

ABSTRACT
Several experiments suggest that in the chronic phase of human immunodeficiency virus type 1 (HIV-1) infection, CD8+ cytotoxic T lymphocytes (CTL) contribute very little to the death of productively infected cells. First, the expected life span of productively infected cells is fairly long, i.e., about 1 day. Second, this life span is hardly affected by the depletion of CD8+ T cells. Third, the rate at which mutants escaping a CTL response take over the viral population tends to be slow. Our main result is that all these observations are perfectly compatible with killing rates that are much faster than one per day once we invoke the fact that infected cells proceed through an eclipse phase of about 1 day before they start producing virus. Assuming that the major protective effect of CTL is cytolytic, we demonstrate that mathematical models with an eclipse phase account for the data when the killing is fast and when it varies over the life cycle of infected cells. Considering the steady state corresponding to the chronic phase of the infection, we find that the rate of immune escape and the rate at which the viral load increases following CD8+ T cell depletion should reflect the viral replication rate, ρ. A meta-analysis of previous data shows that viral replication rates during chronic infection vary between 0.5 ≤ ρ ≤ 1 day−1. Balancing such fast viral replication requires killing rates that are several times larger than ρ, implying that most productively infected cells would die by cytolytic effects.
IMPORTANCE Most current data suggest that cytotoxic T cells (CTL) mediate their control of human immunodeficiency virus type 1 (HIV-1) infection by nonlytic mechanisms; i.e., the data suggest that CTL hardly kill. This interpretation of these data has been based upon the general mathematical model for HIV infection. Because this model ignores the eclipse phase between the infection of a target cell and the start of viral production by that cell, we reanalyze the same data sets with novel models that do account for the eclipse phase. We find that the data are perfectly consistent with lytic control by CTL and predict that most productively infected cells are killed by CTL. Because the killing rate should balance the viral replication rate, we estimate both parameters from a large set of published experiments in which CD8+ T cells were depleted in simian immunodeficiency virus (SIV)-infected monkeys. This confirms that the killing rate can be much faster than is currently appreciated.
INTRODUCTION
The role that cytotoxic T cells (CTL) play in controlling human immunodeficiency virus type 1 (HIV-1) infection is poorly understood (1, 2). Genetic associations with a limited number of protective human leukocyte antigen (HLA) alleles (3) suggest that they can control the infection to very low viral loads in a small subset of patients called “elite controllers.” The fact that, during acute infection, HIV-1 tends to evolve several immune escape mutations suggests that in this early phase, there is a strong selection pressure to evade the CTL responses (4–7; but see Roberts et al. [8]). Finally, the depletion of CTL with monoclonal antibodies to CD8 leads to marked increases in the viral load (9–15). CTL can protect by killing infected cells and/or by various nonlytic mechanisms, including the secretion of gamma interferon (IFN-γ) and macrophage inflammatory protein 1α (MIP-1α) and MIP-1β (16, 17, 18). The relative contributions of these two mechanisms in controlling HIV-1 infection are debated (11, 18–26).
Several lines of evidence suggest that CTL hardly kill CD4+ T cells that are productively infected with HIV-1. First, the death rate of productively infected cells was estimated by the initial downslope of the viral load during successful antiretroviral treatment (ART) (27, 28); this downslope, δ, is remarkably independent of the viral load and the CD4+ T cell count (29) and is currently estimated to be about δ = 1 day−1 (30). If this downslope indeed reflects the rate at which productively infected cells die, the killing rate would have to be slower than one per day (31, 32). Second, and even more striking, it was shown that the prior depletion of CD8+ T cells by monoclonal antibodies hardly affects the downslope of the viral load during ART (11, 12). Hence the death rate, δ, of productively infected cells is hardly influenced by the absence of CD8+ T cells, which suggests that CTL hardly kill, and that the major effect of CTL is nonlytic (11, 22, 24). Similarly, during acute infection, the downslope following the peak in the viral load is hardly affected by the presence of many cognate CD8+ T cells (33, 34), and it is puzzling why the peak viral load, which is a measure of poor immune control, correlates positively with the downslope (35). Third, the very low rate at which most viral mutants escaping a chronic CTL response take over the viral quasispecies (8, 36) suggests that the CTL response kills only a small fraction of the productively infected cells (36). Additionally, cells infected with virus that have escaped a CD8+ T cell response do not live longer than cells infected with wild-type virus (20). These data have typically been analyzed with the conventional model of HIV-1 infection, and this modeling confirms that CTL-mediated killing rates have to be much slower than one per day. Since CTL also have nonlytic effects (16–18), these slow killing rates could be true and be consistent with an important role of CTL in the control of HIV-1 infection.
Our main result is that these interpretations need to be revised when the conventional model is extended with an eclipse phase preceding the stage during which infected cells actively produce virus particles. Whenever cells in the eclipse phase and cells actively producing virus differ in their susceptibility to killing by CTL, this extended model requires much faster killing rates. To compensate for the slow killing in the unsusceptible phase, the killing in the other phase has to be relatively fast. In the extended model, neither the viral downslopes, δ, nor the immune escape rates are expected to reflect the killing rate. We show that the rapid upslopes of the viral loads that were observed following the depletion of CD8+ T cells (9–12, 14, 15) should reflect the effective replication rate, ρ, of the virus at the chronic set point. Since the killing rate should balance this replication rate during the chronic steady state, we can compute the killing rates from the observed upslopes of the viral loads. The rapid killing rates that we obtain are perfectly consistent with the apparently contradictory lines of evidence discussed above. Because the extended model with an eclipse phase is more realistic than the conventional model and readily explains why the observed viral downslopes are independent of the viral load and the CD4+ T cell count (29), our conjecture is that, if CTL protect by killing infected cells, these killing rates are much faster than is currently appreciated.
MATERIALS AND METHODS
Conventional one-stage model.
Consider the general model for an HIV-1 infection, with CD4+ target cells (T), productively infected cells (I), virus particles (V), and n clones of cognate CD8+ T cells (Ei), i.e.,
| (1) |
| (2) |
| (3) |
| (4) |
for i = 1,2,…,n, and where F(T) and G(Ei,V) are functions defining the production of target cells and activation of immune effector cells. The parameter b is the infection rate, f is the fraction of cells that become productively infected, dI is the normal death rate of productively infected cells, ki is a mass action killing rate, p is the rate at which productively infected cells produce virus, dV is the rate at which virus is cleared, and dE is the death rate of cognate CD8+ T cells. After making the conventional quasi–steady-state assumption (QSSA), dV/dt = 0, we study the behavior of this system around its steady states by considering the core viral replication cycle defined by equation 2. By assuming that the dynamics of target cells and immune effector cells are slower than those of the productively infected cells, we replace T and in equation 2 by the presumed slow “constants” T̄ and K, respectively, and observe that the core of viral replication becomes a linear system obeying
| (5) |
where λ = f βT̄ − dI − K, β = bp/dV, and V = (β/b)I.
We define the effective replication rate of the virus as ρ = f βT̄ − dI and observe that steady state requires that the killing rate balances replication, i.e., K = ρ. The fact that the killing rate should reflect the viral replication rate in this very basic model generalizes several earlier papers arguing that the magnitude of the cellular immune response need not be reflected in the death rate of productively infected cells (19, 37–39). The replication rate of the virus during the initial phase of an acute infection is defined by ρ(0) = fβT(0) − dI, and the initial downslope of the viral load during an effective antiretroviral therapy (ART) setting β = 0 is δ = dI + K. Both have been estimated previously. Two recent studies in acutely infected patients estimate that ρ(0) ≃ 1 day−1 (40, 41), whereas an early study based on a limited number of patients and time points estimated that ρ(0) ≃ 1.6 day−1 (42). The latter resembles the replication rate found in macaques (34, 43–45). Since we will also consider the viral replication rate following the depletion of CD8+ T cells, which cannot be done in patients, we will parameterize our model on macaques by considering 1 ≤ ρ(0) ≤ 1.5 day−1 and discuss how the results translate to the probably somewhat slower viral replication in humans. The initial downslope of the viral load, δ, has been estimated in humans (27, 28, 46), and more recent estimates using combinations of drugs that better suppress residual viral replication (30) suggest that this downslope varies around δ ≃ 1 day−1. Very similar downslopes have been found in monkeys (11, 12, 45). Following the depletion of CD8+ T cells, which sets K = 0, the viral load will increase at its effective replication rate, ρ, which in this model reflects the killing rate before depletion because ρ = K (22). The initial replication rate of a viral mutant escaping 1 out of n equal immune responses and experiencing a fitness cost, c, in the chronic steady state is
| (6) |
The mutant is therefore only expected to take over whenever the fitness cost is smaller than the inverse breadth of the immune response, i.e., c < 1/n. The rate, λ′, at which immune escape mutants are expected to replace the wild type should therefore decrease when the breadth, n, of the immune response increases over the course of an infection (6, 36, 38, 39).
Two-stage model.
Several of these well-known kinetic properties of the one-stage model change when we allow for an eclipse phase of the infected cells. Let us therefore split the infected cell population of equation 2 into a subpopulation of recently infected cells that are not yet translating viral mRNA or producing virus (I1) and a subpopulation of productively infected cells (I2) that are actively producing virus, i.e., dV/dt = pI2 − dVV. By the same QSSA, we now obtain that β = bp/dV and that V = (β/b)I2, which leads to the model
| (7a,b) |
| (8a,b) |
In this model, 1/γ defines the average length of the eclipse phase, which is about 1 day (Table 1), and d1 and d2 represent the normal death rates of infected cells. The breadth of the immune response is again defined by n clones of CTL, and by setting the killing rates, k1i and k2i, we can define whether an immune response acts early (k1i > 0) and/or late (k2i > 0). We again study the behavior of this system around its steady states by considering the core viral replication cycle defined by equation 8. Replacing T and the two summation terms in equations 8a and b by the presumed slow “constants” T̄, K1, and K2, respectively, the core again becomes a linear system, now obeying
| (9) |
Since the core model of equation 8 is linear and has no feedbacks, the stable chronic steady state of the full model is established by the feedbacks in equations 7a and b, approaching a target cell availability, T̄, and killing rates, K1 and K2, that perfectly balance the effective viral replication rate.
TABLE 1.
Parameter setting of the two-stage modela
| Parameter | Value (day−1, unless otherwise noted) | Description |
|---|---|---|
| dT | 0.1 | Death rate of CD4+ target cells (range, 0.01–1; see Fig. A1) |
| s | 0.1 | Daily production of target cells (scaled, T̄ = s/dT = 1) |
| dE | 0.01 | Death rate of CD8+ effector (and/or memory) T cells |
| p | 1.01 | Proliferation of CD8+ effector T cells (p − dE = 1 day−1 [44]) |
| γ | 1 | 1/γ is the avg length of the eclipse phase [59, 60, 49] |
| ki | 1–100b | Maximum killing rate |
| ρ(0) | 1.5 | Initial viral replication rate (see equation 12) |
| hi | 0.001–0.01b | Half-saturation constant of the immune response |
| f | 1b | Fraction of cells surviving initial infection |
| d1 | 1 or 0.1 | Death rate of cells in the eclipse phase (early or late) |
| d2 | 1 or 2 | Death rate of productively infected cells (early or late) |
| β | 8.75 or 9.1 | Infection rate: equation 13 for ρ(0) = 1.5 day−1 (early or late) |
To remain consistent with the observed viral downslopes, δ ≃ 1 day−1, during ART, we use equations 14a and b to parametrize the model slightly differently when the killing is largely early or mostly late (see the text). For simplicity, all cells survive the initial infection in the two stage-model (i.e., f = 1); we consider abortive infections (52, 53), i.e., f = 0.1, in the three-stage model.
Not per day.
The general solution of equation 9 obeys where λ1,2 are the eigenvalues of the matrix in equation 9, v1,2 are the corresponding eigenvectors, and C1,2 are integration constants. Since the viral load remains proportional to the density of productively infected cells, V = (β/b)I2, this solution also provides the viral load following a perturbation of the steady state as where the constants c1,2 are complicated combinations of the parameters and the initial conditions, obeying c1 + c2 = V(0). The two eigenvalues are defined as
| (10) |
where Δ = d1 + γ + K1 − d2 − K2. Both eigenvalues are real, the positive root, λ1, is the dominant eigenvalue, and λ2 is negative. The eigenvector associated with the dominant eigenvalue is defined as
| (11) |
The intrinsic replication rate of the virus is now defined by the dominant eigenvalue, λ1, for the case where K1 = K2 = 0, i.e.,
| (12) |
which, apart from the constant parameters, still depends on the actual target cell density, T̄. This equation can be simplified by writing β in terms of ρ,
| (13) |
which we will use later to calculate the infection rate required for obtaining a desired replication rate. Note that the same expression was obtained previously by assuming that the ratio I1(t)/I2(t) rapidly approaches a quasi–steady state (47). The downslope of the viral load during ART is defined by both eigenvalues for the case where β = 0:
| (14a,b) |
which are both negative.
The steady state of equation 9 implies that the determinant of the matrix equals zero, which leads to the solution
| (15) |
(which can also be obtained by setting dI1/dt + dI2/dt = 0, or by setting λ1 = 0). We study the influence of CTL-mediated killing of infected target cells, I1 and I2, for three “extreme” cases: “equal” killing (K1 = K2), “early” killing (K2 = 0), and “late” killing (K1 = 0). This simplifies equation 15 into
| (16a,b,c) |
respectively. This confirms the intuition that, in the two-stage model, the total killing rate also balances viral replication. For K1 = K2 = ρ, this is obvious. For the other two cases, the f βT̄γ term is the rate at which new productively infected cells are formed per productively infected cell, which is divided by the loss rate of infected cells at the stage that is not killed, to deliver a net growth rate. This net growth rate has to be balanced by the killing and the natural loss of infected cells at the stage that is being killed.
Equation 15 can be written in terms of the replication rate, ρ, by substituting equation 13 for β:
| (17a,b) |
corresponding to
| (18a,b) |
for the early and late killing regimes. The latter two expressions demonstrate that it is most efficient to kill during the eclipse phase, when d2 > d1 + γ. Instead, when d2 < d1 + γ, killing during the production phase, K2, requires a smaller immune response than early killing (a similar observation was made by Althaus and De Boer [32]).
Three-stage model.
Realistic models of the HIV-1 life cycle probably require more than just two stages (48–51). The very early events after the virus has infected a target cell seem to be very important, because a large fraction of the cells dies by abortive infection (52, 53) and proteins from the incoming virus particle(s) trigger CD8+ T cell responses on a time scale of a few hours (54, 55). One could argue that most of the CTL-mediated killing during the eclipse phase that we considered in the two-stage model actually occurs in the first few hours after the infection of a target cell. For that reason, we here extend the model to three stages, where the first, I0, stage is thought to be short because cells rapidly enter the abortive program (at rate d0), rapidly proceed to the eclipse phase (at rate γ0), or are killed rapidly (at rate K0):
| (19a,b) |
| (20) |
| (21a,b) |
where equations 19a and b are identical to equations 7a and b, and equation 21b is identical to equation 8b. Again, n is the total number of immune responses, and we can set some of the killing rates to zero to allow for epitopes that are expressed at particular stages only.
The core viral replication cycle is now a linear model defined by a 3-by-3 matrix. One can again find the eigenvalues of this matrix (not shown), define the dominant eigenvalue for K0 = K1 = K2 = 0 as the effective replication rate, ρ, and write this in the same form as above, i.e.,
| (22) |
During ART, we set β = 0 and now find the following three eigenvalues:
| (23a,b,c) |
If d0 and γ0 are large (52, 53), this third eigenvalue will never be dominant, and the predicted downslope during ART will depend on the same two eigenvalues defined by equations 14a and b. The steady state of the linear core defined by equation 20 and equation 21 would require that
| (24) |
and if the early killing were the only form of killing, this would simplify into
| (25) |
Equation 24 can again be written in terms of the replication rate by substituting equation 22 to obtain
| (26) |
We add three notes to this equation. First, the scenario K0 = K1 = K2 = ρ is again a solution. Second, remember that d0 and γ0 are large (52, 53) and observe that K0 will have hardly any effect when K0 ≪ d0 + γ0. Similarly, and third, whenever (d0 + γ0 + ρ)/(d0 + γ0 + K0) ≃ 1, equation 26 will approach equation 17b.
Thus, when d0 and γ0 are large, the three-stage model makes predictions similar to those of the two-stage model. An alternative way to see this is to make the QSSA dI0/dt = 0, which seems valid because many cells are lost at an early stage (52, 53) and the early killing process is fast (54, 55), and define
| (27) |
to observe that the γ0I0 term in equation 21a becomes the fβTI2 of equation 8a, which simplifies the three-stage model into the two-stage model defined above. We will first analyze the two-stage model with f = 1 to show that our results do not depend on having abortive infections. At the last stage, where we aim to explain recent data, we use the more-realistic three-stage model with γ0/(γ0 + d0) = 0.1, concomitantly demonstrating that our general results would also be obtained when most of the infections are abortive.
Parameter values.
In the numerical simulations, we use the function F(T) = s – dTT to complete equation 7a for the target cells. Because not all CD4+ T cells are proper target cells for HIV-1 and true target cell densities are not known, we scale the maximum target cell density to 1 by setting s = dT. We typically set a relatively fast turnover of s = dT = 0.1 day−1 (Table 1), because activated CD4+ T cells are the best target cells (56) and these cells probably live for a shorter time than the average effector memory cell in HIV-1-infected patients, which have a turnover of about 0.02 per day (57, 58). The expected length of the eclipse phase is 1 day, i.e., γ = 1 day−1 (49, 59, 60). In the late killing regime, the model will only be consistent with the observed viral downslope of δ ≃ 1 day−1 when cells stay in the eclipse phase for about 1 day (see Results). Hence, the average life span of I1 cells should be more than a day, and we simply assume that they have the same expected life span of 10 days as target cells and set d1 = 0.1 day−1. In the early killing regime, we have to assume that cells in the eclipse phase die faster, and we set d1 = 1 day−1 (see Results and Table 1). The natural death rate of productively infected cells is not known (61), and we set it to d2 = 1 day−1 in the early killing regime and to d2 = 2 day−1 in the late killing scenario (see Results and Table 1). In the three-stage model, we take into account that most infected cells die rapidly by abortive infection (52, 53) and set d0 = 54 and γ0 = 6 day−1, such that f = 0.1, and in the two-stage model we set f = 1 for reasons of simplicity (which does affect the dynamics, but not our conclusions). The infection rate β is parameterized by requiring an initial replication rate ρ(0) = 1.5 day−1, using equation 13. In macaques, CD8+ effector T cells proliferate at a rate of one per day (44) and have an expected life span of several months to a year (62, 63). We give them an expected life span of 100 days, dE = 0.01 day−1, and correspondingly, set p = 1.01 day−1 (Table 1). Using the function
| (28) |
with V = I2 to complete equation 7b of the CTL (39, 47), we have tuned the saturation constants, hi, such that the immune responses approach their steady state in a few weeks (Fig. 1). For the low saturation constants that we use, the steady-state immune response of equation 7b, i.e., Ēi = V̄(p/dE − 1) − hi = 100V̄ − hi, implies that the magnitude of each individual immune response is more or less proportional to the viral load, V̄, and fairly independent of its saturation constant, hi, whenever hi < 100V̄ (47).
FIG 1.

Acute infection followed by ART at day 150 in the two-stage model of equation 8. The behaviors of the early killing regime (a and c) and of the late-killing scenario (b and d) are shown. Data are shown for 1 immune response, with k1 = 10 (a and b), and 10 immune responses, with ki = 1 for i = 1,2,…,10 (c and d). The rate at which the viral load, V ∝ I2, increases during acute infection is 1.5 day−1 in both cases, and the rate at which it decreases during ART is approximately δ = 1 day−1. We use the function F(T) = s − dTT to complete equation 7a for the target cells and set s = dT to scale the uninfected steady state to one and the function G(Ei,V) = pVEi/(hi + V + Ei) with V = I2 to complete equation 7b of the CTL (39, 47). We here set k1 = 10 day−1 or ki = 1 day−1, hi = i/1,000, and Ei(0) = 0.001/i for i = 1,2,…,n. For the early killing scenario, we set β = 8.75 and d1 = d2 = 1, and for the late-killing regime, we set β = 9.1, d1 = 0.1, and d2 = 2 day−1 (Table 1). By equation 13, both settings account for a realistic initial replication rate in macaques of ρ(0) = 1.5 day−1. See Table 1 for all other parameter values.
RESULTS
Downslopes of the viral load during ART.
In monkeys and patients treated with potent ART, the viral load in the peripheral blood decreases at a rate of δ ≃ 1 day−1 (30) for about a week. The mere fact that monkeys that were treated with ART following the depletion of their CD8+ T cells also had downslopes of δ ≃ 1 day−1 seems difficult to reconcile with the notion that the major protective effect of CTL is cytolytic in the one-stage model (11, 12). Two decades ago, Klenerman et al. (31) developed a model where the HIV-1-infected cells progress through several stages before they commence to produce viral particles (i.e., a model similar to equation 8) and demonstrated that the slope, δ, with which the viral load declines during ART is defined by the slowest time scale of the various stages of infected cells. Since we now know that the average length of the eclipse phase, 1/γ in our model, is about 1 day (49, 59, 60), their result is in excellent agreement with the general observation that δ ≃ 1 day−1 (30), that δ is independent of the viral load and CD4+ T cell count (29), and that δ remains similar in CD8+ T cell-depleted monkeys (11, 12).
Fifteen years later Althaus and De Boer (32) extended these results by showing that rapid killing during the early stage (K1 ≫ K2) is also compatible with all these observations if the expected life span of productively infected cells, 1/d2, is the slowest time scale of the model. The invariant downslope, δ, should then reflect the intrinsic death rate of productively infected cells, implying that d2 ≃ 1 and K2 ≪ d2. Rapid killing at the early stage could be due to the immune responses to proteins of the incoming virus (54, 55), and slow killing at the late stage could be due to downregulation of MHC expression by the late expression of Nef (32). Importantly, both killing regimes imply that the downslope, δ, during ART fails to provide information on the rate at which infected cells are killed (31, 32).
Both results are readily confirmed by equations 14a and b, providing the two negative eigenvalues of equation 10 for β = 0 and illustrating that the observed downslope of the viral load, V(t), during ART will be δ ≃ d1 + γ + K1 whenever |λ1| ≫ |λ2|, whereas the downslope will reflect δ ≃ d2 + K2 whenever |λ1| ≪ |λ2|. For simplicity, consider the three cases of equal killing, early killing, and late killing, i.e., K = K1 = K2, K2 → 0, and K1 → 0, respectively. For equal killing, i.e., |λ1| = d2 + K and |λ2| = d1 + γ + K, we would obtain either that δ ≃ d1 + γ + K or that δ ≃ d2 + K, and since both slopes depend on the killing rate, this is not in agreement with the observation that the downslope is unaffected by the depletion of CD8+ T cells (11, 12). Thus, a two-stage model with similar killing rates at both stages is—like the one-stage model—not compatible with the data (if CTL are considered to be killers).
Next consider late killing (31) by setting K1 = 0. Whenever d1 + γ ≪ d2 + K2, the observed downslope of the viral load, V(t), during ART will be δ ≃ d1 + γ, which is independent of the killing rate and would be in agreement with the general observation δ ≃ 1 day−1 (11, 12, 29, 30) when d1 + γ ≃ 1 day−1. Since δ ≃ 1 after CD8+ T cell depletion (11, 12), this late killing regime requires that d1 + γ ≪ d2 (which we obtain by setting d2 = 2 day−1, implying fairly rapid death of cells that produce virus; Table 1). Finally, consider early killing (32) by setting K2 = 0. Whenever d1 + γ ≫ d2, we obtain that δ ≃ d2. To be consistent with all data, this early killing regime therefore requires d2 ≃ 1 day−1, again implying rapid death of productively infected cells, and to be consistent with the CD8+ T cell depletion, we require d1 + γ > 1 day−1, suggesting fairly rapid death during the eclipse phase (52, 53). We realized this by setting d1 = d2 = 1 day−1 (Table 1).
A numerical confirmation of these results is depicted in Fig. 1, where an infection with n = 1 or n = 10 immune responses is treated with perfect ART (i.e., we set β = 0 at day 150). Although the killing rates at the pretreatment steady state are relatively fast and somewhat different, i.e., K1 ≃ 4.3 day−1 (Fig. 1a), K2 ≃ 4 day−1 (Fig. 1b), K1 ≃ 6.3 day−1 (Fig. 1c), and K2 ≃ 5.8 day−1 (Fig. 1d), the downslope of the viral load [here I2(t)] reflects the rate at which productively infected cells die, δ ≃ d2 = 1, in Fig. 1a and c, whereas it reflects the rate at which the early infected cells, I1(t), depart from the eclipse phase, i.e., δ ≃ d1 + γ ≃ 1.1 day−1, in Fig. 1b and d. If one were to add lines with an exponential downslope of 1 per day in Fig. 1a and c, or 1.1 per day in Fig. 1b and d, these lines would almost perfectly coincide with the straight green lines depicting the downslope of the viral load (I2) from day 150 onwards. Thus, the downslopes in the model are in excellent agreement with the observed downslopes of the viral load. Note that the 10 immune responses approach similar magnitudes (see the subsection on parameter values above) and that 10 independent immune responses with a 10-fold-lower killing rate, ki = 1, control somewhat better than a single response with k1 = 10 (this is due to the absence of direct competition between clones and the presence of intraspecific competition among CTL of the same specificity [39]). Finally note that the ratio I1/I2 is about 1 in the early killing regime and larger than 1 in the late killing regime, which can be understood from the killing of I1 cells in the early regime and of I2 cells in the late killing scenario.
To summarize, the downslopes of the viral load during ART are not expected to provide reliable information on the rate at which productively infected cells are killed by CD8+ T cells (26, 31, 32). Rapid killing is expected to be masked by the slower phases of the viral life cycle.
Downslope after the peak viral load.
Acute infections with HIV-1, simian immunodeficiency virus (SIV), and simian-human immunodeficiency virus (SHIV) are characterized by an initial phase of rapid viral replication [at rate ρ(0)] that ends with a peak viral load, which is followed by a phase during which the viral load declines fairly rapidly until it slowly approaches the viral set point (Fig. 1). The downslope following the peak (sometimes called α [40]) is typically estimated to vary around α = 1 day−1 (33, 35, 40) and, in vaccinated monkeys, is not significantly affected by the presence of a large cellular immune response (33, 34). The maximum downslope that can be obtained in our model is achieved when target cells are completely depleted and the immune responses are maximal. Since setting T = 0 in equation 10 is the same as setting β = 0, we can reuse equations 14a and b to predict the maximum downslope that would be approached shortly after complete target cell depletion. Similar to the situation after the onset of ART, in the early and late killing regime, the maximum downslope following the peak viral load is not expected to depend on the killing rates, K1 or K2, and would be d2 or d1 + γ, respectively. Thus, it is not surprising that similar downslopes were obtained in control and vaccinated monkeys (47), and an eclipse phase of about 1 day (49, 59, 60) would readily explain that the observed downslopes are less than α = 1 (47).
Petravic and Davenport (35) have studied the peak viral load, the nadir of CD4+ T cell numbers, and the downslope, α, of the viral load after the peak in macaques acutely infected with CXCR4-tropic SHIV (which should be able to infect all CD4+ T cells). Combining data from unvaccinated and vaccinated monkeys, they report a negative correlation between the nadir of the CD4+ T cells and the peak viral load and a positive correlation between the peak viral load and the downslope α. Because high viral loads are expected to reduce target cell numbers, the negative correlation seems a natural result (which nevertheless was not confirmed in a recent human study [41], probably because CD4+ T cell numbers are a poor measure of target cell availability for CCR5-tropic HIV-1). The positive correlation is more interesting because high viral loads should be associated with poor immune control and, hence, a longer life span of productively infected cells and not with a faster downslope. This positive correlation has therefore been used as evidence against cytolytic control by the CD8+ T cells (35). A positive correlation is indeed expected when the cellular immune response is not affecting the life span of productively infected cells, because a good immune response should be associated with a higher nadir of the target cells, enabling faster replication of the virus (35). By mathematical modeling, Petravic and Davenport (35) show that the correlation between the peak viral load and the downslope, α, should always be positive in models where the cellular immune control is nonlytic, whereas this relation should be nonmonotonic in models where the CTL are cytolytic. In the latter case, the correlation will be negative when the target cells are severely depleted, because then the downslope, α, reflects the death rate, δ, of productively infected cells. Although Petravic and Davenport (35) demonstrate that a strictly positive correlation is also expected when infected cells are killed during the eclipse phase (i.e., our early killing regime), they nonetheless suggest that vaccine-induced CD8+ T cells control SHIV infection by noncytolytic means.
We have seen in the discussion above that, even in the late killing regime, the downslope, α, need not represent the rate at which productively infected cells are killed and, instead, should reflect the time cells spend in the eclipse phase. We therefore repeat the analysis of Petravic and Davenport (35) for the in silico acute infection depicted in Fig. 1b. To account for vaccination, we vary the number of effector cells, E(0), at the onset of the infection and record the peak viral load, nadir of the target cells, and the downslope of the viral load after the peak (Fig. 2). These results agree with the observed negative correlation between the nadir of the target cells and the peak in the viral load (Fig. 2c) and the predicted nonmonotonic relation between the downslope and the peak viral load (Fig. 2a). Note that the downslope is largely determined by the availability of target cells, as the data for different rates of target cell turnover, s = dT, more or less fall on the same line in Fig. 2b. Although the relation between peak viral load and the downslope is nonmonotonic in our model, the region where this correlation is negative is small (on a log scale), has little effect on the downslope, and is confined to cases with low initial numbers of effector cells, i.e., E(0) ≤ 0.001 (Fig. 2a). This suggests that the correlation should be positive among vaccinated animals and when vaccinated and control animals are mixed but should be small or ambiguous in control animals (with severe target cell depletion and high downslopes). We reanalyzed the data in the study of Petravic and Davenport (35) by performing the same Spearman correlations on these subsets of the data and confirmed this prediction; i.e., r = 0.5 with P = 0.03 in vaccinated monkeys and r = 0.29 with P = 0.31 in control animals. To summarize, the two-stage model with late killing also accounts for an overall positive correlation between peak viral load and the downslope, α, and observing a positive correlation provides little evidence on how CD8+ T cells control SHIV infection.
FIG 2.

Relationships between the peak viral load, nadir of target cells, and downslope of the viral load after the peak. To mimic the effect of vaccination, we study acute infection in the late killing regime (as in Fig. 1b, with n = 1, h1 = 0.001, and k1 = 10), now varying the initial number of effector cells from E(0) = 0.0001, 0.0005, 0.001, 0.005, … , 0.01, 0.05, 0.1, 0.2, 0.3, 0.4 to E(0) = 0.5. These initial conditions can be recognized in the figure because the peak viral load (i.e., I2max) decreases with the E(0) (a, arrows) and the nadir of the target cells (i.e., Tmin) increases with E(0). During the first 2 weeks of the infection, we record the peak viral load and the nadir of the target cells, and we estimate the downslope of the viral load after the peak by linear regression on the natural logarithms of the I2 values (over the first 4 days following the peak). We repeat this procedure for different turnover rates of the target cells (i.e., s = dT = 0.01, 0.1, and 1 day−1; see the key). The darkness of the symbols depicts the nadir of the target cells (where bright means Tmin = 1 and black means Tmin = 0; in panels b and c, the nadir can also be read from the x or y axis). (a) The correlation between the peak viral load and the downslope of the viral load after the peak is positive over a domain of peak viral load values [and is only negative for E(0) ≤ 0.001, i.e., in “unvaccinated” systems]. (b) The correlation between the nadir of the target cells and the downslope is largely negative, which was to be expected because higher availability of target cells decreases the downslope. (c) The peak viral load and the nadir of the target cells are negatively correlated. Similar results are obtained for k = 1 and k = 100 (not shown).
Similarly, the fact that wild-type SHIV and immune escape SHIV mutants decline at similar rates following the peak viral load (20) should not be used as evidence against cytolytic control. Even if cells productively infected with wild-type SHIV are killed much faster than those infected by a mutant that has escaped from a cytolytic CD8+ T cell response, their corresponding viral load would have a very similar rate of decline when the killing is either largely early or largely late (equations 14a and b).
Upslopes of the viral loads.
Given the fact that the downslopes of the two-stage model cannot be used to estimate killing rates, we now turn to the upslopes of the viral loads observed during acute infection, following CD8+ T cell depletion (11, 12), and during immune escape (36). The rate at which the viral load increases in the two-stage model of equation 8 is determined by the dominant eigenvalue defined in equation 10, which depends on the availability of target cells, T̄, and the total killing rates, K1 and K2.
(i) Viral replication during the acute phase of the infection.
The initial rate of expansion of the viral load during the acute phase of the infection before the onset of immune responses is defined by ρ(0), i.e., equation 12 with T̄ = T(0). In Materials and Methods, we estimate that ρ(0) ≃ 1 day−1 in humans (40, 41) and that ρ(0) ≃ 1.5 day−1 in macaques (34, 43–45). Equation 13 allows us to define the effective infection rate, β, delivering the required expansion rate (47). For instance, to obtain the observed expansion rate in macaques of ρ(0) ≃ 1.5 day−1 in the late-killing model, we substitute its parameters d1 = 0.1, d2 = 2, and γ = 1 into equation 13 to find that β = 9.1/[fT(0)] = 9.1 day−1 [because fT(0) = 1]. Similarly, for the early killing model, with d1 = d2 = γ = 1, we find that β = 8.75/[fT(0)] = 8.75 day−1. Since the eigenvector associated with the dominant eigenvalue defines the ratio I1(t)/I2(t), we can also substitute T̄ = T(0) and K1 = K2 = 0 into equation 11 and use the same two sets of parameters to obtain, in the acute phase of the infection, the ratios I1(t)/I2(t) = 2.5 and I1(t)/I2(t) = 3.5 for the early and late killing models, respectively. The four numerical examples depicted in Fig. 1 indeed have an initial expansion rate of ρ ≃ 1.5 day−1 until the viral load approaches its peak value.
(ii) Viral replication during the chronic phase of the infection.
The effective replication rate, ρ, during chronic infection will not be the same as the ρ(0) computed above, because the availability of target cells, T̄, is no longer defined by T(0). T̄ could be lower, due to target cell depletion, or higher, due to immune activation. The chronic viral replication rate can be estimated by a perturbation of the chronic steady state, setting the CTL killing rate to 0, which has been achieved in several experiments by depleting the CD8+ T cells (9–15). The viral load increases steeply in these experiments over a period of several days, and in all three models, this slope should reflect the effective replication rate, ρ, during the chronic steady state. In the Appendix, we perform a meta-analysis of these data to estimate the rate at which the viral load increases. We find that, typically, ρ ≃ 0.5 day−1 (9–15) and that ρ ≃ 1 day−1 in monkeys with very low viral loads (13–15).
(iii) Estimating the killing rates from the observed effective replication rate.
Knowing that during chronic infection, 0.5 ≤ ρ ≤ 1 day−1, we can use equation 17 to compute the killing rates that are required to keep the infection at steady state (Fig. 3). For the maximum effective replication rate of ρ = 1 day−1, equation 17 would predict killing rates of K1 = K2 = 1, K1 = 4, and K2 = 3.7 day−1, for the equal, early, and late killing regimes, respectively (Fig. 3a). For the minimum replication rate, ρ = 0.5 day−1, these would be K1 = K2 = 0.5, K1 = 1.75, and K2 = 1.6 day−1, respectively.
FIG 3.

Killing rates predicted by equation 17. (a) The red line depicts K1 for K2 = 0 as a function of ρ, the blue line K2 for K1 = 0 (see equations 18a and b, respectively), and the black line is just the line K = K1 = K2 = ρ. (b) K2 is depicted as a function of K1 for ρ = 1, using equation 17b for the early killing parameters, d1 = γ = d2 = 1 (in red), and for the late killing parameters, d1 = 0.1, γ = 1, and d2 = 2 day−1 (in blue). Note that we indeed predict that the lines cross at K1 = K2 = ρ = 1. The lines are solid when 0.5 < δ < 1.5 and dashed otherwise. (c) We make small steps through the parameter space for 0.01 ≤ d1, d2 ≤ 5, and 0.5 ≤ ρ ≤ 1, drawing a random value for either K1 or K2 from a uniform distribution, 0 < K1, K2 < 10, and use equation 17a or b to compute the other killing rate. This analysis is performed for γ = 1, and we increase the parameters by 10% when we step through the parameter space. Here, we are more strict and only accept solutions when 0.75 < δ < 1.25 (in the presence and absence of CTL).
Equation 17 enables us to compute the early and late killing rates for any value of the effective replication rate (Fig. 3b). However, not every combination of K1 and K2 will be in agreement with the observed downslope, δ = 1 day−1, during ART (32). Since δ is defined by the dominant eigenvalue of equations 14a and b, i.e., δ = min(|λ1|, |λ2|), we can check which combinations of K1 and K2 obey the observation δ ≃ 1. Points obeying the condition 0.5 < δ < 1.5 are therefore depicted by the heavy lines in Fig. 3b, and all “incorrect” points on the lines defined by equations 17a and b are shown as dashed lines. We observe that the combination of the condition that δ ≃ 1 during ART (in the presence and absence of CTL) and the highest observed effective replication rate, ρ ≃ 1 day−1, strongly constrains the estimated killing rates. First, the killing has to be either mostly early or largely late, as similar killing rates fail to satisfy the δ ≃ 1 condition. Second, we see that for ρ = 1 day−1, the required killing rates are fairly large, i.e., K1 > 2 day−1 and K2 > 2.3 (Fig. 3b).
We perform a similar analysis by stepping through the parameter space with small increments for d1, d2, and ρ, drawing a random value for either K1 or K2 from a uniform distribution, 0 < K1, K2 < 10, and using equation 17 to compute the other killing rate (Fig. 3c). Drawing 2.6 × 106 parameter combinations, we find that about 1% of these are in agreement, with a slope of 0.75 < δ < 1.25 during ART (in the presence and absence of CTL). The fact that most symbols are located along the axes of Fig. 3c shows that the killing rates, K1 and K2, cannot both be high when one selects points in agreement with the observed downslopes, In the early and late killing regimes, the killing rates can be quite high, i.e., 1 < K1, K2 < 6 day−1 (Fig. 3c). Note that equation 17 is independent of the functions F(T) and G(Ei,V) and that these estimates should therefore be relatively generic. Finally, the highest killing rates in Fig. 3c correspond to the parameter settings in which the virus is replicating fast (large ρ) and target cells are dying fast (large d1 and/or d2), such that the window of opportunity for the CTL to kill a target cell is short. Since we require the behavior of the model to be consistent with the observed downslopes, such high killing rates cannot be excluded. However, if the eclipse phase and the production phase both take about a day, the killing rates should vary between 1.5 < K1, K2 < 4 day−1 (Fig. 3a and b).
(iv) Revisiting the upslope following CD8 depletion.
Elemans et al. (22) analyzed the data from CD8 depletion experiments with a variety of models, arguing that the initial rate of increase of viral load reflects the rate of killing prior to the removal of the CTL. For a single-stage infection, this is true, but for the two-stage infection model, this is more complicated. For the late-stage killing scenario, one readily observes from the steady state of dI2/dt in equation 8 that γI1 − d2I2 = K2I2 before CD8+ T cell depletion and, hence, that dI2/dt = γI1 − d2I2 = K2I2 immediately afterwards. Thus, in the late-stage killing scenario, the initial upslope of the viral load is indeed expected to reflect the total killing rate. For the early-stage killing scenario, one solves from dI1/dt = 0 that Ī1 = fβTI2/(γ + d1 + K1). Substitution, and setting K1 = 0 to account for the CD8 depletion, give
| (29) |
which defines a growth rate that is not equal to K1 (equation 16b) and not equal to ρ (equation 18a). Actually, the term between the brackets is equal to K2 defined by equation 16c, showing that the upslope in the early killing regime reflects what the killing rate would have been if the killing were “late only.”
However, in a two-stage model, the upslope is defined by two eigenvalues, defining both an initial transient and the ultimate upslope defined by the dominant eigenvalue. The two upslopes calculated above define the short initial transient before the upslope approaches the dominant eigenvalue, ρ. The characteristic length of this initial transient is determined by the negative eigenvalue of equation 10, and at time zero in Fig. 4a and b, we compute numerically that λ2 = −4.2 and λ2 = −4.4 day−1, respectively. The numerical simulations in Fig. 4 confirm this and show that the regime where the viral load increases at a rate of ρ ≃ 1 day−1 is approached in about half a day and lasts for a few days, until the target cells levels start dropping. The rate at which the viral load increases over a period of a few days will therefore not reflect the killing rate but will rapidly approach the replication rate, ρ, in the chronic steady state. Fortuitously, this means that the CD8 depletion data provide estimates for ρ (see the meta-analysis in the Appendix). The simulations depicted in Fig. 4 look very similar to the CD8 depletion experiments fitted by Elemans et al. (22). We have previously fitted the two-stage model to the same data sets and found that the model can describe the data well for a wide range of parameter values (26, 64). Statistically speaking, fitting such a complicated model to such sparse data is not informative because most parameters turned out to be unidentifiable (not shown).
FIG 4.

CD8+ T cell depletion at day zero is followed by ART at day three (11, 12). We start the model in the chronic states from Fig. 1 (using the same functions and parameters). The dashed line provides the rate of increase of the productively infected cells, dI2/dt/I2, and confirms that the initial rate at which the viral load increases reflects the steady-state killing rate in the late killing regime (K = 4.0) (b) but not in the early killing regime (a). In both regimes, the rate at which the viral load increases rapidly approaches the effective replication rate of ρ ≃ 1 day−1, until the target cell numbers decline. Due to our design of the parameters d1 and d2, the downslope of the viral load during ART remains δ ≃ 1 day−1 in both regimes (see Table 1).
Estimating killing rates from the observed immune escape rates.
HIV-1 readily escapes from CD8+ T cell immune responses by mutations in the major histocompatibility complex (MHC) class I restricted epitopes. The rate at which viral variants carrying one or several immune escape mutations take over the viral quasispecies has been used to estimate the selective pressure imposed by the immune response the virus is escaping from (reviewed by Regoes et al. [65]). These replacements are typically fast during acute infection (6, 7, 66) but tend to be slow during chronic infection (8, 36, 67). Since in our model, the immune responses approach a similar magnitude (Fig. 1), the killing rate of a viral variant that just escaped one immune response is approximately the fraction (n − 1)/n of that of the wild-type virus. The initial selection coefficient of such a variant is defined by its replication rate in the chronic steady state that is still largely determined by the wild-type virus experiencing n immune responses. Since the replication rate is defined by the dominant eigenvalue of equation 10, we again use equation 13 to replace the fβT̄γ term and obtain for the effective replication rate of a viral strain escaping one immune response during chronic infection
| (30) |
where with K1 and K2 defined by equations 18a and b (if the epitope was expressed both early and late).
The initial expansion of the mutant obeys M(t) = M(0)eλ′t, and since the wild type virus initially remains at steady state (i.e., λ ≃ 0), λ′ provides the rate at which the mutant replaces the wild type, i.e., the escape rate. This means that we can predict the escape rate as a function of ρ, and the breadth of the immune response n, in a manner that is independent of the form of the functions F(T) and G(Ei,V) (Fig. 5). Even for a rapid effective replication rate requiring a strong immune response, e.g., ρ = 1 requiring K1 = 4 or K2 = 3.72 day−1 (Fig. 3), we predict escape rates around λ′ = 0.1 day−1 for immune responses with a breadth of n > 5 in both the early and late killing regimes (Fig. 5). Unlike the simple λ′ ≃ ρ/n = K/n of the single-stage model (equation 6), with equation 30, we obtain escape rates that are much smaller than K1/n or K2/n and even somewhat smaller than ρ/n (Fig. 5). Thus, slow immune escapes are perfectly consistent with rapid killing rates because the escape rate is not reflecting the breadth-weighted killing rate, K/n, but on average defines a lower bound on the breadth-weighted replication rate, ρ/n. Given that viral replication is fairly slow, i.e., 0.5 < ρ < 1 day−1, it is no longer surprising that immune escape rates observed during the chronic phase of the infection tend to be slow. Note that we have previously argued that estimating the killing rate by a combination of immune escape and reversion data also depends on the viral replication rate (68) and that rapid viral replication during reversion may lead to an overestimation of the killing rate at a later phase with slower replication. These problems do not apply here because we consider the steady state of a chronic infection.
FIG 5.
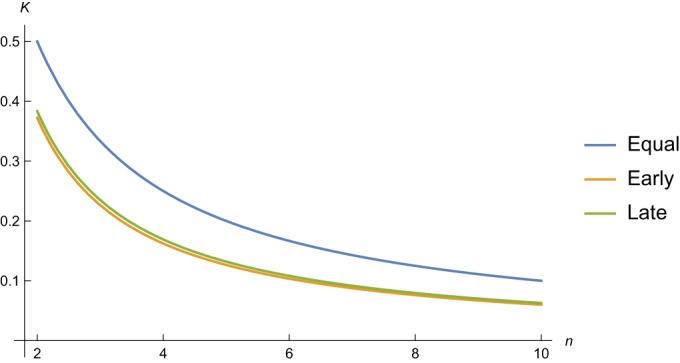
Initial rate at which a mutant escaping one immune response takes over the viral quasispecies. We use equation 30 to predict the escape rate as a function of the breadth of the immune response, n, for a wild-type replication rate of ρ = 1 day−1. Note that these results are independent of the functions F(T) and G(Ei,V) in equations 7a and b. The parameters are as described in Table 1.
Three-stage model.
Chowdhury et al. (15) demonstrate that the upslope of the viral load following CD8+ T cell depletion is about twofold faster in controller monkeys with low viral loads than in “progressor” monkeys with high viral loads (i.e., ρ ≃ 1 and ρ ≃ 0.5 day−1, respectively; see the Appendix). They suggest that the CD8+ T cells exert a better control in monkeys with a low viral load (15). Our results confirm this because a better immune control, e.g., a killing rate, K, should indeed correspond to faster viral replication (equation 18). In our model, this faster viral replication is realized by a better preservation of target cells when the killing is faster (Fig. 1). Our results therefore suggest that target cell levels should be higher in controllers than in progressors, which at least correlates well with the higher CD4+ T cell counts in controllers (1, 15).
To confirm these results in a quantitative manner, we simulate these CD8+ T cell depletion experiments in our most realistic three-stage model, where we allow most CD4+ T cells to die from abortive infection (52, 53). Setting d0 = 54 day−1 and γ0 = 6 day−1, we let about 10% of cells that become infected survive into the eclipse phase, in about 4 h [i.e., f = γ0/(γ0 + d0) = 0.1 in equation 27]. Interestingly, this leads to a steady state with realistically low fractions of infected CD4+ T cells (Fig. 6) (69, 70). We make controller and progressor monkeys by varying the breadth of the immune response (Fig. 6) and pick a regime where most of the killing is late (early killing gives similar results [not shown]). Making a progressor monkey by allowing for just 1 immune response, we obtain a realistic upslope, following CD8+ T cell depletion, of ρ = 0.43 day−1 with a killing rate of K2 = 1.4 day−1 (Fig. 6a). A controller monkey with n = 10 immune responses approaches a replication rate of ρ = 1.02 day−1 (with a killing rate of K2 = 3.8 day−1) and, indeed, has higher target cell availability (Fig. 6b) than the progressor monkey in Fig. 6a. In addition to increasing the viral replication rate by elevating the killing rate, we can also increase ρ more directly by increasing the rate of target cell production, s. A fivefold increase in target cell production indeed increases both the replication rate and the killing rate (Fig. 6c and d).
FIG 6.

Examples of progressor and controller monkeys simulated with the quasi–steady-state version of the three-stage model of equation 21. Progressors are made by giving them just one immune response (with k21 = 10), whereas controllers are given 10 immune responses (with k2i = 10 for i = 1,2,…,10), Performing CD8+ T cell depletion at day 150, we confirm the findings of Chowdhury et al. (15), showing that the viral load increases faster in monkeys that control better. (a) s = dT = 0.1, n = 1: ρ = 0.43 and K2 = 1.4 day−1. (b) s = dT = 0.1, n = 10: ρ = 1.02 and K2 = 3.8 day−1. (c) s = dT = 0.5, n = 1: ρ = 0.88 and K2 = 3.2 day−1. (d) s = dT = 0.5, n = 10: ρ = 1.35 and K2 = 5.4 day−1. (e) Confirmation that all combinations fall on the line predicted by equation 18b. Parameters are for the late killing regime with k2i = 10, and hi = i/1,000 for i = 1,2,…,n, β = 91, d0 = 54, and γ0 = 6 day−1.
Note that all four combinations of ρ and K2 in Fig. 6 fall on the line predicted by equation 18b, suggesting an almost linear increase of the killing rate with the replication rate. In combination, these results predict that patients with a high production of target cells should have fast viral replication and high viral loads with rapid killing, whereas patients with a broad immune response should have low viral loads and high target cell levels and, hence, also have rapid viral replication (see the Appendix for a generalization of these results). Finally, note that the viral set point is approached monotonically in Fig. 6c and d, which is realistic (40, 41) but difficult to achieve with this type of model (47).
DISCUSSION
Assuming that the major protective effect of CTL during HIV-1 infection is to kill infected cells fast, and considering mathematical models with an eclipse phase, we have shown that the total killing rate has to be considerably faster than one per day and that the killing rate should vary over the viral life cycle. Since the killing rate has to balance the viral replication rate, we were able to estimate the killing rate from the observed upslope of the viral load following CD8+ T cell depletion. Because alternative models in which the major protective effect of CTL is nonlytic are also consistent with all data (11, 22, 24), we obviously cannot prove that the killing rates are this fast. Our main result is to demonstrate that the current data provide no evidence that the killing rate is slow (in contrast to the intuitive interpretation that they do).
The killing rates that we estimate for SIV infection in macaques, i.e., 1.6 < Ki < 4 day−1, remain conservative. For instance, if virus-producing cells were to die (or burst) more rapidly than we assumed for the late killing parameters (i.e., in about half a day, d2 = 2 day−1), the corresponding killing rate, K2, should be even larger to balance the effective viral replication (equation 18b). Similarly, if killing during the first few hours after the infection of a cell would indeed be important (32, 54, 55), it would require a killing rate, K0, that is at least an order of magnitude larger than the 4 day−1 estimated above (equation 25; also references 52 and 53). The predicted killing rates in patients infected with HIV-1 could be somewhat lower, however, because the initial replication rates of the virus in humans [ρ(0) ≃ 1 day−1 (40, 41)] are estimated to be lower than those in monkeys [ρ(0) ≃ 1.5 day−1 (34, 43–45)]. A rough estimate for the rate of viral replication during the chronic phase in patients would be that this is also about one third lower than that in monkeys, i.e., 0.3 < ρ < 0.7 day−1. Because the relation between the required rate of killing and the rate of viral replication is almost linear (Fig. 6E), this would mean that the killing rates in humans should be about one-third lower than those reported in Results.
In a recent paper, Halle et al. employed two-photon microscopy in mice to enumerate the number of virus-infected target cells that were killed by CTL per day (71). Using transgenic and normal CTL, they estimated that one CTL kills 2 to 16 target cells per day (71). This estimate of a few dead targets per CTL per day is very similar to our previous estimates on the in vivo killing of splenocytes pulsed with lymphocytic choriomeningitis virus (LCMV) epitopes (72). An HIV-1-infected patient having about 1% cognate CTL in a total pool of approximately 5 × 1010 CD8+ T cells would therefore be able to kill about 109 HIV-1-infected cells per day. Since the total number of productively infected cells is estimated to be 108 cells (70, 73) and these cells have an expected life span of about 1 day (30), it seems perfectly feasible that most productively infected cells die from cytotoxic activity.
In our models, the killing rate increases only marginally if the breadth, n, or the quality, ki, of the immune response is increased (Fig. 1 and 6). An intuitive explanation for this is that the total killing rate ultimately has to balance the viral replication rate, which largely depends on the availability of target cells. Whenever target cell availability is high and the viral replication rate approaches its maximum [ρ → ρ(0)], any further increases in breadth or quality hardly increase the killing rate (38, 39), and each immune response would on average contribute a killing rate of less than ρ(0)/n (Fig. 5). Increasing the breadth would therefore hardly improve the control of the viral load but would provide much better resistance against the evolution of immune escapes, because each escape would provide only a minor selective advantage [i.e., less than ρ(0)/n] (38, 39, 74).
Why is the virus not rejected by the strong immune reactions in our models? It is typically argued that HIV is not rejected because it escapes from immune responses and because it forms latently infected cells. Both mechanisms are absent from our equations. During the acute phase of the infection, the sizes of the immune responses are only limited by the availability of viral antigen, and the immune responses are expanding exponentially (albeit at a rate lower than that of the virus [47, 75]). The immune responses continue to expand (albeit even more slowly) after the peak in the viral load and when the viral load is rebounding after an initial phase of decline (Fig. 1 and 6). Since the total killing rate is monotonically increasing during the first weeks of the infection, the reason for the rebound in the viral load is that target cell levels recover following the peak in the viral load, allowing the virus to increase its effective replication rate.
Generally, CTL express high levels of the proteins associated with cytolytic activity (76). CD8+ T cells from the blood of HIV-infected patients also express high levels of perforin and form conjugates with autologous CD4+ T cells in ex vivo experiments, which in most cases leads to apoptosis of the CD4+ T cells (77). The in vitro cytotoxic capacity of CD8+ T cells on autologous HIV-1-infected CD4+ T cells is one of the best correlates with low HIV-1 loads in humans (78–80). Our modeling study confirms these experimental findings by showing that, if the immune pressure exerted by CD8+ T cells is largely cytolytic, the killing rate has to be much faster than is currently appreciated and that CTL should indeed play a major role in the death of infected cells. Another prediction of our modeling is that most of the killing should then either be early or late. This would be very important to know, but it is unclear how one could test this experimentally in vivo. Using in vitro cultures, it has been established that CTL that are specific for epitopes that are expressed early control HIV-1 replication better than CTL specific for a late protein (81). This suggests that early killing is beneficial (here because the CTL have more time to kill). Nowadays, time-lapse video imaging of target cells and CTL in vitro seems to be the most authoritative approach to establish when in the viral life cycle most infected cells are killed. Such experiments would need markers to know the status of the infected cells, as well as several clonotypes of CTL responding to epitopes that are expressed either early or late.
Like current experimental approaches, our modeling study fails to provide direct evidence that CD8+ T cells control HIV-1 infection by the killing of infected CD4+ T cells. We have only shown here that all experiments that were previously taken to favor nonlytic control by CD8+ T cells are perfectly consistent with a pure cytotoxic mechanism of control. Likewise, the excellent cytotoxic capacity of CD8+ T cells in patients with a very low viral load could be the consequence rather than the cause of low load (78–80). Therefore, we think it remains a crucial open question whether or not the immune control by CTL is largely lytic or nonlytic (and/or that CTL accrue nonlytic affects following killing a target [82]) and that one should not be convinced by the current circumstantial evidence, and we conclude with this important new insight that the current evidence is not conclusive.
ACKNOWLEDGMENTS
We thank Becca Asquith, Ruy Ribeiro, and Alan Perelson for extensive discussions.
APPENDIX
Estimating the increase of the viral slope after CD8+ T cell depletion.
The viral replication rate, ρ, at steady state can be estimated from various sets of data on the increase in the viral load in chronically SIV-infected monkeys that are treated with antibodies depleting CD8+ T cells (9–15, 83, 84). Depending on the number of available time points, the slope, ρ, is estimated by linear regression of the log-transformed data or just from the ratios of pairs of log-transformed data points divided by their time interval. One complication in estimating ρ is that the antibody treatments were given over multiple days, and we start on the first day of depletion, taking it as the most significant depletion event. For the sake of clarity, the day corresponding to the first CD8+ T cell depletion treatment is here consistently referred to as day zero. The data from the Klatt et al. (11) and Chowdhury et al. (15) papers were kindly shared with us by the authors. The other data sets were included by digitizing the figures in the respective papers (using PlotDigitizer).
(i) The Schmitz et al. (9) data.
Already in 1999, CD8 depletion experiments were performed on rhesus macaques chronically infected with SIVmac (9). We digitized the data for the rhesus macaques denoted as A, B, and C. Measurements from days 0, 3, and 6 were available (Table A1). The average replication rate is ρ = 0.57 day−1.
TABLE A1.
Slopes of the viral load in three chronically SIV-infected rhesus macaques in the data of Schmitz et al. (9)
| Animal | Slope of viral load for days: |
Linear regression | |
|---|---|---|---|
| 0–3 | 3–6 | ||
| A | 0.31 | 1.49 | 0.90 |
| B | 0.74 | 0.18 | 0.46 |
| C | 0.67 | 0.02 | 0.35 |
| Mean | 0.57 | 0.56 | 0.57 |
(ii) The Jin et al. (10) data.
In 1999 as well, similar experiments were performed on five rhesus macaques infected with SIVmac251 (animals AT-02, AR-68, AR-71, and AR-93) or SIVmac239 (animal AT-22) (10). Viral load data were collected on days 0, 1, and 2, and the corresponding slopes are presented in Table A2. The mean value of the slopes of the linear regressions is ρ = 0.58 day−1.
TABLE A2.
Slopes estimated from the data of Jin et al. (10)
| Animal | Slope for days: |
Linear regression | |
|---|---|---|---|
| 0–1 | 1–2 | ||
| AT-22 | 0.29 | 0.51 | 0.40 |
| AR-68 | 0.48 | 1.12 | 0.80 |
| AR-71 | 0.010 | 0.57 | 0.33 |
| AR-93 | 0.037 | 0.40 | 0.39 |
| AT-02 | 1.51 | 0.41 | 0.96 |
| Mean | 0.55 | 0.60 | 0.58 |
(iii) The Klatt et al. (11) data.
Two groups of SIVmac239-infected rhesus macaques were denoted as group A (animals RRf6, RAj7, RLi6, RPp6, and RZl5) and group B (animals RSq8, RUe7, RWf7, and XHB). Because there were no measurements of the viral load on day zero, we used its latest measurement prior to the CD8 depletion, which was day −2 in group A and day −6 in group B. Upslope data were available for day 1 and day 4 in group A and for day 1 and day 5 in group B. The estimates for the upslopes for the individual macaques and their means can be found in Table A3. The slopes obtained with linear regression seem the most reliable and have an average of ρ = 0.45 day−1.
TABLE A3.
Estimates of the viral replication rate, ρ, from the data of Klatt et al. (11)
| Group and animal | Upslope value of ρ for days: |
Linear regression | |
|---|---|---|---|
| 0–1 | 1–4 | ||
| A | |||
| RRf6 | 1.59 | 0.73 | 0.85 |
| RAj7 | 0.63 | 0.108 | 0.18 |
| RLi6 | 2.05 | 0.31 | 0.56 |
| RPp6 | −0.14 | 0.16 | 0.12 |
| RZl5 | 2.27 | −0.13 | 0.21 |
| Mean of group A | 1.28 | 0.23 | 0.39 |
| B | 0–1 |
1–5 |
|
| RSq8 | 2.55 | 0.11 | 0.46 |
| RUe7 | 1.58 | 0.57 | 0.72 |
| RWf7 | 1.97 | −0.27 | 0.047 |
| XHB | 2.03 | 0.76 | 0.95 |
| Mean of group B | 2.04 | 0.29 | 0.54 |
| Mean of both groups | 1.62 | 0.26 | 0.45 |
(iv) The Wong et al. (12) data.
Similar experiments were performed on eight rhesus macaques infected with SIVmac251 (12). We digitized the data for the three monkeys (MMU32906, MMU27562, and MMU33580) that approached full depletion of their CD8+ T cells prior to ART. Measurements were available for days 0 and 5, and we take the ratios of the log-transformed values, divide by 5, and obtain a very similar average of ρ = 0.54 day−1 (Table A4).
TABLE A4.
Viral loads and slopes in three CD8-depleted rhesus macaques, from the data of Wong et al. (12)
| Animal | lnV for day: |
Slope | |
|---|---|---|---|
| 0 | 5 | ||
| MMU32906 | 15.20 | 17.04 | 0.37 |
| MMU27562 | 13.01 | 14.92 | 0.38 |
| MMU33580 | 8.80 | 13.13 | 0.87 |
| Mean | 0.54 | ||
(v) The Pandrea et al. (13) data.
After a normal and vigorous acute infection, rhesus macaques infected with SIVagm control the infection and approach undetectable viral loads. Depleting the CD8+ T cells leads to rapid rebound of the virus. The upslope can only be estimated for two monkeys. Monkey BA38 had 3 consecutive time points above the level of detection and before the peak, and we estimate ρ = 0.7 day−1 from these data. Monkey V492 had two time points above the level of detection in the expansion phase, and we estimate ρ = 1.7 day−1 from these data.
(vi) The Fukazawa et al. (14) data.
As part of a larger study on B cell follicle sanctuaries, measurements were taken of the SIV RNA count in the plasma of seven elite-controller rhesus macaques after treatment with anti-CD8 antibodies (14). These monkeys were chronically infected with SIVmac239 or SIVmac251. The depletion treatment was not followed by ART, and remarkably, the monkeys were able to regenerate their SIV-specific CTL population and keep the virus in check. This is most likely due to the fact these monkeys were elite controllers, which is a vital difference with the four previous data sets. Data were taken on days 0, 3, 7, and 10 after the depletion treatments, and viral loads went down after day 10. We could only obtain data on the average values for all monkeys (days 0 to 3, ρ = 0.99; days 3 to 7, ρ = 0.94; and days 7 to 10, ρ = 0.56). As the last interval (days 7 to 10) showed a smaller increase than the first two intervals, linear regression was performed with and without this last interval, yielding slopes of ρ = 0.85 day−1 for 0 to 10 and ρ = 0.96 day−1 for days 0 to 7.
(vii) The Chowdhury et al. (15) data.
Different upslopes were observed following CD8 depletion in progressor and controller monkeys (15), with a significantly greater increase of the plasma viral load in controllers than in progressors. The means of the slopes for all intervals and the linear regression are higher in controllers than in progressor rhesus macaques (Tables A5 and A6). Linear regressions were obtained for days 0 to 2 and days 0 to 3 because the mean value of the slope between day 2 and day 3 is lower than for the other two intervals.
TABLE A5.
Estimates for ρ from the controller monkeys in the study of Chowdhury et al. (15)
| Controller animal | ρ for days: |
Regression value for days: |
|||
|---|---|---|---|---|---|
| 0–1 | 1–2 | 2–3 | 0–3 | 0–2 | |
| RAk9 | 2.60 | 1.85 | −1.11 | 1.18 | 2.22 |
| RJm7 | 0.78 | −0.80 | 1.16 | 0.26 | −0.08 |
| RLf8 | 4.39 | 0.97 | −0.31 | 1.61 | 2.68 |
| RSk8 | 2.84 | 2.62 | 1.50 | 2.35 | 2.73 |
| RZc9 | 1.43 | 1.47 | 1.02 | 1.32 | 1.45 |
| Mean | 2.41 | 1.22 | 0.45 | 1.35 | 1.82 |
TABLE A6.
Estimates for ρ from the progressor monkeys in the study of Chowdhury et al. (15)
| Progressor animal | ρ for days: |
Regression value for days: |
|||
|---|---|---|---|---|---|
| 0–1 | 1–2 | 2–3 | 0–3 | 0–2 | |
| RWm8 | −0.14 | NAa | 0.01 | −0.07 | NA |
| RKv7 | 1.34 | 1.52 | −0.35 | 0.91 | 1.43 |
| RNw10 | 0.92 | 0.46 | −0.07 | 0.44 | 0.69 |
| RAy8 | 1.80 | 0.29 | 1.46 | 1.09 | 1.04 |
| RLw10 | 1.32 | 0.37 | −3.47 | −0.49 | 0.85 |
| RFn7 | 0.67 | 0.72 | −0.39 | 0.37 | 0.70 |
| RHd10 | −0.07 | 0.33 | 0.11 | 0.14 | 0.13 |
| RIc9 | 1.76 | 0.91 | 0.61 | 1.08 | 1.34 |
| RDa10 | 1.92 | 1.43 | 0.19 | 1.21 | 1.68 |
| RBc9 | 1.83 | 0.57 | 0.24 | 0.85 | 1.20 |
| Mean | 1.13 | 0.73 | −0.18 | 0.55 | 1.00 |
NA, not available.
To investigate whether this difference in upslopes between progressor and controller monkeys is significant, we performed a nonparametric Mann-Whitney U test. On the linear regressions based upon days 0 to 2, the test proved to be nonsignificant (P = 0.10). However, monkey RJm7 seemed to corrupt the data, since its viral load hardly increased upon CD8 depletion. If taken out, the results of the test indicated a very significant difference (P = 0.005). The same test was done for the slopes of the linear regression on interval days 0 to 3, and both with and without monkey RJm7, the test indicated a significant difference (P = 0.044 with RJm7 and P = 0.004 without RJm7).
(viii) The Metzner et al. (83) data.
Rhesus monkeys infected with SIVmac239Δnef for over 2 years underwent CTL depletion by treatment with OKT8F (anti-CD8) antibodies (83). We performed linear regression over the time points in which the CTL levels were undetectable, i.e., from day 0 to day 9 (but note that taking day 7, 8, or 10 as an endpoint hardly affects the estimates). Most of these monkeys have a very rapid increase in viral load during the first day after treatment, followed by a small decrease and a subsequent slower increase. The reasons for these fluctuations are unknown to us, and averaging by linear regression, we arrived at an estimate of ρ = 0.41 day−1. See Table A7 for all the values and means of the linear regressions performed.
TABLE A7.
Estimates of the viral replication rate, ρ, from the data of Metzner et al. (83)
| Monkey | Linear regression of ρ for days: |
|||
|---|---|---|---|---|
| 0–7 | 0–8 | 0–9 | 0–10 | |
| A | 0.35 | 0.41 | 0.44 | 0.43 |
| B | 0.31 | 0.37 | 0.42 | 0.41 |
| C | 0.55 | 0.56 | 0.48 | 0.46 |
| D | 0.41 | 0.32 | 0.32 | 0.30 |
| Mean | 0.41 | 0.42 | 0.41 | 0.40 |
(ix) The Friedrich et al. (84) data.
A study was specifically conducted on six elite controller rhesus monkeys that had been chronically infected with SIVmac239 for between 1 and 5 years (84). The monkeys were depleted of their CTL, and a rapid increase in viral load in the blood plasma was observed. By performing linear regressions on the amounts of viral particles in the blood between treatment and 7 days after for the different monkeys, we arrived at an average regression of ρ = 0.96 day−1. One point in the data (for monkey AJ11 on day 3) could not be precisely determined from the figure, and hence, this monkey was not taken into account while estimating the value of the viral replication rate.
(x) Summary.
The average replication rates estimated from the studies of Schmitz et al. (9), Jin et al. (10), Klatt et al. (11), and Wong et al. (12) and from the progressors in the Chowdhury et al. (15) study vary in a narrow window between 0.45 ≤ ρ ≤ 0.6 day−1. Our conservative estimates for the elite controllers in the Fukazawa et al. (14), the Friedrich et al. (84), and the Chowdhury et al. (15) studies are about twofold larger and vary between 0.85 ≤ ρ ≤ 1.35 day−1. The two estimates from rhesus monkeys controlling SIVagm (13) to undetectable set point levels also tend to be high, i.e., ρ = 0.7 and ρ = 1.7.
Target cell availability, viral load and replication, and killing rate.
If the two-stage model is extended with explicit functions for the production of target cells and immune effector cells, its steady state predicts correlations between viral replication, killing rates (equation 17), target cell availability, and viral load. In Fig. A1, we vary the production rate of target cells [s, while constraining T(0) = 1 by keeping s = dT] and the late killing rate k21 (setting K1 = 0) to study these correlations. (Similar results were obtained with the early killing regime [not shown]). The effective replication rate, ρ, increases with the killing rate and the renewal rate of the target cells (Fig. A1b), because a better immune control and a faster production of target cells increases the availability of target cells, T̄, which allows for a faster viral replication. The total killing rate, K2, also increases with the killing rate and the renewal rate of the target cells (Fig. A1b). The first is obvious, and the latter is natural, because a faster production of target cells needs to be compensated by a higher rate of killing at the steady state. The set point viral load increases with the rate of target cell production, s, and decreases with the killing rate k21 (Fig. A1c). Obviously, when the observed values of K2 and ρ are plotted as a function of each other (Fig. A1d), the six lines in Fig. 1a to c collide, as is predicted by equation 18b. Interestingly, markedly different viral loads can share identical values of ρ and K2 (Fig. A1e): for one x value, i.e., one combination of ρ and K2, we obtain 10-fold different viral loads with 10-fold changes in the killing parameter k21.
FIG A1.

The realized killing rate K2 and replication rate ρ as a function of the s and ki parameters in the late-killing model. Panels d and e combine the graphs from panels a to c by plotting K2 or the set point viral load, Ī2, as a function of ρ. The fact that all lines fall on top of each other in panel d numerically confirms equation 18b. We use the function F(T) = s − dTT to complete dT/dt in equation 7a and the competitive monotonic saturation function G(Ei,V) = pVEi/(hi + V + Ei) with V = I2 to complete dEi/dt in equation 7b.
In biological terms, this can be understood by starting the argument either with the virus or with the immune response. First, patients infected with a virus that replicates fast, i.e., high ρ (due to viral properties and/or higher target cell availability by high immune activation), will approach a high set point viral load and, hence, elicit a large immune response that will ultimately balance the rapid viral replication. Second, patients mounting a good and broad immune response will control the virus well, which allows the target cells to recover, which in turn allows for rapid viral replication. Thus, rapid replication and killing are expected in controllers with a low viral load (15, 78–80) and in progressors with a high viral load (Fig. A1e). The fact that CTL appear to be less cytolytic in the latter group (78–80) could be indicative of exhaustion of CTL at high viral loads (85), which we could implement using a nonmonotonic proliferation function, G(Ei,V); e.g., see Conway and Perelson (86). However, exhaustion of CTL would not change our general conclusion that all current evidence is consistent with rapid killing, and for reasons of clarity, we have used a monotonic proliferation function for the immune effector cells.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Deeks SG, Walker BD. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27:406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Miedema F, Hazenberg MD, Tesselaar K, Van Baarle D, De Boer RJ, Borghans JA. 2013. Immune activation and collateral damage in AIDS pathogenesis. Front Immunol 4:298. doi: 10.3389/fimmu.2013.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, Cozzi-Lepri A, De Luca A, Easterbrook P, Francioli P, Mallal S, Martinez-Picado J, Miro JM, Obel N, Smith JP, Wyniger J, Descombes P, Antonarakis SE, Letvin NL, McMichael AJ, Haynes BF, Telenti A, Goldstein DB. 2007. A whole-genome association study of major determinants for host control of HIV-1. Science 317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MB, Shaw GM. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med 3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 5.Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med 10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 6.Ganusov VV, Goonetilleke N, Liu MK, Ferrari G, Shaw GM, McMichael AJ, Borrow P, Korber BT, Perelson AS. 2011. Fitness costs and diversity of the cytotoxic T lymphocyte (CTL) response determine the rate of CTL escape during acute and chronic phases of HIV infection. J Virol 85:10518–10528. doi: 10.1128/JVI.00655-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu MK, Hawkins N, Ritchie AJ, Ganusov VV, Whale V, Brackenridge S, Li H, Pavlicek JW, Cai F, Rose-Abrahams M, Treurnicht F, Hraber P, Riou C, Gray C, Ferrari G, Tanner R, Ping LH, Anderson JA, Swanstrom R, Cohen M, Karim SS, Haynes B, Borrow P, Perelson AS, Shaw GM, Hahn BH, Williamson C, Korber BT, Gao F, Self S, McMichael A, Goonetilleke N. 2013. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J Clin Invest 123:380–393. doi: 10.1172/JCI65330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts HE, Hurst J, Robinson N, Brown H, Flanagan P, Vass L, Fidler S, Weber J, Babiker A, Phillips RE, McLean AR, Frater J. 2015. Structured observations reveal slow HIV-1 CTL escape. PLoS Genet 11:e1004914. doi: 10.1371/journal.pgen.1004914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, Ghrayeb J, Forman MA, Montefiori DC, Rieber EP, Letvin NL, Reimann KA. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 10.Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, Irwin CE, Safrit JT, Mittler J, Weinberger L, Kostrikis LG, Zhang L, Perelson AS, Ho DD. 1999. Dramatic rise in plasma viremia after CD8+ t cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med 189:991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klatt NR, Shudo E, Ortiz AM, Engram JC, Paiardini M, Lawson B, Miller MD, Else J, Pandrea I, Estes JD, Apetrei C, Schmitz JE, Ribeiro RM, Perelson AS, Silvestri G. 2010. CD8+ lymphocytes control viral replication in SIVmac239-infected rhesus macaques without decreasing the lifespan of productively infected cells. PLoS Pathog 6:e1000747. doi: 10.1371/journal.ppat.1000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong JK, Strain MC, Porrata R, Reay E, Sankaran-Walters S, Ignacio CC, Russell T, Pillai SK, Looney DJ, Dandekar S. 2010. In vivo CD8+ T-cell suppression of SIV viremia is not mediated by CTL clearance of productively infected cells. PLoS Pathog 6:e1000748. doi: 10.1371/journal.ppat.1000748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pandrea I, Gaufin T, Gautam R, Kristoff J, Mandell D, Montefiori D, Keele BF, Ribeiro RM, Veazey RS, Apetrei C. 2011. Functional cure of SIVagm infection in rhesus macaques results in complete recovery of CD4+ T cells and is reverted by CD8+ cell depletion. PLoS Pathog 7:e1002170. doi: 10.1371/journal.ppat.1002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, Hagen SI, Shoemaker R, Deleage C, Lucero C, Morcock D, Swanson T, Legasse AW, Axthelm MK, Hesselgesser J, Geleziunas R, Hirsch VM, Edlefsen PT, Piatak M Jr, Estes JD, Lifson JD, Picker LJ. 2015. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med 21:132–139. doi: 10.1038/nm.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chowdhury A, Hayes TL, Bosinger SE, Lawson BO, Vanderford T, Schmitz JE, Paiardini M, Betts M, Chahroudi A, Estes JD, Silvestri G. 2015. Differential impact of in vivo CD8+ T lymphocyte depletion in controller versus progressor simian immunodeficiency virus-infected macaques. J Virol 89:8677–8686. doi: 10.1128/JVI.00869-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeVico AL, Gallo RC. 2004. Control of HIV-1 infection by soluble factors of the immune response. Nat Rev Microbiol 2:401–413. doi: 10.1038/nrmicro878. [DOI] [PubMed] [Google Scholar]
- 17.Saunders KO, Ward-Caviness C, Schutte RJ, Freel SA, Overman RG, Thielman NM, Cunningham CK, Kepler TB, Tomaras GD. 2011. Secretion of MIP-1β and MIP-1α by CD8+ T-lymphocytes correlates with HIV-1 inhibition independent of coreceptor usage. Cell Immunol 266:154–164. doi: 10.1016/j.cellimm.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy JA. 2015. Discovery of another anti-HIV protein in the search for the CD8+ cell anti-HIV factor. Proc Natl Acad Sci U S A 112:7888–7889. doi: 10.1073/pnas.1509324112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganusov VV. 2003. The role of the cytotoxic T-lymphocyte response and virus cytopathogenicity in the virus decline during antiviral therapy. Proc Biol Sci 270:1513–1518. doi: 10.1098/rspb.2003.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balamurali M, Petravic J, Loh L, Alcantara S, Kent SJ, Davenport MP. 2010. Does cytolysis by CD8+ T cells drive immune escape in HIV infection? J Immunol 185:5093–5101. doi: 10.4049/jimmunol.1002204. [DOI] [PubMed] [Google Scholar]
- 21.Elemans M, Thiebaut R, Kaur A, Asquith B. 2011. Quantification of the relative importance of CTL, B cell, NK cell, and target cell limitation in the control of primary SIV-infection. PLoS Comput Biol 7:e1001103. doi: 10.1371/journal.pcbi.1001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elemans M, Seich Al Basatena NK, Klatt NR, Gkekas C, Silvestri G, Asquith B. 2011. Why don't CD8+ T cells reduce the lifespan of SIV-infected cells in vivo? PLoS Comput Biol 7:e1002200. doi: 10.1371/journal.pcbi.1002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elemans M, Seich Al Basatena NK, Asquith B. 2012. The efficiency of the human CD8+ T cell response: how should we quantify it, what determines it, and does it matter? PLoS Comput Biol 8:e1002381. doi: 10.1371/journal.pcbi.1002381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seich Al Basatena NK, Chatzimichalis K, Graw F, Frost SD, Regoes RR, Asquith B. 2013. Can non-lytic CD8+ T cells drive HIV-1 escape? PLoS Pathog 9:e1003656. doi: 10.1371/journal.ppat.1003656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elemans M, Florins A, Willems L, Asquith B. 2014. Rates of CTL killing in persistent viral infection in vivo. PLoS Comput Biol 10:e1003534. doi: 10.1371/journal.pcbi.1003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gadhamsetty S, Beltman JB, De Boer RJ. 2015. What do mathematical models tell us about killing rates during HIV-1 infection? Immunol Lett 168:1–6. doi: 10.1016/j.imlet.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 28.Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH, Saag MS, Shaw GM. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 29.Bonhoeffer S, Funk GA, Gunthard HF, Fischer M, Muller V. 2003. Glancing behind virus load variation in HIV-1 infection. Trends Microbiol 11:499–504. doi: 10.1016/j.tim.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Markowitz M, Louie M, Hurley A, Sun E, Di Mascio M, Perelson AS, Ho DD. 2003. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J Virol 77:5037–5038. doi: 10.1128/JVI.77.8.5037-5038.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klenerman P, Phillips RE, Rinaldo CR, Wahl LM, Ogg G, May RM, McMichael AJ, Nowak MA. 1996. Cytotoxic T lymphocytes and viral turnover in HIV type 1 infection. Proc Natl Acad Sci U S A 93:15323–15328. doi: 10.1073/pnas.93.26.15323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Althaus CL, De Boer RJ. 2011. Implications of CTL-mediated killing of HIV-infected cells during the non-productive stage of infection. PLoS One 6:e16468. doi: 10.1371/journal.pone.0016468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davenport MP, Ribeiro RM, Chao DL, Perelson AS. 2004. Predicting the impact of a nonsterilizing vaccine against human immunodeficiency virus. J Virol 78:11340–11351. doi: 10.1128/JVI.78.20.11340-11351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davenport MP, Zhang L, Bagchi A, Fridman A, Fu TM, Schleif W, Shiver JW, Ribeiro RM, Perelson AS. 2005. High-potency human immunodeficiency virus vaccination leads to delayed and reduced CD8+ T-cell expansion but improved virus control. J Virol 79:10059–10062. doi: 10.1128/JVI.79.15.10059-10062.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petravic J, Davenport MP. 2010. Vaccination-induced noncytolytic effects in the acute phase of SHIV infection. PLoS One 5:e15083. doi: 10.1371/journal.pone.0015083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asquith B, Edwards CT, Lipsitch M, McLean AR. 2006. Inefficient cytotoxic T lymphocyte-mediated killing of HIV-1-infected cells in vivo. PLoS Biol 4:e90. doi: 10.1371/journal.pbio.0040090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arnaout RA, Nowak MA, Wodarz D. 2000. HIV-1 dynamics revisited: biphasic decay by cytotoxic T lymphocyte killing? Proc Biol Sci 267:1347–1354. doi: 10.1098/rspb.2000.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Boer RJ. 2012. Which of our modeling predictions are robust? PLoS Comput Biol 8:e1002593. doi: 10.1371/journal.pcbi.1002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Deutekom HW, Wijnker G, De Boer RJ. 2013. The rate of immune escape vanishes when multiple immune responses control an HIV infection. J Immunol 191:3277–3286. doi: 10.4049/jimmunol.1300962. [DOI] [PubMed] [Google Scholar]
- 40.Ribeiro RM, Qin L, Chavez LL, Li D, Self SG, Perelson AS. 2010. Estimation of the initial viral growth rate and basic reproductive number during acute HIV-1 infection. J Virol 84:6096–6102. doi: 10.1128/JVI.00127-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ndhlovu ZM, Kamya P, Mewalal N, Kloverpris HN, Nkosi T, Pretorius K, Laher F, Ogunshola F, Chopera D, Shekhar K, Ghebremichael M, Ismail N, Moodley A, Malik A, Leslie A, Goulder PJ, Buus S, Chakraborty A, Dong K, Ndung'u T, Walker BD. 2015. Magnitude and kinetics of CD8+ T cEll activation during hyperacute HIV infection impact viral set point. Immunity 43:591–604. doi: 10.1016/j.immuni.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Little SJ, McLean AR, Spina CA, Richman DD, Havlir DV. 1999. Viral dynamics of acute HIV-1 infection. J Exp Med 190:841–850. doi: 10.1084/jem.190.6.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nowak MA, Lloyd AL, Vasquez GM, Wiltrout TA, Wahl LM, Bischofberger N, Williams J, Kinter A, Fauci AS, Hirsch VM, Lifson JD. 1997. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J Virol 71:7518–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davenport MP, Ribeiro RM, Perelson AS. 2004. Kinetics of virus-specific CD8+ T cells and the control of human immunodeficiency virus infection. J Virol 78:10096–10103. doi: 10.1128/JVI.78.18.10096-10103.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pandrea I, Ribeiro RM, Gautam R, Gaufin T, Pattison M, Barnes M, Monjure C, Stoulig C, Dufour J, Cyprian W, Silvestri G, Miller MD, Perelson AS, Apetrei C. 2008. Simian immunodeficiency virus SIVagm dynamics in African green monkeys. J Virol 82:3713–3724. doi: 10.1128/JVI.02402-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 47.De Boer RJ. 2007. Understanding the failure of CD8+ T-cell vaccination against simian/human immunodeficiency virus. J Virol 81:2838–2848. doi: 10.1128/JVI.01914-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grossman Z, Feinberg M, Kuznetsov V, Dimitrov D, Paul W. 1998. HIV infection: how effective is drug combination treatment? Immunol Today 19:528–532. doi: 10.1016/S0167-5699(98)01353-X. [DOI] [PubMed] [Google Scholar]
- 49.Mohammadi P, Desfarges S, Bartha I, Joos B, Zangger N, Munoz M, Gunthard HF, Beerenwinkel N, Telenti A, Ciuffi A. 2013. 24 hours in the life of HIV-1 in a T cell line. PLoS Pathog 9:e1003161. doi: 10.1371/journal.ppat.1003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Althaus CL, Joos B, Perelson AS, Gunthard HF. 2014. Quantifying the turnover of transcriptional subclasses of HIV-1-infected cells. PLoS Comput Biol 10:e1003871. doi: 10.1371/journal.pcbi.1003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noecker C, Schaefer K, Zaccheo K, Yang Y, Day J, Ganusov VV. 2015. Simple mathematical models do not accurately predict early SIV dynamics. Viruses 7:1189–1217. doi: 10.3390/v7031189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, Hebbeler AM, Greene WC. 2010. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143:789–801. doi: 10.1016/j.cell.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sacha JB, Chung C, Rakasz EG, Spencer SP, Jonas AK, Bean AT, Lee W, Burwitz BJ, Stephany JJ, Loffredo JT, Allison DB, Adnan S, Hoji A, Wilson NA, Friedrich TC, Lifson JD, Yang OO, Watkins DI. 2007. Gag-specific CD8+ T lymphocytes recognize infected cells before AIDS-virus integration and viral protein expression. J Immunol 178:2746–2754. doi: 10.4049/jimmunol.178.5.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sacha JB, Buechler MB, Newman LP, Reed J, Wallace LT, Loffredo JT, Wilson NA, Watkins DI. 2010. Simian immunodeficiency virus-specific CD8+ T cells recognize Vpr- and Rev-derived epitopes early after infection. J Virol 84:10907–10912. doi: 10.1128/JVI.01357-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reilly C, Wietgrefe S, Sedgewick G, Haase A. 2007. Determination of simian immunodeficiency virus production by infected activated and resting cells. AIDS 21:163–168. doi: 10.1097/QAD.0b013e328012565b. [DOI] [PubMed] [Google Scholar]
- 57.Mohri H, Perelson AS, Tung K, Ribeiro RM, Ramratnam B, Markowitz M, Kost R, Hurley A, Weinberger L, Cesar D, Hellerstein MK, Ho DD. 2001. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J Exp Med 194:1277–1288. doi: 10.1084/jem.194.9.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vrisekoop N, Drylewicz J, Van Gent R, Mugwagwa T, Van Lelyveld SF, Veel E, Otto SA, Ackermans MT, Vermeulen JN, Huidekoper HH, Prins JM, Miedema F, De Boer RJ, Tesselaar K, Borghans JA. 2015. Quantification of naive and memory T-cell turnover during HIV-1 infection. AIDS 29:2071–2080. doi: 10.1097/QAD.0000000000000822. [DOI] [PubMed] [Google Scholar]
- 59.Nelson PW, Mittler JE, Perelson AS. 2001. Effect of drug efficacy and the eclipse phase of the viral life cycle on estimates of HIV viral dynamic parameters. J Acquir Immune Defic Syndr 26:405–412. doi: 10.1097/00042560-200104150-00003. [DOI] [PubMed] [Google Scholar]
- 60.Dixit NM, Markowitz M, Ho DD, Perelson AS. 2004. Estimates of intracellular delay and average drug efficacy from viral load data of HIV-infected individuals under antiretroviral therapy. Antivir Ther 9:237–246. [PubMed] [Google Scholar]
- 61.De Boer RJ, Ribeiro RM, Perelson AS. 2010. Current estimates for HIV-1 production imply rapid viral clearance in lymphoid tissues. PLoS Comput Biol 6:e1000906. doi: 10.1371/journal.pcbi.1000906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ogg GS, Jin X, Bonhoeffer S, Moss P, Nowak MA, Monard S, Segal JP, Cao Y, Rowland-Jones SL, Hurley A, Markowitz M, Ho DD, McMichael AJ, Nixon DF. 1999. Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. J Virol 73:797–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Casazza JP, Betts MR, Picker LJ, Koup RA. 2001. Decay kinetics of human immunodeficiency virus-specific CD8+ T cells in peripheral blood after initiation of highly active antiretroviral therapy. J Virol 75:6508–6516. doi: 10.1128/JVI.75.14.6508-6516.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Althaus CL, De Vos AS, De Boer RJ. 2009. Reassessing the human immunodeficiency virus type 1 life cycle through age-structured modeling: life span of infected cells, viral generation time, and basic reproductive number, R0. J Virol 83:7659–7667. doi: 10.1128/JVI.01799-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Regoes RR, Yates A, Antia R. 2007. Mathematical models of cytotoxic T-lymphocyte killing. Immunol Cell Biol 85:274–279. doi: 10.1038/sj.icb.7100053. [DOI] [PubMed] [Google Scholar]
- 66.Mandl JN, Regoes RR, Garber DA, Feinberg MB. 2007. Estimating the effectiveness of simian immunodeficiency virus-specific CD8+ T cells from the dynamics of viral immune escape. J Virol 81:11982–11991. doi: 10.1128/JVI.00946-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kadolsky UD, Asquith B. 2010. Quantifying the impact of human immunodeficiency virus-1 escape from cytotoxic T-lymphocytes. PLoS Comput Biol 6:e1000981. doi: 10.1371/journal.pcbi.1000981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ganusov VV, De Boer RJ. 2006. Estimating costs and benefits of CTL escape mutations in SIV/HIV infection. PLoS Comput Biol 2:182–187. doi: 10.1371/journal.pcbi.0020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pantaleo G, Graziosi C, Demarest JF, Butini L, Montroni M, Fox CH, Orenstein JM, Kotler DP, Fauci AS. 1993. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 70.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 71.Halle S, Keyser KA, Stahl FR, Busche A, Marquardt A, Zheng X, Galla M, Heissmeyer V, Heller K, Boelter J, Wagner K, Bischoff Y, Martens R, Braun A, Werth K, Uvarovskii A, Kempf H, Meyer-Hermann M, Arens R, Kremer M, Sutter G, Messerle M, Forster R. 2016. In vivo killing capacity of cytotoxic T cells is limited and involves dynamic interactions and T cell cooperativity. Immunity 44:233–245. doi: 10.1016/j.immuni.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ganusov VV, De Boer RJ. 2008. Estimating in vivo death rates of targets due to CD8 T-cell-mediated killing. J Virol 82:11749–11757. doi: 10.1128/JVI.01128-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haase AT, Henry K, Zupancic M, Sedgewick G, Faust RA, Melroe H, Cavert W, Gebhard K, Staskus K, Zhang ZQ, Dailey PJ, Balfour HH Jr, Erice A, Perelson AS. 1996. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274:985–989. doi: 10.1126/science.274.5289.985. [DOI] [PubMed] [Google Scholar]
- 74.Yue L, Pfafferott KJ, Baalwa J, Conrod K, Dong CC, Chui C, Rong R, Claiborne DT, Prince JL, Tang J, Ribeiro RM, Cormier E, Hahn BH, Perelson AS, Shaw GM, Karita E, Gilmour J, Goepfert P, Derdeyn CA, Allen SA, Borrow P, Hunter E. 2015. Transmitted virus fitness and host T cell responses collectively define divergent infection outcomes in two HIV-1 recipients. PLoS Pathog 11:e1004565. doi: 10.1371/journal.ppat.1004565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pilyugin SS, Antia R. 2000. Modeling immune responses with handling time. Bull Math Biol 62:869–890. doi: 10.1006/bulm.2000.0181. [DOI] [PubMed] [Google Scholar]
- 76.Hukelmann JL, Anderson KE, Sinclair LV, Grzes KM, Murillo AB, Hawkins PT, Stephens LR, Lamond AI, Cantrell DA. 2016. The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat Immunol 17:104–112. doi: 10.1038/ni.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chorin E, Gal-Garber O, Yagel Y, Turner D, Avidor B, Berke G, Hassin D. 2014. PBMC of HIV-infected patients contain CD8 T cells that form conjugates with and kill HIV-infected autologous CD4 T cells. Immunology 144:412–421. doi: 10.1111/imm.12385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saez-Cirion A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, Barre-Sinoussi F, Delfraissy JF, Sinet M, Pancino G, Venet A. 2007. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci U S A 104:6776–6781. doi: 10.1073/pnas.0611244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Migueles SA, Mendoza D, Zimmerman MG, Martins KM, Toulmin SA, Kelly EP, Peterson BA, Johnson SA, Galson E, Poropatich KO, Patamawenu A, Imamichi H, Ober A, Rehm CA, Jones S, Hallahan CW, Follmann DA, Connors M. 2015. CD8+ T-cell cytotoxic capacity associated with human immunodeficiency virus-1 control can be mediated through various epitopes and human leukocyte antigen types. EBioMedicine 2:46–58. doi: 10.1016/j.ebiom.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Migueles SA, Connors M. 2015. Success and failure of the cellular immune response against HIV-1. Nat Immunol 16:563–570. doi: 10.1038/ni.3161. [DOI] [PubMed] [Google Scholar]
- 81.van Baalen CA, Guillon C, Van Baalen M, Verschuren EJ, Boers PH, Osterhaus AD, Gruters RA. 2002. Impact of antigen expression kinetics on the effectiveness of HIV-specific cytotoxic T lymphocytes. Eur J Immunol 32:2644–2652. doi:. [DOI] [PubMed] [Google Scholar]
- 82.Foley MH, Forcier T, McAndrew E, Gonzalez M, Chen H, Juelg B, Walker BD, Irvine DJ. 2014. High avidity CD8+ T cells efficiently eliminate motile HIV-infected targets and execute a locally focused program of anti-viral function. PLoS One 9:e87873. doi: 10.1371/journal.pone.0087873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Metzner KJ, Jin X, Lee FV, Gettie A, Bauer DE, Di Mascio M, Perelson AS, Marx PA, Ho DD, Kostrikis LG, Connor RI. 2000. Effects of in vivo CD8(+) T cell depletion on virus replication in rhesus macaques immunized with a live, attenuated simian immunodeficiency virus vaccine. J Exp Med 191:1921–1931. doi: 10.1084/jem.191.11.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Friedrich TC, Valentine LE, Yant LJ, Rakasz EG, Piaskowski SM, Furlott JR, Weisgrau KL, Burwitz B, May GE, Leon EJ, Soma T, Napoe G, Capuano SV, Wilson NA, Watkins DI. 2007. Subdominant CD8+ T-cell responses are involved in durable control of AIDS virus replication. J Virol 81:3465–3476. doi: 10.1128/JVI.02392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khaitan A, Unutmaz D. 2011. Revisiting immune exhaustion during HIV infection. Curr HIV/AIDS Rep 8:4–11. doi: 10.1007/s11904-010-0066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Conway JM, Perelson AS. 2015. Post-treatment control of HIV infection. Proc Natl Acad Sci U S A 112:5467–5472. doi: 10.1073/pnas.1419162112. [DOI] [PMC free article] [PubMed] [Google Scholar]