

ABSTRACT
While commonplace in clinical settings, DNA-based assays for identification or enumeration of drinking water pathogens and other biological contaminants remain widely unadopted by the monitoring community. In this study, shotgun metagenomics was used to identify taste-and-odor producers and toxin-producing cyanobacteria over a 2-year period in a drinking water reservoir. The sequencing data implicated several cyanobacteria, including Anabaena spp., Microcystis spp., and an unresolved member of the order Oscillatoriales as the likely principal producers of geosmin, microcystin, and 2-methylisoborneol (MIB), respectively. To further demonstrate this, quantitative PCR (qPCR) assays targeting geosmin-producing Anabaena and microcystin-producing Microcystis were utilized, and these data were fitted using generalized linear models and compared with routine monitoring data, including microscopic cell counts, sonde-based physicochemical analyses, and assays of all inorganic and organic nitrogen and phosphorus forms and fractions. The qPCR assays explained the greatest variation in observed geosmin (adjusted R2 = 0.71) and microcystin (adjusted R2 = 0.84) concentrations over the study period, highlighting their potential for routine monitoring applications. The origin of the monoterpene cyclase required for MIB biosynthesis was putatively linked to a periphytic cyanobacterial mat attached to the concrete drinking water inflow structure. We conclude that shotgun metagenomics can be used to identify microbial agents involved in water quality deterioration and to guide PCR assay selection or design for routine monitoring purposes. Finally, we offer estimates of microbial diversity and metagenomic coverage of our data sets for reference to others wishing to apply shotgun metagenomics to other lacustrine systems.
IMPORTANCE Cyanobacterial toxins and microbial taste-and-odor compounds are a growing concern for drinking water utilities reliant upon surface water resources. Specific identification of the microorganism(s) responsible for water quality degradation is often complicated by the presence of co-occurring taxa capable of producing these undesirable metabolites. Here we present a framework for how shotgun metagenomics can be used to definitively identify problematic microorganisms and how these data can guide the development of rapid genetic assays for routine monitoring purposes.
INTRODUCTION
Cyanobacterial harmful algal blooms (CyanoHABs) appear to be increasing globally in frequency, duration, and magnitude in response to human-accelerated eutrophication and climate change (1, 2). From a drinking water perspective, large accumulations of cyanobacteria pose a significant health concern due to their ability to produce a variety of bioactive metabolites, some of which are potent liver toxins or neurotoxins (3, 4). Additionally, cyanobacteria are widely considered the most common source of the taste-and-odor (T&O) compounds geosmin (4,8a-dimethyl-decahydroaphthalen-4a-ol) and MIB (2-methylisoborneol), which impart unpalatable earthy and musty flavors to drinking water (5). Geosmin and MIB represent the most common ecology-related esthetic problem provoking customer complaints to drinking water utilities (6, 7). In response to CyanoHAB-associated water quality impairment, utilities are forced to augment their facilities with expensive treatment modifications in order to cope with these issues. For example, the city of Toledo, Ohio, spends ∼$150,000/month on advanced treatment (activated carbon and ozone) when microcystins are in the city's raw water supply (8), the city of Waco, Texas, spent an estimated $70.4 million over a 10-year period (2002 to 2012) to address T&O problems (8), and the city of Wichita, Kansas, installed an $8.5 million ozone treatment facility as a direct result of T&O issues (9).
Knowledge of the microorganisms responsible for T&O and cyanotoxin production in drinking water is critically important for early warning detection and for developing predictive models that can be used to forecast impending water quality impairment. While many studies have focused on characterizing cyanotoxin producers (10–12), fewer tools are available for assessing geosmin- and MIB-producing taxa. A number of filamentous cyanobacteria, both N2-fixing (Nostocales) and nondiazotrophic (Oscillatoriales), are potential geosmin and/or MIB producers (13). Complicating matters, these compounds may also be produced by a variety of noncyanobacterial microorganisms, including Actinobacteria (e.g., Streptomyces spp. and Nocardia spp.), ascomycetous fungi (e.g., Penicillium spp. and Aspergillus spp.), liverworts (e.g., Symphyogyna spp.), and heterotrophic amoebae (e.g., Vannella spp.) (5). A significant challenge in managing taste-and-odor events arises from the fact that these metabolites are detectable by human olfactory senses at low concentrations in the range of 5 to 10 ng/liter (14), which is near the analytical detection limit for gas chromatography-mass spectrometry (GC-MS) of ∼1 ng/liter. When provided with sufficient advanced warning, typically 24 to 36 h, many utilities can enact countermeasures to prevent malodorous metabolites from reaching customer drinking water taps.
We used shotgun metagenomics to reveal the T&O producers in a drinking water reservoir (Cheney Reservoir, Kansas, USA) that has experienced periodic geosmin and MIB events since 1990 (15). Because cyanotoxins tend to co-occur with taste-and-odor compounds (91% of cyanobacterial blooms contained both in a study of Midwestern reservoirs [16]), we also sought to definitively identify which organisms in the reservoir were capable of toxin biosynthesis. The use of shotgun metagenomics for identifying the sources of water quality impairment requires that the genes involved in the biosynthesis of the metabolites are known or can be inferred from recognized homologs in other organisms. While shotgun metagenomics is unlikely to be used for routine water quality monitoring in the near future, we demonstrate that it can be highly useful when applied in a targeted manner, such as during periods of water quality impairment when the causative organism(s) is unknown and as a starting point for designing real-time quantitative PCR (RT-qPCR) assays to be used for routine monitoring purposes.
MATERIALS AND METHODS
Sample collection and processing.
Cheney Reservoir (surface area, 31 km2), situated on the North Fork Ninnescah River, is a shallow (mean depth, ∼5.3 m), continuously mixed (rarely stratifying) eutrophic water body that serves as the principal water supply of Wichita, Kansas (USA), servicing nearly 400,000 people. Photic zone depth-integrated samples (n = 28) were collected from U.S. Geological Survey (USGS) routine monitoring site 07144790 (designated routine monitoring [RM] site), located ∼10 m from the reservoir dam (Fig. 1). Samples were collected biweekly in the summer (June to September) and approximately monthly at other times of the year throughout 2013 and 2014 (see Table S1 in the supplemental material). Sample collection and routine water quality analyses were performed in accordance with U.S. Geological Survey protocols (17) and are described by Stone and colleagues (18). For each sample, a full physicochemical analysis was conducted, measuring more than 50 different water quality variables. All water quality data are available through the USGS National Water Information System at http://dx.doi.org/10.5066/F7P55KJN. Of direct relevance to this study, geosmin and MIB were measured using gas chromatography-mass spectrometry (GC-MS) by Engineering Performance Solutions (Jacksonville, FL) (19). Phytoplankton analyses were performed by BSA Environmental Services, Inc. (Beachwood, OH). Where possible, the phytoplankton community was enumerated microscopically to the species level using membrane-filtered slides (20) with a minimum of 400 natural units counted per sample. Total microcystins/nodularins were measured by the USGS Organic Geochemistry Research Lab (Lawrence, KS) using polyclonal enzyme-linked immunosorbent assays (ELISA) (Abraxis).
FIG 1.

Map of Cheney Reservoir sampling sites; the square symbol indicates where routine monitoring occurs, and red circles indicate transect sample collection sites. The inset graph displays an unusual geosmin event that occurred across the reservoir on 30 August 2013. Maps were created using Inkscape v0.91.
Three reservoir sampling transects were conducted, one in 2013 and two in 2014. Each transect consisted of six discrete, photic zone depth-integrated samples collected across a longitudinal gradient of the reservoir (n = 18) (Fig. 1). These were collected as part of a separate remote sensing study, and only a subset of the routine physicochemical and phycological analyses (temperature, dissolved oxygen, pH, specific conductance, chlorophyll a [Chl a], turbidity, phycocyanin, geosmin, MIB, microcystin, and nutrients) were analyzed for these samples. One of the transects (30 August 2013) was conducted shortly after a large rainfall/inflow event and corresponded with a period of unusually high concentrations of geosmin across the reservoir (Fig. 1). Lastly, scrapings of periphytic algae were collected from the concrete drinking water intake structure and adjacent reservoir dam wall on 4 August 2015 in order to assess for the occurrence of taste-and-odor producers that would not be captured by only sampling from the water column. These samples were subjected to microscopic enumeration, GC-MS analysis for geosmin and MIB, and endpoint PCR targeting MIB and geosmin synthase genes from cyanobacteria and heterotrophic bacteria (see Table S2 in the supplemental material). The samples were mixed and then allowed to settle in the refrigerator overnight before 10 ml of each sample was collected from the surface or the bottom (sedimented fraction) of the bottle, centrifuged for 5 min at 6,000 rpm, and decanted, and the DNA was extracted. All of the amplified PCR products were dideoxy (Sanger) sequenced.
DNA sequencing.
A subset of each sample was collected using a churn splitter and shipped overnight on wet ice to Oregon State University in 1-liter sterile polycarbonate bottles. Upon receipt, as much volume as possible without clogging was individually (not sequentially) concentrated onto 25-mm diameter, 1.2-μm pore size glass fiber filters (VWR) and 0.2-μm pore size polyethersulfone membrane filters (Supor; Pall) under vacuum filtration. Replicates were collected for all samples, although not all replicates were analyzed. We analyzed 20% of the sample replicates using qPCR and one of the sample replicates (21 October 2013) by shotgun sequencing (see Fig. S1 in the supplemental material). Sample filters were stored at −80°C until further processing. Total DNA was extracted from filters using GeneRite DNA-EZ RW01 extraction kits, which employ both chemical and physical (bead-beating) treatments to ensure complete cell lysis and silica columns to bind and wash DNA. Not every sample collected was shotgun sequenced. From the routine monitoring site, there were nine samples that were sequenced (seven from 0.2-μm filters and two from 1.2-μm filters) and six samples from the 30 August 2013 transect (all 1.2-μm filters) (n = 15 total metagenomes). Both filters will capture most cyanobacteria, but the 1.2-μm glass fiber filters have been shown to only retain 10 to 55% of small-celled heterotrophic bacteria (21). DNA sequencing was carried out at the Center for Genome Research and Biocomputing (CGRB) at Oregon State University. One nanogram of DNA per sample was prepared using a Nextera XT library prep kit; each DNA library was pooled at equimolar concentrations (∼25 ng prepared DNA/sample) and sequenced using an Illumina HiSeq 2000 instrument with 101 bp, paired-end reads and ∼450 bp insert sizes.
Metagenome assembly.
All sequencing reads were quality screened using Trimmomatic (22); only those with Phred scores of >30 were retained. The sequences were interleaved using khmer, and only those with mate pairs and a minimum length of 50 bp were further retained. All sequencing reads were pooled and randomly subsampled to 375 million reads and assembled using IDBA-UD (23), an assembler specifically designed to handle highly uneven read depths characteristic of metagenome data sets. Assembled contigs were binned to the lowest assignable taxonomic level using PhyloPythiaS+ (24) and mmgenome (25); these programs utilize a combination of approaches for assigning contigs to organisms, including Basic Local Alignment Search Tool (BLAST), tetranucleotide frequency, percent GC content, kmer length, read coverage depth, and essential gene content. The sequencing reads from each individual sample were randomly subsampled to ∼29.6 million reads and then were mapped to the concatenated multisample assembly using Burrows-Wheeler Aligner (BWA) (26) in order to determine the read coverage of each contig (average number of sequencing reads per nucleotide).
Bioinformatic analyses.
All contigs were screened for functional genes involved in geosmin, MIB, or cyanotoxin biosynthesis with antiSMASH 2.0 (27) and by using a combination of local alignment searches (both nucleotide and amino acid) and hidden Markov models implemented through Prokka (28). The resulting contigs and genes of interest were compared to the reference database (NCBI GenBank) in order to assign genes to organisms. The nucleotide information was used to validate existing assays or to develop new real-time qPCR oligonucleotide primers and probes. Each qPCR assay utilized a positive-control synthetic gene standard (gBlock; Integrated DNA Technologies) that consisted of double-stranded DNA identical to the genes of interest identified from the metagenomes (see Table S2 in the supplemental material). The gBlock was used as the positive control for all taste-and-odor PCR analyses and contained three different amplicon targets corresponding to an Anabaena geosmin synthase (geoA) identified in the present study, a heterotrophic bacterium-specific geosmin synthase (29) and a universal cyanobacterial MIB synthase (30). A second gBlock contained part of a Microcystis-specific microcystin synthetase E (mcyE) gene that has been adapted for TaqMan qPCR (see Table S2) (31, 32).
In order to account for low-abundance DNA that did not assemble into contigs, all trimmed sequencing reads were individually mapped using BWA (26) to local reference databases containing all geosmin and MIB synthases from all known producers identified in the NCBI GenBank (as of July 2015). The appropriateness of the sequencing effort to uncover rare but ecologically relevant organisms was assessed using Nonpareil (33), which computes the abundance-weighted average coverage profile for each whole-genome shotgun (WGS) metagenome.
Quantitative PCR.
All TaqMan real-time qPCR analyses were run on an Applied Biosystems ABI 7500 Fast cycler using Fermentas Maxima qPCR probe mix (Thermo Fisher) in 25-μl reaction volumes consisting of 12.5 μl 2× master mix, forward primer (750 nM), reverse primer (750 nM), probe (200 nM), 5 μl DNA template diluted 1:10, and 4.5 μl nuclease-free distilled water (dH2O). Two-step PCRs were utilized for all reactions, consisting of a 2-min denaturation step at 95°C, followed by 40 cycles at 95°C for 15 s and 56.5°C (mcyE assay) or 61°C (geoA assay) for 45 s. Standard curves were generated using serial dilutions of the gBlocks (108 to 100 copies/reaction); reaction efficiencies were 104.9% (geoA assay) and 98.4% for the mcyE assay. DNA extraction efficiencies were ∼40% from the glass fiber filters and ∼23% from the membrane filters. Therefore, all final qPCR gene estimates were multiplied by a factor of 2.5× or 4.3×, respectively. The DNA extraction efficiency was estimated by concentrating a known number of cells from cyanobacterial cultures (counted using a light microscope and hemocytometer) onto filters or by pelleting by centrifugation, after which DNA was extracted using GeneRite DNA-EZ RW01 extraction kits and quantified by qPCR; near-quantitative DNA recovery was observed from centrifugally collected cells (i.e., near 1:1 relationship between cells and gene copies) (34).
Statistical analyses.
Linear regression models were developed for total microcystin and geosmin using routine monitoring data collected from Cheney Reservoir during 2013 to 2014. Models used data collected via water quality sondes (temperature, dissolved oxygen, specific conductance, pH, turbidity, and chlorophyll) or data collected discretely using USGS methods (total Kjeldahl nitrogen, ammonia [NH3], nitrate [NO3], NO3-nitrite [NO2], orthophosphate [PO4], dissolved phosphate, total phosphorus, etc.). Nondetect values were excluded from the analysis for microcystin and geosmin; explanatory variables were excluded if the nondetect values comprised more than 10% of the data set. A variety of data transformations were used in the models, including Fourier transformation for seasonality (35). Models with and without transformations were compared using predicted residual error sum of squares (PRESS) and adjusted R2. The PRESS statistic is used as an indication of the predictive power of a model and is calculated by repeatedly fitting a model while leaving out a subset of the data (36). The best models were those exhibiting the lowest PRESS values and the highest correlation coefficients (adjusted R2). To assess the statistical power of the linear regression models, the following equation was applied to estimate the minimum sample size required:
| (1) |
where N is the minimum number of observations, p is the proportion of positive cases (e.g., dates when microcystin, geosmin, or MIB was detected), and k is the number of independent variables (37). Because nondetects were excluded from the analysis, p = 1. Due to the very large range of cell or gene concentrations characteristic of cyanobacterial bloom events, all models were checked for outliers, unusual observations, and overleveraged points based on calculations of standardized residuals and Cook's distance. Standardized residuals with absolute values of >2 or observations (N) with leverage values of >4/N were considered influential. The x and y log-transformed data generally did not violate these conditions, although there were five data points that slightly exceeded the Cook's distance threshold from the geosmin qPCR assay and two data points from the mcyE qPCR assay. Removal of these points had little effect on the geosmin qPCR assay (adjusted R2 = 0.71 versus 0.66) and were retained in the model. The mcyE qPCR assay outliers had a greater effect (adjusted R2 = 0.77 versus 0.66) and were removed from the data set.
Accession number(s).
All contigs were submitted to the U.S. Department of Energy (DOE) Joint Genome Institute (JGI) for annotation (IMG submission no. 68648), and the raw reads were deposited into the National Center for Biotechnology Information (NCBI) Sequence Read Archive under BioProject no. PRJNA294203.
RESULTS
Shotgun metagenomics.
Time series and spatial shotgun metagenomics were employed to identify the specific microorganisms capable of producing nuisance (taste-and-odor) and noxious (cyanotoxins) compounds in Cheney Reservoir during 2013. A total of 15 samples were shotgun sequenced, generating ∼651 million reads after quality assurance/quality control (QA/QC) (minimum, maximum, and mean of 29.6, 72.8, and 43.1 million reads/sample, respectively). As has been done in other longitudinal metagenomic investigations (38), the sequencing reads were pooled in order to increase the probability of assembling lower-abundance taxa present across multiple time points. A random subset of 375 million reads was assembled, of which ∼40% of the reads were assembled into 383,370 contigs spanning 668 million base pairs in length (maximum contig length, 292,210 bp; mean contig length, 1,737 bp; N50, 2,241 bp). Assembled contigs were screened for metabolic pathways of interest, which included monoterpene cyclases that might be involved in geosmin and MIB biosynthesis and all known and putative cyanobacterial nonribosomal peptide synthetases (NRPS) and polyketide synthases (PKS) that may be involved in cyanotoxin synthesis.
An attempt to classify all contigs down to the lowest taxonomic level was made using mmgenome and PhyloPythiaS+, with the latter being designed to only identify bacteria and archaea, leaving contigs from eukaryotic organisms unassigned. Of the total contigs, 186,601 were binned as bacteria, 3,502 were binned as archaea, and 195,551 were left unassigned. Overall, 12,228 essential genes were identified, which aided in contig assignment. The relative abundance of key strains was inferred from the covariance in contig read coverage across time points. Contig read coverage is an important metric as it should reflect the amount of each organism's DNA present in a sample relative to that of other taxa, and, as such, it is a proxy for an organism's relative abundance (25). For illustrative purposes, Fig. 2 displays the relative coverage depths of the assembled contigs for two samples collected at opposite ends of the reservoir during a transect on 30 August 2013 (S41 is located at the inflow, and S9 is near the outflow). Clusters of contigs corresponding to an Anabaena sp. strain (also referred to as Dolichospermum) and a Microcystis sp. strain are circled.
FIG 2.

Visual representation of assembled contigs and their relative read coverage depths (logarithmic scale, ∼0.1 to 300) for two samples collected at opposite ends of the reservoir (S41, inflow; S9, near outflow) on 30 August 2013. Contigs containing geosmin or microcystin synthase genes are highlighted. Contig colors denote taxonomic groupings at the level of order, with larger circles representing longer contigs (scaffolds). Contig clusters that represent the genomes of Anabaena and Microcystis are circled.
The ability to survey all members of a microbial community by metagenomics is directly related to the number of unique individuals comprising the community (i.e., species richness) and the sequencing effort applied (i.e., high-diversity samples require more sequencing effort than low-diversity samples). Since less than half of the sequencing reads assembled into contigs, we used the raw sequencing reads to generate estimates of microbial community composition. These estimates were made for reservoir samples concentrated onto 0.2-μm pore size filters, a size expected to retain almost all living organisms in the water column. After quality screening, all sequencing reads were translated into amino acid sequences and screened for homologous proteins in the NCBI nonredundant short read archive using RAPSearch (39). Normalized subsets (∼233,000/sample) of protein-encoding reads were taxonomically assigned to the lowest common ancestor with MEGAN (40). Rarefaction analysis was done on these assignments, with most curves reaching their asymptote by this depth (see Fig. S2 in the supplemental material). Using this approach, between 750 and 938 genera were identified from samples collected from June to October 2013. Estimates of community richness (Shannon's diversity index [H′]) from these assignments ranged from 1.05 to 3.22 and those of community evenness ranged from 0.16 to 0.49 (Table 1). The average diversity of 2.20 was similar to that estimated for other temperate and tropical freshwater lakes (mean H′ of ∼2.75) by 16S rRNA sequencing (55).
TABLE 1.
Estimates of community diversity and metagenomic coveragea
| Date | Genera | H′ | Evenness | % coverage | Effort (Gbp) |
|---|---|---|---|---|---|
| 6 June 2013 | 832 | 2.48 | 0.37 | 78.7 | 2.12 |
| 8 July 2013 | 938 | 1.85 | 0.27 | 86.5 | 2.50 |
| 23 July 2013 | 834 | 2.21 | 0.33 | 75.3 | 1.94 |
| 6 August 2013 | 796 | 1.05 | 0.16 | 75.5 | 1.93 |
| 19 August 2013 | 858 | 2.37 | 0.35 | 66.6 | 2.18 |
| 21 October 2013 | 730 | 3.22 | 0.49 | 57.6 | 1.50 |
Estimates of total genera (all kingdoms: Bacteria, Archaea, and Eukaryota), Shannon's diversity index (H′), and evenness were based on analysis of a normalized subset (233,000 reads/sample) of protein-encoding sequences binned to the lowest common ancestor using MEGAN (40) . The estimated percentage of the community covered by each sequencing effort (gigabases of data generated) was calculated using Nonpareil (33). Only samples collected onto membrane filters (0.2 μm) are presented.
Estimating appropriate sequencing effort.
A metagenome can be defined as all of the genetic material present in an environmental sample, and, as such, metagenome coverage per sequencing effort (e.g., total base pairs sequenced) will be directly influenced by community evenness and species richness. Table 1 displays Shannon's diversity index values (H′) based on operational taxonomic units (OTU) delineated by individual sequence binning (i.e., reads preassembly) to the lowest common ancestor using RAPSearch and MEGAN and estimated metagenome coverage for each sequencing effort as inferred from the Nonpareil curves (33). Nonpareil curves are generated by calculating the redundancy of individual reads (nonpaired) from a metagenome data set and fitting these data with a log gamma function, from which extrapolations are made to estimate the sequencing effort needed to reach a given level of metagenome coverage (e.g., 95 or 100% coverage). This approach was used to estimate the percentage of each individual metagenome covered by our sequencing effort and to extrapolate how much more sequencing would be needed to uncover the total diversity within each sample. For this analysis, only water samples collected onto 0.2-μm-pore-size filters were included, since the GF/C filters do not truly represent the bacterial community structure. The coverage estimates ranged from 57.6 to 86.5%, with higher-diversity samples trending to lower average coverage (Fig. 3A). To achieve ≥95% metagenome coverage, it is estimated that ≥10 Gbp per sample would be needed for this system, which is 3 to 4 times greater than the only other similarly analyzed lake metagenome set (Lake Lanier, GA, USA) (33, 41).
FIG 3.

(A) Nonpareil curves (33) estimating the average coverage (completeness) of metagenomic data sets (taxa detected relative to estimated number of taxa present) obtained from sequencing samples collected onto 0.2-μm filters. Empty circles denote the actual sequencing effort (base pairs of sequence recovered); dashed red lines indicate 95% and 100% coverage levels. (B) Relationship between gene abundance (copies per milliliter) determined by qPCR and shotgun metagenome coverage depth of the target gene (geoA and mcyE) from samples collected onto glass fiber (1.2-μm pore size) or membrane (0.2-μm pore size) filters; each metagenome was normalized to 29.6 million reads. Adj. R2, adjusted R2.
Since reads from both the 1.2-μm and 0.2-μm filters were pooled and assembled, we also computed the estimated coverage of the combined data set and estimated it to be 100% for a 30.5-Gbp sequencing effort. While this is the best approach that we are aware of for estimating metagenome coverage relative to sequencing effort, this metric still likely underestimated the sequencing depth required for complete representation of the community. The Nonpareil approach was designed to estimate metagenomic data set completeness, and it is not an estimate of the sequencing depth required to assemble all taxa comprising a metagenome. For genome assembly, typically higher coverage depth results in better assembly. At the very least, in order to correct for sequencing errors, redundancy at each nucleotide position is required; assuming a 1% error rate in sequencing reads, 8× read depth corresponds to an associated error rate of 10−16 (42) and represents a practical lower limit to strive to exceed when genome assembly from metagenomic data sets is desired.
Identification of a geosmin producer.
In August 2013, an unusual taste-and-odor event impacted the entire reservoir, with geosmin concentrations ranging from 8,022 ng/liter at the inflow to 84 ng/liter at the outflow (Fig. 1); this event followed a 1-week period of heavy precipitation (∼18.5 cm) that raised the reservoir water height by ∼2.7 m (an increase of ∼1.1E8 m3) (see Fig. S3 in the supplemental material). The shotgun metagenomic data indicated an obvious abundance of Anabaena at the sites with high geosmin concentrations, especially S41 (Fig. 2). Analysis of the assembled contigs identified a single geosmin synthase gene present within a 20.3-kbp-long contig (no. 1655) that contained 18 other predicted coding sequences (CDS) whose closest similarities in the NCBI GenBank database were to Anabaena species sequences (data not shown). The geosmin synthase on this contig was 97% identical to that of an Anabaena ucrainica isolate (GenBank accession no. HQ404997); coverage depths of this contig across the data set indicated that it covaried with other contigs annotated as Anabaena (Fig. 2). The assembly of only a single geosmin synthase suggested that Anabaena was the principal producer over the study period. To screen for other geosmin producers that failed to assemble and were therefore only present in low abundance, all ∼651 million sequencing reads were mapped to all known geosmin synthases recovered from NCBI GenBank. This process identified 465 sequencing reads with homology to known germacradienol/geosmin synthases. However, all reads recruited corresponded to the Anabaena ucrainica reference sequence, suggesting that other geosmin-producing taxa were rare or absent.
Previous studies have reported taste-and-odor events that were linked to periods of heavy rainfall and surface runoff that introduce geosmin and/or MIB-producing soil bacteria such as actinomycetes into aquatic systems (43, 44). To evaluate the relationship between geosmin synthase-containing Anabaena spp. and the metabolite itself, a TaqMan quantitative PCR assay (geoA) was developed with perfect complementarity to the Anabaena geosmin synthase gene identified from the assembly. The assay was used to quantify Anabaena geosmin synthase gene copies from all GC-MS-analyzed depth-integrated samples collected in 2013 and 2014 (n = 35), and the resulting gene equivalent estimates were related to total geosmin by linear regression (Fig. 4B). The robust fit (adjusted R2 = 0.71; P < 0.001) indicated that Anabaena was the principal producer of geosmin during the study period as it singly explained the majority of the variance in the data. Interestingly, microscopic estimates of Anabaena cell concentrations exhibited no correlation to geosmin (adjusted R2 = 0.0; P = 0.39) (Fig. 4C), highlighting the value of screening for only those cells capable of geosmin biosynthesis. Endpoint PCR targeting geosmin synthases from heterotrophic bacteria was performed for all samples as described elsewhere (see Table S2 in the supplemental material) (29). Only the positive controls were amplified by this highly sensitive technique, suggesting that geosmin-producing heterotrophic bacteria were absent or present at undetectable concentrations in the water column throughout 2013 to 2014.
FIG 4.
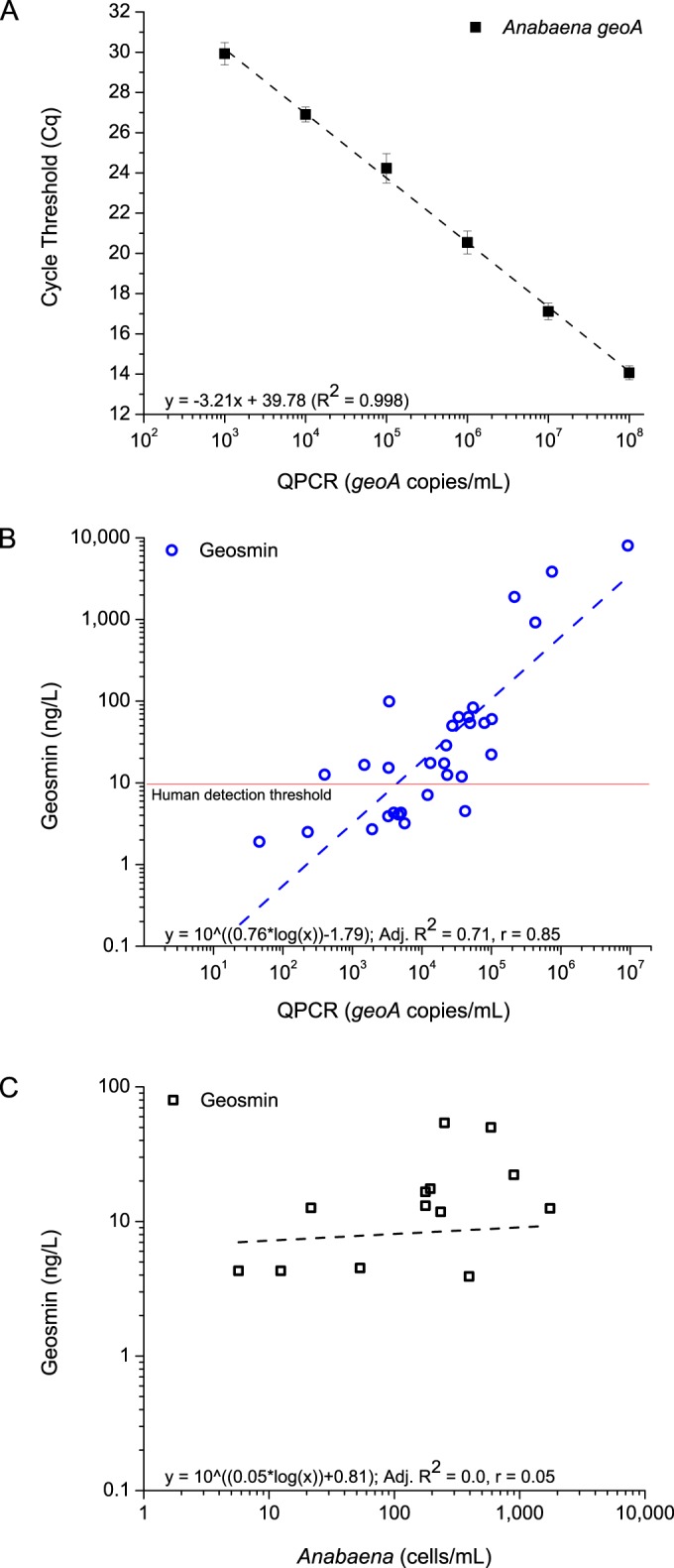
Development of a quantitative PCR assay for enumerating geosmin-producing Anabaena in Cheney Reservoir. (A) Standard curve used for the qPCR assay. (B) Relationship between geosmin concentrations measured by GC-MS and qPCR estimates of Anabaena geosmin synthase genes (geoA) for all sites in 2013 and 2014 transects. (C) Relationship between geosmin concentrations and Anabaena cell concentrations counted microscopically for the routine monitoring site in 2013 and 2014.
Identification of an MIB producer.
In Cheney Reservoir, MIB is periodically detected at moderately low concentrations (1 to 10 ng/liter range) with no clear seasonal patterns (18). During 2013 to 2014, MIB concentrations peaked at 9.7 ng/liter on 9 September 2013 and again at 5.2 ng/liter on 14 April 2014. Shotgun metagenomic analyses conducted on the 9 September 2013 sample and on a 25 September 2013 sample with 8.1 ng/liter MIB failed to identify any MIB synthase genes in the assembled contigs. However, by mapping individual sequencing reads to all known MIB synthases obtained from NCBI GenBank, five reads corresponding to an Oscillatoria sp. or Pseudanabaena sp. MIB synthase were revealed. The recruited sequencing reads indicated that recently described cyanobacterial MIB primers (13, 30) should be able to amplify this MIB synthase from Cheney Reservoir cyanobacteria (see Table S2 in the supplemental material). Endpoint PCR incorporating both primer sets was applied to all study samples, with only two samples amplifying, corresponding to the 9 September and 25 September 2013 dates. The resulting amplicons were Sanger sequenced and aligned with the closest homolog (Oscillatoria limosa) identified in GenBank (HQ630885.1) (see Fig. S4 in the supplemental material), revealing 96% nucleotide similarity.
The 25 September 2013 sample contained 8.1 ng/liter MIB, although no known cyanobacterial MIB producers (e.g., Oscillatoriales) were observed by microscopic cell counts in this sample. The weak detection of MIB DNA in the water column suggested that the causative agent might be a benthic or epiphytic strain of cyanobacteria. To test if periphyton may produce MIB in Cheney Reservoir, scrapings of algae from the concrete drinking water intake structure and adjacent reservoir dam wall were collected on 4 August 2015. GC-MS analyses of these samples revealed total MIB concentrations of 17 ng/liter and >6,800 ng/liter for the dam and intake structure, respectively, although these values were only semiquantitative because the surface area of the scrapings was not measured. Each sample was PCR amplified using the cyanobacterial MIB primers as before (see Table S2 in the supplemental material) (13). An MIB synthase gene was amplified from the sedimented fraction of the offtake structure periphyton sample. Sanger sequencing again revealed the producer to be most closely related to Oscillatoria limosa (98% nucleotide identity across 742 nucleotides [nt]), although there were a number of mismatches between the 2013 and 2015 sequences, suggesting that more than one related MIB producer may be present (see Fig. S4 in the supplemental material). These results provide evidence for a benthic/periphytic MIB producer growing on the intake structure of the reservoir. Notably, no Oscillatoria sp. or Planktothrix sp. was microscopically observed in the intake scrapings, although 53% of the phytoplankton biomass was represented by Pseudanabaena sp., raising the possibility that it was the MIB producer rather than Oscillatoria. This possibility is in line with the results of other studies that have documented Pseudanabaena strains as being prolific producers of MIB (45, 46). To further validate which Oscillatoriales is responsible for MIB production, additional sequencing beyond the MIB synthase gene by primer walking or shotgun metagenomics will be required.
Geosmin was also detected in the periphyton scrapings at concentrations of 21 ng/liter and 109 ng/liter for the dam and intake structure, respectively, although these values are again only semiquantitative. Endpoint PCR and Sanger sequencing were used to identify geosmin producers in these samples. Geosmin synthases were recovered from the benthic fractions of both scrapings belonging to Anabaena and another uncharacterized heterotrophic bacterium sharing 85% nucleotide identity to an Archangium gephyra isolate (GenBank accession no. CP011509), which is a type of biofilm-producing myxobacteria (“slime bacteria”).
Identification of a microcystin producer.
The assembled contigs from the shotgun metagenomes revealed only microcystin synthetase genes from Microcystis (Fig. 2). Other potential microcystin-producing genera, such as Anabaena or Planktothrix, possess gene rearrangements and deletions/insertions within the mcy operon that can be used to distinguish specific taxa (47). From the sequencing data, a previously developed TaqMan qPCR assay targeting mcyE in Microcystis (32) was determined to be a perfect match to the strain(s) present in Cheney Reservoir. Even after removal of highly leveraged points, the qPCR results exhibited a strong, positive relationship (n = 18; adjusted R2 = 0.66; P < 0.001) between Microcystis mcyE gene copy numbers and total microcystin concentrations, providing strong evidence that Microcystis is the principal microcystin producer in Cheney Reservoir. Figure S5A to C in the supplemental material displays log-log regressions between total microcystin concentrations (ELISA) and cell counts of Microcystis (displayed as cells per milliliter) (see Fig. S5A), qPCR estimates of potentially toxigenic Microcystis (mcyE gene copies per milliliter) (see Fig. S5B), and contig read coverage for the assembled mcy genes determined from metagenomes (see Fig. S5C). Microcystin concentrations were most strongly correlated to mcyE gene copy numbers, followed by mcy operon read coverage from the assemblies (n = 13; adjusted R2 = 0.60; P < 0.001). Notably, the qPCR results explained more variance than the microscopic cell counts (n = 13; adjusted R2 = 0.15; P = 0.10). While these data demonstrate the increased accuracy of qPCR over microscopy, this relationship may be further refined with more observations.
Generalized linear models for predicting water quality impairment.
In order to evaluate the usefulness of qPCR for predicting taste-and-odor events or microcystins, the qPCR and metabolite data were modeled using linear regression and compared to multiple linear regression models that incorporated routine physicochemical data from sondes (temperature, pH, dissolved oxygen, conductivity, chlorophyll, and turbidity) or a combination of sonde and laboratory measurements (total Kjeldahl nitrogen, ammonia [NH3], nitrate [NO3], NO3-nitrite [NO2], orthophosphate [PO4], dissolved phosphorus, total phosphorus, and total cyanobacteria) for estimating microcystin and geosmin (Table 2). This comparison is highly relevant because many water quality monitoring programs rely upon indirect sonde and water quality measurements to predict CyanoHAB risks. The analyses utilized all combinations of discrete and continuous data collected throughout 2013 to 2014 as part of the USGS Cheney Reservoir long-term monitoring program. The best mcyE qPCR model used square root-transformed values, and it performed better than the physicochemical models for estimating microcystin concentrations (adjusted R2 = 0.84 compared to 0.82 and 0.48) (Table 2). Similarly, the geoA qPCR model outperformed the physicochemical models for estimating geosmin concentrations (adjusted R2 = 0.71 versus adjusted R2 = 0.51 and 0.63, respectively) (Table 2). However, power analysis indicated that only three of the models had sufficient observations to reliably estimate metabolite concentrations: the single-variable mcyE and geoA qPCR models and the sonde-only geosmin model. The geosmin sonde model had a relatively poor adjusted R2 value (0.63). In contrast, the qPCR assays both met the minimum observation criteria (N) and were the strongest predictors of geosmin and microcystin concentrations as evidenced by the low PRESS values and high correlations between geosmin synthase gene abundance and geosmin concentration and between mcyE gene abundance and microcystin concentration. Physiochemical models were not developed for MIB because only 29% (n = 28) of the samples collected had detectable MIB concentrations and only two of the samples could be amplified by endpoint PCR.
TABLE 2.
Results of multiple regression analysis identifying the strongest (lowest predicted residual error sum of squares and highest adjusted R2) physicochemical or biological predictors for microcystins or geosmin over the study period (2013 to 2014)a
| DV | Model | y int | nvars | N | n | Adj. R2 | PRESS | Data |
|---|---|---|---|---|---|---|---|---|
| Microcystin | MC1/2 ∼ mcyE1/2 · 0.0007 | 0.27 | 1 | 10 | 18 | 0.84 | 0.13 | qPCR |
| Microcystin | MC1/3 ∼ pH1/3 · 4.2 + Cond−1 · 0.92 + PO4 · 4.3 + TP · 1.7 | −17.0 | 4 | 40 | 26 | 0.82 | 0.78 | Sonde + lab |
| Microcystin | MC1/3 ∼ Temp1/3 · −0.53 + Turb · 0.02 + DO · 0.07 + sin · −0.38 + cos · −0.87 | 0.90 | 4 | 40 | 36 | 0.47 | 1.7 | Sonde |
| Geosmin | log10(GEO) ∼ log10(geoA) · 0.76 | −1.79 | 1 | 10 | 31 | 0.71 | 10.9 | qPCR |
| Geosmin | GEO−1 ∼ DO · −0.11 + DP−1 · −0.01 + TP−1 · 0.04 + sin · 0.47 + cos · 0.29 | 1.20 | 4 | 40 | 28 | 0.51 | 1.2 | Sonde + lab |
| Geosmin | GEO−1 ∼ Temp · −0.04 + pH−1 · 10.6 + Cond−1 · 605.6 + sin · 0.10 + cos · −0.63 | −1.08 | 4 | 40 | 41 | 0.63 | 1.1 | Sonde |
DV, dependent variable; y int, y intercept; nvars, number of independent variables; N, minimum number of observations required (37); n, number of observations analyzed; PRESS, predicted residual sum of squares; Adj. R2, adjusted R2; Cond, specific conductance (microsiemens per meter); DO, dissolved oxygen (milligrams per liter); DP, dissolved phosphorus (milligrams per liter); geoA, qPCR result (geoA gene copies per milliliter); GEO, geosmin (nanograms per liter); MC, microcystin; mcyE, qPCR result (mcyE gene copies per milliliter); PO4, orthophosphate (milligrams per liter); Temp, temperature (degrees Centigrade); TP, total phosphorus (milligrams per liter); Turb, turbidity (formazin nephelometric units [FNU]).
DISCUSSION
The application of shotgun metagenomics for identification of the microorganism(s) responsible for water quality impairment relies upon several key assumptions: (i) in order for an organism to impair water quality it must be present in numerically relevant concentrations; (ii) sequencing depth must be sufficient to assemble functional genes from ecologically relevant organisms or to detect their presence by recruiting unassembled reads to reference gene sequences; and (iii) organisms must be present in the water column, as opposed to being of benthic/epiphytic or terrestrial origin. Previous studies have implicated planktonic cyanobacterial genera, Anabaena spp. and Oscillatoria spp., as the likely principal producers of geosmin and MIB in Cheney Reservoir (14). However, there remained significant uncertainty regarding the causative agents because geosmin has been at times detected when cyanobacteria were absent from the water column, suggesting that other sources (e.g., actinobacteria or benthic cyanobacteria) may be involved. Even though Anabaena has historically been the dominant cyanobacterium present in the water column during taste-and-odor events, cell abundance of this organism has tended to correlate poorly (R2 < 0.3) with geosmin and MIB concentrations (15).
In this study, time series shotgun metagenomics was used to survey the microbial community throughout the summer; this provided samples from periods before, during, and after taste-and-odor and cyanotoxin events. The sequencing reads and assemblies implicated Anabaena as a geosmin producer, Microcystis as a microcystin producer, and Oscillatoria and/or Pseudanabaena as MIB producers. The sequencing data were used to guide selection and development of TaqMan qPCR assays targeting Anabaena geosmin synthases and Microcystis microcystin synthetase (mcyE) genes. Estimates of Anabaena geosmin gene copy numbers by qPCR were strongly and positively correlated with GC-MS measurements of geosmin over a 2-year period, indicating that Anabaena was the principal geosmin producer in the water column during this time (Fig. 4). The subsequent identification of a geosmin synthase with similarity to Archangium gephyra recovered from the drinking water intake structure in 2015 indicated that there are other potential producers in the reservoir. However, no other geosmin synthase genes were identified in the metagenome data sets or by endpoint PCR on water column samples, suggesting that other producers are rare or only occurred attached to physical substrates during the study period. We acknowledge the possibility that reverse transcriptase real-time PCR assays measuring geosmin synthase expression could provide better estimates of geosmin concentrations than the DNA-based assays we used; however, at least one study suggests that geoA expression in Anabaena spp. is constitutive (48), indicating that there may be limited value in using RT-qPCR. Additionally, for practical applications such as routine water quality monitoring, DNA-based methods are generally simpler and are therefore considered more appropriate in this context.
Microcystis mcyE gene copy estimates were also strongly and positively correlated with ELISA measurements of total microcystins, indicating that Microcystis spp., but not other cyanobacteria, were the principal—and likely only—microcystin producers in the reservoir over the study period (see Fig. S5 in the supplemental material). Both the reads and the assembled contigs from the shotgun metagenomes were screened for the presence of other cyanotoxin genes; no recognized cyanotoxin biosynthesis genes were detected. However, several uncharacterized NRPS and PKS pathways responsible for the synthesis of unknown metabolites were observed, and these may be worthy of further study (see Table S4 in the supplemental material).
Application of metagenomics to water quality management.
Toxins and taste-and-odor compounds produced by bacteria are strain-specific traits. As a result, the causative organisms for water quality impairment are not easily determined by classical microbiological approaches such as culturing or microscopic enumeration. Shotgun metagenomics can be a highly useful tool for assessing the causative organisms responsible for water quality impairment. Assembled contigs containing the genes for secondary metabolite synthesis can be used to identify potential producers in a semiquantitative way, as contig coverage depth was significantly correlated with metabolite concentrations. Sequence information can also be used to validate existing qPCR primer and probe sequences or to develop novel qPCR assays specific to the system of study. Although studies have described primer sets intended to be “general” or “universal” across all or a subset of organisms, by comparing shotgun metagenome read sets to published primer sets, primer-template mismatches are often observed. In this study, a number of heterogeneities were observed between the assembled Cheney geosmin synthase and 15 previously published primers (13, 48–50) designed to amplify geosmin synthases from cyanobacteria (see Table S3 in the supplemental material). The number of mismatches ranged from 0 to 7 (mean, 2.14), with the 3139F and 3245R primer set developed by Tsao and colleagues (50) being the only perfectly matching set. Based on the sequences currently deposited in NCBI GenBank, it seems unlikely that a perfect, universal primer set targeting all geosmin synthases will be attainable. Thus, selection of qPCR assays should be guided by direct knowledge of the nucleotide content of desired target genes in each system of study. For qPCR to provide absolute quantification of gene targets, the assays must be fully optimized, and primer mismatches should be avoided as they reduce target specificity, lower amplification efficiency, and, as a result, artificially depress gene copy estimates (34). For example, 3′ terminal mismatches on either primer have been shown to exhibit effects ranging from a 2-fold underestimation of target abundance to the complete abolishment of amplification (i.e., false negative) (51).
Log-log regression analysis was utilized to further explore the relationship between absolute quantification of gene targets by qPCR (mcyE and geoA) and normalized metagenomic read coverage depths (Fig. 3B). Water samples that were collected onto 1.2-μm or 0.2-μm pore size filters were individually analyzed since 1.2-μm (GF/C) filters will enrich for larger-celled and colonial organisms, skewing the relationship between sequencing depth and community diversity. For both size classes, strong, positive correlations were observed between the qPCR estimates of geosmin and microcystin synthase genes and their relative read coverage (adjusted R2 = 0.80; P < 0.001 and adjusted R2 = 0.83; P < 0.001 for 0.2-μm and 1.2-μm filters, respectively). For studies focused on the phytoplankton community, deeper coverage per sequencing effort can be attained by working from 1.2-μm filters, which effectively exclude a large percentage of smaller heterotrophic bacteria.
Although metagenomics is powerful for making unbiased assessments of microbial community structure, composition, and gene content, there are limitations in detecting low-abundance organisms whose genes are unlikely to assemble at practical sequencing depths. In this study, geosmin genes were detected by read recruitment for all samples with geosmin concentrations of ≥3.9 ng/liter, which corresponded to Anabaena concentrations of ∼1,900 geoA copies/ml or greater. Microcystin genes were observed in the metagenomes for all samples with ≥0.3 μg/liter microcystin and mcyE gene concentrations of ≥2,800 copies/ml. This represents usefully sensitive detection of producers in samples with geosmin and microcystin present near the low end of the concentration range of concern to water managers. Beyond mere detection, the DNA-based molecular tools (both qPCR and shotgun metagenomics) were found to better estimate levels of taste-and-odor compounds and microcystins than microscopic cell counts or other physicochemical measurements (Fig. 4; Table 2; see also Fig. S5 in the supplemental material). The strong correlation between mcyE gene copy numbers and microcystin concentrations shows that qPCR, a sensitive and readily accessible high-throughput technique, can be useful in predicting periods of cyanotoxin impairment. Although environmental sondes are less predictive, water quality managers may also wish to remotely deploy them to guide sample collection frequency for qPCR analysis (e.g., in response to increases in chlorophyll a). Our findings are contrary to those of a recent study on four Wisconsin lakes that reported a very poor correlation between qPCR estimates of mcyA and microcystin (52). The discrepancy in the relationship in the aforementioned study could be due to template-primer mismatches in the qPCR assays or underestimation of total microcystin concentrations since only four congeners were assessed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
In conclusion, the results reported here demonstrate that future studies of lakes and reservoirs impaired by cyanobacterial blooms would benefit from periodic metagenomic assessments of the microbial taxa present and their gene content. This will enable specific management decisions analogous to personalized medicine treatment regimes that take into account the genome content of individual patients (53). Lake microbial communities are continually in flux (54), and water bodies historically not impacted may begin experiencing cyanobacterial blooms in response to human activities and global climate change (1, 2). Periodic assessments of the microbial consortia in source waters will provide unbiased inventories of potential constituents of concern and will guide assay optimization and management practices.
Supplementary Material
ACKNOWLEDGMENTS
We thank the USGS Wichita field office for collection of routine water quality samples from Cheney Reservoir and Guy Foster, USGS Kansas Water Science Center, for assistance with the design and execution of spatial data collection. We also thank Nathan Brown for assistance in establishing the metagenome bioinformatic analysis pipeline. We thank an anonymous USGS reviewer for helpful comments on the manuscript.
This research was supported by U.S. Geological Survey grant 2012OR127G and the U.S. Geological Survey Kansas Water Science Centers. Additional support was provided by the Oregon State University Agricultural Experiment Station and the Mabel E. Pernot Trust.
Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. government.
Funding Statement
This work was funded by U.S. Geological Survey (USGS) grant 2012OR127G to Theo W. Dreher (principal investigator [PI]) and Jennifer L. Graham (co-PI).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01334-16.
REFERENCES
- 1.Paerl HW, Paul VJ. 2012. Climate change: links to global expansion of harmful cyanobacteria. Water Res 46:1349–1363. doi: 10.1016/j.watres.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Paerl HW, Otten TG. 2013. Harmful cyanobacterial blooms: causes, consequences, and controls. Microb Ecol 65:995–1010. doi: 10.1007/s00248-012-0159-y. [DOI] [PubMed] [Google Scholar]
- 3.Carmichael WW. 2001. Health effects of toxin-producing cyanobacteria: “the cyanoHABs”. Hum Ecol Risk Assess 7:1393–1407. [Google Scholar]
- 4.Otten TG, Paerl HW. 2015. Health effects of toxic cyanobacteria in U.S. drinking and recreational waters: our current understanding and proposed direction. Curr Environ Health Rep 2:75–84. doi: 10.1007/s40572-014-0041-9. [DOI] [PubMed] [Google Scholar]
- 5.Jüttner F, Watson SB. 2007. Biochemical and ecological control of geosmin and 2-methylisoborneol in source waters. Appl Environ Microbiol 73:4395–4406. doi: 10.1128/AEM.02250-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dietrich AM. 2006. Aesthetic issues for drinking water. J Water Health 4:11–16. [PubMed] [Google Scholar]
- 7.Peter A, Gunten UV. 2007. Oxidation kinetics of selected taste and odor compounds during ozonation of drinking water. Environ Sci Technol 41:626–631. doi: 10.1021/es061687b. [DOI] [PubMed] [Google Scholar]
- 8.Dunlap CR, Sklenar K, Blake LA. 2015. Costly endeavor: addressing algae problems in a water supply. J AWWA 107:E255–E262. doi: 10.5942/jawwa.2015.107.0055. [DOI] [Google Scholar]
- 9.Kansas Department of Health and Environment. 2011. Water quality standards white paper: chlorophyll—a criteria for public water supply lakes and reservoirs. http://www.kdheks.gov/water/download/tech/Chlorophylla_final_Jan27.pdf.
- 10.Rantala A, Rajaniemi-Wacklin P, Lyra C, Lepisto L, Rintala J, Mankiewicz-Boczek J, Sivonen K. 2006. Detection of microcystin-producing cyanobacteria in Finnish lakes with genus-specific microcystin synthetase gene E (mcyE) PCR and associations with environmental factors. Appl Environ Microbiol 72:6101–6110. doi: 10.1128/AEM.01058-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasmussen JP, Giglio S, Monis PT, Campbell RJ, Saint CP. 2007. Development and field testing of a real-time PCR assay for cylindrospermopsin-producing cyanobacteria. J Appl Microbiol 104:1503–1515. doi: 10.1111/j.1365-2672.2007.03676.x. [DOI] [PubMed] [Google Scholar]
- 12.Rantala-Ylinen A, Käna S, Wang H, Rouhiainen L, Wahlsten M, Rizzi E, Berg K, Gugger M, Sivonen K. 2011. Anatoxin—a synthetase gene cluster of the cyanobacterium Anabaena sp. strain 37 and molecular methods to detect potential producers. Appl Environ Microbiol 77:7271–7278. doi: 10.1128/AEM.06022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suurnäkki S, Gomez-Saez GV, Rantala-Ylinen A, Jokela J, Fewer DP, Sivonen K. 2015. Identification of geosmin and 2-methylisoborneol in cyanobacteria and molecular detection methods for the producers of these compounds. Water Res 68:56–66. doi: 10.1016/j.watres.2014.09.037. [DOI] [PubMed] [Google Scholar]
- 14.Smith VH, Sieber-Denlinger J, de Noyelles F, Campbell S, Pan S, Randtke SJ, Blain GT, Strasser VA. 2002. Managing taste and odor problems in a eutrophic drinking water reservoir. Lake Res Manage 18:310–323. [Google Scholar]
- 15.Christensen VG, Graham JL, Milligan CR, Pope LM, Ziegler AC. 2006. Water quality and relation to taste-and-odor compounds in North Fork Ninnescah River and Cheney Reservoir, south-central Kansas, 1997−2003. USGS No. 2006-5095. US Geological Survey, Washington, DC. [Google Scholar]
- 16.Graham JL, Loftin KA, Meyer MT, Ziegler AC. 2010. Cyanotoxin mixtures and taste-and-odor compounds in cyanobacterial blooms from the Midwestern United States. Environ Sci Technol 44:7361–7368. doi: 10.1021/es1008938. [DOI] [PubMed] [Google Scholar]
- 17.Graham JL, Loftin KA, Ziegler AC, Meyer MT. 2008. Cyanobacteria in lakes and reservoirs: toxin and taste-and-odor sampling guidelines (version 1.0). In Techniques of water-resources investigations, book 9, chapter A7, section 7.5. US Geological Survey, Washington, DC: http://pubs.water.usgs.gov/twri9A/. [Google Scholar]
- 18.Stone ML, Graham JL, Gatotho JW. 2013. Model documentation for relations between continuous real-time and discrete water-quality constituents in Cheney Reservoir near Cheney, Kansas, 2001−2009. Open-File Report 2013-1123. US Geological Survey, Washington, DC. [Google Scholar]
- 19.Zimmerman LR, Ziegler AC, Thurman EM. 2002. Method of analysis and quality-assurance practices by the U.S. Geological Survey Organic Geochemistry Research Group; determination of geosmin and methylisoborneol in water using solid-phase microextraction and gas chromatography/mass spectrometry. Open-File Report 2002-337. US Geological Survey, Washington, DC. [Google Scholar]
- 20.McNabb CD. 1960. Enumeration of freshwater phytoplankton concentrated on the membrane filter. Limnol Oceanogr 5:57–61. doi: 10.4319/lo.1960.5.1.0057. [DOI] [Google Scholar]
- 21.Morán XAG, Gasol JM, Arin L, Estrada M. 1999. A comparison between glass fiber and membrane filters for the estimation of phytoplankton POC and DOC production. Mar Ecol Prog Ser 187:31–41. doi: 10.3354/meps187031. [DOI] [Google Scholar]
- 22.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng Y, Leung HC, Yiu SM, Chin FY. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 24.Gregor I, Dröge J, Schirmer M, Quince C, McHardy AC. 2014. PhyloPythiaS+: a self-training method for the rapid reconstruction of low-ranking taxonomic bins from metagenomes. PeerJ 4:e1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albertsen M, Hugenholtz P, Skarshewski A, Tyson GW, Nielson KL, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T. 2013. antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41:W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seemann T. 2014. Prokka: rapid prokaryote genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 29.Auffret M, Pilote A, Proulx É, Proulx D, Vandenberg G, Villemur R. 2011. Establishment of a real-time PCR method for quantification of geosmin-producing Streptomyces spp. in recirculating aquaculture systems. Water Res 45:6753–6762. doi: 10.1016/j.watres.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Song G, Shao J, Tan W, Li Y, Li R. 2016. Establishment and field applications of real-time PCR methods for the quantification of potential MIB-producing cyanobacteria in aquatic systems. J Appl Phycol 28:325−333. doi: 10.1007/s10811-015-0529-1. [DOI] [Google Scholar]
- 31.Vaitomaa J, Rantala A, Halinen K, Rouhiainen L, Tallberg P, Mokelke L, Sivonen K. 2003. Quantitative real-time PCR for determination of microcystin synthetase E copy numbers for Microcystis and Anabaena in lakes. Appl Environ Microbiol 69:7289–7297. doi: 10.1128/AEM.69.12.7289-7297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otten TG, Xu H, Qin B, Zhu G, Paerl HW. 2012. Spatiotemporal patterns and ecophysiology of toxigenic Microcystis blooms in Lake Taihu, China: implications for water quality management. Environ Sci Technol 46:3480–3488. doi: 10.1021/es2041288. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-R LM, Konstantinidis KT. 2014. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 30:629–635. doi: 10.1093/bioinformatics/btt584. [DOI] [PubMed] [Google Scholar]
- 34.Otten TG, Crosswell JR, Mackey S, Dreher TW. 2015. Application of molecular tools for microbial source tracking and public health risk assessment of a Microcystis bloom traversing 300 km of the Klamath River. Harmful Algae 46:71–81. doi: 10.1016/j.hal.2015.05.007. [DOI] [Google Scholar]
- 35.Helsel DR, Hirsch RM. 2002. Statistical methods in water resources. In Techniques of water-resources investigations, book 4, chapter A3. US Geological Survey, Washington, DC. [Google Scholar]
- 36.Chen S, Hong X, Harris CJ, Sharkey PM. 2004. Sparse modeling using orthogonal forward regression with PRESS statistic and regularization. IEEE Trans Syst Man Cybern B Cybern 34:898–911. doi: 10.1109/TSMCB.2003.817107. [DOI] [PubMed] [Google Scholar]
- 37.Peduzzi P, Concato J, Kemper E, Holford TR, Feinstein AR. 1996. A simulation study of the number of events per variable in logistic regression analysis. J Clin Epidemiol 49:1373–1379. doi: 10.1016/S0895-4356(96)00236-3. [DOI] [PubMed] [Google Scholar]
- 38.Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. 2013. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res 23:111–120. doi: 10.1101/gr.142315.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye Y, Choi JH, Tang H. 2011. RAPSearch: a fast protein similarity search tool for short reads. BMC Bioinformatics 12:159. doi: 10.1186/1471-2105-12-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res 17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oh S, Caro-Quintero A, Tsementzi D, DeLeon-Rodriguez N, Luo C, Poretsky R, Konstantinidis KT. 2011. Metagenomic insights into the evolution, function, and complexity of the planktonic microbial community of Lake Lanier, a temperate freshwater ecosystem. Appl Environ Microbiol 77:6000–6011. doi: 10.1128/AEM.00107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, Plichta DR, Gautier L, Pedersen AG, Le Chatelier E, Pelletier E, Bonde I, Nielsen T, Manichanh C, Arumugam M, Batto JM, Quintanilha Dos Santos MB, Blom N, Borruel N, Burgdorf KS, Boumezbeur F, Casellas F, Doré J, Dworzynski P, Guarner F, Hansen T, Hildebrand F, Kaas RS, Kennedy S, Kristiansen K, Kultima JR, Léonard P, Levenez F, Lund O, Moumen B, Le Paslier D, Pons N, Pedersen O, Prifti E, Qin J, Raes J, Sørensen S, Tap J, Tims S, Ussery DW, Yamada T, Renault P, Sicheritz-Ponten T, Bork P, Wang J, Brunak S, Ehrlich SD, MetaHIT Consortium . 2014. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol 32:822–828. doi: 10.1038/nbt.2939. [DOI] [PubMed] [Google Scholar]
- 43.Zaitlin B, Watson SB, Dixon J, Steel D. 2003. Actinomycetes in Lake Ontario: habitats, and geosmin and MIB production. J Am Water Works Assoc 95:113–118. [Google Scholar]
- 44.Jüttner F. 1990. Monoterpenes and microbial metabolites in the soil. Environ Pollut 68:377–382. doi: 10.1016/0269-7491(90)90039-F. [DOI] [PubMed] [Google Scholar]
- 45.Izaguirre G, Taylor WD, Pasek J. 1999. Off-flavor problems in two reservoirs, associated with planktonic Pseudanabaena species. Water Sci Technol 40:85–90. doi: 10.1016/S0273-1223(99)00542-9. [DOI] [Google Scholar]
- 46.Kakimoto M, Ishikawa T, Miyagi A, Saito K, Miyazaki M, Asaeda T, Yamaguchi M, Uchimiya H, Kawai-Yamada M. 2014. Culture temperature affects gene expression and metabolic pathways in the 2-methylisoborneol-producing cyanobacterium Pseudanabaena galeata. J Plant Physiol 17:292–300. [DOI] [PubMed] [Google Scholar]
- 47.Welker M, von Döhren H. 2006. Cyanobacterial peptides—nature's own combinatorial biosynthesis. FEMS Microbiol Lett 30:530–563. doi: 10.1111/j.1574-6976.2006.00022.x. [DOI] [PubMed] [Google Scholar]
- 48.Giglio S, Saint CP, Monis PT. 2011. Expression of the geosmin synthase gene in the cyanobacterium Anabaena circinalis AWQC318. J Phycol 47:1338–1343. doi: 10.1111/j.1529-8817.2011.01061.x. [DOI] [PubMed] [Google Scholar]
- 49.Giglio S, Jiang J, Saint CP, Cane D, Monis PT. 2008. Isolation and characterization of the gene associated with geosmin production in cyanobacteria. Environ Sci Technol 42:8027–8032. doi: 10.1021/es801465w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsao H-W, Michinaka A, Yen H-K, Giglio S, Hobson P, Monis P, Lin T-F. 2014. Monitoring of geosmin producing Anabaena circinalis using quantitative PCR. Water Res 49:416–425. doi: 10.1016/j.watres.2013.10.028. [DOI] [PubMed] [Google Scholar]
- 51.Stadhouders R, Pas SD, Anber J, Voermans J, Mes THM, Schutten M. 2010. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5′ nuclease assay. J Mol Diagn 12:109–117. doi: 10.2353/jmoldx.2010.090035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beversdorf LJ, Chaston SD, Miller TR, McMahon KD. 2015. Microcystin mcyA and mcyE gene abundances are not appropriate indicators of microcystin concentrations in lakes. PLoS One 10:e0125353. doi: 10.1371/journal.pone.0125353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hamburg MA, Collins FS. 2010. The path to personalized medicine. N Engl J Med 363:301–304. doi: 10.1056/NEJMp1006304. [DOI] [PubMed] [Google Scholar]
- 54.Logares R, Lindström ES, Langenheder S, Logue JB, Paterson H, Laybourn-Parry J, Rengefors K, Tranvik L, Bertilsson S. 2013. Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J 7:937–948. doi: 10.1038/ismej.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Humbert J-F, Dorigo U, Cecchi P, Le Berre B, Deroas D, Bouvy M. 2009. Comparison of the structure and composition of bacterial communities from temperate and tropical freshwater ecosystems. Environ Microbiol 11:2339–2350. doi: 10.1111/j.1462-2920.2009.01960.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.