

Abstract
Carbohydrate, lipid, and protein metabolism are largely controlled by the interplay of various hormones, which includes those secreted by the pancreatic islets of Langerhans. While typically representing only 1–2% of the total pancreatic mass, the islets have a remarkable ability to adapt to disparate situations demanding a change in hormone release, such as peripheral insulin resistance. There are many different routes to the onset of insulin resistance, including obesity, lipodystrophy, glucocorticoid excess, and the chronic usage of atypical anti-psychotic drugs. All of these situations are coupled to an increase in pancreatic islet size, often with a corresponding increase in insulin production. These adaptive responses within the islets are ultimately intended to maintain glycemic control and to promote macronutrient homeostasis during times of stress. Herein, we review the consequences of specific metabolic trauma that lead to insulin resistance and the corresponding adaptive alterations within the pancreatic islets.
Keywords: Diabetes, Insulin Resistance, Metabolic Disease, Metabolism, Obesity, Proliferation
Introduction
The islets of Langerhans are specialized clusters of endocrine tissue within a larger housing of pancreatic exocrine tissue. Collectively, the islets make up only 1–2% of total pancreatic mass, but remarkably are able to provide fine-tuned control of metabolic events through secretion of the polypeptide hormones insulin and glucagon. These protein hormones regulate storage and utilization of various nutritional fuels from carbohydrate, lipid, and protein sources. For mice, rats, and humans, the pancreatic β-cell, which synthesizes and secretes insulin, is the predominant cell type within the pancreatic islets (1).
Insulin and glucagon (secreted from islet alpha cells) are notable examples of counter-regulatory hormones, with insulin driving metabolic processes associated with the fed state (e.g., glucose utilization and lipid storage) while glucagon typically promotes metabolic outcomes attributed to the fasting state (e.g., gluconeogenesis and lipolysis). Together, these endocrine functions allow for efficient utilization of simple metabolic fuels, such as carbohydrate and fatty acids, to maintain blood glucose levels in a narrow range. Insulin and glucagon ratios also carefully coordinate tissue nutrient storage and utilization via receptor-mediated signals. The necessity of this hormonal interplay is highlighted by development of diabetes when these processes fail (2).
There are numerous conditions requiring the hormonal production capacity of the pancreatic islets to be altered. For example, in situations of caloric surplus, such as occurs during progression to obesity, pancreatic β-cells are required to produce and secrete much more insulin to maintain blood glucose levels within a healthy range (~70–100 mg/dL in humans). In addition, there are clinical situations, such as chronic glucocorticoid regimens, that while necessary to produce a desired therapeutic outcome, also put stress on islet β-cell insulin production due to substantial decreases in peripheral insulin sensitivity.
Obesity and glucocorticoid excess are examples of two specific instances discussed herein that generate metabolic trauma. For the purposes of this review, we define metabolic trauma as a homeostatic disruption severe enough to require systemic signaling adaptations. For example, lipid accumulation in lean tissues, which leads to a reduction in the normal (i.e., physiological) responses to insulin (3). As delineated herein, metabolic trauma can arise via ectopic lipid deposition due to overnutrition (e.g., obesity), defects in nutrient availability (e.g., reduced glucose transport), hormonal excess or deficiency, or side effects of drug therapy.
The extraordinary ability of the pancreatic islet to respond to disparate metabolic alterations by increasing both numbers of β-cells within islets, and enhanced production of insulin, permits control of blood glucose levels during many discrete physiological and pathophysiological scenarios. As we will discuss in each section below, resistance to insulin action by separate and distinct origins produces a common outcome: dynamic changes in pancreatic islet function and mass. Regardless of the primary pathology, insulin resistance is characterized by a reduced ability of insulin to: a) decrease glucose production in the liver and b) stimulate glucose uptake into skeletal muscle and adipose tissue [for reviews, see refs. (4–6)]. The goal of this review is to highlight specific situations that drive adaptive increases in pancreatic islet size, as well as islet β-cell function, and to discuss some of the similarities and differences associated with these metabolic states. A continuum of enhanced islet β-cell function dictated by metabolic need that eventually pushes the islet β-cell into a dysfunctional state is also considered.
Obesity
Obesity defines a state of excess adipose tissue and is correlated with adverse human health outcomes, including increased risk for cardiovascular disease, cancer, and diabetes (7). Many of the negative aspects of obesity are due to both endocrine and immunological changes associated with the obese state (8, 9). Insulin resistance is a major consequence of obesity in mice, rats, and humans. Thus, obesity-induced ectopic lipid deposition contributes to metabolic trauma in a wide variety of tissues (3).
Islet size is increased in human obesity, an observation put forth more than 80 years ago [see ref. (10) and Figure 1], and confirmed in recent studies (11). From an experimental perspective, mice rendered obese by specific genetic mutations also display increased islet size and corresponding morphological alterations that are similar to the human condition (12, 13). Diet-induced metabolic trauma, resulting in insulin resistance and decreased energy expenditure, is observed in mice and rats (14, 15). Indeed, caloric surplus, coupled with sedentary lifestyle, are two key factors that drive obesity and produce insulin resistance; reduced insulin action in target tissues pre-disposes mice, rats, and humans to eventually develop T2DM (16). The determining factor in the development of T2DM is a decrease in function and mass of the pancreatic islet β-cells (17, 18).
Figure 1. Pancreatic islet size is increased in obese humans when compared with lean control groups.
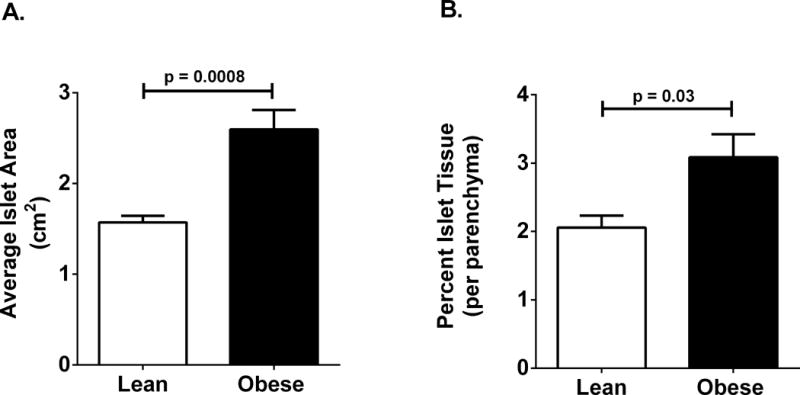
(A and B)
Data are plotted from tabled values originally reported in ref. (10), revealing significant increases in both average islet area (A) and percent islet tissue (B) in obese human subjects (n = 19 per group). Similar data can be obtained using mouse and rat models of obesity.
In an effort to gain further insights into the underlying pathology of insulin resistance and T2DM, rodent genetic models of obesity, such as ob/ob and db/db mice, as well as Zucker Diabetic Fatty (ZDF) rats, are often used to study mechanisms of tissue dysfunction relevant to the obese, insulin resistant state (19). The ob/ob mouse is obese due to a gene mutation preventing production of the leptin protein (20, 21). Leptin is a hormone that controls food intake by signaling satiety via receptor based input into the neuronal circuitry (22) and is also a regulator of intracellular lipid content in multiple tissues (23).
The db/db mouse is deficient in leptin receptor signaling due to a point mutation within the leptin receptor gene (24, 25). Thus, db/db mice are a model of leptin resistance because they have high circulating levels of the hormone, but little to no hormone action in target tissues. Both ob/ob and db/db mice display peripheral insulin resistance and compensatory increases in pancreatic islet size [see refs. (26, 27)]. An important observation noted in the ob/ob mouse model is the increase in islet volume, but an unchanged total number of pancreatic islets (26). The phenotype of increased islet size, due to enhancement of insulin-positive cell area, of these mice is similar to that of the high-fat fed mouse [ref. (28) and SJB, MDK, and JJC, unpublished data]. In addition, the expansion of islets appears to be due to increases in the number of β-cells per islet rather than an increase in the total number of islets (26, 29).
Similar to db/db mice, the ZDF rat has a leptin receptor mutation that causes obesity secondary to deficiencies in leptin receptor signaling. ZDF rats, which are homozygous for the leptin receptor mutation, are insulin resistant, hyperlipidemic, and have larger pancreatic islets than either heterozygous littermates or standard control laboratory rats (e.g., Wistar, Sprague-Dawley, etc.). All of these rodent models share many phenotypic similarities to the human metabolic syndrome, such as obesity, hyperlipidemia, insulin resistance, and hyperinsulinemia. Moreover, once insulin production and release from the pancreatic β-cells are unable to meet metabolic needs in these rodent models, overt hyperglycemia ensues (18). These observations are similar to obese humans that progress to T2DM (30).
A key component of most, if not all, of the diet-induced or genetically-driven obese rodent models is the compensatory expansion of pancreatic islets and resulting hyperinsulinemia. This enhancement in pancreatic islet size appears to arise via an increase in the proliferation of pancreatic β-cells. Most striking is that the induction of islet β-cell proliferation in response to obesigenic signals is rapid, often occurring concurrently with or prior to overt insulin resistance (31, 32). One interpretation of these results is that islet β-cells have evolved acute “sensory” mechanisms that allow the organism to deal with a developing problem of existing or impending homeostatic imbalance, which includes the onset of insulin resistance and related metabolic trauma.
Thus, in obese mice, rats, and humans, prevention of diabetes depends on greater amounts of insulin secretion to compensate for reduced insulin action in peripheral tissues (e.g., liver and skeletal muscle). The unique findings of larger pancreatic islets first put forth by Ogilvie in 1933 (10) have since been confirmed and extended in more recent studies comparing lean and obese humans subjects. These newer studies have further revealed that insulin resistance is directly associated with the observed augmentation in islet size (11, 33). Moreover, the expansion of islet area is most likely due to increased numbers of β-cells as an adaptive response to meet the enhanced metabolic demands associated with the obese state. By contrast, in the absence of functional islet β-cells, the endocrine disease diabetes mellitus develops due to insulin deficiency (16, 17, 34) and parallel hyperglucagonemia (35). Collectively, the use of both genetic and diet-induced rodent models has helped reveal specific features of islet biology that explain particular components of the human disease.
Lipodystrophy
Caloric excess leading to obesity, followed by the increased eventual storage of lipid in lean tissues during chronic overnutrition, is associated with insulin resistance, hyperlipidemia, cardiomyopathy, hepatic steatosis, and T2DM (36). Strikingly, a similar scenario in which lipids accumulate in lean tissues (e.g., liver, muscle, pancreatic islets, etc.) is lipodystrophy, a medical condition characterized by abnormal or degenerative reductions in adipose tissue depots (37). While most heritable lipodystrophies are rare syndromes, a progressively prevalent type of acquired lipodystrophy occurs due to prolonged protease inhibitor therapy, such as needed to control HIV (38). Regardless of the mechanisms underlying lipodystrophy, severe defects in lipid and glucose homeostasis occur as a result of the lack of adipose tissue.
Mice with generalized PPARgamma (PPARγ) knockout (MORE-PGKO) display lipodystrophy, insulin resistance, and increased area of the pancreatic islets. Male and female MORE-PGKO mice have impaired insulin sensitivity; however, only the male mice were hyperglycemic despite elevated plasma insulin levels relative to lean controls (39). Female mice were likely protected from hyperglycemia due to an even 5-fold higher circulating insulin level relative to the male MORE-PGKO mice. Both male and female MORE-PGKO mice displayed enlarged pancreatic islets, consistent with their hyperinsulinemia.
A separate model of lipodystrophy in the mouse was generated by insertion of the Tet activator (tTA) and a tTA-regulated Flag-tagged PPARγ transgene in place of the endogenous PPARγ gene (40). These mice, termed PPARγldi, allow for conditional generation of the lipodystrophic phenotype, with the metabolic sequelae typical of the lack of adipose tissue, including increases in circulating triglycerides levels, cholesterol, and free fatty acids. The PPARγldi /+ mice are glucose intolerant despite hyperinsulinemia, with marked increases in the size of the pancreatic islets (40). This model is consistent with other studies that show insulin resistance correlating with enhanced pancreatic islet area.
In a third mouse model of lipodystrophy, which used fat-specific PPARγ deletion (PPARγ FKO) to drive marked reductions in brown and white adipose tissue, a phenotype of extreme insulin resistance secondary to fat loss occurred (41). This lipodystrophic model also displays hyperglycemia despite striking hyperinsulinemia. Furthermore, a massive expansion of pancreatic islets was present, ostensibly to maintain the higher insulin output required to counterbalance the extreme insulin resistant state of these mice. Indeed, the authors note that circulating insulin is >60-fold higher in the PPARγ FKO mice relative to controls (41), showcasing the amazing ability of the pancreatic islets to adapt to severe alterations in metabolic homeostasis.
In summary, after onset of lipodystrophy in three distinct mouse models, it is clear that the lack of adipose tissue, which is both a lipid storage depot and an endocrine organ, reproduces many of the symptoms associated with the human metabolic syndrome. These phenotypes are remarkably reproducible in mice, rats, and humans, emphasizing three key points:
The importance of storing lipids within adipose tissue to prevent adverse metabolic events (i.e., metabolic trauma).
The significance of adipose tissue derived hormones (e.g., leptin, etc.) for appropriate regulation of glucose and lipid homeostasis.
The ability of the pancreatic islets to adapt to metabolic trauma (e.g., insulin resistance induced by lipid accumulation in lean tissues) via increases in β-cell mass and heightened insulin secretion.
Glucocorticoid Excess
The corticosteroid hormones (e.g., glucocorticoids) are synthesized in the adrenal glands and signal through specific nuclear hormone receptors (e.g., glucocorticoid receptor aka NR3C1). The glucocorticoid receptor (GR) is present in nearly all vertebrate animal cells, indicating widespread organismal tissue sensitivity to glucocorticoid hormones. Glucocorticoids play important roles in regulating tissue nutrient processing, thus contributing to overall maintenance of glucose homeostasis (42). However, glucocorticoid excess and deficiency each produce specific clinical conditions, illustrating the importance of precise control of the synthesis, release, and activity of these endogenous steroid hormones.
Cushing’s syndrome describes excess glucocorticoid exposure, regardless of the primary causative agent (43). Cushing’s disease is a specific situation involving adrenocorticotropic hormone (ACTH) excess due to pituitary adenoma and will therefore often include specific symptoms unrelated to glucocorticoid overexposure. Currently, however, glucocorticoid excess is most commonly caused by clinical administration of these steroids to treat diseases with inflammatory components. For that reason, we will restrict the current discussion solely to glucocorticoid excess induced via pharmacologically relevant means to illustrate how this situation produces insulin resistance resulting in adaptive responses within the pancreatic islets.
The predominant acute metabolic effects of glucocorticoids are to promote increases in blood glucose by stimulating hepatic gluconeogenesis. This metabolic outcome is driven by alterations in specific metabolic enzyme genes, such as enhanced transcription of the gene encoding phosphoenolpyruvate carboxykinase, an enzyme widely viewed as rate controlling for gluconeogenesis (44, 45). In addition, breakdown of skeletal muscle provides free alanine, one of the most important amino acids contributing to gluconeogenesis. While acute endogenous glucocorticoid production will promote lipolysis to support fasting state needs, chronic glucocorticoid excess, especially due to pharmacologic administration, often promotes enhanced fat deposition. Clinical administration of glucocorticoids typically produces insulin resistance within 48h in human subjects (46), with a compensatory increase in insulin secretion (47). This phenotype can be reproduced in rats given daily injections of dexamethasone (48, 49), a synthetic glucocorticoid often used clinically to treat diseases with inflammatory components. Thus, glucocorticoids have both rapid and sustained effects that influence overall glucose homeostasis.
In an effort to model the human conditions associated with chronic glucocorticoid excess, a mouse model of oral corticosterone delivery was developed by Karatsoreos and coworkers (50). This in vivo experimental system has several advantages, including that it is non-invasive, does not require adrenalectomy, and is relatively inexpensive to conduct. In this model, corticosterone is delivered over several weeks via drinking water at 100 μg/mL, which is sufficient to increase deposition of white adipose tissue, promote overall weight gain, and impair glucose tolerance (50). The mice receiving corticosterone also display elevated lipids in the blood as well as hyperleptinemia and hyperinsulinemia.
In a separate study, the metabolic trauma associated with oral corticosterone delivery (100 μg/mL) was extended to include increased lipid storage in liver and skeletal muscle (51). Furthermore, pancreatic islet size was increased in mice receiving oral corticosterone relative to their vehicle control counterparts. Insulin-positive cell area was enlarged by 2.3-fold in mice exposed to corticosterone versus mice given the vehicle control. These results are consistent with the 3-fold increase in pancreatic islet volume and are likely explained by the 2.7-fold increase in markers of islet β-cell proliferation (51). Because glucocorticoid regimens can induce rapid insulin resistance in humans (46), it is plausible that the increase in pancreatic islet size and insulin output in mice and humans are adaptive responses to address the need for more circulating insulin to compensate for reduced insulin action in liver and skeletal muscle.
Genetic Deletion of Fibroblast Growth Factor 21 (FGF21)
Human FGF21 is a 181 amino acid protein with ~75% identity to mouse FGF21. It is secreted predominantly by the liver but also by other tissues involved in glucose and lipid homeostasis [e.g., adipose tissue and pancreas; see ref. (52)]. FGF21 serves as an endocrine signal and influences insulin sensitivity by signaling through FGF receptors, where β-Klotho serves as a cofactor (53). There are documented alterations in FGF21 production and circulation during many stress and disease settings, with increased abundance observed in coronary heart disease (54), Cushing’s syndrome (55), and obesity (56), but decreased abundance in anorexia nervosa (57) and autoimmune diabetes (58). In addition, FGF21 protects mice from toxicity induced by lipopolysaccharide, a model of sepsis (59).
Mice with genetic deletion of FGF21 gain more weight and have increased adipose tissue and liver mass relative to their control counterparts (60). In addition, FGF21 KO mice store more lipid in hepatic tissue (60). The FGF21 KO mice are insulin resistant, but exhibit largely normal blood glucose levels (61). The prevention of hyperglycemia in the FGF21 KO mice is most likely due to their enhanced circulating levels of insulin (61). Upon examination, FGF21 KO mice have larger islets with more production of insulin transcripts when compared with littermate control mice. Moreover, there is an increase in Ki-67 positive cells, an index of proliferation, within the islets of FGF21 KO mice. This Ki-67 data is consistent with the increase in islet size and is most likely explained by augmented β-cell mass through cellular replication.
There are more glucagon positive alpha cells within the islets of FGF21 KO mice, although no increases in glucagon mRNA or circulating hormone were reported. Moreover, the islets isolated from FGF21 deficient mice display blunted responses to a glucose challenge (61), similar to what is observed in islets isolated from obese, insulin-resistance mice (62). Thus, pancreatic islets from discrete models of insulin resistance, such as the db/db and FGF21 KO mice, often display a common phenotype of enhanced insulin secretion at low glucose but diminished insulin secretory output in response to a high glucose challenge. The most likely explanation for these experimental phenotypes is the necessity for sustained production and secretion of insulin to offset chronically reduced hormonal action in peripheral tissues.
Heterozygous Deletion of GLUT4
One of the major functions of insulin is to stimulate glucose uptake into adipose tissue and skeletal muscle. Normally, distributed control of glucose uptake occurs via recruitment of the facilitative glucose transporter GLUT4 to the cell surface coupled to phosphorylation of the sugar by hexokinase II in response to insulin receptor activation by the insulin hormone (63). These processes are disrupted or severely diminished in the obese state, where a decreased action of insulin is an early link to the metabolic syndrome (64).
To determine the contribution of GLUT4 to overall glucose homeostasis in vivo, mice with one copy of the GLUT4 gene were generated (65). Not surprisingly, skeletal muscle glucose uptake was impaired due to this genetically-induced reduction in the major insulin-stimulated glucose transport protein. As a result, these mice are insulin resistant without being obese. The majority of the GLUT4+/− mice display elevated circulating levels of insulin in the fed state, which is sustained throughout their lifespan. Since the GLUT4+/− mice maintain lifelong insulin production, the remarkable ability of the pancreatic β-cells to compensate for reduced glucose uptake into skeletal muscle (and adipose tissue) over the mouse life span exemplifies the highly adaptable nature of the endocrine pancreas.
Not surprisingly, the pancreatic islets in the GLUT4+/− mice are much larger when compared to their GLUT4+/+ control counterparts (66). It is plausible that insulin resistance induced by heterozygous deletion of GLUT4 protein in mice is a major driver of the adaptive expansion in islet β-cell mass. This is interesting because the GLUT4+/− mice, while not displaying an obese phenotype, have islets as large as those from mice fed a high-fat diet (66). In addition, Dai, et al. found that in multiple mouse models of insulin resistance, there is an increased dilation of blood vessels in the pancreatic islets, but no change in angiogenesis (66). Thus, the vascular adaptation that occurs in the pancreatic islets of insulin-resistant animals may be one way to increase insulin output in an attempt to counteract the reduced actions of insulin in peripheral tissues.
Atypical Anti-psychotic Drug Therapy
The atypical antipsychotics, also known as second-generation antipsychotics (SGAs), include clozapine, olanzapine, and risperidone, and are used worldwide as pharmacological interventions for psychiatric conditions. These drugs are deemed essential by the clinical guidelines for their use in the treatment of schizophrenia, particularly in forms of the disease that have shown resistance to first generation antipsychotic drugs. For example, clozapine is often used in the management of early onset and treatment-resistant schizophrenia (67), with reduced motor system side effects when compared with other neuroleptic drugs (68). Clozapine and other SGAs are classified as atypical antipsychotic drugs due to their ability to engage serotonin, dopamine, and other receptors related to psychiatric disorders. However, these atypical antipsychotic drugs often produce a variety of metabolically relevant side effects, which include obesity and insulin resistance (69).
Patients with mental illness have increased risk of metabolic syndrome correlating with reduced life span, primarily due to premature cardiovascular events (70). The prolonged use of SGAs to treat psychiatric conditions (e.g., schizophrenia) promotes weight gain, insulin resistance, hyperlipidemia, and risk of hyperglycemia. The use of mouse models to study the consequences associated with SGA therapy have yielded insights into these metabolic effects, including the impact of drug administration on the pancreatic islets.
In C57BL/6 mice, administration of discrete atypical antipsychotic drugs impairs glucose transport, leading to hyperglycemia (71). In addition, clozapine dose-dependently increases blood glucose levels within 3 h after administration, demonstrating potent acute metabolic effects of this drug. Chronic administration (21 days) of clozapine to rats induces marked changes in islet morphology, including increases in islet size (72). This observation is similar to that observed in the GLUT4+/− mice (see above), where genetically-induced decreases in glucose transport (i.e., resistance to insulin action) augments insulin secretion and pancreatic islet size.
The risk of diabetes is much higher in humans on an atypical antipsychotic drug regimen (73). This marked increased in diabetes risk is most likely due to the acute decrease in glucose transport, promoting early stage hyperglycemia. If this hyperglycemia is not corrected, and is followed by a later stage failure of insulin output to meet the demand for peripheral insulin resistance, diabetes will manifest. Data from rodent models support this interpretation. Because glucose serves a natural mitogen for β-cell replication (74, 75), early hyperglycemic episodes, which can be induced by psychotropic medications (71), likely support increases in β-cell number and thus overall islet expansion.
Moreover, acute clozapine exposure in isolated mouse islets enhance both insulin and glucagon release. These islet secretory responses show a clear deviation from the usual islet physiology where elevated insulin secretion normally suppresses glucagon release (76). Additionally, seven days of clozapine exposure in isolated rat pancreatic islets decreases stimulus-secretion coupling responses necessary for fuel-mediated insulin release (77). Thus, the overall increase in risk of diabetes due to SGA drug regimens arises through both peripheral effects of the drugs and via major alterations in pancreatic islet physiology.
Relevance to Shock, Trauma and Critical Illness
While the focus of this review centers on the functional and structural adaptations of the endocrine pancreas to chronic conditions of metabolic trauma, we note that it is not completely understood how pancreatic islets specifically respond to, or compensate for, the acute metabolic demands associated with specific traumatic events, such as hemorrhagic, septic, burn or cardiogenic shock. Literature reports indicate that critical illness and other forms of trauma, including burn injury, are associated with a decrease in glucose utilization and peripheral insulin resistance (78–81). Consequently, blockade of the renin-angiotensin system (RAS) may improve glucose utilization in diabetic subjects and thus conceivably decrease the onset or severity of type 2 diabetes (82). This is interesting because plasma angiotensin II is elevated during sepsis, trauma and burn injury (83–85), situations that create whole body insulin resistance. The insulin resistant state associated with these events can be partially ameliorated by immediate post-trauma intervention with an angiotensin II type 1 (AT1) receptor blocker in a rat model of burn injury (78, 79). The adaptive responses of the pancreatic islets in these situations have, in many cases, not been thoroughly examined.
In a recent study, it was reported that a 4 week chronic infusion of angiotensin II impaired glucose tolerance in vivo and abolished glucose stimulated insulin secretion independent of changes in blood pressure (86). Angiotensin II also had deleterious effects on pancreatic islet mitochondrial function and insulin secretion due to IL-1β and NF-κB mediated inflammation in islets (86). Since components of the RAS, including angiotensin-converting enzyme 2 (ACE2), are found in rodent and human pancreatic islets (87–89), and overexpression of ACE2 in pancreas of diabetic mice improve glycemic control (89), the efficacy of RAS blockade to ameliorate insulin resistance in burn injury may also be due to improvements in pancreatic islet function.
Angiotensin II also reduces pancreatic blood flow and a reduction in first phase of glucose-stimulated insulin secretion (90). This is consistent with the regulation of blood flow to the pancreas being under the control of a variety of neural and hormonal mediators as well as other changes in circulatory conditions (91). Under conditions of hemorrhagic stress and hypotension, intravital fluorescence microscopy has shown that nutritive perfusion of pancreatic tissue is reduced in a stepwise fashion with reductions in blood pressure (92). However, it is not known if a change in local perfusion of the endocrine pancreas under conditions of hemorrhagic hypotension has long-term compensatory consequences on islet beta-cell mass and/or stimulus-secretion coupling that controls insulin secretion.
In addition, in a sepsis model, parental glucose infusion impairs the insulin secretion capability (93). This appears to be due to the combination of a glucose challenge on top of severe systemic inflammation, because LPS or glucose alone do not create metabolic dysfunction. A key component of this model is that induction of hyperglycemia appears to require the presence of inflammation and reduced insulin secretion (94). These studies, using a clinically relevant sepsis model in C57BL/6 mice, are consistent with previous work by our group and others showing that pro-inflammatory cytokine exposure impairs glucose-stimulated insulin secretion (95, 96). Thus, inflammation-based dysfunction in the pancreatic islets may be a critical factor promoting hyperglycemia in disease models.
On a final note, we suspect that obese individuals or patients on specific drug regimens that influence insulin sensitivity (e.g., atypical anti-psychotics) would fare worse during septic shock or burn trauma. The rationale is that metabolic dysregulation may arise through a multifactorial process that can include a traumatic event (e.g., sepsis), coupled with a potential drug therapy, or existing condition (e.g., obesity), that promotes adverse metabolic side effects. The combination of these factors would ostensibly put undue stress on the pancreatic islets and other tissues to maintain glucose homeostasis. This extra stress may directly promote insulin insufficiency or predispose the islets to pathological alterations in fuel-mediated insulin release. Studies directly exploring these possibilities are warranted.
Summary and Discussion
In this review, we have attempted to highlight and summarize the response of pancreatic islets to a variety of adverse metabolic conditions, many of which arise through dissimilar origins (Figure 2). Other specific scenarios mandating an increase in islet size, such as pregnancy, have been reviewed elsewhere (97), and was not considered here because it is not a pathological condition. The transcriptional responses and cell cycle regulatory mechanisms that facilitate islet development and growth have also been reviewed previously (98, 99).
Figure 2. Insulin resistance arises through many discrete possibilities, all of which are capable of inducing increases in pancreatic islet mass and function.
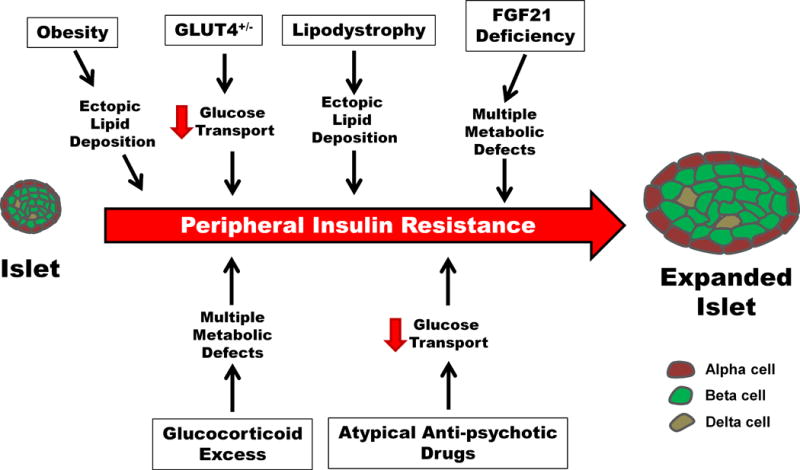
Enhancements in pancreatic islet mass are often linked with the induction of insulin resistance. Insulin resistance can be induced by a variety of distinct events and can also be temporally sustained. The impact of insulin resistance on insulin secretion is often an increase in basal insulin release with a reduced ability of the islet to respond to a glucose challenge. The eventual failure of pancreatic β-cells to produce and secrete sufficient quantities of insulin results in adverse metabolic events, most notably the onset of diabetes mellitus.
It is noteworthy that there are other experimental situations, such as tissue-specific states of insulin resistance, which trigger increases in pancreatic islet mass and function. For example, when insulin signaling is specifically disrupted in the liver, glucose homeostasis and other metabolic outcomes in the whole animal are perturbed (100–102). As a result of this hepatic insulin resistance, a massive increase in pancreatic islet mass (up to 10-fold enhancement) ensues, producing pronounced hyperinsulinemia (102). The striking expansion in pancreatic islet mass and function that occurs upon induction of hepatic insulin resistance relies, at least in part, on the transcriptional activity of Pdx-1 (103). Pdx-1 is a transcription factor involved in pancreas development and in the maintenance of adult β-cell identity (104).
Additional transcription factors, such as Nkx6.1 and MafA, which are also required to maintain the adult β-cell phenotype (105, 106), likely play key roles during β-cell mass expansion, enhanced islet β-cell function, or both processes throughout situations of metabolic trauma. The absence of these key transcriptional regulatory proteins is linked to the diminutions of islet β-cell mass and function associated with overt diabetes (95, 107). Collectively, these findings connect basic and clinical research, because an overall reduction in islet β-cell mass and function is the ultimate driver of diabetes onset.
Two separate biological mechanisms have been put forth to explain the reduction in islet β-cell quantity and overall insulin output that precede the development of diabetes mellitus. These include activation of specific death mechanisms, such as apoptosis, and a more recently described process of cellular de-differentiation. Evidence for apoptosis has largely arisen through histological analysis of pancreatic tissue, often using TUNEL staining as a marker (108). However, TUNEL staining only indicates that DNA damage is present but is incapable of identifying a specific death mechanism (109). Because β-cells have the ability to repair damaged DNA (110), the link between loss of β-cell mass and a specific death pathway (e.g., apoptosis, necrosis, etc.) leading to T2DM is still an unresolved issue.
On the other hand, the process of de-differentiation is an exciting and compelling new viewpoint from which to explain losses in β-cell mass and function (111, 112). In this scenario, the islet β-cell loses the ability to maintain a mature phenotype, defined by insulin production and stimulus-secretion coupling. This is typically due to diminished expression of key transcription factors required to support β-cell growth and function (107, 113). Importantly, this description of β-cell loss is not associated with any evidence of apoptosis (114), which is consistent with the idea that the “immature” cells can also regain mature β-cell identity (i.e., re-differentiation). Thus, it is entirely possible that the continuum of mature β-cell → “immature” de-differentiated β-cell allows for protection against inflammatory or other damaging signals. Once the offending stimuli are withdrawn, re-differentiation, such as with insulin therapy, could restore β-cell mass and function (114). Further studies will be required to fully address these outstanding issues.
In summary, the function and mass of the pancreatic islets are altered in states of metabolic disturbance, showcasing the dynamic ability of the endocrine pancreas to compensate for peripheral abnormalities. For diabetes to occur, the function and/or mass of the pancreatic β-cells must be compromised (115–118). Importantly, attempts to understand the dynamic changes in islet mass expansion, as well as inflammatory responses within β-cells, have benefitted from the establishment of novel “omics” technologies (119–122). Our goal herein was to illustrate the outstanding ability of the endocrine pancreas to respond to various, albeit distinct, scenarios that require an increase in insulin production and release. As outlined in the disparate situations presented above, the responses of the pancreatic islets have evolved to combat multiple situations of reduced peripheral insulin action and related metabolic trauma. Whether the increase in pancreatic islet size is secondary to insulin resistance, arises in parallel with insulin resistance, or is independently a marker of insulin resistance is not completely understood. In addition, the specific growth mechanisms required for increased islet size, enhanced β-cell mass, and heightened insulin secretion are all active research areas.
Acknowledgments
We thank Dr. Robert C. Noland for useful discussions.
Support: Research in the Collier laboratory is supported by NIH grants P20 GM103528 and R44 GM099207, a grant from the Edward G. Schlieder Foundation, and funds provided via the Physicians’ Medical Education Research Foundation, Knoxville, TN.
Research in the Karlstad laboratory is supported by NIH grant R44 GM099207 and through the Physicians’ Medical Education Research Foundation, Knoxville, TN.
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Bonner-Weir S, Sullivan BA, Weir GC. Human Islet Morphology Revisited: Human and Rodent Islets Are Not So Different After All. J Histochem Cytochem. 2015;63(8):604–612. doi: 10.1369/0022155415570969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab. 2003;14(9):398–403. doi: 10.1016/j.tem.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Cefalu WT. Insulin resistance: cellular and clinical concepts. Exp Biol Med (Maywood) 2001;226(1):13–26. doi: 10.1177/153537020122600103. [DOI] [PubMed] [Google Scholar]
- 5.Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15(5):635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haslam DW, James WP. Obesity. Lancet. 2005;366(9492):1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 8.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89(6):2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 9.Grant RW, Dixit VD. Adipose tissue as an immunological organ. Obesity (Silver Spring) 2015;23(3):512–518. doi: 10.1002/oby.21003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogilvie RF. The Islands of Langerhans in 19 Cases of Obesity. The Journal of Pathology and Bacteriology. 1933;37(3):473–481. [Google Scholar]
- 11.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. beta-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36(1):111–117. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hellerstrom C, Hellman B. The Islets of Langerhans in yellow obese mice. Metabolism. 1963;12:527–536. [PubMed] [Google Scholar]
- 13.Gonet AE, Stauffacher W, Pictet R, Renold AE. Obesity and diabetes mellitus with striking congenital hyperplasia of the islets of langerhans in spiny mice (Acomys Cahirinus): I. Histological findings and preliminary metabolic observations. Diabetologia. 1966;1(3–4):162–171. doi: 10.1007/BF01257907. [DOI] [PubMed] [Google Scholar]
- 14.Storlien LH, James DE, Burleigh KM, Chisholm DJ, Kraegen EW. Fat feeding causes widespread in vivo insulin resistance, decreased energy expenditure, and obesity in rats. Am J Physiol. 1986;251(5 Pt 1):E576–583. doi: 10.1152/ajpendo.1986.251.5.E576. [DOI] [PubMed] [Google Scholar]
- 15.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37(9):1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 16.Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. 2014;383(9922):1068–1083. doi: 10.1016/S0140-6736(13)62154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kahn BB. Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance. Cell. 1998;92(5):593–596. doi: 10.1016/s0092-8674(00)81125-3. [DOI] [PubMed] [Google Scholar]
- 18.Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(3):193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 19.Unger RH, Orci L. Diseases of liporegulation: new perspective on obesity and related disorders. FASEB J. 2001;15(2):312–321. doi: 10.1096/fj.00-0590. [DOI] [PubMed] [Google Scholar]
- 20.Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Hered. 1950;41(12):317–318. doi: 10.1093/oxfordjournals.jhered.a106073. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 22.Munzberg H, Morrison CD. Structure, production and signaling of leptin. Metabolism. 2015;64(1):13–23. doi: 10.1016/j.metabol.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimabukuro M, Koyama K, Chen G, Wang MY, Trieu F, Lee Y, Newgard CB, Unger RH. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc Natl Acad Sci U S A. 1997;94(9):4637–4641. doi: 10.1073/pnas.94.9.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379(6566):632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 25.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84(3):491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 26.Bock T, Pakkenberg B, Buschard K. Increased islet volume but unchanged islet number in ob/ob mice. Diabetes. 2003;52(7):1716–1722. doi: 10.2337/diabetes.52.7.1716. [DOI] [PubMed] [Google Scholar]
- 27.Boquist L, Hellman B, Lernmark A, Taljedal IB. Influence of the mutation “diabetes” on insulin release and islet morphology in mice of different genetic backgrounds. J Cell Biol. 1974;62(1):77–89. doi: 10.1083/jcb.62.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hull RL, Andrikopoulos S, Verchere CB, Vidal J, Wang F, Cnop M, Prigeon RL, Kahn SE. Increased dietary fat promotes islet amyloid formation and beta-cell secretory dysfunction in a transgenic mouse model of islet amyloid. Diabetes. 2003;52(2):372–379. doi: 10.2337/diabetes.52.2.372. [DOI] [PubMed] [Google Scholar]
- 29.Dalboge LS, Almholt DL, Neerup TS, Vassiliadis E, Vrang N, Pedersen L, Fosgerau K, Jelsing J. Characterisation of age-dependent beta cell dynamics in the male db/db mice. PLoS One. 2013;8(12):e82813. doi: 10.1371/journal.pone.0082813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA. Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care. 2008;31(Suppl 2):S161–164. doi: 10.2337/dc08-s243. [DOI] [PubMed] [Google Scholar]
- 31.Mosser RE, Maulis MF, Moulle VS, Dunn JC, Carboneau BA, Arasi K, Pappan K, Poitout V, Gannon M. High-fat diet-induced beta-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am J Physiol Endocrinol Metab. 2015;308(7):E573–582. doi: 10.1152/ajpendo.00460.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stamateris RE, Sharma RB, Hollern DA, Alonso LC. Adaptive beta-cell proliferation increases early in high-fat feeding in mice, concurrent with metabolic changes, with induction of islet cyclin D2 expression. Am J Physiol Endocrinol Metab. 2013;305(1):E149–159. doi: 10.1152/ajpendo.00040.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mezza T, Muscogiuri G, Sorice GP, Clemente G, Hu J, Pontecorvi A, Holst JJ, Giaccari A, Kulkarni RN. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes. 2014;63(3):994–1007. doi: 10.2337/db13-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward WK, Johnston CL, Beard JC, Benedetti TJ, Porte D., Jr Abnormalities of islet B-cell function, insulin action, and fat distribution in women with histories of gestational diabetes: relationship to obesity. J Clin Endocrinol Metab. 1985;61(6):1039–1045. doi: 10.1210/jcem-61-6-1039. [DOI] [PubMed] [Google Scholar]
- 35.Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A. 2010;107(37):16009–16012. doi: 10.1073/pnas.1006639107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Unger RH. Lipotoxic diseases. Annu Rev Med. 2002;53:319–336. doi: 10.1146/annurev.med.53.082901.104057. [DOI] [PubMed] [Google Scholar]
- 37.Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta. 2010;1801(3):209–214. doi: 10.1016/j.bbalip.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Carr A, Samaras K, Burton S, Law M, Freund J, Chisholm DJ, Cooper DA. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12(7):F51–58. doi: 10.1097/00002030-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Duan SZ, Ivashchenko CY, Whitesall SE, D’Alecy LG, Duquaine DC, Brosius FC, 3rd, Gonzalez FJ, Vinson C, Pierre MA, Milstone DS, Mortensen RM. Hypotension, lipodystrophy, and insulin resistance in generalized PPARgamma-deficient mice rescued from embryonic lethality. J Clin Invest. 2007;117(3):812–822. doi: 10.1172/JCI28859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S, Huang LW, Snow KJ, Ablamunits V, Hasham MG, Young TH, Paulk AC, Richardson JE, Affourtit JP, Shalom-Barak T, Bult CJ, Barak Y. A mouse model of conditional lipodystrophy. Proc Natl Acad Sci U S A. 2007;104(42):16627–16632. doi: 10.1073/pnas.0707797104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang F, Mullican SE, DiSpirito JR, Peed LC, Lazar MA. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. Proc Natl Acad Sci U S A. 2013;110(46):18656–18661. doi: 10.1073/pnas.1314863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Granner DK, Wang JC, Yamamoto KR. Regulatory Actions of Glucocorticoid Hormones: From Organisms to Mechanisms. Adv Exp Med Biol. 2015;872:3–31. doi: 10.1007/978-1-4939-2895-8_1. [DOI] [PubMed] [Google Scholar]
- 43.Nieman LK. Cushing’s syndrome: update on signs, symptoms and biochemical screening. Eur J Endocrinol. 2015;173(4):M33–38. doi: 10.1530/EJE-15-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collier JJ, Scott DK. Sweet changes: glucose homeostasis can be altered by manipulating genes controlling hepatic glucose metabolism. Mol Endocrinol. 2004;18(5):1051–1063. doi: 10.1210/me.2003-0357. [DOI] [PubMed] [Google Scholar]
- 45.O’Brien RM, Printz RL, Halmi N, Tiesinga JJ, Granner DK. Structural and functional analysis of the human phosphoenolpyruvate carboxykinase gene promoter. Biochim Biophys Acta. 1995;1264(3):284–288. doi: 10.1016/0167-4781(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 46.Beard JC, Halter JB, Best JD, Pfeifer MA, Porte D., Jr Dexamethasone-induced insulin resistance enhances B cell responsiveness to glucose level in normal men. Am J Physiol. 1984;247(5 Pt 1):E592–596. doi: 10.1152/ajpendo.1984.247.5.E592. [DOI] [PubMed] [Google Scholar]
- 47.Ludvik B, Clodi M, Kautzky-Willer A, Capek M, Hartter E, Pacini G, Prager R. Effect of dexamethasone on insulin sensitivity, islet amyloid polypeptide and insulin secretion in humans. Diabetologia. 1993;36(1):84–87. doi: 10.1007/BF00399099. [DOI] [PubMed] [Google Scholar]
- 48.Rafacho A, Marroqui L, Taboga SR, Abrantes JL, Silveira LR, Boschero AC, Carneiro EM, Bosqueiro JR, Nadal A, Quesada I. Glucocorticoids in vivo induce both insulin hypersecretion and enhanced glucose sensitivity of stimulus-secretion coupling in isolated rat islets. Endocrinology. 2010;151(1):85–95. doi: 10.1210/en.2009-0704. [DOI] [PubMed] [Google Scholar]
- 49.Rafacho A, Cestari TM, Taboga SR, Boschero AC, Bosqueiro JR. High doses of dexamethasone induce increased beta-cell proliferation in pancreatic rat islets. Am J Physiol Endocrinol Metab. 2009;296(4):E681–689. doi: 10.1152/ajpendo.90931.2008. [DOI] [PubMed] [Google Scholar]
- 50.Karatsoreos IN, Bhagat SM, Bowles NP, Weil ZM, Pfaff DW, McEwen BS. Endocrine and physiological changes in response to chronic corticosterone: a potential model of the metabolic syndrome in mouse. Endocrinology. 2010;151(5):2117–2127. doi: 10.1210/en.2009-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fransson L, Franzen S, Rosengren V, Wolbert P, Sjoholm A, Ortsater H. beta-Cell adaptation in a mouse model of glucocorticoid-induced metabolic syndrome. J Endocrinol. 2013;219(3):231–241. doi: 10.1530/JOE-13-0189. [DOI] [PubMed] [Google Scholar]
- 52.Woo YC, Xu A, Wang Y, Lam KS. Fibroblast growth factor 21 as an emerging metabolic regulator: clinical perspectives. Clin Endocrinol (Oxf) 2013;78(4):489–496. doi: 10.1111/cen.12095. [DOI] [PubMed] [Google Scholar]
- 53.Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem. 2011;149(2):121–130. doi: 10.1093/jb/mvq121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin Z, Wu Z, Yin X, Liu Y, Yan X, Lin S, Xiao J, Wang X, Feng W, Li X. Serum levels of FGF-21 are increased in coronary heart disease patients and are independently associated with adverse lipid profile. PLoS One. 2010;5(12):e15534. doi: 10.1371/journal.pone.0015534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Durovcova V, Marek J, Hana V, Matoulek M, Zikan V, Haluzikova D, Kavalkova P, Lacinova Z, Krsek M, Haluzik M. Plasma concentrations of fibroblast growth factors 21 and 19 in patients with Cushing’s syndrome. Physiol Res. 2010;59(3):415–422. doi: 10.33549/physiolres.931801. [DOI] [PubMed] [Google Scholar]
- 56.Zhang X, Yeung DC, Karpisek M, Stejskal D, Zhou ZG, Liu F, Wong RL, Chow WS, Tso AW, Lam KS, Xu A. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes. 2008;57(5):1246–1253. doi: 10.2337/db07-1476. [DOI] [PubMed] [Google Scholar]
- 57.Dostalova I, Kavalkova P, Haluzikova D, Lacinova Z, Mraz M, Papezova H, Haluzik M. Plasma concentrations of fibroblast growth factors 19 and 21 in patients with anorexia nervosa. J Clin Endocrinol Metab. 2008;93(9):3627–3632. doi: 10.1210/jc.2008-0746. [DOI] [PubMed] [Google Scholar]
- 58.Xiao Y, Xu A, Law LS, Chen C, Li H, Li X, Yang L, Liu S, Zhou Z, Lam KS. Distinct changes in serum fibroblast growth factor 21 levels in different subtypes of diabetes. J Clin Endocrinol Metab. 2012;97(1):E54–58. doi: 10.1210/jc.2011-1930. [DOI] [PubMed] [Google Scholar]
- 59.Feingold KR, Grunfeld C, Heuer JG, Gupta A, Cramer M, Zhang T, Shigenaga JK, Patzek SM, Chan ZW, Moser A, Bina H, Kharitonenkov A. FGF21 is increased by inflammatory stimuli and protects leptin-deficient ob/ob mice from the toxicity of sepsis. Endocrinology. 2012;153(6):2689–2700. doi: 10.1210/en.2011-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. 2009;150(11):4931–4940. doi: 10.1210/en.2009-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.So WY, Cheng Q, Xu A, Lam KS, Leung PS. Loss of fibroblast growth factor 21 action induces insulin resistance, pancreatic islet hyperplasia and dysfunction in mice. Cell Death Dis. 2015;6:e1707. doi: 10.1038/cddis.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Do OH, Low JT, Gaisano HY, Thorn P. The secretory deficit in islets from db/db mice is mainly due to a loss of responding beta cells. Diabetologia. 2014;57(7):1400–1409. doi: 10.1007/s00125-014-3226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wasserman DH. Four grams of glucose. Am J Physiol Endocrinol Metab. 2009;296(1):E11–21. doi: 10.1152/ajpendo.90563.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Utzschneider KM, Van de Lagemaat A, Faulenbach MV, Goedecke JH, Carr DB, Boyko EJ, Fujimoto WY, Kahn SE. Insulin resistance is the best predictor of the metabolic syndrome in subjects with a first-degree relative with type 2 diabetes. Obesity (Silver Spring) 2010;18(9):1781–1787. doi: 10.1038/oby.2010.77. [DOI] [PubMed] [Google Scholar]
- 65.Stenbit AE, Tsao TS, Li J, Burcelin R, Geenen DL, Factor SM, Houseknecht K, Katz EB, Charron MJ. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat Med. 1997;3(10):1096–1101. doi: 10.1038/nm1097-1096. [DOI] [PubMed] [Google Scholar]
- 66.Dai C, Brissova M, Reinert RB, Nyman L, Liu EH, Thompson C, Shostak A, Shiota M, Takahashi T, Powers AC. Pancreatic islet vasculature adapts to insulin resistance through dilation and not angiogenesis. Diabetes. 2013;62(12):4144–4153. doi: 10.2337/db12-1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hrdlicka M, Dudova I. Atypical antipsychotics in the treatment of early-onset schizophrenia. Neuropsychiatr Dis Treat. 2015;11:907–913. doi: 10.2147/NDT.S82185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kurz M, Hummer M, Oberbauer H, Fleischhacker WW. Extrapyramidal side effects of clozapine and haloperidol. Psychopharmacology (Berl) 1995;118(1):52–56. doi: 10.1007/BF02245249. [DOI] [PubMed] [Google Scholar]
- 69.Rojo LE, Gaspar PA, Silva H, Risco L, Arena P, Cubillos-Robles K, Jara B. Metabolic syndrome and obesity among users of second generation antipsychotics: A global challenge for modern psychopharmacology. Pharmacol Res. 2015;101:74–85. doi: 10.1016/j.phrs.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 70.Newcomer JW. Antipsychotic medications: metabolic and cardiovascular risk. J Clin Psychiatry. 2007;68(Suppl 4):8–13. [PubMed] [Google Scholar]
- 71.Dwyer DS, Donohoe D. Induction of hyperglycemia in mice with atypical antipsychotic drugs that inhibit glucose uptake. Pharmacol Biochem Behav. 2003;75(2):255–260. doi: 10.1016/s0091-3057(03)00079-0. [DOI] [PubMed] [Google Scholar]
- 72.Abdelrahim EA. Histopathological change of the endocrine pancreas in male albino rat treated with the atypical antipsychotic clozapine. Rom J Morphol Embryol. 2013;54(2):385–394. [PubMed] [Google Scholar]
- 73.Lambert BL, Chou CH, Chang KY, Tafesse E, Carson W. Antipsychotic exposure and type 2 diabetes among patients with schizophrenia: a matched case-control study of California Medicaid claims. Pharmacoepidemiol Drug Saf. 2005;14(6):417–425. doi: 10.1002/pds.1092. [DOI] [PubMed] [Google Scholar]
- 74.Alonso LC, Yokoe T, Zhang P, Scott DK, Kim SK, O’Donnell CP, Garcia-Ocana A. Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes. 2007;56(7):1792–1801. doi: 10.2337/db06-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamashita S, Tobinaga T, Ashizawa K, Nagayama Y, Yokota A, Harakawa S, Inoue S, Hirayu H, Izumi M, Nagataki S. Glucose stimulation of protooncogene expression and deoxyribonucleic acid synthesis in rat islet cell line. Endocrinology. 1988;123(4):1825–1829. doi: 10.1210/endo-123-4-1825. [DOI] [PubMed] [Google Scholar]
- 76.Greenbaum CJ, Havel PJ, Taborsky GJ, Jr, Klaff LJ. Intra-islet insulin permits glucose to directly suppress pancreatic A cell function. J Clin Invest. 1991;88(3):767–773. doi: 10.1172/JCI115375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sasaki N, Iwase M, Uchizono Y, Nakamura U, Imoto H, Abe S, Iida M. The atypical antipsychotic clozapine impairs insulin secretion by inhibiting glucose metabolism and distal steps in rat pancreatic islets. Diabetologia. 2006;49(12):2930–2938. doi: 10.1007/s00125-006-0446-6. [DOI] [PubMed] [Google Scholar]
- 78.Kasper SO, Castle SM, Daley BJ, Enderson BL, Karlstad MD. Blockade of the renin-angiotensin system improves insulin sensitivity in thermal injury. Shock. 2006;26(5):485–488. doi: 10.1097/01.shk.0000230302.24258.9f. [DOI] [PubMed] [Google Scholar]
- 79.Kasper SO, Phillips EE, Castle SM, Daley BJ, Enderson BL, Karlstad MD. Blockade of the Renin-Angiotensin system improves insulin receptor signaling and insulin-stimulated skeletal muscle glucose transport in burn injury. Shock. 2011;35(1):80–85. doi: 10.1097/SHK.0b013e3181e762da. [DOI] [PubMed] [Google Scholar]
- 80.Sugita H, Kaneki M, Sugita M, Yasukawa T, Yasuhara S, Martyn JA. Burn injury impairs insulin-stimulated Akt/PKB activation in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288(3):E585–591. doi: 10.1152/ajpendo.00321.2004. [DOI] [PubMed] [Google Scholar]
- 81.Ikezu T, Okamoto T, Yonezawa K, Tompkins RG, Martyn JA. Analysis of thermal injury-induced insulin resistance in rodents. Implication of postreceptor mechanisms. J Biol Chem. 1997;272(40):25289–25295. doi: 10.1074/jbc.272.40.25289. [DOI] [PubMed] [Google Scholar]
- 82.Scheen AJ. Prevention of type 2 diabetes mellitus through inhibition of the Renin-Angiotensin system. Drugs. 2004;64(22):2537–2565. doi: 10.2165/00003495-200464220-00004. [DOI] [PubMed] [Google Scholar]
- 83.Bucher M, Ittner KP, Hobbhahn J, Taeger K, Kurtz A. Downregulation of angiotensin II type 1 receptors during sepsis. Hypertension. 2001;38(2):177–182. doi: 10.1161/01.hyp.38.2.177. [DOI] [PubMed] [Google Scholar]
- 84.Hilton JG, Marullo DS. Trauma induced increases in plasma vasopressin and angiotensin II. Life Sci. 1987;41(19):2195–2200. doi: 10.1016/0024-3205(87)90515-7. [DOI] [PubMed] [Google Scholar]
- 85.Liu D, Yang Z, Li A. Plasma renin activity (PRA), angiotensin II (AII), atrial natriuretic peptide (ANP) and AII/ANP ratio in severely burned patients. Zhonghua Zheng Xing Shao Shang Wai Ke Za Zhi. 1994;10(2):117–120. [PubMed] [Google Scholar]
- 86.Sauter NS, Thienel C, Plutino Y, Kampe K, Dror E, Traub S, Timper K, Bedat B, Pattou F, Kerr-Conte J, Jehle AW, Boni-Schnetzler M, Donath MY. Angiotensin II Induces Interleukin-1beta-Mediated Islet Inflammation and beta-Cell Dysfunction Independently of Vasoconstrictive Effects. Diabetes. 2015;64(4):1273–1283. doi: 10.2337/db14-1282. [DOI] [PubMed] [Google Scholar]
- 87.Lau T, Carlsson PO, Leung PS. Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia. 2004;47(2):240–248. doi: 10.1007/s00125-003-1295-1. [DOI] [PubMed] [Google Scholar]
- 88.Tahmasebi M, Puddefoot JR, Inwang ER, Vinson GP. The tissue renin-angiotensin system in human pancreas. J Endocrinol. 1999;161(2):317–322. doi: 10.1677/joe.0.1610317. [DOI] [PubMed] [Google Scholar]
- 89.Shoemaker R, Yiannikouris F, Thatcher S, Cassis L. ACE2 deficiency reduces beta-cell mass and impairs beta-cell proliferation in obese C57BL/6 mice. Am J Physiol Endocrinol Metab. 2015;309(7):E621–631. doi: 10.1152/ajpendo.00054.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carlsson PO, Berne C, Jansson L. Angiotensin II and the endocrine pancreas: effects on islet blood flow and insulin secretion in rats. Diabetologia. 1998;41(2):127–133. doi: 10.1007/s001250050880. [DOI] [PubMed] [Google Scholar]
- 91.Ballian N, Brunicardi FC. Islet vasculature as a regulator of endocrine pancreas function. World J Surg. 2007;31(4):705–714. doi: 10.1007/s00268-006-0719-8. [DOI] [PubMed] [Google Scholar]
- 92.Vollmar B, Franke K, Menger MD. Nutritive perfusion of pancreatic endocrine tissue during hemorrhagic hypotension: how differ islets in situ from islet isografts? Shock. 2008;30(4):428–433. doi: 10.1097/SHK.0b013e31816736b. [DOI] [PubMed] [Google Scholar]
- 93.Watanabe Y, Singamsetty S, Zou B, Guo L, Stefanovski D, Alonso LC, Garcia-Ocana A, O’Donnell CP, McVerry BJ. Exogenous glucose administration impairs glucose tolerance and pancreatic insulin secretion during acute sepsis in non-diabetic mice. PLoS One. 2013;8(6):e67716. doi: 10.1371/journal.pone.0067716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Singamsetty S, Shah FA, Guo L, Watanabe Y, McDonald S, Sharma R, Zhang Y, Alonso LC, O’Donnell CP, McVerry BJ. Early initiation of low-level parenteral dextrose induces an accelerated diabetic phenotype in septic C57BL/6J mice. Appl Physiol Nutr Metab. 2016;41(1):12–19. doi: 10.1139/apnm-2015-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burke SJ, Stadler K, Lu D, Gleason E, Han A, Donohoe DR, Rogers RC, Hermann GE, Karlstad MD, Collier JJ. IL-1beta reciprocally regulates chemokine and insulin secretion in pancreatic beta-cells via NF-kappaB. Am J Physiol Endocrinol Metab. 2015;309(8):E715–726. doi: 10.1152/ajpendo.00153.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Corbett JA, McDaniel ML. Intraislet release of interleukin 1 inhibits beta cell function by inducing beta cell expression of inducible nitric oxide synthase. J Exp Med. 1995;181(2):559–568. doi: 10.1084/jem.181.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab. 2010;21(3):151–158. doi: 10.1016/j.tem.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocana A, Vasavada R, Stewart AF. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev. 2006;27(4):356–370. doi: 10.1210/er.2006-0004. [DOI] [PubMed] [Google Scholar]
- 99.Sachdeva MM, Stoffers DA. Minireview: Meeting the demand for insulin: molecular mechanisms of adaptive postnatal beta-cell mass expansion. Mol Endocrinol. 2009;23(6):747–758. doi: 10.1210/me.2008-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Aleman JO, Suzuki R, Scapa EF, Agarwal C, Carey MC, Stephanopoulos G, Cohen DE, King GL, Ginsberg HN, Kahn CR. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7(2):125–134. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Biddinger SB, Haas JT, Yu BB, Bezy O, Jing E, Zhang W, Unterman TG, Carey MC, Kahn CR. Hepatic insulin resistance directly promotes formation of cholesterol gallstones. Nat Med. 2008;14(7):778–782. doi: 10.1038/nm1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6(1):87–97. [PubMed] [Google Scholar]
- 103.Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest. 2004;114(6):828–836. doi: 10.1172/JCI21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oliver-Krasinski JM, Stoffers DA. On the origin of the beta cell. Genes Dev. 2008;22(15):1998–2021. doi: 10.1101/gad.1670808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Taylor BL, Liu FF, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013;4(6):1262–1275. doi: 10.1016/j.celrep.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schisler JC, Jensen PB, Taylor DG, Becker TC, Knop FK, Takekawa S, German M, Weir GC, Lu D, Mirmira RG, Newgard CB. The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc Natl Acad Sci U S A. 2005;102(20):7297–7302. doi: 10.1073/pnas.0502168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, Robertson RP, Powers AC, Stein R. Inactivation of specific beta cell transcription factors in type 2 diabetes. J Clin Invest. 2013;123(8):3305–3316. doi: 10.1172/JCI65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 109.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21(5):1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 110.Hughes KJ, Meares GP, Chambers KT, Corbett JA. Repair of nitric oxide-damaged DNA in beta-cells requires JNK-dependent GADD45alpha expression. J Biol Chem. 2009;284(40):27402–27408. doi: 10.1074/jbc.M109.046912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pajvani UB, Accili D. The new biology of diabetes. Diabetologia. 2015;58(11):2459–2468. doi: 10.1007/s00125-015-3722-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weir GC, Aguayo-Mazzucato C, Bonner-Weir S. beta-cell dedifferentiation in diabetes is important, but what is it? Islets. 2013;5(5):233–237. doi: 10.4161/isl.27494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150(6):1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014;19(5):872–882. doi: 10.1016/j.cmet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.DeFronzo RA, Abdul-Ghani MA. Preservation of beta-cell function: the key to diabetes prevention. J Clin Endocrinol Metab. 2011;96(8):2354–2366. doi: 10.1210/jc.2011-0246. [DOI] [PubMed] [Google Scholar]
- 116.Burke SJ, Collier JJ. Insulitis and Diabetes: A Perspective on Islet Inflammation. Immunome Research. 2014;10 Special Issue: Cytokine Biology. [Google Scholar]
- 117.Kahn SE, Zraika S, Utzschneider KM, Hull RL. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia. 2009;52(6):1003–1012. doi: 10.1007/s00125-009-1321-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Doria A, Patti ME, Kahn CR. The emerging genetic architecture of type 2 diabetes. Cell Metab. 2008;8(3):186–200. doi: 10.1016/j.cmet.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gooding JR, Jensen MV, Newgard CB. Metabolomics applied to the pancreatic islet. Arch Biochem Biophys. 2015 doi: 10.1016/j.abb.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Collier JJ, Burke SJ, Eisenhauer ME, Lu D, Sapp RC, Frydman CJ, Campagna SR. Pancreatic beta-Cell Death in Response to Pro-Inflammatory Cytokines Is Distinct from Genuine Apoptosis. PLoS One. 2011;6(7):e22485. doi: 10.1371/journal.pone.0022485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.El Ouaamari A, Zhou JY, Liew CW, Shirakawa J, Dirice E, Gedeon N, Kahraman S, De Jesus DF, Bhatt S, Kim JS, Clauss TR, Camp DG, 2nd, Smith RD, Qian WJ, Kulkarni RN. Compensatory Islet Response to Insulin Resistance Revealed by Quantitative Proteomics. J Proteome Res. 2015;14(8):3111–3122. doi: 10.1021/acs.jproteome.5b00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Horn S, Kirkegaard JS, Hoelper S, Seymour PA, Rescan C, Nielsen JH, Madsen OD, Jensen JN, Kruger M, Gr Nborg M, Ahnfelt RNJ. Research resource: A dual proteomic approach identifies regulated islet proteins during beta cell mass expansion in vivo. Mol Endocrinol. 2015:me20151208. doi: 10.1210/me.2015-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]