

Abstract
Degenerative ataxias are a common form of neurodegenerative disease that affect about 20 individuals per 100,000. The autosomal dominant spinocerebellar ataxias (SCAs) are caused by a variety of protein coding mutations (single nucleotide changes, deletions and expansions) in single genes. Affected genes encode plasma membrane and intracellular ion channels, membrane receptors, protein kinases, protein phosphatases and proteins of unknown function. Although SCA‐linked genes are quite diverse they share two key features: first, they are highly, although not exclusively, expressed in cerebellar Purkinje neurons (PNs), and second, when mutated they lead ultimately to the degeneration of PNs. In this review we summarize ataxia‐related changes in PN neurophysiology that have been observed in various mouse knockout lines and in transgenic models of human SCA. We also highlight emerging evidence that altered metabotropic glutamate receptor signalling and disrupted calcium homeostasis in PNs form a common, early pathophysiological mechanism in SCAs. Together these findings indicate that aberrant calcium signalling and profound changes in PN neurophysiology precede PN cell loss and are likely to lead to cerebellar circuit dysfunction that explains behavioural signs of ataxia characteristic of the disease.
Abbreviations
- CF
climbing fibre
- DCN
deep cerebellar nucleus
- GPCR
G‐protein‐coupled glutamate receptor
- IP3
inositol trisphosphate
- IP3R
inositol trisphosphate receptor
- LTD
long‐term depression
- LTP
long‐term potentiation
- mGluR
metabotropic glutamate receptor
- MF
mossy fibre
- PF
parallel fibre
- PN
Purkinje neuron
- SCA
spinocerebellar ataxia
Introduction
The autosomal dominant class of spinocerebellar ataxias (SCAs) comprises disorders caused by single gene mutations in more than 30 distinct genetic loci. The consequences of these genetic alterations encompass single amino acid changes, deletions, and repeat expansions in protein sequence. The diverse set of genes implicated is enriched in signalling proteins and proteins that interact with them such as ion channels, kinases, phosphatases and growth factor receptors. However, there are also several genes that encode proteins of undetermined function, as well as proteins involved in histone regulation. Even mutations in the TATA binding protein can cause a form of SCA. Thus, there is no unifying theme for how alterations in these genes lead to a cerebellar disorder characterized at least at the earliest stages by dysfunction of, and at later stages eventual loss of cerebellar Purkinje neurons (PNs).
A clearly central question is why these genetic insults with such diverse gene products all lead to PN vulnerability. Relatedly, it remains unclear whether there are any common pathophysiological mechanisms in SCAs. This short review will not address the considerable progress that has been made in elucidating molecular and cell biological aetiologies of the different forms of SCA. We direct readers to several excellent reviews focusing on such topics, particularly those involving details of RNA processing and post‐translational protein modifications in disease pathology (Carlson et al. 2009; Paulson, 2009; Orr, 2013). Rather, we will focus on changes in PN physiology that are common to several mouse models of SCA and that occur early in the disease (i.e. prior to significant PN loss). These pathophysiological changes are likely to explain ataxic behaviour observed at these points in disease progression and more importantly could serve as a point of leverage in developing treatments for these disorders.
Alterations in spontaneous action potential firing in PNs in SCAs
A hallmark of the physiology of PNs is that they are highly, intrinsically active. This sustained activity does not require excitatory synaptic input but arises as a result of the concerted activity of a unique set of ion channels expressed by PNs (Raman & Bean, 1999; Khaliq et al. 2003). Together these channels prompt PNs to generate their own regular spiking activity such that enzymatically dissociated PNs fire at approximately 40 Hz with striking, almost metronomic regularity (Raman & Bean, 1999). Under these conditions, coefficients of variation in inter‐spike instantaneous frequencies are typically less than 10% over long periods of time (Hausser & Clark, 1997; Smith & Otis, 2003). Thus, individual PNs appear to have a characteristic baseline firing rate or set point, and across a population of PNs, firing rates are much more variable than within any single PN over time. In vivo, PN firing varies systematically across different regions with baseline firing rates ranging from 40 to 100 Hz (Zhou et al. 2014). Of course, the interaction between this intrinsically generated spiking and excitatory and inhibitory synaptic inputs generate the more variable and complex patterns of spiking observed in vivo. Nonetheless, we use the term ‘pacemaking’ to refer to two features of the robust intrinsic excitability: PNs fire at high spontaneous rates, and, in the absence of synaptic inputs, this firing is remarkably regular.
Such sustained, regular PN firing is degraded in at least six mouse models of SCA and in a number of other non‐SCA‐related transgenic mouse models that exhibit behavioural ataxia. In a PN‐specific, human transgene model of SCA2 (pcp2‐ Atxn2127Q), the progressive dysregulation of transcriptional expression patterns and the severity of behavioural ataxia track the reduction of mean PN firing rates over an 8 month time course of disease progression (Hansen et al. 2013). This model recapitulates basic features of the human disease such as intact motor behaviour and normal cerebellar morphology at birth and in early adulthood followed by progression of motor dysfunction and PN loss in later life (Pulst et al. 1996; Hansen et al. 2013). Another PN‐specific SCA2 model with a smaller CAG repeat length (pcp2‐ATXN2Q58) similarly shows impaired firing rates and less regular firing (Kasumu et al. 2012 a,b). Progressive reductions in pacemaking have also been described in PN‐specific models of SCA 1 (pcp2‐ATXN1Q82) (Hourez et al. 2011), in a global YAC transgenic for SCA3 (ATXN3Q84) (Shakkottai et al. 2011), and in a β‐III spectrin knockout mouse model of SCA5 (Perkins et al. 2010). Tellingly, the SCA5 model exhibited reductions in a resurgent component of voltage‐gated Na+ current known to play a key role in pacemaking (Perkins et al. 2010). Lastly, a PN‐specific SCA6 transgenic mouse line expressing a C‐terminal fragment, corresponding to exon 47, of the P‐type Ca2+ channel containing 27 polyglutamine repeats has been characterized (Mark et al. 2015). PN firing rates in this mouse are reduced and firing becomes markedly irregular. Importantly, in all of these mouse lines utilizing different transgenes, promoters and SCA subtypes, the loss of pacemaking ability tightly correlates with behavioural ataxia. Moreover, degradation in the physiological output of PNs precedes overt loss of PNs. These findings suggest that reduced PN spiking output could be a common pathophysiological feature of SCAs and that it may contribute to the ataxic symptoms characteristic of the disease class.
Consistent with the hypothesis that reduced PN pacemaking contributes to ataxia, several transgenic mouse lines in which cerebellar genes are deleted also show slowed PN firing rates and an ataxic phenotype. The moonwalker mouse line, characterized by a point mutation in the TRPC3 ion channel causing its constitutive activation, shows profound reductions in PN firing frequencies (Sekerkova et al. 2013). A PN‐specific TSC1 knockout mouse (pcp2‐TSC1−/−) shows significant reductions in firing with loss of one copy and more severe reductions with loss of both copies of the TSC1 gene (Tsai et al. 2012). Finally, both global (Rbfox1+/loxP/Rbfox2loxP/loxP/Nestin‐Cre+/–) and PN‐specific (RbfoxloxP/loxP/Rbfox2loxP/loxP/pcp2‐Cre+/–) lines in which copies of the RNA splicing genes Rbfox1 and ‐2 have been deleted show ataxic behaviour and reduced PN firing (Gehman et al. 2012). Here, similar to the SCA5 mouse model described above, there is evidence for disordered expression of splice variants of the resurgent Na+ channel isoform NaV1.6 encoded by the gene Scn8a (Gehman et al. 2012). Although these mouse models do not recapitulate genetic forms of SCAs, the correspondence between reduced PN intrinsic excitability and the behavioural ataxia is supportive of the key role this electrophysiological feature plays in normal motor behaviour.
More subtle changes in PN pacemaking have also been linked to ataxia. In mutant mouse lines containing mutations in P‐type calcium channel genes, Walter and colleagues describe changes in the regularity of PN firing but not the average firing rate that correlate with behavioural ataxia. They find that treatment of mice with EBIO‐1, a positive modulator of SK‐type calcium‐activated potassium channels, reduces the ataxia and causes PN firing to become more regular in the mutant animals (Walter et al. 2006). These findings imply that subtle changes in the pattern of PN output can also lead to ataxic behaviour.
Circuit consequences of reduced PN output
PN output is directed exclusively to deep cerebellar nucleus (DCN) and vestibular nucleus neurons (Fig. 1). PNs are GABAergic and so their tonic activity provides a potent baseline inhibition of these downstream target neurons, some of which function as premotor neurons, for example in the descending rubrospinal motor pathway. Assuming there is no compensation in downstream pathways, loss of tonic inhibition from PNs would be expected to lead to consequences at a circuit level, such as increased cerebellar nuclear neuron excitability and increased motor drive. Confirming this prediction, optogenetic experiments in which PN output is transiently silenced show that cerebellar nucleus neurons burst and that this drives rapid movements (Heiney et al. 2014; Lee et al. 2015). In addition, optogenetic stimuli affecting PN firing are very effective at driving associative learning at a behavioural level (Lee et al. 2015). Such stimuli may drive activity‐dependent synaptic plasticity; in PNs, this would result in parallel fibre (PF)–PN long‐term depression (LTD) and thus reduced excitatory drive from PFs (the red burst in Fig. 1), while in DCN, it would result in mossy fibre (MF)–DCN long‐term potentiation (LTP) leading to increased excitatory drive onto DCN neurons (the blue burst in Fig. 1). All of these circuit changes would be expected to promote the sort of ectopic movements observed after optogenetic training (Lee et al. 2015).
Figure 1. Simplified schematic diagram of the cerebellar circuit .
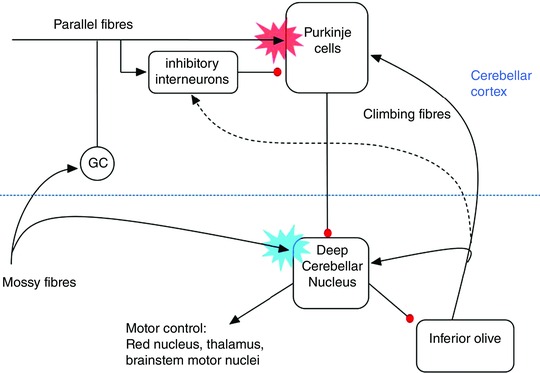
Black arrowheads represent excitatory and red circles denote inhibitory connections. The dashed arrow indicates an unconventional connection between climbing fibres and interneurons (Szapiro & Barbour, 2007; Mathews et al. 2012). The red and blue starbursts indicate hypothesized sites of climbing fibre‐instructed, associative forms of synaptic plasticity required for associative motor learning (LTD of Purkinje fibre inputs and LTP of mossy fibre inputs, respectively). These forms of plasticity may be saturated during the SCA disease process.
So, given SCA‐associated alterations in PN firing, why are ectopic movements, dystonia and/or chorea not observed in SCA? Our working hypothesis is that neurons downstream of PNs, e.g. cerebellar nucleus neurons, red nucleus neurons and thalamic neurons (see Fig. 1) compensate for the increased excitability that may result from the reduction in PN inhibition. Future experiments will be required to explore these possibilities and their functional contributions to the ataxic phenotype.
Metabotropic glutamate receptors and intracellular calcium mobilization in SCAs
Other changes that have been observed in PN physiology as a consequence of SCA involve inositol trisphosphate receptor (IP3R)‐linked calcium signalling networks. PNs express IP3Rs at extremely high levels and these IP3‐gated intracellular calcium channels are downstream of the metabotropic glutamate receptor type I (mGluR1), a G‐protein‐coupled glutamate receptor (GPCR). Glutamate released from either of the two excitatory synaptic inputs to PNs (see Fig. 1), PFs or climbing fibres (CFs), activates glutamate‐gated ion channels (AMPA receptors; AMPARs), which are responsible for the fast electrical signals, but also mGluR1 (Batchelor & Garthwaite, 1997; Brasnjo & Otis, 2001; Dzubay & Otis, 2002). These GPCRs are then coupled to various biochemical pathways mainly via heterotrimeric G proteins containing the Gαq subunit (Offermanns et al. 1997; Tanaka, 2000; Hartmann et al. 2004). A principal set of biochemical pathways activated by Gαq involves phospholipase Cβ (PLCβ), which generates IP3 and triggers intracellular Ca2+ release from ER stores that sit at the base of dendritic spines (Finch & Augustine, 1998; Takechi et al. 1998). Another, more direct limb of the Gαq pathway activates TRPC3 ion channels on the plasma membrane, leading to a slow synaptic current. Activation of TRPC3 requires Gαq (Hartmann et al. 2004) but is independent of PLCβ and of the IP3R (Dzubay & Otis, 2002; Hartmann et al. 2008, 2011). This suggests a model in which signalling diverges from Gαq to TRPC3 channel and to PLCβ–IP3R limbs as indicated in Fig. 2. A third pathway stimulates local protein synthesis regulated by the Fragile X protein (Huber, 2006) (not shown in Fig. 2).
Figure 2. Schematic diagram of the mGluR–Ca2+ hypothesis .
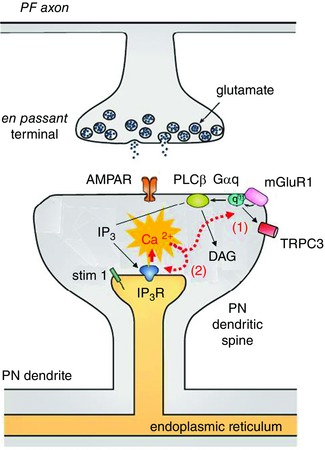
Glutamate release from PFs activates both AMPARs and mGluR1 GPCRs. As indicated by the black arrows, mGluR1 is coupled to PLCβ, which leads to release of Ca2+ from IP3Rs on the endoplasmic reticulum. Ca2+ exerts positive feedback on mGluR1 transduction at a step early in the cascade (1) as well as at the IP3R (2). Thus, elevations in Ca2+ will exacerbate the IP3R hyperactivity observed in SCA2. Modified from Hartmann et al. (2011). DAG, diacylglycerol.
A central role for mGluR dysregulation in cerebellar ataxia is supported by abundant evidence from mouse and human genetics, implicating each of the signalling proteins in the cascade from mGluR1 to the IP3R. Importantly, data from dozens of mouse models show that ataxia can result from reductions or increases in several elements of the mGluR signalling cascade, presenting a complex picture of the pathophysiological role of this key feature of PN biology. Below we summarize evidence and speculate on a possible unified view of how this signalling mechanism could be central to this class of disorders.
Evidence suggesting that reduced mGluR signalling plays a role in SCA
Genetic reductions in mGluR signalling have been studied using knockout mice for mGluR1 (Aiba et al. 1994; Conquet et al. 1994), Gαq (Offermanns et al. 1997; Hartmann et al. 2004), PLCβ (Kano et al. 1998; Miyata et al. 2001), IP3R (Matsumoto et al. 1996; van de Leemput et al. 2007) and TRPC3 (Hartmann et al. 2008); in addition a conditional mGluR1 knockout and PN‐specific mGluR1 rescue mouse line have been generated (Ichise et al. 2000; Nakao et al. 2007). All of the genetic deletion mice show ataxia and deficits in cerebellum‐dependent forms of motor learning while the rescue mouse restores these cerebellar functions. In humans, loss of function alleles of some of these genes are known to cause specific ataxias. These include SCA15, which is caused by a mutation in the gene for the IP3R (van de Leemput et al. 2007), as well as a recessive form of congenital ataxia caused by mutations in the mGluR1 gene (Guergueltcheva et al. 2012). Finally, SCA14 is caused by constitutively activating mutations in PKCγ, a protein kinase downstream of mGluR/IP3R activation (Yabe et al. 2003).
There are also several mouse models of SCA in which it is reported that elements of the mGluR signalling/cytoplasmic calcium homeostasis cascades are downregulated. This has perhaps been studied most thoroughly in SCA1, where mRNAs for the type I IP3R, PKCγ, homer 3, the EAAT4 glutamate transporter, and the SERCA3 calcium pump are reduced (Lin et al. 2000; Serra et al. 2004, 2006). mRNA for mGluR1 is also reduced in SCA 82Q lines (Serra et al. 2006). At the protein level things are more complex as mGluR1 levels drop overall as assessed by quantitative Western blot (Zu et al. 2004), but such reductions are accompanied by a loss of dendritic complexity and spine density. At remaining spines mGluR protein levels appear unchanged (Skinner et al. 2001; Zu et al. 2004), raising the possibility that reductions in mGluR protein reflect disease‐related alterations in excitatory signalling but are not on their own a driver of pathological symptoms.
Other models also report indirect reductions in mGluR signalling due to changes in protein localization. An SCA3 mouse line expressing a truncated SCA3 transgene with a 69 poly Q repeat exhibits internalization of mGluR protein and a reduction in mGluR‐mediated cannabinoid release (Konno et al. 2014). Similarly, in an SCA5 mouse model expressing a mutant form of β‐III spectrin there is an apparent mislocalization of dendritic mGluR protein and degraded mGluR‐mediated physiology (Armbrust et al. 2014). These data along with those described for SCA1 above suggest that various subtypes of SCA lead to concerted pathophysiology of mGluR1 signalling and calcium homeostasis in PNs. Also consistent with this picture is the finding that acute pharmacological enhancement of mGluR1 with a positive allosteric modulator improves ataxic behaviour in an SCA1 154Q mouse line (Notartomaso et al. 2013).
Evidence suggesting that increased mGluR signalling plays a role in SCA
In the pcp2‐ATXN2Q58 mouse model, work has suggested that mGluR‐triggered, IP3R‐mobilized calcium elevations are enhanced in PNs (Liu et al. 2009) due to specific protein–protein interaction between the expanded repeat protein (ataxin 2) and the IP3R. Subsequent work showed that viral delivery of the IP3 degradation enzyme inositol 1,4,5‐phosphatase to PNs led to improvement in motor behaviour and neuropathology in the SCA2 58Q mouse (Kasumu et al. 2012 a).
Although there are no mouse lines that overexpress mGluR1 or downstream signalling elements, there is ample evidence that excessive mGluR signalling and elevated calcium levels can also lead to ataxia. One of the more striking examples comes from the moonwalker mouse line mentioned earlier. Here a point mutation in TRPC3 constitutively activates this cation channel, which sits downstream from mGluR1 (Fig. 2). This results in a severe ataxia in mice and concomitant loss of specific types of cerebellar neurons (Becker et al. 2009; Sekerkova et al. 2013). As mentioned above, this defect also alters PN firing. Interestingly, one adult onset case of ataxia has been described with a gain of function point mutation in TRPC3 and it will be interesting to see whether this mutation results in enhanced or constitutive channel activity (Fogel et al. 2015).
Genetic deletion of various other molecules required for normal calcium homeostasis, such as the calcium buffering proteins parvalbumin and calbindin D28K, results in altered PN physiology and calcium signalling, and behavioural ataxia (Airaksinen et al. 1997; Vecellio et al. 2000). There has also been work showing that impairment of calcium homeostasis exacerbates SCA pathology. Genetic deletion of one copy of the calcium buffering protein calbindin D28K enhances the ataxic phenotype in an SCA1 mouse model (Vig et al. 2012).
Loss of a single copy of the gene encoding the plasma membrane calcium pump PMCA also results in ataxia in mice (Empson et al. 2010), and rare mutations in this gene have been found in human ataxia patients (Zanni et al. 2012; Cali et al. 2015). Taken together, such findings strongly suggest that elevated calcium levels, one of the possible outcomes of increased activity in the mGluR signalling cascade, can compromise the health of PNs and lead to ataxia.
Is it possible to reconcile increases and decreases in mGluR signalling as a cause of ataxia?
Taken together the findings summarized above strongly implicate dysfunction in mGluR1 and calcium signalling pathways as causative for genetic forms of ataxia in mice and humans. However, the data also paradoxically indicate that either reductions or increases in these signalling cascades can lead to ataxia. While it is certainly possible that distinct SCA subtypes are associated with different changes in mGluR1 signalling and calcium homeostasis, we speculate that there could be common pathophysiological mechanisms in this broad class of disease.
A starting point is the assumption that, although the different genetic insults cause disease by different molecular mechanisms (e.g. transcriptional dysregulation, protein aggregation, impaired calcium homeostasis, altered excitability), as PNs sicken they gradually lose the ability to regulate intracellular calcium. Given the extensive intracellular calcium stores and various pathways for calcium entry across the plasma membrane of PNs, this assumption is almost certainly true at end stages of all types of SCA.
If PNs suffer from elevated basal calcium this could trigger a common disease process characterized by two potent positive feedback mechanisms (the mGluR–Ca2+ excitotoxicity hypothesis of SCA outlined in Fig. 2). Batchelor & Garthwaite (1997) first demonstrated that there is a robust form of positive feedback regulation exerted by intracellular Ca2+ on the mGluR signalling cascade. This finding, confirmed by subsequent studies, indicates that slightly elevated (200–300 nm) levels of intracellular Ca2+ strongly potentiate mGluR‐mediated signals such as TRPC3 currents (Dzubay & Otis, 2002) and IP3R‐initiated Ca2+ transients (Wang et al. 2000), suggesting that Ca2+ potentiates mGluR function at a PLCβ‐ and IP3R‐independent step early in the signal transduction cascade (see (1) in Fig. 2). It is also known that Ca2+ directly interacts with the IP3R (see (2) in in Fig. 2). At steady state, there is a bell‐shaped potentiation/inhibition exerted by Ca2+ on the IP3R such that 200–500 nm Ca2+ markedly potentiates while higher concentrations inhibit IP3R function (Bezprozvanny et al. 1991; Finch et al. 1991). In more dynamic circumstances, such as during physiological activity, an order dependence has been described in which IP3 followed by Ca2+ gives the largest enhancements (Sarkisov & Wang, 2008); interestingly, this positive feedback has been proposed to contribute to learning rules for PF LTD (Wang et al. 2000).
These two forms of Ca2+‐mediated positive feedback (on TRPC3 currents and on IP3R function) in Fig. 2 are likely to be independent of one another implying a strong and multifaceted potentiation exerted by Ca2+ on mGluR signalling. In PNs and in cerebellar nucleus neurons, changes in Ca2+ are required for forms of synaptic plasticity such as PF LTD (the red burst in Fig. 1) and MF LTP (the blue burst in Fig. 1 ) that are implicated in associative cerebellar learning. The mGluR–Ca2+ excitotoxicity hypothesis of SCA suggests that these forms of synaptic plasticity could become saturated as part of the SCA disease process. Moreover, the reductions in expression of mGluR1 signalling elements observed in some SCA models could reflect a compensatory mechanism driven by excessive signalling in this pathway.
Conclusions
It of course remains to be demonstrated whether the highly varied set of molecular alterations seen across SCAs share the two physiological mechanisms discussed here, slowed PN pacemaking, and dysregulated intracellular Ca2+ and alterations in the mGluR signalling cascade. Arguing in favour of this possibility is the fact that such mechanisms, pacemaking and IP3‐mediated Ca2+ signalling, are defining features of PN physiology, setting PNs apart from most other neuronal types. This could explain why, despite the ubiquity of expression of many of the SCA‐related gene products, these diseases, at least at their outset, selectively impact PNs. Although the root cause of the calcium dysregulation could be different for each of the SCAs, this hypothesis could also explain why the varied molecular lesions all result in PN pathophysiology and eventually degeneration. A mechanism involving slowly rising calcium levels reinforced by the positive feedback elements described here provides a potential explanation for the slowly progressive nature of the disease. However, experiments in the numerous SCA mouse models that are available will be required to settle the generality of this mechanism.
How might dysregulated intracellular Ca2+ and PN spontaneous firing interact? Ca2+‐activated SK potassium channels provide a critical brake on PN excitability; thus, the chronically elevated basal Ca2+ hypothesized to occur as part of SCA pathology could lead directly to the slowed pacemaking. It is also reasonable to speculate that chronically elevated Ca2+ could dysregulate the expression and or post‐translational modulation of the various ion channels required for pacemaking. Indeed, recent results have implicated changes in BK‐type, Ca2+‐activated and A‐type voltage‐gated potassium channels (Hourez et al. 2011; Dell'Orco et al. 2015). Finally, as mentioned above, the slowed PN firing and elevated Ca2+ could together lead to saturated forms of circuit plasticity, which would impair motor learning and lead to imbalances in the output of cerebellar circuitry. Finally, both of the putative pathophysiological mechanisms offer potential therapeutic targets by which one might normalize circuit activity and in this way treat the ataxic symptomology.
Additional information
Competing interests
None declared.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA & Tonegawa S (1994). Deficient cerebellar long‐term depression and impaired motor learning in mGluR1 mutant mice. Cell 79, 377–388. [PubMed] [Google Scholar]
- Airaksinen MS, Eilers J, Garaschuk O, Thoenen H, Konnerth A & Meyer M (1997). Ataxia and altered dendritic calcium signalling in mice carrying a targeted null mutation of the calbindin D28k gene. Proc Natl Acad Sci USA 94, 1488–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbrust KR, Wang X, Hathorn TJ, Cramer SW, Chen G, Zu T, Kangas T, Zink AN, Oz G, Ebner TJ & Ranum LP (2014). Mutant β‐III spectrin causes mGluR1α mislocalization and functional deficits in a mouse model of spinocerebellar ataxia type 5. J Neurosci 34, 9891–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor AM & Garthwaite J (1997). Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature 385, 74–77. [DOI] [PubMed] [Google Scholar]
- Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM & Davies KE (2009). A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc Natl Acad Sci USA 106, 6706–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J & Ehrlich BE (1991). Bell‐shaped calcium‐response curves of Ins(1,4,5)P3‐ and calcium‐gated channels from endoplasmic reticulum of cerebellum. Nature 351, 751–754. [DOI] [PubMed] [Google Scholar]
- Brasnjo G & Otis TS (2001). Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long‐term depression. Neuron 31, 607–616. [DOI] [PubMed] [Google Scholar]
- Cali T, Lopreiato R, Shimony J, Vineyard M, Frizzarin M, Zanni G, Zanotti G, Brini M, Shinawi M & Carafoli E (2015). A novel mutation in isoform 3 of the plasma membrane Ca2+ pump impairs cellular Ca2+ homeostasis in a patient with cerebellar ataxia and laminin subunit 1α mutations. J Biol Chem 290, 16132–16141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson KM, Andresen JM & Orr HT (2009). Emerging pathogenic pathways in the spinocerebellar ataxias. Curr Opin Genes Dev 19, 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz‐Bacon K, Reggiani A, Matarese V, Conde F, et al (1994). Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature 372, 237–243. [DOI] [PubMed] [Google Scholar]
- Dell'Orco JM, Wasserman AH, Chopra R, Ingram MA, Hu YS, Singh V, Wulff H, Opal P, Orr HT & Shakkottai VG (2015). Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J Neurosci 35, 11292–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzubay JA & Otis TS (2002). Climbing fibre activation of metabotropic glutamate receptors on cerebellar purkinje neurons. Neuron 36, 1159–1167. [DOI] [PubMed] [Google Scholar]
- Empson RM, Turner PR, Nagaraja RY, Beesley PW & Knopfel T (2010). Reduced expression of the Ca2+ transporter protein PMCA2 slows Ca2+ dynamics in mouse cerebellar Purkinje neurones and alters the precision of motor coordination. J Physiol 588, 907–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA & Augustine GJ (1998). Local calcium signalling by inositol‐1,4,5‐trisphosphate in Purkinje cell dendrites. Nature 396, 753–756. [DOI] [PubMed] [Google Scholar]
- Finch EA, Turner TJ & Goldin SM (1991). Calcium as a coagonist of inositol 1,4,5‐trisphosphate‐induced calcium release. Science 252, 443–446. [DOI] [PubMed] [Google Scholar]
- Fogel BL, Hanson SM & Becker EB (2015). Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov Disord 30, 284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Meera P, Stoilov P, Shiue L, O'Brien JE, Meisler MH, Ares M Jr, Otis TS & Black DL (2012). The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev 26, 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guergueltcheva V, Azmanov DN, Angelicheva D, Smith KR, Chamova T, Florez L, Bynevelt M, Nguyen T, Cherninkova S, Bojinova V, Kaprelyan A, Angelova L, Morar B, Chandler D, Kaneva R, Bahlo M, Tournev I & Kalaydjieva L (2012). Autosomal‐recessive congenital cerebellar ataxia is caused by mutations in metabotropic glutamate receptor 1. Am J Hum Genet 91, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen ST, Meera P, Otis TS & Pulst SM (2013). Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet 22, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Henning HA & Konnerth A (2011). mGluR1/TRPC3‐mediated synaptic transmission and calcium signaling in mammalian central neurons. Cold Spring Harb Perspect Biol 3, a006726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Blum R, Kovalchuk Y, Adelsberger H, Kuner R, Durand GM, Miyata M, Kano M, Offermanns S & Konnerth A (2004). Distinct roles of Gαq and Gα11 for Purkinje cell signaling and motor behavior. J Neurosci 24, 5119–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L & Konnerth A (2008). TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 59, 392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser M & Clark BA (1997). Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron 19, 665–678. [DOI] [PubMed] [Google Scholar]
- Heiney SA, Kim J, Augustine GJ & Medina JF (2014). Precise control of movement kinematics by optogenetic inhibition of Purkinje cell activity. J Neurosci 34, 2321–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d'Exaerde A, Cheron G, Orr HT, Pandolfo M & Schiffmann SN (2011). Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci 31, 11795–11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM (2006). The fragile X‐cerebellum connection. Trends Neurosci 29, 183–185. [DOI] [PubMed] [Google Scholar]
- Ichise T, Kano M, Hashimoto K, Yanagihara D, Nakao K, Shigemoto R, Katsuki M & Aiba A (2000). mGluR1 in cerebellar Purkinje cells essential for long‐term depression, synapse elimination, and motor coordination. Science 288, 1832–1835. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Watanabe M, Kurihara H, Offermanns S, Jiang H, Wu Y, Jun K, Shin HS, Inoue Y, Simon MI & Wu D (1998). Phospholipase cβ4 is specifically involved in climbing fiber synapse elimination in the developing cerebellum. Proc Natl Acad Sci USA 95, 15724–15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasumu AW, Hougaard C, Rode F, Jacobsen TA, Sabatier JM, Eriksen BL, Strobaek D, Liang X, Egorova P, Vorontsova D, Christophersen P, Ronn LC & Bezprozvanny I (2012. b). Selective positive modulator of calcium‐activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol 19, 1340–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasumu AW, Liang X, Egorova P, Vorontsova D & Bezprozvanny I (2012. a). Chronic suppression of inositol 1,4,5‐triphosphate receptor‐mediated calcium signaling in cerebellar purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J Neurosci 32, 12786–12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW & Raman IM (2003). The contribution of resurgent sodium current to high‐frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci 23, 4899–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno A, Shuvaev AN, Miyake N, Miyake K, Iizuka A, Matsuura S, Huda F, Nakamura K, Yanagi S, Shimada T & Hirai H (2014). Mutant ataxin‐3 with an abnormally expanded polyglutamine chain disrupts dendritic development and metabotropic glutamate receptor signaling in mouse cerebellar Purkinje cells. Cerebellum 13, 29–41. [DOI] [PubMed] [Google Scholar]
- Lee KH, Mathews PJ, Reeves AM, Choe KY, Jami SA, Serrano RE & Otis TS (2015). Circuit mechanisms underlying motor memory formation in the cerebellum. Neuron 86, 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Antalffy B, Kang D, Orr HT & Zoghbi HY (2000). Polyglutamine expansion down‐regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci 3, 157–163. [DOI] [PubMed] [Google Scholar]
- Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP, Pulst SM & Bezprozvanny I (2009). Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci 29, 9148–9162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark MD, Krause M, Boele HJ, Kruse W, Pollok S, Kuner T, Dalkara D, Koekkoek S, De Zeeuw CI & Herlitze S (2015). Spinocerebellar ataxia type 6 protein aggregates cause deficits in motor learning and cerebellar plasticity. J Neurosci 35, 8882–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews PJ, Lee KH, Peng Z, Houser CR & Otis TS (2012). Effects of climbing fiber driven inhibition on Purkinje neuron spiking. J Neurosci 32, 17988–17997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K & Noda T (1996). Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5‐trisphosphate receptor. Nature 379, 168–171. [DOI] [PubMed] [Google Scholar]
- Miyata M, Kim HT, Hashimoto K, Lee TK, Cho SY, Jiang H, Wu Y, Jun K, Wu D, Kano M & Shin HS (2001). Deficient long‐term synaptic depression in the rostral cerebellum correlated with impaired motor learning in phospholipase C β4 mutant mice. Eur J Neurosci 13, 1945–1954. [DOI] [PubMed] [Google Scholar]
- Nakao H, Nakao K, Kano M & Aiba A (2007). Metabotropic glutamate receptor subtype‐1 is essential for motor coordination in the adult cerebellum. Neurosci Res 57, 538–543. [DOI] [PubMed] [Google Scholar]
- Notartomaso S, Zappulla C, Biagioni F, Cannella M, Bucci D, Mascio G, Scarselli P, Fazio F, Weisz F, Lionetto L, Simmaco M, Gradini R, Battaglia G, Signore M, Puliti A & Nicoletti F (2013). Pharmacological enhancement of mGlu1 metabotropic glutamate receptors causes a prolonged symptomatic benefit in a mouse model of spinocerebellar ataxia type 1. Mol Brain 6, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, Inoue Y, Kano M & Simon MI (1997). Impaired motor coordination and persistent multiple climbing fibre innervation of cerebellar Purkinje cells in mice lacking Gαq. Proc Natl Acad Sci USA 94, 14089–14094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT (2013). Toxic RNA as a driver of disease in a common form of ALS and dementia. Proc Natl Acad Sci USA 110, 7533–7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL (2009). The spinocerebellar ataxias. J Neuroophthalmol 29, 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins EM, Clarkson YL, Sabatier N, Longhurst DM, Millward CP, Jack J, Toraiwa J, Watanabe M, Rothstein JD, Lyndon AR, Wyllie DJ, Dutia MB & Jackson M (2010). Loss of β‐III spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J Neurosci 30, 4857–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes‐Cendes I, Pearlman S, Starkman S, Orozco‐Diaz G, Lunkes A, DeJong P, Rouleau GA, Auburger G, Korenberg JR, Figueroa C & Sahba S (1996). Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 14, 269–276. [DOI] [PubMed] [Google Scholar]
- Raman IM & Bean BP (1999). Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci 19, 1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisov DV & Wang SS (2008). Order‐dependent coincidence detection in cerebellar Purkinje neurons at the inositol trisphosphate receptor. J Neurosci 28, 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekerkova G, Kim JA, Nigro MJ, Becker EB, Hartmann J, Birnbaumer L, Mugnaini E & Martina M (2013). Early onset of ataxia in moonwalker mice is accompanied by complete ablation of type II unipolar brush cells and Purkinje cell dysfunction. J Neurosci 33, 19689–19694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY & Orr HT (2004). Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet 13, 2535–2543. [DOI] [PubMed] [Google Scholar]
- Serra HG, Duvick L, Zu T, Carlson K, Stevens S, Jorgensen N, Lysholm A, Burright E, Zoghbi HY, Clark HB, Andresen JM & Orr HT (2006). RORα‐mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 127, 697–708. [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, do Carmo Costa M, Dell'Orco JM, Sankaranarayanan A, Wulff H & Paulson HL (2011). Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci 31, 13002–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner PJ, Vierra‐Green CA, Clark HB, Zoghbi HY & Orr HT (2001). Altered trafficking of membrane proteins in purkinje cells of SCA1 transgenic mice. Am J Pathol 159, 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SL & Otis TS (2003). Persistent changes in spontaneous firing of Purkinje neurons triggered by the nitric oxide signaling cascade. J Neurosci 23, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szapiro G & Barbour B (2007). Multiple climbing fibers signal to molecular layer interneurons exclusively via glutamate spillover. Nat Neurosci 10, 735–742. [DOI] [PubMed] [Google Scholar]
- Takechi H, Eilers J & Konnerth A (1998). A new class of synaptic response involving calcium release in dendritic spines. Nature 396, 757–760. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Nakagawa S, Kushiya E, Yamasaki M, Fukaya M, Iwanaga T, Simon MI, Kano M & Watanabe M (2000). Gq protein alpha subunits Galphaq and Galpha11 are localized at postsynaptic extra‐junctional membrane of cerebellar Purkinje cells and hippocampal pyramidal cells. Eur J Neurosci 12, 781–792. [DOI] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene‐Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG & Sahin M (2012). Autistic‐like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR, Houlden H, Gwinn‐Hardy K, Fung HC, Lin X, Hernandez D, Simon‐Sanchez J, Wood NW, Giunti P, Rafferty I, Hardy J, Storey E, Gardner RJ, Forrest SM, Fisher EM, Russell JT, Cai H & Singleton AB (2007). Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet 3, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecellio M, Schwaller B, Meyer M, Hunziker W & Celio MR (2000). Alterations in Purkinje cell spines of calbindin D‐28 k and parvalbumin knock‐out mice. Eur J Neurosci 12, 945–954. [DOI] [PubMed] [Google Scholar]
- Vig PJ, Wei J, Shao Q, Lopez ME, Halperin R & Gerber J (2012). Suppression of calbindin‐D28k expression exacerbates SCA1 phenotype in a disease mouse model. Cerebellum 11, 718–732. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C & Khodakhah K (2006). Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci 9, 389–397. [DOI] [PubMed] [Google Scholar]
- Wang SS, Denk W & Hausser M (2000). Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci 3, 1266–1273. [DOI] [PubMed] [Google Scholar]
- Yabe I, Sasaki H, Chen DH, Raskind WH, Bird TD, Yamashita I, Tsuji S, Kikuchi S & Tashiro K (2003). Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C γ. Arch Neurol 60, 1749–1751. [DOI] [PubMed] [Google Scholar]
- Zanni G, Cali T, Kalscheuer VM, Ottolini D, Barresi S, Lebrun N, Montecchi‐Palazzi L, Hu H, Chelly J, Bertini E, Brini M & Carafoli E (2012). Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X‐linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc Natl Acad Sci USA 109, 14514–14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Lin Z, Voges K, Ju C, Gao Z, Bosman LW, Ruigrok TJ, Hoebeek FE, De Zeeuw CI & Schonewille M (2014). Cerebellar modules operate at different frequencies. eLife 3, e02536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB & Orr HT (2004). Recovery from polyglutamine‐induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci 24, 8853–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]