DNA – genetics and maintenance
Changes in the mitochondrial genome have been implicated in physiological ageing of the majority of organs. In muscle, mtDNA content is inversely related to age (Short et al. 2005) and the remaining mtDNA copies acquire rearrangements in an age‐related manner (Meissner et al. 2006). Large‐scale mtDNA deletions are most frequently found (Kraytsberg & Khrapko, 2005; Bua et al. 2006; Meissner et al. 2006), but there have been reports of duplications and triplications detected in muscle from elderly individuals (Tengan & Moraes, 1998).
The origin of mtDNA deletions is still unclear, but it is likely that they are either inherited at low levels from the maternal germline, or acquired somatically early in life. Somatic mutations are formed as a consequence of either replication errors or defective repair of double‐strand breaks (Bua et al. 2006; Krishnan et al. 2008). At the beginning of this process there are very few mutated molecules present amongst masses of healthy mtDNA counterparts in the cell, but with time, the mutated copies replicate and begin to outnumber normal ones. This process, termed clonal expansion, manifests only in a limited number of single muscle fibres. As a result, the ageing muscle becomes a mosaic of deletion‐loaded and deletion‐free cells (Murphy et al. 2012). Interestingly, due to the multiple‐copy nature of mtDNA, the cells are fully respiratory‐functional until they cross a certain threshold of mutated‐to‐healthy molecules. This threshold is tissue and cell type dependent but often only 10% of healthy mtDNA molecules is enough for the cells to maintain normal respiratory functions (Stewart & Chinnery, 2015) (Fig. 3).
Figure 3. Proposed mechanism of skeletal muscle mitochondria involvement in sarcopenia .
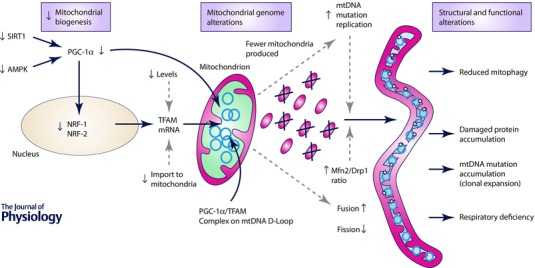
Mitochondrial biogenesis declines with age, dependent and independent of PGC‐1α. Proteins essential for PGC‐1α post‐translational modification, namely SIRT1 and AMPK, are either down‐regulated (SIRT1) or less responsive to activation (AMPK), resulting in depletion of active PGC‐1α pool. Consequently, the downstream nuclear factors NRF‐1 and NRF‐2 are down‐regulated, leading to a reduction in TFAM transcription, and transport of TFAM protein into mitochondria. TFAM and PGC‐1α form a complex, localised to the D‐loop region of mtDNA, which regulates replication and transcription of the mitochondrial genome. Age‐related depletion of both causes reduced nucleic acid and protein turnover, which leads to not only reduced mitochondrial mass but also accumulation of mtDNA mutations and damaged proteins. This induces fusion as a compensatory mechanism to reduce the detrimental consequences of the accumulated defects. Fused mitochondria escape mitophagy and continue accumulating mtDNA (clonal expansion) and protein damage, which ultimately leads to respiratory deficiency.
Mechanisms behind clonal expansion are still unclear. What is certain is that mitochondrial deletions accumulate in longitudinal segments of individual muscle fibres. Studies on ageing rats have elegantly demonstrated that there is a functional relationship between mitochondrial dysfunction and muscle phenotype. Muscle fibre segments of complete respiratory deficiency due to a high mutation load showed atrophy, splitting and, in some cases, rupturing (Bua et al. 2002). However, this phenomenon is relatively rare in other species and certainly does not extend to humans, where mutation load does not correlate with muscle fibre size (Bua et al. 2006; Picard et al. 2012). Interestingly, in inclusion body myositis, an age‐related inflammatory myopathy, respiratory‐deficient myofibres are more prone to atrophy than their unaffected counterparts (Rygiel et al. 2014). It is therefore difficult to conclude what the true relationship between respiratory chain dysfunction and muscle fibre atrophy is.
How prevalent are respiratory‐deficient myofibres in ageing muscle? The most comprehensive study that quantified the level of the respiratory deficiency in ageing human muscle reported up to 5% deficient fibres in 60‐ to 90‐year‐old participants (Brierley et al. 1996). Preliminary data obtained in our laboratory for a large cohort of 70‐year‐old participants showed 6–14% of respiratory‐deficient fibres in the most affected cases (unpublished data). The proportion of the respiratory‐deficient fibres may not appear high and their significance is questionable. It is worth stressing, however, that the deficiency is segmental and can affect any fragment of the muscle fibre along its length. The proportion of respiratory‐deficient cells is typically derived from assessment of a single or a small number of muscle sections, which may significantly underestimate the true deficiency in the entire muscle.
The next fundamental question is whether mitochondrial dysfunction has a functional consequence for muscle wasting in sarcopenia. Patients with mitochondrial disease tend to develop myopathy, involving selected muscle groups, which is mostly associated with weakness (McFarland & Turnbull, 2009). Data from animals are somewhat puzzling. A mouse model with a skeletal muscle conditional knock‐out of the COX10 gene (encoding a subunit of mitochondrial respiratory chain complex IV) showed virtually no reduction in maximal contractile force for the first 2.5 months of age despite complex IV activity being reduced by over 95% (Diaz et al. 2005). Additionally, there was only a 10% increase in fatigability and no signs of oxidative damage or apoptosis in young mice, suggesting that the relationship between mitochondrial respiratory chain function and muscle phenotype is not direct (Diaz et al. 2005). However, the COX10‐induced myopathy worsened with time, consistent with progressive muscle degeneration in humans.
Mitochondrial dynamics and degradation
In the ageing muscle, abnormal mitochondrial dynamics of fusion/fission could contribute to muscle dysfunction via two major mechanisms. The first relates to the fact that mitochondrial dynamics is essential to mitophagy and quality control processes. Fission is necessary for subsequent mitophagy to remove defective proteins or mtDNA molecules from the mitochondrial network. In this case, inhibition of fission and thus mitophagy may promote accumulation of dysfunctional mitochondria within skeletal muscle fibres (Masiero et al. 2009; Grumati et al. 2010). The down‐regulation of specific Parkin machinery responsible for culling and degrading dysfunctional mitochondria may also be down‐regulated in aged skeletal muscle (Gouspillou et al. 2014). Secondly, dysfunctional mitochondria may promote atrophy via multiple different pathways discussed above, including pro‐apoptotic signalling (Gouspillou et al. 2014), energy deficiency and impacting nuclear gene expression.
With ageing, mitochondrial dynamics is altered and mitochondria undergo structural remodelling. Electron microscopy performed on muscle samples from old mice revealed changes in both subsarcolemmal and intermyofibrillar mitochondria: in aged muscle exhibiting a 30% reduction in myofibre size, subsarcolemmal mitochondria were larger and more elongated whereas intermyofibrillar mitochondria were longer and more branched than in young muscle (Leduc‐Gaudet et al. 2015). Together with the up‐regulated protein ratio of Mfn2/Drp1 and larger mitochondrial volumes, this strongly suggested increased fusion in both mitochondrial subpopulations (Leduc‐Gaudet et al. 2015) (Fig. 3). In support of these findings, a different mouse study found up‐regulated Mfn1 and Mfn2 and down‐regulated Fis1 (Joseph et al. 2013), and mitochondrial elongation was observed in fibroblasts of aged individuals (Allen et al. 2015). Apparently in contrast to these findings, mRNA levels for Mfn2 were found to be down‐regulated in elderly human skeletal muscle (Crane et al. 2010). Keeping in mind that mRNA levels do not directly translate into corresponding protein levels, and that mitochondrial dynamics proteins are heavily post‐translationally regulated (Shutt et al. 2012), this discrepancy could be attributable to different factors and be without relevance to mitochondrial dynamics. Overall, atrophying skeletal muscles may exhibit excess mitochondrial fusion, which consists in a normal response to mild stress (Shutt & McBride, 2013), and may thus represent a compensatory response to an intrinsic functional defect within aged organelles.
Because mitochondria have to undergo fragmentation in order to allow autophagosome formation and engulfment, mitochondrial elongation via increased fusion in aged muscle could prevent degradation via autophagy (Rambold et al. 2011). In line with this, a general decrease in proteolysis pathways has been reported in various ageing tissues from senescent mice, rats and humans (Cuervo & Dice, 2000; Ferrington et al. 2005; Wohlgemuth et al. 2010). Data from the skeletal muscle, however, is limited and not conclusive, particularly with regard to the proteasome and mitophagy pathways. Lon protease‐associated degradation is the only system that has been clearly shown to be affected in ageing skeletal muscle (Bota & Davies, 2002). Lon protein levels are significantly down‐regulated in the muscle of old mice and cabonylated proteins accumulate within the muscle mitochondria (Bota et al. 2002).
Mitochondrial biogenesis
Skeletal muscle mitochondrial mass tends to decrease with age (e.g. Short et al. 2005), although this may be specific to certain muscles and species (Picard et al. 2011). This is believed to be primarily due to reduced mitochondrial biogenesis, i.e. the production of new mitochondria. As mitochondrial proteins are encoded in both mitochondrial and nuclear genomes, biosynthesis of new organelles requires transcription factors and molecular regulators that act on them both. Peroxisome proliferator‐activated receptor γ coactivator (PGC) 1α is considered a master regulator of mitochondrial biogenesis and has been shown to be decreased at both mRNA and protein level in aged skeletal muscle (Ling et al. 2004; Rossi et al. 2009). PGC‐1α lies downstream from metabolic sensors AMP‐activated protein kinase (AMPK), sirtuin 1 (SIRT1), and mitogen‐associated protein kinase (p38MAPK), which synergize to activate PGC‐1α in the cytoplasm. This causes its nuclear and mitochondrial translocation, where it initiates expression of mitochondrial proteins by binding to the nDNA and mtDNA (Safdar et al. 2011). Because PGC‐1α co‐regulates several genes and its expression alone is sufficient to increase mitochondrial mass (Wu et al. 1999), it is considered to play an important role in skeletal muscle mitochondrial biogenesis.
However, this remains controversial since mitochondrial biogenesis can still happen in the absence of PGC‐1α. A mouse model of skeletal muscle‐specific knock‐out of PGC‐1α does not prevent exercise‐induced mitochondrial biogenesis (Rowe et al. 2012). Similarly, removal of PGC‐1α from cultured myoblasts using siRNA technology fails to inactivate genes involved in oxidative metabolism and mitochondrial biogenesis following forced contractile activity (Uguccioni & Hood, 2011). Currently, pathways other than PGC‐1α‐dependent are under investigation. PGC‐1β, a close homologue of PGC‐1α, has been shown to stimulate mitochondrial biogenesis in an equally robust manner, and deletion of both PGC‐1α and PGC‐1β from muscle down‐regulates mitochondrial function much more dramatically than deletion of either alone (Arany et al. 2007; Zechner et al. 2010). Another pathway reported to be critical for exercise‐induced biogenesis is mediated by p38MAPK (Pogozelski et al. 2009). For a more detailed discussion on this topic, we refer the reader to comprehensive reviews (Hawley et al. 2014; Drake et al. 2015).
PGC‐1α also up‐regulates mtDNA replication, transcription and stability. This is achieved by a twofold mechanism involving its direct association with the mitochondrial transcription factor A (TFAM) at the D‐loop region of mtDNA, and activation of nuclear respiratory factor 1 and 2 (NRF‐1 and NRF‐2, respectively), which stimulate expression of TFAM and its import into mitochondria, thus promoting mtDNA transcription and replication (Campbell et al. 2012) (Fig. 3).
Another interesting possibility beyond mitochondrial biogenesis stipulates that down‐regulation of PGC‐1α with ageing may also contribute to skeletal muscle atrophy by promoting destabilization of the neuromuscular junction and ‘denervation’ in aged skeletal muscle (Gouspillou et al. 2013). In parallel with ageing, PGC‐1α levels are reduced in sedentary persons compared with physically active individuals (Lanza et al. 2008). But endurance exercise can counteract the decline in both PGC‐1α expression and mitochondrial biogenesis, with data indicating that trained elderly individuals may even maintain the PGC‐1α mRNA content at a stable level exceeding that of their sedentary young counterparts (Lanza et al. 2008). This suggests a molecular avenue by which exercise could be an effective countermeasure to prevent sarcopenia by affecting both skeletal muscle and the NMJ.
Exercise as a countermeasure for sarcopenia
Exercise has classically been divided into two major categories, endurance and resistance, representing two ends of a spectrum. Both exercise modalities offer benefits in terms of ageing muscle, as discussed elsewhere in detail (Barbieri et al. 2015). Briefly, to summarize their major effects, resistance exercise mainly leads to muscle hypertrophy, and increase in muscle mass and, in most cases, strength and power output (Cadore et al. 2014), whereas endurance exercise, or aerobic training, mainly improves cardiorespiratory fitness (maximal oxygen consumption – ), muscle oxidative capacity and overall physical performance (Cadore et al. 2014). Another form of physical activity is high‐intensity interval training (HIT, or HIIT), which involves intermittent short bouts of maximum activity interspersed with periods of low intensity. This type of training provides similar benefits to endurance training but volume and time of exercise are markedly reduced (Little et al. 2010). More importantly in the context of exercise adaptations is that HIT induce molecular adaptations that trigger mitochondrial biogenesis. For instance, 2 weeks of HIT was sufficient to increase the amount of PGC‐1α in the nucleus, as well as overall SIRT1 and TFAM content, resulting in increased muscle mitochondrial mass and exercise performance, demonstrating that HIT elicits robust adaptive mitochondrial and physiological responses opposite to those associated with ageing (Little et al. 2010).
Exercise‐induced adaptations involve proportional changes in mitochondrial biogenesis and content. For example, across various exercise modalities exercise‐induced increases in occur in proportion to increase in mitochondrial content indexed by citrate synthase activity (Vigelso et al. 2014). This represents additional evidence that exercise‐induced physiological adaptations are linked, and could be dependent upon mitochondrial biogenesis.
Aerobic training can improve mitochondrial function irrespective of age, although the mechanisms may differ between young and older, and between male and female individuals (Vigelso et al. 2014). An acute endurance exercise programme up‐regulated proteins involved in activation of the electron transport chain components such as mitochondrial SIRT3, as well as mitochondrial antioxidant capacity, in older adults (>65 years) (Johnson et al. 2014). Nevertheless, elevated protein degradation and reduced oxidative damage were only observed in the young, suggesting age‐specific effects (Johnson et al. 2014). In contrast, 12 weeks of aerobic exercise intervention in 20‐ and 70‐year‐old participants resulted in increased aerobic capacity, skeletal muscle size and markers of mitochondrial biogenesis and dynamics in both groups (Konopka et al. 2014). Transcriptome (mRNA levels) analysis showed down‐regulation of mitochondrial genes and pathways engaged in oxidative phosphorylation with ageing, whereas redox homeostasis genes were up‐regulated in older sedentary adults. Interestingly, those differences were not present between chronically endurance‐trained older adults and their young counterparts (Johnson et al. 2014), suggesting that exercise is an effective countermeasure against ageing‐associated transcriptional remodelling.
Endurance exercise may also impact muscle function via epigenetic effects, consisting of modifications of DNA‐associated proteins and the DNA itself (i.e. the chromatin) that promote stable changes in gene expression (Bird, 2007). One such modification is DNA cytosine methylation, which was found to be decreased overall in skeletal muscle samples from sedentary men and women following acute exercise (Barres et al. 2012). Exercise induced a dose‐dependent expression of key metabolism regulator genes such as PGC‐1α, PDK4 and PPAR‐δ, with corresponding hypomethylation of their respective promoter regions, consistent with exercise‐induced epigenetic regulation of gene transcription (Barres et al. 2012).
Perhaps the most valuable data on the effects of life‐long endurance training on muscle function come from a study on over‐80‐year‐old athletes (Trappe et al. 2013). Muscle biopsy analyses revealed a markedly higher oxidative profile consisting of elevated mitochondrial markers in athletes compared with their untrained peers. Moreover, the magnitude of differences in mitochondrial function between the trained and untrained octogenarians was comparable to the differences between trained and untrained young adults (Burgomaster et al. 2008). Importantly, commencing endurance training after the age of 80 did not improve the age‐related decline in mitochondrial function, suggesting that only life‐long regular exercise results in the metabolic flexibility necessary to maintain a healthy muscular system (Trappe et al. 2013).
In light of the above, it appears difficult to discriminate between the role of ageing and inactivity on skeletal muscle deterioration. It is apparent that sedentary behaviour accelerates age‐related decline in muscle structure and function (Booth et al. 2011). Evidence collected so far suggests that a life‐long regular exercise can significantly delay the onset of sarcopenia (Barbieri et al. 2015).
Conclusion
The human motor system deteriorates with age, leaving no one untouched. However, the rate of this deterioration varies from person to person. The underlying mechanisms are still unclear, but it has become clear that changes among both the nervous and the muscular systems are implicated in this degenerative process. It is likely that individual factors predispose to sarcopenia, for malfunction of one or the other, which makes our neuromuscular system age differently. There are also important inter‐individual differences in levels of physical activity and sedentary behaviour, diet and other factors, making experimental discrimination of various contributing factors difficult. Most likely, there are different pathways or ‘trajectories’ of ageing that are impacted by a variety of personal and environmental factors (Picard, 2011).
Mitochondria, as essential powerhouses and signalling organelles, are implicated in sarcopenia. Experimentally, mitochondrial abnormalities have been identified in both neurons and muscle fibres in elderly and sedentary subjects, and known mechanisms exist whereby abnormal mitochondrial functions can promote neuromuscular disorders. However, it is still unclear whether mitochondrial dysfunction, at a level reported for these two tissues in normal human ageing, is a primary cause of the phenotypic and functional changes seen in sarcopenia. Nevertheless, together with other dysregulated processes involved in sarcopenia, mitochondrial abnormalities are likely to contribute to loss of skeletal muscle mass and function with age (Hepple, 2014). Technical advances to probe mitochondria at a molecular level, longitudinal studies in humans, and comprehensive hypotheses involving both nerve and muscle factors should enhance our ability to understand and prevent sarcopenia.
Additional information
Competing interests
None declared.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The authors’ work is supported by the Newcastle University Centre for Ageing and Vitality (supported by the Biotechnology and Biological Sciences Research Council and Medical Research Council [G016354/1]), Wellcome Trust Centre for Mitochondrial Research [G906919], MRC Centre for Neuromuscular Disease [G000608‐1], The MRC Centre for Translational Research in Neuromuscular Disease Mitochondrial Disease Patient Cohort (UK) [G0800674], The Lily Foundation, and the UK NIHR Biomedical Research Centre in Age and Age Related Diseases award to the Newcastle upon Tyne Hospitals NHS Foundation Trust.
Biographies
Karolina Rygiel has been working in Doug Turnbull's Mitochondrial Research Group at Newcastle University as a research associate for the last six years. She investigates the role of mitochondria in sarcopenia as well as age‐related myopathies, with a particular interest in inclusion body myositis. During her PhD training, Karolina studied epithelial to mesenchymal transition in autoimmune liver disease in John Kirby's laboratory. Apart from science, she loves cycling, skiing, traveling and Pixar, and she bakes her own bread from a 300 year old French sour starter named Monsieur Hugo.

Martin Picard trained as a physiologist in mitochondrial biology (Taivassalo/Hepple Labs, McGill University) and genetics (Wallace Lab, UPenn), and is Assistant Professor at Columbia University where he studies mitochondria at the interface of biological and psychosocial sciences. His work combines molecular biology, mitochondrial bioenergetics, high‐resolution quantitative microscopy, and omics technologies to understand how stressors of different nature translate into variable ageing trajectories in cells, animals and humans. Apart from science, he loves cycling, photography and visiting friends.
Doug Turnbull is Professor of Neurology at Newcastle University. He is Director of the MRC/BBSRC Centre for Ageing and Vitality and Director of the Wellcome Trust Centre for Mitochondrial Research. He has a long‐standing interest in the role of mitochondria in disease and ageing.
This is an Editor's Choice article from the 15 August 2016 issue.
References
- Adalbert R & Coleman MP (2013). Review: Axon pathology in age‐related neurodegenerative disorders. Neuropathol Appl Neurobiol 39, 90–108. [DOI] [PubMed] [Google Scholar]
- Albert V & Hall MN (2015). mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol 33, 55–66. [DOI] [PubMed] [Google Scholar]
- Allen SP, Duffy LM, Shaw PJ & Grierson AJ (2015). Altered age‐related changes in bioenergetic properties and mitochondrial morphology in fibroblasts from sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging 36, 2893–2903. [DOI] [PubMed] [Google Scholar]
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH & Neufer PD (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, Lebrasseur N, Morris C, Smith E, Yang W, Ma Y, Chin S & Spiegelman BM (2007). The transcriptional coactivator PGC‐1β drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab 5, 35–46. [DOI] [PubMed] [Google Scholar]
- Archer SL (2013). Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med 369, 2236–2251. [DOI] [PubMed] [Google Scholar]
- Barbieri E, Agostini D, Polidori E, Potenza L, Guescini M, Lucertini F, Annibalini G, Stocchi L, De Santi M & Stocchi V (2015). The pleiotropic effect of physical exercise on mitochondrial dynamics in aging skeletal muscle. Oxid Med Cell Longev 2015, 917085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, Caidahl K, Krook A, O'Gorman DJ & Zierath JR (2012). Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab 15, 405–411. [DOI] [PubMed] [Google Scholar]
- Bird A (2007). Perceptions of epigenetics. Nature 447, 396–398. [DOI] [PubMed] [Google Scholar]
- Boirie Y, Short KR, Ahlman B, Charlton M & Nair KS (2001). Tissue‐specific regulation of mitochondrial and cytoplasmic protein synthesis rates by insulin. Diabetes 50, 2652–2658. [DOI] [PubMed] [Google Scholar]
- Bonaldo P & Sandri M (2013). Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth FW, Laye MJ & Roberts MD (2011). Lifetime sedentary living accelerates some aspects of secondary aging. J Appl Physiol (1985) 111, 1497–1504. [DOI] [PubMed] [Google Scholar]
- Bota DA & Davies KJ (2002). Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP‐stimulated mechanism. Nat Cell Biol 4, 674–680. [DOI] [PubMed] [Google Scholar]
- Bota DA, Van Remmen H & Davies KJ (2002). Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett 532, 103–106. [DOI] [PubMed] [Google Scholar]
- Brierley EJ, Johnson MA, James OF & Turnbull DM (1996). Effects of physical activity and age on mitochondrial function. QJM 89, 251–258. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW & Sheu SS (2004). Calcium, ATP, and ROS: a mitochondrial love‐hate triangle. Am J Physiol Cell Physiol 287, C817–C833. [DOI] [PubMed] [Google Scholar]
- Brown WF, Strong MJ & Snow R (1988). Methods for estimating numbers of motor units in biceps‐brachialis muscles and losses of motor units with aging. Muscle Nerve 11, 423–432. [DOI] [PubMed] [Google Scholar]
- Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S & Aiken JM (2006). Mitochondrial DNA‐deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet 79, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bua EA, McKiernan SH, Wanagat J, McKenzie D & Aiken JM (2002). Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J Appl Physiol 92, 2617–2624. [DOI] [PubMed] [Google Scholar]
- Burgomaster KA, Howarth KR, Phillips SM, Rakobowchuk M, Macdonald MJ, McGee SL & Gibala MJ (2008). Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J Physiol 586, 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadore EL, Pinto RS, Bottaro M & Izquierdo M (2014). Strength and endurance training prescription in healthy and frail elderly. Aging Dis 5, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CT, Kolesar JE & Kaufman BA (2012). Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim Biophys Acta 1819, 921–929. [DOI] [PubMed] [Google Scholar]
- Carlson B (2014). The biology of long‐term denervated skeletal muscle. Eur J Transl Myol 24, 3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46, 265–287. [DOI] [PubMed] [Google Scholar]
- Chen H & Chan DC (2006). Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol 18, 453–459. [DOI] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM & Chan DC (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JD, Devries MC, Safdar A, Hamadeh MJ & Tarnopolsky MA (2010). The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J Gerontol A Biol Sci Med Sci 65, 119–128. [DOI] [PubMed] [Google Scholar]
- Cruz‐Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, Martin FC, Michel JP, Rolland Y, Schneider SM, Topinkova E, Vandewoude M & Zamboni M (2010). Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 39, 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM & Dice JF (2000). Age‐related decline in chaperone‐mediated autophagy. J Biol Chem 275, 31505–31513. [DOI] [PubMed] [Google Scholar]
- Detmer SA & Chan DC (2007). Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 8, 870–879. [DOI] [PubMed] [Google Scholar]
- Diaz F, Thomas CK, Garcia S, Hernandez D & Moraes CT (2005). Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet 14, 2737–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JC, Wilson RJ & Yan Z (2015). Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J 30, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington DA, Husom AD & Thompson LV (2005). Altered proteasome structure, function, and oxidation in aged muscle. FASEB J 19, 644–646. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Fernandez A & Solas MT (2013). Mitochondria, motor neurons and aging. J Neurol Sci 330, 18–26. [DOI] [PubMed] [Google Scholar]
- Gouspillou G, Picard M, Godin R, Burelle Y & Hepple RT (2013). Role of peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α) in denervation‐induced atrophy in aged muscle: facts and hypotheses. Longev Healthspan 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Kapchinsky S, Purves‐Smith F, Norris B, Pion CH, Barbat‐Artigas S, Lemieux F, Taivassalo T, Morais JA, Aubertin‐Leheudre M & Hepple RT (2014). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J 28, 1621–1633. [DOI] [PubMed] [Google Scholar]
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, Maraldi NM, Bernardi P, Sandri M & Bonaldo P (2010). Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16, 1313–1320. [DOI] [PubMed] [Google Scholar]
- Hawley JA, Hargreaves M, Joyner MJ & Zierath JR (2014). Integrative biology of exercise. Cell 159, 738–749. [DOI] [PubMed] [Google Scholar]
- Hepple RT (2014). Mitochondrial involvement and impact in aging skeletal muscle. Front Aging Neurosci 6, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A & Ciechanover A (1992). The ubiquitin system for protein degradation. Annu Rev Biochem 61, 761–807. [DOI] [PubMed] [Google Scholar]
- Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A & Van Remmen H (2010). Increased superoxide in vivo accelerates age‐associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 24, 1376–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC & Van Remmen H (2011). Age‐associated alterations of the neuromuscular junction. Exp Gerontol 46, 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ML, Irving BA, Lanza IR, Vendelbo MH, Konopka AR, Robinson MM, Henderson GC, Klaus KA, Morse DM, Heppelmann C, Bergen HR 3rd, Surendra D, Schimke JM, Jakaitis DR & Nair KS (2014). Differential effect of endurance training on mitochondrial protein damage, degradation, and acetylation in the context of aging. J Gerontol A Biol Sci Med Sci 70, 1386–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph AM, Adhihetty PJ, Wawrzyniak NR, Wohlgemuth SE, Picca A, Kujoth GC, Prolla TA & Leeuwenburgh C (2013). Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS One 8, e69327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Rodriguez‐Enriquez S & Lemasters JJ (2007). Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462, 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka AR, Suer MK, Wolff CA & Harber MP (2014). Markers of human skeletal muscle mitochondrial biogenesis and quality control: effects of age and aerobic exercise training. J Gerontol A Biol Sci Med Sci 69, 371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraytsberg Y & Khrapko K (2005). Single‐molecule PCR: an artifact‐free PCR approach for the analysis of somatic mutations. Expert Rev Mol Diagn 5, 809–815. [DOI] [PubMed] [Google Scholar]
- Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN & Turnbull DM (2008). What causes mitochondrial DNA deletions in human cells? Nat Genet 40, 275–279. [DOI] [PubMed] [Google Scholar]
- Landi F, Liperoti R, Russo A, Giovannini S, Tosato M, Capoluongo E, Bernabei R & Onder G (2012). Sarcopenia as a risk factor for falls in elderly individuals: results from the ilSIRENTE study. Clin Nutr 31, 652–658. [DOI] [PubMed] [Google Scholar]
- Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP & Nair KS (2008). Endurance exercise as a countermeasure for aging. Diabetes 57, 2933–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc‐Gaudet JP, Picard M, Pelletier FS, Sgarioto N, Auger MJ, Vallee J, Robitaille R, St‐Pierre DH & Gouspillou G (2015). Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6, 17923–17937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexell J (1997). Evidence for nervous system degeneration with advancing age. J Nutr 127, 1011S–1013S. [DOI] [PubMed] [Google Scholar]
- Lexell J & Downham DY (1991). The occurrence of fibre‐type grouping in healthy human muscle: a quantitative study of cross‐sections of whole vastus lateralis from men between 15 and 83 years. Acta Neuropathol 81, 377–381. [DOI] [PubMed] [Google Scholar]
- Lexell J & Taylor CC (1991). Variability in muscle fibre areas in whole human quadriceps muscle: effects of increasing age. J Anat 174, 239–249. [PMC free article] [PubMed] [Google Scholar]
- Li M, Schroder R, Ni S, Madea B & Stoneking M (2015). Extensive tissue‐related and allele‐related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc Natl Acad Sci USA 112, 2491–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C, Poulsen P, Carlsson E, Ridderstrale M, Almgren P, Wojtaszewski J, Beck‐Nielsen H, Groop L & Vaag A (2004). Multiple environmental and genetic factors influence skeletal muscle PGC‐1α and PGC‐1β gene expression in twins. J Clin Invest 114, 1518–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JP, Safdar A, Wilkin GP, Tarnopolsky MA & Gibala MJ (2010). A practical model of low‐volume high‐intensity interval training induces mitochondrial biogenesis in human skeletal muscle: potential mechanisms. J Physiol 588, 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S & Sandri M (2009). Autophagy is required to maintain muscle mass. Cell Metab 10, 507–515. [DOI] [PubMed] [Google Scholar]
- McFarland R & Turnbull DM (2009). Batteries not included: diagnosis and management of mitochondrial disease. J Intern Med 265, 210–228. [DOI] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM & Fon EA (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner C, Bruse P & Oehmichen M (2006). Tissue‐specific deletion patterns of the mitochondrial genome with advancing age. Exp Gerontol 41, 518–524. [DOI] [PubMed] [Google Scholar]
- Murphy JL, Ratnaike TE, Shang ES, Falkous G, Blakely EL, Alston CL, Taivassalo T, Haller RG, Taylor RW & Turnbull DM (2012). Cytochrome c oxidase‐intermediate fibres: Importance in understanding the pathogenesis and treatment of mitochondrial myopathy. Neuromuscul Disord 22, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Isobe K, Nakada K & Hayashi JI (2001). Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet 28, 272–275. [DOI] [PubMed] [Google Scholar]
- Picard M (2011). Pathways to aging: the mitochondrion at the intersection of biological and psychosocial sciences. J Aging Res 2011, 814096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Jung B, Liang F, Azuelos I, Hussain S, Goldberg P, Godin R, Danialou G, Chaturvedi R, Rygiel K, Matecki S, Jaber S, Des Rosiers C, Karpati G, Ferri L, Burelle Y, Turnbull DM, Taivassalo T & Petrof BJ (2012). Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am J Respir Crit Care Med 186, 1140–1149. [DOI] [PubMed] [Google Scholar]
- Picard M, Ritchie D, Thomas MM, Wright KJ & Hepple RT (2011). Alterations in intrinsic mitochondrial function with aging are fiber type‐specific and do not explain differential atrophy between muscles. Aging Cell 10, 1047–1055. [DOI] [PubMed] [Google Scholar]
- Picard M, Shirihai OS, Gentil BJ & Burelle Y (2013). Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol 304, R393–R406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O'Hearn S, Levy S, Potluri P, Lvova M, Davila A, Lin CS, Perin JC, Rappaport EF, Hakonarson H, Trounce IA, Procaccio V & Wallace DC (2014). Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci USA 111, E4033–E4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogozelski AR, Geng T, Li P, Yin X, Lira VA, Zhang M, Chi JT & Yan Z (2009). p38γ mitogen‐activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS One 4, e7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh TD, Conklin MW, Evans TD, Polewski MA, Barbian HJ, Pass R, Anderson BD, Colman RJ, Eliceiri KW, Keely PJ, Weindruch R, Beasley TM & Anderson RM (2013). A shift in energy metabolism anticipates the onset of sarcopenia in rhesus monkeys. Aging Cell 12, 672–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N & Lippincott‐Schwartz J (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108, 10190–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan SL, Rygiel K, Purves‐Smith FM, Solbak NM, Turnbull DM & Hepple RT (2012). Denervation causes fiber atrophy and myosin heavy chain co‐expression in senescent skeletal muscle. PLoS One 7, e29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe GC, El‐Khoury R, Patten IS, Rustin P & Arany Z (2012). PGC‐1α is dispensable for exercise‐induced mitochondrial biogenesis in skeletal muscle. PLoS One 7, e41817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rygiel KA, Grady JP & Turnbull DM (2014). Respiratory chain deficiency in aged spinal motor neurons. Neurobiol Aging 35, 2230–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rygiel KA, Miller J, Grady JP, Rocha MC, Taylor RW & Turnbull DM (2014). Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol Appl Neurobiol 41, 288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M & Tarnopolsky MA (2011). Exercise increases mitochondrial PGC‐1α content and promotes nuclear‐mitochondrial cross‐talk to coordinate mitochondrial biogenesis. J Biol Chem 286, 10605–10617. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Scelsi R, Marchetti C & Poggi P (1980). Histochemical and ultrastructural aspects of m. vastus lateralis in sedentary old people (age 65–89 years). Acta Neuropathol 51, 99–105. [DOI] [PubMed] [Google Scholar]
- Short KR, Bigelow ML, Kahl J, Singh R, Coenen‐Schimke J, Raghavakaimal S & Nair KS (2005). Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci USA 102, 5618–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutt T, Geoffrion M, Milne R & McBride HM (2012). The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep 13, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutt TE & McBride HM (2013). Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta 1833, 417–424. [DOI] [PubMed] [Google Scholar]
- Stalberg E & Fawcett PR (1982). Macro EMG in healthy subjects of different ages. J Neurol Neurosurg Psychiatry 45, 870–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauch KL, Purnell PR & Fox HS (2014). Quantitative proteomics of synaptic and nonsynaptic mitochondria: insights for synaptic mitochondrial vulnerability. J Proteome Res 13, 2620–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JB & Chinnery PF (2015). The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet 16, 530–542. [DOI] [PubMed] [Google Scholar]
- Stump CS, Short KR, Bigelow ML, Schimke JM & Nair KS (2003). Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci USA 100, 7996–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Qiao H, Pan PY, Chen Y & Sheng ZH (2013). Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep 4, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnopolsky MA, Rennie CD, Robertshaw HA, Fedak‐Tarnopolsky SN, Devries MC & Hamadeh MJ (2007). Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol Regul Integr Comp Physiol 292, R1271–R1278. [DOI] [PubMed] [Google Scholar]
- Taylor RW & Turnbull DM (2005). Mitochondrial DNA mutations in human disease. Nat Rev Genet 6, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengan CH & Moraes CT (1998). Duplication and triplication with staggered breakpoints in human mitochondrial DNA. Biochim Biophys Acta 1406, 73–80. [DOI] [PubMed] [Google Scholar]
- Tessari P, Biolo G, Inchiostro S, Sacca L, Nosadini R, Boscarato MT, Trevisan R, De Kreutzenberg SV & Tiengo A (1990). Effects of insulin on whole body and forearm leucine and KIC metabolism in type 1 diabetes. Am J Physiol Endocrinol Metab 259, E96–E103. [DOI] [PubMed] [Google Scholar]
- Tomlinson BE & Irving D (1977). The numbers of limb motor neurons in the human lumbosacral cord throughout life. J Neurol Sci 34, 213–219. [DOI] [PubMed] [Google Scholar]
- Trappe S, Hayes E, Galpin A, Kaminsky L, Jemiolo B, Fink W, Trappe T, Jansson A, Gustafsson T & Tesch P (2013). New records in aerobic power among octogenarian lifelong endurance athletes. J Appl Physiol (1985) 114, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uguccioni G & Hood DA (2011). The importance of PGC‐1α in contractile activity‐induced mitochondrial adaptations. Am J Physiol Endocrinol Metab 300, E361–E371. [DOI] [PubMed] [Google Scholar]
- Vigelso A, Andersen NB & Dela F (2014). The relationship between skeletal muscle mitochondrial citrate synthase activity and whole body oxygen uptake adaptations in response to exercise training. Int J Physiol Pathophysiol Pharmacol 6, 84–101. [PMC free article] [PubMed] [Google Scholar]
- Visser M & Schaap LA (2011). Consequences of sarcopenia. Clin Geriatr Med 27, 387–399. [DOI] [PubMed] [Google Scholar]
- Vos M, Lauwers E & Verstreken P (2010). Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci 2, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2013). A mitochondrial bioenergetic etiology of disease. J Clin Invest 123, 1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenz T, Rossi SG, Rotundo RL, Spiegelman BM & Moraes CT (2009). Increased muscle PGC‐1α expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA 106, 20405–20410. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Westermann B (2010). Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11, 872–884. [DOI] [PubMed] [Google Scholar]
- Wohlgemuth SE, Seo AY, Marzetti E, Lees HA & Leeuwenburgh C (2010). Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life‐long exercise. Exp Gerontol 45, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC & Spiegelman BM (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC‐1. Cell 98, 115–124. [DOI] [PubMed] [Google Scholar]
- Youle RJ & van der Bliek AM (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, Collia D, Chen Z, Wozniak DF, Leone TC & Kelly DP (2010). Total skeletal muscle PGC‐1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab 12, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]