

ABSTRACT
1,2-Dichloroethane (DCA) is a problematic xenobiotic groundwater pollutant. Bacteria are capable of biodegrading DCA, but the evolution of such bacteria is not well understood. In particular, the mechanisms by which bacteria acquire the key dehalogenase genes dhlA and dhlB have not been well defined. In this study, the genomic context of dhlA and dhlB was determined in three aerobic DCA-degrading bacteria (Starkeya novella strain EL1, Xanthobacter autotrophicus strain EL4, and Xanthobacter flavus strain EL8) isolated from a groundwater treatment plant (GTP). A haloalkane dehalogenase gene (dhlA) identical to the canonical dhlA gene from Xanthobacter sp. strain GJ10 was present in all three isolates, and, in each case, the dhlA gene was carried on a variant of a 37-kb circular plasmid, which was named pDCA. Sequence analysis of the repA replication initiator gene indicated that pDCA was a member of the pTAR plasmid family, related to catabolic plasmids from the Alphaproteobacteria, which enable growth on aromatics, dimethylformamide, and tartrate. Genes for plasmid replication, mobilization, and stabilization were identified, along with two insertion sequences (ISXa1 and ISPme1) which were likely to have mobilized dhlA and dhlB and played a role in the evolution of aerobic DCA-degrading bacteria. Two haloacid dehalogenase genes (dhlB1 and dhlB2) were detected in the GTP isolates; dhlB1 was most likely chromosomal and was similar to the canonical dhlB gene from strain GJ10, while dhlB2 was carried on pDCA and was not closely related to dhlB1. Heterologous expression of the DhlB2 protein confirmed that this plasmid-borne dehalogenase was capable of chloroacetate dechlorination.
IMPORTANCE Earlier studies on the DCA-degrading Xanthobacter sp. strain GJ10 indicated that the key dehalogenases dhlA and dhlB were carried on a 225-kb linear plasmid and on the chromosome, respectively. The present study has found a dramatically different gene organization in more recently isolated DCA-degrading Xanthobacter strains from Australia, in which a relatively small circular plasmid (pDCA) carries both dhlA and dhlB homologs. pDCA represents a true organochlorine-catabolic plasmid, first because its only obvious metabolic phenotype is dehalogenation of organochlorines, and second because acquisition of this plasmid provides both key enzymes required for carbon-chlorine bond cleavage. The discovery of the alternative haloacid dehalogenase dhlB2 in pDCA increases the known genetic diversity of bacterial chloroacetate-hydrolyzing enzymes.
INTRODUCTION
Organochlorine chemicals have been widely used as pesticides, solvents, and for plastic manufacture. Many of these compounds are xenobiotic, and, as a result, their environmental breakdown is often slow (1). The environmental persistence of organochlorines is a problem, since many of these chemicals are damaging to human and environmental health (2). One organochlorine of concern is 1,2-dichloroethane (DCA), which is often present as a groundwater contaminant (3, 4). DCA is not known to exist in nature and was first produced industrially as a solvent in the 1920s (5), then later found use as a lead scavenger in petrol (3). Currently, DCA is primarily used as the precursor for polyvinyl chloride plastics (6).
Biodegradation of DCA has been demonstrated both at lab scale (7) and at field scale (8). There are at least four distinct physiological types of bacteria that can degrade DCA: first, anaerobes that reductively dehalogenate DCA and use it as an electron acceptor (9–11); second, aerobes containing broad-spectrum monooxygenases that attack DCA by oxidative cometabolism (12–14); third, aerobes that grow on DCA as a carbon and energy source after an initial oxidative reaction (15, 16); and finally, aerobes that grow on DCA as a carbon and energy source after an initial hydrolytic reaction (17, 18). These four distinct physiological modes of DCA biodegradation encapsulate the majority of well-studied isolates, but they do not represent an exhaustive list; for example, some anaerobes can use DCA as a carbon and energy source while respiring nitrate (19). It is likely that further metabolic novelty among DCA-degrading bacteria will be discovered as modern approaches, such as metagenomics, are increasingly applied to contaminated sites (20).
The aerobic hydrolytic DCA degraders (Xanthobacter and Ancylobacter spp.) (7, 18, 21–24) appear to be very widespread, and, due to their ease of growth and favorable kinetics, they are excellent candidates for bioremediation of DCA-contaminated sites. The DCA degradation pathway in these bacteria begins with hydrolytic removal of a chlorine atom by a haloalkane dehalogenase (DhlA), yielding 2-chloroethanol. This first metabolite is sequentially oxidized by alcohol and aldehyde dehydrogenases (Mox, AldA) to give chloroacetic acid, and then a hydrolytic haloacid dehalogenase (DhlB) converts chloroacetate to glycolic acid, which can enter core metabolism via the glyoxylate cycle. All of the aerobic hydrolytic DCA degraders isolated to date are facultative methylotrophs; this seems to be related to the fact that the methanol dehydrogenase Mox is an effective 2-chloroethanol dehydrogenase (18). Since AldA is not unique in its ability to metabolize chloroacetaldehyde (25–27), it is likely that acquisition of just DhlA and DhlB would be sufficient to enable many types of bacteria to grow on DCA.
The DhlA and DhlB enzymes of Xanthobacter autotrophicus GJ10 are very well characterized (28–32), but less is known about the evolutionary history and genomic context of the dhlA and dhlB genes. Identical copies of dhlA exist in 12 independent bacterial isolates from diverse locations (18, 28, 32, 33), and this gene seems to be uniquely associated with growth on DCA. The sequence for dhlB is only available for Xanthobacter sp. strain GJ10 (30, 34), although this gene is presumed to be present in other aerobic hydrolytic DCA degraders. In strain GJ10, dhlA and aldA are associated with a 225-kb linear plasmid, while the dhlB and mox genes are chromosomal (26, 35). To date, only small regions of flanking DNA sequences are known for the dhlA and dhlB dehalogenases; there are no whole genomes or plasmid genomes available.
The dhlA and dhlB genes are likely to have been mobilized within and between bacterial cells by mobile genetic elements to yield bacteria that can productively utilize DCA (35, 36), but the details of these processes are unknown. It is unclear to what extent different aerobic hydrolytic degraders may have different evolutionary histories and different arrangements of dehalogenase genes, or to what extent (if at all) other types of dehalogenases may substitute for the canonical forms of dhlA and dhlB. Here, we aim to answer these questions, using a set of related aerobic DCA-degrading bacteria isolated from a groundwater treatment plant (24).
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions. (i) DCA degraders.
The DCA-degrading strains Starkeya novella strain EL1, Xanthobacter autotrophicus strain EL4, and Xanthobacter flavus strain EL8 were isolated from samples obtained from the Botany groundwater treatment plant (GTP) (Sydney, Australia). Strains EL1 and EL4 were from a granular activated carbon filtration unit, while strain EL8 was from a membrane bioreactor. The details of this GTP, and a preliminary report on the isolation of DCA degraders from it, can be found in a previous study (24). Enrichments on DCA were done in mineral salts medium (MSM) (37) using DCA (100 mg/liter) as the sole added carbon and energy source, and then strains were isolated and purified on R2A agar (38). DCA-degrading cultures were grown aerobically at 30°C, with broths shaken at 200 rpm. Growth of DCA-containing liquid cultures was done in serum bottles that were crimp sealed with Teflon-faced butyl rubber stoppers. Growth on agar with DCA was done by incubating the inoculated MSM plates in a gas-tight glass jar (2.5 liters) alongside a 2-ml crimp-sealed vial containing 200 μl DCA, pierced with a 25-gauge needle. Biodegradation of DCA was monitored by gas chromatography and chloride analysis, as described previously (39).
(ii) Escherichia coli.
Escherichia coli strains TOP10 (Thermo Fisher Scientific), BL21(DE3)(pLysS) (Thermo Fisher Scientific), and EPI300 (Epicentre) were grown in LB medium (40) aerobically at 37°C, with broths shaken at 200 rpm, unless otherwise indicated. Strain TOP10 was used for construction of plasmids, strain BL21(DE3)(pLysS) was used for protein expression, and strain EPI300 was used for maintaining fosmids. Antibiotics were added to LB medium as appropriate for the different cloning vectors used: ampicillin (Ap) was added at 100 μg/ml to select pET15+ derivatives, chloramphenicol (Cm) was added at 25 μg/ml to select pLysS and induced fosmids, and Cm was added at 12.5 μg/ml to select uninduced fosmids.
Genomic DNA extraction.
Genomic DNA was extracted from strains EL1, EL4, and EL8 according to the protocol of Wilson (40), with modifications to enhance cell lysis, as follows: after resuspension in TE, cells were exposed to three freeze-thaw cycles using a dry ice-ethanol bath and a 65°C water bath. Prior to addition of SDS, resuspended cells were incubated with lysozyme (15 mg/ml) for 2 h at 37°C, and the proteinase K incubation was increased to 2 h.
PCR.
PCR was done with a Mastercycler ep gradient S machine (Eppendorf). The typical thermocycling protocol included an initial denaturation (94°C, 5 min) and then 30 cycles of 94°C (30 s), 55 to 60°C (depending on primers) (30 s), 72°C (1 min per kb of product), and a final extension (72°C, 5 min). Taq polymerase (New England BioLabs) was used for routine PCRs, such as screening E. coli clones on agar plates, while Phusion polymerase (New England BioLabs) was used for amplifying whole genes for cloning and expression. The extension time was reduced to 20 s per kb of product in PCR using Phusion polymerase. DCA-degrading strains were identified by PCR amplification and sequencing of partial 16S rRNA gene fragments using primers 27f and 519r (41), followed by BLAST search of the NCBI nucleotide database. The sequences were deposited in GenBank as JX515582 to JX515584. The presence of dhlA was tested in each strain via PCR using primers DHLA-319F (CTTGCACTAATCGAACGGCTTG) and DHLA-603R (AGCTTCGGTCAGTGTGGGCG).
Fosmid library construction.
The CopyRight v2.0 fosmid cloning kit (Lucigen) was used for construction of fosmid libraries according to the manufacturer's instructions. Briefly, genomic DNA from DCA-degrading isolates was sheared by repeated pipetting, then separated by pulsed-field gel electrophoresis (1% low-melting-point agarose, 0.5 × Tris-borate-EDTA buffer, switch time of 1 to 6 s, run time of 16 h, pulse angle of 120°, 5 V/cm, 4°C). DNA fragments between 30 and 45 kb were excised with reference to the Lambda mono-cut ladder (New England BioLabs) and extracted from the agarose by beta agarase (New England BioLabs) treatment and ethanol precipitation. The DNA was then ligated into the vector pSMART-FOS (Lucigen) and packaged using MaxPlax lambda packaging extracts (Epicentre). Packaged fosmids were transduced into EPI300 cells and selected on LB-sucrose (5%, wt/vol)-chloramphenicol (12.5 μg/ml) plates. Fosmid-carrying Cm-resistant (Cmr) clones of EPI300 were picked into 96-well plates and grown for 16 h in 150 μl LB-Cm broth. For each genomic library (EL1, EL4, and EL8), 288 clones were screened.
Fosmid library screening for dhlA and dhlB.
Fosmids containing dhlA were detected via PCR, as follows: 10 μl of culture from each well in a single column (8 wells) was combined, and then 1 μl of the mixture was assayed by PCR, using the DHLA-319F and DHLA-603R primers. Individual wells containing dhlA-positive clones were identified via a second round of dhlA PCRs, using 1 μl of culture from each of the 8 wells in a positive column as the template. Columns and wells with dhlB-containing fosmids were detected by a similar strategy, except that Southern hybridization (dot blot) was used instead of PCR, as follows: 10 μl of culture from each well in a column was combined, and then 10 μl of the mixture was spotted onto an LB-Cm agar plate (12 spots per plate) and grown overnight. Hybridization was performed as described below, and then the experiment was repeated with 10 μl culture from individual wells from any positive columns.
A digoxigenin (DIG)-labeled probe was generated via PCR, by including dioxigenin-11-dUTP at 200 μM in the master mix and performing a PCR with a doubled extension time. The probe consisted of 403 bp of the dhlB gene from Xanthobacter strain GJ10, amplified with primers JM12 (CAGAAGCAGCTGGAATACAGCT) and JM13 (CGTTGGAGGACACGAACAGC). Southern hybridization was performed using the following protocol (adapted from the Amersham Biosciences product booklet): heat-denatured (99°C, 10 min) DNA (approximately 200 ng) was spotted onto a Hybond N+ nylon membrane (Amersham) and allowed to dry. Cross-linking was performed by UV exposure, and the membrane was immediately submerged in a solution of DIG Easy Hyb (Roche) (200 μl/cm2 of membrane). The following steps were performed with constant agitation in a hybridization oven (Hybaid), with solutions from the Easy Hyb kit used at 200 μl/cm2: prehybridization (50°C, 1 h), hybridization (50°C, 16 h, probe at 25 ng/μl), low-stringency wash (20°C, 5 min), two medium stringency washes (50°C, 15 min), water rinse (20°C, 30 s), blocking (1 h, 20°C), anti-DIG antibody addition (20 U/liter of reagent at 20°C, 30 min), two washes (wash buffer, 20°C, 15 min), and then equilibration with detection buffer (20°C, 30 s). The membrane was then placed in an autoradiography cassette, detection reagent was added (CDP star; Roche), and luminescence was captured using X-ray film (Kodak) with an exposure time of 5 min. Film was developed in an automatic processor (Agfa).
Fosmid sequencing and annotation.
Fosmids that contained dhlA or dhlB genes were purified from arabinose-induced EPI300 cells using the PlasmidMax DNA isolation kit (Epicentre) according to the manufacturer's instructions. Fosmids identified as containing dhlB genes from isolates EL1 and EL8 were sequenced by the primer walking approach (Sanger sequencing; Australian Genome Research Facility) until the full dhlB gene sequence was obtained. Fosmids containing dhlA genes from all three isolates, and the fosmid containing dhlB from isolate EL4, were completely sequenced using a mix of Illumina Hi-Seq (Beijing Genomics Institute [BGI], Hong Kong), and Ion Torrent (Research and Testing Labs [RTL], Lubbock, Texas, USA) sequencing. Assembly was carried out using SOAPdenovo (BGI) and NGEN (RTL). Gaps in the resultant draft sequences were manually filled by PCR from genomic DNA of the corresponding DCA-degrading isolates and sequencing of the products. Gene prediction was carried out by GeneMark.hmm (42), and the functions of putative coding regions were inferred by BLASTp queries against the NCBI nonredundant database. Annotations were completed and visualized using Sequin (NCBI), Geneious (Biomatters, Ltd.), and Snapgene (GSL Biotech).
Cloning of dhlb2 in pET15b+.
The dhlB2 gene of Xanthobacter strain EL4 was synthesized as an 809-bp GBlock (Integrated DNA Technologies), which contained 30-bp sequences at the ends that match the ends of a XhoI/BamHI-cut pET15b+ plasmid. pET15b+ (Novagen) was digested with 20 U XhoI and 20 U BamHI-HF and blunted with 20 U T4 DNA polymerase (all enzymes from New England BioLabs). Isothermal assembly of the plasmid and GBlock was conducted with the Gibson Assembly cloning kit (New England BioLabs) according to the manufacturer's instructions. The ligation mix was transformed by heat shock into chemically competent E. coli TOP10, and resultant ampicillin-resistant colonies were screened for the presence of a recombinant DNA junction by PCR with primer pair VMC15 (CTCACTATAGGGGAATTGTGAGCGG) and MAL92 (CTAAGGGGCAGTTCTACATC). Plasmid was purified from one positive clone and named pET-dhlB2. The correct structure of this plasmid was confirmed by sequencing of the insert region and HindIII digestion.
Expression and purification of the DhlB2 protein.
pET-dhlB2 was transformed into E. coli BL21(DE3)(pLysS), and the resultant transformants were grown in LB broth containing 100 μg/ml ampicillin and 25 μg/ml chloramphenicol to mid-log phase (optical density at 600 nm [OD600], 0.6); then, 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) was added, and incubation continued for a further 6 h at 25°C (final OD600, 3.75). Cells were centrifuged (8,000 × g, 10 min), resuspended in 20 ml of binding buffer (50 mM sodium phosphate [pH 8], 0.5 M NaCl, 20 mM imidazole), and stored at −20°C until required. Cells were thawed on ice and lysed by sonication (Branson sonifier 250) (50% pulse, output control 6, 3 × 30 s cycles, with 1 min rest on ice between each cycle). The cell lysate was centrifuged (3,500 × g, 20 min, 4°C), and the supernatant was filtered through a 0.2-μm-pore-size filter. The resultant cell extract was pushed through an equilibrated 1-ml His-trap column (GE Healthcare) at a flow rate of 2.5 ml/min, followed by 5 ml of wash buffer (50 mM sodium phosphate [pH 8], 0.5 M NaCl, 80 mM imidazole), then 5 ml of elution buffer (50 mM sodium phosphate [pH 8], 0.5 M NaCl, 400 mM imidazole), and 5 × 1-ml elution fractions were collected. Fractions containing overexpressed DhlB2, as determined by SDS-PAGE, were pooled, and 3 ml was injected in a hydrated dialysis cassette (3.5-kDa cutoff). The sample was dialyzed with stirring at 4°C in 1 liter of high-salt dialysis buffer (50 mM sodium phosphate [pH 7.5], 50 mM NaCl, 5% glycerol, and 1 mM EDTA) for 2 h, then 1 liter of medium-salt dialysis buffer (50 mM sodium phosphate [pH 7.5], 5 mM NaCl, 5% glycerol, and 1 mM EDTA) for 2 h, then 1 liter low-salt dialysis buffer (50 mM sodium phosphate [pH 7.5], 0.5 mM NaCl, 5% glycerol, and 1 mM EDTA) for 16 h. The amount of purified DhlB2 was measured spectrophotometrically using the estimated extinction coefficient for this protein sequence at 280 nm (44,265 M−1 · cm−1).
Chloride release assay.
The haloacid dehalogenase activity of DhlB2 was measured using chloroacetate as the substrate. A set of six reactions was set up containing purified DhlB2 protein (2 μg) in 1 ml of 20 mM Tris-sulfate buffer (pH 7.5) containing 2 mM chloroacetate and incubated at 20°C. At 15-min intervals, one reaction was sacrificed for analysis by colorimetric chloride assay. The assay of Bergmann and Sanik (43) was used, with modifications as described previously (39). The entire experiment was repeated three times, and the specific activity was determined as micromoles of chloride released per minute per microgram of protein, based on rates determined as straight lines of best fit through the chloride data. Purified DhlB2 protein that had been heated at 90°C for 30 min was used as a negative control in the assays.
Accession number(s).
The 16S rRNA gene sequences were deposited in GenBank under accession numbers JX515582 to JX515584. Complete fosmid sequences were deposited in GenBank under accession numbers KU922118 to KU922121.
RESULTS
Isolation and genetic characterization of DCA-degrading bacteria.
Four aerobic bacteria that could biodegrade DCA were isolated from a groundwater treatment plant (GTP), which was treating DCA-contaminated groundwater (24). The isolates were identified by partial 16S rRNA gene sequencing. Isolate EL1 was Starkeya novella (100% nucleotide identity to strain DSM506T), isolate EL4 was Xanthobacter autotrophicus (99% nucleotide identity to strain JCM1202T), isolate EL5 was a Leifsonia sp. (97% nucleotide identity to Leifsonia xyli subsp. cynodontis DSM46306T), and isolate EL8 was Xanthobacter flavus (100% identity to strain NBRC 144759T). All of the isolates could biodegrade DCA as a sole carbon and energy source (see Fig. S1 in the supplemental material), with release of chloride (see Fig. S2 in the supplemental material). Due to clumping of cells and the low growth yield on DCA, it was difficult to confirm the OD600 or viable count increases linked to DCA biodegradation, but total protein in cultures increased by 5 to 10 μg/ml after degradation of 4 mM DCA. While DCA-degrading Xanthobacter spp. have been reported (17, 22), Starkeya and Leifsonia were not previously known as DCA degraders.
Based on review of the GenBank database and previous literature (44), it appeared that dhlA genes were very highly conserved (>98%) at the nucleotide level among different DCA-degrading isolates, but dhlB genes had many diverse homologs, making PCR primer design easy in the case of dhlA but difficult in the case of dhlB. Therefore, a PCR strategy was used for detection of dhlA and a Southern hybridization strategy was used for detection of dhlB in the new isolates from the GTP. Strains EL1, EL4, and EL8 were found to contain both dhlA and dhlB dehalogenase genes, while EL5 did not yield products of the expected size in the dhlA PCR and did not hybridize to the dhlB probe (see Fig. S3 and S4 in the supplemental material). While the situation in EL5 was intriguing, we decided to leave further investigation of this strain for a future study and instead focused here on identifying the genomic context of dhlA and dhlB in the Xanthobacteraceae isolates EL1, EL4, and EL8.
Fosmid libraries (288 clones each) constructed from genomic DNA of strains EL1, EL4, and EL8 were screened for PCR for dhlA genes, resulting in three positive results for EL4, two for EL1, and one for EL8. One dhlA-positive fosmid clone derived from each strain was completely sequenced. Screening by hybridization for dhlB resulted in one positive result for EL1, seven for EL4, and four for EL8. It was notable that none of the fosmid clones which were positive for dhlA were also positive for dhlB, apparently confirming previous reports (22, 35) that these genes are present at different genomic loci in DCA-degrading bacteria. One fosmid containing dhlB from each strain was partially sequenced by primer walking, until the complete sequence of the dhlB gene was obtained. In addition, the EL4 dhlB-containing fosmid was completely sequenced.
A typical dhlB gene (dhlB1) is present in strains EL1, EL4, and EL8.
The fosmid sequence data revealed that the dhlB genes in all three Botany isolates were nonidentical but similar to each other and similar to the dhlB gene of strain GJ10 (86 to 94% inferred amino acid [aa] identity in DhlB protein). The GJ10-like dhlB sequences in the Botany isolates are here referred to as dhlB1. As seen previously with Xanthobacter strain GJ10 (34), the dhlB1 gene in strain EL4 was clustered with the dhlC and dhlR genes (Fig. 1), which are likely to code for a haloacid transporter and a regulator of haloacid metabolism, respectively, although neither function has yet been experimentally confirmed. The dhlB1 gene in strain EL8 was found to be at the same genomic locus as that of EL4, as far as could be ascertained from the sequence available (1,260 bp). In contrast, based on a similar amount of sequence information (1,301 bp), the dhlB1 gene of EL1 appeared to be in a different genomic locus, and was not colocated with dhlC (Fig. 1).
FIG 1.

Comparison of genomic locations of dhlB1 homologs in Xanthobacter strains EL1, EL4, EL8, GJ10, and Py2. The location of dhlB1 is highlighted with a blue star. A conserved 4-gene cluster, present in both EL4 and Py2, is boxed. The open reading frame color scheme is as follows: blue, metabolism; yellow, regulators; orange, transporters; light gray, hypothetical proteins; dark gray, transposases; maroon, integrase. Maps prepared in SnapGene.
Intriguingly, BLAST analysis revealed that a dhlB1 homolog (60% inferred aa identity to GJ10 DhlB) was found in the chromosome of Xanthobacter strain Py2, a bacterium isolated on propene as a carbon and energy source (45). Although the genomic context of the Py2 dhlB gene is not identical to that of the GJ10 and EL4 dhlB1 genes, there are similarities (Fig. 1), notably the presence of a conserved cluster of genes nearby (araC family regulator, NADH:flavin oxidoreductase, membrane protein, and acetate transporter) which share 81% DNA identity over 4.6 kb to the corresponding EL4 genes. Therefore, it can be inferred that, first, the dhlB1 gene of EL4 is most likely chromosomally located, and, second, haloacid dehalogenases are not specific just to Xanthobacter strains isolated on halogenated substrates. The presence of a phage-like integrase gene and an IS110-family transposase gene (see Fig. S5 in the supplemental material) near the dhlB1 gene of EL4 may explain the changes in this genomic region of EL4 relative to Xanthobacter strain Py2. Due to the very high sequence identity of the dhlB1 haloacid dehalogenases in the Botany isolates to dhlB of strain GJ10, we did not biochemically characterize their gene products and initially assumed that the DhlB1 enzymes were responsible for chloroacetate metabolism in strains EL1, EL4, and EL8.
The dhlA gene is found on a catabolic plasmid, pDCA.
One dhlA-containing fosmid from each strain was completely sequenced. When the final assemblies were obtained and analyzed, it became clear that all three dhlA-containing fosmids were similar, and, within each fosmid, the complete sequence of a dhlA-containing plasmid of approximately 37 kb had been cloned; these three variant plasmids are here referred to as pDCA-EL1, pDCA-EL4, and pDCA-EL8 (Fig. 2 and 3). All three variants of the pDCA plasmid had very similar overall gene organization, which could be classified broadly into a replication and stability region, a mobilization region, a catabolism region, and a junkyard region, full of insertion sequences (IS) and fragments thereof (Fig. 3). The dhlA genes on all three variant pDCA plasmids were 100% identical to each other and to the dhlA genes of Xanthobacter strain GJ10 (28), Ancylobacter spp. (18), and Xanthobacter strain UE15 (22). The DNA sequences immediately upstream of dhlA in the Botany isolates contained a putative ribosome binding site (TACGGAGG) and two overlapping putative sigma-70 promoters (TTGACA-n17-TACTTT and TTGTCA-n18-TAACTT); these regulatory signals were also 100% identical to those previously identified in strain GJ10 (28). Due to the very strong sequence identity of the dhlA genes and their regulatory sequences in the Botany isolates to those in strain GJ10, and the fact that dhlA has to date only been found in bacteria capable of growth on DCA, the dhlA genes detected on the pDCA plasmids were assumed to be responsible for the initial dehalogenation of DCA in isolates EL1, EL4, and EL8.
FIG 2.

Circular map of plasmid pDCA-EL1 from Starkeya novella strain EL1. Genes are color coded as follows: red for critical functions (replication, partitioning, multimer resolution), purple for stability-enhancing systems (toxin/antitoxin pairs and restriction/modification systems), green for mobilization functions, yellow for transcriptional regulators, blue for metabolic genes, orange for transporters, dark gray for transposases and transposon-associated resolvases, and light gray for hypothetical proteins.
FIG 3.

Alignment of open reading frames in the three variant pDCA plasmids obtained from isolates EL1, EL4, and EL8. The color scheme is the same as in Fig. 2. Note that the functional categories assigned to each region of the plasmid are generalizations based on the function of the majority of genes in each region, but there are exceptions (e.g., dhlB2 is within the mobilization region). The locations of a few key features (repA, dhlB2, traA, dhlA, IS66) are shown to assist comparisons with Fig. 2.
A second dhlB homolog (dhlB2) is found on pDCA.
Surprisingly, during analysis of the pDCA sequences, a haloacid dehalogenase was found in the sequence of all three variant plasmids. This gene, named dhlB2, was 100% identical across all three plasmids. The dhlB2 gene clearly had not hybridized to the strain GJ10 dhlB gene probe used for fosmid clone screening, although this probe successfully detected the dhlB1 genes in other fosmids in these libraries. The predicted DhlB2 protein had only 29% amino acid identity to DhlB1 of GJ10, but, based on BLAST analysis and sequence alignment (Fig. 4), DhlB2 was clearly part of the HAD-II haloacid dehalogenase family. DhlB2 was 100% identical to an uncharacterized protein encoded in the genome of the acetochlor-degrading Sphingobium sp. strain DC2 (46) and possessed 40 to 60% identity to cryptic HAD-II dehalogenases that are present in hundreds of genome sequences from the Proteobacteria.
FIG 4.

Alignment of DhlB proteins from Xanthobacter and Starkeya spp. Note the high sequence divergence of DhlB2 from all the other DhlB proteins. The alignment was generated in ClustalX (58) and then visualized with GeneDoc (59). Shading indicates sequence conservation at that position, as follows: black, 100%; dark gray, 80%; light gray, 60%. Bars below and above the alignment indicate residues known to be essential for catalysis in other haloacid dehalogenases and residues forming the hydrophobic pocket, respectively (60).
Due to the low amino acid identity of DhlB2 to previously characterized dehalogenases, this gene was expressed in E. coli to confirm the activity of the encoded enzyme. Plasmid pET-DhlB2 was constructed, which fused a 6× His tag to the N terminus of DhlB2, and protein expression was examined in IPTG-induced cells of BL21(DE3)(pLysS)(pET-DhlB2). Overexpression of a protein of the expected size (27 kDa) was seen in induced cells, and, although much of this protein was in the insoluble fraction, at least some could be successfully purified by nickel affinity chromatography, giving a preparation of approximately 80% purity (see Fig. S6 in the supplemental material). The biochemical activity of the purified 6× His-DhlB2 protein was examined by assaying for inorganic chloride release from chloroacetate. A specific activity of 8.9 μmol/min/mg protein was determined (see Fig. S7 in the supplemental material), which was similar to the purified DhlB from strain GJ10 (30). Thus, despite the high sequence divergence from DhlB1, the DhlB2 enzyme is clearly a chloroacetate dehalogenase and thus is likely to contribute to the growth of strains EL1, EL4, and EL8 on DCA.
pDCA is a member of the pTAR family of Alphaproteobacteria plasmids.
The putative RepA protein of the pDCA plasmids was related to replication initiators from the pTAR plasmid family (Fig. 5; see also Table S1 in the supplemental material), which are widespread in the Alphaproteobacteria. There are several uncharacterized close homologs (80 to 100% inferred aa identity) of the pDCA RepA gene product in draft genome sequences from Ochrobactrum, Paracoccus, Agrobacterium, Sphingobium, and Mesorhizobium strains. The closest homolog to the pDCA RepA protein that is known to be plasmid associated is RepA from the aromatic hydrocarbon catabolic plasmid pSKYE1 (88% inferred aa identity), derived from an uncultured bacterium (47). The closest homolog that is experimentally characterized is RepA from the tartrate catabolic plasmid pTAR, from Agrobacterium tumefaciens (34% inferred aa identity). Other pDCA RepA homologs of note exist in the agrocin84 biosynthetic plasmid pAgK84 from Agrobacterium radiobacter (53% inferred aa identity) and in the dimethylformamide catabolic plasmid pAMI2 from Paracoccus aminophilus (79% inferred aa identity). The gene organization of the pDCA backbone (replication, stability, mobilization functions) was very similar to that of pAMI2 and less similar to that of pAgK84 (see Fig. S8 in the supplemental material). Given the large number (>200) of uncharacterized pDCA RepA homologs in GenBank, it can be surmised that this plasmid family is a more important player in the evolution of the Alphaproteobacteria than has previously been appreciated.
FIG 5.
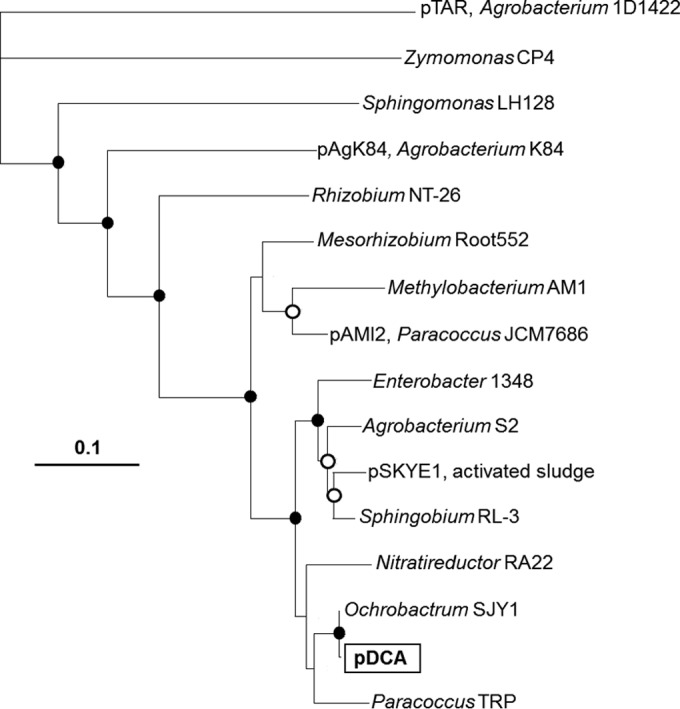
Phylogenetic tree showing relationships among RepA proteins in the pTAR plasmid family. Plasmid names are shown where known. In other cases (draft genomes), the genus and strain names are shown. The tree was prepared from a 538-aa alignment (including gaps), generated in ClustalX (59), which gave a consensus tree via the neighbor-joining method. The tree was visualized with TreeView (61) and manually annotated using GIMP (https://www.gimp.org/). Filled circles at nodes indicate bootstrap values >95%, open circles indicate bootstrap values >75%, and nodes with no symbol have bootstrap values <75%. More detail on these representative RepA homologs is available in Table S1 in the supplemental material.
Stability and mobilization functions of pDCA.
A 3-kb putative replication and stability region containing the repA gene along with partitioning genes parA and parB, and a toxin-antitoxin gene pair (relE-relB family), was absolutely conserved (100% nucleotide identity) among all three variants of the pDCA plasmid (Fig. 3), suggesting that this region is important for plasmid maintenance. Many homologs of the inferred pDCA ParA protein were detected in GenBank, with most in the Alphaproteobacteria, but the vast majority of these have not been characterized. The closest homolog of the inferred pDCA ParA protein with experimentally confirmed function is the MipZ chromosome partitioning protein of Caulobacter crescentus, at 20% aa identity (36% aa similarity). Despite the low overall protein identity, both pDCA ParA and Caulobacter MipZ contained identical Walker A motifs near the N terminus (KGGAGK), consistent with an ATP-binding function and characteristic of active DNA partitioning systems (48).
The toxin-antitoxin gene pair related to the relE-relB family found on all three variant pDCA plasmids has homologs in plasmids and chromosomes of many members of the Bacteria and Archaea, with the closest matches (67 to 99% inferred aa identity) found to uncharacterized gene products in Alphaproteobacteria draft genomes. The predicted pDCA RelE toxin has 14% aa identity and 33% aa similarity to the chromosomal E. coli RelE protein, which acts as a translation inhibitor (49). pDCA-EL1 and pDCA-EL8 contain three additional stability systems compared to pDCA-EL4, namely, a toxin-antitoxin pair in the higB-higA family (34% inferred aa identity to HigB killer protein of E. coli plasmid Rts), a type III restriction-modification system (10% inferred aa identity and 30% aa similarity to EcoPI Res of phage P1), and a serine recombinase-type resolvase (75% inferred aa identity to the resolvase of Pseudomonas plasmid pVS1). It appears that plasmid pDCA-EL4 has suffered a 6.3-kb deletion of stability-associated genes relative to the other two plasmids, possibly mediated by the Tn3-like elements in the neighboring junkyard region of the plasmid; however, this deletion does not seem to have critically impacted the maintenance of this plasmid in Xanthobacter strain EL4.
An identical three-component plasmid mobilization system was encoded in all three pDCA variants, consisting of mobC, traA, and traG homologs. There are hundreds of close homologs to these mobilization genes in genome-sequenced members of the Alphaproteobacteria, but all of these relatively close matches (>50% inferred aa identity) are uncharacterized. The MobC protein in pDCA has 48% inferred aa identity to MobC of the resistance plasmid RSF1010, while TraA and TraG of pDCA have 47% and 41% inferred aa identity, respectively, to TraA and TraG of the genomic island pKLC102 of Pseudomonas aeruginosa. Based upon the presence of mobC (relaxosome protein), traA (relaxase), and traG (conjugative coupling protein) genes, and the absence of any pilus-encoding genes, it seems likely that pDCA is a mobilizable, nonconjugative plasmid (50), which depends upon other mobile genetic elements to provide conjugative pili to enable its horizontal transfer between bacteria.
Transposons and insertion sequences in pDCA.
An insertion sequence from the ISAs1 family was located just downstream of the dhlA gene in all three variants of the pDCA plasmid. The transposase has 100% aa identity to the gene in the corresponding location in Xanthobacter strain GJ10 (28) but has no other close homologs in GenBank (all <50% aa identity). The closest homolog with an experimentally confirmed function was the transposase of ISPmar4 from Paracoccus marcusii (51), at 38% aa identity. The dhlA-associated IS element in pDCA has identical 16-bp inverted repeats (IR-L and IR-R: CAGGGCAATCGCACTA) and is flanked by a 10-bp direct repeat (ACCCCAACCT); the latter indicates that this is an active mobile element, capable of transposition. This insertion sequence has previously been named ISXa1, since it was first identified in X. autotrophicus GJ10. The present study has enabled the complete characterization of this element, and its details have been updated in the ISFinder database (www-is.biotoul.fr).
An insertion sequence from the IS1380 family was found immediately adjacent and upstream of the dhlB2 gene in all three variants of the pDCA plasmid. The transposase of this element has 99.8% amino acid identity to the transposase of ISPme1 (52), which is found in the catabolic plasmids pMTH1 and pMTH4 of Paracoccus methylutens. The dhlB2-associated IS element has 16-bp inverted repeats that are identical to those of ISPme1 (IR-L: CCCACATTCTCCAAGT; IR-R: CCGACATTCCCCAAGT); thus, the element in pDCA was also named ISPme1. The transposase of ISPme1 is very closely related (92% amino acid identity) to that of IS1247, which has previously been found upstream of the dhlB gene in mutants of Xanthobacter strain GJ10 with enhanced haloacid dehalogenase activity (36). A remarkable feature of the ISPme1 and IS1247 elements is that they are capable of mobilizing the 3′ adjacent DNA regions in the absence of a composite transposon structure (36, 52), so it seems very likely that ISPme1 was responsible for recruiting the dhlB2 gene into pDCA. ISPme1 is also likely to play a role in activating the transcription of dhlB2. Two putative sigma-70 promoters were identified at the 3′ end of the ISPme1 sequence; one of these was wholly contained within the IS, while the other was formed at the junction of the IS with the downstream DNA sequence (Fig. 6).
FIG 6.
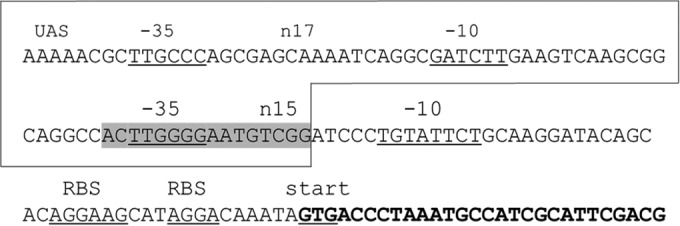
DNA region at the junction of ISPme1 and dhlB2. DNA corresponding to the insertion sequence is boxed, and the inverted repeat of the IS element is shaded gray. Two putative sigma-70 promoters are indicated, including upstream activator sequences (UAS), −35 and −10 signals, and the spacing between the −35 and −10 signals (17 or 15 nucleotides). Note that the second promoter has an extended −10 motif, including a TG at −12. Two possible ribosome binding sites are shown for dhlB2. The dhlB2 open reading frame is shown in bold.
The junkyard region (Fig. 3) was the most variable part of the pDCA plasmids and offers a window into the recent evolutionary processes impacting these elements. pDCA-EL1 contained two different Tn3 homologs in this region (19% aa identity and 38% aa similarity in the inferred transposase protein). It appears that one complete Tn3-like element (including both transposase and resolvase) has transposed into a preexisting Tn3-like element. The newer acquisition remains intact, with identical 38-bp IR-L and IR-R sequences (GGGGGCACCTCAAGGTTTTACCGCCAGAATACGCTAAG) and flanked by 5-bp direct repeats (TTCTT); all of these features were consistent with an active Tn3 family transposon. The older Tn3 element present in pDCA-EL1 did not have an identifiable associated resolvase or IR-R sequence, but a possible IR-L sequence was identified (GGGGAAGTTTTTGGGTAACAGCCCCAAACAGAGGCGCT).
The junkyard region of pDCA-EL4 differed from pDCA-EL1 in that the older Tn3 element had been more completely deleted, and a small gene encoding a hypothetical protein was also missing. The corresponding region in pDCA-EL8 was different again; in this plasmid variant, an IS66-family insertion sequence had been inserted within the complete Tn3 element, and again the small hypothetical protein-encoding gene was missing. The differences observed in the IS and transposon content in the three pDCA variants first provided confidence that these really were independent plasmids, derived from three different host bacteria; second, they provided hints to the amount of evolutionary time that has elapsed since these plasmids had a common ancestor. While it is difficult to put an exact figure on this, clearly this amount of time is sufficient to allow large-scale changes to nonessential regions of the plasmid but not long enough to allow sequence divergence of the core plasmid backbone regions, which, as noted above, have 100% nucleotide identity.
Other metabolic, regulatory, and transporter genes on pDCA.
The region of pDCA containing the dhlA gene also carries several other predicted metabolic genes, regulators and transporters. Immediately upstream of dhlA is a gene encoding a predicted TetR family regulator, which has 100% identity to a gene in the corresponding location in Xanthobacter strain GJ10. The gene in GJ10 has not been experimentally characterized, and, thus, this homology alone does not provide strong evidence that it is indeed a haloalkane-responsive regulator. However, the case for this is strengthened by BLAST analysis, which revealed at least 10 homologs of this predicted regulatory gene in marine bacteria (Vibrio, Pseudoalteromonas, Ruegeria); these have 46 to 53% inferred aa identity to the dhlA-associated regulator, and are all immediately adjacent to haloalkane dehalogenases with 60 to 65% inferred aa identity to DhlA (see Fig. S9 in the supplemental material). Notably, the dehalogenases retrieved from GenBank by this approach are the same as the top matches if the DhlA protein is directly analyzed by BLAST, indicating that these dehalogenases from marine bacteria are the closest relatives to DhlA in terms of both amino acid sequence and gene organization.
Also present upstream of dhlA in all three pDCA variants is a conserved cluster of genes, including catabolic genes, regulators, and transporters. These include homologs of phytanoyl-coenzyme A dioxygenase and acyl-coenzyme A thioesterase, regulators from the TetR, AraC, and MarR families, and a major facilitator superfamily (MFS) family transporter. The very high conservation of this gene region between all pDCA homologs (100% nucleotide identity) suggests these genes are encoding a useful phenotype for the host bacteria, but, at this stage, it is unknown what this might be or whether it has any link to biodegradation of DCA.
DISCUSSION
Our study has highlighted the fact that xenobiotic-degrading bacteria may contain novel catabolic genes and novel gene arrangements, even when they are phylogenetically and phenotypically similar to earlier isolates. Our study had two major unexpected findings: first, that the Xanthobacteriaceae isolated from the Botany GTP carried the dhlA gene not on a large linear plasmid but instead on a smaller circular plasmid; second, that a new type of dhlB gene had been recruited onto this plasmid, while a traditional dhlB gene was still maintained in the chromosome. It is hard to know whether our isolates are descendants of GJ10-like bacteria, or whether they represent an independent line of evolution. It is tempting to speculate that these recent Australian isolates are more highly evolved than the bacteria isolated in Europe in the 1980s, in the sense that both dehalogenase genes are now present on the same mobile element, potentially facilitating the spread of the DCA-degrading phenotype among other environmental bacteria. It could also be argued that the Botany isolates represent an advance in the genome organization of DCA-degrading bacteria, moving toward a colocated and coregulated operon structure for the DCA catabolic genes, as is seen in more established bacterial metabolic pathways. It should be noted that we cannot rule out the presence of a large linear plasmid, such as pXAU1, in our isolates, and we have not localized the other two essential DCA biodegradation genes, aldA and mox; further work is needed to determine the locations of these dehydrogenases, both in our isolates and in earlier DCA degraders.
Our findings offer new insights into the relationships of dehalogenase genes with mobile genetic elements. This study provides a new link between organochlorine biodegradation genes and plasmids from the pTAR family and reaffirms the importance of two particular IS element families (ISAs1 and IS1380) in the movement of the dhlA and dhlB dehalogenases. It is remarkable that, given the distance in time and space between the isolation of Xanthobacter strain GJ10 and our Botany GTP isolates (27 years and 16,500 km), and despite the change in replicon that carries the dhlA gene, dhlA is still tightly associated with the ISXa1 element. Yet, in a different study (22), dhlA was seen to be associated with an IS1380 family element (IS1247), which was nearly identical to an element originally seen next to dhlB in strain GJ10. The link between insertion sequences and dhlB is notable in that IS1380 family elements (either IS1247 or ISPme1) appear to be very active in moving dhlB homologs (both dhlB1 and dhlB2) among different genomic locations, for example, enabling the evolution of bromoacetate-assimilating mutants of strain GJ10 (36) and recruiting dhlB2 to pDCA (this study).
We have shed new light on the diversity of dehalogenase genes in aerobic DCA-degrading bacteria. To date, DhlA remains the only hydrolytic dehalogenase that is known to be associated with DCA biodegradation; this is not due to the lack of haloalkane dehalogenases in bacterial communities (these are both numerous and diverse [53]) but is more likely because other enzymes in this family, such as DhaA (54), LinB (55), and DatA (56), lack sufficient activity on DCA to enable bacterial growth. On the other hand, our work here indicates that the relationship between haloacid dehalogenases and DCA degraders is much more flexible and that many different variants of the archetypal DhlB enzyme of Xanthobacter strain GJ10 could be involved in chloroacetate metabolism in DCA-degrading bacteria. Further evidence that supports this hypothesis is found in the cis-dichloroethene-degrading Polaromonas sp. strain JS666, which can also grow on DCA (16) and contains a dhlB homolog (57) that is very different from both the dhlB1 and dhlB2 genes described in the present study. In the case of the DCA-degrading isolates from the Botany GTP, it remains to be determined what the relative roles of DhlB1 and DhlB2 are, but, based on sequence analysis in the former case and heterologous expression in the latter case, it appears likely that both enzymes are active chloroacetate dehalogenases and that both contribute to growth on DCA.
There are still many unanswered questions concerning the biology of the pDCA plasmid. Most notably, what is the host range of this element for transfer and replication? Based on BLAST analysis of the plasmid backbone genes, it appears that these pTAR family plasmids are almost exclusively associated with the Alphaproteobacteria, suggesting a relatively narrow host range. However, to the best of our knowledge, no representatives from this plasmid family have been systematically tested for their host ranges for replication or transfer. A related question concerns the mechanism by which the pDCA plasmid and its relatives can move between different bacteria, given that there are no pilus-formation genes present; this implies that such plasmids are mobilized by other conjugative elements, but which conjugative elements are responsible? Answering these questions will assist in understanding how the DCA-degradative phenotype can be acquired and passed on by bacteria at contaminated sites, which in turn will aid the management and prediction of DCA bioremediation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Orica, Ltd., and the NSW Environmental Trust for funding our work on DCA-degrading bacteria. We thank Dick Janssen, who provided plasmids containing Xanthobacter strain GJ10 genes.
Funding Statement
This work was funded by the NSW Environmental Trust (2009/RD/0051) and Orica, Ltd. (consultancy agreement Sydnovate 14270).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01373-16.
REFERENCES
- 1.Janssen DB, Dinkla IJT, Poelarends GJ, Terpstra P. 2005. Bacterial degradation of xenobiotic compounds: evolution and distribution of novel enzyme activities. Environ Microbiol 7:1868–1882. doi: 10.1111/j.1462-2920.2005.00966.x. [DOI] [PubMed] [Google Scholar]
- 2.Weber R, Gaus C, Tysklind M, Johnston P, Forter M, Hollert H, Heinisch E, Holoubek I, Lloyd-Smith M, Masunaga S, Moccarelli P, Santillo D, Seike N, Symons R, Torres JPM, Verta M, Varbelow G, Vijgen J, Watson A, Costner P, Woelz J, Wycisk P, Zennegg M. 2008. Dioxin- and POP-contaminated sites: contemporary and future relevance and challenges. Environ Sci Pollut Res 15:363–393. doi: 10.1007/s11356-008-0024-1. [DOI] [PubMed] [Google Scholar]
- 3.Falta RW. 2004. The potential for ground water contamination by the gasoline lead scavengers ethylene dibromide and 1,2-dichloroethane. Ground Water Monitor Remed 24:76–87. [Google Scholar]
- 4.Grady SJ, Casey GD. 2001. Occurrence and distribution of methyl tert-butyl ether and other volatile organic compounds in drinking water in the Northeast and Mid-Atlantic regions of the United States, 1993-1998. US Department of the Interior, US Geological Survey, Washington, DC. [Google Scholar]
- 5.Fife H, Reid E. 1930. New industrial solvents: ethylene dichloride, dichloroethyl ether, and isopropyl ether. Ind Eng Chem 22:513–515. doi: 10.1021/ie50245a024. [DOI] [Google Scholar]
- 6.Go KS, Kim Y, Real Son S, Kim SD. 2010. 1,2-Dichloroethane production by two-step oxychlorination reactions in a fluidized bed reactor. Chem Eng Sci 65:499–503. doi: 10.1016/j.ces.2009.02.052. [DOI] [Google Scholar]
- 7.Stucki G, Krebser U, Leisinger T. 1983. Bacterial growth on 1,2-dichloroethane. Experientia 39:1271–1273. doi: 10.1007/BF01990365. [DOI] [PubMed] [Google Scholar]
- 8.Stucki G, Thuer M. 1995. Experiences of a large scale application of 1,2-dichloroethane degrading microorganisms for groundwater treatment. Environ Sci Technol 29:2339–2345. doi: 10.1021/es00009a028. [DOI] [PubMed] [Google Scholar]
- 9.Maymó-Gatell X, Anguish T, Zinder SH. 1999. Reductive dechlorination of chlorinated ethenes and 1,2-dichloroethane by “Dehalococcoides ethenogenes” 195. Appl Environ Microbiol 65:3108–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grostern A, Edwards EA. 2009. Characterization of a Dehalobacter coculture that dechlorinates 1,2-dichloroethane to ethene and identification of the putative reductive dehalogenase gene. Appl Environ Microbiol 75:2684–2693. doi: 10.1128/AEM.02037-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moe WM, Yan J, Nobre MF, da Costa MS, Rainey FA. 2009. Dehalogenimonas lykanthroporepellens gen. nov., sp nov., a reductively dehalogenating bacterium isolated from chlorinated solvent-contaminated groundwater. Int J Syst Evol Microbiol 59:2692–2697. doi: 10.1099/ijs.0.011502-0. [DOI] [PubMed] [Google Scholar]
- 12.Oldenhuis R, Vink RL, Janssen DB, Witholt B. 1989. Degradation of chlorinated aliphatic hydrocarbons by Methylosinus trichosporium OB3b expressing soluble methane monooxygenase. Appl Environ Microbiol 55:2819–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ely RL, Williamson KJ, Hyman MR, Arp DJ. 1997. Cometabolism of chlorinated solvents by nitrifying bacteria: kinetics, substrate interactions, toxicity effects, and bacterial response. Biotech Bioeng 54:520–534. [DOI] [PubMed] [Google Scholar]
- 14.Kim Y, Arp DJ, Semprini L. 2000. Chlorinated solvent cometabolism by butane-grown mixed culture. J Environ Eng 126:934–942. doi: 10.1061/(ASCE)0733-9372(2000)126:10(934). [DOI] [Google Scholar]
- 15.Hage JC, Hartmans S. 1999. Monooxygenase-mediated 1,2-dichloroethane degradation by Pseudomonas sp. strain DCA1. Appl Environ Microbiol 65:2466–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishino SF, Shin KA, Gossett JM, Spain JC. 2013. Cytochrome p450 initiates degradation of cis-dichloroethene by Polaromonas sp. strain JS666. Appl Environ Microbiol 79:2263–2272. doi: 10.1128/AEM.03445-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssen DB, Scheper A, Dijkhuizen L, Witholt B. 1985. Degradation of halogenated aliphatic compounds by Xanthobacter autotrophicus GJ10. Appl Environ Microbiol 49:673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Wijngaard AJ, van der Kamp KW, van der Ploeg J, Pries F, Kazemier B, Janssen DB. 1992. Degradation of 1,2-dichloroethane by Ancylobacter aquaticus and other facultative methylotrophs. Appl Environ Microbiol 58:976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dinglasan-Panlilio MJ, Dworatzek S, Mabury S, Edwards E. 2006. Microbial oxidation of 1,2-dichloroethane under anoxic conditions with nitrate as electron acceptor in mixed and pure cultures. FEMS Microbiol Ecol 56:355–364. doi: 10.1111/j.1574-6941.2006.00077.x. [DOI] [PubMed] [Google Scholar]
- 20.Vilchez-Vargas R, Junca H, Pieper DH. 2010. Metabolic networks, microbial ecology and ‘omics’ technologies: towards understanding in situ biodegradation processes. Environ Microbiol 12:3089–3104. doi: 10.1111/j.1462-2920.2010.02340.x. [DOI] [PubMed] [Google Scholar]
- 21.Janssen D, Scheper A, Witholt B. 1984. Biodegradation of 2-chloroethanol and 1,2-dichloroethane by pure bacterial cultures. Prog Ind Microbiol 20:169–178. [Google Scholar]
- 22.Song JS, Lee DH, Lee K, Kim CK. 2004. Genetic organization of the dhlA gene encoding 1,2-dichloroethane dechlorinase from Xanthobacter flavus UE15. J Microbiol 42:188–193. [PubMed] [Google Scholar]
- 23.Janssen DB, van der Ploeg JR, Pries F. 1995. Genetic adaptation of bacteria to halogenated aliphatic compounds. Environ Health Perspect 103(Suppl 5):29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munro JE, Liew EF, Coleman NV. 2013. Adaptation of a membrane bioreactor to 1,2-dichloroethane revealed by 16S rDNA pyrosequencing and dhlA qPCR. Environ Sci Technol 47:13668–13676. doi: 10.1021/es403292s. [DOI] [PubMed] [Google Scholar]
- 25.Mena-Benitez GL, Gandia-Herrero F, Graham S, Larson TR, McQueen-Mason SJ, French CE, Rylott EL, Bruce NC. 2008. Engineering a catabolic pathway in plants for the degradation of 1,2-dichloroethane. Plant Physiol 147:1192–1198. doi: 10.1104/pp.108.119008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergeron H, Labbe D, Turmel C, Lau PC. 1998. Cloning, sequence and expression of a linear plasmid-based and a chromosomal homolog of chloroacetaldehyde dehydrogenase-encoding genes in Xanthobacter autotrophicus GJ10. Gene 207:9–18. doi: 10.1016/S0378-1119(97)00598-2. [DOI] [PubMed] [Google Scholar]
- 27.Dijk JA, Gerritse J, Schraa G, Stams AJM. 2004. Degradation pathway of 2-chloroethanol in Pseudomonas stutzeri strain JJ under denitrifying conditions. Arch Microbiol 182:514–519. doi: 10.1007/s00203-004-0737-6. [DOI] [PubMed] [Google Scholar]
- 28.Janssen D, Pries F, Van der Ploeg J, Kazemier B, Terpstra P, Witholt B. 1989. Cloning of 1,2-dichloroethane degradation genes of Xanthobacter autotrophicus GJ10 and expression and sequencing of the dhlA gene. J Bacteriol 171:6791–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verschueren KHG, Seljee F, Rozeboom HJ, Kalk KH, Dijkstra BW. 1993. Crystallographic analysis of the catalytic mechanism of haloalkane dehalogenase. Nature 363:693–698. doi: 10.1038/363693a0. [DOI] [PubMed] [Google Scholar]
- 30.van der Ploeg J, van Hall G, Janssen DB. 1991. Characterization of the haloacid dehalogenase from Xanthobacter autotrophicus GJ10 and sequencing of the dhlB gene. J Bacteriol 173:7925–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridder IS, Rozeboom HJ, Kalk KH, Janssen DB, Dijkstra BW. 1997. Three-dimensional structure of L-2-haloacid dehalogenase from Xanthobacter autotrophicus GJ10 complexed with the substrate-analogue formate. J Biol Chem 272:33015–33022. doi: 10.1074/jbc.272.52.33015. [DOI] [PubMed] [Google Scholar]
- 32.Keuning S, Janssen DB, Witholt B. 1985. Purification and characterization of hydrolytic haloalkane dehalogenase from Xanthobacter autotrophicus GJ10. J Bacteriol 163:635–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Govender A, Pillay B. 2011. Characterization of 1,2-dichloroethane (DCA) degrading bacteria isolated from South African waste water. African J Biotech 10:11567–11573. [Google Scholar]
- 34.van der Ploeg J, Janssen DB. 1995. Sequence analysis of the upstream region of dhlB, the gene encoding haloalkanoic acid dehalogenase of Xanthobacter autotrophicus GJ10. Biodegradation 6:257–263. doi: 10.1007/BF00700465. [DOI] [PubMed] [Google Scholar]
- 35.Tardif G, Greer CW, Labbe D, Lau PC. 1991. Involvement of a large plasmid in the degradation of 1,2-dichloroethane by Xanthobacter autotrophicus. Appl Environ Microbiol 57:1853–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Ploeg J, Willemsen M, van Hall G, Janssen DB. 1995. Adaptation of Xanthobacter autotrophicus GJ10 to bromoacetate due to activation and mobilization of the haloacetate dehalogenase gene by insertion element IS1247. J Bacteriol 177:1348–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coleman NV, Mattes TE, Gossett JM, Spain JC. 2002. Biodegradation of cis-dichloroethene as the sole carbon source by a β-proteobacterium. Appl Environ Microbiol 68:2726. doi: 10.1128/AEM.68.6.2726-2730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reasoner DJ, Geldreich EE. 1985. A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol 49:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le N, Coleman N. 2011. Biodegradation of vinyl chloride, cis-dichloroethene and 1,2-dichloroethane in the alkene/alkane-oxidizing Mycobacterium strain NBB4. Biodegradation 22:1095–1108. doi: 10.1007/s10532-011-9466-0. [DOI] [PubMed] [Google Scholar]
- 40.Wilson K. 1987. Preparation of genomic DNA from bacteria, p 2.4.1–2.4.5 In Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (ed), Current protocols in molecular biology. John Wiley & Sons, New York, NY. [DOI] [PubMed] [Google Scholar]
- 41.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175. In Stackebrandt E, Goodfellow M (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Inc., New York, NY. [Google Scholar]
- 42.Lukashin AV, Borodovsky M. 1998. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res 26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bergmann J, Sanik J Jr. 1957. Determination of trace amounts of chlorine in naphtha. Anal Chem 29:241–243. doi: 10.1021/ac60122a018. [DOI] [Google Scholar]
- 44.Coleman NV. 2015. Primers: functional genes for aerobic chlorinated hydrocarbon-degrading microbes. In McGenity TJ, Timmis KN, Nogales Fernández B (ed), Hydrocarbon and lipid microbiology protocols: primers. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 45.van Ginkel CG, de Bont JAM. 1986. Isolation and characterization of alkene-utilizing Xanthobacter spp. Arch Microbiol 145:403–407. doi: 10.1007/BF00470879. [DOI] [Google Scholar]
- 46.Li Y, Chen Q, Wang CH, Cai S, He J, Huang X, Li SP. 2013. Degradation of acetochlor by consortium of two bacterial strains and cloning of a novel amidase gene involved in acetochlor-degrading pathway. Bioresour Technol 148:628–631. doi: 10.1016/j.biortech.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 47.Suenaga H, Koyama Y, Miyakoshi M, Miyazaki R, Yano H, Sota M, Ohtsubo Y, Tsuda M, Miyazaki K. 2009. Novel organization of aromatic degradation pathway genes in a microbial community as revealed by metagenomic analysis. ISME J 3:1335–1348. doi: 10.1038/ismej.2009.76. [DOI] [PubMed] [Google Scholar]
- 48.Ebersbach G, Gerdes K. 2005. Plasmid segregation mechanisms. Annu Rev Genet 39:453–479. doi: 10.1146/annurev.genet.38.072902.091252. [DOI] [PubMed] [Google Scholar]
- 49.Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. 2003. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112:131–140. doi: 10.1016/S0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- 50.Alvarez-Martinez CE, Christie PJ. 2009. Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev 73:775–808. doi: 10.1128/MMBR.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szuplewska M, Bartosik D. 2009. Identification of a mosaic transposable element of Paracoccus marcusii composed of insertion sequence ISPmar4 (ISAs1 family) and an IS1247a-driven transposable module (TMo). FEMS Microbiol Lett 292:216–221. doi: 10.1111/j.1574-6968.2009.01495.x. [DOI] [PubMed] [Google Scholar]
- 52.Bartosik D, Putyrski M, Dziewit L, Malewska E, Szymanik M, Jagiello E, Lukasik J, Baj J. 2008. Transposable modules generated by a single copy of insertion sequence ISPme1 and their influence on structure and evolution of natural plasmids of Paracoccus methylutens DM12. J Bacteriol 190:3306–3313. doi: 10.1128/JB.01878-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koudelakova T, Bidmanova S, Dvorak P, Pavelka A, Chaloupkova R, Prokop Z, Damborsky J. 2013. Haloalkane dehalogenases: biotechnological applications. Biotechnol J 8:32–45. doi: 10.1002/biot.201100486. [DOI] [PubMed] [Google Scholar]
- 54.Schindler JF, Naranjo PA, Honaberger DA, Chang CH, Brainard JR, Vanderberg LA, Unkefer CJ. 1999. Haloalkane dehalogenases: steady-state kinetics and halide inhibition. Biochemistry 38:5772–5778. doi: 10.1021/bi982853y. [DOI] [PubMed] [Google Scholar]
- 55.Nagata Y, Miyauchi K, Damborsky J, Manova K, Ansorgova A, Takagi M. 1997. Purification and characterization of a haloalkane dehalogenase of a new substrate class from a gamma-hexachlorocyclohexane-degrading bacterium, Sphingomonas paucimobilis UT26. Appl Environ Microbiol 63:3707–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hasan K, Fortova A, Koudelakova T, Chaloupkova R, Ishitsuka M, Nagata Y, Damborsky J, Prokop Z. 2011. Biochemical characteristics of the novel haloalkane dehalogenase DatA, isolated from the plant pathogen Agrobacterium tumefaciens C58. Appl Environ Microbiol 77:1881–1884. doi: 10.1128/AEM.02109-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mattes TE, Alexander AK, Richardson PM, Munk AC, Han CS, Stothard P, Coleman NV. 2008. The genome of Polaromonas sp. strain JS666: insights into the evolution of a hydrocarbon- and xenobiotic-degrading bacterium, and features of relevance to biotechnology. Appl Environ Microbiol 74:6405–6416. doi: 10.1128/AEM.00197-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nicholas KB, Nicholas HB, Deerfield DW. 1997. GeneDoc: analysis and visualization of genetic variation. https://www.psc.edu/index.php/user-resources/software/genedoc.
- 60.Hill KE, Marchesi JR, Weightman AJ. 1999. Investigation of two evolutionarily unrelated halocarboxylic acid dehalogenase gene families. J Bacteriol 181:2535–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Page RDM. 1996. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.