

ABSTRACT
The symbiotic relationship between vestimentiferan tubeworms and their intracellular chemosynthetic bacteria is one of the more noteworthy examples of adaptation to deep-sea hydrothermal vent environments. The tubeworm symbionts have never been cultured in the laboratory. Nucleotide sequences from the small subunit rRNA gene suggest that the intracellular symbionts of the eastern Pacific vent tubeworms Oasisia alvinae, Riftia pachyptila, Tevnia jerichonana, and Ridgeia piscesae belong to the same phylotype of gammaproteobacteria, “Candidatus Endoriftia persephone.” Comparisons of symbiont genomes between the East Pacific Rise tubeworms R. pachyptila and T. jerichonana confirmed that these two hosts share the same symbionts. Two Ridgeia symbiont genomes were assembled from trophosome metagenomes from worms collected from the Juan de Fuca Ridge (one and five individuals, respectively). We compared these assemblies to those of the sequenced Riftia and Tevnia symbionts. Pangenome composition, genome-wide comparisons of the nucleotide sequences, and pairwise comparisons of 2,313 orthologous genes indicated that “Ca. Endoriftia persephone” symbionts are structured on large geographical scales but also on smaller scales and possibly through host specificity.
IMPORTANCE Remarkably, the intracellular symbionts of four to six species of eastern Pacific vent tubeworms all belong to the same phylotype of gammaproteobacteria, “Candidatus Endoriftia persephone.” Understanding the structure, dynamism, and interconnectivity of “Ca. Endoriftia persephone” populations is important to advancing our knowledge of the ecology and evolution of their host worms, which are often keystone species in vent communities. In this paper, we present the first genomes for symbionts associated with the species R. piscesae, from the Juan de Fuca Ridge. We then combine these genomes with published symbiont genomes from the East Pacific Rise tubeworms R. pachyptila and T. jerichonana to develop a portrait of the “Ca. Endoriftia persephone” pangenome and an initial outline of symbiont population structure in the different host species. Our study is the first to apply genome-wide comparisons of “Ca. Endoriftia persephone” assemblies in the context of population genetics and molecular evolution.
INTRODUCTION
A defining characteristic of hydrothermal vent ecosystems is the diversity and ubiquity of mutualistic partnerships between metazoa (multicellular organisms) and chemolithoautotrophic bacteria. Among these associations, one of the most remarkable is the well-studied model symbiosis between the giant tubeworm Riftia pachyptila and its unique sulfide-oxidizing gammaproteobacterial partner, “Candidatus Endoriftia persephone” (1). These intracellular symbionts are hosted within the specialized cells (bacteriocytes) of an organ known as the trophosome, which occupies most of the space in the cœlomic cavity of the animal's trunk. In this mutualistic association, the worm supplies the bacteria with the inorganic compounds necessary for coupling sulfide oxidation to CO2 fixation: dioxygen, carbon dioxide, and hydrogen sulfide (mostly as its sulfhydric anion HS−). These substances diffuse across the gills into the blood of the animal and are then transported to the trophosome. In return, the endosymbionts provide the tubeworm with the organic molecules necessary for growth and metabolism, either by excreting those molecules or by being digested directly (2, 3). The symbiotic bacteria are transmitted horizontally, that is to say, acquired de novo from the environment at each generation (4). The symbionts penetrate the worm tissues through the epidermis and migrate to a region between the dorsal blood vessel and the foregut to form the prototrophosome. As the metatrochophore larva develops into an adult, its digestive tract atrophies in favor of the trophosome (5). The vestimentiferan adult thus becomes completely dependent on its bacteria for nutrition. For the symbionts, however, this association seems to be facultative. Free-living “Ca. Endoriftia persephone” symbionts have been detected in biofilms and seawater surrounding R. pachyptila aggregations (4), and it has been demonstrated recently that the Riftia symbionts can return to their free-living stage upon the death of the worm (6), thereby maintaining/sustaining environmental populations.
In addition to having a viable free-living stage, the symbionts exhibit low partner fidelity. “Ca. Endoriftia persephone” is also associated with three to five vent tubeworm species other than R. pachyptila: Tevnia jerichonana, Ridgeia piscesae, Oasisia alvinae, and possibly Escarpia spicata and some Lamellibrachia spp. (7), as evidenced by sequence analyses of the 16S rRNA gene marker along with the internal transcribed spacer (ITS) gene. This is somewhat surprising, given that these host species can be separated by thousands of kilometers of fragmented habitat and can colonize very different hydrothermal vent habitats (8).
For example, R. pachyptila and T. jerichonana can both inhabit the same general vent locations in the East Pacific Rise (EPR) but thrive under contrasting venting conditions. Tevnia is typically found at sites of high hydrothermal discharge, characterized by low oxygen and high sulfide concentrations, while Riftia flourishes at sites with more-diffuse flow, with higher oxygen and lower sulfide concentrations (9). Further north, in the northeast Pacific Ocean, at the hydrothermal vents of the Explorer Ridge, Juan de Fuca Ridge (JdFR), and Gorda Ridge, the tubeworm species R. piscesae can be found at temperatures ranging from 2 to 30°C (10, 11) and sulfide concentrations at their branchial plumes ranging from <0.1 μM to 200 μM (11–13).
The symbionts' broad geographic distribution and the wide range of vent habitats occupied by their tubeworm hosts raise questions about the connectivity and population structure of “Ca. Endoriftia persephone.” The limited data available on “Ca. Endoriftia persephone” populations indicate significant strain-level differences between symbionts from different geographical locations (7; M. Perez and S. K. Juniper, submitted for publication); in particular, the symbionts associated with Ridgeia tubeworms might belong to a population different from those found in tubeworms from the EPR (14). However, these studies were based on comparison of only a few conserved genetic sequences.
Since the advent of accessible, high-throughput sequencing, several “Ca. Endoriftia persephone” draft genomes have been reconstructed from metagenomic sequences of Riftia and Tevnia trophosome extracts (1, 15). Recently, deep sequencing of the R. piscesae trophosome microflora confirmed that “Ca. Endoriftia persephone” was the sole symbiotic partner of these tubeworms (16), although there is evidence for multiple “Ca. Endoriftia persephone” strains or lineages within single hosts (Perez and Juniper, submitted). Hence, assuming that the worm's trophosome is not monoclonal, an assembly essentially represents a consensus genome of the symbiont population inhabiting the host's trophosome.
In this study, we sequenced and assembled two consensus genomes representing the symbiont populations inhabiting the trophosomes of one and five individual R. piscesae worms, respectively. Upon confirmation that the R. piscesae symbionts indeed belonged to the same species as “Ca. Endoriftia persephone,” we compared our genome assemblies to those published previously by Gardebrecht et al. (15) with the goals of characterizing the pangenome and population structure of “Ca. Endoriftia persephone” in the different host species. For the latter goal, we undertook (i) genome-wide comparisons of the nucleotide sequences of the core genome, (ii) characterization of the composition of the accessory genomes, and (iii) pairwise comparisons of 2,313 putative orthologous genes.
These new genomic comparisons both support the hypothesis advanced by Nelson and Fisher in 2000 (14), that the “Ca. Endoriftia persephone” symbionts associated with Ridgeia tubeworms belong to a population different from those on the EPR, and suggest that symbiont population structure may have habitat-specific or larger-scale spatial features within regions and may be shaped by host selection.
MATERIALS AND METHODS
Ridgeia symbiont genome assembly. (i) Sample collection and symbiont genome sequencing.
Specimens of R. piscesae were collected from the Axial Volcano and the Main Endeavor Field, on the Juan de Fuca Ridge, during a remotely operated vehicle (ROV) cruise on the R/V Thomas G. Thompson in July 2010. As described by Forget et al. (16), the worms were recovered and brought to the ship in sealed bioboxes. Once on board, individual worms were carefully removed from their tubes, and those that showed no visible tissue damage were rinsed with 70% ethyl alcohol (EtOH) and were flash frozen at −80°C until further processing. In our laboratory, the contents of the worms' trunks (which include the trophosome) were removed by dissection and rinsed with 70% EtOH. Finally, the DNA from each dissected trunk was extracted using the Qiagen DNeasy blood and tissue kit. DNA extracts from six individual worms were sequenced and assembled at Genome Quebec, Montreal, Canada. Samples were prepared using standard protocols and were sequenced on the Illumina HiSeq 2000 platform. A subset of these samples was also sequenced on the Illumina MiSeq platform. The resulting metagenomes were assembled de novo into two high-quality symbiont genome assemblies essentially free of host and bacterial contaminant sequences (see the supplemental material for a description of data quality assessment, curation to remove contaminant sequences, and de novo assembly). The first genome (referred to as the Ridgeia 1 symbiont) was assembled from the symbiont metagenome of one individual tubeworm, while the second genome (the Ridgeia 2 symbiont) was assembled from the pooled data of five other individual worms in order to increase the depth of coverage (Table 1).
TABLE 1.
Metagenomic samples
| Sample name | Vent site | Flow regimea | Depth (m) | Plume/base max temp (°C) | Sequencing platform(s) | No. of symbiont readsb (% of total reads) | De novo assembly name |
|---|---|---|---|---|---|---|---|
| Ind 11 | Main Endeavour Field (Hulk) | High flow | 2,190 | 14.0/51.0 | HiSeq | 6.94 (33) | Ridgeia 1 symbiont |
| Ind 13 | Main Endeavour Field (Hulk) | High flow | 2,190 | 14.0/51.0 | HiSeq, MiSeq | 0.18 (<1) | Ridgeia 2 symbiont |
| Ind 15 | Main Endeavour Field (Hulk) | Low flow | 2,191 | 2.5/2.5 | HiSeq, MiSeq | 0.11 (<1) | Ridgeia 2 symbiont |
| Ind 10 | Axial Volcano (Hot Spot 2) | Low flow | 1,517 | 2.0/3.4 | HiSeq | 0.24 (<1) | Ridgeia 2 symbiont |
| Ind 12 | Axial Volcano (Hot Spot 2) | High flow | 1,516 | 4.1/30.3 | HiSeq, MiSeq | 0.13 (<1) | Ridgeia 2 symbiont |
| Ind 14 | Axial Volcano (Hot Spot 2) | Low flow | 1,517 | 2.0/3.4 | HiSeq, MiSeq | 0.29 (<1) | Ridgeia 2 symbiont |
Associated with tubeworm morphotypes. High flow is associated with a short-fat morphotype; low flow, with a long-skinny morphotype.
In millions; based on alignment rates of reads mapped to “Candidatus Endoriftia persephone” (from the Ridgeia 1 assembly).
(ii) Gene annotations.
Gene calling for the Ridgeia 1 and Ridgeia 2 symbiont assemblies was performed using the IMG-ER platform of the Joint Genome Institute.
Genome-wide comparisons of Ridgeia symbionts with all other published vestimentiferan genomes. (i) Genome alignments.
The genomes of the Ridgeia 1 and Ridgeia 2 symbionts were aligned with the closely related Riftia 1, Riftia 2, and Tevnia symbionts (15) using progressiveMauve (17). This anchored alignment algorithm finds so-called locally colinear blocks (LCBs)—genome segments that appear free of chromosomic rearrangement—and outputs the aligned sequences of each LCB in XMFA multiple alignment format as well as in a file containing the positions of the LCBs in each of the genomes (backbone file).
(ii) Pangenome composition.
The pangenome composition was determined by the presence or absence and sizes of LCB sequences in each genome. Since colinear blocks have been found previously to be informative for phylogenomic analysis (18), we further used the presence or absence of individual LCBs (>100 bp) to compute Jaccard distances with the vegan package in R (19) and built a neighbor-joining tree using Populations software, version 1.2.32 (http://bioinformatics.org/∼tryphon/populations/). Bootstrap values were obtained from 100 bootstrap subsamples by use of the boot.phylo function from the ape package in R. Finally, a custom Python script was used to extract the annotations of all genes within the LCBs of interest from GenBank files. These genes were further annotated through visual inspection against the Mauve-generated multiple-genome alignment in order to record additional information, such as the representation of neighboring LCBs across the assemblies and nucleotide conservation.
(iii) Core genome nucleotide heterogeneity.
Our analysis of core genome nucleotide heterogeneity for symbionts from the three tubeworm species also included the assembly for the Riftia symbionts (1). This first published assembly was not used in subsequent, more in-depth analyses because of its lower quality, as explained below.
We used the stripSubsetLCBs command to extract the large (>100-bp) LCBs represented in all of the assemblies from the XMFA file. For each LCB, a FASTA file was generated, and the sequences were aligned with MAFFT (20). Subsequently, all the resulting alignments were concatenated to form a single genome-wide alignment of 2,580,528 bp with 75,472 variable sites. Finally, we used SeaView (21) to calculate the pairwise genetic distances using the Hasegawa-Kishino-Yano (HKY) model (22) and to generate a 100-bootstrap neighbor-joining tree.
(iv) Pairwise comparison of homologous genes.
A file containing a table of all the homologous protein-coding genes was obtained using the Export Positional Homologs command from Mauve's menu and the following parameters: minimum identity, 80; minimum coverage, 50. This table was then curated to keep only the entries of genes present in all of the genomes. Subsequently, we generated local protein and nucleic acid Blast databases of all of the coding sequences of the pangenome, from which we extracted, in FASTA format, the nucleotide and amino acid sequences of these genes by use of the blastdbcmd tool of BLAST++ (23). Then we aligned the amino acid sequences and generated protein sequence identity matrices with Clustal Omega (24). Subsequently, protein alignments were converted into codon-based nucleotide alignments with PAL2NAL (25). Finally, the nucleotide sequence identity matrices and the ratios of nonsynonymous to synonymous substitution rates (dN/dS ratios) were calculated using Clustal Omega (24) and the YN00 program of the PAML package (26, 27), respectively. Genome-wide dN/dS ratios were generated from the concatenated codon-based alignments.
Mauve's transitive algorithm identifies positionally homologous sequences. In closely related genomes, these positional homologs are also orthologs, but the algorithm could still mistakenly catch recently duplicated genes (paralogs). To prevent comparisons between paralogs, the protein sequences of all homologs with nucleotide identities lower than 50% were subjected to reciprocal BLAST searches against the five reference genomes. The homologous associations were then adjusted to include the true orthologs or were removed from the data set if orthologous sequences were missing in at least one genome. Fewer than a dozen homologous genes among the 2,324 identified were thereby curated (excluded from further analyses). We believe that the remaining cases of paralogous associations are limited to just a few extra genes and do not significantly affect our results.
Accession number(s).
The Ridgeia 1 and Ridgeia 2 symbiont assemblies are published on the Joint Genome Institute IMG system with genome IDs 2651869500 and 2643221413, respectively, as well as in GenBank under accession numbers LDXT00000000 and LMXI00000000, respectively. The versions described in this paper are LDXT01000000 and LMXI01000000.
RESULTS
Metagenome assemblies of Ridgeia symbionts. (i) Assembly quality.
The quality of a genome assembly depends on its completeness and coverage. These and other features of all available “Ca. Endoriftia persephone” genome assemblies are compared in Table 2. Completeness can be estimated from the number and size distribution (N50) of contigs, while coverage, calculated as the average per-base sequencing depth, is a measure of the sampling effort. Higher sequencing depth results in higher sequence accuracy but also improves the completeness of isolate genomes.
TABLE 2.
Overview of vestimentiferan symbiont metagenomes
| Symbionta | Genome size (Mbp) | No. of contigs | N50 | Coverage (fold) | No. of reads | GC% | No. of genes |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Protein coding | rRNA | tRNA | |||||||
| Ridgeia 1 symbiont | 3.44 | 97 | 83.9 | 180 | 7,436,749 | 58.9 | 3,188 | 3,132 | 3 | 47 |
| Ridgeia 2 symbiont | 3.42 | 693 | 7.6 | 17 | 993,690 | 58.9 | 3,698 | 3,641 | 3 | 43 |
| Tevnia symbiont | 3.64 | 184 | 92.7 | 15 | 212,833 | 58.2 | 3,277 | 3,230 | 3 | 44 |
| Riftia 1 symbiont | 3.48 | 197 | 28.4 | 25 | 467,070 | 58.2 | 3,254 | 3,209 | 3 | 42 |
| Riftia 2 symbiont | 3.71 | 414 | 24.6 | 13 | 205,880 | 58.2 | 3,566 | 3,515 | 4 | 47 |
| “Candidatus Endoriftia persephone” | 3.20 | 2,170 | 1.9 | 11 | 130,000 | 57.9 | 6,450 | 6,414 | 4 | 32 |
The Ridgeia 1 symbiont assembly was of high quality (Table 2). It contained fewer and longer contigs than the Riftia 1 symbiont assembly, and its coverage was 7 to 16 times greater than those of all previously published assemblies of “Ca. Endoriftia persephone” (1, 15). Yet even with such high sequencing depth, we were not able to close the genome. We suspect that chromosome rearrangement within the symbiont population might be the cause of this fragmentation, because it can create ambiguous links during the scaffolding step of the assembly.
The “pooled” Ridgeia 2 symbiont genome was generally lower in quality than the assemblies of Gardebrecht et al. (15) but still notably superior in completeness and coverage to the assembly of Robidart et al. (1) (Table 2). Because of the lower overall quality and considerable differences in gene annotations, the assembly of Robidart et al. (1) was not used in our analyses.
(ii) Confirmation that the Ridgeia symbionts are “Ca. Endoriftia persephone.”
The Ridgeia 1 and Ridgeia 2 16S rRNA, 23S rRNA, and ITS sequences were 100% identical to each other and differed from the Tevnia and Riftia 1 and 2 symbiont sequences by 1, 0, and 3 nucleotides, respectively. This is consistent with the hypothesis that the same species of symbionts, “Ca. Endoriftia persephone,” is associated with Riftia, Tevnia, and Ridgeia tubeworms.
This hypothesis is further supported by the fact that the majority of “Ca. Endoriftia persephone” genes had homologs in the Ridgeia symbiont assemblies (see Fig. 3; also Data Set S1 in the supplemental material).
FIG 3.

(A and B) Distribution of heterogeneity between pairs of homologous genes based on nucleotide sequences (A) and amino acid sequences (B). Only heterogeneities of <5% are represented (>90% of data). (C) Negative correlation of the dN/dS ratio with divergence between individuals from different metapopulations based on the concatenated alignments of 2,313 homologous gene sequences (1,926,255 bp). See Data Set S1 in the supplemental material for details.
Like the symbionts associated with Riftia and Tevnia tubeworms, the Ridgeia symbionts have a diverse metabolism and possess genes for sulfide oxidation, carbon fixation through the Calvin-Benson-Bassham and reverse tricarboxylic acid (rTCA) cycles, denitrification, motility, and chemotaxis (see Table S5 in the supplemental material).
Pangenome composition of “Ca. Endoriftia persephone.”
Figure 1 shows the composition of the pangenome of “Ca. Endoriftia persephone” based on the nucleotide sequences of the five most recent “Ca. Endoriftia persephone” assemblies. A core genome, representing 89% of the pangenome of “Ca. Endoriftia persephone,” was shared across all of the assemblies of Riftia, Tevnia, and Ridgeia symbionts. In addition, 4% of the pangenome was region specific, i.e., found in and shared among symbionts from the same geographical region only (the JdFR symbionts associated with R. piscesae or the EPR symbionts associated with R. pachyptila and T. jerichonana) (Fig. 1). Symbionts from the same geographical region shared as much as 98% of their genomes.
FIG 1.
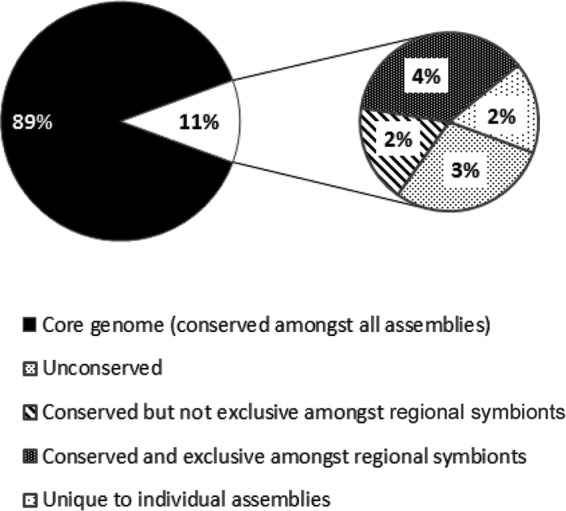
Pangenome of “Candidatus Endoriftia persephone” based on the relative sizes of the locally colinear blocks shared by five “Ca. Endoriftia persephone” assemblies from two distinct geographical regions. The Ridgeia 1 and Ridgeia 2 symbionts are from the Juan de Fuca Ridge, and the Tevnia, Riftia 1, and Riftia 2 symbionts are from the East Pacific Rise. The five genome assemblies were aligned with progressiveMauve (17).
Finally, we found that 0.7 to 2.9% of the pangenome was unique to the specific assemblies and was in part composed of contaminant sequences and/or exogenous genetic material acquired recently through horizontal transfer. This is supported by the fact that the GC content of the unique genome for some assemblies was notably different from that of the core genome. In the Tevnia symbiont, for example, the GC content of the unique genome (96 kbp) was 42%, while that of the core genome was 60%.
The relatively large size of the unconserved genome (3% of the “Ca. Endoriftia persephone” pangenome) was likely the result of gaps in the genome assemblies and the small sample size. We expect that increasing the quality of the data and the number of samples would reduce the relative importance of the unconserved genome in favor of the conserved pangenome or the regional core genome.
Genes encoded in the accessory genome. (i) Region-specific genome.
The LCBs that were exclusive to the JdFR or EPR symbiont genomes both carried unique genes coding for transposases, integrases, and other phage-associated proteins, as well as a few genes involved in cell wall/membrane/envelope biogenesis (see Tables S1 and S2 in the supplemental material).
Interestingly, two clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein (Cas) systems (28) were found in all of the genome assemblies. The first was well conserved, but the spacers were notably different in the two genomes for which the CRISPR locus was successfully assembled (the Ridgeia 1 and Tevnia symbionts). In the second CRISPR-Cas system, the cas operon was not conserved across symbionts from the JdFR and the EPR; half of the cas genes were not homologous (see Tables S1 and S2 in the supplemental material).
Finally, a 17-kbp scaffold with genes encoding a type VI secretion system was found uniquely in the Ridgeia symbionts (see Table S1 in the supplemental material).
(ii) Vent site- versus species-specific genomes at the EPR.
The LCBs unique to Riftia symbiont assemblies were limited to three contiguous scaffolds (see Table S3 in the supplemental material). The first was about 60 kbp long and contained genes typically found in fertility factors, i.e., the gene coding for the OmpA/MotB domain-containing protein, tra genes, IS200-like genes coding for transposases, and two genes coding for nucleotide-binding proteins. The other two scaffolds were smaller (about 10 kbp) and contained, respectively, six genes of the CRISPR-Cas3 system and four genes: two unannotated genes, one gene encoding a transcriptional regulator, and one encoding a putative relaxase. Because of the incompleteness of the Tevnia symbiont assembly, we could not rule out the possibility that these differences resulted from a biased sampling of the Tevnia symbiont's metagenome. However, given the large size of the missing scaffolds, we suggest that this was unlikely.
Among the LCBs found exclusively in the assemblies of symbionts from the vent site at 9°N, however, many seemed to have resulted from a poor sampling of the fragmented genome of the Riftia 1 symbiont. They represented stretches of DNA of a few thousand base pairs, often located at the extremities of conserved contigs. In contrast, other unique sequences seemed to represent real chromosomic differences, tended to be larger (up to 16.3 kbp), were flanked by regions of low nucleotide conservation, and contained unique mobile elements, toxin/antitoxin genes, and transcriptional regulator genes typically found in phage genomes (see Table S4 in the supplemental material).
Population structure of “Ca. Endoriftia persephone.” (i) Cluster analyses.
The first sequenced metagenome of “Ca. Endoriftia persephone” (1) clustered apart from the more recent assemblies. This is probably a result of sequencing errors due to the overall lower quality of reads associated with the sequencing methods used at the time.
The Ridgeia symbionts cluster apart from the EPR symbionts both in terms of nucleotide distances in the core genome and in terms of the compositions of their accessory genomes (Fig. 2).
FIG 2.
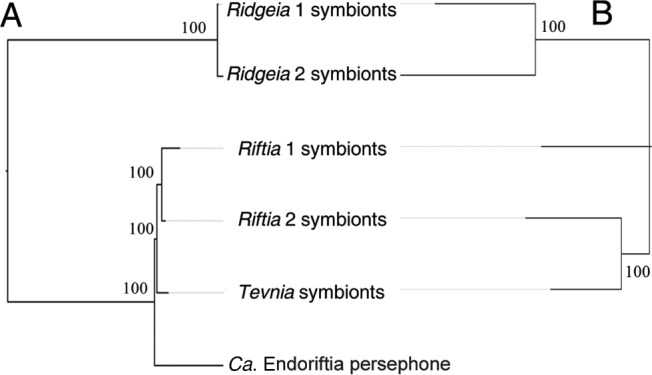
Neighbor-joining trees of “Candidatus Endoriftia persephone” based on the genetic distances (HKY model) between nucleotide sequences of the core genome (A) and the presence or absence of sequences of the accessory genome (B). (A) The six assemblies were aligned with Mauve and the locally colinear blocks extracted. Of these, only the LCBs of >100 bp that were represented in all assemblies were kept. The sequences within each LCB were aligned with MAAFT and were concatenated to form a genome-wide alignment of 2,580,528 bp containing 75,472 variable sites. (B) The first assembly of “Ca. Endoriftia persephone” (1) was not included in this analysis because of high genome fragmentation. Assemblies were aligned with Mauve, and the presence or absence of LCBs of >100 bp was used to generate a distance matrix (Jaccard index) from which a neighbor-joining tree was constructed using Populations, version 1.2.32. Bootstrap values are indicated at the branches.
Within the EPR, the symbionts cluster by host species when classified by the nucleotide sequences of the core genome, while they cluster by vent site when classified by the composition of the accessory genome. Thus, at the area of the EPR (near 9°N) for which there are genome assemblies for both Riftia and Tevnia symbionts, the symbionts from these two hosts shared more exclusive LCBs than did Riftia symbionts collected from different EPR areas (9°N versus 13°N). Interestingly, the accessory genome exclusive to the vent site at 9°N was composed of shorter LCBs, and was slightly smaller overall, than the symbiont genome exclusive to the Riftia host species (70 kbp and 80 kbp, respectively) (see Tables S3 and S4 in the supplemental material).
Finally, Tevnia symbionts seemed to be closer to Ridgeia than to Riftia symbionts in terms of nucleotide identity.
(ii) Comparisons of orthologous genes.
Nucleotide heterogeneity was <1% within the EPR tubeworm symbionts and within the Ridgeia symbionts from the JdFR but ca. 2% between symbionts from the two different regions (Fig. 3A). Yet many homologous proteins were highly conserved across the assemblies from the JdFR and the EPR, indicating strong purifying selection acting on “Ca. Endoriftia persephone” (Fig. 3B).
The Ridgeia symbionts appeared more homogeneous than those from the EPR. Eighty-nine percent of the genes in the two Ridgeia symbiont assemblies had identical nucleotide sequences, compared to 54% on the EPR. These results were corroborated with the overall synonymous substitution rates (dS) and the ratio of nonsynonymous to synonymous substitution rates (dN/dS ratio) between pairs of symbiont assemblies (Fig. 3C).
The synonymous substitution rate is the ratio of the number of synonymous substitutions to the number of synonymous sites. Because synonymous substitutions tend to be selectively neutral, they accumulate over time and thus can be used as a proxy for divergence between genomes (29, 30). Assuming allopatric divergence, we can then derive a molecular clock for the substitution rate (r) for the “Ca. Endoriftia persephone” symbionts by use of the equation r = dS/2T, where dS is the divergence observed between the vicariant populations at the synonymous sites and T is the time of last contact between the East Pacific Rise and northeast Pacific ridge systems. Following the work of Vrijenhoek (31), we used a T of 28.5 million years ago and obtained a substitution rate of 0.14% (±0.01%) per million years.
This substitution rate is lower than the rates observed for Escherichia coli in culture (0.45%) (32) and for the host vestimentiferan tubeworms themselves (∼0.2%) (33). However, both of the latter substitution rates were based on comparisons of isolate genomes, while our rate was determined from comparisons of genome assemblies that resulted from the concatenation of multiple symbionts with potentially multiple genotypes. Thus, we may have underestimated the genetic diversity within and across the symbiont populations and therefore the rate of divergence between the two populations. Furthermore, the divergence between two populations depends on their respective reproduction rates and on the parameters that affect their respective genetic diversity over generations (i.e., their underlying biological mutation rate, effective size, and clonality [34, 35]). Current knowledge of doubling times and genetic diversity for these symbionts does not permit confident estimation of most of these parameters.
Our data can be used for an initial consideration of effective symbiont population sizes for the three host tubeworm species considered here. The genome-wide dN/dS ratio of “Ca. Endoriftia persephone” symbionts falls into the upper range of what has been observed in closely related obligate symbionts (36). For closely related genomes, the dN/dS ratio is also intrinsically dependent on the time since divergence and the effective population size. More-closely related lineages or lineages with smaller population sizes tend to have higher dN/dS ratios due to a time lag or delay in the curation of slightly deleterious mutations (37, 38). “Ca. Endoriftia persephone” symbiont populations showed this pattern in that the dN/dS ratios were negatively correlated with the divergence between genome pairs. The highest divergence with the lowest dN/dS ratio was seen in the comparison between Ridgeia and EPR symbionts (dS, ∼0.08), and the lowest divergence and the highest dN/dS ratio were between the two Ridgeia symbiont assemblies (Fig. 3C).
Interestingly, while the divergences between EPR symbionts were quite similar (0.0101 < dS < 0.0136), the dN/dS ratio for the two Riftia assemblies was notably higher than those for the other pairs. He et al. (39) and Luo et al. (40) made similar observations for the pathogen Clostridium difficile and lineages of the marine alphaproteobacterium Roseobacter, respectively. This suggests that the symbionts in association with Riftia tubeworms have a smaller effective population size than the overall EPR “Ca. Endoriftia persephone” symbionts, and thus, that the latter might be further structured either spatiotemporally, according to environmental conditions, or through host specificity.
DISCUSSION
Divergence of JdFR and EPR symbionts.
We used five high-quality genome assemblies of “Ca. Endoriftia persephone” to analyze the structure of the “Ca. Endoriftia persephone” population though pairwise comparisons of (i) the composition of the pangenome, (ii) the nucleotide identity within the core genome, and (iii) the synonymous and nonsynonymous substitution rates for a large subsample of the core genome genes. Our results were consistent with those obtained from phylogenetic analyses based on 16S rRNA gene (41) and ITS sequences, as well as repetitive element palindromic PCR (rep-PCR) fingerprints (7), and indicated that the population of “Ca. Endoriftia persephone” symbionts in association with R. piscesae on the Juan de Fuca Ridge (JdFR) was distinct from the “Ca. Endoriftia persephone” population on the East Pacific Rise (EPR), which is associated with R. pachyptila and T. jerichonana.
(i) Allopatry.
Comparisons of the composition of vent-associated macrofauna communities (42) and the genetic structure of vestimentiferan worms (33) provide evidence that the northeast Pacific and EPR vent communities have been isolated by the development of discontinuities along the Pacific midocean ridge caused by the tectonic fracturing of the Farallon Plate about 30 million years ago (42, 43). Similar dichotomies attributed to later plate fragmentation events were observed in populations of various invertebrate species spanning multiple ridge systems in the northeast Pacific (44–46). It is therefore reasonable to assume that “Ca. Endoriftia persephone” populations were similarly affected by the emergence of these geographical barriers. Our results indicate that the divergence of the JdFR and EPR symbionts was dominated by passive processes/genetic drift. On the one hand, the core genome was characterized by overall low dN/dS ratios and a conserved codon bias (data not shown), suggesting that the same selective constraints acted on both populations. Additionally, when we compared the functional distribution of core genome genes with median dN/dS values to that of genes with extreme dN/dS values (5% highest dN/dS ratios), no Clusters of Orthologous Groups (COGs) or KEGG Orthology (KO) categories appeared to be overrepresented in the outliers (P, >0.05 by the chi-square test of independence). On the other hand, the accessory genome of each population of symbionts was composed of many mobile elements and selfish sequences, as well as unique CRISPR spacers, all of which suggest two distinct histories of interactions that have independently modified the EPR and JdFR symbiont genomes.
(ii) Adaptations to viral predation.
The presence of phage DNA as well as two to three (for Ridgeia 1 symbionts) CRISPR operons can be seen as evidence that viruses are an important “enemy” of free-living and/or intracellular “Ca. Endoriftia persephone” and that the symbiont genomes carry these markers of phage infections.
Although little considered until recently, there is accumulating evidence for a viable and presumably metabolically active free-living stage of “Ca. Endoriftia persephone” (4, 6). Viruses are known to be abundant at deep-sea hydrothermal vents and are likely an important cause of mortality for free-living bacteria (47). Alternatively, the trophosome might also be a favorable environment for the proliferation of phages among the dense and fast-growing intracellular symbiont population.
The presence of CRISPR spacers that differ between the JdFR and EPR symbiont populations could suggest the existence of different “Ca. Endoriftia persephone”-specific viruses on these two midocean ridges, although we have also found CRISPR spacer variability within the symbiont population of a single worm (M. Perez and S. K. Juniper, submitted for publication).
(iii) Host adaptation.
Some genes possibly involved in the symbiosis had relatively high dN/dS ratios (e.g., the chemotaxis protein CheY, the cell division protein DamX, an outer membrane protein), but the divergence between the two populations was too small for detection of the signature of positive selection (48). Nevertheless, the large scaffolds containing genes associated with a type VI secretion system, found only in Ridgeia symbionts, could be part of a mechanism of host adaptation. Indeed, the type VI secretion system can act as a virulence factor against eukaryotic cells or competing bacteria (49, 50). It has also been found to be key in determining host specificity in Rhizobium leguminosarum (51). Other genes involved in cell wall/membrane biogenesis could be involved in the expression of microbe-associated molecular patterns (MAMPs), hypothesized to be critical in mediating host-symbiont interactions (52). Genomic and proteomic comparisons with a sympatric population of “Ca. Endoriftia persephone” symbionts associated with a different host species (e.g., a Lamellibrachia sp. [7]) might tell us more about host specificity.
EPR symbionts are further structured into populations that might be relatively isolated spatially or temporally.
Our results show little evidence for geographic differentiation of symbionts from the two sites on the EPR for which genome sequence data are available. Symbionts from a single vent site, at 9°N, were no more similar to each other than symbionts from two different sites, at 9°N and 13°N. In contrast, when symbionts from the two EPR host tubeworms were compared, the nucleotide sequences of symbionts hosted by the same tubeworm species were more homogeneous and had a higher dN/dS ratio, suggesting that Riftia symbionts formed a subpopulation within the EPR. Additionally, Riftia symbionts carried scaffolds with genes typically found in F-type conjugative plasmids. These genes have been speculated to play a role in horizontal gene transfer (15) and might allow for a high degree of genetic exchange between Riftia symbionts, thus keeping this population homogeneous.
While free-living symbionts can probably disperse on large scales and colonize new surfaces/vents independently of their hosts, small-scale spatial or temporal differences in environmental conditions could favor particular strains of symbionts, resulting in population partitioning. This local increase in homogeneity might be amplified or maintained in the presence of the tubeworm hosts through pseudovertical transfer of symbionts (6).
Molecular mechanisms controlling host specificity might also exist, but a higher resolution of genetic diversity would be needed to clearly characterize differences in the symbionts' accessory genomes.
Toward a better characterization of “Ca. Endoriftia persephone” populations.
Whereas previous studies presenting “Ca. Endoriftia persephone” genomes focused on the metabolism of the symbiont (1, 15), this study was the first to apply genome-wide comparisons of “Ca. Endoriftia persephone” assemblies in the context of population genetics and molecular evolution. These comparisons underline the importance of viruses and genetic drift in shaping the genetic makeup of the symbionts and defining populations. Our findings suggest that, as with vent animal species, midocean ridge discontinuities in the eastern Pacific Ocean have resulted in allopatric divergence of symbiont populations on the Juan de Fuca Ridge and the East Pacific Rise. Furthermore, within a single ridge system, the symbiont populations are not panmictic and are possibly structured according to environmental conditions or host specificity, or both. Finally, genome-wide comparisons revealed that the population-specific functional genes are likely encoded in the accessory genome and potentially in plasmids.
While the number and quality of our samples were limited, we are confident that further population genetic studies, using rapidly advancing sequencing platforms, will provide further insight into the symbionts' evolutionary history and adaptation to their hosts and environment.
We suggest that future studies focus on assessing the number and diversity of “Ca. Endoriftia persephone” genotypes. To this end, we propose that CRISPR spacers and extrachromosomic genetic material may have the potential to be used for high-resolution differentiation of populations of symbionts. For example, “CRISPR typing” has been used for genotyping human bacterial pathogens (53–57) and aquatic bacteria (58–60). In the meantime, sequencing of the complete genomes of individual Endoriftia cells would allow us to detect chromosomic differences.
Understanding the structure, dynamism, and interconnectivity of “Ca. Endoriftia persephone” populations is important to advancing our knowledge of the ecology and evolution of their host worms, which are often keystone species in vent communities.
Supplementary Material
ACKNOWLEDGMENTS
We thank the crews of the R/V Atlantis and R/V Thomas G. Thompson, as well as the pilots of the submersibles Alvin and ROPOS. This research was enabled in part by computing assistance provided by Belaid Moa, WestGrid (westgrid.ca), and Compute Canada/Calcul Canada (computecanada.ca). We also thank Nathalie Forget, Steve Perlman, Sebastien Duperron, the members of Verena Tunicliffe's lab, Lee Katz, Diana Varela, Real Roy, Francis Nano, and all of the contributors to seqanswers.com and biostars.org.
We declare no conflict of interest.
Funding Statement
This research was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant and a Canadian Healthy Oceans Network (NSERC Canada) grant to S.K.J.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00953-16.
REFERENCES
- 1.Robidart JC, Bench SR, Feldman RA, Novoradovsky A, Podell SB, Gaasterland T, Allen EE, Felbeck H. 2008. Metabolic versatility of the Riftia pachyptila endosymbiont revealed through metagenomics. Environ Microbiol 10:727–737. doi: 10.1111/j.1462-2920.2007.01496.x. [DOI] [PubMed] [Google Scholar]
- 2.Felbeck H, Jarchow J. 1998. Carbon release from purified chemoautotrophic bacterial symbionts of the hydrothermal vent tubeworm Riftia pachyptila. Physiol Biochem Zool 71:294–302. [DOI] [PubMed] [Google Scholar]
- 3.Bright M, Keckeis H, Fisher C. 2000. An autoradiographic examination of carbon fixation, transfer and utilization in the Riftia pachyptila symbiosis. Mar Biol 136:621–632. doi: 10.1007/s002270050722. [DOI] [Google Scholar]
- 4.Harmer TL, Rotjan RD, Nussbaumer AD, Bright M, Ng AW, DeChaine EG, Cavanaugh CM. 2008. Free-living tube worm endosymbionts found at deep-sea vents. Appl Environ Microbiol 74:3895–3898. doi: 10.1128/AEM.02470-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nussbaumer AD, Fisher CR, Bright M. 2006. Horizontal endosymbiont transmission in hydrothermal vent tubeworms. Nature 441:345–348. doi: 10.1038/nature04793. [DOI] [PubMed] [Google Scholar]
- 6.Klose J, Polz MF, Wagner M, Schimak MP, Gollner S, Bright M. 2015. Endosymbionts escape dead hydrothermal vent tubeworms to enrich the free-living population. Proc Natl Acad Sci U S A 112:11300–11305. doi: 10.1073/pnas.1501160112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Meo CA, Wilbur AE, Holben WE, Feldman RA, Vrijenhoek RC, Cary SC. 2000. Genetic variation among endosymbionts of widely distributed vestimentiferan tubeworms. Appl Environ Microbiol 66:651–658. doi: 10.1128/AEM.66.2.651-658.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bright M, Lallier FH. 2010. The biology of vestimentiferan tubeworms. Oceanogr Mar Biol Annu Rev 48:213–266. doi: 10.1201/EBK1439821169-c4. [DOI] [Google Scholar]
- 9.Nees HA, Lutz RA, Shank TM, Luther GW III. 2009. Pre- and post-eruption diffuse flow variability among tubeworm habitats at 9°50′ north on the East Pacific Rise. Deep Sea Res Part 2 Top Stud Oceanogr 56:1607–1615. doi: 10.1016/j.dsr2.2009.05.007. [DOI] [Google Scholar]
- 10.Carney SL, Flores JF, Orobona KM, Butterfield DA, Fisher CR, Schaeffer SW. 2007. Environmental differences in hemoglobin gene expression in the hydrothermal vent tubeworm, Ridgeia piscesae. Comp Biochem Physiol B Biochem Mol Biol 146:326–337. doi: 10.1016/j.cbpb.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Urcuyo IA, Massoth GJ, Julian D, Fisher CR. 2003. Habitat, growth and physiological ecology of a basaltic community of Ridgeia piscesae from the Juan de Fuca Ridge. Deep Sea Res Part 1 Oceanogr Res Pap 50:763–780. doi: 10.1016/S0967-0637(03)00061-X. [DOI] [Google Scholar]
- 12.Carney SL, Peoples JR, Fisher CR, Schaeffer SW. 2002. AFLP analyses of genomic DNA reveal no differentiation between two phenotypes of the vestimentiferan tubeworm, Ridgeia piscesae. Cah Biol Mar 43:363–366. [Google Scholar]
- 13.Brand GL, Horak RV, Bris NL, Goffredi SK, Carney SL, Govenar B, Yancey PH. 2007. Hypotaurine and thiotaurine as indicators of sulfide exposure in bivalves and vestimentiferans from hydrothermal vents and cold seeps. Mar Ecol (Berl) 28:208–218. doi: 10.1111/j.1439-0485.2006.00113.x. [DOI] [Google Scholar]
- 14.Nelson K, Fisher C. 2000. Absence of cospeciation in deep-sea vestimentiferan tube worms and their bacterial endosymbionts. Symbiosis 28:1–15. [Google Scholar]
- 15.Gardebrecht A, Markert S, Sievert SM, Felbeck H, Thürmer A, Albrecht D, Wollherr A, Kabisch J, Le Bris N, Lehmann R. 2012. Physiological homogeneity among the endosymbionts of Riftia pachyptila and Tevnia jerichonana revealed by proteogenomics. ISME J 6:766–776. doi: 10.1038/ismej.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forget NL, Perez M, Juniper SK. 2014. Molecular study of bacterial diversity within the trophosome of the vestimentiferan tubeworm Ridgeia piscesae. Mar Ecol 36(S1):35–44. doi: 10.1111/maec.12169. [DOI] [Google Scholar]
- 17.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Lin K. 2012. A phylogenomic analysis of Escherichia coli/Shigella group: implications of genomic features associated with pathogenicity and ecological adaptation. BMC Evol Biol 12:174. doi: 10.1186/1471-2148-12-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara R, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2015. vegan: Community Ecology Package. R package, version 2.2-1 https://cran.r-project.org/web/packages/vegan/index.html.
- 20.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 22.Hasegawa M, Kishino H, Yano T. 1985. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Ooi BC, Tan K-L, Ong T-H, Zhou L. 2003. BLAST++: BLASTing queries in batches. Bioinformatics 19:2323–2324. doi: 10.1093/bioinformatics/btg310. [DOI] [PubMed] [Google Scholar]
- 24.Sievers F, Higgins DG. 2014. Clustal Omega. Curr Protoc Bioinformatics 48:1.25.1–1.25.33. doi: 10.1002/0471250953.bi0313s48. [DOI] [PubMed] [Google Scholar]
- 25.Suyama M, Torrents D, Bork P. 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z, Nielsen R. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol 17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 28.Westra ER, Buckling A, Fineran PC. 2014. CRISPR-Cas systems: beyond adaptive immunity. Nat Rev Microbiol 12:317–326. doi: 10.1038/nrmicro3241. [DOI] [PubMed] [Google Scholar]
- 29.Ochman H, Wilson AC. 1987. Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J Mol Evol 26:74–86. doi: 10.1007/BF02111283. [DOI] [PubMed] [Google Scholar]
- 30.Wielgoss S, Barrick JE, Tenaillon O, Cruveiller S, Chane-Woon-Ming B, Médigue C, Lenski RE, Schneider D. 2011. Mutation rate inferred from synonymous substitutions in a long-term evolution experiment with Escherichia coli. G3 (Bethesda) 1:183–186. doi: 10.1534/g3.111.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vrijenhoek RC. 2013. On the instability and evolutionary age of deep-sea chemosynthetic communities. Deep Sea Res Part 2 Top Stud Oceanogr 92:189–200. doi: 10.1016/j.dsr2.2012.12.004. [DOI] [Google Scholar]
- 32.Ochman H, Elwyn S, Moran NA. 1999. Calibrating bacterial evolution. Proc Natl Acad Sci U S A 96:12638–12643. doi: 10.1073/pnas.96.22.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chevaldonne P, Jollivet D, Desbruyeres D, Lutz R, Vrijenhoek R. 2002. Sister-species of eastern Pacific hydrothermal vent worms (Ampharetidae, Alvinellidae, Vestimentifera) provide new mitochondrial COI clock calibration. Cah Biol Mar 43:367–370. [Google Scholar]
- 34.Shapiro BJ, David LA, Friedman J, Alm EJ. 2009. Looking for Darwin's footprints in the microbial world. Trends Microbiol 17:196–204. doi: 10.1016/j.tim.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 35.Fraser C, Hanage WP, Spratt BG. 2007. Recombination and the nature of bacterial speciation. Science 315:476–480. doi: 10.1126/science.1127573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuo C-H, Moran NA, Ochman H. 2009. The consequences of genetic drift for bacterial genome complexity. Genome Res 19:1450–1454. doi: 10.1101/gr.091785.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rocha EP, Smith JM, Hurst LD, Holden MT, Cooper JE, Smith NH, Feil EJ. 2006. Comparisons of dN/dS are time dependent for closely related bacterial genomes. J Theor Biol 239:226–235. doi: 10.1016/j.jtbi.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 38.Peterson GI, Masel J. 2009. Quantitative prediction of molecular clock and Ka/Ks at short timescales. Mol Biol Evol 26:2595–2603. doi: 10.1093/molbev/msp175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, Holt KE, Seth-Smith HMB, Quail MA, Rance R, Brooks K, Churcher C, Harris D, Bentley SD, Burrows C, Clark L, Corton C, Murray V, Rose G, Thurston S, van Tonder A, Walker D, Wren BW, Dougan G, Parkhill J. 2010. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A 107:7527–7532. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo H, Swan BK, Stepanauskas R, Hughes AL, Moran MA. 2014. Comparing effective population sizes of dominant marine alphaproteobacteria lineages. Environ Microbiol Rep 6:167–172. doi: 10.1111/1758-2229.12129. [DOI] [PubMed] [Google Scholar]
- 41.Feldman R, Black M, Cary C, Lutz R, Vrijenhoek R. 1997. Molecular phylogenetics of bacterial endosymbionts and their vestimentiferan hosts. Mol Mar Biol Biotechnol 6:268. [PubMed] [Google Scholar]
- 42.Tunnicliffe V. 1988. Biogeography and evolution of hydrothermal-vent fauna in the eastern Pacific Ocean. Proc R Soc Lond B Biol Sci 233:347–366. doi: 10.1098/rspb.1988.0025. [DOI] [Google Scholar]
- 43.Atwater T, Stock J. 1998. Pacific-North America plate tectonics of the Neogene southwestern United States: an update. Int Geol Rev 40:375–402. doi: 10.1080/00206819809465216. [DOI] [Google Scholar]
- 44.Johnson SB, Young CR, Jones WJ, Warén A, Vrijenhoek RC. 2006. Migration, isolation, and speciation of hydrothermal vent limpets (Gastropoda; Lepetodrilidae) across the Blanco Transform Fault. Biol Bull 210:140–157. doi: 10.2307/4134603. [DOI] [PubMed] [Google Scholar]
- 45.Plouviez S, Shank TM, Faure B, Daguin-Thiebaut C, Viard F, Lallier FH, Jollivet D. 2009. Comparative phylogeography among hydrothermal vent species along the East Pacific Rise reveals vicariant processes and population expansion in the South. Mol Ecol 18:3903–3917. doi: 10.1111/j.1365-294X.2009.04325.x. [DOI] [PubMed] [Google Scholar]
- 46.Hurtado LA, Lutz RA, Vrijenhoek RC. 2004. Distinct patterns of genetic differentiation among annelids of eastern Pacific hydrothermal vents. Mol Ecol 13:2603–2615. doi: 10.1111/j.1365-294X.2004.02287.x. [DOI] [PubMed] [Google Scholar]
- 47.Ortmann AC, Suttle CA. 2005. High abundances of viruses in a deep-sea hydrothermal vent system indicates viral mediated microbial mortality. Deep Sea Res Part 1 Oceanogr Res Pap 52:1515–1527. doi: 10.1016/j.dsr.2005.04.002. [DOI] [Google Scholar]
- 48.Kryazhimskiy S, Plotkin JB. 2008. The population genetics of dN/dS. PLoS Genet 4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jani AJ, Cotter PA. 2010. Type VI secretion: not just for pathogenesis anymore. Cell Host Microbe 8:2–6. doi: 10.1016/j.chom.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coulthurst SJ. 2013. The Type VI secretion system—a widespread and versatile cell targeting system. Res Microbiol 164:640–654. doi: 10.1016/j.resmic.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 51.Bladergroen MR, Badelt K, Spaink HP. 2003. Infection-blocking genes of a symbiotic Rhizobium leguminosarum strain that are involved in temperature-dependent protein secretion. Mol Plant Microbe Interact 16:53–64. doi: 10.1094/MPMI.2003.16.1.53. [DOI] [PubMed] [Google Scholar]
- 52.Nyholm SV, Song P, Dang J, Bunce C, Girguis PR. 2012. Expression and putative function of innate immunity genes under in situ conditions in the symbiotic hydrothermal vent tubeworm Ridgeia piscesae. PLoS One 7:e38267. doi: 10.1371/journal.pone.0038267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fabre L, Zhang J, Guigon G, Le Hello S, Guibert V, Accou-Demartin M, de Romans S, Lim C, Roux C, Passet V, Diancourt L, Guibourdenche M, Issenhuth-Jeanjean S, Achtman M, Brisse S, Sola C, Weill F-X. 2012. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS One 7:e36995. doi: 10.1371/journal.pone.0036995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cui Y, Li Y, Gorgé O, Platonov ME, Yan Y, Guo Z, Pourcel C, Dentovskaya SV, Balakhonov SV, Wang X, Song Y, Anisimov AP, Vergnaud G, Yang R. 2008. Insight into microevolution of Yersinia pestis by clustered regularly interspaced short palindromic repeats. PLoS One 3:e2652. doi: 10.1371/journal.pone.0002652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lopez-Sanchez M-J, Sauvage E, Da Cunha V, Clermont D, Ratsima Hariniaina E, Gonzalez-Zorn B, Poyart C, Rosinski-Chupin I, Glaser P. 2012. The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol Microbiol 85:1057–1071. doi: 10.1111/j.1365-2958.2012.08172.x. [DOI] [PubMed] [Google Scholar]
- 56.Yin S, Jensen MA, Bai J, DebRoy C, Barrangou R, Dudley EG. 2013. The evolutionary divergence of Shiga toxin-producing Escherichia coli is reflected in clustered regularly interspaced short palindromic repeat (CRISPR) spacer composition. Appl Environ Microbiol 79:5710–5720. doi: 10.1128/AEM.00950-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kovanen SM, Kivistö RI, Rossi M, Hänninen M-L. 2014. A combination of MLST and CRISPR typing reveals dominant Campylobacter jejuni types in organically farmed laying hens. J Appl Microbiol 117:249–257. doi: 10.1111/jam.12503. [DOI] [PubMed] [Google Scholar]
- 58.Held NL, Herrera A, Cadillo-Quiroz H, Whitaker RJ. 2010. CRISPR associated diversity within a population of Sulfolobus islandicus. PLoS One 5:e12988. doi: 10.1371/journal.pone.0012988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuno S, Yoshida T, Kaneko T, Sako Y. 2012. Intricate interactions between the bloom-forming cyanobacterium Microcystis aeruginosa and foreign genetic elements, revealed by diversified clustered regularly interspaced short palindromic repeat (CRISPR) signatures. Appl Environ Microbiol 78:5353–5360. doi: 10.1128/AEM.00626-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuno S, Sako Y, Yoshida T. 2014. Diversification of CRISPR within coexisting genotypes in a natural population of the bloom-forming cyanobacterium Microcystis aeruginosa. Microbiology 160:903–916. doi: 10.1099/mic.0.073494-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.