

ABSTRACT
Murine polyomavirus has repeatedly provided insights into tumorigenesis, revealing key control mechanisms such as tyrosine phosphorylation and phosphoinositide 3-kinase (PI3K) signaling. We recently demonstrated that polyomavirus small T antigen (ST) binds YAP, a major effector of Hippo signaling, to regulate differentiation. Here we characterize YAP as a target of middle T antigen (MT) important for transformation. Through a surface including residues R103 and D182, wild-type MT binds to the YAP WW domains. Mutation of either R103 or D182 of MT abrogates YAP binding without affecting binding to other signaling molecules or the strength of PI3K or Ras signaling. Either genetic abrogation of YAP binding to MT or silencing of YAP via short hairpin RNA (shRNA) reduced MT transformation, suggesting that YAP makes a positive contribution to the transformed phenotype. MT targets YAP both by activating signaling pathways that affect it and by binding to it. MT signaling, whether from wild-type MT or the YAP-binding MT mutant, promoted YAP phosphorylation at S127 and S381/397 (YAP2/YAP1). Consistent with the known functions of these phosphorylated serines, MT signaling leads to the loss of YAP from the nucleus and degradation. Binding of YAP to MT brings it together with protein phosphatase 2A (PP2A), leading to the dephosphorylation of YAP in the MT complex. It also leads to the enrichment of YAP in membranes. Taken together, these results indicate that YAP promotes MT transformation via mechanisms that may depart from YAP's canonical oncogenic transcriptional activation functions.
IMPORTANCE The highly conserved Hippo/YAP pathway is important for tissue development and homeostasis. Increasingly, changes in this pathway are being associated with cancer. Middle T antigen (MT) is the primary polyomavirus oncogene responsible for tumor formation. In this study, we show that MT signaling promotes YAP phosphorylation, loss from the nucleus, and increased turnover. Notably, MT genetics demonstrate that YAP binding to MT is important for transformation. Because MT also binds PP2A, YAP bound to MT is dephosphorylated, stabilized, and localized to membranes. Taken together, these results indicate that YAP promotes MT transformation via mechanisms that depart from YAP's canonical oncogenic transcriptional activation functions.
INTRODUCTION
Middle T antigen (MT) is the primary oncogene of murine polyomavirus, which causes a wide variety of tumors (1–3). When expressed as a transgene, MT causes tumors in virtually any tissue (see references 4 and 5 for reviews). It has repeatedly provided insight into the regulation of cell growth. Tyrosine phosphorylation (6) and phosphoinositide 3-kinase (PI3K) (7) are avenues of cancer research opened by analysis of MT. Recent work pointing out the importance of protein phosphatase 2A (PP2A) Aβ isoforms (8) shows the continuing value of this system. Indeed, results of studies on PI3K isoform dependence in an MT-driven genetically engineered mouse (GEM) model of breast cancer have helped in the design of clinical trials of p110α isoform-specific PI3K inhibitors (9). The ability of MT to transform depends on its association with membranes (10), and it has sometimes been likened to an activated receptor tyrosine kinase (11). Binding of protein phosphatase 2A (12–14) leads to the recruitment of protein tyrosine kinases of the Src family (Src, Yes, and Fyn) (15–18). MT is phosphorylated on three major tyrosine residues, residues 315, 322, and 250 (19–22). Each site represents a connection to a signal generator: residue 315 to PI3K (23, 24), residue 250 to SHC (Src homology 2 domain-containing) (25, 26) and then to Grb2 and SOS, and residue 322 to phospholipase C γ1 (PLC-γ1) (27). All these connections are important for transformation.
The work described here connects MT function to YAP (and TAZ), which is the major effector of the Hippo pathway. Hippo signaling, which was initially studied in Drosophila melanogaster, is currently an area of intense investigation (28–33). The Hippo pathway controls organ size, cell proliferation, and survival. It also regulates stem cell proliferation and maintenance (34–37). Hippo signaling through YAP/TAZ is inextricably linked to cancer (38–40). Colorectal (41), breast (39), ovarian (42), squamous cell (43, 44), liver (45), and lung (46) cancers have all shown key roles for Hippo/YAP/TAZ. Hippo signaling is also involved in the regulation of mechanotransduction (47).
The core of the Hippo pathway involves a kinase cascade (Hippo/Mst1/2 and Lats1/2) (48). Diverse stimuli can activate Mst, the upstream kinase, including polarity, G-protein-coupled receptor (GPCR) signaling, and mechanical cues such as extracellular matrix rigidity (49). The cascade results in the phosphorylation of YAP/TAZ, the major Hippo effectors (50). In the most basic view, YAP and TAZ are regulated by phosphorylation. Unphosphorylated, these effectors control a complex transcriptional program (51) by interactions with transcription factors, including TEAD (52), Runx (53), TBX5 (54), and SMAD (55). The result is cross-regulation of the Hedgehog, Notch, transforming growth factor β (TGF-β), and Wnt signaling pathways (38, 56), which shows how central Hippo signaling is to cellular control. Phosphorylation of YAP by LATS at S127 induces an association with 14-3-3 proteins and relocalization to the cytoplasm (52, 57). From this perspective, Hippo signaling is inactivating, as is YAP/TAZ phosphorylation. However, in the cytoplasm, YAP/TAZ contributes to the regulation of tissue architecture, including apical-basal polarity (via the Crumbs complex [58]), tight junctions (via ZO-2 [59]), and adherens junctions (via α/β-catenin [60]).
We previously demonstrated that polyomavirus small T antigen (ST) binds YAP to affect differentiation (61). The sequence identity between ST and the N terminus of MT, together with the knowledge that MT binds the YAP relative TAZ (62, 63), led us to hypothesize that MT may interact with YAP. Here data from mass spectrometry (MS) and coimmunoprecipitation experiments demonstrate that YAP is indeed a novel MT interactor. Furthermore, soft-agar transformation assays show that YAP is important for cellular transformation by MT. Not only is YAP bound by MT, it also is a target of MT signaling. Wild-type MT is ineffective at transforming YAP knockdown cells, and MT mutants that do not bind YAP are defective in transformation. At the protein level, MT affects the phosphorylation status, stability, and subcellular localization of YAP in a manner that departs from the canonical understanding of YAP-mediated oncogenesis.
MATERIALS AND METHODS
Cells and antibodies.
The normal mouse fibroblast cell line NIH 3T3; the normal mouse mammary gland epithelial cell line NMuMG; the normal rat fibroblast cell line Rat-1; and human 293, 293T, 293FT, and 293 Phoenix cell lines were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). NIH 3T3 cells were propagated in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO) supplemented with 10% calf serum (CS; Invitrogen, Grand Island, NY). NMuMG, Rat-1, 293T, and 293FT cells were propagated in DMEM supplemented with 10% fetal bovine serum (FBS; Invitrogen). All media were supplemented with 1% glutamine and 1% penicillin-streptomycin (Invitrogen). 293FT cells were propagated in the presence of 500 μg/ml G418 (Invitrogen). All cells were propagated at 37°C in a humidified atmosphere composed of 5% CO2. The antibody to the PP2A A subunit that recognizes both Aα and Aβ was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibodies to Src, Shc, phospho-Shc, phospho-Akt, phospho-MEK, phospho-extracellular signal-regulated kinase (ERK), YAP, phospho-YAP, and TAZ were purchased from Cell Signaling Technology (Danvers, MA). MT-1 is an antipeptide antibody that recognizes a region of MT between residues 282 and 300, a region known to be dispensable for virus growth and transformation (64), and monoclonal antibody (MAb) PN116, recognizing the domain common to large T antigen (LT), MT, and ST, was described previously (65). The polyclonal antibody to p85, the PI3K regulatory subunit, used in this study was generated in our laboratories. FLAG-tagged proteins were immunoprecipitated by using anti-FLAG resin (Sigma-Aldrich). The anti-FLAG antibody used for Western blotting was purchased from Clontech (Mountain View, CA). Hemagglutinin (HA)-tagged proteins were immunoprecipitated with anti-HA resin (Sigma-Aldrich). The anti-HA antibody used for Western blotting was purchased from Covance (Princeton, NJ).
Constructs and stable expression.
The MT open reading frame (ORF) was subcloned from a pOZ vector encoding MT doubly tagged at the C terminus with FLAG and HA into the doxycycline-inducible pInducer20 lentiviral vector, a gift from Stephen Elledge (Addgene plasmid 44012) (66). The following primers were used to clone untagged MT into pInducer20: 5′-GCGCGCGGATCCACCATGGATAGAGTTCTGAGCAGA-3′ and 5′-GCGCGCGGATCCTTAGAAATGCCGGGAACGTTTTATTAG-3′. The forward primer described above and the following reverse primer were used to clone MT tagged at the C terminus with FLAG and HA into pInducer20: 5′-GCGCGCGGATCCCTAGGCGTAGTCGGGCACGTC-3′. Amplification of MT with each primer pair generated MT ORFs flanked with Bam restriction sites. The fragments were digested with Bam and inserted into the pENTR1A entry vector, a gift from Eric Campeau (Addgene plasmid 17398) (67). Site-specific recombination into pInducer20 was conducted by using Gateway LR Clonase II (Invitrogen). The reaction product was transformed into Stbl3 competent cells (Invitrogen) propagated at 30°C. After confirmation of the sequence, the purified construct was transfected into 293FT cells (Invitrogen) for lentiviral packaging (66). The virus-containing 293FT cell supernatant was collected at 48 h posttransfection. For infections of NIH 3T3 and NMuMG cells, cells were exposed overnight to a 1:4 dilution of the 293FT supernatant in propagation medium with a final concentration of 8 μg/ml Polybrene (Sigma-Aldrich). The medium was then replaced with fresh propagation medium supplemented with 500 μg/ml G418 (Invitrogen) to generate a stable cell line. MT expression was induced upon exposure to 300 ng/ml doxycycline (Sigma-Aldrich). The MT ORF was cloned into the pWZL retroviral vector (68) and packaged in 293 Phoenix cells (ATCC). The virus-containing cell supernatant was collected at 48 h postinfection. For infection, Rat-1 cells were exposed overnight to the 293 Phoenix supernatant supplemented with 8 μg/ml Polybrene. The medium was then replaced with fresh propagation medium supplemented with blasticidin (20 μg/ml; Santa Cruz) to generate a stable cell line. YAP short hairpin RNA (shRNA) and TAZ/WWTR1 shRNA lentiviral vectors were purchased from Sigma-Aldrich and packaged in 293T cells according to methods reported previously (61). NIH 3T3 cells were infected as described above. Stable expression was achieved by using 5 μg/ml puromycin (Invitrogen).
Transient transfection.
pInducer20-MT; wild-type FLAG-YAP2 (Addgene plasmid 19045), WW1 FLAG-YAP mutant (Addgene 17795), and WW1/WW2 FLAG YAP mutants, gifts from Marius Sudol (69); and HA-TAZ, a gift from Kunliang Guan (Addgene plasmid 32839) (70), were transiently expressed in 293T cells according to a method reported previously (61).
Proteomics.
FLAG-HA-tagged MT was expressed in 293 and NIH 3T3 cells by using the pInducer20 vector. The protein was induced upon exposure to 300 ng/ml doxycycline for 6 h. The cells were trypsinized and washed in phosphate-buffered saline (PBS). The cell pellet was snap-frozen in liquid nitrogen and stored at −80°C until use. Cell lysis, single-step HA immunoprecipitation (IP), elution with HA peptide, and concentration of the eluate by trichloroacetic acid (Sigma) precipitation were conducted according to methods reported previously (71). Sample preparation for mass spectrometry, sequencing, and data analysis using the Comparative Proteomic Analysis Software Suite (CompPASS) were described previously (71, 72).
Immunoprecipitation and Western blotting.
MT was immunoprecipitated by using MT-1, HA-tagged proteins were immunoprecipitated by using anti-HA resin, and FLAG-tagged proteins were immunoprecipitated by using anti-FLAG resin according to methods reported previously (61). SDS-PAGE (10%) analysis was described previously (73). For extended SDS-PAGE experiments, proteins with a molecular mass of 50 kDa were allowed to migrate over 12 cm. Western blotting of MT was carried out by using monoclonal antibody PN116 and methods described previously (65).
Phosphatase treatment.
Calf intestinal alkaline phosphatase (CIP) was purchased from New England BioLabs (Ipswich, MA), and lambda phosphatase was purchased from Calbiochem (San Diego, CA). YAP immunoprecipitates were exposed to a combination of both enzymes for 60 min at 37°C according to the manufacturers' recommendations.
Soft-agar transformation.
NIH 3T3 and NMuMG cells were induced to express the MT protein by exposure to doxycycline for 24 h prior to being suspended in agar, and doxycycline was maintained in the agar for the duration of the assay. The assay was set up in 6-well plates. The bottom of each well was coated with 1 ml propagation medium containing 0.6% agar. Once the bottom coat was polymerized, cells (1 × 105) were suspended in 1 ml propagation medium containing 0.4% agar. Colonies were allowed to form for 3 to 4 weeks. At the completion of the assay, colonies were imaged by using an AlphaImager instrument (Alpha Innotech, San Jose, CA) and quantified by using the open-source software ImageJ (74).
Immunofluorescence.
Cells were seeded in propagation medium in chamber slides (EMS, Hatfield, PA) and allowed to attach overnight in the presence of 300 ng/ml doxycycline to induce MT expression. The cells were washed once in PBS at room temperature, fixed in 3.6% formaldehyde (Sigma-Aldrich) for 30 min at room temperature, washed 3 times in PBS at room temperature, permeabilized in 0.2% Triton X-100 (Sigma-Aldrich) for 30 min at room temperature, washed 3 times in PBS at room temperature, and incubated in PBS supplemented with 1% bovine serum albumin (BSA; Sigma-Aldrich) at room temperature for 1 h. The cells were then incubated at 4°C overnight with primary antibody diluted 1:200 in PBS–1% BSA, washed 3 times in PBS at room temperature, incubated with secondary antibody conjugated to tetramethylrhodamine (Invitrogen) diluted 1:1,000 in PBS–1% BSA for 3 h at room temperature, and washed 3 times in PBS at room temperature. Upon the removal of PBS, mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA) was added to the cells, and a coverslip was fitted onto the slide. Staining was imaged by using a Nikon Olympus microscope and Spot Advanced software (Spot Imaging Solutions, Sterling Heights, MI).
Cell fractionation.
Cytoplasmic and nuclear fractions were isolated from NIH 3T3 cells by using the NE-PER cytoplasmic and nuclear extraction assay (Life Technologies, Grand Island, NY). Membrane fractions were extracted by hypotonic fractionation (75).
Real-time PCR.
Total RNA was extracted by using the Qiagen RNeasy method (Qiagen, Valencia, CA) and reverse transcribed by using the iScript cDNA transcription method (Bio-Rad, Hercules, CA). Real-time PCR was conducted with Sybr green on a LightCycler 480 thermal cycler (Roche, Basel, Switzerland). Primer sequences were as follows: MT forward primer 5′-GTCTGAGTCCATGGAAGGGTCTGATTCTTC-3′, MT reverse primer 5′-GCGCGCGGATCCTTAGAAATGCCGGGAACGTTTTATTAG-3′, YAP forward primer 5′-AGATCCCTGATGATGTACCAC-3′, YAP reverse primer 5′-AGGAACGTTCAGTTGCGA-3′, ANKRD1 forward primer 5′-AAAGGCAAGGGTTGATCCCC-3′, and ANKRD1 reverse primer 5′-TGCCTTCACCTTGGGACATC-3′.
RESULTS
The Hippo effector YAP interacts with MT.
Proteomic experiments were carried out to look for new MT-interacting proteins. We chose to use affinity purification-mass spectrometry (AP-MS) followed by analysis with CompPASS software to identify MT-interacting proteins (72). 293 cells stably transduced with the pInducer20-FlagHA-MT vector were treated with doxycycline for 6 h to induce MT production and then subjected to anti-HA immunoprecipitation. Immunoprecipitates were digested with trypsin, and MT-interacting proteins were identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as previously described (72). High-confidence interacting proteins (HCIP) that bound to MT were distinguished from nonspecific interactors by comparison to a 293 cell-specific statistics table in the CompPASS software. These experiments were followed by similar experiments with NIH 3T3 cells that gave essentially identical results. Most known MT interactors were detected (Table 1). The PP2A A and C subunits; Src family members required for the phosphorylation of MT; the Shc adaptor, which is known to bind at Y250; PI3K, which binds at Y315; PLC-γ1; and the 14-3-3 proteins were all observed. A number of proteins not previously reported to be MT partners were also observed. Lipins, which were previously shown to bind ST (76), were found. The catalytic subunit of protein phosphatase 4 (PP4), Ran GAP, and RanBP1 were also observed. These interactions were confirmed by coimmunoprecipitation (not shown) but not studied further. The Hippo effector TAZ was also detected, as was previously reported (62). Of particular interest, YAP was also found. We recently characterized the association of YAP with polyomavirus ST (61). Recently reported evidence also suggests that other polyomaviruses use their STs to affect YAP (77).
TABLE 1.
YAP is a novel MT-binding partnera
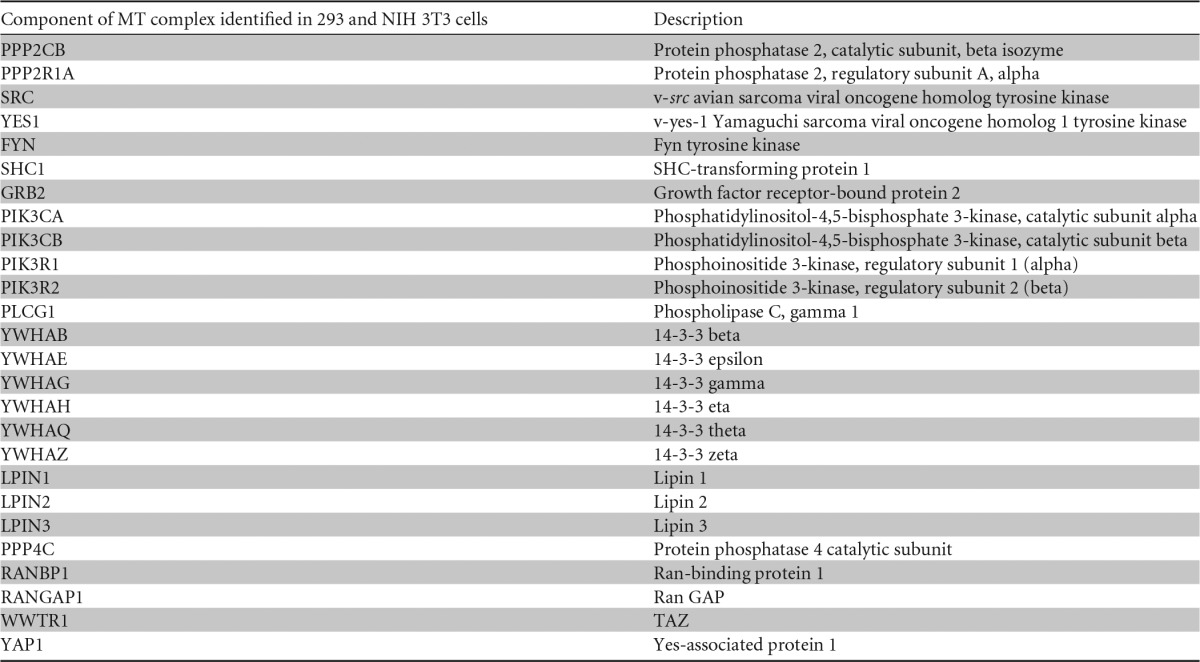
293 cells and NIH 3T3 cells were induced to express EGFP- or HA-tagged full-length wild-type MT by exposure to doxycycline for 6 h. Proteomic analysis of MT complexes was conducted by using affinity purification-mass spectrometry followed by analysis with CompPASS software.
Our previous work with ST identified sites of interaction with YAP to include residues 103 and 182 (61). ST and MT have identical sequences from the N terminus through residue 191, so these sites are also found in MT. Structural models of the ST-PP2A interaction suggested that these two residues are close together on ST and some distance from the PP2A interaction site (61). Wild-type MT or the R103A MT mutant was transiently coexpressed in 293T cells with either FLAG-YAP or HA-TAZ. Western blot analysis of YAP complexes immunoprecipitated with anti-FLAG antibody or TAZ complexes immunoprecipitated with anti-HA antibody were probed for MT. As shown in Fig. 1A, mutation of R103 to A abolishes YAP binding to MT. Figure 1 (right) shows that the R103A MT mutant is also defective in TAZ binding. Tian and colleagues (62) previously identified residues 2 to 4 in the extreme N terminus to be important for TAZ binding. In our hands, such a mutant is also defective in association with PP2A, so the work shown here was performed with the R103A mutant. However, we note that the D182E MT mutant (not shown) seems to behave identically to the R103A mutant.
FIG 1.

MT binding of YAP and TAZ. (A) MT-103A is defective in binding to YAP and TAZ. 293T cells were transiently transfected with vectors encoding MT, FLAG-YAP, or HA-TAZ. After ∼40 h, YAP or TAZ was immunoprecipitated with antibodies to the tag. After SDS-PAGE, Western blotting was carried out to detect YAP, TAZ, and MT. (B) MT and ST have similar affinities for YAP and TAZ. Full-length wild-type (WT) MT and ST were cotransfected in 293T cells in the presence of FLAG-YAP or HA-TAZ. YAP or TAZ was immunoprecipitated from cell extracts 40 h later. After SDS-PAGE, Western blot analysis was carried out to detect MT and ST and compare the relative ratios of MT to ST between cell extracts and YAP or TAZ complexes. WCL, whole-cell lysate. (C) The WW domains of YAP are involved in binding MT. A diagram of the YAP domain structure is shown. Full-length YAP, YAP mutated in the first WW domain (W199A and P202A), YAP mutated in the two WW domains (W199A, P202A, W258A, and P261A), and YAP with a 5-residue C-terminal deletion were each cotransfected with MT in 293T cells. After SDS-PAGE, Western blot analysis of MT complexes was carried out to detect MT and YAP.
Our laboratories previously identified YAP as a binding partner of ST (61). To examine whether there was any substantial difference in the affinity for YAP between ST and MT, wild-type MT and ST were coexpressed in 293T cells with FLAG-YAP or HA-TAZ (Fig. 1B). Western blot analysis of YAP complexes or TAZ complexes showed a small enrichment of ST over MT in the complex compared to the ratio of MT to ST in the whole-cell lysate. This difference suggests that MT binding is modestly weaker than that of ST.
YAP (Fig. 1C, top) and TAZ have multiple domains. Both proteins have a domain that interacts with TEAD transcription factors, a transactivation domain, and a PDZ-binding domain. TAZ has a single WW domain, while YAP has two WW domains. YAP also has an SH3-binding domain and a proline-rich region absent from TAZ (29). Previously reported experiments (62) showed that the WW1 domain was critical for the binding of TAZ to polyomavirus T antigens. Figure 1C shows that this is also true for YAP. A double point mutation (W199A/P202A) that inactivates the WW1 domain of YAP almost completely blocks the association with MT, while simultaneous mutation of both WW (W199A/P202A//W258A/P261A) domains abolished the interaction. As a control, deletion of the 5 carboxy-terminal amino acids to prevent PDZ interactions did not affect binding to MT (Fig. 1C, bottom).
YAP is important for MT transformation.
The consequences of YAP binding by MT for oncogenic transformation were assessed by comparing wild-type MT to the R103A MT mutant in three different cell types. Rat-1 cells were transformed with pWZL constructs stably expressing wild-type MT or the R103A MT mutant. Wild-type MT strongly induced growth in soft agar compared to either uninfected controls or R103A MT (Fig. 2A). Doxycycline-inducible pInducer20 lentiviral constructs were used to stably express enhanced green fluorescent protein (EGFP), wild-type MT, and R103A MT in untransformed NIH 3T3 fibroblasts and normal mouse mammary epithelial (NMuMG) cells. The results showed reduced transformation by R103A MT compared to wild-type MT in these two cell types as well, suggesting that binding to endogenous YAP and/or TAZ contributes to MT transformation (Fig. 2A).
FIG 2.

Role of YAP in MT transformation. (A) MT-103A is defective in transformation. Rat-1 cells stably expressing MT were subjected to colony formation in soft agar. NIH 3T3 or NMuMG cells were induced to express EGFP or the MT protein by exposure to doxycycline for 24 h and subjected to colony formation in soft agar. *, P < 0.05; **, P < 0.01, ***, P < 0.001 (significance versus wild-type [WT] MT). (B) Knockdown of YAP or TAZ inhibits wild-type MT transformation. NIH 3T3 cells stably expressing doxycycline-inducible wild-type MT were infected with YAP shRNA, TAZ shRNA, or control shRNA and selected to achieve stable hairpin expression. After exposure to doxycycline for 24 h to induce the expression of wild-type MT, the cells were transferred to soft agar to assess colony formation. (Left) Data from five YAP shRNA experiments were combined, and data from three TAZ shRNA experiments were combined. ***, P < 0.001 compared to control shRNA per analysis of variance. (Right) Control Western blotting after SDS-PAGE shows MT, YAP, TAZ, and p85 levels. p85 served as a loading control.
An important question is whether MT is using YAP activity to transform or whether it is simply inactivating YAP as a suppressor of growth. Two shRNAs were used to reduce YAP expression in NIH 3T3 cells (Fig. 2B). While these shRNAs did not significantly affect the rate of cell proliferation, both shRNAs dramatically reduced the ability of MT-transformed cells to grow in soft agar. This suggests that MT needs YAP for transformation. TAZ, a YAP family member with structural similarities to YAP, is another effector of the Hippo pathway. There is evidence that TAZ is also important in MT transformation (63). Figure 2B also shows a comparison of YAP and TAZ knockdowns. Like YAP, TAZ seems to contribute to MT transformation.
Interfering with YAP binding does not block MT signaling to Ras and PI3K/Akt.
A series of experiments was performed to compare wild-type MT to the R103A MT mutant to determine the effect of YAP binding on other aspects of MT signaling. The first question was whether MT binding of YAP is independent of associations with other signaling molecules. MT was immunoprecipitated from NIH 3T3 cells induced to express it after exposure to doxycycline for 24 h. Western blot analysis of MT complexes showed that R103A MT was the same as the wild type for the recruitment of PP2A, Src, Shc, and p85 (Fig. 3A). These data suggested that a failure to bind YAP (or TAZ) should not affect MT tyrosine kinase signaling. To ask whether the signaling was affected, levels of Akt phosphorylation, MEK phosphorylation, and p38 phosphorylation were measured after induction of MT for 8 h followed by overnight serum starvation in the continued presence of doxycycline. Mutation of the YAP-binding site at R103 did not impair the ability of MT to induce Akt, MEK, ERK, or p38 phosphorylation (Fig. 3B). For Akt and p38, identical results were obtained in NMuMG or Rat-1 cells (data not shown). In these cell types, no pronounced activation of MEK was observed, but again, wild-type MT and R103A MT were quite similar.
FIG 3.

Ras and PI3K/Akt signaling. (A) MT-103A is wild type for the recruitment of known MT-binding partners. NIH 3T3 cells were exposed to doxycycline for 24 h to induce the expression of EGFP or MT. MT was immunoprecipitated with the MT-1 antibody. After SDS-PAGE, Western blotting was carried out to detect PP2A, Src, Shc, p85, MT, and actin. Actin served as a loading control. (B) MT-103A is wild type for the activation of Ras and PI3K/Akt signaling. NIH 3T3 cells were exposed to doxycycline for 24 h to induce the expression of EGFP or MT, and serum was withdrawn for the last 16 h of induction. After SDS-PAGE, Western blotting was conducted to detect the phosphorylation of Akt S473, MEK S298, Erk1/2 T202/Y204, and p38 T180/Y182 as well as total MT and p85. p85 served as a loading control. (C) Knockdown of YAP or TAZ preserves wild-type MT signaling through Ras and PI3K/Akt. NIH 3T3 cells stably expressing doxycycline-inducible wild-type MT and either control shRNA, YAP shRNA, or TAZ shRNA were induced to express the MT protein by exposure to doxycycline for 24 h, and serum was withdrawn for the last 16 h of induction. After SDS-PAGE, Western blotting was conducted to detect the phosphorylation of Akt S473, Shc Y317, Erk1/2 T202/Y204, and p38 T180/Y182 as well as total YAP, TAZ, MT, p85, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase). p85 and GAPDH served as loading controls.
The ability of wild-type MT to activate the PI3K or Ras pathway in YAP and TAZ knockdown cells was then tested in the same way. As shown in Fig. 3C, no effect was seen on the ability of MT to induce Akt activation measured at S473. The Ras pathway was examined at the levels of both phospho-Shc and phospho-ERK. Again, there was no indication that YAP (or TAZ) was required for MT induction of either of these phosphorylations.
MT causes both phosphorylation and dephosphorylation of YAP.
Because we have shown that ST affects the phosphorylation of YAP, we examined the effect of MT on YAP phosphorylation. After extended electrophoresis, YAP from MT-expressing cells migrated somewhat more slowly than did YAP from control cells. The YAP-binding-deficient R103A MT mutant showed an even greater effect (Fig. 4A). Reduced electrophoretic mobility of YAP results from phosphorylation (61). To confirm this, YAP immunoprecipitates were treated with a combination of calf intestinal and lambda phosphatases (Fig. 4B). The patterns for the control, wild-type MT, and R103A MT were essentially identical after phosphatase treatment. Migration became more rapid in each case, and three distinct bands were resolved. While it is possible that the remaining heterogeneity results from some other modification besides phosphorylation, it is more likely that we are resolving YAP variants such as YAP1 and -2 or other spliced variants (78). The conclusion from data from these two experiments is that wild-type MT promotes the phosphorylation of YAP. When PP2A is inhibited with okadaic acid, YAP mobility is shifted up in wild-type MT cells (not shown), again suggesting an increase in YAP phosphorylation. Because R103A MT has an even greater effect on mobility than the wild type, it appears that MT signaling rather than MT binding is responsible for phosphorylation.
FIG 4.

YAP phosphorylation and dephosphorylation. (A) MT affects YAP mobility. Upon induction of EGFP or MT in NIH 3T3 cells by exposure to doxycycline for 24 h, whole-cell extracts were subjected to extended SDS-PAGE and Western blotting to detect YAP, MT, and p85 (loading control). (B) Phosphorylation controls YAP mobility. Upon induction of EGFP or MT in NIH 3T3 cells by exposure to doxycycline for 24 h, YAP immunoprecipitates were subjected to combined calf intestinal phosphatase (CIP) and lambda phosphatase treatment and subsequent extended SDS-PAGE and Western blotting to detect YAP. The WT MT and MT-103A samples were overloaded to correct for the lower levels of YAP seen in these cells. In doing this, we overloaded them compared to controls. (C) MT induces YAP phosphorylation at S397 and S127. NIH 3T3 cells were induced to express EGFP or MT by exposure to doxycycline for 24 h. After SDS-PAGE, phosphospecific Western blotting was conducted to detect YAP phosphorylation at S397 and S127 as well as total YAP, MT, and GAPDH (loading control) levels. In the bottom three blots, three times the amount of the R103A MT extract was loaded to normalize total YAP. (D) YAP destabilization is a consequence of MT signaling. NIH 3T3 cells were induced to express EGFP or MT by exposure to doxycycline for 24 h, and the Src inhibitor SU6656 was added for the last 16 h of induction. After SDS-PAGE, Western blotting was conducted to detect YAP phosphorylation at S397 as well as total YAP, MT, and p85 (a loading control). The bar graph represents quantification of Western blot data and shows the averages of data from two independent experiments. **, P < 0.01 versus MT-103A with SU6656 or the wild type without SU6656. (E) NIH 3T3 cells were prepared to express the T203E MT (Src-minus) mutant that is defective in binding Src and inducible by doxycycline. Extracts of these cells were compared to those of cells expressing wild-type MT for YAP S397 phosphorylation, total YAP, and MT by Western blotting. (F) Wild-type MT increases the association of YAP with PP2A. 293T cells were transiently transfected with vectors encoding MT and/or FLAG-YAP. After ∼40 h, YAP was immunoprecipitated with antibodies to the tag. After SDS-PAGE, Western blotting was carried out to detect YAP, PP2A, and MT. (G) MT-bound YAP is phosphorylated differently than unbound YAP in cells expressing wild-type MT. NIH 3T3 cells were induced to express MT by exposure to doxycycline for 24 h. MT was immunoprecipitated by using the MT-1 antibody. After extended SDS-PAGE, Western blotting of MT immunoprecipitates and the total cell extract was carried out to detect YAP and MT. The two bands marked “MT bound” represent identical samples loaded onto both sides of MT-bound YAP to show alignment compared to unbound YAP.
YAP phosphorylation is known to be quite complex. LATS phosphorylates YAP at S61, S109, S127, S164, and S381/397 (YAP2/YAP1) (31). In addition, cyclin-dependent kinase 1 (CDK1) (99), Jnk/p38 (100), and protein kinase C zeta (PKC-zeta) (101) have all been reported to phosphorylate YAP. Phosphospecific antibodies permitted the interrogation of some specific sites on YAP. Both S127 and S397 showed increased phosphorylation in wild-type MT cells. A much greater increase in YAP phosphorylation was observed in R103A MT cells (Fig. 4C), even though the total amount of YAP was reduced by degradation (see below). This finding emphasizes that MT signaling rather than its binding to YAP is driving phosphorylation. Two arguments suggest that signaling is mediated by Src. The inhibitor SU6656 inhibits Src family kinases (79). Cells were treated with SU6656 for the last 16 h of the 24-h doxycycline induction. This treatment reduced the level of S397 phosphorylation (Fig. 4D). Second, mutation of MT at T203 to E abrogates Src binding (80). As shown in Fig. 4E, the T203E mutant fails to increase the specific activity of YAP phosphorylation at position 397, even when expressed at a somewhat higher level than the wild type.
Why does wild-type MT have a smaller effect than the R103A MT mutant? As shown in Fig. 4F, MT, by its binding, promotes the formation of a complex of YAP and PP2A. The result of this is that the ratio of more rapidly migrating, underphosphorylated YAP to the more slowly migrating, more phosphorylated upper band is higher for MT-bound YAP (Fig. 4G). In other words, YAP bound to wild-type MT is underphosphorylated compared to unbound YAP. R103A MT would not have the ability to bring together YAP and PP2A to cause dephosphorylation.
MT binding stabilizes YAP.
Phosphorylation of YAP on S397 has been shown to promote the degradation of YAP. Western blotting of endogenous YAP showed that NIH 3T3, NMuMG, or Rat-1 cells expressing the R103A MT mutant always had less YAP than did cells expressing wild-type MT. This effect was especially pronounced in NIH 3T3 cells (Fig. 5A). The mechanism of YAP downregulation by R103A MT was investigated. Total RNA was extracted from NIH 3T3 cells induced to express the MT protein by exposure to doxycycline for 24 h and subjected to reverse transcription and real-time quantitative PCR. The levels of YAP mRNA in cells expressing EGFP, wild-type MT, and R103A MT were similar, suggesting that 103A does not affect YAP transcription (Fig. 5B). To assess whether R103A MT downregulates YAP stability, NIH 3T3 cells induced to express MT by exposure to doxycycline for 24 h were exposed to the protein synthesis inhibitor cycloheximide (Fig. 5C). In the absence of cycloheximide, YAP protein levels were reduced 1.4-fold in wild-type MT-expressing cells compared to the control and were reduced a further 2.25-fold in cells expressing R103A MT compared to wild-type MT. Exposure to cycloheximide for 24 h dramatically increased that difference: YAP protein levels in cells expressing R103A MT were reduced 60-fold compared to those in cells expressing wild-type MT. This reflects a loss of YAP stability in cells expressing R103A MT. Interestingly the loss of YAP in wild-type MT cells was only slightly greater than that in controls. Since phosphorylation of YAP S397 is associated with proteasome-dependent degradation, the effect of the pharmacological proteasome inhibitor bortezomib was examined. Bortezomib treatment increased the levels of both total YAP and S397 YAP phosphorylation in cells expressing R103A MT (Fig. 5D). This suggests that MT signaling drives phosphorylation at S397 and subsequent proteasomal degradation. The above-described Src inhibition experiment (Fig. 4D) also confirms a role for MT signaling. Inhibition of Src and, hence, the MT pathways downstream of Src increased the amount of YAP in cells expressing R103A MT compared to cells not treated with an inhibitor. Inhibition of the proteasome or inhibition of Src signaling had a much smaller effect on YAP levels in wild-type MT cells than in R103A mutant cells. Taken together, these results suggest that MT signaling caused by R103A promotes YAP turnover, but binding of YAP by wild-type MT stabilizes it. This is consistent with the idea that MT signaling causes YAP phosphorylation and destabilization while YAP bound by MT is dephosphorylated and stabilized.
FIG 5.

YAP stability. (A) MT-103A downregulates endogenous YAP. Rat-1 cells stably expressing MT and NIH 3T3 cells and NMuMG cells induced to express EGFP or MT by exposure to doxycycline for 24 h were subjected to SDS-PAGE and Western blotting to detect YAP, MT, and GAPDH. GAPDH served as a loading control. (B) MT-103A does not downregulate YAP at the mRNA level. NIH 3T3 cells were induced to express EGFP (control [Con]) or MT by exposure to doxycycline for 24 h. Total RNA was extracted and subjected to reverse transcription and real-time PCR analysis. (C) MT-103A destabilizes the YAP protein. NIH 3T3 cells were induced to express EGFP or MT by exposure to doxycycline for 48 h, and the last 24 h of induction took place in the presence of the protein synthesis inhibitor cycloheximide. After SDS-PAGE, Western blotting was conducted to detect YAP, MT, and p85. p85 was used as a loading control. The bar graph represents quantification of Western blot data and shows the averages of data from two independent experiments. In the absence of cycloheximide, the difference between WT MT and MT-103A is significant (P < 0.05). In the presence of cycloheximide, the difference between WT MT and MT-103A is more significant (P < 0.001). (D) YAP destabilization is associated with S397 phosphorylation and is proteasome dependent. NIH 3T3 cells were induced to express EGFP or MT by exposure to doxycycline for 24 h, and the proteasome inhibitor bortezomib was added for the last 16 h of induction. After SDS-PAGE, Western blotting was conducted to detect YAP phosphorylation at S397 as well as total YAP, MT, and p85 levels. p85 served as a loading control.
MT promotes loss of YAP from the nucleus and enrichment in membranes.
Immunofluorescence in control Rat-1 cells showed that YAP was found almost exclusively in the nucleus (Fig. 6A). In contrast, Rat-1 cells expressing the MT protein showed a substantial amount of YAP in the cytoplasm. This cytoplasmic localization of YAP appeared to result from MT signaling, since Rat-1 cells expressing the R103A MT mutant, in which the YAP-binding defect is observed (Fig. 6B), also showed cytoplasmic YAP. Subcellular fractionation of NIH 3T3 cells, previously induced to express EGFP or MT by exposure to doxycycline for 24 h, followed by Western blotting for YAP confirmed the results of the immunofluorescence analysis. Both wild-type MT and R103A MT promoted a significant loss of YAP from the nuclear compartment (Fig. 6C). The decrease in the fraction of nuclear YAP seen upon MT expression is opposite of what was observed previously for ST (61). Notably, this relocalization seems to depend on MT signaling rather than upon MT binding of YAP, since R103A MT caused a relocalization similar to that caused by wild-type MT.
FIG 6.
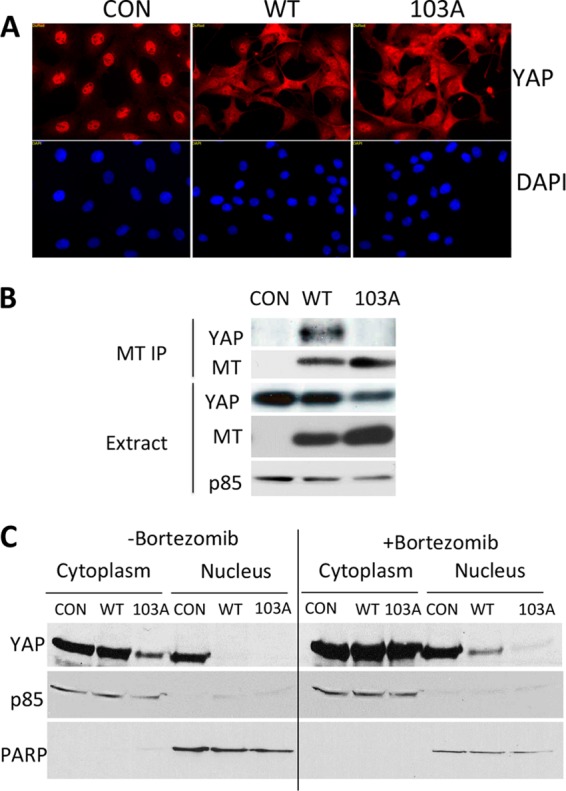
YAP localization. (A) MT induces YAP nuclear exit. Control Rat-1 cells and Rat-1 cells expressing MT were subjected to immunofluorescence staining using an antibody specific to YAP. (B) MT was immunoprecipitated from control Rat-1 cells and Rat-1 cells expressing MT. MT immunoprecipitates were subjected to Western blotting to detect YAP, MT, and p85. p85 served as a loading control. (C) MT induces YAP nuclear exit. NIH 3T3 cells were induced to express EGFP (control [Con]) or MT by exposure to doxycycline for 24 h. A second set of cells was treated with bortezomib to reduce proteasomal breakdown and make YAP in R103A MT cells easier to see. Cellular fractionation, SDS-PAGE, and Western blotting were conducted to detect YAP, poly(ADP-ribose) polymerase (PARP), and p85 levels. PARP served as a loading and nuclear control. p85 served as a loading and nonnuclear control.
The finding that MT promotes YAP nuclear exit (Fig. 6A and C) led us to hypothesize that MT may relocalize YAP to the plasma membrane, where MT itself resides (81, 82). We fractionated cells in hypotonic buffer to isolate membranes and conducted Western blotting after extended SDS-PAGE to determine whether multiple YAP bands could be resolved. The results showed that wild-type MT, but not EGFP or the R103A MT mutant, enriched YAP in membranes but not the cytoplasm or whole-cell extract (Fig. 7A). To look more closely at cells expressing the 103A mutant, bortezomib was used to stabilize YAP. In Fig. 7B, it is obvious that there was a much higher fraction of YAP in membranes from wild-type cells than in those from 103A cells. Furthermore, lengthy electrophoresis showed that membrane-localized YAP from wild-type MT cells was dephosphorylated compared to the small amount of YAP in membranes from 103A mutant cells. YAP in the cytoplasmic fraction of MT cells showed lower mobility, indicating increased phosphorylation, than did YAP in the membranes of MT-expressing cells (Fig. 7C). Furthermore, the shift in the membrane fraction was the same shift seen for YAP in complexes with MT brought down by immunoprecipitation (Fig. 4G).
FIG 7.

Enrichment of membranes in YAP. (A) Wild-type MT enriches membranes in YAP. NIH 3T3 cells were induced to express EGFP or MT by exposure to doxycycline for 24 h. Hypotonic cell fractionation was conducted to isolate the membrane fraction and cytoplasmic fraction. After extended SDS-PAGE, Western blotting was carried out to detect YAP, vimentin, MT, and p85. p85 and vimentin served as loading controls. (B) Control cells (expressing EGFP) or cells expressing wild-type MT or 103A were treated with bortezomib to stabilize YAP in 103A cells. (Left) Total extract; (middle and right) extended electrophoresis to compare wild-type MT to control and 103A membrane extracts at two different concentrations. Blotting was carried out for YAP, MT, p85, and vimentin (loading control). In the right panel, lengthy electrophoresis was performed to compare YAP in wild-type MT cells with twice as much of the 103A sample. (C) Cytoplasmic and membrane fractions prepared as described above for panels A and B were compared by extended electrophoresis.
DISCUSSION
The most important message from this work is that MT has a dual effect on YAP that is important for transformation (Fig. 8). MT signaling through pathways activated by Src leads to YAP phosphorylation that reduces the levels of YAP in the nucleus and increases YAP turnover. MT binding to YAP leads to its dephosphorylation and localization in membranes. It is highly probable that YAP is important for viral replication as well. AP-1 interactions with the polyomavirus enhancer are known to be important for replication (83). YAP and TEAD were recently shown to act at AP-1 enhancers to drive growth (84).
FIG 8.
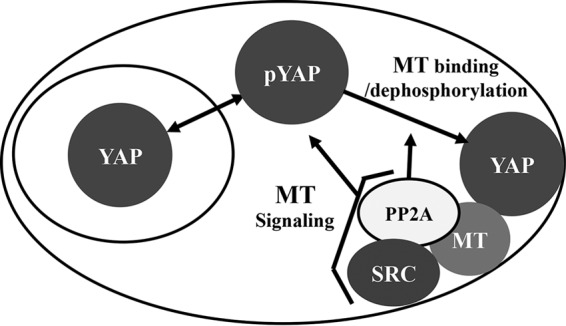
Dual effects of MT on YAP. MT signaling drives phosphorylation of YAP, promoting nuclear exit and turnover. MT binding of YAP promotes the dephosphorylation of YAP by PP2A and localization in membranes.
Originally detected by mass spectrometry, the interaction between YAP and MT has been validated by immunoprecipitation and genetic analyses identifying specific binding sites on each partner. The binding site on MT is the same as the one that we previously characterized for ST (61). This is expected since ST has only 4 amino acids not found in MT. TAZ, a close relative of YAP, was identified previously as a partner for all three T antigens using amino acid residues 2 to 4 (62). Mutations that affect YAP binding also block TAZ binding to MT. MT binds to YAP and TAZ through their WW domains, particularly WW1. The YAP WW domains bind PPXY-containing targets. MT and ST have no PPXY sequence, so they must use a different binding mechanism.
Since knockdown of YAP blocks MT transformation, YAP must contribute to transformation in a positive way. This is counter to the traditional idea that Hippo targeting of YAP is associated with growth suppression. Nonetheless, our results are consistent with recently reported results showing that MT breast tumorigenesis is reduced in YAP knockout mice (85). How this happens is not yet clear. Given that activated Ras or activated PI3K can transform cells, the general feeling has been that the activation of these two pathways by MT is key for transformation and tumorigenesis. Here we show that downregulation of YAP protein levels, either as a consequence of disrupting YAP binding or by introducing YAP shRNAs, is sufficient to inhibit anchorage-independent growth in soft agar despite fully active RAS and PI3K signaling. Certainly interactions of the WW domains of YAP with PPXY motifs could be perturbed by MT binding. These interactions might include targets such as angiomotin (86), involved in cell motility; patched protein homolog 1 (PTCH1), a protein from the Sonic Hedgehog pathway (87); and SMAD1 (88, 89) in the BMP signaling pathway. In the case of SMAD1, phosphorylation by CDK8/9 acts as a switch necessary for the WW interaction, and phosphorylation by glycogen synthase kinase 3 (GSK3) switches it off (89). It is easy to speculate that MT, by bringing PP2A to YAP, alters the operation of these kinds of phosphoswitches.
From our work here and those of others (62, 63), TAZ, which is also a Hippo effector, seems to be required for MT transformation as well. Although a formal possibility, we believe that it is unlikely that YAP and TAZ have identical roles and that knockdown of either one results in a kind of haploinsufficiency. More likely, specific functions unique to YAP and TAZ would explain why both proteins are needed for MT transformation. YAP was identified by its association with the tyrosine kinase Yes through its SH3-binding and proline-rich domains (90). TAZ lacks these elements and thus does not bind Yes. YAP, but not TAZ, binds to heterogeneous ribonuclear protein U (91). TAZ, but not YAP, is localized to nuclear foci (57). Interactions of PDZ domains of YAP and TAZ are also different (57). Not surprisingly, there are notable differences in the YAP and TAZ protein interactomes (92). The relevant targets will have to be identified in future experiments.
The effects of MT on YAP are pleiotropic. Clearly, some effects are caused by MT's activation of signaling pathways. MT signaling results in an alteration of phosphorylation. The specific activity of phosphorylation at S127 and S397 is increased. Intriguingly, the opposite effects are observed for these sites when ST is expressed (61). The reason for this difference is clear. MT signaling via Src is needed to increase phosphorylation at S397. This results in YAP destabilization. MT signaling via Src also increases YAP phosphorylation at S127, which promotes the cytoplasmic localization of YAP. ST, of course, does not activate Src. ST brings PP2A to YAP. This results in the dephosphorylation of YAP at both sites, leading to nuclear localization and stabilization. However, MT also brings PP2A to the fraction of YAP that it binds. The YAP associated with MT is also relatively dephosphorylated. Thus, the overall effect of MT expression on YAP phosphorylation may well depend on the level of expression of MT in a given cell type and the fraction bound. Quantitative sequencing of YAP phosphorylations will tell us how the many YAP phosphorylations may be targeted by PP2A associated with MT.
MT reduces the levels of YAP in the nucleus. This is caused by MT signaling, because it is not affected by the R103A mutation. Such a change in localization might be expected to decrease the expression levels of genes for which YAP acts as a transcriptional coactivator. Transcriptome sequencing (RNA-seq) experiments could shed light on these possibilities. We know from real-time PCR measurements that RNA levels of AnkyrdD1, a known target of TEAD, are reduced in MT cells (data not shown). Again, this decrease does not depend on the binding of YAP by MT. Interestingly, the reduction in the amount of nuclear YAP and the increase in the amount cytoplasmic YAP also do not depend on binding to MT. The known cytoplasmic functions of YAP are tumor suppressive: YAP can bind to the beta-catenin destruction complex and/or DVL2 to restrict beta-catenin and/or DVL2 nuclear localization and Wnt-dependent transcription (93, 94). Similarly, binding of YAP to angiomotin sequesters YAP in the cytoplasm and is associated with decreased lung cancer progression (95).
The binding of YAP to MT brings it to membranes (Fig. 7A), suggesting that this translocation might be important for the MT-transforming phenotype. This meeting of MT and YAP at the membrane could have a number of important outcomes. MT binding would bring YAP into contact with some or all of the other MT-binding partners. The dephosphorylation that is observed in the MT complexes almost certainly comes from PP2A, since PP2A inhibition by okadaic acid shifts the YAP phosphorylation state. MT associates with the Yes tyrosine kinase (96), which was the basis for the original discovery of YAP (90). A recent report identified the gp130 interleukin-6 (IL-6) receptor/Src pathway as being important for inducing proliferation and aberrant differentiation in epithelial cells (97). The membrane-associated Crumbs family of proteins involved in cell polarity uses YAP to control differentiation (98). Working at the membrane, MT could inhibit or enhance such processes. Future effort will be needed to explore how MT directs YAP function either by direct binding or by signaling.
ACKNOWLEDGMENTS
We thank Peter Howley for helpful discussions. We also thank Wade Harper for help with mass spectrometry.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Dawe CJ, Freund R, Mandel G, Ballmer-Hofer K, Talmage DA, Benjamin TL. 1987. Variations in polyoma virus genotype in relation to tumor induction in mice. Characterization of wild type strains with widely differing tumor profiles. Am J Pathol 127:243–261. [PMC free article] [PubMed] [Google Scholar]
- 2.Eddy B. 1969. Polyoma virus. Virol Monogr 7:1–114. [Google Scholar]
- 3.Fluck MM, Haslam SZ. 1996. Mammary tumors induced by polyomavirus. Breast Cancer Res Treat 39:45–56. doi: 10.1007/BF01806077. [DOI] [PubMed] [Google Scholar]
- 4.Schaffhausen BS, Roberts TM. 2009. Lessons from polyoma middle T antigen on signaling and transformation: a DNA tumor virus contribution to the war on cancer. Virology 384:304–316. doi: 10.1016/j.virol.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fluck MM, Schaffhausen BS. 2009. Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Microbiol Mol Biol Rev 73:542–563. doi: 10.1128/MMBR.00009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eckhart W, Hutchinson MA, Hunter T. 1979. An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 18:925–933. doi: 10.1016/0092-8674(79)90205-8. [DOI] [PubMed] [Google Scholar]
- 7.Whitman M, Downes CP, Keeler M, Keller T, Cantley L. 1988. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 332:644–646. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- 8.Hwang JH, Jiang T, Kulkarni S, Faure N, Schaffhausen BS. 2013. Protein phosphatase 2A isoforms utilizing Abeta scaffolds regulate differentiation through control of Akt protein. J Biol Chem 288:32064–32073. doi: 10.1074/jbc.M113.497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Utermark T, Rao T, Cheng H, Wang Q, Lee SH, Wang ZC, Iglehart JD, Roberts TM, Muller WJ, Zhao JJ. 2012. The p110alpha and p110beta isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev 26:1573–1586. doi: 10.1101/gad.191973.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmichael G, Schaffhausen B, Dorsky D, Oliver D, Benjamin T. 1982. The role of the carboxy terminus of middle T antigen of polyoma virus. Proc Natl Acad Sci U S A 79:3579–3583. doi: 10.1073/pnas.79.11.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ichaso N, Dilworth SM. 2001. Cell transformation by the middle T-antigen of polyoma virus. Oncogene 20:7908–7916. doi: 10.1038/sj.onc.1204859. [DOI] [PubMed] [Google Scholar]
- 12.Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, Roberts TM. 1990. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60:167–176. doi: 10.1016/0092-8674(90)90726-U. [DOI] [PubMed] [Google Scholar]
- 13.Walter G, Ruediger R, Slaughter C, Mumby M. 1990. Association of protein phosphatase 2A with polyoma virus medium tumor antigen. Proc Natl Acad Sci U S A 87:2521–2525. doi: 10.1073/pnas.87.7.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogris E, Mudrak I, Mak E, Gibson D, Pallas DC. 1999. Catalytically inactive protein phosphatase 2A can bind to polyomavirus middle tumor antigen and support complex formation with pp60(c-src). J Virol 73:7390–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Courtneidge S, Smith AE. 1983. Polyoma virus transforming protein associates with the product of the c-src cellular gene. Nature 303:435–439. doi: 10.1038/303435a0. [DOI] [PubMed] [Google Scholar]
- 16.Cheng SH, Harvey R, Espino PC, Semba K, Yamamoto T, Toyoshima K, Smith AE. 1988. Peptide antibodies to the human c-fyn gene product demonstrate pp59c-fyn is capable of complex formation with the middle-T antigen of polyomavirus. EMBO J 7:3845–3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horak ID, Kawakami T, Gregory F, Robbins KC, Bolen JB. 1989. Association of p60fyn with middle tumor antigen in murine polyomavirus-transformed rat cells. J Virol 63:2343–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kornbluth S, Sudol M, Hanafusa H. 1987. Association of the polyomavirus middle-T antigen with c-yes protein. Nature 325:171–173. doi: 10.1038/325171a0. [DOI] [PubMed] [Google Scholar]
- 19.Hunter T, Hutchinson MA, Eckhart W. 1984. Polyoma middle-sized T antigen can be phosphorylated on tyrosine at multiple sites in vitro. EMBO J 3:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey R, Oostra BA, Belsham GJ, Gillett P, Smith AE. 1984. An antibody to a synthetic peptide recognizes polyomavirus middle-T antigen and reveals multiple in vitro tyrosine phosphorylation sites. Mol Cell Biol 4:1334–1342. doi: 10.1128/MCB.4.7.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaffhausen B, Benjamin TL. 1981. Comparison of phosphorylation of two polyoma virus middle T antigens in vivo and in vitro. J Virol 40:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmichael G, Schaffhausen BS, Mandel G, Liang TJ, Benjamin TL. 1984. Transformation by polyoma virus is drastically reduced by substitution of phenylalanine for tyrosine at residue 315 of middle-sized tumor antigen. Proc Natl Acad Sci U S A 81:679–683. doi: 10.1073/pnas.81.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM. 1985. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315:239–242. doi: 10.1038/315239a0. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan DR, Whitman M, Schaffhausen B, Raptis L, Garcea RL, Pallas D, Roberts TM, Cantley L. 1986. Phosphatidylinositol metabolism and polyoma-mediated transformation. Proc Natl Acad Sci U S A 83:3624–3628. doi: 10.1073/pnas.83.11.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell KS, Ogris E, Burke B, Su W, Auger KR, Druker BJ, Schaffhausen BS, Roberts TM, Pallas DC. 1994. Polyoma middle T antigen interacts with SHC via the NPTY motif in middle T. Proc Natl Acad Sci U S A 91:6344–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dilworth SM, Brewster CE, Jones MD, Lanfrancone L, Pelicci G, Pelicci PG. 1994. Transformation by polyoma virus middle T-antigen involves the binding and tyrosine phosphorylation of Shc. Nature 367:87–90. doi: 10.1038/367087a0. [DOI] [PubMed] [Google Scholar]
- 27.Su W, Liu W, Schaffhausen BS, Roberts TM. 1995. Association of polyomavirus middle tumor antigen with phospholipase C-gamma 1. J Biol Chem 270:12331–12334. doi: 10.1074/jbc.270.21.12331. [DOI] [PubMed] [Google Scholar]
- 28.Varelas X. 2014. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141:1614–1626. doi: 10.1242/dev.102376. [DOI] [PubMed] [Google Scholar]
- 29.Gumbiner BM, Kim NG. 2014. The Hippo-YAP signaling pathway and contact inhibition of growth. J Cell Sci 127:709–717. doi: 10.1242/jcs.140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsui Y, Lai ZC. 2013. Mutual regulation between Hippo signaling and actin cytoskeleton. Protein Cell 4:904–910. doi: 10.1007/s13238-013-3084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao B, Li L, Lei Q, Guan KL. 2010. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen CG, Moroishi T, Guan KL. 2 June 2015. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol doi: 10.1016/j.tcb.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayashi S, Yokoyama H, Tamura K. 2015. Roles of Hippo signaling pathway in size control of organ regeneration. Dev Growth Differ 57:341–351. doi: 10.1111/dgd.12212. [DOI] [PubMed] [Google Scholar]
- 34.Ramos A, Camargo FD. 2012. The Hippo signaling pathway and stem cell biology. Trends Cell Biol 22:339–346. doi: 10.1016/j.tcb.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao B, Tumaneng K, Guan KL. 2011. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol 13:877–883. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hiemer SE, Varelas X. 2013. Stem cell regulation by the Hippo pathway. Biochim Biophys Acta 1830:2323–2334. doi: 10.1016/j.bbagen.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 37.Imajo M, Ebisuya M, Nishida E. 2015. Dual role of YAP and TAZ in renewal of the intestinal epithelium. Nat Cell Biol 17:7–19. doi: 10.1038/ncb3084. [DOI] [PubMed] [Google Scholar]
- 38.Nishio M, Otsubo K, Maehama T, Mimori K, Suzuki A. 2013. Capturing the mammalian Hippo: elucidating its role in cancer. Cancer Sci 104:1271–1277. doi: 10.1111/cas.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Du YC, Zhou XJ, Liu H, Tang SC. 2014. The dual functions of YAP-1 to promote and inhibit cell growth in human malignancy. Cancer Metastasis Rev 33:173–181. doi: 10.1007/s10555-013-9463-3. [DOI] [PubMed] [Google Scholar]
- 40.Moroishi T, Hansen CG, Guan KL. 2015. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15:73–79. doi: 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Shi S, Guo Z, Zhang X, Han S, Yang A, Wen W, Zhu Q. 2013. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS One 8:e65539. doi: 10.1371/journal.pone.0065539. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Zhang X, George J, Deb S, Degoutin JL, Takano EA, Fox SB, AOCS Study Group, Bowtell DD, Harvey KF. 2011. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene 30:2810–2822. doi: 10.1038/onc.2011.8. [DOI] [PubMed] [Google Scholar]
- 43.Wei Z, Wang Y, Li Z, Yuan C, Zhang W, Wang D, Ye J, Jiang H, Wu Y, Cheng J. 2013. Overexpression of Hippo pathway effector TAZ in tongue squamous cell carcinoma: correlation with clinicopathological features and patients' prognosis. J Oral Pathol Med 42:747–754. doi: 10.1111/jop.12062. [DOI] [PubMed] [Google Scholar]
- 44.Ge L, Smail M, Meng W, Shyr Y, Ye F, Fan KH, Li X, Zhou HM, Bhowmick NA. 2011. Yes-associated protein expression in head and neck squamous cell carcinoma nodal metastasis. PLoS One 6:e27529. doi: 10.1371/journal.pone.0027529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perra A, Kowalik MA, Ghiso E, Ledda-Columbano GM, Di Tommaso L, Angioni MM, Raschioni C, Testore E, Roncalli M, Giordano S, Columbano A. 2014. YAP activation is an early event and a potential therapeutic target in liver cancer development. J Hepatol 61:1088–1096. doi: 10.1016/j.jhep.2014.06.033. [DOI] [PubMed] [Google Scholar]
- 46.Lau AN, Curtis SJ, Fillmore CM, Rowbotham SP, Mohseni M, Wagner DE, Beede AM, Montoro DT, Sinkevicius KW, Walton ZE, Barrios J, Weiss DJ, Camargo FD, Wong KK, Kim CF. 2014. Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J 33:468–481. doi: 10.1002/embj.201386082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 48.Avruch J, Zhou D, Fitamant J, Bardeesy N, Mou F, Barrufet LR. 2012. Protein kinases of the Hippo pathway: regulation and substrates. Semin Cell Dev Biol 23:770–784. doi: 10.1016/j.semcdb.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu FX, Guan KL. 2013. The Hippo pathway: regulators and regulations. Genes Dev 27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong W, Guan KL. 2012. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol 23:785–793. doi: 10.1016/j.semcdb.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang K, Degerny C, Xu M, Yang XJ. 2009. YAP, TAZ, and Yorkie: a conserved family of signal-responsive transcriptional coregulators in animal development and human disease. Biochem Cell Biol 87:77–91. doi: 10.1139/O08-114. [DOI] [PubMed] [Google Scholar]
- 52.Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. 2001. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev 15:1229–1241. doi: 10.1101/gad.888601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. 1999. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J 18:2551–2562. doi: 10.1093/emboj/18.9.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murakami M, Nakagawa M, Olson EN, Nakagawa O. 2005. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc Natl Acad Sci U S A 102:18034–18039. doi: 10.1073/pnas.0509109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hiemer SE, Szymaniak AD, Varelas X. 2014. The transcriptional regulators TAZ and YAP direct transforming growth factor beta-induced tumorigenic phenotypes in breast cancer cells. J Biol Chem 289:13461–13474. doi: 10.1074/jbc.M113.529115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao B, Li L, Guan KL. 2010. Hippo signaling at a glance. J Cell Sci 123:4001–4006. doi: 10.1242/jcs.069070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, Hisaminato A, Fujiwara T, Ito Y, Cantley LC, Yaffe MB. 2000. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J 19:6778–6791. doi: 10.1093/emboj/19.24.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, Rossant J, Wrana JL. 2010. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev Cell 19:831–844. doi: 10.1016/j.devcel.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 59.Oka T, Remue E, Meerschaert K, Vanloo B, Boucherie C, Gfeller D, Bader GD, Sidhu SS, Vandekerckhove J, Gettemans J, Sudol M. 2010. Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem J 432:461–472. doi: 10.1042/BJ20100870. [DOI] [PubMed] [Google Scholar]
- 60.Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD. 2011. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 144:782–795. doi: 10.1016/j.cell.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang JH, Pores Fernando AT, Faure N, Andrabi S, Adelmant G, Hahn WC, Marto JA, Schaffhausen BS, Roberts TM. 13 August 2014. Polyomavirus small T antigen interacts with yes-associated protein to regulate cell survival and differentiation. J Virol doi: 10.1128/JVI.01399-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tian Y, Li D, Dahl J, You J, Benjamin T. 2004. Identification of TAZ as a binding partner of the polyomavirus T antigens. J Virol 78:12657–12664. doi: 10.1128/JVI.78.22.12657-12664.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shanzer M, Ricardo-Lax I, Keshet R, Reuven N, Shaul Y. 3 November 2014. The polyomavirus middle T-antigen oncogene activates the Hippo pathway tumor suppressor Lats in a Src-dependent manner. Oncogene doi: 10.1038/onc.2014.347. [DOI] [PubMed] [Google Scholar]
- 64.Bendig M, Thomas T, Folk W. 1980. Viable deletion mutant in the medium and large T antigen coding sequences of the polyoma virus genome. J Virol 33:1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holman P, Gjoerup O, Davin T, Schaffhausen B. 1994. Characterization of an immortalizing N-terminal domain of polyomavirus large T antigen. J Virol 68:668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, Schmitt EM, Bernardi RJ, Fu X, Bland CS, Cooper TA, Schiff R, Rosen JM, Westbrook TF, Elledge SJ. 2011. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A 108:3665–3670. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. 2009. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4:e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Utermark T, Schaffhausen BS, Roberts TM, Zhao JJ. 2007. The p110alpha isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J Virol 81:7069–7076. doi: 10.1128/JVI.00115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oka T, Mazack V, Sudol M. 2008. Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J Biol Chem 283:27534–27546. doi: 10.1074/jbc.M804380200. [DOI] [PubMed] [Google Scholar]
- 70.Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, Guan KL. 2008. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 28:2426–2436. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan MJ, White EA, Sowa ME, Harper JW, Aster JC, Howley PM. 2012. Cutaneous beta-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc Natl Acad Sci U S A 109:E1473–E1480. doi: 10.1073/pnas.1205991109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sowa ME, Bennett EJ, Gygi SP, Harper JW. 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 74.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Silver J, Schaffhausen B, Benjamin T. 1978. Tumor antigens induced by nontransforming mutants of polyoma virus. Cell 15:485–496. doi: 10.1016/0092-8674(78)90018-1. [DOI] [PubMed] [Google Scholar]
- 76.Andrabi S, Hwang JH, Choe JK, Roberts TM, Schaffhausen BS. 2011. Comparisons between murine polyomavirus and simian virus 40 show significant differences in small T antigen function. J Virol 85:10649–10658. doi: 10.1128/JVI.05034-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nguyen HT, Hong X, Tan S, Chen Q, Chan L, Fivaz M, Cohen SM, Voorhoeve PM. 2014. Viral small T oncoproteins transform cells by alleviating hippo-pathway-mediated inhibition of the YAP proto-oncogene. Cell Rep 8:707–713. doi: 10.1016/j.celrep.2014.06.062. [DOI] [PubMed] [Google Scholar]
- 78.Gaffney CJ, Oka T, Mazack V, Hilman D, Gat U, Muramatsu T, Inazawa J, Golden A, Carey DJ, Farooq A, Tromp G, Sudol M. 2012. Identification, basic characterization and evolutionary analysis of differentially spliced mRNA isoforms of human YAP1 gene. Gene 509:215–222. doi: 10.1016/j.gene.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA. 2000. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol 20:9018–9027. doi: 10.1128/MCB.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Glover HR, Brewster CE, Dilworth SM. 1999. Association between src-kinases and the polyoma virus oncogene middle T-antigen requires PP2A and a specific sequence motif. Oncogene 18:4364–4370. doi: 10.1038/sj.onc.1202816. [DOI] [PubMed] [Google Scholar]
- 81.Ito Y. 1979. Polyoma virus-specific 55K protein isolated from plasma membrane of productively infected cells is virus-coded and important cell transformation. Virology 98:261–266. doi: 10.1016/0042-6822(79)90545-2. [DOI] [PubMed] [Google Scholar]
- 82.Schaffhausen BS, Dorai H, Arakere G, Benjamin TL. 1982. Polyoma virus middle T antigen: relationship to cell membranes and apparent lack of ATP-binding activity. Mol Cell Biol 2:1187–1198. doi: 10.1128/MCB.2.10.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Murakami Y, Satake M, Yamaguchi IY, Sakai M, Muramatsu M, Ito Y. 1991. The nuclear protooncogenes c-jun and c-fos as regulators of DNA replication. Proc Natl Acad Sci U S A 88:3947–3951. doi: 10.1073/pnas.88.9.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. 2015. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 17:1218–1227. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Q, Zhang N, Gray RS, Li H, Ewald AJ, Zahnow CA, Pan D. 2014. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev 28:432–437. doi: 10.1101/gad.233676.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chan SW, Lim CJ, Huang C, Chong YF, Gunaratne HJ, Hogue KA, Blackstock WP, Harvey KF, Hong W. 2011. WW domain-mediated interaction with Wbp2 is important for the oncogenic property of TAZ. Oncogene 30:600–610. doi: 10.1038/onc.2010.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iglesias-Bexiga M, Castillo F, Cobos ES, Oka T, Sudol M, Luque I. 2015. WW domains of the yes-kinase-associated-protein (YAP) transcriptional regulator behave as independent units with different binding preferences for PPxY motif-containing ligands. PLoS One 10:e0113828. doi: 10.1371/journal.pone.0113828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova-Todorova K, Macias MJ, Sapkota G, Pan D, Massague J. 2009. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 139:757–769. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aragon E, Goerner N, Zaromytidou AI, Xi Q, Escobedo A, Massague J, Macias MJ. 2011. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev 25:1275–1288. doi: 10.1101/gad.2060811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sudol M. 1994. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene 9:2145–2152. [PubMed] [Google Scholar]
- 91.Howell M, Borchers C, Milgram SL. 2004. Heterogeneous nuclear ribonuclear protein U associates with YAP and regulates its co-activation of Bax transcription. J Biol Chem 279:26300–26306. doi: 10.1074/jbc.M401070200. [DOI] [PubMed] [Google Scholar]
- 92.Kohli P, Bartram MP, Habbig S, Pahmeyer C, Lamkemeyer T, Benzing T, Schermer B, Rinschen MM. 2014. Label-free quantitative proteomic analysis of the YAP/TAZ interactome. Am J Physiol Cell Physiol 306:C805–C818. doi: 10.1152/ajpcell.00339.2013. [DOI] [PubMed] [Google Scholar]
- 93.Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V, Fassina A, Cordenonsi M, Piccolo S. 2014. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 158:157–170. doi: 10.1016/j.cell.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 94.Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, Kuo CJ, Camargo FD. 2013. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 493:106–110. doi: 10.1038/nature11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hsu YL, Hung JY, Chou SH, Huang MS, Tsai MJ, Lin YS, Chiang SY, Ho YW, Wu CY, Kuo PL. 2015. Angiomotin decreases lung cancer progression by sequestering oncogenic YAP/TAZ and decreasing Cyr61 expression. Oncogene 34:4056–4068. doi: 10.1038/onc.2014.333. [DOI] [PubMed] [Google Scholar]
- 96.Kornbluth S, Cheng SH, Markland W, Fukui Y, Hanafusa H. 1990. Association of p62c-yes with polyomavirus middle t-antigen mutants correlates with transforming ability. J Virol 64:1584–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, Sandborn WJ, Hardiman G, Raz E, Maehara Y, Yoshimura A, Zucman-Rossi J, Guan KL, Karin M. 2015. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 519:57–62. doi: 10.1038/nature14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Szymaniak AD, Mahoney JE, Cardoso WV, Varelas X. 2015. Crumbs3-mediated polarity directs airway epithelial cell fate through the Hippo pathway effector Yap. Dev Cell 34:283–296. doi: 10.1016/j.devcel.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, Dong J. 2013. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res 73:6722–6733. doi: 10.1158/0008-5472.CAN-13-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tomlinson V, Gudmundsdottir K, Luong P, Leung KY, Knebel A, Basu S. 2010. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis 1:e29. doi: 10.1038/cddis.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Llado V, Nakanishi Y, Duran A, Reina-Campos M, Shelton PM, Linares JF, Yajima T, Campos A, Aza-Blanc P, Leitges M, Diaz-Meco MT, Moscat J. 2015. Repression of intestinal stem cell function and tumorigenesis through direct phosphorylation of β-catenin and Yap by PKCζ. Cell Rep 5:740–754. doi: 10.1016/j.celrep.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]