


Abstract
CoinFold (http://raptorx2.uchicago.edu/ContactMap/) is a web server for protein contact prediction and contact-assisted de novo structure prediction. CoinFold predicts contacts by integrating joint multi-family evolutionary coupling (EC) analysis and supervised machine learning. This joint EC analysis is unique in that it not only uses residue coevolution information in the target protein family, but also that in the related families which may have divergent sequences but similar folds. The supervised learning further improves contact prediction accuracy by making use of sequence profile, contact (distance) potential and other information. Finally, this server predicts tertiary structure of a sequence by feeding its predicted contacts and secondary structure to the CNS suite. Tested on the CASP and CAMEO targets, this server shows significant advantages over existing ones of similar category in both contact and tertiary structure prediction.
INTRODUCTION
Protein contact prediction is the problem of predicting whether two residues in a protein are spatially proximal (typically within 8 Å in Cβ atoms) to each other in the 3D structure (1). It is known that protein residue–residue contacts contain important information for protein folding and recent works indicate that one correct long-range contact for every 12 residues in the protein allows accurate topology level modelling (2). However, contact prediction from sequence alone remains very challenging (3).
Co-evolving residues are often found to be spatially proximal in the protein structure due to the evolution pressure (4). Multiple sequence alignment (MSA) of a protein family is widely used to detect residue co-evolution (5). Recently, evolutionary coupling (EC) analysis has made good progress in contact prediction by using global statistical inference (3,4). Representative methods include EVfold (6), PSICOV (7) and pseudo-likelihood approaches (8) such as GREMLIN (9) and CCMpred (10). Nevertheless, all these EC methods analyze an individual protein family independent of the others.
Here, we present CoinFold, a web server predicting protein contact map and 3D structure using a new method (see Figure 1). In particular, CoinFold predicts contacts by joint EC analysis via Group Graphical Lasso (GGL) (11) of multiple (distantly) related protein families which may have divergent sequences but similar folds (i.e. co-evolution patterns) (12). By enforcing co-evolution pattern consistency among a set of related families, we can significantly improve contact prediction accuracy. CoinFold further improves prediction accuracy by integrating supervised learning with this joint EC analysis. Since EC analysis and supervised learning use different types of information, their combination leads to much better prediction. Finally, CoinFold predicts secondary structure using a new in-house tool DeepCNF (13) and then tertiary structure by feeding predicted contacts and secondary structure to the Crystallography & NMR System (CNS) software package (14), but without using any templates (15). Our experiments on CASP and CAMEO datasets show that CoinFold greatly outperforms the other publicly available servers of similar category.
Figure 1.

Illustration of CoinFold workflow. Given an input protein sequence, CoinFold uses HHblits (22) and HHpred (23) to generate sequence profile and search for related protein families. Then CoinFold conducts joint evolutionary coupling analysis and supervised prediction of both contacts and secondary structure. Finally, CoinFold predicts 3D models using CNS.
MATERIALS AND METHODS
The contact prediction method employed by CoinFold has been published in (12). Here, we briefly describe it and please see the paper for more technical details.
Joint evolutionary coupling analysis via group graphical lasso
We model a single protein family using Gaussian Graphical Model (GGM) (7) and jointly infer protein contacts for K related protein families (12). Let X = {X1, X2, ..., XK} denote the set of multiple sequence alignments (MSA), each for one individual family. Assume that each MSA satisfies a Gaussian distribution with a precision matrix Ωk (k = 1,2, ..., K). Let Ω denote the set {Ω1, Ω2, ..., ΩK}. Let L denote the number of columns in a MSA. We align these K MSAs and then group all the column pairs such that each group contains only mutually-aligned column pairs (see Figure 1 in our method paper (12) for an example of two aligned families). Let G ≤ L(L – 1)/2 denote the number of groups. We estimate the K precision matrices by taking into account their correlation using Group Graphical Lasso (GGL) (11) as follows.
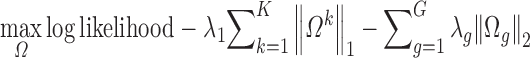 |
where the last penalty item enforces that the column pairs in the same group have similar interaction strength. That is, if a column pair in an MSA has a strong interaction, the other aligned column pairs shall also have strong interactions. The parameter λg is proportional to the conservation level in each group. See our method paper for technical details (12).
Supervised learning via neural network (NN)
In addition to co-evolution information, CoinFold uses the following features for supervised contact prediction: sequence profile (16), contact or distance potential (17), and some non-evolutionary information (18) (see our method paper (12) for their description). To make use of them, we use a supervised neural network (19) to predict the probability of two residues forming a contact and then integrate this predicted probability with joint EC analysis (12).
Tertiary structure construction
We use a similar approach as described in ConFold (15) to build 3D models of a sequence by feeding predicted secondary structure and contacts to the CNS suite (14). In brief, we predict secondary structure using our in-house new tool DeepCNF (13) and then convert it to distance, angle and h-bond restraints. We also convert the top predicted contacts to distance restraints. That is, a pair of residues predicted to form a contact is assumed to have distance between 3.5 and 8.0 Å. Finally, we build 3D structure models using the CNS suite and select top five models by energy function.
RESULT
Servers to compare
There are many methods developed for contact prediction and protein folding. Here, we compare our web server only to those publicly available servers of similar category.
Contact map prediction
We compare our server with EVfold (6), CCMpred (10), PSICOV (7), and metaPSICOV (20). The first three servers use only EC analysis, while the last one combines both EC analysis and supervised learning. Among these four servers, only EVfold yields 3D models.
Tertiary structure prediction
We compare our server with EVfold (6) and ConFold (15). We cannot compare to the RBO aleph server (21) since we failed to obtain any results from it. For each server, we evaluate only the top five predicted models (ranked by their respective scores).
Evaluation criteria
Contact map prediction
To measure the contact prediction, we evaluate the top L/10, L/5 and L/2 predicted contacts where L is the sequence length of the input protein (12). The prediction accuracy is defined as the percentage of native contacts among the top predicted contacts. Contacts are short-, medium- and long-range when the sequence distance between the two residues in a contact falls into three intervals from 6 to 11, from 12 to 23, and ≥24, respectively (1). We consider only medium- and long-range contacts, which are more relevant for protein folding (12), but more challenging to predict.
Tertiary structure prediction
We use TMscore to measure quality of a 3D (24). TMscore ranges from 0 to 1, with 0 indicating the worst quality and 1 the best quality, respectively.
Performance on the CASP and CAMEO datasets
We tested CoinFold using 228 CASP test proteins (123 CASP10 plus 105 CASP11 targets) (25), and 47 CAMEO hard test proteins (from 2015-08-01 to 2015-09-12) (26). To evaluate tertiary structure prediction, we consider 47 CAMEO hard targets and 85 CASP hard targets (36 CASP10 plus 49 CASP11). The reason why we focus on hard targets for evaluating 3D model is due to the fact that template-based modeling may be better for easy and medium-level targets (27,28). Note that all these targets share <25% sequence identity with the training data of our supervised learning method.
Contact map prediction. As shown in Tables 1 and 2, tested on all 228 CASP targets and 47 CAMEO hard targets, our server greatly outperforms EVfold, CCMpred and PSICOV when the top L/10, L/5 and L/2 predicted contacts are evaluated, no matter whether the contacts are medium- and long-range. CoinFold performs also better than the CASP11 winner metaPSICOV, which integrates both EC analysis and supervised learning.
Table 1. Contact prediction result on all 228 CASP test proteins.
| Medium range | Long range | |||||
|---|---|---|---|---|---|---|
| Methods | L/10 | L/5 | L/2 | L/10 | L/5 | L/2 |
| EVfold | 0.42 | 0.35 | 0.24 | 0.43 | 0.38 | 0.30 |
| CCMpred | 0.48 | 0.39 | 0.27 | 0.49 | 0.44 | 0.34 |
| PSICOV | 0.42 | 0.34 | 0.23 | 0.42 | 0.37 | 0.28 |
| metaPSICOV | 0.69 | 0.59 | 0.43 | 0.59 | 0.54 | 0.44 |
| CoinFold | 0.71 | 0.60 | 0.45 | 0.61 | 0.56 | 0.46 |
Table 2. Contact prediction result on all 47 CAMEO hard targets.
| Medium range | Long range | |||||
|---|---|---|---|---|---|---|
| Methods | L/10 | L/5 | L/2 | L/10 | L/5 | L/2 |
| EVfold | 0.33 | 0.28 | 0.22 | 0.49 | 0.44 | 0.35 |
| CCMpred | 0.35 | 0.29 | 0.23 | 0.44 | 0.41 | 0.35 |
| PSICOV | 0.30 | 0.24 | 0.18 | 0.39 | 0.36 | 0.31 |
| metaPSICOV | 0.57 | 0.47 | 0.35 | 0.61 | 0.55 | 0.47 |
| CoinFold | 0.59 | 0.48 | 0.37 | 0.64 | 0.59 | 0.50 |
Tertiary structure prediction
As shown in Figure 2A and C, our server can generate much better Top1 3D models on CASP and CAMEO hard targets than ConFold and EVfold. When the best of Top5 models are evaluated, our server still significantly outperforms the others (see Figure 2B and D). It should be noted that among the 132 hard targets, EVfold failed to produce 3D models for 59 targets (see Figure 2C and D).
Figure 2.

TMscore comparison of the top models generated by CoinFold, ConFold, and EVfold. The top 1 and the best (in terms of TMscore) of top 5 models are evaluated. The 36 CASP10 targets, 49 from CASP11 targets and 47 CAMEO targets are shown in red square, blue diamond, and green triangle, respectively. (A and B) Head-to-head comparison of Top1 and Top5 best models by CoinFold (in X-axis) and ConFold (in Y-axis). (C and D) Head-to-head comparison of Top1 and Top5 best models by CoinFold (in X-axis) and EVfold (in Y-axis).
SERVER IMPLEMENTATION
Overall description
Our server predicts contact map and the tertiary structure of an input protein sequence, without using any templates. Users can submit sequences through our web interface or using a publicly available program curl (see Figure 3). When the web interface is used, users may submit a batch of ≤50 sequences at a time. Our server first predicts the contact map of an input sequence and its secondary structure, then the tertiary structure using the predicted contacts and secondary structure (see Figure 4).
Figure 3.

CoinFold server job submission. (A) The web interface for job submission has fields for job name (1), optional user email address (2), and sequences to be submitted (3). The sequences shall be in FASTA format and can also be submitted in a file (3). User can also click on the example link to see an example. Submit a job by clicking on the submit button (4). (B) An example for submission by a publicly available program Curl. Only ‘sequences’ and the submission URL (shown in underlines) are required and the others are optional. A job URL will be returned on screen after submission.
Figure 4.

CoinFold server result page. The left part shows the predicted contact map (1), where the predicted score is displayed in greyscale, with a higher score represented by a darker color. The middle part shows the job status (the submitted, scheduled, and finished time) (2), as well as two downloading buttons for the predicted contact map (3) and 5 predicted 3D models (4). The right part contains a button to view alternative 3D models (5), a display bar for showing rank and score of the selected 3D model (6) and visualization of the selected 3D model (7).
Input
The only required input to the server is one (or batch of) protein sequence(s). Users may optionally provide a jobname and an email address, which can be used to retrieve the job results.
Output
For each submission, one unique job ID and one URL are assigned to track the job results. When an email is provided in submission, users will be notified by email once the jobs are done. The result web page has three sections. The first section shows the predicted contact map. The second section includes (a) job status, (b) download button for predicted contact map, and (c) download button for 5 predicted 3D models. The third section displays the predicted 3D models by JSmol. The model score mainly reflects the degree of violation of the model against the input constraints of CNS (i.e. predicted secondary structure and contacts). The lower the model score, the more likely the model has a higher quality.
Processing time
The running time depends on three factors: (i) sequence length, (ii) the number of related protein families, and (iii) the number of sequence homologs. Since the joint evolutionary coupling analysis consumes most of the running time (i.e. in proportion to the number of related protein families), we restrict the maximal number of related protein families to three. Typically, for a protein of about 250 residues, it takes 1:30 h to finish, with 1 hour spent on contact prediction, and 30 minutes on constructing 3D models. When there are many submissions, it may take a longer time since a small number of jobs can be scheduled to run right after submission.
Documentation
The documentation of CoinFold is available by the ‘Docs’ link at the web page. It includes some details about the server, descriptions of input and output, explanations of prediction results, and a sample prediction result. There is also an example input at the submission page.
CONCLUSION AND FUTURE WORK
We have presented CoinFold, a web server for ab initio protein contact and tertiary structure prediction without using any templates. It significantly outperforms other servers of similar category in both contact prediction and 3D model prediction, especially for those proteins without very good templates.
Our server has better contact prediction accuracy due to its novel joint evolutionary analysis and supervised learning methods. In the future, we may further improve contact prediction accuracy by Deep Learning (19), and increase the 3D model quality by a procedure similar to RASREC (29). In the future, we could use more computer power or GPU cluster to speed up the prediction.
Acknowledgments
The authors are also grateful to the computing power provided by the UChicago Beagle and RCC allocations.
FUNDING
National Institutes of Health [R01GM0897532 to J.X.]; National Science Foundation [DBI-0960390 to J.X.]. Funding for oen access charge: National Institutes of Health [R01GM0897532 to J.X.]; National Science Foundation [DBI-0960390 to J.X.]; Computer owder provided by UChicago Beagle and RCC allocations .
Conflict of interest statement. None declared.
REFERENCES
- 1.Di Lena P., Nagata K., Baldi P. Deep architectures for protein contact map prediction. Bioinformatics. 2012;28:2449–2457. doi: 10.1093/bioinformatics/bts475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim D.E., DiMaio F., Yu‐Ruei Wang R., Song Y., Baker D. One contact for every twelve residues allows robust and accurate topology‐level protein structure modeling. Proteins: Struct. Funct. Bioinform. 2014;82:208–218. doi: 10.1002/prot.24374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Juan D., Pazos F., Valencia A. Emerging methods in protein co-evolution. Nat. Rev. Genet. 2013;14:249–261. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- 4.Marks D.S., Hopf T.A., Sander C. Protein structure prediction from sequence variation. Nat. Biotechnol. 2012;30:1072–1080. doi: 10.1038/nbt.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Göbel U., Sander C., Schneider R., Valencia A. Correlated mutations and residue contacts in proteins. Proteins: Struct. Funct. Bioinform. 1994;18:309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- 6.Marks D.S., Colwell L.J., Sheridan R., Hopf T.A., Pagnani A., Zecchina R., Sander C. Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011;6:e28766. doi: 10.1371/journal.pone.0028766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones D.T., Buchan D.W., Cozzetto D., Pontil M. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28:184–190. doi: 10.1093/bioinformatics/btr638. [DOI] [PubMed] [Google Scholar]
- 8.Ekeberg M., Lövkvist C., Lan Y., Weigt M., Aurell E. Improved contact prediction in proteins: using pseudolikelihoods to infer Potts models. Phys. Rev. E. 2013;87:012707. doi: 10.1103/PhysRevE.87.012707. [DOI] [PubMed] [Google Scholar]
- 9.Kamisetty H., Ovchinnikov S., Baker D. Assessing the utility of coevolution-based residue–residue contact predictions in a sequence-and structure-rich era. Proc. Natl. Acad. Sci. 2013;110:15674–15679. doi: 10.1073/pnas.1314045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seemayer S., Gruber M., Söding J. CCMpred—fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics. 2014;30:3128–3130. doi: 10.1093/bioinformatics/btu500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danaher P., Wang P., Witten D.M. The joint graphical lasso for inverse covariance estimation across multiple classes. J. Roy. Stat. Soc.: Ser. B (Stat. Methodol.) 2014;76:373–397. doi: 10.1111/rssb.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma J., Wang S., Wang Z., Xu J. Protein contact prediction by integrating joint evolutionary coupling analysis and supervised learning. Bioinformatics. 2015:btv472. doi: 10.1093/bioinformatics/btv472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S., Peng J., Ma J., Xu J. Protein secondary structure prediction using deep convolutional neural fields. Sci. Rep. 2016;6:18962. doi: 10.1038/srep18962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briinger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.-S., Kuszewski J., Nilges M., Pannu N.S. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 15.Adhikari B., Bhattacharya D., Cao R., Cheng J. CONFOLD: residue‐residue contact‐guided ab initio protein folding. Proteins: Struct. Funct. Bioinform. 2015;83:1436–1449. doi: 10.1002/prot.24829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma J., Wang S., Zhao F., Xu J. Protein threading using context-specific alignment potential. Bioinformatics. 2013;29:i257–i265. doi: 10.1093/bioinformatics/btt210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao F., Xu J. A position-specific distance-dependent statistical potential for protein structure and functional study. Structure. 2012;20:1118–1126. doi: 10.1016/j.str.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma J., Wang S. AcconPred: Predicting solvent accessibility and contact number simultaneously by a multitask learning framework under the conditional neural fields model. BioMed Res. Int. 2015;2015:1. doi: 10.1155/2015/678764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LeCun Y., Bengio Y., Hinton G. Deep learning. Nature. 2015;521:436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 20.Jones D.T., Singh T., Kosciolek T., Tetchner S. MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics. 2015;31:999–1006. doi: 10.1093/bioinformatics/btu791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mabrouk M., Putz I., Werner T., Schneider M., Neeb M., Bartels P., Brock O. RBO Aleph: leveraging novel information sources for protein structure prediction. Nucleic Acids Res. 2015;43:W343–W348. doi: 10.1093/nar/gkv357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Remmert M., Biegert A., Hauser A., Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 23.Söding J. Protein homology detection by HMM–HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y., Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins: Struct. Funct. Bioinform. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 25.Kryshtafovych A., Monastyrskyy B., Fidelis K. CASP11 statistics and the prediction center evaluation system. Proteins: Struct. Funct. Bioinform. 2016 doi: 10.1002/prot.25005. doi:10.1002/prot.25005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haas J., Roth S., Arnold K., Kiefer F., Schmidt T., Bordoli L., Schwede T. The Protein Model Portal—a comprehensive resource for protein structure and model information. Database. 2013;2013:bat031. doi: 10.1093/database/bat031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Källberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., Xu J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Källberg M., Margaryan G., Wang S., Ma J., Xu J. RaptorX server: a resource for template-based protein structure modeling. Protein Struct. Predict. 2014:17–27. doi: 10.1007/978-1-4939-0366-5_2. [DOI] [PubMed] [Google Scholar]
- 29.Braun T., Leman J.K., Lange O.F. Combining evolutionary information and an iterative sampling strategy for accurate protein structure prediction. PLoS Comput. Biol. 2015;11:e1004661. doi: 10.1371/journal.pcbi.1004661. [DOI] [PMC free article] [PubMed] [Google Scholar]