

ABSTRACT
Coculturing dark- and photofermentative bacteria is a promising strategy for enhanced hydrogen (H2) production. In this study, next-generation sequencing was used to query the global transcriptomic responses of an artificial coculture of Clostridium cellulovorans 743B and Rhodopseudomonas palustris CGA009. By analyzing differentially regulated gene expression, we showed that, consistent with the physiological observations of enhanced H2 production and cellulose degradation, the nitrogen fixation genes in R. palustris and the cellulosomal genes in C. cellulovorans were upregulated in cocultures. Unexpectedly, genes related to H2 production in C. cellulovorans were downregulated, suggesting that the enhanced H2 yield was contributed mainly by R. palustris. A number of genes related to biosynthesis of volatile fatty acids (VFAs) in C. cellulovorans were upregulated, and correspondingly, a gene that mediates organic compound catabolism in R. palustris was also upregulated. Interestingly, a number of genes responsible for chemotaxis in R. palustris were upregulated, which might be elicited by the VFA concentration gradient created by C. cellulovorans. In addition, genes responsible for sulfur and thiamine metabolism in C. cellulovorans were downregulated in cocultures, and this could be due to a response to pH changes. A conceptual model illustrating the interactions between the two organisms was constructed based on the transcriptomic results.
IMPORTANCE The findings of this study have important biotechnology applications for biohydrogen production using renewable cellulose, which is an industrially and economically important bioenergy process. Since the molecular characteristics of the interactions of a coculture when cellulose is the substrate are still unclear, this work will be of interest to microbiologists seeking to better understand and optimize hydrogen-producing coculture systems.
INTRODUCTION
Microorganisms do not live in isolation in nature; instead, they interact with each other in complex ecological networks within a microbial community in order to function and resist stresses in various environments (1). In general, interspecies interactions define the characteristics and robustness of microbial communities. The concept of microbe-microbe interactions has been harnessed and applied in the bioremediation (2, 3), food and beverage (4–7), and biofuel (8–12) industries. The application of multispecies systems could be a promising alternative bioprocess strategy to the use of single species, which requires extensive genetic engineering before multiple desirable traits are incorporated, whereas these functions are distributed among different organisms in a multispecies system (13). However, multispecies systems can be challenging to control (14); therefore, a detailed understanding of the physiological and molecular mechanisms of microbial interactions is important in engineering robust complex microbial communities.
Studying the interactions that occur in a complex microbial community involving hundreds of species is inherently challenging because of the metabolic complexity of individual species as well as all the potential interactions between species. Hence, there is growing interest in studying artificial coculture or triculture models, in which the interactions are simpler and can thereby be more precisely analyzed and interpreted. For example, He et al. imitated the natural dechlorinating communities and established a coculture to investigate the positive impact of microbial interactions on the dechlorination activity and growth of dechlorinating strains (2). Xie et al. cultivated algae with cobalamin-producing bacteria to study the algal-bacterial mutualistic interaction that resulted in thermal tolerance enhancement in the algae (15). Cheirsilp et al. employed a coculture of a lactic acid bacterium and a lactic acid-assimilating yeast, which led to higher kefiran productivity (7). In the field of biofuels, such as biohydrogen, coculture models have also been established. For example, studies have demonstrated that cocultures containing dark-fermentative bacteria such as Clostridium species and photosynthetic bacteria such as Rhodopseudomonas species can enhance hydrogen (H2) production and substrate consumption (16–22).
The experimental characterizations of the physiology of coculture models have provided some basic understanding of microbial interactions. However, in order to obtain a system understanding of how species interact, it is also essential to probe the genome-wide molecular responses. Sieuwerts et al. have applied physiological and transcriptome profiling approaches to a yogurt fermentation process of a coculture containing two species of lactic acid bacteria. They have identified the molecular basis of the mutual beneficial interactions between the bacteria that influenced their growth and fermentation performance (23). Beliaev et al. have analyzed the photoautotroph-heterotroph interactions between a cyanobacterium and a marine facultative aerobe with transcriptome sequencing. Their results have not only provided insights into the interactions between cyanobacteria and heterotrophs but also allowed the formulation of new hypotheses on cyanobacterial-heterotrophic interactions (24). Furthermore, Men et al. have characterized the interactions of a dechlorinating coculture and triculture by transcriptome and proteome analyses. The molecular data have revealed the mechanisms of the enhanced dechlorination activity observed in the cocultures (3).
In view of the breadth of information that can be gained from advanced molecular tools, the combination of physiological characterization and gene expression profiling could provide novel insights into microbial interactions involved in biofuel production, which could ultimately enhance the yield of biofuels. While H2 production from dark-fermentative and photosynthetic bacteria in cocultures has been intensively investigated in small-scale experiments and large-scale reactors (8, 10, 19, 21, 25–27), the molecular mechanisms governing the bacterial dynamics and interactions remain unclear. In particular, to the best of our knowledge, the use of a high-throughput sequencing (RNA-seq) strategy to query the transcriptomic responses of H2-producing cocultures utilizing cellulose as the sole carbon source has not been previously investigated.
We have previously established and investigated a coculture model containing a cellulose-degrading bacterium, Clostridium cellulovorans 743B, and a photosynthetic bacterium, Rhodopseudomonas palustris CGA009, for enhanced H2 production (26). A mutant strain of R. palustris with constitutively expressed nitrogenases in the presence of ammonium (NH4+) (28) is not investigated in the artificial coculture, as our study intends to mimic a natural microbial community. In the cocultures, the physiology (such as H2 yield, cell growth, and extent and rates of transformation of cellulose degradation, production of volatile fatty acids [VFAs], and pH) and the impacts of different cellulose concentrations have been characterized to obtain a basic understanding of the interactions between the two strains. In the cocultures, significantly higher H2 yield and more-complete cellulose degradation are achieved than in the monocultures. In addition, since the consumption of VFAs by R. palustris can help to control pH, less chemical buffer is required; hence, this coculture strategy has economic advantages for future large-scale industrial applications. The objective of this study is to further investigate the interactions between these two H2 fermenters at the transcription level. Gene expression levels of the cocultures and the respective monocultures at the early, mid-, and late exponential growth phases were analyzed and compared via genome-wide transcriptome sequencing. Furthermore, the transcriptome results were correlated with the physiological characteristics of the cocultures to achieve a more comprehensive understanding of the microbial interactions.
MATERIALS AND METHODS
Culture experimental conditions.
Monocultures of C. cellulovorans and of R. palustris were grown with 3 g/liter of cellulose and 20 mM acetate, respectively, and light in 100 ml of the same defined medium in 160-ml serum bottles as described previously (26). Sodium bicarbonate (30 mM) and N-[Tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid (10 mM) were used as a buffer to maintain the medium pH (initially at pH 7.2); however, C. cellulovorans inherently produces VFAs, leading to a drop in pH. Compared to C. cellulovorans monocultures, cocultures exhibited a strong buffering capacity due to the VFA consumption by R. palustris. In order to prevent repression of the nitrogenases of R. palustris by NH4+, the headspace of the bottles was filled with argon gas, and glutamate instead of NH4+ served as the nitrogen source for both C. cellulovorans and R. palustris (29–31). Cocultures of C. cellulovorans and R. palustris were inoculated at a cell ratio of 1:4 with 3 g/liter of cellulose. All monocultures and cocultures were incubated at 30°C and 100 rpm and under 6,000 lx of illumination from six 15-W Grolux fluorescent tubes. For both monocultures and cocultures, cellulose was the sole carbon and energy source for C. cellulovorans. On the other hand, while light was the energy source for R. palustris in both monocultures and cocultures, various VFAs produced by C. cellulovorans were the carbon sources for R. palustris in cocultures, whereas acetate was the sole carbon source for R. palustris in monocultures. The design of the cultivation experiments was to compare C. cellulovorans and R. palustris in cocultures grown with light and cellulose as the only exogenous substrate with their respective monocultures so that any changes in conditions because of coculturing would be reflected in the transcriptome. Monocultures and cocultures were harvested for transcriptomic sequencing when cells reached the early, mid-, and late exponential growth phases as determined by the amount of H2 produced via a gas chromatograph equipped with a thermal conductivity detector as described previously (26). Two identical monocultures or cocultures were prepared for duplicate transcriptomic analysis at each time point. The effect of NH4+ on repressing the nitrogenases of R. palustris was tested using the culture conditions as described above, except that 11.2 mM NH4+ was added to the growth medium of cocultures and monocultures. Triplicate cultures were set up for the experiment. The obtained physiological results were compared with data derived from cultures without NH4+ (26).
RNA extraction and sequencing.
Cells were harvested from each duplicate culture by centrifugation at 21,100 × g for 3 min at 4°C. The cell pellets were immediately flash frozen in liquid nitrogen and stored at −80°C until further processing within 24 h. Total RNA was extracted using the RNeasy minikit (Qiagen, California, USA) according to the manufacturer's protocol with the addition of lysozyme (7.5 mg/ml final concentration) for cell lysis. Contaminating DNA was removed by on-column and in-solution DNase digestion with an RNase-free DNase set (Qiagen, California, USA) according to the manufacturer's instructions.
In order to examine the completeness of DNA removal, total RNA was first reverse transcribed into cDNA using the Superscript III (Invitrogen, California, USA) reverse transcriptase according to the manufacturer's instructions. For negative reverse transcriptase controls, diethylpyrocarbonate (DEPC)-treated water replaced the SuperScript III reverse transcriptase. The resulting cDNA and the negative controls were used in the subsequent PCR step for amplifying the cellulase gene of C. cellulovorans (GeneID 9607758) and the 16S rRNA genes of R. palustris (GeneID 2690886 and GeneID 2690040) with primers described previously (26). PCR products were visible in the samples with reverse transcriptase while the negative reverse transcriptase controls resulted in no band.
The purity and concentration of the total RNA were determined by a Nano Drop 2000 spectrophotometer (Thermo Fisher Scientific, Massachusetts, USA). For all the samples, the A260/A280 ratios ranged from 2.0 to 2.1 and the concentrations were >149 ng/μl. The integrity and quality of the total RNA were further assessed on a Bioanalyzer 2100 (Agilent, California, USA) with the Agilent RNA 6000 Pico kit (Agilent, California, USA) according to the manufacturer's instructions, and only samples with an RNA integrity number (RIN) of >7.8 were selected for sequencing. The average RIN for all the samples was 9.0. Total RNA was depleted of rRNA using the Ribo-Zero rRNA removal Gram-negative bacteria kit (Epicentre, Wisconsin, USA). A total of 18 samples were submitted to the sequencing facility (BGI Tech Solutions, Hong Kong) for library construction and sequencing. Libraries were sequenced on a HiSeq 2000 platform (Illumina, California, USA) to yield 91 paired-end reads.
RNA-sequencing data analysis.
In order to obtain high-quality reads for downstream analyses, reads with adaptor sequence, reads with unknown bases representing greater than 10% of a read, and low-quality reads (i.e., for which the percentage of low-quality bases was greater than 50% of a read) were removed. The retained high-quality data for each monoculture and coculture sample were ∼0.55 Gb and ∼1.1 Gb, respectively. High-quality reads were mapped to the genomes of C. cellulovorans (GenBank accession no. NC_014393.1) and R. palustris (GenBank accession no. NC_005296.1) using the SOAP aligner (32). By not allowing more than two mismatches in the alignment, the average percentage of total reads mapped to the reference genomes for all the samples is 97.0%.
Normalization of the RNA-sequencing data was conducted using the reads per kilobase per million (RPKM) calculation (33). The transcription data were analyzed using the nonparametric NOIseq method (34) to identify differentially expressed genes (DEGs). Specifically, a q value (differential expression probability) of ≥0.8 (34) and the absolute value of log2 (coculture/monoculture) of ≥1 were used as thresholds to identify genes that exhibited significant differences in expression between cocultures and monocultures at each growth phase. Throughout this study, upregulation refers to a higher relative molar concentration of transcripts of a particular gene of C. cellulovorans or R. palustris detected in cocultures relative to the respective monocultures, and downregulation refers to a lower relative molar concentration of transcripts detected in cocultures. In addition, a positive expression ratio represents upregulation, and a negative one represents downregulation. The genes of C. cellulovorans are designated “Clocel,” and those of R. palustris are “RPA.”
In order to determine the main biological functions that the DEGs represent, all DEGs were subjected to Gene Ontology (GO) (35) functional annotation by using Blast2GO (36) to determine GO terms. In addition, GO functional classification and plotting of DEGs were performed by using WEGO (37) to understand the distribution of gene functions from the macro level (see Fig. S1 and S2 in the supplemental material). DEGs were annotated across the GO subcategories and grouped by biological process, cellular component, or molecular function. Furthermore, in order to identify significantly enriched metabolic pathways and signal transduction pathways related to the identified DEGs, Kyoto Encyclopedia of Genes and Genomes (KEGG) (38) annotation was also performed using Blast2GO. In order to gain an overview of the complete metabolism in the interactions, KEGG-based maps were generated. First of all, the key pathways were identified based on the DEGs. Once the key pathways were identified, all of the DEGs in the same pathway were evaluated to determine whether they were upregulated or downregulated relative to the monocultures. If ≥50% of the DEGs in the pathway possessed the same trend, then the pathway would be designated upregulated or downregulated according to the majority. The cellular pathways are displayed using the iPath 2.0 platform (39). Heat maps were generated using the “ggplot2” package in R for comparing the expression level of genes across different growth phases (40).
RT-qPCR.
In order to verify the RNA-sequencing results, 16 differentially expressed genes (Table 1) spanning a range of expression levels at the mid-exponential growth phase were selected and analyzed by reverse transcription-quantitative PCR (RT-qPCR) using specific primers designed by using Primer Express 3.0 (Applied Biosystems, California, USA). Total RNA from two biological replicates of cocultures and monocultures was reverse transcribed into cDNA using SuperScript III (Invitrogen, California, USA) according to the manufacturer's instructions. Amplification of the synthesized cDNA (two technical replicates per biological replicate) was performed with a StepOne Plus real-time PCR system (Applied Biosystems, California, USA) using the PowerUp SYBR green master mix (Applied Biosystems, California, USA) according to the manufacturer's instructions and the default thermal-cycling conditions. Comparative threshold (CT) differences between cocultures and monocultures were calculated from averages of quadruplicate samples. The fold difference for each target gene was calculated using the 2−ΔΔCT method (41).
TABLE 1.
Primer sequences for RT-qPCR validation against RNA-seq results
| Gene category and primer | Sequence (5′ to 3′) |
|---|---|
| Upregulated | |
| Clocel 1529-F | AAAGGAGGAAGGAAAAAATGTGAA |
| Clocel 1529-R | GAGAGCCAGAACCAGCAAAATT |
| Clocel 3111-F | ATGCCAACTGACCCAGCAA |
| Clocel 3111-R | TCTTCAGCTGTTCCCCAGGTA |
| Clocel 4138-F | CTCCTGTAGACAAGATAGTGGAAGAAAC |
| Clocel 4138-R | GCCTCAGGACTTGGTGCATT |
| Clocel 2295-F | AGAACTCGCGAACAGGTCCTT |
| Clocel 2295-R | AAGCCTTAAAGTGGTCGCTAACA |
| RPA 4628-F | GGGCGACTACAAGCTGTTTCTG |
| RPA 4628-R | TCGGATCGTGGAACATGAAG |
| RPA 4209-F | GCGGCAAGGAGTATTTCGAA |
| RPA 4209-R | AGGACCATACACCGCGATGT |
| RPA 4784-F | TTACGTGTTCCGAGCCTACAAG |
| RPA 4784-R | ACGCTCGCTTTGAGGTTGTC |
| RPA 4618-F | CCGAACGAGTCGATCAACTTC |
| RPA 4618-R | GTGTAATCGACGCCCATCAGT |
| RPA 1435-F | GCGACTTCGTCAAGCATTTCT |
| RPA 1435-R | TCTCGATCTCGAACAACACGTT |
| RPA 0275-F | CCTGCATTATTTGGGCTCTGTAC |
| RPA 0275-R | GCGCAGAAGGTCATCTGGAA |
| Downregulated | |
| Clocel 0527-F | CACTGTCCATTCCCATTTATACATG |
| Clocel 0527-R | CCCATTCCAGCTTCAATAGCTT |
| Clocel 3433-F | ATTGCTGCAACTACCGTTGAAG |
| Clocel 3433-R | AGTCGTGCTTTGGGCCTGTA |
| Clocel 3475-F | GTGCCGGGTTTGTACCAGAT |
| Clocel 3475-R | CTCCTGATGAAATTCCGACCAA |
| Clocel 4097-F | ATCGGCGGAAGTGAGTATAACGT |
| Clocel 4097-R | AGGTTGACCTCCTCCGTTCA |
| RPA 0137-F | AGCTGCGTCGTATTCGGTATG |
| RPA 0137-R | GGTTCAGCGACACAACCTTCTC |
| RPA 1259-F | TGCAGAGCTACGGACCCAAT |
| RPA 1259-R | AGCAGGAAGTAGATCAGCGTGTT |
Accession number(s).
All RNA-sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) (BioProject accession number PRJNA280696).
RESULTS AND DISCUSSION
Validation of transcriptome sequencing results.
To validate the transcriptome sequencing results, RT-qPCR was used to quantify changes in the transcript levels of selected C. cellulovorans and R. palustris genes (Table 1) between the cocultures and monocultures. The transcriptome sequencing and RT-qPCR results for the 16 tested genes (eight each for C. cellulovorans and R. palustris) were strongly correlated (R2 = 0.88; slope = 0.93), and the expected trend in gene expression was obtained (see Fig. S3 in the supplemental material). This indicates that the transcriptome sequencing results can be reliably used to infer the metabolism in the cocultures.
Overview of the transcriptome.
In order to understand the molecular responses of C. cellulovorans and R. palustris in cocultures, the transcriptomes of cells at the early, mid-, and late exponential growth phases were examined and compared with those of the respective monocultures. For C. cellulovorans, a total of 73, 134, and 146 genes (Fig. 1; see also Table S1 in the supplemental material) were differentially expressed at the early, mid-, and late exponential growth phases, respectively. Specifically, 33 and 40 genes at the early exponential growth phase, 58 and 76 genes at the mid-exponential growth phase, and 65 and 81 genes at the late exponential growth phase were up- and downregulated, respectively, with ≥2-fold changes. On the other hand, for R. palustris, a total of 155, 421, and 1,116 genes (Fig. 1; see also Table S2 in the supplemental material) were differentially expressed at the early, mid-, and late exponential growth phases, respectively. Specifically, 93 and 62 genes at the early exponential growth phase, 204 and 217 genes at the mid-exponential growth phase, and 290 and 826 genes at the late exponential growth phase were up- and downregulated, respectively, with ≥2-fold changes. Notably, the number of downregulated R. palustris genes at the late exponential growth phase was significantly greater than the number of upregulated genes. This might be due to the fact that the pH of cocultures at the late exponential growth phase (Fig. 2a) was significantly lower than the constant neutral pH in R. palustris monocultures. The acidic pH likely inhibited cellular metabolism, which resulted in a cell-wide downregulation of transcription. The DEGs of C. cellulovorans represent 1.7% to 3.5% of the 4,220 total predicted protein-encoding genes in its genome, while the DEGs of R. palustris represent 3.2% to 23% of the 4,836 predicted protein-encoding genes in its genome.
FIG 1.
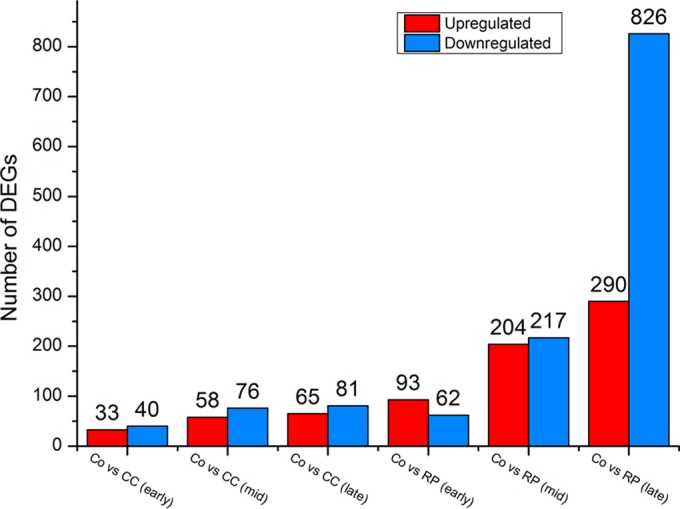
Number of differentially expressed genes (DEGs) at the early, mid-, and late exponential growth phases when comparing C. cellulovorans (CC) or R. palustris (RP) in cocultures (Co) with their respective monocultures.
FIG 2.

Comparison of pH (a), H2 production (b), cellulose degradation (c), and VFA concentrations (d) for cocultures (Co) and monocultures of C. cellulovorans (CC) at a cellulose concentration of 3 g/liter. HAc, acetic acid; HBu, butyric acid; FA, formic acid; LA, lactic acid. Each data point is an average of biological triplicate results, and error bars represent 1 standard deviation. (Modified from reference 26 with permission from Elsevier.)
In order to obtain an overview on how cells responded to cocultivation, the identified DEGs were classified according to Gene Ontology (GO) terms (see Fig. S1 and S2 in the supplemental material). For both C. cellulovorans and R. palustris, the category of “biological process” was the most enriched, containing both up- and downregulated genes of the transcriptome from all three growth phases. Furthermore, the identified DEGs were mapped to the corresponding metabolic pathways to highlight the key cellular metabolism (see Fig. S4 and S5 in the supplemental material). In general, the DEGs were distributed in a number of different pathways in C. cellulovorans and R. palustris.
According to our previous physiological study (26), the mid-exponential growth phase is most representative of the characteristics of the interactions between C. cellulovorans and R. palustris (Fig. 2). Hence, analysis of the interactions at the molecular level here focused mainly on the transcriptomic responses at the mid-exponential growth phase. Genes and pathways that are associated with important cellular metabolic processes and deemed statistically significant are highlighted and discussed in the following sections.
H2 production.
From our previous study (26), cocultures of C. cellulovorans and R. palustris exhibited enhanced H2 production compared to the monocultures of C. cellulovorans (Fig. 2b). Pyruvate:ferredoxin oxidoreductase and hydrogenase are known to play critical roles in catalyzing H2 evolution during dark fermentation by C. cellulovorans. In the dark fermentation, pyruvate is oxidized to acetyl-coenzyme A (acetyl-CoA) by ferredoxin oxidoreductase and simultaneously electrons are donated to convert the oxidized ferredoxin to its reduced form. Subsequently, the reduced ferredoxin is oxidized by hydrogenase to generate H2 by using protons as terminal electron acceptors (42). In this study, coculturing with R. palustris did not lead to an increase in transcript abundance of genes related to H2 evolution in C. cellulovorans. Instead, the transcripts of a pyruvate:ferredoxin oxidoreductase (Clocel 1684) and a hydrogenase (Clocel 4097) were downregulated (Table 2).
TABLE 2.
Selected differentially expressed genes of C. cellulovorans grouped into functional categories in response to cocultivation with R. palustris at mid-exponential growth phase
| Functional category and gene ID | Description | log2 (Coa/C. cellulovorans) |
|---|---|---|
| H2 production (2 genes) | ||
| Clocel 1684 | Pyruvate ferredoxin/flavodoxin oxidoreductase | −1.3 |
| Clocel 4097 | Hydrogenase, Fe only | −2.3 |
| Cellulose degradation (3 genes) | ||
| Clocel 3359 | Cellulase, glycosyl hydrolase family 5 protein | 1.7 |
| Clocel 3111 | Cellulase, glycosyl hydrolase family 5 protein | 2.1 |
| Clocel 2295 | Xylanase, glycosyl hydrolase family 11 protein | 2.5 |
| Fatty acid biosynthesis (3 genes) | ||
| Clocel 4138 | Acetyl-CoA carboxylase, biotin carboxyl carrier protein | 2.7 |
| Clocel 4144 | 3-Oxoacyl-(acyl-carrier-protein) synthase III | 2.6 |
| Clocel 4162 | Beta-ketoacyl-ACP synthase | 1.4 |
| Sulfur metabolism (10 genes) | ||
| Clocel 0525 | Fumarate reductase/succinate dehydrogenase flavoprotein domain-containing protein | −5.2 |
| Clocel 0526 | 4Fe-4S ferredoxin | −13.6 |
| Clocel 2532 | O-acetylhomoserine/O-acetylserine sulfhydrylase | −4.8 |
| Clocel 0527 | Phosphoadenosine phosphosulfate reductase (thioredoxin) | −7.3 |
| Clocel 0528 | Sulfate adenylyltransferase, large subunit | −6.0 |
| Clocel 3475 | Cysteine synthase | −5.4 |
| Clocel 0521 | Sulfate ABC transporter, periplasmic sulfate-binding protein | −3.9 |
| Clocel 0524 | Sulfate ABC transporter ATPase | −4.5 |
| Clocel 0523 | Sulfate ABC transporter, inner membrane subunit CysW | −5.5 |
| Clocel 0522 | Sulfate ABC transporter, inner membrane subunit CysT | −5.9 |
| Thiamine metabolism (6 genes) | ||
| Clocel 2812 | Thiamine biosynthesis protein ThiC | −1.9 |
| Clocel 0684 | Thiazole biosynthesis protein ThiH | −2.0 |
| Clocel 0685 | Thiazole biosynthesis family protein | −2.2 |
| Clocel 0683 | Thiamine biosynthesis protein ThiF | −2.6 |
| Clocel 0682 | Thiamine-phosphate pyrophosphorylase | −2.9 |
| Clocel 3433 | Biotin and thiamine synthesis-associated protein | −3.1 |
| Nitrogen metabolism (5 genes) | ||
| Clocel 1684 | Pyruvate ferredoxin/flavodoxin oxidoreductase | −1.3 |
| Clocel 1284 | Glu/Leu/Phe/Val dehydrogenase | −1.9 |
| Clocel 2836 | Nitrogenase iron protein | 2.0 |
| Clocel 2838 | NADH dehydrogenase ubiquinone 24-kDa subunit | 2.0 |
| Clocel 4147 | 2-Nitropropane dioxygenase | 2.1 |
| Molybdate transport (3 genes) | ||
| Clocel 1529 | Molybdenum ABC transporter periplasmic molybdate-binding protein | 2.5 |
| Clocel 1530 | Molybdate ABC transporter inner membrane subunit | 2.0 |
| Clocel 1531 | ABC transporter | 2.1 |
Co, cocultures of C. cellulovorans and R. palustris.
On the other hand, during photofermentation by R. palustris, H2 evolution is mediated mainly by nitrogenases through nitrogen fixation. In particular, when R. palustris is grown on poor nitrogen sources or under NH4+ deprivation, the proton-reducing activity of nitrogenases can be retained at a high level to actively convert the proton to H2 if provided with sufficient ATP and reducing power (43). In addition to the molybdenum (Mo) cofactor nitrogenases (encoded by nif genes), R. palustris also encodes two alternative functional nitrogenase isozymes: vanadium (V) cofactor (encoded by vnf genes) and iron (Fe) cofactor (encoded by anf genes) nitrogenases. However, reactions involving V and Fe nitrogenases theoretically consume more reducing power than Mo nitrogenases, and they have been reported (44, 45) to serve as alternative routes for nitrogen fixation only when Mo is deficient in the environment. Conversely, the synthesis of alternative nitrogenases can be repressed by Mo (46). From the transcriptome, the nitrogenase genes of R. palustris were upregulated as much as 10-fold in cocultures compared to monocultures. Specifically, 26 of 32 genes in the nif cluster (RPA 4602 to 4633) and 3 of 5 genes in the anf cluster (RPA 1435 to 1439) were upregulated more than 2-fold (Table 3), while no genes in the vnf cluster (RPA 1370 to 1380) were differentially expressed. These results are supported by a previous study aiming to understand the regulation of nitrogenase expression and activity in monocultures of R. palustris. Their results demonstrated that R. palustris preferentially expressed Mo nitrogenase over V and Fe nitrogenases when Mo was present in the growth medium (44).
TABLE 3.
Selected differentially expressed genes of R. palustris grouped into functional categories in response to cocultivation with C. cellulovorans at mid-exponential growth phase
| Functional category and gene ID | Description | log2 (Coa/R. palustris) |
|---|---|---|
| Nitrogen metabolism | ||
| nif cluster (26 genes) | ||
| RPA 4602 | Ferredoxin-like protein FixX | 2.5 |
| RPA 4603 | Electron-transferring-flavoprotein dehydrogenase | 1.4 |
| RPA 4604 | Electron transfer flavoprotein subunit beta | 1.2 |
| RPA 4606 | Nitrogenase-stabilizing/protective protein | 1.9 |
| RPA 4607 | Homocitrate synthase | 1.6 |
| RPA 4608 | Class V aminotransferase | 1.3 |
| RPA 4611 | Nitrogen fixation protein NifQ | 2.7 |
| RPA 4612 | 4Fe-4S ferredoxin | 3.2 |
| RPA 4613 | Hypothetical protein | 2.0 |
| RPA 4614 | Hypothetical protein | 1.8 |
| RPA 4615 | Dinitrogenase iron-molybdenum cofactor biosynthesis | 2.2 |
| RPA 4616 | Nitrogenase molybdenum-cofactor biosynthesis protein NifN | 1.8 |
| RPA 4617 | Nitrogenase molybdenum-cofactor biosynthesis protein NifE | 1.8 |
| RPA 4618 | Nitrogenase molybdenum-iron protein subunit beta | 1.8 |
| RPA 4619 | Nitrogenase molybdenum-iron protein subunit alpha | 1.6 |
| RPA 4620 | Nitrogenase reductase | 1.3 |
| RPA 4621 | Hypothetical protein | 1.8 |
| RPA 4622 | Hypothetical protein | 2.1 |
| RPA 4623 | Hypothetical protein | 1.7 |
| RPA 4624 | Hypothetical protein | 2.6 |
| RPA 4625 | Nitrogen fixation protein NifZ | 2.2 |
| RPA 4626 | Hypothetical protein | 2.1 |
| RPA 4627 | Hypothetical protein | 1.7 |
| RPA 4628 | HesB/YadR/YfhF | 1.5 |
| RPA 4629 | 4Fe-4S ferredoxin | 2.0 |
| RPA 4631 | 4Fe-4S ferredoxin | 1.1 |
| anf cluster (3 genes) | ||
| RPA 1435 | Alternative nitrogenase 3 subunit beta | 2.8 |
| RPA 1437 | Nitrogenase molybdenum-iron protein subunit alpha | 2.1 |
| RPA 1438 | Nitrogenase reductase | 3.3 |
| Reductant-encoding genes (5 genes) | ||
| RPA 1927 | Hypothetical protein | 1.4 |
| RPA 1928 | Ferredoxin | 1.3 |
| RPA 2116 | Hypothetical protein | 1.6 |
| RPA 2117 | Flavodoxin FldA | 1.4 |
| RPA 0732 | Formate dehydrogenase subunit gamma | 1.3 |
| Ntr regulon (4 genes) | ||
| RPA 4209 | Glutamine synthetase | 1.4 |
| RPA 2966 | Nitrogen regulatory protein P-II | 1.4 |
| RPA 0274 | GlnK nitrogen regulatory protein P-II | 1.7 |
| RPA 2592 | Signal transduction histidine kinase, nitrogen specific, NtrB | 1.1 |
| Transporters (5 genes) | ||
| RPA 0275 | Ammonium transporter | 1.5 |
| RPA 4714 | Hypothetical protein | 1.6 |
| RPA 2112 | Nitrate transporter component NrtA | 2.7 |
| RPA 2113 | Nitrate transport system permease | 2.6 |
| RPA 2114 | Nitrate transport system ATP-binding protein | 3.0 |
| Organic compound catabolism | ||
| RPA 2940 | NADH dehydrogenase subunit K | 1.3 |
| Bacterial chemotaxis (18 genes) | ||
| RPA 0137 | Chemotaxis methylesterase CheB1 | 1.2 |
| RPA 4784 | OmpA/MotB domain-containing protein | −1.3 |
| RPA 1884 | Methyl-accepting chemotaxis receptor/sensory transducer | 1.2 |
| RPA 0999 | Hypothetical protein | −2.1 |
| RPA 4639 | Methyl-accepting chemotaxis receptor/sensory transducer | 1.1 |
| RPA 1678 | Chemotaxis methyltransferase CheR3 | 1.2 |
| RPA 3709 | Hemoprotein | 2.2 |
| RPA 1267 | Flagellar motor switch protein | 1.1 |
| RPA 4481 | Methyl-accepting chemotaxis sensory transducer | 1.1 |
| RPA 0139 | Methyl-accepting chemotaxis receptor/sensory transducer | 1.4 |
| RPA 3751 | Hypothetical protein | 1.1 |
| RPA 3185 | Methyl-accepting chemotaxis receptor/sensory transducer | 1.4 |
| RPA 3750 | Methyl-accepting chemotaxis sensory transducer | 1.4 |
| RPA 0138 | Chemotaxis methyltransferase CheR1 | 1.3 |
| RPA 4531 | Histidinol dehydrogenase | −1.1 |
| RPA 0140 | Chemotaxis signal transduction/oligomerization protein CheW1-2 | 1.1 |
| RPA 1774 | Porin | 1.7 |
| RPA 0141 | Chemotaxis signal transduction/oligomerization protein CheW1-1 | 1.4 |
| Photosynthesis | ||
| puc operon (8 genes) | ||
| RPA 2653 | Light harvesting protein b-800-850 alpha subunit A | 3.0 |
| RPA 2654 | Light harvesting protein b-800-850 beta subunit A | 2.5 |
| RPA 3009 | Light harvesting protein b-800-850 beta subunit C | 1.8 |
| RPA 3010 | Pseudo | 2.0 |
| RPA 1491 | Light harvesting protein b-800-850 beta subunit E | 2.1 |
| RPA 1492 | Light harvesting protein b-800-850 alpha subunit E | 1.9 |
| RPA 4291 | Light harvesting protein b-800-850 beta subunit B | 2.3 |
| RPA 4292 | Light harvesting protein b-800-850 alpha subunit B | 1.9 |
| puf operon (3 genes) | ||
| RPA 1525 | Antenna complex alpha/beta subunit | 2.2 |
| RPA 1526 | Light-harvesting complex 1 subunit alpha | 1.5 |
| RPA 1527 | Photosynthetic reaction center subunit l | 1.2 |
Co, cocultures of C. cellulovorans and R. palustris.
Nitrogenase activity not only is an energy-demanding process but also requires a large amount of reducing power. Accordingly, the ferredoxin (RPA 1927 to 1928) and flavodoxin (RPA 2116 to 2117) genes were also upregulated along with the nitrogenase genes (Table 3), likely to generate more electrons for the nitrogenases. In addition, the gene encoding a NAD-dependent formate dehydrogenase gamma subunit (RPA 0732) was also upregulated more than 2-fold, suggesting that R. palustris is seeking more reducing power in cocultures with C. cellulovorans. Furthermore, several genes in the Ntr regulon of R. palustris, such as genes encoding the glutamine synthetase, glnAII (RPA 4209), and the nitrogen regulatory protein P-II, glnB (RPA 2966), the glnK2 gene (RPA 0274), and the gene encoding signal transduction histidine kinase, ntrB (RPA 2592), which controls nitrogenase activity (47, 48), were all upregulated in the cocultures (Table 3).
Besides upregulation of the nif and anf operons, reductant-encoding genes, and the Ntr regulon, a number of genes that encode transport systems for nitrogenous compounds were also upregulated. Transcripts were upregulated for the amtB gene (RPA 0275) encoding NH4+ transporters (Table 3). Meanwhile, a gene (RPA 4714) encoding a hypothetical protein for an ABC transporter for Mo was upregulated (Table 3), suggesting that Mo is transported to support the synthesis of Mo nitrogenases. Furthermore, genes predicted to encode an ABC transporter for nitrate (RPA 2112 to 2114) were all upregulated (Table 3). In the pathway of assimilatory nitrate reduction, nitrate can be reduced to ammonia, which is then incorporated into the amino acids glutamine and glutamate using the glutamine synthetase-glutamate synthase (GS/GOGAT) system. Glutamate and glutamine are the primary nitrogen suppliers for the other nitrogen-containing compounds in cells, and they serve as the amino donors for nucleic acid and amino acid biosynthesis and other reactions (49). Hence, the influx of nitrate and the upregulation of the glutamine synthetase-encoding gene (RPA 4209) suggest that the demand for a nitrogen source is higher for R. palustris in the cocultures.
Overall, the higher transcript levels of the nif and anf operons, reductant-encoding genes, Ntr regulon, and transporter genes for NH4+, Mo, and nitrate in R. palustris indicate that the presence of C. cellulovorans had a stimulatory effect on the expression of nitrogen fixation-related genes. These results, coupled with the observation that the H2 evolution-related genes of C. cellulovorans were downregulated, suggest that the measured enhanced H2 production in the cocultures is most likely from R. palustris alone instead of C. cellulovorans or both. To further examine whether the enhanced H2 production resulted from the activity of nitrogenases, we tested the effect of adding NH4+ in the growth medium for cocultures and monocultures to repress the expression of nitrogenases. While there was no H2 produced in R. palustris monocultures with NH4+, the cocultures with NH4+ produced 46 ml of H2, which was 20 ml less than the cocultures without NH4+ and 6 ml more than C. cellulovorans monocultures with NH4+ (see Fig. S6 in the supplemental material). These results further confirmed that the enhanced H2 production was due mainly to the nitrogenases activity of R. palustris. Although R. palustris cannot produce H2 in the cocultures with NH4+, it enhanced the buffering capacity by consuming some of the VFAs, which stabilized the pH and in turn allowed C. cellulovorans to consume extra cellulose for additional H2 production (see Fig. S7 in the supplemental material).
Cellulose degradation.
Aside from enhanced H2 production, cocultures were also associated with more-complete cellulose degradation (Fig. 2c). Clostridium species are capable of producing cellulosomes, which are multienzyme complexes for efficient degradation of polysaccharides (e.g., cellulose and xylan). In the genome of C. cellulovorans, a total of 57 cellulosomal genes are present (50), including 53 dockerin-containing proteins and four cohesin-containing scaffolding proteins. Of all the cellulosomal genes, 29 are predicted to encode enzymes with cellulolytic, hemicellulolytic, and pectin-degrading functions. In this study, two C. cellulovorans cellulase genes (Clocel 3111 and Clocel 3359) encoding family 5 glycosyl hydrolases (GHs) were upregulated in the cocultures (Table 2), suggesting that cellulolytic activity was enhanced in the presence of R. palustris. Furthermore, one C. cellulovorans xylanase gene (Clocel 2295) encoding family 11 GH was also upregulated in response to cocultivation with R. palustris (Table 2), suggesting that cellulase and hemicellulase might share a regulatory mechanism that can be induced by the presence of cellulose. The coordinated expression of both cellulase and hemicellulase genes has previously been reported when C. cellulovorans was cultivated with cellulose as the carbon source in monoculture (51). The upregulation of a number of cellulosomal genes is consistent with the physiological characterization whereby enhanced cellulolytic activity was observed in cocultures (Fig. 2c). The enhanced cellulose degradation is attributed to the consumption of VFAs by R. palustris, which in turn stabilizes the pH of cocultures, in contrast to the more-acidic pH in the monocultures (Fig. 2a). Acidic pH likely negatively influences the expression of cellulosomal genes in monocultures of C. cellulovorans, as it has previously been shown that cellobiose and cellulose degradation was inhibited in the cellulolytic ruminal bacterium Bacteroides succinogenes under low pH (52).
Fatty acid metabolism.
The fermentative metabolites of C. cellulovorans are H2, carbon dioxide, ethanol, and VFAs, including formate, acetate, butyrate, and lactate (50). In the cocultures, fatty acid biosynthesis of C. cellulovorans is critical to R. palustris since it is obligately dependent on the VFAs from C. cellulovorans as electron donors and carbon sources for growth. During dark fermentation by C. cellulovorans, acetyl-CoA carboxylase is the essential enzyme that functions in the first committed step of fatty acid biosynthesis (53). Acetyl-CoA carboxylase comprises two carboxyl transferase subunits, as well as biotin carboxylase and biotin carboxyl carrier protein (BCCP). Together, these four components of acetyl-CoA carboxylase are encoded by Clocel 4134 to 4136 and Clocel 4138, respectively. In the acetyl-CoA carboxylase reaction, biotin is first coupled to BCCP. Following that, the biotin carboxylase catalyzes the Mn-ATP-dependent carboxylation of biotin to generate CO2−-BCCP. Subsequently, the transcarboxylase transfers the carboxyl group from the biotin moiety of BCCP to acetyl-CoA to form malonyl-CoA, the precursor to all the elongation steps of fatty acid biosynthesis (53). In the cocultures, the gene (Clocel 4138) encoding BCCP in C. cellulovorans was upregulated (Table 2). Although the other three acetyl-CoA carboxylase-encoding genes were not statistically classified as differentially expressed, they were all consistently upregulated.
Moreover, the beta-ketoacyl-acyl-carrier-protein (ACP) synthase and the 3-oxoacyl-ACP synthase III are the key enzymes involved in the initiation of fatty acid biosynthesis. They conduct the condensation of acetyl-CoA with malonyl-ACP to supply the intermediates of short-chain fatty acids (54, 55). In the cocultures, the beta-ketoacyl-ACP synthase gene (Clocel 4162) and 3-oxoacyl-ACP synthase III gene (Clocel 4144) of C. cellulovorans were both upregulated (Table 2). The upregulation of the fatty acid biosynthesis-related genes suggests that C. cellulovorans was attempting to synthesize more VFAs when R. palustris was present (Fig. 2d). This could be due to the consumption of VFAs by R. palustris, which prompted further VFA production. The genome of R. palustris encodes two homologues of NADH dehydrogenase complexes (RPA 2937 to 2952 and RPA 4252 to 4264), which mediate the catabolism of organic compounds (e.g., fatty acids, dicarboxylic acid, and lignin monomers) (56). In R. palustris, the gene nuoK1 (RPA 2940) encoding NADH dehydrogenase subunit K was upregulated (Table 3), suggesting that the VFA catabolism of R. palustris in the cocultures was more active than in the monocultures. This is probably due to the fact that multiple VFAs produced by C. cellulovorans were utilized as the carbon substrates and electron donors for R. palustris, while there was only acetate in the monocultures of R. palustris.
Bacterial chemotaxis.
As a motile bacterium with flagella, R. palustris is able to perform chemotaxis. In general, most bacteria are too small to accurately measure the chemoeffector gradient between the ends of the cell. Therefore, bacteria measure changes in chemoeffector concentrations over time rather than detecting the spatial gradient within the bacterium itself (57). On the other hand, although C. cellulovorans also has peritrichous flagella (58), they are nonmotile. In contrast to R. palustris monocultures, where acetate was initially amended at a relatively high concentration (20 mM), the VFA concentrations in the cocultures began at zero and increased over time as a result of the continuous VFA synthesis by C. cellulovorans during dark fermentation (Fig. 2d). Although it was previously reported that permeant acids such as acetate and benzoate act as repellents and elicit negative chemotaxis (59) in Escherichia coli K-12, another study has demonstrated that Rhodobacter sphaeroides, a nonenteric bacterial model for the chemotaxis pathways, is chemotactic to organic acids such as succinate, pyruvate, propionate, and acetate (60). Since VFAs are the carbon sources for R. palustris, a logical assumption is that R. palustris might move toward VFAs, which is favorable for the growth of R. palustris. Interestingly, this assumption seems to be supported by the transcriptome results of R. palustris in the cocultures.
The genome of R. palustris contains the essential proteins required for chemotaxis. In the chemotaxis process, the periplasmic concentrations of chemoeffectors are first sensed by the transmembrane sensor proteins, which are usually referred to as the methyl-accepting chemotaxis proteins (MCPs). The MCPs then interact with the cytoplasmic signaling proteins (CheW and CheA), resulting in a change in the rate of autophosphorylation of CheA. Subsequently, the phosphoryl group from the phosphorylated CheA is transferred to the response regulator proteins (CheY and CheB). At the end, the phosphorylated CheY in turn interacts with the flagellar motor switch protein (FliM, FliN, FliG) to induce cell movement. Furthermore, CheR is a methyltransferase responsible for the methylation of MCPs, which plays a crucial role in modulating the adaptation to chemoeffectors. In the cocultures, genes predicted to encode MCPs CheW, CheB, CheR, CheY, and FliN were all upregulated at least 2-fold (Table 3), suggesting that the chemotaxis response of R. palustris is elicited during cocultivation with C. cellulovorans, or more likely, moving toward the VFAs produced by C. cellulovorans.
Sulfur metabolism.
In response to cocultivation with R. palustris, a number of C. cellulovorans genes responsible for assimilative sulfur metabolism were downregulated (Table 2). In particular, several genes related to sulfate reduction and conversion of intracellular sulfate to cysteine were expressed at a significantly lower level. For example, the gene for sulfate adenylyltransferase (Clocel 0528), which converts sulfate to adenosine-5′-phosphosulfate (APS) in the first step of assimilatory sulfate reduction, was downregulated nearly 60-fold. Also, the gene for thioredoxin (Clocel 0527), which reduces the phosphoadenosine phosphosulfate (PAPS) to sulfite, was significantly downregulated more than 162-fold. In addition, the gene for cysteine synthase (Clocel 3475), which synthesizes cysteine from O-acetylserine and sulfide, was downregulated more than 41-fold. Previous studies have shown that the gene expression of cysteine synthase is a general stress response to tellurite, hydrogen peroxide, acid, and diamide (61, 62). Corresponding with the downregulation of sulfate reduction genes, genes predicted to encode an ABC transporter for sulfate (Clocel 0521 to 0524) were also downregulated (Table 2).
The downregulation of the genes related to sulfate transport and reduction in the cocultures is physiologically interesting, as the final product of the sulfate reduction pathway is cysteine, an essential amino acid that plays a vital role in the catalytic activity and stress responses. In particular, cysteine is known as a source of sulfur required to repair oxidatively damaged iron-sulfur cluster proteins with essential roles in metabolism (63). Specifically, the cysteine-containing molecules glutathione and thioredoxin are known to play a protective role in maintaining an intracellular reducing environment in response to oxidative stress (64). The higher expression level of sulfate reduction in the C. cellulovorans monocultures (i.e., downregulation in cocultures) seems to suggest an enhanced cellular demand for cysteine, which might be a direct response to oxidative stress. However, here both the coculture and monoculture experiments were conducted under anaerobic conditions. It is unlikely that the assumed oxidative stress was caused by oxygen. Previous studies have reported that acid can cause an imbalance in the thiol redox status of the cytoplasm, and responses to stresses such as oxidative stress, heat shock, and envelope stress have been shown to be strongly connected with pH stress and pH resistance in some bacteria (62, 65). Therefore, the gene expression of sulfate transport and reduction could be linked to pH changes.
As opposed to a more stabilized pH as the VFAs were consumed by R. palustris in the cocultures, the pH of C. cellulovorans monocultures was increasingly becoming more acidic as VFAs accumulated during dark fermentation (Fig. 2a). Our data show that most of the sulfate transport and reduction genes of C. cellulovorans in the cocultures were becoming more downregulated over time from the early to late exponential phases (Fig. 3), when the pH of C. cellulovorans monocultures was becoming more acidic, while the pH of cocultures changed more slowly. Coincidentally, acetate stress has previously been shown to result in upregulation of genes related to the uptake and conversion of sulfate to cysteine in Clostridium acetobutylicum, although the opposite downregulation was observed upon butyrate stress (66). Furthermore, a previous study carried out with Shewanella oneidensis has also shown that genes involved with sulfate transport and assimilative sulfur metabolism were induced upon pH stress (alkaline) (67). Although further investigations are required to fully appreciate the exact physiological role of cysteine under conditions of pH stress, our results suggest that the synthesis of cysteine might be physiologically important for C. cellulovorans to protect against acid stress and the expression of related genes might be a stress response to maintain cellular metabolism.
FIG 3.

(a) Upregulated (red) and downregulated (blue) expression profiles of C. cellulovorans genes related to sulfate transport and reduction at the early, mid-, and late exponential growth phases in response to cocultivation with R. palustris. DEGs (genes differentially expressed at any time point) are marked with an asterisk (*). (b) The genes indicated in panel a are shown in the corresponding metabolic pathways. APS, adenosine-5′-phosphosulfate; PAPS, phosphoadenosine phosphosulfate. A question mark (?) indicates that the gene has not been annotated in the C. cellulovorans genome.
Thiamine metabolism.
The downregulation of C. cellulovorans genes related to the thiamine metabolism pathway was also observed in response to cocultivation with R. palustris (Table 2). These genes include those encoding thiamine-phosphate pyrophosphorylase (Clocel 0682), biotin and thiamine synthesis-associated protein (Clocel 3433), thiamine biosynthesis proteins ThiC and ThiF (Clocel 2812 and Clocel 0683), and thiazole synthase ThiH and ThiG (Clocel 0684 and Clocel 0685), which catalyze the formation of 5-(2-hydroxyethyl)-4-methylthiazole, the precursor to the condensation of thiamine monophosphate. The downregulation of these genes likely influences the biosynthesis of the final products of thiamine metabolism, namely, thiamine and its active form, thiamine pyrophosphate (TPP). The physiological role of TPP has been well characterized as the universal cofactor for enzymes engaged in crucial metabolic pathways, such as transketolase, involved in the pentose phosphate pathway, and pyruvate:ferredoxin oxidoreductase, required for the interconversion of pyruvate and acetyl-CoA in the pyruvate metabolism pathway. Notably, the C. cellulovorans genes (Clocel 1257 and Clocel 1684) annotated to encode these two TPP-dependent enzymes were both downregulated in response to cocultivation with R. palustris.
In addition, studies have shown that thiamine metabolism might play a protective role in defending the cell against adverse conditions. It has been reported that thiamine compounds accumulated in response to amino acid starvation and energy stress conditions in E. coli (68). Also, a recent study (69) carried out with Saccharomyces cerevisiae showed that genes related to TPP biosynthesis were significantly upregulated and the corresponding enzymatic activity levels increased in response to oxidative and osmotic stresses, indicating that thiamine metabolism can partly compensate for damages of the yeast general defense systems. Surprisingly, similar to what is observed with the sulfate transport and reduction genes, a strong correlation of thiamine metabolism gene expression with pH was also observed for C. cellulovorans (Fig. 4). From the early to late exponential phases, the expression level of a number of the thiamine metabolism genes of C. cellulovorans (10 of 22) decreased over time in cocultures (Fig. 4). The downregulation of thiamine metabolism genes in the cocultures reflects the increased transcriptional level of thiamine metabolism genes in C. cellulovorans monocultures, indicating that an enhanced thiamine and TPP biosynthesis was elicited in response to acidic pH. Although the gene expression of thiamine metabolism seems to be an oxidative stress response, it might be induced by acidic pH as well. These results again suggest that R. palustris in the cocultures can play an important role in mitigating the pH-induced oxidative stress response.
FIG 4.

(a) Upregulated (red) and downregulated (blue) expression profiles of C. cellulovorans genes related to thiamine metabolism at the early, mid-, and late exponential growth phases in response to cocultivation with R. palustris. DEGs (genes differentially expressed at any time point) are marked with an asterisk (*). (b) The genes indicated in panel a are shown in the corresponding metabolic pathways. HET, 5-(2-hydroxyethyl)-4-methylthiazole; HET-P, HET phosphate; HMP, 4-amino-5-hydroxymethyl-2-methylpyrimidine; HMP-P, HMP phosphate; HMP-PP, HMP diphosphate; TMP, thiamine monophosphate; TDP, thiamine diphosphate; AIR, 1-(5′-phospho-ribosyl)-5-amino-imidazole; COSH, thiocarboxylate. A question mark (?) indicates that the gene has not been annotated in the R. palustris genome.
Photosynthesis.
Under anaerobic conditions, R. palustris is capable of conserving energy from light via photosynthesis using its photosystem. From an energetic point of view, the photosystem is extremely important because it carries out the main photochemical reactions within a cell, including light absorption and energy and electron transfer. The structure of a photosystem is comprised of two light-harvesting complexes, I and II (encoded by the puc and puf operons, respectively), and the reaction center (encoded by the puh operon) (70). In response to cocultivation with C. cellulovorans, 11 of 14 genes in the puc, puf, and puh operons in R. palustris were upregulated more than 2-fold (Table 3). Although the upregulations of two puc operon genes (RPA 3012 to 3013) and a puh operon gene (RPA 1548) were below 2-fold, these three genes were still upregulated more than 1.7-fold. The overexpression of these photosystem-encoding genes strongly indicates that the photosynthesis of R. palustris was more active in the cocultures than in the monocultures. This most likely reflects the increased metabolic activity and cellular energy demand of R. palustris in the cocultures. For example, the above-mentioned nitrogen fixation, carbon assimilation, and chemotaxis are all energy-demanding processes.
Other genes and pathways of significance.
In response to cocultivation with R. palustris, a number of C. cellulovorans genes responsible for nitrogen metabolism were upregulated (Table 2). For example, the ABC transporters genes for Mo (Clocel 1529 to 1531) were upregulated (Table 2). The upregulation of a few nitrogen metabolism and related genes (Clocel 2836, Clocel 2838, and Clocel 4147) suggests that nitrogen demand was enhanced for C. cellulovorans in cocultures, possibly due to the increased overall metabolic level of C. cellulovorans triggered by the presence of R. palustris. In our previous physiological study (26), cocultivation with R. palustris did not result in significant differences in cell density for C. cellulovorans relative to the monocultures. Consistent with this observation, none of the C. cellulovorans genes related to cell division were differentially expressed in the cocultures. Similarly, the R. palustris genes responsible for cell division were also not differentially expressed. In addition, a number of R. palustris genes exhibited downregulation in response to cocultivation with C. cellulovorans. For example, genes related to nicotinate and nicotinamide metabolism, alanine, aspartate, and glutamate metabolism, and RNA degradation were downregulated.
In summary, a number of physiological studies of H2-producing cocultures (18–21, 26) incorporating dark-fermentative and photosynthetic bacteria have shown that H2 yield and substrate utilization are enhanced during coculturing. However, the detailed molecular mechanisms of how each organism in the coculture responds to the presence of the culturing partner and the changing culturing conditions (e.g., pH) remain unclear. In order to further understand the interactions in an H2-producing coculture, we report a transcriptomic analysis of the H2-producing coculture of C. cellulovorans and R. palustris utilizing cellulose as the sole carbon substrate in this study. By correlating the transcriptomic data to the physiological characteristics of the cocultures, a conceptual model that summarizes the key differentially regulated responses in both strains was developed and is illustrated in Fig. 5. In this coculture model, three cellulosomal genes of C. cellulovorans were upregulated, indicating enhanced cellulolytic activity in response to cocultivation with R. palustris. Meanwhile, genes involved in nitrogen metabolism and fatty acid biosynthesis in C. cellulovorans were also upregulated in the cocultures. The upregulation of fatty acid biosynthesis in C. cellulovorans is likely to increase VFA production, which could be further utilized by R. palustris as the carbon source for growth. Accordingly, a gene involved in VFA consumption in R. palustris was also upregulated. As the VFAs were consumed by R. palustris, the pH in cocultures was stabilized in comparison to the acidic pH in C. cellulovorans monocultures. Surprisingly, a strong correlation of sulfur and thiamine metabolism gene expression with pH was observed for C. cellulovorans, in which these genes were downregulated in the cocultures, which suggests that they function as a stress response against acidic pH in the monocultures.
FIG 5.

The physiological and transcriptome conceptual model of C. cellulovorans and R. palustris in cocultures. The key differentially regulated genes and their pathways of interaction are highlighted. The final products of the metabolic pathways are also shown in order to describe the carbon and cellular metabolism in the coculture model. Red and blue, upregulated and downregulated genes and pathways, respectively. Green, correlations between VFAs, pH, and pathways.
In addition, as a motile bacterium, a number of chemotaxis genes in R. palustris were upregulated, most likely moving toward the VFAs produced by C. cellulovorans. Consistent with the physiological observations of enhanced H2 production, genes related to nitrogen fixation in R. palustris were upregulated. These genes include the nif and anf operons, reductant-encoding genes, Ntr regulon, and transporter genes for NH4+, Mo, and nitrate. However, unexpectedly, genes related to H2 production in C. cellulovorans were downregulated; hence, the enhanced H2 yield most likely was from R. palustris alone instead of C. cellulovorans or both. Moreover, genes involved in photosynthesis in R. palustris were upregulated, reflecting an increased metabolic activity of R. palustris in the cocultures. The derived conceptual model is inferred from results at the transcription level only; hence, future studies should also apply high-throughput shotgun proteomics to examine the expressed proteins so that our understanding of the coculture interactions can be further enhanced. Nevertheless, the global transcriptomic analysis in this study enables a molecular view of the interactions of an H2-producing coculture and provides insights into the observed physiological characteristics, which would benefit the engineering of more-effective consortia for H2 production in industrial processes.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Research Grants Council of Hong Kong through projects 116111 and 11206514 and a grant from the Ability R&D Energy Research Centre.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00789-16.
REFERENCES
- 1.Faust K, Raes J. 2012. Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 2.He J, Holmes VF, Lee PKH, Alvarez-Cohen L. 2007. Influence of vitamin B12 and cocultures on the growth of Dehalococcoides isolates in defined medium. Appl Environ Microbiol 73:2847–2853. doi: 10.1128/AEM.02574-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Men Y, Feil H, Verberkmoes NC, Shah MB, Johnson DR, Lee PKH, West KA, Zinder SH, Andersen GL, Alvarez-Cohen L. 2012. Sustainable syntrophic growth of Dehalococcoides ethenogenes strain 195 with Desulfovibrio vulgaris Hildenborough and Methanobacterium congolense: global transcriptomic and proteomic analyses. ISME J 6:410–421. doi: 10.1038/ismej.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossouw D, Du Toit M, Bauer FF. 2012. The impact of co-inoculation with Oenococcus oeni on the transcriptome of Saccharomyces cerevisiae and on the flavour-active metabolite profiles during fermentation in synthetic must. Food Microbiol 29:121–131. doi: 10.1016/j.fm.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Maligoy M, Mercade M, Cocaign-Bousquet M, Loubiere P. 2008. Transcriptome analysis of Lactococcus lactis in coculture with Saccharomyces cerevisiae. Appl Environ Microbiol 74:485–494. doi: 10.1128/AEM.01531-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendes F, Sieuwerts S, de Hulster E, Almering MJH, Luttik MAH, Pronk JT, Smid EJ, Bron PA, Daran-Lapujade P. 2013. Transcriptome-based characterization of interactions between Saccharomyces cerevisiae and Lactobacillus delbrueckii subsp. bulgaricus in lactose-grown chemostat cocultures. Appl Environ Microbiol 79:5949–5961. doi: 10.1128/AEM.01115-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheirsilp B, Shoji H, Shimizu H, Shioya S. 2003. Interactions between Lactobacillus kefiranofaciens and Saccharomyces cerevisiae in mixed culture for kefiran production. J Biosci Bioeng 96:279–284. doi: 10.1016/S1389-1723(03)80194-9. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Ye X, Finneran KT, Zilles JL, Morgenroth E. 2013. Interactions between Clostridium beijerinckii and Geobacter metallireducens in co-culture fermentation with anthrahydroquinone-2,6-disulfonate (AH2QDS) for enhanced biohydrogen production from xylose. Biotechnol Bioeng 110:164–172. doi: 10.1002/bit.24627. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y. 2011. Development and application of co-culture for ethanol production by co-fermentation of glucose and xylose: a systematic review. J Ind Microbiol Biotechnol 38:581–597. doi: 10.1007/s10295-010-0894-3. [DOI] [PubMed] [Google Scholar]
- 10.Fang HHP, Zhu H, Zhang T. 2006. Phototrophic hydrogen production from glucose by pure and co-cultures of Clostridium butyricum and Rhodobacter sphaeroides. Int J Hydrogen Energy 31:2223–2230. doi: 10.1016/j.ijhydene.2006.03.005. [DOI] [Google Scholar]
- 11.Chang JJ, Chou CH, Ho CY, Chen WE, Lay JJ, Huang CC. 2008. Syntrophic co-culture of aerobic Bacillus and anaerobic Clostridium for bio-fuels and bio-hydrogen production. Int J Hydrogen Energy 33:5137–5146. doi: 10.1016/j.ijhydene.2008.05.021. [DOI] [Google Scholar]
- 12.He Q, Hemme CL, Jiang H, He Z, Zhou J. 2011. Mechanisms of enhanced cellulosic bioethanol fermentation by co-cultivation of Clostridium and Thermoanaerobacter spp. Bioresour Technol 102:9586–9592. doi: 10.1016/j.biortech.2011.07.098. [DOI] [PubMed] [Google Scholar]
- 13.Alper H, Stephanopoulos G. 2009. Engineering for biofuels: exploiting innate microbial capacity or importing biosynthetic potential? Nat Rev Microbiol 7:715–723. doi: 10.1038/nrmicro2186. [DOI] [PubMed] [Google Scholar]
- 14.Brenner K, You L, Arnold FH. 2008. Engineering microbial consortia: a new frontier in synthetic biology. Trends Biotechnol 26:483–489. doi: 10.1016/j.tibtech.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Xie B, Bishop S, Stessman D, Wright D, Spalding MH, Halverson LJ. 2013. Chlamydomonas reinhardtii thermal tolerance enhancement mediated by a mutualistic interaction with vitamin B12-producing bacteria. ISME J 7:1544–1555. doi: 10.1038/ismej.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen CY, Yang MH, Yeh KL, Liu CH, Chang JS. 2008. Biohydrogen production using sequential two-stage dark and photo fermentation processes. Int J Hydrogen Energy 33:4755–4762. doi: 10.1016/j.ijhydene.2008.06.055. [DOI] [Google Scholar]
- 17.Su H, Cheng J, Zhou J, Song W, Cen K. 2009. Combination of dark- and photo-fermentation to enhance hydrogen production and energy conversion efficiency. Int J Hydrogen Energy 34:8846–8853. doi: 10.1016/j.ijhydene.2009.09.001. [DOI] [Google Scholar]
- 18.Liu BF, Ren NQ, Xie GJ, Ding J, Guo WQ, Xing DF. 2010. Enhanced bio-hydrogen production by the combination of dark- and photo-fermentation in batch culture. Bioresour Technol 101:5325–5329. doi: 10.1016/j.biortech.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 19.Ding J, Liu BF, Ren NQ, Xing DF, Guo WQ, Xu JF, Xie GJ. 2009. Hydrogen production from glucose by co-culture of Clostridium butyricum and immobilized Rhodopseudomonas faecalis RLD-53. Int J Hydrogen Energy 34:3647–3652. doi: 10.1016/j.ijhydene.2009.02.078. [DOI] [Google Scholar]
- 20.Liu BF, Ren NQ, Tang J, Ding J, Liu WZ, Xu JF, Cao GL, Guo WQ, Xie GJ. 2010. Bio-hydrogen production by mixed culture of photo- and dark-fermentation bacteria. Int J Hydrogen Energy 35:2858–2862. doi: 10.1016/j.ijhydene.2009.05.005. [DOI] [Google Scholar]
- 21.Jiao Y, Navid A, Stewart BJ, McKinlay JB, Thelen MP, Pett-Ridge J. 2012. Syntrophic metabolism of a co-culture containing Clostridium cellulolyticum and Rhodopseudomonas palustris for hydrogen production. Int J Hydrogen Energy 37:11719–11726. doi: 10.1016/j.ijhydene.2012.05.100. [DOI] [Google Scholar]
- 22.Cheng J, Su H, Zhou J, Song W, Cen K. 2011. Hydrogen production by mixed bacteria through dark and photo fermentation. Int J Hydrogen Energy 36:450–457. doi: 10.1016/j.ijhydene.2010.10.007. [DOI] [Google Scholar]
- 23.Sieuwerts S, Molenaar D, van Hijum SAFT, Beerthuyzen M, Stevens MJA, Janssen PWM, Ingham CJ, de Bok FAM, de Vos WM, van Hylckama Vlieg JET. 2010. Mixed-culture transcriptome analysis reveals the molecular basis of mixed-culture growth in Streptococcus thermophilus and Lactobacillus bulgaricus. Appl Environ Microbiol 76:7775–7784. doi: 10.1128/AEM.01122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beliaev AS, Romine MF, Serres M, Bernstein HC, Linggi BE, Markillie LM, Isern NG, Chrisler WB, Kucek LA, Hill EA, Pinchuk GE, Bryant DA, Steven Wiley H, Fredrickson JK, Konopka A. 2014. Inference of interactions in cyanobacterial-heterotrophic co-cultures via transcriptome sequencing. ISME J 8:2243–2255. doi: 10.1038/ismej.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu BF, Xie GJ, Wang RQ, Xing DF, Ding J, Zhou X, Ren HY, Ma C, Ren NQ. 2015. Simultaneous hydrogen and ethanol production from cascade utilization of mono-substrate in integrated dark and photo-fermentative reactor. Biotechnol Biofuels 8:8. doi: 10.1186/s13068-014-0191-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu H, Lee PKH. 2015. Effects of cellulose concentrations on the syntrophic interactions between Clostridium cellulovorans 743B and Rhodopseudomonas palustris CGA009 in coculture fermentation for biohydrogen production. Int J Hydrogen Energy 40:11800–11808. doi: 10.1016/j.ijhydene.2015.05.135. [DOI] [Google Scholar]
- 27.Pawar S, Vongkumpeang T, Grey C, van Niel E. 2015. Biofilm formation by designed co-cultures of Caldicellulosiruptor species as a means to improve hydrogen productivity. Biotechnol Biofuels 8:19. doi: 10.1186/s13068-015-0201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adessi A, McKinlay JB, Harwood CS, De Philippis R. 2012. A Rhodopseudomonas palustris nifA* mutant produces H2 from NH4+-containing vegetable wastes. Int J Hydrogen Energy 37:15893–15900. doi: 10.1016/j.ijhydene.2012.08.009. [DOI] [Google Scholar]
- 29.Sun Q, Xiao W, Xi D, Shi J, Yan X, Zhou Z. 2010. Statistical optimization of biohydrogen production from sucrose by a co-culture of Clostridium acidisoli and Rhodobacter sphaeroides. Int J Hydrogen Energy 35:4076–4084. doi: 10.1016/j.ijhydene.2010.01.145. [DOI] [Google Scholar]
- 30.Hillmer P, Gest H. 1977. H2 metabolism in the photosynthetic bacterium Rhodopseudomonas capsulata: H2 production by growing cultures. J Bacteriol 129:724–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hillmer P, Gest H. 1977. H2 metabolism in the photosynthetic bacterium Rhodopseudomonas capsulata: production and utilization of H2 by resting cells. J Bacteriol 129:732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. 2009. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 33.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 34.Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. 2011. Differential expression in RNA-seq: a matter of depth. Genome Res 21:2213–2223. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene Ontology: tool for the unification of biology. Nat Genet 25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 37.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J. 2006. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. 2008. KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamada T, Letunic I, Okuda S, Kanehisa M, Bork P. 2011. iPath2.0: interactive pathway explorer. Nucleic Acids Res 39:W412–W415. doi: 10.1093/nar/gkr313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wickham H. (ed). 2009. ggplot2: elegant graphics for data analysis. Springer, New York, NY. [Google Scholar]
- 41.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 42.Mathews J, Wang G. 2009. Metabolic pathway engineering for enhanced biohydrogen production. Int J Hydrogen Energy 34:7404–7416. doi: 10.1016/j.ijhydene.2009.05.078. [DOI] [Google Scholar]
- 43.Harwood CS. 2008. Nitrogenase-catalyzed hydrogen production by purple non-sulfur photosynthetic bacteria, p 259–272. In Wall JD, Harwood CS, Demain A (ed), Bioenergy. ASM Press, Washington, DC. [Google Scholar]
- 44.Oda Y, Samanta SK, Rey FE, Wu L, Liu X, Yan T, Zhou J, Harwood CS. 2005. Functional genomic analysis of three nitrogenase isozymes in the photosynthetic bacterium Rhodopseudomonas palustris. J Bacteriol 187:7784–7794. doi: 10.1128/JB.187.22.7784-7794.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kutsche M, Leimkuehler S, Angermueller S, Klipp W. 1996. Promoters controlling expression of the alternative nitrogenase and the molybdenum uptake system in Rhodobacter capsulatus are activated by NtrC, independent of σ54, and repressed by molybdenum. J Bacteriol 178:2010–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lehman LJ, Roberts GP. 1991. Identification of an alternative nitrogenase system in Rhodospirillum rubrum. J Bacteriol 173:5705–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drepper T, Groß S, Yakunin AF, Hallenbeck PC, Masepohl B, Klipp W. 2003. Role of GlnB and GlnK in ammonium control of both nitrogenase systems in the phototrophic bacterium Rhodobacter capsulatus. Microbiology 149:2203–2212. doi: 10.1099/mic.0.26235-0. [DOI] [PubMed] [Google Scholar]
- 48.Leigh JA, Dodsworth JA. 2007. Nitrogen regulation in bacteria and archaea. Annu Rev Microbiol 61:349–377. doi: 10.1146/annurev.micro.61.080706.093409. [DOI] [PubMed] [Google Scholar]
- 49.Reitzer L. 2003. Nitrogen assimilation and global regulation in Escherichia coli. Annu Rev Microbiol 57:155–176. doi: 10.1146/annurev.micro.57.030502.090820. [DOI] [PubMed] [Google Scholar]
- 50.Tamaru Y, Miyake H, Kuroda K, Nakanishi A, Matsushima C, Doi RH, Ueda M. 2011. Comparison of the mesophilic cellulosome-producing Clostridium cellulovorans genome with other cellulosome-related clostridial genomes. Microb Biotechnol 4:64-73. doi: 10.1111/j.1751-7915.2010.00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han SO, Cho HY, Yukawa H, Inui M, Doi RH. 2004. Regulation of expression of cellulosomes and noncellulosomal (hemi)cellulolytic enzymes in Clostridium cellulovorans during growth on different carbon sources. J Bacteriol 186:4218-4227. doi: 10.1128/JB.186.13.4218-4227.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Russell JB. 1987. Effect of extracellular pH on growth and proton motive force of Bacteroides succinogenes, a cellulolytic ruminal bacterium. Appl Environ Microbiol 53:2379–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Magnuson K, Jackowski S, Rock CO, Cronan JE Jr. 1993. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol Rev 57:522–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taguchi F, Ogawa Y, Takeuchi K, Suzuki T, Toyoda K, Shiraishi T, Ichinose Y. 2006. A homologue of the 3-oxoacyl-(acyl carrier protein) synthase III gene located in the glycosylation island of Pseudomonas syringae pv. tabaci regulates virulence factors via N-acyl homoserine lactone and fatty acid synthesis. J Bacteriol 188:8376–8384. doi: 10.1128/JB.00763-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lai C-Y, Cronan JE. 2003. β-Ketoacyl-acyl carrier protein synthase III (FabH) is essential for bacterial fatty acid synthesis. J Biol Chem 278:51494–51503. doi: 10.1074/jbc.M308638200. [DOI] [PubMed] [Google Scholar]
- 56.Larimer FW, Chain P, Hauser L, Lamerdin J, Malfatti S, Do L, Land ML, Pelletier DA, Beatty JT, Lang AS, Tabita FR, Gibson JL, Hanson TE, Bobst C, Torres JL, Peres C, Harrison FH, Gibson J, Harwood CS. 2004. Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat Biotechnol 22:55–61. doi: 10.1038/nbt923. [DOI] [PubMed] [Google Scholar]
- 57.Macnab RM, Koshland DE Jr. 1972. Gradient-sensing mechanism in bacterial chemotaxis. Proc Natl Acad Sci U S A 69:2509–2512. doi: 10.1073/pnas.69.9.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sleat R, Mah RA, Robinson R. 1984. Isolation and characterization of an anaerobic, cellulolytic bacterium, Clostridium cellulovorans sp. nov. Appl Environ Microbiol 48:88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maurer LM, Yohannes E, Bondurant SS, Radmacher M, Slonczewski JL. 2005. pH regulates genes for flagellar motility, catabolism, and oxidative stress in Escherichia coli K-12. J Bacteriol 187:304–319. doi: 10.1128/JB.187.1.304-319.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeziore-Sassoon Y, Hamblin PA, Bootle-Wilbraham CA, Poole PS, Armitage JP. 1998. Metabolism is required for chemotaxis to sugars in Rhodobacter spheroides. Microbiology 144:229–239. doi: 10.1099/00221287-144-1-229. [DOI] [PubMed] [Google Scholar]
- 61.Vorobjeva L, Leverrier P, Zinchenko A, Boyaval P, Khodjaev E, Varioukhina S, Ponomareva G, Gordeeva E, Jan G. 2004. Anti-stress activity of Propionibacterium freudenreichii: identification of a reactivative protein. Antonie Van Leeuwenhoek 85:53–62. doi: 10.1023/B:ANTO.0000020276.18127.99. [DOI] [PubMed] [Google Scholar]
- 62.Lithgow JK, Hayhurst EJ, Cohen G, Aharonowitz Y, Foster SJ. 2004. Role of a cysteine synthase in Staphylococcus aureus. J Bacteriol 186:1579–1590. doi: 10.1128/JB.186.6.1579-1590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frazzon J, Fick JR, Dean DR. 2002. Biosynthesis of iron-sulphur clusters is a complex and highly conserved process. Biochem Soc Trans 30:680–685. doi: 10.1042/bst0300680. [DOI] [PubMed] [Google Scholar]
- 64.Carmel-Harel O, Storz G. 2000. Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and Saccharomyces cerevisiae responses to oxidative stress. Annu Rev Microbiol 54:439–461. doi: 10.1146/annurev.micro.54.1.439. [DOI] [PubMed] [Google Scholar]
- 65.Storz G, Hengge-Aronis R. 2000. Microbial responses to acid stress. ASM Press, Washington, DC. [Google Scholar]
- 66.Alsaker KV, Paredes C, Papoutsakis ET. 2010. Metabolite stress and tolerance in the production of biofuels and chemicals: gene-expression-based systems analysis of butanol, butyrate, and acetate stresses in the anaerobe Clostridium acetobutylicum. Biotechnol Bioeng 105:1131–1147. doi: 10.1002/bit.22628. [DOI] [PubMed] [Google Scholar]
- 67.Leaphart AB, Thompson DK, Huang K, Alm E, Wan XF, Arkin A, Brown SD, Wu L, Yan T, Liu X, Wickham GS, Zhou J. 2006. Transcriptome profiling of Shewanella oneidensis gene expression following exposure to acidic and alkaline pH. J Bacteriol 188:1633–1642. doi: 10.1128/JB.188.4.1633-1642.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gigliobianco T, Lakaye B, Wins P, El Moualij B, Zorzi W, Bettendorff L. 2010. Adenosine thiamine triphosphate accumulates in Escherichia coli cells in response to specific conditions of metabolic stress. BMC Microbiol 10:148. doi: 10.1186/1471-2180-10-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kowalska E, Kujda M, Wolak N, Kozik A. 2012. Altered expression and activities of enzymes involved in thiamine diphosphate biosynthesis in Saccharomyces cerevisiae under oxidative and osmotic stress. FEMS Yeast Res 12:534–546. doi: 10.1111/j.1567-1364.2012.00804.x. [DOI] [PubMed] [Google Scholar]
- 70.Jagannathan B, Golbeck JH. 2009. Photosynthesis: microbial, p 325–341. In Schaechter M. (ed), Encyclopedia of microbiology, 3rd ed Academic Press, Oxford, England. doi: 10.1016/B978-012373944-5.00352-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.