

Abstract
The aim of this work is to better understand the genetic mechanisms determining two complex traits affecting porcine meat quality: intramuscular fat (IMF) content and its fatty acid (FA) composition. With this purpose, expression Genome-Wide Association Study (eGWAS) of 45 lipid-related genes associated with meat quality traits in swine muscle (Longissimus dorsi) of 114 Iberian × Landrace backcross animals was performed. The eGWAS identified 241 SNPs associated with 11 genes: ACSM5, CROT, FABP3, FOS, HIF1AN, IGF2, MGLL, NCOA1, PIK3R1, PLA2G12A and PPARA. Three expression Quantitative Trait Loci (eQTLs) for IGF2, ACSM5 and MGLL were identified, showing cis-acting effects, whereas 16 eQTLs had trans regulatory effects. A polymorphism in the ACSM5 promoter region associated with its expression was identified. In addition, strong candidate genes regulating ACSM5, FOS, PPARA, PIK3R1, PLA2G12A and HIF1AN gene expression were also seen. Notably, the analysis highlighted the NR3C1 transcription factor as a strong candidate gene involved in the regulation of the 45 genes analysed. Finally, the IGF2, MGLL, MC2R, ARHGAP6, and NR3C1 genes were identified as potential regulators co-localizing within QTLs for fatness and growth traits in the IBMAP population. The results obtained increase our knowledge in the functional regulatory mechanisms involved in these complex traits.
Pork-meat cuts and their derived products are paid according to the lean percentage in pork carcasses and meat quality, since they determine better acceptance for consumers1. A high amount of backfat content is a less desirable trait; meanwhile, meat with high intramuscular fat (IMF) is considered to have better taste, conferring juiciness to the meat. Additionally, fatty acid (FA) composition of IMF affects meat nutritional and sensory quality parameters. Monounsaturated FAs (MUFA) confer more oxidative stability than do polyunsaturated FAs (PUFA), improving meat taste and colour2. Furthermore, PUFA decreases the risk of suffering cardiovascular diseases, being healthier than saturated FAs (SFA)3. Therefore, there is a consumer requirement for porcine meat with high IMF with a balanced FA composition.
Some pig breeds, such as Landrace, have undergone an intense selection process towards efficient meat production with rapid growth and a leaner carcass; however, its resulting meat is less appreciated by consumers due to having low IMF composed of a high proportion of PUFA content4. In contrast, rustic pigs, such as the Iberian, are fatter, with more IMF with higher SFA and MUFA content and lower PUFA concentrations, which yields high quality and tasty meat, especially for dry-cured products. For instance, when comparing Iberian pigs with commercial breeds, Iberian pigs show higher palmitoleic acid (C16:1(n-7); 4.08 vs. 3.31; p < 0.001), oleic acid (C18:1(n-9); 44.96 vs. 40.56; p < 0.001), and general MUFAs (49.08 vs. 40.25; p < 0.001), and approximately half of the linoleic acid (C18:2(n-6); 7.52 vs. 16.11; p < 0.001) proportion, which is the most abundant PUFA4. Moreover, Iberian pigs have more than twice the amount of IMF, when compared to Landrace pigs (1.6% vs. 3.7%)5. The differences in the genetic background of these breeds determine the IMF and its FA composition, affecting meat quality5.
The experimental IBMAP population6, a cross between Iberian x Landrace pigs, which clearly differs in growth, carcass and meat quality, such as the IMF content and FA composition, was generated to obtain a population with large phenotypic differences, in which alleles from both parental breeds are segregating and can be tested. Strategies such as QTL and GWAS have been useful to highlight many genes, for instance determining intramuscular fatty acid (IMFA) composition in the IBMAP population6,7,8,9,10,11,12,13. Despite this, the underlying physio-genetic complex mechanisms of IMF deposition and its FA composition have not been clarified. The difficulty in the detection of QTLs for complex traits may be influenced by the pleiotropic nature of these traits and the use of multiple-tests correction methods14. Integrating other types of data such as intermediate molecular phenotypes to identify associations can overcome these limitations.
The detection of expression Quantitative Trait Loci (eQTLs) has recently been proposed as a good strategy to deepen the study of the genetic architecture of complex traits15. This technique allows for the identification of genetic variants associated with gene transcription levels, which may determine the phenotypic differences of complex traits. With the objective to better understand the mechanisms affecting IMF content and FA composition, we performed expression Genome-Wide Association Study (eGWAS) of 45 strong candidate genes for these traits, identified in previous studies by our group12,16,17,18, in 114 BC1_LD animals.
Results and Discussion
Gene-expression profiling
In the present study, 114 BC1_LD animals generated from the experimental IBMAP population and showing a wide range of IMF content and FA composition values were used for RNA extraction and RT-qPCR to perform eGWAS (Supplementary Table S1).
A significant sex effect (p-value ≤ 0.05) between gene-expression levels was detected in 20 out of the 45 genes analysed (44%): ACSS1, ACSS2, ATF3, CREG1, DGAT2, ETS1, FABP5, HIF1AN, IGF2, NCOA2, NCOA6, PLA2G12A, PPARA, PPARG, PPARGC1A, PRKAA1, PEX2, SCD, SP1 and SREBF1 (Fig. 1). Interestingly, some of the sex-biased genes identified are key regulators of lipid metabolism, such as PPARA, PPARG, PPARGC1A and SREBF1. Note that within the sex-biased genes, several lipogenic genes were more expressed in females (DGAT2, NCOA2, NCOA6, PPARG, PRKAA1, SCD, SP1 and SREBF1), whereas more lipolytic genes were over-expressed in males (ATF3, PPARA and PPARGC1A). Sexually dimorphic gene expression of genes involved in lipid metabolism has already been described in muscle as well as in other tissues such as liver19,20. Consistent with our results, PPARA and SREBF1 presented sex-biased gene expression in human skeletal muscle21. Furthermore, PPARGC1A and PPARA gene expression in muscle are affected under diet supplementation with 17β-estradiol (E2)21. In addition, the SCD and PPARG mRNA expressions have been observed to be lower in male than in female human muscle cell cultures22. To assess the relationship between muscle gene-expression levels and phenotypes, a hierarchical cluster analysis of the correlation values between the gene expression levels of the 45 genes and the fatty acid content in muscle was performed (Fig. 2). The hierarchical cluster analysis showed that genes mainly related to lipogenic pathways (DGAT2, PPARG, SCD, MGLL, NCOA1, NCOA2, NCOA6, PRKAA1, SP1, SREBF1) clustered together, whereas genes mainly related to lipolytic pathways (ATF3, CPT1B, PPARA, PPARD, PPARGC1A) joined together (Fig. 2). Genes within the lipogenic-related cluster showed, in general, positive correlations with palmitoleic (C16:1(n-7)) and octadecenoic (C18:1(n-7)) FAs, while the lipolytic-related group showed a positive correlation with PUFAs in general, specifically with the linoleic (C18:2(n-6)) FA. Overall, these results are in agreement with a previous muscle RNA-Seq transcriptome study of animals which were extreme for IMFA composition performed by our group16, where a higher expression of genes related to lipogenic pathways was observed in the muscle of animals with high MUFA and SFA content in muscle.
Figure 1. Comparison between males and females of muscle gene-expression levels of 45 lipid-related genes.

Data represent means ± standard error of the means (SEM). Significant differences between sexes are indicated as *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Figure 2. Hierarchical cluster of correlations among gene-expression levels (RQ) of the 45 genes and fatty acid content in muscle.

The highest correlations for gene-expression values (p-value < 1.00 × 10−16) were observed for PPARG and SCD (r = 0.78), DGAT2 and PPARG (0.85), SCD and DGAT2 (r = 0.77), FABP3 and PLIN5 (r = 0.83), FABP3 and AQP7 (r = 0.80), ACAA2 and FABP3 (r = 0.78) and ELF1 and PPAP2A (r = 0.75). Interestingly, the strong correlation between PPARG and SCD mRNA expression (r = 0.78; p-value < 1.00 × 10−16) suggests a transcriptional regulation of SCD by the PPARG nuclear factor.
Here, PPARG, DGAT2 and SCD, which are involved in the triacylglycerol synthesis, were highly correlated. Similarly, FABP3, AQP7 and PLIN5, which encode for proteins responsible for lipid and glucose transport, were also highly correlated. Remarkably, these three genes were over-expressed in animals with higher MUFA and SFA content in muscle, when compared with animals having more PUFA in a RNA-Seq study16.
eQTL identification
An eGWAS was performed using a total of 40,586 SNPs and the mRNA expression values of the 45 lipid-related genes of 114 BC1_LD animals. A total of 241 SNPs located in 18 Sus scrofa chromosomal regions of SSC1, SSC2, SSC3, SSC6, SSC8, SSC9, SSC10, SSC11, and SSC13 were identified for 11 genes: ACSM5, CROT, FABP3, FOS, HIF1AN, PIK3R1, PLA2G12A, MGLL, IGF2, NCOA1 and PPARA (FDR < 0.05; Table 1). Five genes (ACSM5, IGF2, MGLL, PLA2G12A and PPARA) presented more than one associated eQTL (Table 1). Three out of 18 eQTLs were identified as cis-acting, for ACSM5, IGF2 and MGLL gene expression (Fig. 3), suggesting the presence of a mutation in the same gene directly affecting its expression, whereas 16 eQTLs had trans regulatory effects. The majority of eQTLs (8 of 18) were located on SSC2 and SSC6 (Table 1).
Table 1. Significant eQTLs identified.
| No. | Gene | Most significant SNPs in eQTL | p-value | q-value | eQTL intervala | No. SNPsb | Type of eQTL |
|---|---|---|---|---|---|---|---|
| 1 | ACSM5 | ASGA0090088, ASGA0105223, SIRI0001454 | 7.12E-14 | 9.29E-10 | 3:16487448-27941321 | 43 | CIS |
| 2 | ACSM5 | ALGA0020170 | 8.58E-11 | 3.73E-07 | 3:100347076 | 1 | TRANS |
| 3 | ACSM5 | ASGA0090778 | 2.55E-08 | 3.44E-05 | 10:175359 | 1 | TRANS |
| 4 | CROT | ALGA0046590 | 4.01E-07 | 9.77E-03 | 8:15870120-15912410 | 3 | TRANS |
| 5 | FABP3 | ALGA0063896 | 1.26E-06 | 4.47E-02 | 11:81364018-82160789 | 2 | TRANS |
| 6 | FOS | H3GA0031293 | 1.22E-09 | 4.95E-05 | 11:6736568-11968518 | 6 | TRANS |
| 7 | HIF1AN, PLA2G12A | H3GA0028012, ASGA0044215, ALGA0117195 | 2.91E-07 | 3.92E-03 | 9:117742788-117851340 | 3 | TRANS |
| 4.48E-06 | 2.74E-02 | ||||||
| 8 | IGF2 | ASGA0101159, DIAS0000846 | <1.0E-25 | <1.0E-25 | 2:16416-11175095 | 17 | CIS* |
| 9 | IGF2 | ASGA0085784 | 6.63E-09 | 2.45E-05 | 2:162088043-162298086 | 2 | TRANS |
| 10 | MGLL | ASGA0057854 | 7.06E-08 | 6.28E-05 | 1:287348708 | 1 | TRANS |
| 11 | MGLL | ASGA0093606 | 2.20E-09 | 2.61E-05 | 13:27954256-82589660 | 147 | CIS |
| 12 | NCOA1 | ALGA0016576, MARC0045025, MARC0087200 | 2.60E-06 | 3.49E-02 | 2:146195530-146722998 | 3 | TRANS |
| 13 | PIK3R1 | ALGA0117336 | 1.24E-06 | 3.02E-02 | 6:143952619-145605378 | 3 | TRANS |
| 14 | PLA2G12A | ALGA0113789 | 1.96E-08 | 7.84E-04 | 6:10020702 | 1 | TRANS |
| 15 | PLA2G12A | ALGA0103867 | 3.41E-06 | 2.74E-02 | 6:79924228 | 1 | TRANS |
| 16 | PLA2G12A | ASGA0039774 | 1.34E-06 | 2.68E-02 | 8:128899782-136113975 | 3 | TRANS |
| 17 | PPARA | M1GA0003328 | 3.25E-06 | 3.30E-02 | 2:150634202 | 1 | TRANS |
| 18 | PPARA | MARC0074986, DIAS0004325, CASI0006620 | 6.17E-07 | 6.17E-07 | 6:89986075-90352248 | 3 | TRANS |
aChromosomal location is given according to the Sscrofa10.2 assembly coordinates. Positions are relative to the significant eQTL interval start and end. Lengths are given in base-pairs.
bNumber of significant SNPs within the eQTL interval.
*Approximate location of the gene described by Fontanesi et al.26.
Figure 3. GWAS plot of ACSM5, IGF2 and MGLL gene expression in muscle tissue.
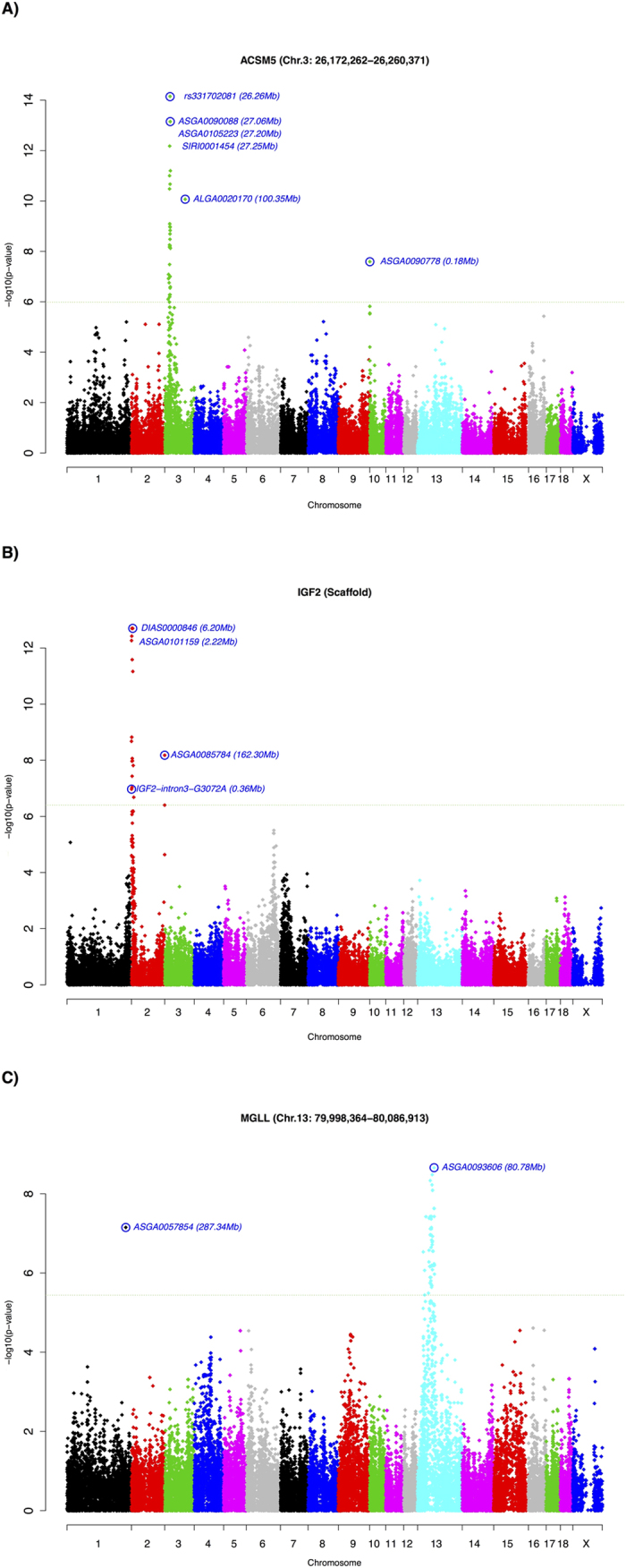
Positions in Mb are relative to Sscrofa10.2 assembly of the pig genome. Horizontal dashed lines indicate the genome-wide significance level (FDR-based q-value ≤ 0.001). Plot of eGWAS for (A) ACSM5 gene expression in muscle (B) IGF2 gene expression in muscle (C) MGLL gene expression in muscle.
From the associated SNPs (n = 241), 215 SNPs were successfully annotated with the Variant Effect Predictor (VEP) of Ensembl (Sscrofa 10.2 annotation release 80) of which 54% (117 SNPs) were located in intergenic regions. The remaining 46% (98) of SNPs were mapped within 76 genes: 69 (32%) in introns, 10 in the 5′ flanking region, 13 in the 3′ flanking region, three in the 3′UTR region, two in the coding regions of genes and determining synonymous mutations, and one missense mutation (Supplementary Table S2). Twelve out of the 98 intragenic SNPs were located within a gene exerting a lipid metabolism function (ABCA3, ACSM3, LRP5, LHFPL4, BRPF1, HRH1, PPARG, ACAD9, COPG1, MGLL, ACAD11, ARHGAP26) (Supplementary Table S2), but only six of them were expressed in muscle (ACSM3, PPARG, ACAD9, MGLL, ACAD11 and ARHGAP26), according to the Gene Expression Atlas23 and the RNA-Seq experiment performed in individuals from the BC1_LD16 (Supplementary Table S2). Only one of the intragenic SNPs mapping within genes with a lipid-related function was identified in a trans eQTL: the ARHGAP26 gene located in a trans-eQTL for the PPARA gene. This gene is activated via lipid interaction, however, its role has not been well-defined24.
cis-eQTL identified
The Insulin-Like Growth Factor 2 (IGF2) gene is not mapped in the current Sscrofa10.2 assembly25. However, it has been located by linkage map in the telomeric end of the p arm of SSC226. An intronic IGF2 mutation (IGF2-intron3-G3072A) was described by Van Laere et al.27 to have a major effect on muscle growth in pigs. Although this mutation only segregated in a small family in the IBMAP F2 population28, the cis-eQTL for IGF2 suggests that the intronic mutation identified by Van Laere et al.27 may segregate in the IBMAP BC1_LD, having an important effect on the traits analysed. However, after genotyping the IGF2-intron3-G3072A mutation in the BC1_LD animals, we observed that this SNP was not the most associated (p-value = 1.07 × 10−7) with the IGF2 mRNA expression levels (Fig. 3B), suggesting that other mutations in the IGF2 gene may be the responsible for the mRNA variation in the BC1_LD population. Further analysis will be conducted to validate our hypothesis.
Concerning the Monoglyceride Lipase (MGLL) eGWAS results, one of the annotated cis-SNPs (ASGA0103932) was mapped within an intronic region of the MGLL gene (Supplementary Table S2). Nevertheless, this SNP was not the most significant, associated SNP (ASGA0103932; p-value = 2.34 × 10−8), suggesting the presence of another polymorphism within or near this gene as the causative mutation affecting MGLL gene expression levels. The most significant cis-SNP for MGLL (ASGA0093606; p-value = 2.20 × 10−9) was located less than 0.69 Mb downstream of the MGLL gene. In addition, an SNP (ISU10000701) in the MGLL cis-eQTL was annotated in the upstream region of the PPARG gene, more than 4 Mb from the MGLL gene (Supplementary Table S2). PPARG and MGLL were reported to be co-associated in a previous study of our group for growth and fatness traits, where PPARG was described as a major regulator17. Additionally, the literature-based analysis with Genomatix also identified an interaction between these two genes (Supplementary Fig. S1), and a chromatin immunoprecipitation (ChiP) experiment performed in epithelial cultured cells revealed PPARG binding sites in the distal MGLL promoter29. Therefore, it is likely that this region, initially considered as a single cis-acting eQTL, in fact comprises two eQTLs. The correlation between PPARG and MGLL gene expression in muscle was moderate (r = 0.48; p-value = 8.28 × 10−8), reinforcing the hypothesis that at least two factors may determine the different levels of MGLL mRNA. In this regard, the MGLL gene expression would be affected by a variant present in the same gene (MGLL), and also by PPARG. However, both markers (ASGA0093606 and ISU10000701) were in strong LD (R2 = 0.99), not being able to assess whether MGLL gene expression is affected by cis-, trans- or both cis-/trans- factors on SSC13. Further investigation with other populations is required to reveal this. Remarkably, the significantly associated SNP inside the PPARG gene (ISU10000701) was one of the three main co-associated SNPs in the network identified in Puig-Oliveras et al.17 involved in the determination of growth and fatness traits. Nonetheless, no association was found between the SNP within PPARG (ISU10000701) and the PPARG gene expression.
Finally, three SNPs (ASGA0090088, ASGA0105223 and SIRI0001454) in complete linkage disequilibrium (R2 = 1) were the most significant ones in the cis-eQTL (p-value = 7.12 × 10−14) associated with the mRNA levels of the Acyl-CoA Synthetase Medium-Chain Family Member 5 (ACSM5) (Table 1). The ASGA0090088 marker was the closest cis-SNP, mapping at approximately 798 kb from the upstream region of the ACSM5 gene (Supplementary Table S2). Notably, after amplifying and sequencing the proximal promoter region of the ACSM5 gene in ten BC1_LD animals, three polymorphisms were identified at positions g26260919C > T (rs323520560), g26260505G > A (rs339587799) and g26260422G > A (rs331702081), according to the Ensembl ENSSSCG00000026453 sequence, which seem to be in linkage disequilibrium. The most proximal 5′ mutation (g26260422G > A) was genotyped in the BC1_LD animals, and the association analysis of this polymorphism with the ACSM5 mRNA expression values revealed that this SNP was the most associated one (p-value = 7.11 × 10−15; Fig. 3A). Further analyses are necessary to determine the role of these polymorphisms in the transcriptional regulation of the ACSM5 gene.
trans-eQTL identified
Table 2 summarizes all the relevant lipid-related genes mapped in the trans-eQTL regions associated with the genes analysed.
Table 2. Trans-eQTL annotation.
| Gene | Chromosome | Candidate genes within the eQTL |
|---|---|---|
| ACSM5 | SSC3 | CALM1, PRKCE |
| SSC10 | COG7, GGA2, NDUFAB1 | |
| FOS | SSC11 | ALOX5AP, SPG20, STARD13 |
| PPARA | SSC2 | ARHGAP26, FGF1, NR3C1 |
| SSC6 | CIDEA, IMPA2, MC2R, MC5R, MPPE1, PPT | |
| PIK3R1 | SSC6 | DAB1, DHCR24, LRP8, PCSK9, PPAP2B, PRKAA2 |
| PLA2G12A | SSC6 | MECR, SESN2, WWOX |
| SSC8 | ADH4, ADH5, ADH7, HPDGS, MTTP | |
| SSC9 | PIK3CG, DLD | |
| IGF2 | SSC2 | SIRT3 |
ACSM5 gene expression was also associated in trans with two chromosomal regions on SSC3 and SSC10. The most associated SNP on SSC10 was an intronic polymorphism (ASGA0090778; p-value = 2.55 × 10−8) in the Component of Oligomeric Golgi Complex 7 (COG7) gene (Supplementary Table S2). Although the COG7 gene has not been described to have a direct relationship with lipid metabolism function, it is reported that members of the COG complex are involved in intra-golgi trafficking and glycosylation of proteins and lipids30. Other lipid-related genes were identified within this trans-eQTL on SSC10 such as the NADH Dehydrogenase (Ubiquinone) 1, Alpha/Beta Subcomplex, 1, 8 kDa (NDUFAB1) gene and the Golgi-associated, Gamma Adaptin Ear Containing, ARF Binding Protein 2 (GGA2) gene (Supplementary Table S3). Within the second trans-eQTL for ACSM5 at 100.35 Mb on SSC3, two genes that may affect lipid metabolism were identified: Protein Kinase C, Epsilon (PRKCE) and Calmodulin 1 (Phosphorylase Kinase, Delta) (CALM1) (Supplementary Table S3).
For FBJ Murine Osteosarcoma Viral Oncogene Homolog (FOS) gene expression, three lipid-related genes in a SSC11 trans-eQTL were identified: StAR-Related Lipid Transfer (START) Domain Containing 13 (STARD13), Spastic Paraplegia 20 (Troyer Syndrome) (SPG20), and Arachidonate 5-Lipoxygenase-Activating Protein (ALOX5AP) (Supplementary Table S3). The StAR gene family encodes for globular proteins that form cavities where lipids and lipid hormones bind to be exchanged between biological membranes31. Supporting STARD13 as a regulator of FOS gene expression, transgenic mice with pancreas STARD13 ablation showed no detectable mRNA expression of the FOS gene32.
Two regions on SSC2 and SSC6 were associated in trans with the Peroxisome Proliferator-Activated Receptor Alpha (PPARA) gene expression. In SSC2, several genes related to lipid metabolism were identified: Rho GTPase Activating Protein 26 (ARHGAP26), Fibroblast Growth Factor 1 (Acidic) (FGF1) and Nuclear Receptor Subfamily 3, Group C, Member 1 (glucocorticoid receptor) (NR3C1) (Supplementary Table S3). ARHGAP26 is activated via lipid interaction24 and may play a role in adipogenesis, as well as the transcription factor NR3C1, which maps at approximately 0.5 Mb of the ARHGAP26 gene. The FGF1 gene has been identified as being differentially expressed in animals phenotypically extreme for FA composition in muscle16. Being noteworthy, FGF1 is involved in preadipocyte differentiation and has been suggested to be acting in the PPARG system, however, the mechanisms remain unclear33. In accordance with this hypothesis, it is described that FGF1 can be trans-located to the nucleus, exerting a growth regulatory activity34. Hence, it would be important to study the FGF1-PPARA relationship. In the SSC6, PPARA trans-eQTL mapped the Palmitoyl-Protein Thioesterase 1 (PPT1) gene, Metallophosphoesterase 1 (MPPE1), Inositol(Myo)-1(or 4)-Monophosphatase 2 (IMPA2), and Cell Death-Inducing DFFA-Like Effector A (CIDEA). Interestingly, CIDEA-null mice showed a decreased PPARA, PPARG and SREBF1 gene expression and a decreased de novo FA synthesis in the liver35. Furthermore, the CIDEA-PPARA interaction identified in the eQTL analysis was also captured by Genomatix literature-based analysis (Supplementary Fig. S1). In addition, two melanocortin receptor genes (MC2R and MC5R) mapped in the PPARA SSC6 trans-eQTL. In adipocyte cells of MC2R knockdown mice alterations in fatty acid composition were observed: a reduction in the C16:1/C16:0 and C18:1/C18:0 ratios and an increase in the arachidonic acid content36. On the other hand, MC5R has been associated with obesity phenotypes such as body mass index and fat mass in humans37.
The Phosphoinositide-3-Kinase, Regulatory Subunit 1 Alpha (PIK3R1) eGWAS revealed a trans-eQTL on SSC6 at 145.60 Mb, where several interesting genes involved in lipid metabolism were mapped: Low Density Lipoprotein Receptor-Related Protein 8, Apolipoprotein E Receptor (LRP8), 24-Dehydrocholesterol Reductase (DHCR24), Dab Reelin Signal Transducer Homolog 1 (DAB1), Protein Kinase, AMP-Activated, Alpha 2 Catalytic Subunit (PRKAA2), Phosphatidic Acid Phosphatase Type 2B (PPAP2B), and Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) (Supplementary Table S3). Interestingly, the PRKAA2 gene identified within the SSC6 trans-eQTL for PIK3R1 is a catalytic subunit of the AMP-Activated Protein Kinase (AMPK) which is in charge of regulating lipid synthesis by phosphorylating lipid metabolic enzymes such as ACACA, ACACB, ACC, GYS1, HMGCR, HSL and LIPE to inactivate them. Therefore, it acts regulating key enzymes of fatty acid uptake, esterification, lipolysis and oxidation38. In the same way, PRKAA2 knockdown affects Akt activation. Therefore, PIK3R1 and PRKAA2 are both associated with the PI3K-Akt signalling pathway. Supporting these results, recently published studies suggest that AMPK activates Akt via regulating PI3K39. These results highlight PRKAA2 as a strong candidate gene to explain the variation in the mRNA levels of PIK3R1.
Four chromosomal regions (two regions on SSC6, one region on SSC8, and one on SSC9) were associated in trans with the Phospholipase A2, Group XIIA (PLA2G12A) gene expression. Specifically, in the SSC8 eQTL, at approximately 128 to 136 Mb, the Microsomal Triglyceride Transfer Protein (MTTP), the Hematopoietic Prostaglandin D Synthase (HPGDS), and the Alcohol Dehydrogenases 4, 5 and 7 (ADH7, ADH4, and ADH5) genes involved in lipid metabolism (Supplementary Table S3) were mapped. Interestingly, a polymorphism in the porcine MTTP gene has been associated with the lipid transfer activity of the MTTP protein and with the fatty acid composition of fat40. ADH5 was the most expressed gene in pig muscle; meanwhile, the other ADH members (ADH7 and ADH4), jointly with MTTP and HPGDS genes, showed very low expression levels in muscle16, which is in concordance with what was reported in human muscle23. Two potential regulators of PLA2G12A were identified within the trans-eQTL at the 79.9 Mb position on SSC6: Mitochondrial Trans-2-Enoyl-CoA Reductase (MECR) and Sestrin 2 (SESN2) (Supplementary Table S3). Finally, only one lipid-related gene was identified within the second trans-eQTL at 9.7 Mb, the WW Domain Containing Oxidoreductase (WWOX) gene, which has recently been described to play a role in cholesterol homeostasis and triglyceride biosynthesis41.
On SSC9, an eQTL at around 117 Mb affected the expression of genes PLA2G12A and the Hypoxia Inducible Factor 1, Alpha Subunit Inhibitor (HIF1AN) (Table 1). Their mRNA expression was highly correlated (r = 0.60; p-value = 1.98 × 10−12), suggesting a common element regulating their transcriptional levels. The three most significant SNPs (H3GA0028012, ASGA0044215, ALGA0117195; p-value = 4.48 × 10−6) within this eQTL were in complete linkage disequilibrium (R2 = 1) (Table 1). In this region PIK3CG was mapped, which encodes a Phosphatidylinositol-4,5-Bisphosphate 3-Kinase which is known to participate in different functions, including the insulin-signalling pathway and lipid metabolism42, and the DLD gene which encodes a Dihydrolipoamide Dehydrogenase involved in acetyl-coA biosynthesis (Supplementary Table S3). The PIK3CG gene, unlike other PI3K family members, is activated by interaction with G-protein-coupled receptors and by silencing this gene the PI3K-Akt signalling pathway is inhibited43. Accordingly, studies in cell lines have suggested that HIF1AN gene expression is repressed by a mechanism involving PI3K signalling44. Altogether, these results suggest that PRKAA2 may regulate the Class I PI3K Regulatory Subunit 1 (PIK3R1), which is able to form a heterodimer with the PIK3CG catalytic subunit, activate the Akt pathway and inhibit HIF1AN gene expression.
For IGF2 gene expression, apart from the SSC2 cis-eQTL, a trans eQTL located approximately at 162 Mb in the same chromosome was identified, and the Sirtuin 3 (SIRT3) gene mapped in this region (Supplementary Table S3). It has been described that SIRT3 knockout mice exhibited decreased oxygen consumption and increased oxidative stress in skeletal muscle45. Moreover, SIRT3 knockout mice showed a down-regulation of Akt phosphorylation45 (Supplementary Fig. S2). Members of the SIRT gene family have been described as being relevant genes controlling lipolysis and promoting fat mobilization in white adipose tissue46, and the SIRT1 member has been identified as one of the most central genes in a liver co-association network for intramuscular FA composition in IBMAP animal material18. Recently, the SIRT3 member, which can be activated by the AMPK protein, has been suggested as playing a major role in obesity-related diseases47.
Finally, within the trans-eQTLs identified for CROT, FABP3, MGLL, and NCOA1 gene expression, no strong candidate gene exerting a known lipid metabolism function could be detected.
Functional network analysis of genes mapping in eQTLs
For trans-eQTLs, all genes located within a ± 1 Mb interval were selected for gene annotation. Conversely, for cis-eQTLs, only the candidate gene studied was considered (ACSM5, IGF2, and MGLL) for further analyses. In the 18 eQTLs, a total of 292 protein-coding genes, 13 miRNA, one miscRNA, six pseudogenes, one rRNA, eleven snoRNAs and four snRNAs were annotated (Supplementary Table S4). From the 292 protein-coding genes with Ensembl Gene ID, 256 had at least one human orthologous gene and were submitted to IPA to perform a functional categorization (Supplementary Table S4).
The main networks identified by IPA analysis were: (i) energy production, small molecule biochemistry and drug metabolism (score = 44); (ii) organismal injury and abnormalities, cancer and haematological disease (score = 42); and (iii) connective tissue disorders, inflammatory disease, skeletal and muscular disorders (score = 37) (Supplementary Table S4).
Focusing on the first network comprising energy production, small molecule biochemistry and drug metabolism functions, we identified the Serine/Threonine Kinase Effector (Akt) complex as central in the network (Supplementary Fig. S2). Remarkably, the Akt complex, which is involved in glucose transport and lipogenesis, was also identified in the muscle transcriptome study of 12 BC1_LD animals which were extreme for intramuscular FA composition16. In agreement with these results, several genes (PIK3CG, PPAP2B, PRKAA2, PTPN2, and SIRT3), identified as potential regulators within the eQTLs of the 45 lipid-related genes, are strongly related to the Akt pathway.
Identification of master regulators
A total of 298 genes, including (1) the 253 genes annotated in the trans-eQTL intervals and, (2) the 45 genes studied of the present study were analysed with iRegulon Cytoscape plugin48. We observed that the EP300 gene was the most enriched transcription factor motif (enrichment score threshold for the motif, NES = 4.737). What was noteworthy was the EP300 gene was identified as a key regulator of FA composition and IMF traits in the same material using a gene co-association network18. On the other hand, five of the 45 genes studied were identified as strong regulators of several genes analysed with iRegulon: NR1H3 (NES = 5.348; 10 target genes), MLXIPL (NES = 4.988; 29 target genes), PPARA (NES = 4.785; 32 target genes), NFKB1 (NES = 3.855; 6 target genes) and PPARG (NES = 3.536; 10 target genes). The PPARA motif was present in the highest number of target genes (32 out of 45 genes). Interestingly, the NR3C1 gene identified within the PPARA trans-eQTL on SSC2 was also identified with iRegulon among the regulators for the 45 genes analysed and MatInspector (Genomatix software) predicted a binding site for this transcription factor in the promoter of PPARA. The NR3C1 gene may play a negative role in adipogenesis, regulating the lipolytic and anti-lipogenic gene expression. In human studies, polymorphisms in the NR3C1 gene have been suggested as contributing to obesity49. Afterwards, by using the Genomatix interface, it was observed that NR3C1 interacts with NFKB, CREB, NCOA1 and NCOA2 genes, and may be affected by the Corticotropin-Releasing Hormone (CRH) and Insulin (INS) (Supplementary Fig. S3). Accordingly, CRH was identified in both network analysis for fatness and growth traits17 and the insulin-signalling pathway in a RNA-Seq study comparing animals extreme for their intramuscular FA composition16, both studies performed with IBMAP animal material. Thus, we hypothesize that NR3C1 may be a master regulator of lipid metabolism through the regulation of PPARA gene expression. However, further studies are necessary to corroborate this hypothesis.
QTL and eQTL co-localization
Transcriptomic data can be used to identify candidate genes underlying QTLs through the co-localization with eQTLs. Thus, our group looked for the overlapping of the 241 SNPs with the QTLs described in PigQTLDB50. A total of 234 SNPs (97%) co-localized in 132 QTLs for fatness traits and 157 SNPs (65%) within 10 different QTLs for FA composition (Supplementary Table S5), confirming a high co-localization of eQTLs and fat-related QTLs. The large number of QTLs described for these traits, covering a large extension of the porcine genome, provides evidence for a complex genetic pleiotropic regulation basis of these traits.
Notably, five genes identified within the eQTLs (i.e. ARHGAP6, IGF2, MC2R, MGLL, NR3C1) overlapped with QTLs described in the IBMAP population. For instance, the IGF2 gene, for which a cis-eQTL was detected, was identified within a confidence interval of one of the epistatic regions affecting muscle fibre traits51. The MGLL gene, showing a cis-acting eQTL, was located close to a GWAS interval affecting FA composition traits: palmitic (C16:0) and oleic (C18:1(n-9)) FAs, the polyunsaturated/saturated ratio (PUFA/SFA ratio), SFA, and unsaturated index (UI)12. Furthermore, MGLL maps within a QTL interval affecting growth11. The MC2R gene, identified in a PPARA trans-eQTL, maps within an IBMAP QTL region for IMF content and backfat thickness52. Moreover, ARHGAP6 and NR3C1 genes also identified in a trans-eQTL for PPARA gene expression are located within a QTL for growth traits in the IBMAP population11.
Conclusions
In the present study, we provide a list of potential candidate genes and genetic variants that may be regulating the transcriptional level of relevant lipid-related metabolism genes. Combined assessment of the results obtained from eGWAS and GWAS may provide complementary information of genes and variants determining the IMF and FA composition traits. Therefore, the present results identify potentially key genes and variants affecting pork-meat quality. However, more efforts should be made to validate our results, for instance, the involvement of the NR3C1 gene as a major regulator in muscle FA metabolism.
Methods
Animal samples and phenotypes
The IBMAP population was obtained by crossing three Guadyerbas Iberian boars with 31 Landrace sows6. In the present study, 114 animals were used (65 females and 49 males) belonging to the BC1_LD generation of the IBMAP population obtained by crossing five F1 boars with 26 Landrace sows. Animals were fed ad libitum with a cereal-based commercial diet and slaughtered at 179.8 days ±2.6 days. Animal care and procedures were performed following national and institutional guidelines for the Good Experimental Practices and approved by the Ethical Committee of the Institution (IRTA- Institut de Recerca i Tecnologia Agroalimentàries). Samples of the Longissimus dorsi muscle were collected, snap-frozen in liquid nitrogen and stored at −80 °C until further RNA isolation. Genomic DNA was extracted from blood samples according to the phenol-chloroform method53.
Selection of lipid-related metabolism genes in muscle
In previous studies of our group, strong candidate genes affecting IMFA content and FA composition of the Longissimus dorsi muscle of the IBMAP BC1_LD, 25% Iberian and 75% Landrace, were identified by using GWAS, RNA-Seq and co-association network approaches12,16,17,18. In the present study, a list of 45 genes functionally related with lipid metabolism was selected, prioritizing candidate genes for FA composition (Supplementary Table S6).
We included: (1) candidate genes differentially expressed (ACAA2, AQP7, ALB, ANGPT1, ATF3, MLXIPL, FOS, HIF1AN, PIK3R1, PLIN5, PPARG, SCD, SLC2A4) in the Longissimus dorsi muscle of two phenotypically extreme groups of animals for intramuscular FA composition from the IBMAP cross and their potential regulators (NFKB1, PPARGC1A)16; (2) candidate functional and positional genes (FABP5, PIK3R1, PLA2G12A, PPAP2A) identified in a GWAS study for intramuscular FA composition in the same animal material12; and (3) genes related to lipid metabolism identified in gene co-association networks for FA composition18 (ACSM5, ANGPT1, FABP3, FABP5, MGLL, NCOA2, PEX2, PPARG, SETD7) and fatness and growth- related traits17 (ALB, CREG1, ELF1, FABP5, MGLL, PPARG). Finally, in order to complete the set of genes, we chose genes which have been described in the literature to play different roles (fatty acid synthesis, transport, storage and oxidation) in muscle lipid metabolism, prioritizing those genes which are expressed in muscle, such as transcriptional factors, co-factors and nuclear receptors (ETS1, LPIN1, NR1H3, NCOA1, NCOA6, PPARA, PPARD, PRKAA1, RXRG, SP1, SREBF1)54 (freely available at http://www.bioguo.org/AnimalTFDB/), enzymes (ACSS1, ACSS2, CPT1B, CROT, DGAT1, DGAT2, PDHX), as well as the IGF2 gene, which has been described as the causal factor of the imprinted QTL for muscle growth and fat deposition in a Meishan × Large White intercross27 (Supplementary Table S6). Moreover, polymorphisms in the IGF2 gene have been significantly associated with muscle FA composition in swine55.
Genotyping
A total of 197 animals from the BC1_LD (160 backcrossed individuals and their respective parents) were genotyped with the Porcine SNP60 Beadchip (Illumina), following the Infinium HD Assay Ultra protocol (Illumina)56. Raw data were visualized with GenomeStudio software (Illumina) and trimmed for high genotyping quality (call rate >0.99). Markers with minor allele frequency (MAF) >5% and animals with missing genotypes <5% were retained. After the quality control filter, a subset of 40,586 SNPs and 197 animals remained.
Furthermore, the BC1_LD animals were genotyped with two SNPs within the ACSM5 (rs331702081) and IGF2 (IGF2-intron3-G3072A) genes using Taqman OpenArrayTM genotyping plates custom-designed in a QuantStudioTM 12K flex Real-Time PCR System (ThermoFisher Scientific) (Supplementary Table S7) and the pyrosequencing protocol previously described27, respectively.
Gene-expression profiling
Total RNA was isolated from the Longissimus dorsi muscle of 114 samples with RiboPure kit (Ambion; Austin, TX, USA). Total RNA was quantified in a NanoDrop ND-1000 spectrophotometer (NanoDrop Products; Wilmington, DE, USA). The RNA was converted to cDNA using the High-Capacity cDNA Reverse Transcription (Applied Biosystems). The cDNA samples were loaded into a Dynamic Array 48.48 chip in a BioMark system (Fluidigm; San Francisco, CA, USA) through a NanoFlex controller following a protocol previously described18. For this experiment, the expressed levels of 48 genes were analysed, and a total of 45 target genes were normalized using the two most stable housekeeping genes (ACTB and TBP). Primers used for the analyses are detailed in Supplementary Table S8. Data were collected using the Fluidigm Real-Time PCR analysis software 3.0.2 (Fluidigm) and analysed using the DAG expression software 1.0.4.1157, applying the relative standard curve method (See Applied Biosystems user bulletin #2). Analyses were performed using the normalized gene-expression levels of each sample and assay. The animals showing abnormal gene expression levels (outliers) were removed and data obtained were normalized if necessary by performing log2 transformation of the Normalized Quantity (NQ) value. The sex effect was also tested by using a linear model with R programme58.
Gene-expression association analysis
An eGWAS was also performed using the genotypes of BC1_LD animals and the expression values from muscle. A mixed model was employed implemented on Qxpak 5.059:
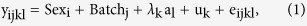 |
in which yijkl was the kth individual record, sex (two levels) and batch (five levels) were fixed effects, λk was a −1, 0, +1 indicator variable depending on the kth individual genotype for the lth SNP, al represents the additive effect associated with the lth SNP, uk is the infinitesimal genetic effect treated as random and distributed as N(0, Aσu2), where A is the numerator of the pedigree-based relationship matrix and eijkl the residual.
The same mixed model was applied to perform the association analyses with the ACSM5 (rs331702081) and IGF2 (IGF2-intron3-G3072A) polymorphisms and the ACSM5 and IGF2 mRNA expressions, respectively.
To correct for multiple testing, the false discovery rate (FDR) was calculated with the q-value library of the R package setting the threshold at q-value ≤ 0.0558,60. Given that cis-acting eQTLs tend to have larger magnitudes of effect on gene expression than do trans-acting eQTLs61, and several SNPs are affected by strong LD, a more restrictive threshold to avoid false positives was applied at p-value ≤ 0.001 for those eGWAS showing a cis-eQTL (see Q-Q plot in Supplementary Fig. S4).
The SNPs identified were classified as cis when they were located within 1 Mb from the gene analysed and as trans when they were located elsewhere in the genome. The number of significant SNPs belonging to the same interval was considered among associated SNPs less than 10 Mb apart.
Gene annotation and functional analysis
The significantly associated SNPs were mapped in the Sscrofa10.2 assembly and were annotated with the Ensembl Genes 80 Database using VEP software62. The genomic eQTL intervals considering ±1 Mb around the candidate chromosomal regions were annotated using BioMart software63.
The Core Analysis function included in the Ingenuity Pathway Analysis software (IPA 4.0, Ingenuity Systems Inc., http://www.ingenuity.com; Accessed 8 July 2015) and the Genomatix Pathway System (GePS) (Release 2.8.0) in the Genomatix software suite (Genomatix Software GmbH; Munich, Germany; available at: http://www.genomatix.de; accessed 8 October 2015) were used to perform the functional analysis of genes mapped in the 18 eQTLs regions. Specifically, the IPA software was used for data interpretation in the context of biological processes, pathways, networks and upstream regulators. All information generated in this software is derived from the Ingenuity Pathway Knowledge Base (IPKB), which is based on functions and interactions of genes published in the literature. Genomatix was used to retrieve additional information of gene functions, interactions and upstream regulators based on the literature. Furthermore, information from Mouse Genome Database64 and Genecards65 was used to identify gene functions affecting the phenotypes analysed. For the lipid-related genes, the Gene Expression Atlas23 was used to determine whether they were expressed in muscle or not.
Finally, an in-silico identification of transcription-factor binding sites in the promoter region of the 256 annotated genes was performed. For this analysis, iRegulon48 was used, which relies on the analysis of the motif enrichment for a transcription factor in the gene set using databases of nearly 10,000 TF motifs and 1,000 ChIP-seq data sets or “tracks”.
Correlation of gene expression and phenotypes
Correlations were performed among gene expression of the 45 genes to explore the relationship between genes. Furthermore, pairwise correlations among gene expression and FA composition percentages in muscle12 were carried out to explore the relationships between gene expression and phenotypes. All values were normalized applying the log2 of raw data if necessary. Afterwards, gene expression was corrected by sex (two levels) and batch (five levels) effects, whereas FA composition percentages were corrected by sex, batch and carcass weight. The remaining residuals of the phenotypes and gene-expression values corrected for the corresponding effects were used to obtain the pairwise correlations. The hierarchical clustering option of PermutMatrix software66 was used to visualize the results of both traits and genes.
Additional Information
How to cite this article: Puig-Oliveras, A. et al. Expression-based GWAS identifies variants, gene interactions and key regulators affecting intramuscular fatty acid content and composition in porcine meat. Sci. Rep. 6, 31803; doi: 10.1038/srep31803 (2016).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge J.L. Noguera (Institut de Recerca i Tecnologia Agroalimentàries; IRTA) for the animal material contribution. This work was funded by the Ministerio de Economía y Competitividad (MINECO AGL2011-29821-C02 and AGL2014-56369-C2). A. Puig-Oliveras was funded by a “Personal Investigador en Formació” (PIF) grant from Universitat Autònoma de Barcelona (458-01-1/2011). M. Revilla was funded by a Formació i Contractació de Personal Investigador Novell (FI-DGR) Ph.D grant from the Generalitat de Catalunya (ECO/1639/2013). M. Ballester is financially supported by a Ramon y Cajal contract (RYC-2013-12573) from the Spanish Ministry of Economy and Competitiveness.
Footnotes
Author Contributions J.M.F., M.B. and A.I.F. conceived of and designed the experiment. J.M.F. was the principal investigator of the project. A.I.F. and J.M.F. collected the samples. M.B., A.P.-O. and J.M.F. chose genes, M.B. and A.C. designed and tested the efficiency of the primers, and M.B. carried out the Real-Time PCR analyses. M.B. and M.R. analysed the data. M.R. and A.P.-O. performed the eGWAS. M.B. carried out the functional analysis. A.P.-O., M.B. and J.M.F. drafted the manuscript. All authors read and approved the final manuscript.
References
- Schwab C. R., Baas T. J., Stalder K. J. & Mabry J. W. Effect on long-term selection for leanness on meat and eating quality traits in Duroc swine. J. Anim. Sci. 84, 1577–1583 (2006). [DOI] [PubMed] [Google Scholar]
- Wood J. D. et al. Fat deposition, fatty acid composition and meat quality: A review. Meat Sci. 78, 343–358 (2008). [DOI] [PubMed] [Google Scholar]
- Michas G., Micha R. & Zampelas A. Dietary fats and cardiovascular disease: putting together the pieces of a complicated puzzle. Atherosclerosis 234, 320–328 (2014). [DOI] [PubMed] [Google Scholar]
- Estévez M., Morcuende D. & Cava R. Physico-chemical characteristics of M. Longissimus dorsi from three lines of free-range reared Iberian pigs slaughtered at 90 kg live-weight and commercial pigs: a comparative study. Meat Sci. 64, 499–506 (2003). [DOI] [PubMed] [Google Scholar]
- Serra X. et al. A comparison of carcass meat quality and histochemical characteristics of Iberian (Guadyerbas line) and Landrace pigs. Livest. Prod. Sci. 56, 215–223 (1998). [Google Scholar]
- Pérez-Enciso M. et al. A QTL on pig chromosome 4 affects fatty acid metabolism: evidence from an Iberian by Landrace intercross. J. Anim. Sci. 78, 2525–2531 (2000). [DOI] [PubMed] [Google Scholar]
- Óvilo C. et al. Test for positional candidate genes for body composition on pig chromosome 6. Genet. Sel. Evol. 34, 465–479 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clop A. et al. Detection of QTL affecting fatty acid composition in pig. Mamm. Genome 14, 650–656 (2003). [DOI] [PubMed] [Google Scholar]
- Mercadé A. et al. On growth fatness and form: A further look at porcine Chromosome 4 in an Iberian×Landrace cross. Mamm. Genome 16, 374–382 (2005). [DOI] [PubMed] [Google Scholar]
- Muñoz M. et al. Survey of SSC12 regions affecting fatty acid composition of intramuscular fat using high-density SNP data. Front. Genet. 2, 101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández A. I. et al. Genome-wide linkage analysis of QTL for growth and body composition employing the Porcine SNP60 BeadChip. BMC genetics 13, 41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramayo-Caldas Y. et al. Genome-wide association study for intramuscular fatty acid composition in an Iberian×Landrace cross. J. Anim. Sci. 90, 2883–2893 (2012). [DOI] [PubMed] [Google Scholar]
- Muñoz M. et al. Genome-wide analysis of porcine backfat and intramuscular fat fatty acid composition using high-density genotyping and expression data. BMC Genomics 14, 845 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao D. C. An overview of the genetic dissection of complex traits. Adv.Genet. 60, 3–34 (2008). [DOI] [PubMed] [Google Scholar]
- Gilad Y., Rifkin S. A. & Pritchard J. K. Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. 24, 408–415 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Oliveras A. et al. Differences in muscle transcriptome among pigs phenotypically extreme for fatty acid composition. Plos One 9, e103668 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Oliveras A. et al. A co-association network analysis of the genetic determination of pig conformation growth and fatness. Plos One 9, e114862 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramayo-Caldas Y. et al. From SNP co-association to RNA co-expression: Novel insights into gene networks for intramuscular fatty acid composition in porcine. BMC Genomics 15, 232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. et al. Skeletal muscle gene expression in response to resistance exercise: sex specific regulation. BMC Genomics 11, 659 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. Transcriptional profiling of human liver identifies sex-biased genes associated with polygenic dyslipidemia and coronary artery disease. Plos One 6, e23406 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M. H., Maher A. C., Hamadeh J. M., Ye C. & Tarnopolsky M. A. Exercise, sex, menstrual cycle phase, and 17β-estradiol influence metabolism-related genes in human skeletal muscle. Physiol. Genomics 40, 34–47 (2009). [DOI] [PubMed] [Google Scholar]
- Rune A. et al. Evidence against a sexual dimorphism in glucose and fatty acid metabolism in skeletal muscle cultures from age-matched men and post-menopausal women. Acta Physiol. 197, 207–215 (2009). [DOI] [PubMed] [Google Scholar]
- Kapushesky M. et al. Gene expression atlas at the European Bioinformatics Institute. Nucleic Acids Res. 38, D690–D698 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlmann P. et al. DLC1 activation requires interaction through a polybasic region preceding the RhoGAP domain. Mol. Biol. Cell. 15, 4400–4411 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin C. J. et al. Strong signatures of selection in the domestic pig genome. PNAS 109, 19529–19536 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanesi L. et al. The insulin-like growth factor 2 (IGF2) gene intron3-g.3072G >A polymorphism is not the only Sus scrofa chromosome 2p mutation affecting meat production and carcass traits in pigs: evidence from the effects of a cathepsin D (CTSD) gene polymorphism. J. Anim. Sci. 88, 2235–2245 (2010). [DOI] [PubMed] [Google Scholar]
- Van Laere A. S. et al. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 425, 832–836 (2003). [DOI] [PubMed] [Google Scholar]
- Estellé J. et al. Effect of the porcine IGF2-intron3-G3072A substitution in an outbred Large White population and in an Iberian × Landrace cross. J. Anim. Sci. 83, 2723–2728 (2005). [DOI] [PubMed] [Google Scholar]
- Harmon G. S. et al. Pharmacological correction of a defect in PPAR-γ signaling ameliorates disease severity in Cftr-deficient mice. Nat. Med. 16, 313–318 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. D. & Lupashin V. V. Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydr. Res. 343, 2024–2031 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A. G. et al. Comparative structural analysis of lipid binding START domains. Plos One 6, e19521 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold K. M., Naumann H. & Spagnoli F. M. Rho signalling restriction by the Rho GAP Stard13 integrates growth and morphogenesis in the pancreas. Development 140, 126–135 (2013). [DOI] [PubMed] [Google Scholar]
- Hutley L. et al. Fibroblast Growth Factor 1 a key regulator of human adipogenesis. Diabetes 53, 3097–3106 (2004). [DOI] [PubMed] [Google Scholar]
- Zhen Y. et al. Nuclear import of exogenous FGF1 requires the ER-Protein LRRC59 and the importins Kpnα1 and Kpnβ1. Traffic 13, 650–664 (2012). [DOI] [PubMed] [Google Scholar]
- Zhou L. et al. Cidea promotes hepatic steatosis by sensing dietary fatty acids. Hepatology 65, 96–107 (2012). [DOI] [PubMed] [Google Scholar]
- Betz M. J. et al. Mc2 receptor knockdown modulates differentiation and lipid composition in adipocytes. Horm. Metab. Res. 44, 670–675 (2012). [DOI] [PubMed] [Google Scholar]
- Chagnon Y. C. et al. Linkage and association studies between the melanocortin receptors 4 and 5 genes and obesity-related phenotypes in the Quebec Family Study. Mol. Med. 3, 663–673 (1997). [PMC free article] [PubMed] [Google Scholar]
- O’Neill H. M., Holloway G. P. & Steinberg G. R. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: Implications for obesity. Mol. Cell. Endocrinol. 366, 135–151 (2013). [DOI] [PubMed] [Google Scholar]
- Tao R. et al. AMPK exerts dual regulatory effects on the PI3K pathway. J. Mol. Signal. 5, 1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estellé J. et al. A non-synonymous mutation in a conserved site of the MTTP gene is strongly associated with protein activity and fatty acid profile in pigs. Anim. Genet. 40, 813–820 (2009). [DOI] [PubMed] [Google Scholar]
- Iatan I. et al. The WWOX gene modulates high-density lipoprotein and lipid metabolism. Circ. Cardiovasc. Genet. 7, 491–504 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N. et al. Blockade of class IB phosphoinositide-3 kinase ameliorates obesity-induced inflammation and insulin resistance. PNAS 108, 5753–5758 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba S. et al. Down-regulation of PIK3CG, a catalytic subunit of phosphatidylinositol 3-OH kinase, by CpG hypermethylation in human colorectal carcinoma. Clin. Cancer Res. 8, 2824–3831 (2002). [PubMed] [Google Scholar]
- Datta K. et al. Protein kinase C ζ transactivates hypoxia-inducible factor a by promoting its association with p300 in renal cancer. Cancer Res. 64, 456–462 (2004). [DOI] [PubMed] [Google Scholar]
- Jing E. et al. Sirtuin-3 (SIRT3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. PNAS 108, 14608–14613 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug T. T. & Li X. Sirtuin 1 in lipid metabolism and obesity. Ann. Med. 43, 198–211 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsom S. A., Boyle K. E. & Friedman J. Sirtuin 3: A major control point for obesity-related metabolic diseases? Drug Discov. Today Dis. Mech. 10, e35–e40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janky R. et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 10, e1003731 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson M. G., Redfern C. P. F., Unwin N. & Weaver J. U. The N363S polymorphism of the glucocorticoid receptor: potential contribution to central obesity in men and lack of association with other risk factors of coronary heart disease and diabetes mellitus. J. Clin. Endocrinol. Metab. 86, 2270–2274 (2001). [DOI] [PubMed] [Google Scholar]
- Hu Z.-L., Park C. A., Wu X.-L. & Reecy J. M. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 41, D871–D879 (2013). Available at: http://www.animalgenome.org/cgi-bin/QTLdb/SS/index. (Accessed: 29 November 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estellé J. et al. A quantitative trait locus genome scan for porcine muscle fiber traits reveals overdominance and epistasis. J. Anim. Sci. 86, 3290–3299 (2008). [DOI] [PubMed] [Google Scholar]
- Óvilo C. et al. A QTL for intramuscular fat and backfat thickness is located on porcine chromosome 6. Mamm. Genome 11, 344–346 (2000). [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. E. & Maniatis T. In Molecular Cloning:A Laboratory Manual 2nd edn., Vol 3, E3–E4 (Cold Spring Harbor Laboratory Press, 1989). [Google Scholar]
- Zhang H. M. et al. Animal TFDB: a comprehensive animal transcription factor database. Nucl. Acids Res. 40, D144–D149 (2012). Available at: http://www.bioguo.org/AnimalTFDB/. (Accessed: 8 July 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J. et al. Effects of genetic variants for the swine FABP3, HMGA1, MC4R, IGF2, and FABP4 genes on fatty acid composition. Meat Sci. 110, 46–51 (2015). [DOI] [PubMed] [Google Scholar]
- Ramos A. M. et al. Design of a high-density SNP genotyping assay in the pig using SNPs identified and characterized by next-generation sequencing technology. Plos One 4, e6524 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester M., Cordón R. & Folch J. M. DAG Expression: high-throughput gene expression analysis on real-time PCR data using standard curves for relative quantification. Plos One 8, e80385 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R. & Gentleman R. R: A language for data analysis and graphics. J. Comp. Graph. Stat. 5, 299–314 (1996). [Google Scholar]
- Pérez-Enciso M. & Misztal I. Qxpak.5: Old mixed model solutions for new genomics problems. BMC Bioinformatics 12, 202 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J. D. & Tibshirani R. Statistical significance for genomewide studies. PNAS 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt E. E. et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat. Genet. 37, 710–717 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010). Available at: http://www.ensembl.org. (Accessed: 7 July 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D. et al. The BioMart community portal. An innovative alternative to large, centralized data repositories. Nucl. Acids Res. 43, W589–W598 (2015). Available at: http://www.biomart.org. (Accessed: 7 July 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppig J. T., Blake J. A., Bult C. J., Kadin J. A., Richardson J. E. & The Mouse Genome Database Group. The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucl. Acids Res. 43, D726–D736 (2015). Available at: http://www.informatics.jax.org. (Accessed 8 October 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safran M. et al. GeneCards 2002: towards a complete, object-oriented, human gene compendium. Bioinformatics 18, 11 (2002). Available at: http://www.genecards.org. (Accessed 15 October 2015). [DOI] [PubMed] [Google Scholar]
- Caraux G. & Pinloche S. PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 21, 1280–1281 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.