

ABSTRACT
The template for Ebola virus (EBOV) transcription and replication is the helical viral nucleocapsid composed of the viral negative-sense (−) RNA genome, which is complexed by the nucleoprotein (NP), VP35, polymerase L, VP24, and VP30. While viral replication is exerted by polymerase L and its cofactor VP35, EBOV mRNA synthesis is regulated by the viral nucleocapsid protein VP30, an essential EBOV-specific transcription factor. VP30 is a homohexameric phosphoprotein containing a nonconventional zinc finger. The transcriptional support activity of VP30 is strongly influenced by its phosphorylation state. We studied here how RNA binding contributed to VP30's function in transcriptional activation. Using a novel mobility shift assay and the 3′-terminal 154 nucleotides of the EBOV genome as a standard RNA substrate, we detected that RNA binding of VP30 was severely impaired by VP30 mutations that (i) destroy the protein's capability to form homohexamers, (ii) disrupt the integrity of its zinc finger domain, (iii) mimic its fully phosphorylated state, or (iv) alter the putative RNA binding region. RNA binding of the mutant VP30 proteins correlated strongly with their transcriptional support activity. Furthermore, we showed that the interaction between VP30 and the polymerase cofactor VP35 is RNA dependent, while formation of VP30 homohexamers and VP35 homotetramers is not. Our data indicate that RNA binding of VP30 is essential for its transcriptional support activity and stabilizes complexes of VP35/L polymerase with the (−) RNA template to favor productive transcriptional initiation in the presence of termination-active RNA secondary structures.
IMPORTANCE Ebola virus causes severe fevers with unusually high case fatality rates. The recent outbreak of Ebola virus in West Africa claimed more than 11,000 lives and threatened to destabilize a whole region because of its dramatic effects on the public health systems. It is currently not completely understood how Ebola virus manages to balance viral transcription and replication in the infected cells. This study shows that transcriptional support activity of the Ebola virus transcription factor VP30 is highly correlated with its ability to bind viral RNA. The interaction between VP30 and VP35, the Ebola virus polymerase cofactor, is dependent on the presence of RNA as well. Our data contribute to the understanding of the dynamic interplay between nucleocapsid proteins and the viral RNA template in order to promote viral RNA synthesis.
INTRODUCTION
The nonsegmented, negative-sense RNA viruses Ebola virus (EBOV), Lloviu virus (LLOV), and Marburg virus (MARV) constitute the family Filoviridae in the order Mononegavirales (1). Filoviruses cause severe hemorrhagic fever in humans and nonhuman primates with fatality rates of up to 90% in humans (2). The recent EBOV outbreak in West Africa underlines the emerging potential of filoviruses and the consequences that those outbreaks can have for public health and the stability of a whole region (3). Filoviruses are classified as biosafety level 4 (BSL4) pathogens that need to be handled under the highest biosafety conditions (4).
The 19-kb genome of EBOV encodes seven structural proteins and is flanked by two regulatory regions at the genome ends, the 3′ leader and the 5′ trailer (5). Both regions contain essential signals for replication and encapsidation. The transcription start site of the first gene, encoding the nucleoprotein (NP), is located in the 3′-leader, 56 nucleotides (nt) upstream of its 3′ end (6–8).
The template for Ebola viral transcription and replication is the helical viral nucleocapsid, composed of the viral genome, which is complexed by the nucleoprotein, NP, viral protein 35 (VP35), and VP24 (9, 10). While viral replication is exerted by polymerase L and its cofactor VP35, viral transcription further requires the participation of the EBOV transcription factor VP30 (8). The presence of VP30 also downregulates viral replication (11). So far, it is largely unclear how VP30 commits the polymerase to its transcriptional mode. Previous studies revealed different functional elements within VP30, including those for homodimerization and hexamerization, NP binding, phosphorylation, and RNA binding (Fig. 1D) (11–15). The transcriptional activation function of VP30 is regulated by phosphorylation at six N-proximal serine residues (S29 to S31, S42, S44, S46) (Fig. 1D) (16). Viral transcription is supported by non- or weakly phosphorylated VP30, whereas extensive phosphorylation of the serine residues inhibits viral transcription (16). Moreover, phosphorylation of VP30 enhances its affinity for NP and negatively affects the interaction with the polymerase cofactor VP35, which may result in a release of VP30 from the viral transcription complex (11).
FIG 1.

Effect of VP30 phosphorylation on RNA binding. (A) SDS-PAGE and Coomassie blue staining of purified MBP-VP308–272 before (−TEV) and after (+TEV) cleavage, exemplarily illustrated for VP30 arginine mutants. VP30 mutants were N- and C-terminally truncated (lacking aa 1 to 7 and 273 to 288) to increase the solubility of recombinant VP30. (B) Transcriptional support activity of VP30_wt, VP308–272, and MBP-VP308–272 in producer cells. The assay was performed as described in the legend to Fig. 2D. (C) (Left) Representative EMSA illustrating the separation of the free RNA probe (extended genomic 3′ leader, nt 154 to 1) from two types of complexes (C1 and CHMW). Whereas C1 was assumed to mimic functional complexes representing hexameric VP30 and RNA, CHMW complexes are high-molecular-weight complexes whose functional significance is unclear. (Right) Binding curves of VP30 to two different RNA substrates (taken from reference 25): the genomic RNA substrate (including the first 154 nt of the EBOV genome) and the antigenomic RNA substrate (representing the mRNA start region of the first gene, nt 56 to 158); error bars are standard errors of the mean (SEM). For derived appKd values, see Tables 1 and 2. (D) Schematic presentation of VP30 functional domains/motifs, including the region involved in RNA binding and phosphorylation (white box), the zinc finger domain (hatched with vertical lines), the hexamerization motif (black box), and the region mediating VP30 dimerization and binding to NP (diagonally hatched box; for details, see the text). The enlarged box below the scheme details the two serine clusters that are phosphorylation targets and specifies the VP30 mutants that were constructed to mimic the (un)phosphorylated state of the protein. Serine residues 29 to 31, 42, 44, and 46 were mutated either to uncharged alanines (VP30_AA, mimicking unphosphorylated VP30) or to aspartates (VP30_DD, mimicking the fully phosphorylated state). (E) RNA binding curves and derived appKd values for VP30, VP30_AA, and VP30_DD based on C1 complex formation (mean values from 3 to 5 independent experiments; error bars are given as SEM). Data for VP30 are taken from reference 25. For more details, see Table 1 and Materials and Methods.
VP30-driven EBOV transcription is also regulated by RNA hairpin structure formation close to the genomic 3′ end (nt 56 to 78) (17). The involved nucleotide positions have the potential to promote hairpin formation in the genomic 3′-leader (−) RNA as well as on the complementary (+) RNA level, where the hairpin forms directly at the 5′ end of the NP mRNA transcript. It is not yet fully clear if hairpin formation on the genomic and/or mRNA level is important for productive transcription. Previous data indicated that VP30 also acts as a transcription reinitiation factor, which prevents the L/VP35 polymerase complex from falling off the template at the gene ends (17–20). In addition, VP30 was reported to contain an unconventional zinc-binding Cys3-His motif (amino acids [aa] 72 to 90) that is crucial for transcriptional activation and is thought to contribute to VP30's RNA binding activity (14, 15). Bioinformatic analyses identified an arginine-rich putative RNA-binding region of VP30 comprising residues R26, 28, 32, 36, 40, and 43; UV cross-linking and nitrocellulose filter binding assays using truncated and mutated VP30 suggested that these arginine residues may contribute to RNA binding (15). Interestingly, the putative RNA-binding region overlaps the N-proximal phosphorylation region of VP30 (Fig. 1D).
Here, we addressed the correlation between VP30's RNA-binding capacity and its functional role in transcription activation. Using a tailored electrophoretic mobility shift assay (EMSA), we found that binding of VP30 to the genomic EBOV 3′-leader RNA depends on its phosphorylation state and the capability to form homohexamers as well as the integrity of its zinc (Zn)-finger domain (12, 14). Furthermore, we provide evidence for the contribution of arginine residues in the N-proximal arginine-rich cluster to RNA binding and the balancing between viral transcription and replication. Finally, we show by coimmunoprecipitation experiments that the interaction of VP30 with the polymerase cofactor VP35 is mediated by RNA and depends on the double-stranded RNA (dsRNA)-binding capacity of VP35. In contrast, self-oligomerization of VP30 as well as VP35 is RNA independent. Taken together, our data substantiate the essential role of VP30's RNA-binding activity in the regulation of transcription and replication.
MATERIALS AND METHODS
Cell culture.
HEK-293 (human embryonic kidney) and HuH-7 (human hepatoma) cells were cultivated in Dulbecco's modified Eagle medium, additionally containing 100 U/ml penicillin, 100 mg/ml streptomycin, 5 mM l-glutamine, and 10% fetal bovine serum (all from Gibco Thermo Fisher Scientific), at 37°C and 5% CO2 in a humidified atmosphere.
Plasmids.
All plasmids coding for wild-type (wt) EBOV proteins (pCAGGS VP30, -NP, -VP35, -L, -VP24, -VP40, and -GP) as well as the EBOV-specific minigenome (pANDY 3E5E) and pCAGGS T7 polymerase have been described earlier (11, 21). Cloning of VP30_5LA, VP30_R26A, VP30_R28A, VP30_R40A, VP30_3RA, and VP35_ R305A/K309A/R312A into pCAGGS was performed with the multisite-directed mutagenesis kit (Agilent) according to the manufacturer's recommendations. pCAGGS VP308–272 and pCAGGS MBP/VP308–272 were generated by standard cloning strategies utilizing EcoRI and NotI restriction sites. All pCAGGS VP30 constructs contained a C-terminal FLAG epitope for immunological detection. For VP30 expression in bacteria, the coding sequence of a minimally truncated version of VP30 (amino acids 8 to 272) was cloned into the pUC-derivative plasmid pBADM41 using unique NcoI and NotI restriction sites. This resulted in a fusion protein consisting of an N-terminally His-tagged maltose-binding protein (MBP) followed by a linker with a tobacco etch virus (TEV) protease cleavage site and the C-terminal VP308–272 moiety. For producing the 3′-leader RNA of the EBOV wt genome (nucleotides 154 to 1, 5′-gGU GAU GUG GCU CUG AAA CAA ACC AGG UGU GAU UAC AGU AAC AAU UUC AAU UUA AAU UCC GAU AUA AAU UUC AAU GAG AGG AAA AUU AUU AAU CUU CCU CAU AGU UAU UCG CAC ACA AAA GAU CCU AAA AAU UCU UCU UUC UUU UUG UGU GUC CGGUC-3′) and the antigenomic RNA representing the mRNA start region of the NP gene (nucleotides 56 to 158, 5′-gGA GGA AGA UUA AUA AUU UUC CUC UCA UUG AAA UUU AUA UCG GAA UUU AAA UUG AAA UUG UUA CUG UAA UCA CAC CUG GUU UGU UUC AGA GCC ACA UCA CAA AGg aau u-3′), T7 transcription cassettes were cloned into pUC19 encoding the T7 promoter, the desired substrate sequence (nucleotides 154 to 1 [genome] or 56 to 158 [antigenome] with an additional G at their 5′ ends, indicated above by small letters in italics) and a 3′-cis-hammerhead ribozyme in case of the genomic substrate (inserted via BamHI and EcoRI restriction sites), resulting in the addition of three extra nucleotides (5′-GUC-3′) at the 3′ end. T7 transcripts of RNA nt 56 to 158 were 3′ extended by 5′-GAAUU-3′ (indicated by small letters above) owing to linearization of transcription templates with EcoRI. Detailed cloning strategies and primer sequences are available on request. All constructs were verified by sequencing.
Expression and purification of MBP fusion proteins.
VP308–272 (termed VP30 throughout) and mutants thereof were expressed as MBP fusion proteins (see above) in Escherichia coli BL21(DE3) cells. Cells were cultured at 37°C in LB medium. When cells reached an optical density at 600 nm (OD600) of 0.6 to 0.7, protein expression was induced by adjusting the medium to 0.02% (wt/vol) arabinose and further incubation for 3 h. Cells were then harvested by centrifugation (2,000 × g, 10 min, 4°C), and the supernatant was discarded. Cell lysis was performed by sonication in 5 ml of lysis buffer (50 mM Tris-HCl [pH 8.0], 200 mM NaCl, 0.1% [wt/vol] Triton X-100, one tablet of cOmplete protease inhibitor cocktail [EDTA-free; Roche]). Subsequently, insoluble material was removed by centrifugation (17,000 × g, 30 min, 4°C). An amylose matrix equilibrated with lysis buffer was added to the supernatant, and the suspension was rotated for 2 h at 8°C. After centrifugation (2,000 × g, 10 min, 4°C), the supernatant was discarded and the matrix was washed three times with buffer A (50 mM Tris-HCl [pH 8.0], 200 mM NaCl). Bound material was eluted stepwise with increasing amounts of maltose in buffer B (50 mM Tris-HCl [pH 8.0], 200 mM NaCl): 1 mM (step 1), 5 mM (step 2), and 10 mM maltose (step 3). Elution steps were performed by rotating the matrix at each step for 30 min at 8°C. Eluted fractions from steps 2 and 3 (buffer B with 5 mM and 10 mM maltose) were mixed with glycerol (50%, vol/vol), rapidly frozen in liquid nitrogen, and stored at −20°C. The purity of the protein was analyzed using SDS-PAGE. The purification of the arginine-to-alanine mutant proteins (see Fig. 4A) was conducted at pH 7.5 (instead of pH 8.0).
FIG 4.
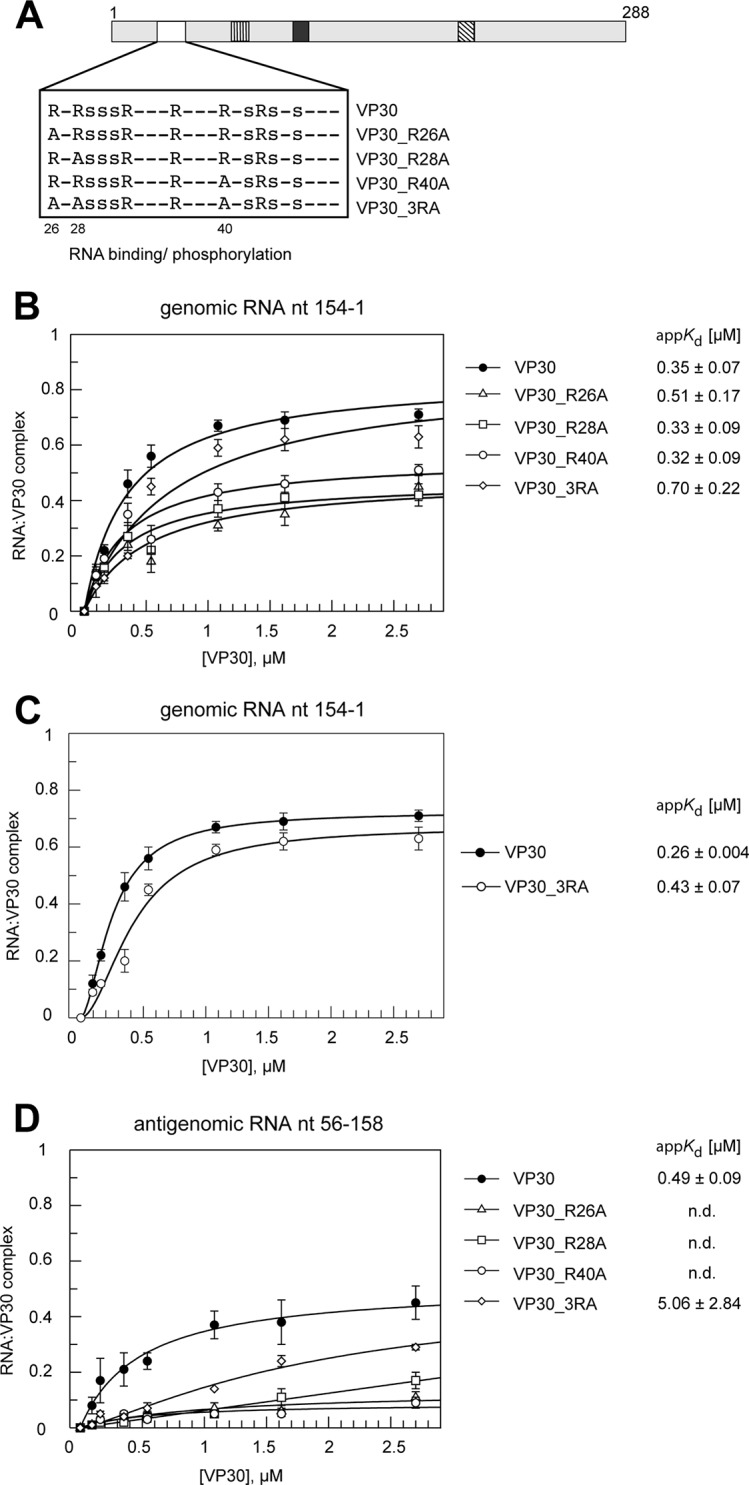
Effects of mutations in the N-proximal arginine cluster on RNA binding. (A) Schematic presentation of the N-proximal VP30 arginine/serine cluster subjected to mutation. Arginine residues in the proposed RNA binding region were mutated to uncharged alanines: R26A, R28A, R40A, or a combination of all three mutations (VP30_3RA). (B) RNA binding curves and derived appKd values (± SD) for VP30 and mutants R26A, R28A, R40A, and 3RA using the genomic RNA nt 154 to 1 (mean values from 3 to 7 independent experiments; error bars are standard deviations of the means). Curves were fitted according to a one-ligand-binding-site model. AppKd values were determined at pH 7.5 for all mutants except for VP30, measured at pH 8.0. (C) Curve fit for VP30 and VP30_3RA according to a cooperative binding model (multiple binding sites of differing affinities); n, cooperativity factor; n = 1.69 ± 0.04 (mean ± SD) for VP30 and 1.87 ± 0.46 for VP30_3RA (for details, see Materials and Methods); this model significantly improved the fit for VP30_3RA relative to the one-site model shown in panel B. (D) RNA binding curves (measured at pH 7.5) and derived appKd values as in panel B but using the antigenomic RNA substrate nt 56 to 158. Data points are based on 3 independent experiments (error bars are standard deviations of the means); curves were fitted according to a one-ligand-binding-site model; n.d., no determination of appKd values due to very low complex formation. Data for VP30 are taken from reference 25.
TEV cleavage.
For binding studies, the fusion proteins were cleaved with TEV protease (Invitrogen, Life Technologies) to remove the MBP moiety according to the manufacturer's protocol. For this purpose, 20 μg protein was mixed with 2.5 μl 20× TEV buffer (50 mM Tris-HCl [pH 8.0], 0.5 mM EDTA), 0.5 μl 0.1 M dithiothreitol (DTT), 1 μl TEV protease (10 U), and glycerol-buffer A (50%–50%) to an end volume of 50 μl. As a control, 20 μg MBP was treated in the same manner.
In vitro transcription and 32P-end labeling of RNA.
Plasmids were linearized with EcoRI followed by runoff transcription using bacteriophage T7 RNA polymerase. For transcription, 80 mM HEPES (pH 7.5), 40 mM MgCl2, 5 mM DTT, 1 mM spermidine, 4 mM each nucleoside triphosphate (NTP), 9 mM GMP (initiator nucleotide), 50 μg linearized plasmid DNA, and T7 RNA polymerase (∼30 U/μl; Institute of Pharmaceutical Chemistry, Marburg, Germany) were mixed and incubated at 37°C for 4 h in a total volume of 5 × 200 μl. After phenol and chloroform extraction, the RNA was purified using denaturing 10% PAGE. The RNA was visualized by UV shadowing, excised, and eluted from the gel using 1 M sodium acetate (NaOAc) solution (pH 4.9). RNA precipitation was performed with isopropanol. The RNA pellet was washed with ethanol (75%, vol/vol), air dried, and dissolved in double-distilled water (ddH2O). 5′-32P-end labeling was performed using T4 polynucleotide kinase (Thermo Scientific), and labeled products were purified using Illustra Microspin G-25 columns (GE Healthcare).
Electrophoretic mobility shift assay.
The 5′-32P-end-labeled RNA (adjusted to 24 nM RNA in 50 mM Tris-HCl [pH 8.0], 200 mM NaCl) was refolded by first heating the sample to 95°C for 1 min followed by cooling on ice for 2 min. The RNA was then incubated at room temperature for 20 min before 5 μl of the RNA solution was mixed with an equal volume of TEV-cleaved MBP-VP30 protein (final concentrations, 0.1 to 2.7 μM VP30 and 12 nM RNA) in buffer A (50 mM Tris-HCl [pH 8.0], 200 mM NaCl, 25% glycerol) and incubated for 10 min at room temperature. For the control reaction (0 μM VP30), MBP alone (4 μM) was incubated with the RNA. Reaction mixtures were immediately loaded (without addition of sample buffer) onto a native polyacrylamide (PAA) gel, consisting of two layers (lower part, 10% PAA; upper part, 4% PAA; casting buffer, 50 mM Tris-HCl, 200 mM NaOAc, pH 8.0). The electrophoresis buffer contained 25 mM Tris-HCl and 100 mM NaOAc (pH 8.0). Two different types of VP30-RNA complexes (C1 and CHMW) were observed in addition to the free RNA. Complex C1 migrated into the gel, whereas CHMW (where HMW is high molecular weight) remained in the gel wells. For quantification, the intensity of the radioactive signal associated with C1 and CHMW was calculated relative to the total intensity (free RNA + C1 + CHMW) after subtraction of the background and the intensity of C1 and CHMW for the MBP control. For calculating apparent dissociation constant (Kd) values (appKd), data for C1 complex formation were fitted to the following one-site binding model unless noted otherwise: complex = (capacity × [VP30])/(Kd + [VP30]). In the case of suboptimal fitting results, a model involving more than one site and cooperative binding was employed: complex = ([VP30]n × capacity)/(Kdn + [VP30]n). EMSA conditions for the arginine-to-alanine mutant proteins (see Fig. 4) were adjusted to pH 7.5 to reduce the fraction of CHMW complexes in favor of C1 complexes. Mobility shift gels were scanned with a Bio-Imaging Analyzer FLA3000-2R (Fujifilm), and 32P-end-labeled bands were marked by boxes and quantified using the analysis software PCBAS/AIDA (Raytest, Straubenhardt, Germany). The software Grafit (version 5.0.139) was used for the curve fitting. For pairwise comparison of appKd values, two-tailed P values were determined using the unpaired t test (GraphPad); the mean appKd ± standard deviation (SD) and the number of independent experiments (N) were used as input.
EBOV-specific trVLP assay.
The EBOV-specific transcription- and replication-competent virus-like particle (trVLP) assay was performed as described previously (21). Briefly, plasmids encoding all viral proteins, a T7 promoter-driven EBOV-specific minigenome coding for Renilla luciferase, and a plasmid encoding T7 RNA polymerase were transfected into HEK-293 cells (producer cells) using TransIT (Mirus). Additional transfection of pGL4.70 (Promega) encoding a firefly luciferase was performed for normalization of transfection efficiencies. VP30_wt (i.e., VP301–288) was replaced with VP30 variants as indicated. Reporter activity in producer cells was measured 72 h posttransfection using the Renilla and firefly luciferase assays (both purchased from pjk GmBH, Germany). Released trVLPs were purified from the supernatant via ultracentrifugation (∼220,000 × g) over a 20% sucrose cushion and used for infection of naive Huh-7 cells (indicator cells). An aliquot of trVLPs was analyzed for incorporation of VP30 mutant variants using the proteinase K digestion assay as described previously (22) and subjected to Western blot analysis. Sixty hours postinfection, indicator cells were lysed and the Renilla luciferase assay (purchased from pjk GmbH, Germany) was performed. Results obtained with VP30_wt were set to 100%.
Strand-specific RT-qPCR for the detection of cRNA/mRNA and vRNA in producer cells.
Producer cells (HEK-293) were transfected with the components of an EBOV-specific minigenome system (11). Cells were harvested at 48 h posttransfection, and RNA was isolated using the RNeasy minikit (Qiagen) according to the manufacturer's protocol, including on-column digestion with the RNase-Free DNase set (Qiagen) to avoid plasmid contamination. The RNA was eluted in 50 μl RNase-free water, and 10 μl was used for reverse transcription (RT) of either negative-strand RNA (vRNA) or positive-strand cRNA/mRNA using the Omniscript RT kit (Qiagen) in the presence of 10 U RiboLock RNase inhibitor (Thermo Fisher Scientific Fermentas) according to the manufacturer's instructions. The specific primer for reverse transcription of vRNA was luc(+) (5′-GGC CTC TTC TTA TTT ATG GCG A-3′), and that for cRNA/mRNA was luc(−) (5′-AGA ACC ATT ACC AGA TTT GCC TGA-3′). Subsequently, quantitative real-time PCR (qPCR) was performed on a StepOne real-time PCR system (Applied Biosystems) using the Platinum Taq Polymerase kit (Thermo Fisher Scientific Invitrogen), both primers luc(+) and luc(−), and 5 μl cDNA. A probe specific for the Renilla luciferase reporter (5′-CCA CAT ATT GAG CCA GTA GCG CGG-3′) containing 5′ FAM (6-carboxyfluorescein) and 3′ DDQ-I (Deep Dark Quencher-I; Biomers) was used for detection of amplified products. Serial 10-fold dilutions (103 to 108) of the minigenome plasmid were used as standards to determine the amount of minigenome copies in the samples. PCR conditions were as follows: initial denaturation of cDNA was performed at 95°C for 2 min, followed by 40 cycles with denaturation at 95°C for 15 s and annealing/elongation at 60°C for 30 s, and a single final step at 95°C for 15 s. Results are mean values from three independent experiments (n = 3), error bars are standard deviations of the means.
EBOV-specific replication-deficient minigenome assay.
The assay was performed as described above. Instead of the classical EBOV-specific minigenome, we used a previously described replication-deficient minigenome with a deletion of the 5′-trailer region (23). VP30_wt was replaced with VP30 mutants as indicated. Reporter activity in producer cells was measured 72 h posttransfection using the Renilla and firefly luciferase assays (pjk GmbH, Germany). Results are mean values (with standard deviations) based on three independent experiments (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001). Here, reporter gene activity reflects viral transcription in the absence of any viral replication.
Co-IP experiments.
Coimmunoprecipitation (co-IP) analyses were done as previously described (11). Briefly, HEK-293 cells (8 × 105 cells per well) were transfected using TransIT (Mirus) with pCAGGS VP30FLAG or pCAGGS VP35HA (0.5 μg each). DNA amounts were adjusted, if necessary, with empty pCAGGS vector. Cells were lysed at 48 h posttransfection for 20 min at room temperature in ice-cold co-IP buffer (20 mM Tris-HCl [pH 7.5], 100 mM NaCl, 1% [vol/vol] Nonidet P-40, 17.5 mM EDTA) supplemented with 0.1% (vol/vol) Triton X-100. Cell debris was removed by centrifugation (10 min at 8,000 × g, 4°C). For RNase treatment, the cleared supernatant was incubated with 10 μl RNase A/T1 mix (1,000 U/ml; Thermo Fisher Scientific). An aliquot was taken for expression control (input). The supernatant was added to 35 μl equilibrated mouse antihemagglutinin (anti-HA) or anti-FLAG M2 affinity gel agarose (both from Sigma-Aldrich). Precipitation was performed for 2 h at 4°C. Precipitated protein complexes were washed three times with co-IP buffer via centrifugation at 1,500 × g for 1 min. The pellet was resuspended in 45 μl 1× sample loading buffer (100 mM Tris-HCl [pH 6.8], 0.2% bromophenol blue, 20% glycerol, 10% 2-mercaptoethanol, 4% SDS) and subjected to SDS-PAGE and Western blot analysis.
Electrophoresis and Western blot analysis.
Sodium dodecyl sulfate (SDS) gel electrophoresis (12%) and Western blotting were performed as described previously (24). Proteins were transferred onto nitrocellulose membranes (Whatman) for 40 min (at a constant 20 V) using a Bio-Rad semidry blot chamber. First antibodies (rabbit anti-FLAG, biotinylated mouse anti-FLAG M2, rabbit anti-hemagglutinin [anti-HA]; all from Sigma-Aldrich) were diluted 1:500 in deficient phosphate-buffered saline (PBSdef; i.e., PBS lacking MgCl2) containing 0.1% Tween and 1% milk powder. Alexa Fluor 680-conjugated secondary antibodies were purchased from Invitrogen Molecular Probes (Thermo Fisher Scientific), and IRDye 800-conjugated antibodies came from Rockland (all diluted 1:5,000). Detection of antibodies was performed using the LiCor Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE, USA).
RESULTS
Establishment of a tailored EMSA to analyze RNA-VP30 binding.
To analyze the RNA-binding activity of EBOV VP30, we established a tailored electrophoretic mobility shift assay (EMSA) using purified VP30 and truncated EBOV RNA. We used a truncated form of VP30 (VP308–272) since deletion of the N-terminal 7 and C-terminal 16 aa of VP30_wt was previously shown to enhance stability and solubility of the recombinant protein (13). For recombinant expression, VP308–272 was N-terminally fused to the maltose binding protein (MBP). Purity of the fusion protein MBP-VP30 before and after separation of MBP and VP308–272 moieties by TEV protease cleavage was verified by Western blotting and Coomassie blue staining, as shown in the example of Fig. 1A.
Importantly, VP308–272 supports viral transcription in producer cells as proficiently as VP30_wt; the MBP-VP308–272 fusion protein was functional as well, although 2-fold less efficiently than VP30_wt (Fig. 1B). To exclude possible steric hindrance by the MBP moiety during RNA binding, RNA binding experiments were conducted after TEV cleavage of MBP-VP308–272, but without any further chromatographic separation of MBP and VP308–272 moieties (which we tried, but which was unsuccessful, leading to dilution and loss of VP308–272). Samples containing MBP alone, mock treated with TEV protease, were used as controls. For simplicity, VP308–272 is termed VP30 and VP301–288 is termed VP30_wt in the following.
As potential substrates for RNA binding to VP30, we employed the 3′-terminal 154 nt of the EBOV genome (GenBank accession number AF086833; this RNA fragment comprises the signals essential for replication and encapsidation as well as the transcription start site of the first viral gene [6, 17]) and an antigenomic substrate (mRNA start region, nt 56 to 158).
Since attempts to obtain gel-resolvable VP30-RNA complexes under standard native PAGE conditions, such as the Tris-borate-EDTA (TBE) buffer system, were unsuccessful, we changed the buffer system to 25 mM Tris-HCl and 100 mM NaOAc (pH 8.0), which resembled the conditions used for recombinant VP30 purification and storage (13). This was combined with a discontinuous gel system (4 and 10% PAA; for details, see Materials and Methods) to better resolve larger complexes. Comparison of the binding affinities of VP30 to the genomic versus antigenomic substrate revealed a preference for the genomic substrate (Fig. 1C). The apparent Kd (appKd) value was 0.35 ± 0.07 μM (mean ± SD) for genomic RNA nt 154 to 1 and 0.49 ± 0.09 μM for the antigenomic RNA nt 56 to 158 (25) (P < 0.05, based on unpaired t test analysis; see Materials and Methods). The antigenomic RNA additionally showed a reduced F2.7μM value (fraction of C1 complexes at the highest feasible VP30 concentration of 2.7 μM) (Fig. 1C, right; Tables 1 and 2). Since we could not rule out that these differences included contributions from the different lengths of the two probes, we also tested an antigenomic substrate fully complementary to RNA nt 154 to 1; this RNA still had a higher appKd of 0.49 ± 0.07 μM (P < 0.05 relative to RNA 154 to 1) (25), although the 5′-terminal extension increased F2.7μM roughly to that of the genomic RNA probe (∼0.7) (25). These results, however, argue against the possibility that VP30 has a substantial preference for interaction with the 5′-terminal portion of the mRNA relative to the genomic 3′ leader, although it is not yet clear to which extent NP is displaced from the genomic RNA during transcription to allow access by VP30. Nevertheless, all further analyses were performed with the genomic substrate. A fully double-stranded hybrid of the genomic leader RNA nt 154 to 1 and its antigenomic complement served as control RNA, which showed no binding to VP30. A detailed analysis of RNA molecules with VP30 binding capacity is described elsewhere (25). EMSAs revealed two types of RNA-VP30 complexes, one migrating into the gel (C1; assumed to represent hexameric VP30 bound to the RNA, as outlined below) and a second, apparently high-molecular-weight complex (CHMW) barely migrating into the gel (Fig. 1C, left). Such HMW complexes were described previously for EBOV VP30 and most likely represent higher-order oligomeric complexes of VP30 and RNA (15). appKd values were derived from the amount of C1 complexes (for details, see Materials and Methods). The highest VP30 concentration used here was 2.7 μM because the protein started to aggregate at higher concentrations. F2.7μM, i.e., the fraction of RNA forming C1 complexes at the experimental endpoint (2.7 μM VP30), substantially varied between VP30 and its mutants and was therefore also taken as a quality feature in the assessment of VP30-RNA interactions.
TABLE 1.
Binding data for the tested VP30 mutants and the genomic RNA nt 154 to 1, as inferred from EMSAsa
| Protein | appKd/μM | F2.7μM (C1) | calc. EP C1 | F2.7μM (CHMW) |
|---|---|---|---|---|
| VP30 | 0.35 ± 0.07 | 0.71 ± 0.05 | 0.85 ± 0.05 | 0.18 ± 0.01 |
| VP30_AA | 0.17 ± 0.03 | 0.66 ± 0.03 | 0.70 ± 0.03 | 0.20 ± 0.07 |
| VP30_DD | 0.73 ± 0.15 | 0.47 ± 0.08 | 0.61 ± 0.05 | 0.39 ± 0.06 |
| VP30_5LA | ND | 0.08 ± 0.03 | 0.02 ± 0.007 | |
| VP30_R26A | 0.51 ± 0.17 | 0.45 ± 0.12 | 0.48 ± 0.06 | 0.10 ± 0.05 |
| VP30_R28A | 0.33 ± 0.09 | 0.42 ± 0.09 | 0.47 ± 0.04 | 0.12 ± 0.06 |
| VP30_R40A | 0.32 ± 0.09 | 0.51 ± 0.06 | 0.55 ± 0.05 | 0.09 ± 0.04 |
| VP30_3RA | 0.70 ± 0.22 | 0.63 ± 0.08 | 0.86 ± 0.11 | 0.29 ± 0.09 |
| VP30_C72S | ND | 0.18 ± 0.03 | 0.03 ± 0.008 |
Data for VP30 are taken from reference 25. AppKd values are based on the quantification of C1 complexes (Fig. 1C; see also Materials and Methods), derived from the fit to a one-ligand-binding-site model. F2.7μM (C1) (termed F2.7μM in the text), fraction of the RNA probe in complex C1 at 2.7 μM VP30, i.e., at the highest protein concentration used in the gel shift assay; F2.7μM (CHMW), fraction of the RNA probe in complex CHMW at 2.7 μM VP30; ND, not determined, due to either no or very low complex formation or because no reasonable fit to the “one-ligand-binding-site” model could be obtained; calc. EP C1, endpoint (capacity, saturation of binding) from the fit to a one-ligand-binding-site model. Values are based on 5 independent experiments for VP30, 4 for VP30_AA, 3 for VP30_DD, 4 for VP30_5LA, 6 for VP30_R26A, 5 for VP30_R28A, 7 for VP30_R40A, 4 for VP30_3RA, and 3 for VP30_C72S. Values are means ± SD.
TABLE 2.
Binding data for VP30 and its arginine mutants (VP30_R26A, _R28A, _R40A, and _3RA) acting on the antigenomic substrate RNA nt 56 to 158a
| Protein | appKd/μM | F2.7μM (C1) | calc. EP C1 | F2.7μM (CHMW) |
|---|---|---|---|---|
| VP30 | 0.49 ± 0.09 | 0.45 ± 0.1 | 0.51 ± 0.03 | 0.11 ± 0.05 |
| VP30_R26A | ND | 0.11 ± 0.03 | ND | 0.05 ± 0.03 |
| VP30_R28A | ND | 0.17 ± 0.06 | ND | 0.10 ± 0.03 |
| VP30_R40A | ND | 0.09 ± 0.03 | ND | 0.05 ± 0.02 |
| VP30_3RA | 5.06 ± 2.84 | 0.29 ± 0.02 | 0.86 ± 0.35 | 0.22 ± 0.02 |
All values are based on 3 independent experiments and are means ± SD. Data for VP30 are taken from reference 25. AppKd values are based on the quantification of C1 complexes (Fig. 1C; see also Materials and Methods). ND, no curve fitting applied owing to low F2.7μM (C1) values. For further details, see Table 1. The P value for appKd of VP30 versus VP30_3RA is 0.0495.
As a control, we analyzed RNA-binding activity of the C-terminal domain of VP30 (VP30CTD, aa 142 to 272), whose crystal structure was previously solved (13). VP30CTD lacks the arginine-rich sequence (region of aa 26 to 46) and the zinc-binding motif (around aa 80; see below) as well as the hexamerization motif (aa 100 to 103). VP30CTD showed only extremely weak RNA binding (50% complex formation with RNA nt 154 to 1 at ∼40 μM protein; data not shown) compared with VP30 (50% complex formation at ∼0.35 μM [Fig. 1C]). These findings led us to focus on structural elements within the N-terminal half of VP30.
Phosphorylation of VP30 impairs RNA binding.
The N-proximal phosphorylation sites of VP30 (S29 to 31, S42, S44, S46) overlap its putative arginine-rich RNA-binding pocket (Fig. 1D). We thus asked whether the phosphorylation status may impair RNA binding of VP30 due to increased electrostatic repulsion. It was demonstrated before that only mutants of VP30 mimicking a non- or weakly phosphorylated state support viral transcription, whereas those representing a fully phosphorylated state fail to do so (11, 16, 18, 26).
To address the effect of phosphorylation on RNA binding, we used a mutant of VP30 with serine (S)-to-aspartate (D) exchanges at all six serine positions (VP30_DD, Fig. 1D), which mimicked the phosphorylated state. We further included the mutant VP30_AA with S-to-alanine (A) mutations at the same positions, representing the unphosphorylated state (Fig. 1D) (16). VP30_DD bound the viral RNA substrate with about 2-fold lower affinity than VP30 (P = 0.0024), while VP30_AA even showed an increased affinity relative to VP30 (Fig. 1E and Table 1), resulting in a 4.3-fold affinity difference between VP30_AA and VP30_DD (P = 0.0003). For VP30_DD, we also observed a reduction in the fraction of RNA that was present in C1 complexes at saturation (Fig. 1E). This could be explained by increased CHMW formation (data not shown). We also determined appKd values based on total complex formation (CT = C1 + CHMW). This yielded very similar appKd values of 0.35 ± 0.07 μM for VP30, 0.15 ± 0.03 μM for VP30_AA, and 0.81 ± 0.11 μM for VP30_DD. Our results show that an increase of negative charges in the putative RNA binding region of VP30 (∼aa 26 to 46) impairs the protein's ability to bind RNA and modifies the migration pattern of the complex in the gel.
Hexamerization of VP30 is essential for RNA binding.
VP30 self-interacts into hexamers, and this capacity is essential for its transcriptional activation function (12, 13). So far, it has been unclear if VP30 homo-oligomerization is a prerequisite for RNA binding or, vice versa, whether RNA binding is involved in mediating VP30 homo-oligomerization. In order to investigate the latter question, we performed coimmunoprecipitation (co-IP) experiments using differently epitope-tagged VP30 in the presence of RNases (Fig. 2A). Self-interaction of VP30 was not affected by RNase treatment of cell extracts before precipitation (Fig. 2A, lanes 3 and 6), indicating that hexamerization of VP30 is independent of RNA binding.
FIG 2.

VP30 hexamerization and RNA binding. (A) Co-IP analysis of FLAG-tagged VP30 with MYC-tagged VP30 after or without RNase treatment. HEK-293 cells expressing VP30FLAG and/or VP30MYC were lysed 48 h posttransfection and subjected to co-IP using anti-FLAG agarose (Sigma-Aldrich). Mock lane, untransfected cells (no expression of VP30). An aliquot was taken from cell lysates as a control for protein expression levels (input). Western blot analysis was performed using a mouse anti-FLAG M2 biotinylated antibody and Alexa Fluor 680-conjugated streptavidin (shown in red). MYC-tagged VP30 was stained by rabbit anti-myc and IRDye 800-conjugated goat anti-rabbit (shown in green). Detection of proteins was obtained with the LiCor Odyssey Imaging System. The quantification of Western blot signals for VP30MYC coprecipitated with VP30FLAG after or without RNase treatment (bar diagram on the right) was based on four independent experiments. (B) Amino acid exchanges (four L-to-A and one I-to-A mutation) in the hexamerization-deficient variant VP30_5LA. For further details, see legend to Fig. 1D. (C) (Left) Representative EMSA; for details, see legend to Fig. 1C. (Right) RNA binding curves for VP30 and VP30_5LA based on C1 complex formation (mean values from 3 to 5 independent experiments; error bars are standard deviations of the mean). Data for VP30 are taken from reference 25. (D) Transcription/replication-competent virus-like particle (trVLP) assay. HEK-293 producer cells were transfected with plasmids encoding all viral proteins and a plasmid for expression of phage T7 RNA polymerase, as well as an EBOV-specific minigenome coding for the Renilla luciferase reporter under the control of a T7 promoter. trVLPs released into the supernatant were used to infect naive Huh-7 indicator cells or indicator cells that have been transfected beforehand with VP30_wt. (E) Reporter gene activity in producer cells was measured 72 h posttransfection via the Renilla luciferase assay and normalized to firefly luciferase levels reflecting cellular transcription activity (black bars). Reporter gene activity in naive (dotted bars) or pretransfected (striped bars) indicator cells was measured 60 h postinfection via the Renilla luciferase assay. Luciferase activity obtained with VP30_wt was set to 100%. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
To investigate whether VP30 homo-oligomerization is required for RNA binding, we made use of a previously described mutant of VP30 with a destroyed hexamerization motif. Upon exchange of four leucines (aa 100 to 103) and the adjacent isoleucine to alanines (Fig. 2B, VP30_5LA), homohexamerization of VP30 is blocked while dimerization via a C-terminal basic cluster and NP-binding can still occur (12, 13). EMSAs revealed an almost complete loss of RNA binding for VP30_5LA (Fig. 2C), indicating that the hexameric state of VP30 is a prerequisite for efficient interaction with RNA substrates. This finding also indicated that C1 complexes (Fig. 1C) consist of RNA bound to hexameric VP30.
We next analyzed VP30_5LA for its transcriptional activation function in an EBOV-specific trVLP assay (21) (Fig. 2D; see Materials and Methods). This assay is based on plasmid-driven expression of all viral proteins together with an EBOV-specific minigenome encoding Renilla luciferase as reporter in producer cells. Generated trVLPs were used to infect indicator cells to measure primary transcription in the absence of excess viral proteins. In this assay, the VP30_5LA variant was as defective in supporting viral transcription as control trVLPs lacking VP30 (Fig. 2E, dotted bars), which is in line with the finding that already single leucine-to-alanine substitutions in the hexamerization motif completely abrogated transcriptional activation by VP30 (12).
To exclude that VP30_5LA negatively influenced replication in producer cells and thus caused reduced amounts of genome-containing trVLPs, we used the trVLPs to infect indicator cells that were transfected beforehand with VP30_wt. Under these conditions, minigenome activity can be restored from trVLPs containing no or inactive VP30 (11). Using this setup, reporter gene activity could be rescued with trVLPs containing VP30_5LA (Fig. 2E, striped bars). This result indicated that the inability of VP30_5LA to support primary transcription was not the result of impaired replication in producer cells.
Together, these results suggest that VP30 hexamerization is a prerequisite for binding to RNA and consequently for VP30's function in viral transcription.
Integrity of the zinc-binding motif is essential for RNA binding and transcriptional activation by VP30.
John et al. (15) observed a reduction in VP30-RNA cross-links upon EDTA treatment of VP30, suggesting a role of divalent cations in RNA binding. We therefore investigated whether the integrity of the Zn-binding motif in VP30 is a prerequisite for RNA binding. A cysteine-to-serine mutation at position 72 (VP30_C72S) (Fig. 3A) of this motif abolished transcriptional activation by VP30 in a minigenome system (14). We tested VP30_C72S for binding to the genomic RNA nt 154 to 1. This single mutation of the first cysteine of the Zn-binding motif largely impaired RNA binding (Fig. 3B), comparable in its effect to the VP30_5LA variant (Fig. 2C). The loss in RNA binding correlates with the observed loss of transcriptional activation caused by the C72S mutation (14).
FIG 3.
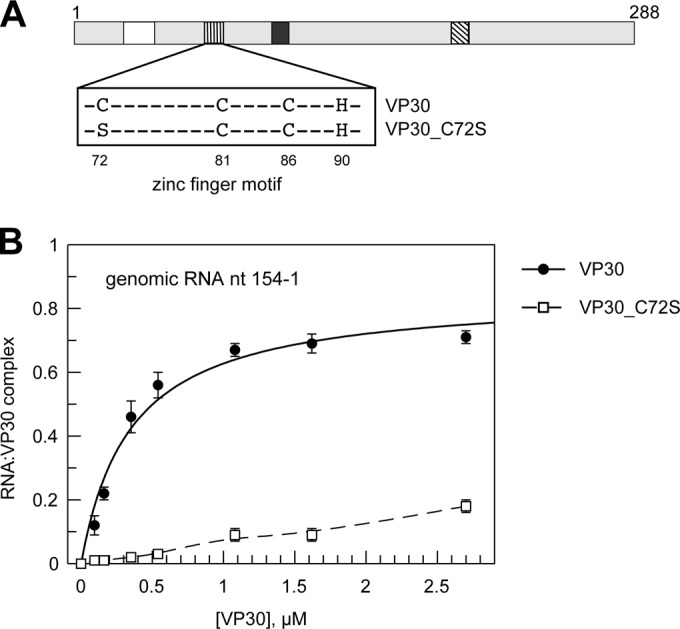
Role of the zinc-binding domain in RNA binding. (A) Scheme depicting the mutation of the first cysteine to serine in the zinc-binding motif (VP30_C72S). (B) RNA binding curves for VP30 in comparison to VP30_C72S based on C1 complex formation (mean values from 3 to 5 independent experiments; error bars are standard deviations of the mean). Data for VP30 are taken from reference 25.
Role of R26, R28, and R40 in RNA binding.
A previous study provided first evidence that the arginine-rich sequence (∼aa 26 to 46) (Fig. 1D) of VP30 is an interaction site for RNA substrates (15). Replacement of arginine residues by alanine showed no significant RNA-binding defect for the R28A and R36A mutants, a 1.5-fold affinity reduction for the R32A mutant, and a >4-fold-increased Kd for the R40A mutant (15). We also analyzed mutants of VP30 containing single R26A, R28A, or R40A exchanges, as well as a mutant carrying R-to-A exchanges at all three positions (VP30_3RA) (Fig. 4A) for their RNA binding properties. The appKd values presented in Fig. 4B and Table 1 revealed <1.5-fold appKd increases for the single mutants, including VP30_R40A, which contrasts with the >4-fold Kd increase reported in reference 15. However, the single R-to-A mutants showed reduced F2.7μM values relative to VP30. For VP30_3RA, the appKd increase was 2-fold relative to that of VP30 (P = 0.0138), but the fit to the one-ligand-binding-site model was suboptimal. Thus, we additionally fitted the binding curve for VP30_3RA according to a cooperative binding model (multiple binding sites of differing affinities; for details, see Materials and Methods). This clearly improved the fit for VP30_3RA (Fig. 4C), while the binding curve for VP30 was less sigmoidal and gave reasonable fits with both models (Fig. 4B and C). Using the cooperative binding model, the calculated appKd ratios for VP30_3RA (appKd = 0.43 μM) and VP30 (appKd = 0.26 μM) were similar (1.7-fold) (Fig. 4C; P = 0.0012) compared with the values obtained from fitting to the one-ligand-binding-site model (Fig. 4B). Cooperative binding of RNA by VP30 would not be too surprising, considering that the VP30 hexamer may expose multiple RNA binding patches rather than a single composite RNA-binding interface, similar to the VP30-related tetrameric M2-1 transcription factor of respiratory syncytial virus (RSV), which presents several positively charged patches that are spread over an extended surface area (27).
As the aforementioned previous study (15) reported a substantially increased Kd for the R40A mutant using an RNA substrate derived from the antigenomic strand (nt 36 to 80), we additionally tested binding of the VP30 arginine mutants to the antigenomic RNA (nt 56 to 158) probe. Compared with VP30, the arginine mutants also showed impaired binding to the antigenomic RNA (Fig. 4D; Table 2), and we refrained from providing appKd values for the R26A, R28A, and R40A variants because of low F2.7μM values. With both probes, the 3RA mutant gave higher F2.7μM values than any of the single arginine mutants (Fig. 4B and D), suggesting an intricate mode of VP30-RNA interaction that is poorly understood at present. In summary, all arginine mutants gave rise to impaired RNA binding based on either appKd values, F2.7μM values, and/or changes in the binding mode (VP30_3RA).
Impact of R26, R28, and R40 mutations on transcriptional activation.
We next analyzed mutants of VP30 with single R-to-A exchanges as well as VP30_3RA for their transcriptional activation function in an EBOV-specific trVLP assay (21) (Fig. 2D; see Materials and Methods). Similar to VP30_wt, all mutants with single R-to-A mutations were able to support transcriptional activity in producer cells. The triple mutant VP30_3RA, however, activated viral transcription only slightly less efficiently than VP30_wt (Fig. 5A, black bars). Viral transcription in the (plasmid-transfected) producer cells mimics a late stage of viral infection, when viral proteins and genomic template RNA are highly abundant. It was therefore of interest to determine if the arginine mutations have substantial effects on primary transcription, i.e., when the available amount of viral proteins for RNA synthesis is restricted to the proteins associated with the incoming VLP. To this end, we purified trVLPs from the supernatant of producer cells and confirmed the incorporation of VP30_wt and arginine mutants into trVLPs by proteinase K (PK) digestion and Western blot analysis (Fig. 5A, right, cf. VLPs and VLPs + PK). Since the incorporation of VP30 into trVLPs is dependent on its interaction with NP, these data also indicated that the VP30-NP interaction was not affected by the mutations (11). We then infected naive target cells (indicator cells) with the trVLPs containing either wild-type or mutant VP30. While single R-to-A exchanges did not affect the ability to support primary transcription, simultaneous mutation of all three arginine residues almost completely abolished primary transcription (Fig. 5A, dotted bars).
FIG 5.

Role of VP30 arginine residues for activation of viral transcription. (A) (Left) Reporter gene activity in producer cells was measured 72 h posttransfection via the Renilla luciferase assay and normalized to firefly luciferase levels reflecting cellular transcription activity (black bars). Reporter gene activity in naive (dotted bars) or pretransfected (striped bars) indicator cells was measured 60 h postinfection via the Renilla luciferase assay. Luciferase activity obtained with VP30_wt was set to 100%. VP30 was replaced with the respective VP30 arginine mutant(s) as indicated. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. (Right) Expression and incorporation of VP30 arginine mutant proteins into trVLPs. Purified trVLPs were incubated with proteinase K (PK) to analyze specific incorporation of VP30 into trVLPs. Activity of proteinase K was controlled by addition of the detergent Triton X-100 to wild-type trVLPs (lane 7, + PK and + Triton X-100); Triton X-100 disrupts the trVLPs without impairing proteinase K activity, such that VP30 becomes accessible to proteolytic degradation. Western blotting of cell lysates and purified trVLPs was performed using a rabbit anti-FLAG antibody and Alexa Fluor 680 goat anti-rabbit antibody for VP30 staining. NP was stained with a chicken anti-NP and Alexa Fluor 680 donkey anti-chicken antibody. Detection was obtained using the LiCor Odyssey Imaging System. (B) Influence of VP30 arginine mutants on viral replication. Replication of the minigenome (vRNA) and cRNA/mRNA levels were analyzed using a strand-specific two-step RT quantitative real-time PCR as described previously (11). Briefly, producer cells were transfected with the components of an EBOV-specific minigenome system (2, 3). Cells were harvested at 48 h posttransfection, and RNA was isolated and subjected to RT and qPCR. Results are mean values from three independent experiments (n = 3); error bars are standard deviations of the means. (C) Replication-deficient minigenome assay. The assay was performed as described for Fig. 2D. Instead of the classical minigenome (transcription and replication competent), a replication-deficient minigenome was used for measuring exclusively viral transcription in producer cells (23). Reporter gene activity was measured 72 h posttransfection using the Renilla luciferase assay and normalizing to firefly luciferase levels as a measure of cellular transcription activity. Luciferase activity obtained with VP30_wt was set to 100%. VP30 was replaced with the respective VP30 arginine mutant(s) as indicated. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
To exclude the possibility that the transcriptional defect of trVLPs containing VP30_3RA in indicator cells was due to inefficient incorporation of functional nucleocapsids into VLPs (and, as a consequence, decreased delivery of functional nucleocapsids into indicator cells), we pretransfected indicator cells with VP30_wt prior to infection with the variant trVLPs (11, 21). Under these conditions, minigenome activity is no longer dependent on the presence of VP30 in trVLPs or on mutant versions of VP30 associated with trVLPs, as VP30 expressed in trans can functionally compensate for the lack of VP30 activity of the trVLPs. Indeed, this setup rescued transcription for trVLPs harboring VP30_3RA to levels similar to those seen for VP30_wt and the single arginine mutants, indicating that VP30_3RA does not impair the delivery of functional trVLPs into target cells (Fig. 5A, striped bars). We conclude that the defect of VP30_3RA is indeed on the level of transcriptional activation.
Since the minigenome system does not distinguish between replication and transcription and enhanced replication gives rise to more templates for transcription, we wanted to test if the VP30 arginine mutants affect transcription exclusively or whether they also impact viral replication. To this end, we analyzed the levels of replicated vRNA as well as cRNA/mRNA in a strand-specific RT-qPCR employing RT primers that exclusively recognized either genomic RNA or cRNA/mRNA (11). As previously reported (11), EBOV replication is upregulated in the absence of VP30, suggesting that transcriptional inactivity favors replication. Levels of cRNA/mRNA in the presence of arginine mutants of VP30 (Fig. 5B, gray bars) showed a pattern similar to that detected in reporter assays (Fig. 5A, black bars). In the presence of the arginine mutants, replication (vRNA) was slightly increased in comparison to VP30_wt (Fig. 5B, striped bars), although these effects did not reach statistical significance. However, as the trend was toward synthesis of more genome copies in the presence of VP30_3RA, our findings indicate that the defect of VP30_3RA observed in indicator cells (Fig. 5A) was not due to decreased replication but resulted from impaired transcriptional support activity. To further substantiate this notion, we made use of a previously described replication-deficient minigenome that allows one to exclusively analyze viral transcription, owing to deletion of the 5′-trailer region of the minigenome (23). Using this experimental setup, the defect of VP30_3RA in producer cells was more pronounced and, in addition, a defect in transcriptional support activity became evident for VP30_R40A as well (Fig. 5C). Since the reporter gene activities obtained with VP30_3RA and VP30_R40A were comparable, the result suggests that in particular arginine 40 is important for the transcription support activity of VP30.
Interaction of VP30 with the polymerase cofactor VP35 is RNA dependent.
Phosphorylation of VP30 impairs RNA binding and its interaction with the polymerase cofactor VP35 (11). Since both proteins, VP30 and VP35, are RNA-binding proteins, we wanted to clarify if their interaction may also be mediated by RNA. This was addressed by co-IP experiments using FLAG-tagged VP30 (VP30FLAG) and HA-tagged VP35 (VP35HA) expressed in HEK-293 cells. Before co-IP using an anti-HA antibody matrix, cell extracts were incubated in the absence and presence of an RNase A/T1 mixture (Fig. 6A). Treatment with RNases resulted in complete loss of VP30FLAG coprecipitation (Fig. 6A, co-IP, lane 3 versus lane 6), demonstrating that the interaction of VP30 and VP35 is mediated by RNA.
FIG 6.

Interaction of VP30 and VP35 is RNA dependent. (A) Co-IP analysis of FLAG-tagged VP30_wt with HA-tagged VP35 after RNase treatment. HEK-293 cells expressing VP30FLAG and/or VP35HA were lysed 48 h posttransfection. An aliquot was taken from cell lysates as control for protein expression levels (input). Two additional aliquots from each extract sample were processed in parallel by adding an RNase A/T1 mix to the first aliquot (+ RNase), but not to the second (− RNase), followed by precipitation of protein complexes for 2 h at 4°C using mouse anti-HA agarose (Sigma-Aldrich). Western blot analysis was performed using a mouse anti-FLAG M2 biotinylated antibody and Alexa Fluor 680-conjugated streptavidin (shown in red). HA-tagged VP35 was stained by rabbit anti-HA and IRDye 800-conjugated goat anti-rabbit (shown in green). Detection of proteins was obtained with the LiCor Odyssey Imaging System. The quantification of Western blot signals for VP30 coprecipitated by VP35 after or without RNase treatment (bar diagram on the right) was based on five independent experiments. VP30FLAG amounts coimmunoprecipitated without RNase were set to 100%. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. (B) Co-IP analysis of FLAG-tagged VP35 with HA-tagged VP35 after or without RNase treatment. The procedure was identical to that described for panel A, except that anti-FLAG agarose (Sigma-Aldrich) was used for the co-IP, a mouse anti-HA biotinylated antibody and Alexa Fluor 680-conjugated streptavidin (shown in red) were used for Western blot detection of VP35HA, and a rabbit anti-FLAG and IRDye 800-conjugated goat anti-rabbit (shown in green) antibodies were used for detection of VP35FLAG. The quantification of Western blot signals for VP35FLAG coprecipitated with VP35HA after or without RNase treatment (bar diagram on the right) was based on five independent experiments. (C) Interaction of VP30 with VP35 is dependent on both proteins' ability to bind to RNA. Co-IP analysis of VP30 or VP35 with corresponding mutants that lack RNA binding activity. For VP35, we used a mutant (VP35mutHA) lacking dsRNA binding activity owing to mutations R305A, K309A, and R312A. VP30_5LAFLAG was used as a negative RNA binding mutant to evaluate its interaction with VP35. Co-IP of protein complexes was performed as described for panel B. Single expression of the proteins used as control is shown in lanes 1 to 4. Lane 5, coexpression of VP30FLAG with VP35HA; lane 6, coexpression of VP30FLAG with VP35mutHA; lane 7, coexpression of VP30_5LAFLAG with VP35HA. Western blot signals of VP30FLAG precipitated by VP35HA versus VP35mutHA were quantified based on three independent experiments (upper bar diagram on the right); Western blot signals of VP30FLAG or VP30_5LAFLAG precipitated by VP35HA were quantified based on four independent experiments (lower bar diagram on the right). Amounts of precipitated wild-type proteins were set to 100%. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. Mock lanes, untransfected cells.
Both VP30 and VP35 form homo-oligomers as a prerequisite for their specific functions in viral transcription and replication. VP35 was shown to form trimers via an N-terminal coiled-coil motif (28). For Marburg virus VP35, it was shown that homo-oligomerization is important for the protein's interaction with polymerase L (29). To investigate whether the observed RNA dependency of the interaction between VP35 and VP30 is due to RNA-mediated self-interaction of VP35, we performed co-IP analyses with differently tagged VP35 mutants. Pretreatment of cellular extracts with RNases did not affect the amount of VP35FLAG coprecipitated with VP35HA (Fig. 6B, co-IP, lane 3 versus lane 7) but prevented the co-IP of VP30FLAG with VP35HA, used here as a control (Fig. 6B, co-IP, lane 4 versus lane 8).
To further substantiate our finding that the interaction of VP30 and VP35 is bridged by RNA, we utilized in our co-IP experiments two mutant proteins defective in RNA binding: VP30_5LA (Fig. 2C) and a previously described VP35 mutant (VP35mut) lacking dsRNA binding activity due to mutations within the basic cluster of the VP35 C-terminal interferon inhibitory domain (IID) (R305A, K309A, R312A) (30–32). We employed HA-tagged wt VP35 or VP35mut together with FLAG-tagged wt VP30 or VP30_5LA in the co-IP experiments (Fig. 6C). In contrast to VP35, VP35mut was largely impaired in its capacity to coprecipitate VP30 (Fig. 6C, lanes 6 versus 5), indicating that the dsRNA binding activity of VP35 is crucial for the formation of VP35/VP30 complexes (Fig. 6C, upper graph on the right). Also, smaller amounts of VP30_5LA (Fig. 6C, lane 7) were coprecipitated with VP35 than with VP30_wt (Fig. 6C, lane 5; summarized in the lower graph on the right), indicating that VP30's RNA binding ability is required as well for the interaction of VP30 and VP35.
While it is theoretically possible that unspecific binding to the same cellular RNA molecules leads to the interaction of VP30 and VP35, we expect a large excess of cellular RNA molecules over VP30 and VP35, such that binding of the two proteins to different RNAs, rather than to the same RNAs, is more likely. Future investigations will address this question.
Taken together, our data indicate that RNA mediates the complex formation between VP30 and VP35. Interestingly, the ability of VP35 to bind dsRNA is crucial in this process.
DISCUSSION
The template for viral transcription of viruses within the order Mononegavirales is the nucleocapsid complex consisting of the N (NP)-encapsidated genomic RNA (33). Usually, the viral polymerase complex composed of the catalytic L subunit and its cofactor P (VP35) recognizes and transcribes viral genomic RNA. Filoviruses as well as pneumoviruses make an exception by encoding additional transcriptional support factors (VP30 and M2-1, respectively) (8, 18, 34). Recent publications identified and characterized several functional domains and posttranslational modifications of EBOV VP30 that are required for viral transcription (11, 16–18). However, the mechanism of how VP30 supports viral transcription remained unclear.
The present study establishes that the previously described functional domains of VP30 and posttranslational modifications, which were found to be crucial for transcriptional support activity of the protein, simultaneously affect its capacity to bind to viral RNA. This includes the hexamerization domain of VP30, the Zn-binding motif, the phosphorylation status of VP30, and the N-proximal RNA-binding interface. Thus, transcriptional support activity of VP30 is closely correlated with efficient binding of viral RNA. VP30 was also able to bind antigenomic RNA mimicking the 5′ end of viral NP mRNA. This complex had a 1.4-fold-higher appKd than genomic leader RNA.
The almost complete loss of RNA binding affinity observed for the hexamerization-defective VP30_5LA mutant indicated that a VP30 hexamer is required to assemble a composite interaction array for RNA substrates, similar to the functionally related tetrameric M2-1 protein exposing an array of positively charged patches spread over the protein's surface (27). Examples of similar architectural concepts may include the bacterial Hfq hexamer and eukaryal Lsm proteins that “chaperone” RNA-RNA and RNA-protein interactions (35). Another feature demonstrated here to be critical for RNA binding by VP30 is the integrity of the Zn finger, which may directly contribute to RNA binding and/or may influence RNA binding by VP30 indirectly. The first possibility is likely, as the corresponding Zn-binding domain of M2-1 indeed contacts RNA residues (36). It is worth noting that the Zn-binding domain of M2-1 of RSV was suggested to contribute to the interaction of M2-1 monomers by maintaining the protein's authentic structure (27). Although the sequence homology of VP30 and M2-1 is low, the two proteins share the architectural feature of a Zn finger domain tethered to an entirely α-helical C-terminal domain (13, 27). It is therefore also possible that the Zn finger domain of VP30 supports hexamer formation, which, in turn, is a prerequisite for RNA binding by VP30.
To deepen the insight into RNA binding by VP30, we have established a novel mobility shift assay tailored to the analysis and visualization of VP30-RNA complexes that allowed a more detailed investigation of the protein-RNA complexes formed. For example, we detected that some VP30 mutants tend to form high-molecular-weight complexes (CHMW) in addition to the C1 complexes that migrate readily into the PAA gel. The formation of those complexes had an impact on the measured appKd values and thus needed to be taken into account. We observed an increased fraction of CHMW complexes for VP30_3RA at pH 8.0, and this fraction could be reduced in favor of C1 complexes by lowering the pH of the buffer system to 7.5 (data not shown). Similarly, the C1/CHMW ratio was shifted toward CHMW for VP30_DD. While the substantial changes in the surface charge of VP30_DD and VP30_3RA might have contributed to their transcriptional phenotypes, similar mutations in M2-1 did not change the overall folding of the protein (11, 37).
Mutations in the N-proximal arginine cluster resulted in differential effects. All tested mutants showed a tendency toward increased genome replication (Fig. 5B) and impaired RNA binding in vitro. VP30 mutants R26A, R28A, and R40A showed substantial decreases in F2.7μM (Tables 1 and 2). Simultaneous mutations of all three arginines in VP30_3RA led to a substantial increase in appKd (Tables 1 and 2) and changed 3′-leader RNA binding toward a more cooperative mode, as inferred from EMSAs (Fig. 4C). The 3RA mutation also profoundly impaired the function of VP30 in primary transcription (Fig. 5). Less severe effects of the single R26A, R28A, or R40A mutations may suggest that the arginines of the cluster have redundant functions in RNA binding.
The arginine cluster in VP30 is intertwined with phosphorylated serines (aa 29 to 31, 42, 44, and 46). Arginines in the vicinity of serine residues are often part of kinase recognition motifs. This putative effect on phosphorylation is conceivably most prominent with the triple arginine-to-alanine mutant VP30_3RA (Fig. 5A, dotted bars). Reduced phosphorylation of VP30 does not play an important role in producer cells since the VP30_AA mutant is also perfectly functional under these conditions (16). However, recently published data revealed that reversible phosphorylation of VP30 is essential under conditions of primary transcription (38). Therefore, effects of the arginine mutations on phosphorylation might be negligible in the producer cells (secondary transcription) and become functionally important only during primary viral transcription in indicator cells (Fig. 5A, dotted bars) (38). Observed differences between the single arginine mutants and the 3RA mutant in primary transcription might be caused by differences in phosphorylation.
Analysis of the RNA binding activity of the functionally related M2-1 of RSV showed that simultaneous exchange of two arginines to alanines in the putative RNA binding region decreased RNA binding affinity of M2-1 less than 3-fold, that is, to an extent similar to that seen for VP30_3RA (26, 36). Two double mutant variants (M2-1_R3A/R4A and M2-1_K150A/R151A) decreased reporter transcription to 20 to 30% already in producer cells. We observed a similar effect in producer cells when using a replication-deficient minigenome (Fig. 5C) and saw a strong transcription defect of the 3RA mutant in primary transcription (Fig. 5A, dotted bar). These observations point to similar overall contributions of positively charged amino acid side chains to M2-1 and VP30 function. However, it should be noted that RNA binding by VP30 and M2-1 is difficult to compare, as M2-1 lacks the N-terminal ∼65-aa domain harboring the analyzed arginine cluster of VP30 (Fig. 4A), such that only the Zn finger and core domain confer RNA binding in the case of M2-1 (27, 36).
With respect to the VP30_R40A variant, John et al. (15) reported a >4-fold Kd increase for RNA binding. In our study, this mutation did not change the appKd for RNA binding to a significant extent, similar to the other two single R-to-A mutants (Fig. 4B; Table 1). However, VP30_R40A showed a very low fraction of C1 complexes at the highest VP30 concentration (F2.7μM) using antigenomic RNA as the substrate (Fig. 4D; Table 2). Combined with the transcriptional defect of VP30_R40A in producer cells transfected with a replication-deficient minigenome (Fig. 5C), the available data shown here and reported previously (15) suggest a key role for R40 in VP30 function.
VP30_DD and VP30_3RA mutants showed similar RNA binding affinities (appKd, ∼0.7 μM) (Table 1). However, the DD mutant essentially lacked any transcriptional support activity in producer cells transfected with replication- and transcription-competent minigenomes (11, 18), whereas VP30_3RA retained substantial activity under identical conditions (Fig. 5A). The more severe phenotype of the DD mutant may be explained by the influence of phosphorylation on several of the protein's functions. On the one hand, VP30 phosphorylation impairs binding to VP35 and, on the other hand, favors binding to NP. While we have shown here that interaction of VP30 and VP35 is RNA dependent, the interaction of VP30 with NP is not (13). It is thus conceivable that enhanced binding of VP30 to NP in its phosphorylated or phosphomimetic state (VP30_DD) acts as a trap to withdraw VP30 from transcription start sites and transcription complexes. This may exacerbate the phenotype of VP30_DD relative to VP30_3RA, although both mutant proteins display similar RNA-binding defects.
The two N-proximal serine clusters (aa 29 to 31 and aa 42, 44, and 46) (Fig. 1D) have been shown to undergo dynamic phosphorylation (16). Phosphorylation of VP30 downregulates RNA binding (this study) and transcriptional support activity (11). This is similar to findings for M2-1 of RSV, for which phosphomimetic mutants showed comparable impairments in RNA binding and transcriptional activation (27). In contrast to the EBOV system, where VP30_AA supported transcription as strongly as wild-type VP30 (11, 18), an M2-1 mutant (M2-1_S58AS61A) mimicking the unphosphorylated state showed decreased transcriptional activation. This finding led the authors to conclude that constitutively charged or uncharged M2-1 mutants are unable to sustain optimal levels of viral transcription (27) and indicated that dynamic phosphorylation might be important for efficient transcription. While dynamic phosphorylation might also be important for VP30, the difference between RSV and EBOV with respect to the effect of a permanently unphosphorylated state likely reports structural and functional differences of the two proteins. First, the VP30 phosphorylation domain and the overlapping arginine cluster are located in the protein's N-terminal extension, which is absent from M2-1 proteins. Thus, the spatial organization of phosphorylation sites is different in VP30 and M2-1 proteins. Second, RSV and related Paramyxoviridae encode a second protein, M2-2, involved in regulating transcription. M2-2 promotes a regulatory switch from transcription to replication (39).
Phosphorylation of VP30 has a negative effect not only on RNA binding (this study) but also on interaction with the polymerase cofactor VP35 (11). Here, we were able to show that association of VP30 and VP35 is sensitive to RNase treatment (Fig. 6A), suggesting that their interaction is mediated by viral RNA. Remarkably, we were able to demonstrate that complex formation of VP30 and VP35 is also dependent on the ability of VP35 to bind dsRNA (Fig. 6C). So far, dsRNA binding of VP30 has been primarily assigned to the protein's role as an interferon antagonist (30, 32, 40). While the natural dsRNA substrate(s) of VP35 is currently unknown, it is tempting to speculate that both VP30 and VP35 concertedly interact with the viral RNA during the formation of transcription complexes. Further experiments are needed to investigate this hypothesis.
Interestingly, the interaction domains of M2-1 for the viral RNA and the P protein (VP35 analog) overlap, and both ligands compete for binding to M2-1 (37, 41). Despite commonalities (see above), M2-1 and VP30 seem to differ at this point, since complex formation of VP30 and VP35 requires the presence of RNA, making it unlikely that binding of RNA to VP30 and binding of VP35 to VP30 are mutually exclusive. This also implies different mechanisms of VP30 and M2-1 function in viral transcription. M2-1 of human metapneumovirus (hMPV) was found (36) to form an asymmetric tetramer with three subunits in a closed conformation and with the fourth subunit in an open conformation (defined by the relative orientation of the Zn finger and core domains). The proposed model suggests that one protomer of the tetramer can bind RNA (which stabilizes the closed conformation) while the other three protomers interact with the P protein to tether M2-1 to hMPV transcription complexes.
Based on the data presented here, we propose a refined model of VP30-driven activation of EBOV transcription by binding to the negative-sense vRNA (Fig. 7). We are aware that it is not yet fully clear if VP30 binds the genomic template RNA and/or nascent transcripts. However, at present we favor the idea of VP30 binding to the genomic RNA, for several reasons: first, we do not see any preferential binding of VP30 to positive-sense mRNA substrates (Fig. 1C and 4B and D); second, VP30 and VP35 interact in an RNA-dependent manner and VP35 is an integral part of the viral polymerase, which is more easy to reconcile with a model that invokes VP30 binding to the template RNA. Also, VP30 binding to mRNAs would require to newly recruit the protein (after its release from the template RNA together with the mRNA) at every internal transcription initiation site of the viral genome (18), which would compromise the efficiency of the transcription process.
FIG 7.

Model of EBOV transcription initiation. Nonphosphorylated (transcriptionally active) VP30 hexamers form a ternary complex with the viral leader RNA (black line) and VP35. The VP30-VP35 interaction depends on RNA binding as shown in this study. We propose that VP30 clamps the RNA template and VP35/L together, thereby increasing the polymerase's affinity for the (−) RNA template to increase the rate of productive transcription initiation events despite the presence of RNA secondary structures. In the absence of VP30 or upon VP30 phosphorylation (below), VP35/L polymerase complexes bind to the RNA template without VP30, but binding is weaker (more prone to dissociation) and may occur in a nonproductive manner (stalled complexes). As a consequence, polymerases predominantly fail to initiate productive transcription in the presence of stable RNA secondary structures. VP35 is shown here as a trimer but may also form functional tetramers (28, 42).
According to our model, VP30 hexamers with no phosphorylation in the serine cluster form a ternary complex with the viral leader RNA and VP35, the latter being part of the polymerase complex (VP35/L). This stabilizes transcription initiation complexes that are otherwise destabilized or stalled by the formation of secondary structures at the transcriptional start site (8, 17). We propose that VP30 clamps the RNA template and VP35/L together to increase the polymerase's affinity for the (−) RNA template, either to prevent the enzyme from dissociation or to stabilize a conformation of the enzyme that allows productive transcriptional initiation in the presence of secondary structures in the template or at the transcript 5′ end (Fig. 7, top). In the absence of VP30 or in the presence of phosphorylated VP30, the fraction of polymerases that successfully initiate transcription is largely decreased (Fig. 7, bottom). This model is supported by the findings of Weik et al. (17), who showed that mutations in the viral 3′ leader weakening the secondary structure at the transcriptional start site enabled VP35/L to transcribe viral mRNA in the absence of VP30. The same or a similar mechanism might be important to reduce the fraction of polymerase complexes that dissociate from the template at each successive gene upon termination of mRNA synthesis instead of reinitiating transcription (18). Thus, we seem to be faced with a polymerase/template complex that is prone to lose its grip on the RNA template in its transcription mode. With VP30, EBOV evolved a transcriptional support factor that keeps the polymerase on “track” through concerted interaction with the template RNA and VP35. It appears plausible in such a context that transcriptional blocks can be cleared by destabilizing the RNA secondary structures that otherwise inhibit the polymerase from productive transcriptional initiation, which then obviates the absolute need for the participation of VP30, as demonstrated in reference 17.
ACKNOWLEDGMENTS
We acknowledge experimental and technical support from Yodita Asfaha, Daniel Langfermann, Jens Dorna, Astrid Herwig, and Michael Krybus.
This work was supported by the German Research Foundation (DFG), grant CRC 1021, and the German Centre for Infection Research (DZIF) to N.B. and S.B.
REFERENCES
- 1.Bukreyev AA, Chandran K, Dolnik O, Dye JM, Ebihara H, Leroy EM, Mühlberger E, Netesov SV, Patterson JL, Paweska JT, Saphire EO, Smither SJ, Takada A, Towner JS, Volchov VE, Warren TK, Kuhn JH. 2014. Discussions and decisions of the 2012-2014 International Committee on Taxonomy of Viruses (ICTV) Filoviridae Study Group, January 2012–June 2013. Arch Virol 159:821–830. doi: 10.1007/s00705-013-1846-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahanty S, Bray M. 2004. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect Dis 4:487–498. doi: 10.1016/S1473-3099(04)01103-X. [DOI] [PubMed] [Google Scholar]
- 3.WHO Ebola Response Team, Agua-Agum J, Ariyarajah A, Aylward B, Blake IM, Brennan R, Cori A, Donnelly CA, Dorigatti I, Dye C, Eckmanns T, Ferguson NM, Formenty P, Fraser C, Garcia E, Garske T, Hinsley W, Holmes D, Hugonnet S, Iyengar S, Jombart T, Krishnan R, Meijers S, Mills HL, Mohamed Y, Nedjati-Gilani G, Newton E, Nouvellet P, Pelletier L, Perkins D, Riley S, Sagrado M, Schnitzler J, Schumacher D, Shah A, Van Kerkhove MD, Varsaneux O, Wijekoon Kannangarage N. 2015. West African Ebola epidemic after one year—slowing but not yet under control. N Engl J Med 372:584–587. doi: 10.1056/NEJMc1414992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falzarano D, Geisbert TW, Feldmann H. 2011. Progress in filovirus vaccine development: evaluating the potential for clinical use. Expert Rev Vaccines 10:63–77. doi: 10.1586/erv.10.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanchez A, Kiley MP, Holloway BP, Auperin DD. 1993. Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res 29:215–240. doi: 10.1016/0168-1702(93)90063-S. [DOI] [PubMed] [Google Scholar]
- 6.Weik M, Enterlein S, Schlenz K, Mühlberger E. 2005. The Ebola virus genomic replication promoter is bipartite and follows the rule of six. J Virol 79:10660–10671. doi: 10.1128/JVI.79.16.10660-10671.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volchkov VE, Volchkova VA, Chepurnov AA, Blinov VM, Dolnik O, Netesov SV, Feldmann H. 1999. Characterization of the L gene and 5′ trailer region of Ebola virus. J Gen Virol 80:355–362. doi: 10.1099/0022-1317-80-2-355. [DOI] [PubMed] [Google Scholar]
- 8.Mühlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. 1999. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol 73:2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mühlberger E. 2007. Filovirus replication and transcription. Future Virol 2:205–215. doi: 10.2217/17460794.2.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bharat TA, Noda T, Riches JD, Kraehling V, Kolesnikova L, Becker S, Kawaoka Y, Briggs JA. 2012. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc Natl Acad Sci U S A 109:4275–4280. doi: 10.1073/pnas.1120453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biedenkopf N, Hartlieb B, Hoenen T, Becker S. 2013. Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: impact on viral transcription and replication. J Biol Chem 288:11165–11174. doi: 10.1074/jbc.M113.461285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartlieb B, Mordof J, Mühlberger E, Klenk HD, Becker S. 2003. Oligomerization of Ebola virus VP30 is essential for viral transcription and can be inhibited by a synthetic peptide. J Biol Chem 278:41830–41836. doi: 10.1074/jbc.M307036200. [DOI] [PubMed] [Google Scholar]
- 13.Hartlieb B, Muziol T, Weissenhorn W, Becker S. 2007. Crystal structure of the C-terminal domain of Ebola virus VP30 reveals a role in transcription and nucleocapsid association. Proc Natl Acad Sci U S A 104:624–629. doi: 10.1073/pnas.0606730104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Modrof J, Becker S, Mühlberger E. 2003. Ebola virus transcription activator VP30 is a zinc-binding protein. J Virol 77:3334–3338. doi: 10.1128/JVI.77.5.3334-3338.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.John SP, Wang T, Steffen S, Longhi S, Schmaljohn CS, Jonsson CB. 2007. Ebola virus VP30 is an RNA binding protein. J Virol 81:8967–8976. doi: 10.1128/JVI.02523-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Modrof J, Mühlberger E, Klenk HD, Becker S. 2002. Phosphorylation of VP30 impairs Ebola virus transcription. J Biol Chem 227:33099–33104. [DOI] [PubMed] [Google Scholar]
- 17.Weik M, Modrof J, Klenk HD, Becker S, Mühlberger E. 2002. Ebola virus VP30-mediated transcription is regulated by RNA secondary structure formation. J Virol 76:8532–8539. doi: 10.1128/JVI.76.17.8532-8539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martínez MJ, Biedenkopf N, Volchkova V, Hartlieb B, Alzard-Dany N, Reynard O, Becker S, Volchkov V. 2008. Role of Ebola virus VP30 in transcription reinitiation. J Virol 82:12569–12573. doi: 10.1128/JVI.01395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abraham G, Banerjee AK. 1976. Sequential transcription of the genes of vesicular stomatitis virus. Proc Natl Acad Sci U S A 73:1504–1508. doi: 10.1073/pnas.73.5.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ball LA, White CN. 1976. Order of transcription of genes of vesicular stomatitis virus. Proc Natl Acad Sci U S A 73:442–446. doi: 10.1073/pnas.73.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoenen T, Groseth A, Kolesnikova L, Theriault S, Ebihara H, Hartlieb B, Bamberg S, Feldmann H, Ströher U, Becker S. 2006. Infection of naive target cells with virus-like particles: implications for the function of Ebola virus VP24. J Virol 80:7260–7264. doi: 10.1128/JVI.00051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wenigenrath J, Kolesnikova L, Hoenen T, Mittler E, Becker S. 2010. Establishment and application of an infectious virus-like particle system for Marburg virus. J Gen Virol 91:1325–1334. doi: 10.1099/vir.0.018226-0. [DOI] [PubMed] [Google Scholar]
- 23.Hoenen T, Jung S, Herwig A, Groseth A, Becker S. 2010. Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription. Virology 403:56–66. doi: 10.1016/j.virol.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Kolesnikova L, Berhöfer B, Bamberg S, Becker S. 2004. Multivesicular bodies as a platform for formation of the Marburg virus envelope. J Virol 78:12277–12287. doi: 10.1128/JVI.78.22.12277-12287.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schlereth J, Grünweller A, Biedenkopf N, Becker S, Hartmann RK. RNA binding specificity of Ebola virus transcription factor VP30. RNA Biol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez MJ, Volchkova VA, Raoul H, Alazard-Dany N, Reynard O, Volchkov VE. 2011. Role of VP30 phosphorylation in the Ebola virus replication cycle. J Infect Dis 204(Suppl 3):S934–S940. doi: 10.1093/infdis/jir320. [DOI] [PubMed] [Google Scholar]
- 27.Tanner SJ, Ariza A, Richard CA, Kyle HF, Dods RL, Blondot ML, Wu W, Trincao J, Trinh CH, Hiscox JA, Carroll MW, Silman NJ, Eléouet JF, Edwards TA, Barr JN. 2014. Crystal structure of the essential transcription antiterminator M2-1 protein of human respiratory syncytial virus and implications of its phosphorylation. Proc Natl Acad Sci U S A 111:1580–1585. doi: 10.1073/pnas.1317262111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reid SP, Cárdenas WB, Basler CF. 2005. Homo-oligomerization facilitates the interferon-antagonist activity of the ebolavirus VP35 protein. Virology 341:179–189. doi: 10.1016/j.virol.2005.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Möller P, Pariente N, Klenk HD, Becker S. 2005. Homo-oligomerization of Marburgvirus VP35 is essential for its function in replication and transcription. J Virol 79:14876–14886. doi: 10.1128/JVI.79.23.14876-14886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cárdenas WB, Loo YM, Gale M Jr, Hartman AL, Kimberlin CR, Martínez-Sobrido L, Saphire EO, Basler CF. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung DW, Prins KC, Borek DM, Farahbakhsh M, Tufariello JM, Ramanan P, Nix JC, Helgeson LA, Otwinowski Z, Honzatko RB, Basler CF, Amarasinghe GK. 2010. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat Struct Mol Biol 17:165–172. doi: 10.1038/nsmb.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zinzula L, Esposito F, Pala D, Tramontano E. 2012. dsRNA binding characterization of full length recombinant wild type and mutants Zaire ebolavirus VP35. Antiviral Res 93:354–363. doi: 10.1016/j.antiviral.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol 283:61–119. [DOI] [PubMed] [Google Scholar]
- 34.Fearns R, Collins PL. 1999. Role of the M2-1 transcription antitermination protein of respiratory syncytial virus in sequential transcription. J Virol 73:5852–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilusz CJ, Wilusz J. 2013. Lsm proteins and Hfq: life at the 3′ end. RNA Biol 10:592–601. doi: 10.4161/rna.23695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM. 2014. Drastic changes in conformational dynamics of the antiterminator M2-1 regulate transcription efficiency in Pneumovirinae. Elife 3:e02674. doi: 10.7554/eLife.02674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blondot ML, Dubosclard V, Fix J, Lassoued S, Aumont-Nicaise M, Bontems F, Eléouet JF, Sizun C. 2012. Structure and functional analysis of the RNA- and viral phosphoprotein-binding domain of respiratory syncytial virus M2-1 protein. PLoS Pathog 8(5):e1002734. doi: 10.1371/journal.ppat.1002734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biedenkopf N, Lier C, Becker S. 2016. Dynamic phosphorylation of VP30 is essential for Ebola virus life cycle. J Virol 90:4914–4925. doi: 10.1128/JVI.03257-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bermingham A, Collins PL. 1999. The M2-2 protein of human respiratory syncytial virus is a regulatory factor involved in the balance between RNA replication and transcription. Proc Natl Acad Sci U S A 96:11259–11264. doi: 10.1073/pnas.96.20.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prins KC, Binning JM, Shabman RS, Leung DW, Amarasinghe GK, Basler CF. 2010. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J Virol 84:10581–10591. doi: 10.1128/JVI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tran TL, Castagné N, Dubosclard V, Noinville S, Koch E, Moudjou M, Henry C, Bernard J, Yeo RP, Eléouet JF. 2009. The respiratory syncytial virus M2-1 protein forms tetramers and interacts with RNA and P in a competitive manner. J Virol 83:6363–6374. doi: 10.1128/JVI.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luthra P, Jordan DS, Leung DW, Amarasinghe GK, Basler CF. 2015. Ebola virus VP35 interaction with dynein LC8 regulates viral RNA synthesis. J Virol 89:5148–5153. doi: 10.1128/JVI.03652-14. [DOI] [PMC free article] [PubMed] [Google Scholar]