

Abstract
Dissolved organic nitrogen (DON) supports a significant amount of heterotrophic production in the ocean. Yet, to date, the identity and diversity of microbial groups that transform DON are not well understood. To better understand the organisms responsible for transforming high molecular weight (HMW)-DON in the upper ocean, isotopically labeled protein extract from Micromonas pusilla, a eukaryotic member of the resident phytoplankton community, was added as substrate to euphotic zone water from the central California Current system. Carbon and nitrogen remineralization rates from the added proteins ranged from 0.002 to 0.35 μmol C l−1 per day and 0.03 to 0.27 nmol N l−1 per day. DNA stable-isotope probing (DNA-SIP) coupled with high-throughput sequencing of 16S rRNA genes linked the activity of 77 uncultivated free-living and particle-associated bacterial and archaeal taxa to the utilization of Micromonas protein extract. The high-throughput DNA-SIP method was sensitive in detecting isotopic assimilation by individual operational taxonomic units (OTUs), as substrate assimilation was observed after only 24 h. Many uncultivated free-living microbial taxa are newly implicated in the cycling of dissolved proteins affiliated with the Verrucomicrobia, Planctomycetes, Actinobacteria and Marine Group II (MGII) Euryarchaeota. In addition, a particle-associated community actively cycling DON was discovered, dominated by uncultivated organisms affiliated with MGII, Flavobacteria, Planctomycetes, Verrucomicrobia and Bdellovibrionaceae. The number of taxa assimilating protein correlated with genomic representation of TonB-dependent receptor (TBDR)-encoding genes, suggesting a possible role of TBDR in utilization of dissolved proteins by marine microbes. Our results significantly expand the known microbial diversity mediating the cycling of dissolved proteins in the ocean.
Introduction
Dissolved organic nitrogen (DON) is the second most abundant form of fixed nitrogen in the ocean, with a pool size of 7.7 (±2.3) × 104 Tg N (Karl et al., 2001; Bronk, 2002; Aluwihare and Meador, 2009). The total DON pool consists of numerous low and high molecular weight (LMW, HMW) compounds and compound classes of varying concentration and bioavailability including amino acids, N-acetyl amino polysaccharides (for example, chitin and peptidoglycan), dissolved proteins and uncharacterizable proteinaceous matter (Tanoue et al., 1995; McCarthy et al., 1997, 1998; Aluwihare et al., 2005). Microbial heterotrophy can control the flux and composition of DON (Carlson and Ducklow, 1995), yet the specific organisms responsible for DON transformations and their biochemical mechanisms are poorly understood (Aluwihare and Meador, 2009).
Phytoplankton are a major DON source (Nguyen and Harvey, 1997; Bronk and Steinberg, 2009). Release of DON results from a variety of forms of phytoplankton cell death including viral lysis (Fuhrman, 1999) and grazing (Worden et al., 2015), and can also be released via passive diffusion (Fogg, 1971). The liberated dissolved proteins and oligopeptides are initially regarded as labile HMW-DON compounds (Keil and Kirchman, 1993; Keil et al., 2000), but become more refractory because of processes such as abiotic complexation with existing dissolved organic matter (DOM) (Keil and Kirchman, 1994). Dissolved proteins are recycled faster relative to the bulk HMW-DON pool, as evidenced by their lower δ15N values (Meador et al., 2007) and more modern Δ14C values (Loh et al., 2004) relative to bulk HMW-DON.
Protein in dissolved and particulate fractions can be readily degraded in seawater by microbial activity (Sizemore and Stevenson, 1974; Smith et al., 1982), and turnover rates of proteins can be the same order of magnitude as turnover rates of dissolved free amino acids (DFAA) (Hollibaugh and Azam, 1983). LMW-DON such as DFAA released by protein hydrolysis (Billen and Fontigny, 1987) are readily assimilated by bacteria in aquatic systems (Hobbie et al., 1968) and can support up to ~50% of bacterial production in the oceans (Kirchman, 2000).
Studies linking some microbial groups to LMW-DON (Alderkamp et al., 2006; Nikrad et al., 2012; Liu et al., 2013) and HMW-DON (Nagata et al., 1998; Cottrell and Kirchman, 2000) uptake have demonstrated important spatiotemporal variability in DON utilization, but have been limited in their ability to understand the full diversity of bacteria and archaea involved in DON cycling by the need for a priori selection of target groups. Nevertheless, a relatively large number of bacterial taxa in the ocean have been implicated in HWM-DOM turnover, either through experimental additions or following phytoplankton blooms, and some overlap should be expected between these taxa and those involved in HWM-DON cycling. These include the Alteromonads (for example, McCarren et al., 2010; Sharma et al., 2014; Mayali et al., 2015), Flavobacteria (for example, Kirchman, 2002; Rinta-Kanto et al., 2012; Teeling et al., 2012; Sharma et al., 2014; Mayali et al., 2015) and the gammaproteobacterial clades NOR5/OM60, SAR92 (Teeling et al., 2012; Sharma et al., 2014) and SAR86 (Dupont et al., 2012). Bacterial groups specifically implicated in the cycling of HMW-DON and proteins include the Flavobacteria (Cottrell and Kirchman, 2000; Kirchman, 2002), the γ-Proteobacteria subgroups Arctic96B16, Ant4D3 and SAR86 (Nikrad et al., 2014), and the α-Proteobacteria clade SAR11 (Malmstrom et al., 2005).
The uncultivated Marine Group II Euryarchaeaota (MGII) are also predicted to have a role in DON transformations, based on the recovery of protein degradation pathways in metagenomic data (Iverson et al., 2012; Orsi et al., 2015). MGII are thought to be heterotrophs, through their utilization of LMW-DON (Alderkamp et al., 2006) and available metagenomic data (Frigaard et al., 2006; Zhang et al., 2015). However, direct evidence of their role in utilizing dissolved proteins has not been demonstrated, and interactions between archaea and HWM organic matter are largely unexplored. Several of the aforementioned groups, including the MGII, have been found in association with particulate organic matter (Delong et al., 1993), where they have an enhanced genomic capacity for HMW substrate utilization (Orsi et al., 2015).
We sought to determine whether microbial taxa previously implicated in HWM-DOM turnover might specifically be involved in HMW-DON (protein) cycling. We further hypothesized that MGII archaea are actively involved in protein turnover, and that this activity would be enhanced in association with particles (Orsi et al., 2015). To this end, we performed DNA stable-isotope probing (SIP) using isotopically labeled (13C and 15N) proteins from the picoeukaryotic phytoplankter Micromonas pusilla. We coupled SIP with high-throughput Illumina sequencing of 16S small subunit ribosomal RNA (rRNA) genes (Hungate et al., 2015; Morando and Capone, in review) in three different size fractions to identify protein assimilation by individual bacterial and archaeal operational taxonomic units (OTUs). We performed the incubations at a station within the coastal transition zone of the Central California Current, where there is a rich contextual background on biogeochemistry (for example, Collins et al., 2003), and where M. pusilla is a resident of the phytoplankton community (Thomsen and Buck, 1998). Free-living (0.2–0.8 μm) and particle-associated (>3 μm) fractions contained a diversity of newly implicated, uncultivated bacterial and archaeal taxa mediating the cycling of dissolved protein. Our results suggest that when dissolved proteins become available, highly diverse microbial populations with the genomic capacity to transport HMW-DON can acquire it rapidly. These results refine our understanding of DOM turnover in the ocean and indicate a strong affinity of several uncultivated clades for dissolved protein, and suggest that diverse particle-attached microbial communities can act as a DON sink.
Methods
Sampling
Seawater was collected aboard the R/V Western Flyer during Monterey Bay Aquarium Research Institute cruise CN13ID, 7–17 October 2013, along California Cooperative Fisheries Investigations (CalCOFI) Line 67 (Collins et al., 2003; Pennington et al., 2010). Sampling and hydrographic profiling were conducted using a conductivity-temperature-depth rosette sampler (Sea-Bird Electronics) equipped with (12) 10 l Niskin sample bottles. In addition to the standard conductivity-temperature-depth measurements, the profiling rosette was equipped with the following sensors: WETstar fluorometer (Wetlabs, Philomath, OR, USA), transmissometer (SeaTech, Bellevue, WA, USA), SBE43 dissolved oxygen sensor (Sea-Bird Electronics, Bellevue, WA, USA) and a radiometer (Biospherical, Inc., San Diego, CA, USA). Chlorophyll a (Chl a) pigment concentrations were determined by fluorometry using established protocols (Pennington and Chavez, 2000). Major phytoplankton groups (Prochlorococcus, Synechococcus and picoeukaryotic phytoplankton) were quantified at sea by flow cytometry as described previously (Cuvelier et al., 2010).
Stable isotope probing incubation
M. pusilla CCMP1545 was grown for 10 days at 21 °C under a light:dark cycle of 14 h:10 h using cool white light (150 μmol Q m−2 s−1) in L1 medium (Guillard and Hargraves, 1993) based on artificial seawater amended with 2.5 mM 99 atm% 13C-labeled sodium bicarbonate and 882 μM 98 atm% 15N-labeled sodium nitrate (Cambridge Isotope Laboratories, Andover, MA, USA). The growth rate was 0.47 per day (s.d.: 0.09). Isotopic enrichment of the cells was obtained by mixing the culture in unlabeled media 1:1 with labeled medium, incubating for 2 days, followed by another 1:1 mixture with labeled media. This was repeated five times. The isotopic composition of whole cells was measured at the UC Davis Stable Isotope Facility using online combustion coupled to a mass spectrometer; values were 16 atm% for 13C and 42 atm% for 15N. It should be noted, however, that particulate C isotopic composition was determined for cells that had been in stationary phase for several days relative to the cells harvested for protein extraction, potentially leading to an underestimation of the starting atm% 13C.
Proteins from M. pusilla were extracted according to the method by Santoro et al., 2015. In brief, cell pellets were incubated at room temperature for 15 min in an extraction buffer, followed by heating at 95 °C for 10 min and shaking at 350 r.p.m. for 1 h. Protein extract was removed into a new sterile tube and centrifuged for 30 min at 4000 r.p.m. at room temperature, followed by filtration through a 5-μm pore size sterile filter. Filtered samples were then concentrated using 6 ml 5000 MWCO Vivaspin columns (Sartorius, Göttingen, Germany) down to 400 μl, split in half, and precipitated with 1.6 ml of cold (4 °C) solution of 1:1 MeOH and acetone, and 0.5 mM HCl for 3 days at −20 °C. Precipitated samples were spun down for 30 min at 4 °C at 14 500 r.p.m., air dried in a laminar flow hood and resuspended in 1 ml extraction buffer prior to quantification. Protein extracts were quantified fluorometrically. No DNA was measureable in the extracts using fluorometric measurements (detection limit=2 ng μl−1) suggesting that co-extraction of nucleic acids with the proteins was minor. However, we cannot exclude the possibility that a small amount of labeled nucleic acids were co-extracted with the proteins. There were no visible particles in the extracts suggesting that the protein was mostly dissolved.
Incubation experiments (SIP and remineralization rates) were conducted at CalCOFI station 67–70 (36.74 °N, 122.02 °W), approximately 170 km from shore, within the coastal transition zone between the coastal upwelling and oligotrophic waters offshore (Collins et al., 2003). The site was selected because of the historic presence of a significant picoeukaryote population M. pusilla (Thomsen and Buck, 1998), and previous characterization of particle-associated taxa (Orsi et al., 2015). Water for the SIP incubation was acquired from 20 m depth. M. pusilla protein extract was added to 4 l of seawater to a final concentration of 28 μg l−1 in trace metal-clean polycarbonate bottles. Bottles were amended with dual isotope-labeled proteins (13C and 15N), unlabeled proteins or no proteins, in triplicate. Bottles were incubated for 24 h in the dark in coolers that were halfway filled with surface seawater to maintain in situ temperature. The bottles were subsampled every 4 h (except for the 16 h timepoint) to determine the concentration of ammonium and DFAA, and the nitrogen isotopic composition of NO3−+NO2− (δ15NNOx). The carbon isotopic composition of dissolved inorganic carbon (DIC) was determined in samples taken at 24 h (see details below). At the end of the incubation 1–2 l of seawater was filtered sequentially through 3 μm (Pall Versapor-3000 T, Port Washington, NY, USA), 0.8 μm (Pall Supor-800, Port Washington, NY, USA) and 0.2 μm (Pall Supor-200, Port Washington, NY, USA) filters using a peristaltic pump, and DNA was extracted as previously described (Santoro et al., 2010). Catalyzed reporter deposition—fluorescence in situ hybridization for Archaea and Euryarchaeota, DAPI staining and epifluorescence microscopy followed the protocol of Orsi et al. (2015). This protocol utilized the Eury806 probe that is specific for Euryarchaeota (Tiera et al., 2004) and the Arch915 probe that targets the domain Archaea (Stahl and Amann, 1991), but also binds non-specifically to some pelagic marine bacteria (Pernthaler et al., 2002).
Ammonium, DFAA and DCAA measurements
Water column [NH4+] samples were collected in 125 ml acid-cleaned polyethylene bottles and incubation samples were collected in 27 ml acid-cleaned polyethylene vials and refrigerated. Samples were analyzed within hours using a fluorometric method with a single working reagent (orthophthaldialdehyde, sodium sulfite and sodium borate) (Kerouel and Aminot, 1997; Holmes et al., 1999). The system was automated with a peristaltic pump-based flow injection analysis. Detection limits, estimated as 1% of full scale, gave +/− 3 nM with the high sensitivity injection loop. An estimate of the precision from a cast where samples were analyzed from 12 bottles tripped at the same location gave 25 +/− 1 nM.
Depth profiles of DFAA and total dissolved amino acids (TDAA) were collected from the rosette into pre-combusted (450 °C, 5 h) glass scintillation vials after filtering through a sterile 0.2-μm pore size filter and analyzed as described below. DFAA were analyzed by high performance liquid chromatography (Shimadzu Prominence, Kyoto, Japan) with fluorescence detection after pre-column o-phthaldialdehyde derivatization (Lee et al., 2000). TDAA were analyzed in the same way as DFAA but after hydrolysis by 6 N HCl under nitrogen at 110 °C for 20 h (Kuznetsova and Lee, 2002). Dissolved combined amino acids (DCAA) were calculated as the difference between TDAA and DFAA. The coefficient of variation for replicate DFAA and DCAA samples was 10–20%. The concentrations of individual amino acids were calculated on the basis of an amino acid external standard mixture (Amino Acid Standard H, Thermo Scientific, Waltham, MA, USA).
In situ array
Water for measurements of 13C, 15N-labeled protein remineralization and 15NH4+ oxidation rates was collected from depths of 2, 5, 10, 20, 30 and 50 m using the conductivity-temperature-depth rosette sampling system described above. Upon return to deck, water from each depth was drained directly into clear, acid-cleaned, 280 ml polycarbonate bottles. Following addition of isotopically labeled substrates to a final concentration of 50 nmol l−1 and 28 μg l−1 for 15NH4+ and 13C, 15N-proteins, respectively, the incubation bottles were loaded into hand-made nylon mesh bags, hung at their depth of origin on a 6.3 mm double-braided line attached to a custom GPS-enabled Surface Temperature Experimental Longitude Latitude Asset (STELLA) drifter (http://www.mbari.org/bog/drifterdata/) with a 60 cm surface float, and deployed in situ from dusk to dawn (ca. 12 h). Following recovery of the array, samples were immediately filtered through combusted GF/F filters housed in Swinnex filter holders (EMD Millipore, Billerica, MA, USA), into acid-washed 60 ml HDPE bottles and kept frozen until analysis. The δ15NNOx was determined at the University of Connecticut Avery Point campus using the denitrifier method (Sigman et al., 2001) and a GasBench and PreCon trace gas concentration system interfaced to a Delta VPLUS mass spectrometer (ThermoFinnigan, Bremen, Germany). Nitrate isotope reference materials USGS32, USGS34 and USGS35 were analyzed in parallel to calibrate δ15N values. δ15N precision, determined by repeat analysis of the reference materials, was found to be 0.15‰ for the entire data set. Resultant δ15NNOx data were used to calculate rates of NH4+ oxidation or labeled protein remineralization using previously described methods (Smith et al., 2014) derived from the original equations laid out by Dugdale and Goering (1967).
Protein remineralization rates
Protein-derived C and N remineralization rates were determined in bottle incubations on the in situ array and the on-deck SIP experiment by measuring changes in stable-isotope ratios in the DIC (δ13C-DIC) and inorganic nitrogen (δ15NNOx) pools after 24 h. δ13C-DIC was measured in acidified samples using a GasBench II system interfaced to a Delta VPLUS isotope ratio mass spectrometer (ThermoFinnigan) at the UC Davis Stable Isotope Facility. δ15NNOx was measured and rates of remineralization calculated as described in the previous section. A range of initial isotopic enrichment values for the starting 13C atm% enrichment in the labeled proteins was used in the rate calculations, because of potential effects of the delay in measuring the starting 13C atm% of the M. pusilla cells (see above). We used a lower value of 16 atm% 13C based on the whole cell measurement and an upper value of 95 atm% 13C based on the calculated atm% 13C for the number of cell doublings that occurred assuming uptake of fully labeled medium. The starting ambient DCAA pool was assumed to have a δ13C of −20‰ (McCarthy et al., 2004) and δ15N of 8‰ (McCarthy et al., 2007). Limitations to this method include an incomplete characterization of both the isotopically labeled protein preparation and the ambient DCAA pool, and considerable uncertainties in the starting concentrations of both (Lipschultz, 2008).
Density gradient centrifugation and gradient fractionation
At the 24 h timepoint, one of the triplicate experimental (13C, 15N protein addition) and control (unlabeled protein addition) bottles (see above) was sampled for DNA-SIP. DNA was extracted from the experimental and control bottles after 24 h according to the method of Santoro et al. (2010). In brief, 850 μl of lysis buffer and 100 μl of 10% SDS were added to 2 ml gasketed bead-beating tubes containing the filters and 0.1 mm sterile glass beads (Biospec, Kyoto, Japan). Bead beating was performed for 1 min and samples heated for 2 min at 99 °C. After heating, 25 μl of 20 mg ml−1 proteinase K was added and tubes were incubated over night at 55 °C. DNA was then purified from the lysate with the DNeasy Blood and Tissue Kit (Qiagen, Venlo, Netherlands).
The protocol for density gradient centrifugation and gradient fractionation followed previously described methods for DNA-SIP (Neufeld et al., 2007; Dunford and Neufeld, 2010), with some minor modifications. In brief, density gradient centrifugation was performed in a TV90 vertical rotor at 20 °C for 40 h at 177 000 g in an Optima XL-90 ultracentrifuge (Beckman Coulter, Brea, CA, USA). DNA was spun in 4 ml polyallomer Optiseal tubes (Beckman Coulter) in cesium chloride (CsCl) gradients with an average density of 1.725 g ml−1. Centrifuged gradients were fractionated into 15 equal fractions via careful manual pipetting from the top of the polyallomer tube. DNA was precipitated with 2 volumes of polyethylene glycol for 2 h at room temperature and pelleted by centrifugation (30 min, 13 000 g). Pellets were washed once with 70% ethanol, resuspended in molecular grade water, and quantified fluorometrically using a Qubit (Life Technologies, Carlsbad, CA, USA).
PCR and qPCR
16S rRNA genes from each fraction were amplified using primers equipped for dual-indexed barcoded sequencing on an Illumina (San Diego, CA, USA) MiSeq (Kozich et al., 2013), which target the V4 hypervariable region (515F/806 R; (Caporaso et al., 2012)). We used a version of the 515F primer with a single-base modification (in bold) to increase coverage of the SAR11 clade (515F: 5′ – GTGCCAGCMGCCGCGGTAA – 3′ Apprill et al., 2015). PCR reactions were carried out in 25 μl volumes with 2 μl of template added to each reaction for 35 cycles. Each gradient fraction was amplified using a different barcode combination and amplicons were gel-extracted (QIAquick Kit, Qiagen) and pooled in equimolar concentrations prior to high-throughput paired-end Illumina sequencing (MiSeq, 2 × 250 bp reads) at the University of Illinois Keck Center for Comparative and Functional Genomics. Each density fraction was also screened for the major subgroups of marine archaea using quantitative PCR (qPCR). qPCR for MGII Euryarchaeaota followed a previously published assay (Orsi et al., 2015), while the qPCR assay for Marine Group I ammonia-oxidizing archaea using the ammonia monooxygenase subunit A (amoA) gene followed that of Santoro et al. (2010).
Bioinformatic analysis
Processing of the paired-end MiSeq data was performed according to Kozich et al. (2013) using MOTHUR (Schloss et al., 2009), and pair-wise OTU clustering was performed in QIIME (Caporaso et al., 2010). A total of 12.5 million quality-checked 16S rRNA gene sequences corresponding to 2722 OTUs were used for downstream analysis. Sequences were clustered at 97% sequence identity using UCLUST (Edgar, 2010). OTUs were identified to taxonomic groups through BLASTn searches against the SILVA database (Pruesse et al., 2007). OTU tables were rarified per size fraction, to the sample with the least number of sequences (0.2–0.8 μm=27,327; 0.8–3 μm=57,110; >3 μm: 34,970). Rarified counts for each OTU in the control and SIP gradients were normalized to the maximal abundance of that OTU across density fractions in unlabeled or SIP-labeled bottles. Only OTUs with >10 sequences that were detected in >50% of the fractions were used in downstream analysis. After these quality control criteria, a total of 4.1 million 16S rRNA gene sequences corresponding to 702 OTUs were used for downstream analysis. Sequence data have been deposited in the NCBI Short Read Archive under BioProject ID PRJNA287804.
We defined DNA-SIP incorporation similar to Nelson and Carlson (2012) and Hungate et al. (2015) on the basis of a comparison of the relative distribution of an OTU across the CsCl density gradient in a control (12C and 14N substrate added) versus an experimental (13C and 15N substrate added) incubation. OTUs incorporating the label were defined as those OTUs that exhibited a relatively unimodal distribution in the CsCl gradient and whose peak buoyant density (BD) was shifted >0.012 g ml−1 between the experiment and the control (similar to Buckley et al., 2007a). In this case, a relatively higher number of sequences from an OTU retrieved in the heavy part of the gradient compared with its distribution in the control is a function of isotopically labeled protein assimilation. Thus, throughout the manuscript, we refer to OTUs meeting the criteria defined above as those assimilating protein.
Results
Cruise setting and vertical biogeochemical profiles
Oceanographic conditions during the cruise were characterized by a general decreasing gradient in phytoplankton biomass with distance from shore, with upwelling and higher concentrations of Chl a along the coast (Supplementary Figure S1). The site for the DNA-SIP incubations, station 67–70, had lower surface Chl a concentrations compared with neighboring stations to the east and west (1.5–2 versus >3 μg l −1) (Supplementary Figure S1) and exhibited a broad subsurface chlorophyll maximum spanning the depths of 17–40 m (Figure 1a). The phytoplankton community (quantified by flow cytometry) was dominated by Prochlorococcus (48% of phytoplankton cells) and Synechococcus (37% of phytoplankton cells). Eukaryotic phytoplankton comprised 15% of total phytoplankton cells (Supplementary Figure S2). NH4+ concentrations ranged from below detection limits to 260 nM, with a maximum of 260 nM at 45 m (Figure 1a). Vertical profiles of DFAA and TDAA (the combination of DFAA and peptides) at 67–70 show a peak in DFAA and TDAA at 40 m (Figure 1b), which coincides with the subsurface NH4+ maximum at 40 m (Figure 1a). Concentrations of most individual DFAA peak at 40 m and progressively decrease in deeper water (down to 500 m), with threonine, valine, beta-alanine and methionine being exceptions to this trend (Supplementary Figure S3). The composition of DFAA between 0–40 m is significantly different from the DFAA pool found in waters 60–500 m (analysis of similarity: P=0.001).
Figure 1.

(a) Vertical profiles of Chl a and NH4+ at station 67–70, error bars represent s.d. of Chl a measurements over a 3-day period at station 67–70. (b) Vertical profiles of DFAA and TDAA at station 67–70. (c) In situ remineralization rates of protein C and N, and NH4+ oxidation rates. Protein N remineralization rates are the result of ammonification plus nitrification.
Protein remineralization rates from the in situ array ranged from 0.002 to 0.35 μmol C l−1 per day with a maximum rate at 20 m (Figure 1c). The maximum protein C remineralization rate at 20 m coincides with the subsurface chlorophyll maximum (Figure 1a) and was the depth sampled for the SIP experiment. The protein N remineralization rates (ammonification plus nitrification) measured on the in situ array ranged from 0.03 to 0.27 nmol N l−1 per day, with a maximal rate at 5 m. Protein N remineralization rates (±s.d.) in the SIP incubation were similar to those measured on the array under in situ light conditions (0.11 versus 0.14 nmol N l−1 per day. In comparison, protein C remineralization rates in the SIP incubation were slightly higher than the rates measured on the array under in situ light conditions (0.55 (±0.09) μmol C l−1 per day). NH4+ oxidation rates ranged from 0.2 nmol l−1 per day at the surface to a maximum of 29.8 nmol l−1 per day at 50 m (Figure 1c).
Timecourse measurements
There was no significant difference in net NH4+ production between control (no protein added) and experimental (protein added) bottles at 0 and 24 h (t test: P>0.05) (Supplementary Figures S4A and B). Furthermore, there was no increase in DFAA concentration after 24 h between bottles that did, or did not, receive proteins. Depletion of aspartate, glycine, histidine, alanine, valine, serine and leucine was faster in bottles amended with proteins, relative to unamended control (Supplementary Figure S5). There were no significant differences in total free-living DAPI-stained cells between treatments after 24 h (t test: P>0.05; Supplementary Figure S4C). Total DAPI-stained cells attached to particles also did not increase significantly over the course of the experiment (t test; P>0.05; Supplementary Figure S4D). However, free-living euryarchaea increased significantly after 24 h (Supplementary Figure S4C). Interestingly, no archaea were observed on diatomaceaous particles, whereas a high number of bacteria were found attached to these particles (Supplementary Figure S6).
Free-living and particle-attached bacteria and archaea utilizing dissolved proteins
The microbial community in the free-living fraction was dominated by the α-Proteobacteria and γ-Proteobacteria, whereas the particle-associated fractions were dominated by Verrucomicrobia and the Flavobacteria (Supplementary Figure S7). The most abundant free-living taxa were affiliated with the SAR11 clade followed by the SAR86 clade (Figure 2), whereas the dominant taxa in the particle-associated size fractions were affiliated with Synechococcus, Roseibacillus, Fluviicola, Formosa and uncultivated Flavobacteria (Figures 2). There was a shift in the BD of peak DNA quantity between the experimental and control bottles in all size fractions (Supplementary Figures S8A–C). Moreover, the taxonomic composition of density fractions varied significantly as a function of BD between experimental and control bottles (analysis of similarity: P<0.05; Supplementary Figures S8D–F). No discernable difference in community structure was observed between control (no protein added) and experimental (protein added) incubations after 24 h (Supplementary Figure S7). Post incubation, communities in the 0.8–3 μm and >3 μm fractions were similar to each other and contained relatively higher percentages of Flavobacteria, Planctomycetes and Verrucomicrobia compared with free-living fractions (Supplementary Figure S7).
Figure 2.

Relative abundance of the most abundant OTUs in each of the three size fractions after 24 h, asterisks indicate OTUs that assimilated protein.
Seventy-seven bacterial and archaeal OTUs assimilated the amended protein after 24 h (Supplementary Table S1), which exhibit high variability in relative abundance (Table 1). OTUs affiliated with γ-Proteobacteria (SAR86, KI89A, OM60 and SAR92 clades), Verrucomicrobia, Flavobacteria and MGII had the highest numbers of OTUs assimilating protein, and had the highest percentages of OTUs utilizing proteins compared with other groups (Supplementary Table S1). The dominant taxa in the free-living fraction assimilating protein were affiliated with the α-Proteobacteria (Roseobacter, Rhodobacteraceae), γ-Proteobacteria (SAR86 clade), and MGII (Figure 2). There was little overlap in protein-utilizing taxa between the size fractions (Figure 3). All of the Bacteroidetes-affiliated OTUs assimilating protein belonged to the Flavobacteria. One SAR86 OTU that assimilated protein was the third most abundant OTU (Figure 2) detected in the free-living fraction (Figure 4) and was affiliated with the SAR86 clade III (Figure 5b).
Table 1. A summary of taxa assimilating protein after the 24 h incubation.
| Microbial group |
0.2–0.8 μm |
0.8–3 μm |
>3 μm |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Average BD shift (g ml−1) | OTUs with BD shift (% total) | Sequences with % total | BD shift % total per taxon | Average BD shift (g ml−1) | OTUs with BD shift (% total) | Sequences with % total | BD shift % total per taxon | Average BD shift (g ml−1) | OTUs with BD shift (% total) | Sequences with % total | BD shift % total per taxon | |
| Flavobacteria | 0.018 | 2 (0.3%) | 319 (0.04%) | 0.3% | 0.027 | 9 (1.2%) | 29 035 (2%) | 3% | 0.020 | 16 (2.4%) | 105 527 (13%) | 41% |
| Verrucomicrobia | 0.016 | 3 (0.4%) | 2524 (0.34%) | 51% | 0.018 | 4 (0.5%) | 18 835 (1.3%) | 10% | 0.014 | 8 (1.2%) | 33 880 (4.3%) | 46% |
| Euryarchaeota (MGII) | 0.014 | 5 (0.6%) | 9776 (1.3%) | 52% | 0.012 | 1 (0.2%) | 704 (0.05%) | 0.6% | 0 | 0 | 0 | 0 |
| Gammaproteobacteria | 0.02 | 3 (0.5%) | 44 354 (6%) | 22% | 0.017 | 4 (0.5%) | 315 (0.02%) | 0.07% | 0 | 0 | 0 | 0 |
| Alphaproteobacteria | 0.016 | 2 (0.3%) | 5755 (0.8%) | 2% | 0.014 | 3 (0.4%) | 79 (0.005%) | 0.01% | 0.012 | 2 (0.3%) | 122 (0.01%) | 0.1% |
| Actinobacteria | 0.012 | 1 (0.2%) | 350 (0.04%) | 0.7% | 0 | 0 | 0 | 0 | 0.012 | 1 (0.2%) | 357 (0.04%) | 2% |
| Planctomycetes | 0 | 0 | 0 | 0 | 0.021 | 2 (0.3%) | 1678 (0.11%) | 10% | 0.01 | 3 (0.4%) | 2,041 (0.3%) | 10% |
| Deltaproteobacteria | 0 | 0 | 0 | 0 | 0.016 | 6 (0.3%) | 4619 (0.3%) | 21% | 0.012 | 2 (0.3%) | 684 (0.1%) | 2% |
| Cyanobacteria | 0 | 0 | 0 | 0 | 0.018 | 1 (0.3%) | 21 (0.001%) | 0.006% | 0 | 0 | 0 | 0 |
| Total | 16 (3%) | 63 078 (8%) | 30 (3.7%) | 55 286 (3.8%) | 31 (4.6%) | 142 254 (18%) | ||||||
Abbreviations: BD, buoyant density; OTU, operational taxonomic unit. For all OTUs exhibiting a BD shift, see Supplementary Table S1 for abundance and shifts in BD. The ‘% total' refers to the percent of total sequences or percent of total OTUs per size fraction. ‘% total per taxon' refers to percent of total sequences per taxonomic group.
Figure 3.
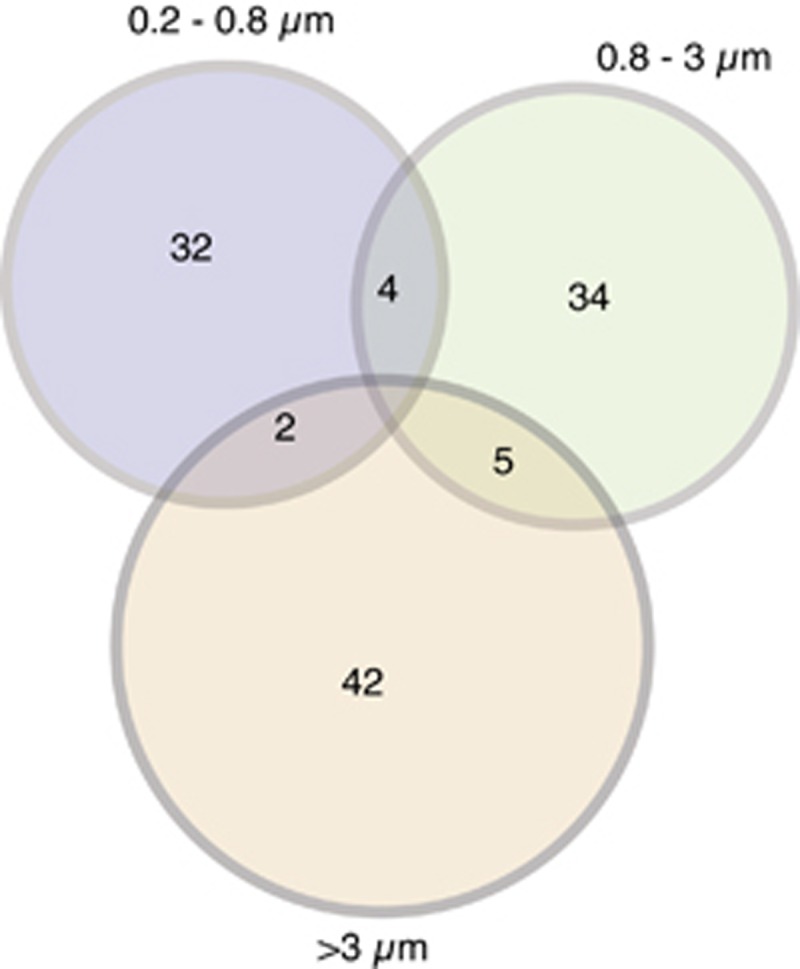
The number of overlapping and non-overlapping OTUs between size fractions that assimilated protein. Note that most OTUs assimilated protein in only one size fraction.
Figure 4.

OTUs exhibiting the greatest degree of protein assimilation. Lines represent the distribution of OTUs in the control (unlabeled proteins, dashed lines) and experiment (labeled proteins, solid lines). The y axis represents the relative abundance of each OTU normalized to its maximal abundance across all density fractions. See Supplementary Table S1 for relative abundance and BD shifts for all OTUs.
Figure 5.

Maximum likelihood phylogenies (PhyML) of MGII (a), SAR86 (b) and Flavobacteria (c) 16S rRNA genes sharing >97% sequence identity with OTUs, which assimilated (red font), or did not assimilate (bold black font), protein after a 24 h incubation. Colored squares indicate the size fraction where protein assimilation was observed. Black circles at nodes represent bootstrap support of >90% and white circles represent bootstrap support of >50%. Numbers represent NCBI GI accession IDs.
Microbial populations that assimilated protein in particle-associated size fractions are markedly different from free-living populations. For example, OTUs affiliated with Planctomycetes, Fluviicola (Flavobacteria), Formosa (Flavobacteria), Roseibacillus (Verrucomicrobia) and OM27 clade (δ-Proteobacteria) were the dominant populations assimilating proteins in the particle-attached fractions (Table 1, Supplementary Table S1). Most of these bacterial taxa were not observed to assimilate protein in the free-living fraction and were not abundant in the free-living fraction (Figure 2), although some were detected in relatively low abundance (data not shown). Moreover, marine Euryarchaeota affiliated with the MGII.A clade assimilated protein in both the 0.2–0.8 and 0.8–3 μm size fractions, whereas the MGII.C clade only assimilated protein in the 0.2–0.8 μm fraction (Figure 5a).
The assimilation of protein by marine euryarchaea was further validated by qPCR (Figure 6a). MGII incubated with labeled protein exhibit higher quantities of rRNA genes in gradient fractions >1.71 g ml−1 in every size fraction relative to the control (Figure 6a). This assimilation of protein by heterotrophic euryarchaea was markedly larger compared with the ammonia-oxidizing thaumarchaea, which did not meet our criteria for protein assimilation (Figure 6b).
Figure 6.

(a) Quantification (qPCR) of MGII Euryarchaeota 16S rRNA genes in density gradient fractions from the control and experiment, in the three size fractions. In all size fractions, higher quantities of MGII rRNA genes are detected in density fractions >1.71 g ml−1 in DNA extracted from bottles amended with labeled proteins, relative to the control. (b) Quantification (qPCR) of MGI (Thaumarchaeota) amoA genes in density gradient fractions from the control and experiment, in the 0.2–0.8 μm size fraction.
Discussion
Phytoplankton biomass supports significant secondary production by heterotrophic prokaryotes, and is central to organic matter cycling in the ocean. Phytoplankton-derived proteins likely have an important role in this process. Here, we investigated the specific microbial taxa involved in the uptake of dissolved proteins, both on and off particles, and found that although some protein-assimilating taxa were previously identified in the utilization of phytoplankton-derived organic matter, others are newly implicated.
As expected, we detected protein assimilation by free-living α-Proteobacteria, γ-Proteobacteria and Flavobacteria previously implicated in this process. The relative percentage of α-Proteobacteria assimilating protein (<1%, Table 1) is markedly lower than observed in a study from coastal waters, where 13% of α-Proteobacteria cells assimilated protein (Cottrell and Kirchman, 2000). In contrast, roughly 6% of total SAR86 (γ-Proteobacteria) sequences assimilated protein in the free-living fraction (Supplementary Table S1), which is comparable with a MAR-FISH study that found 8% of all SAR86 cells assimilated protein in the surface ocean (Nikrad et al., 2014). Methodological differences preclude a direct comparison between the fraction of labeled cells by MAR-FISH and the fraction of a clade with SIP-labeled OTUs (carried out here), but the indirect comparison here shows that the same groups are found to assimilate protein with both methods. The ability of SAR86 to acquire protein may be facilitated by their genomic capacity for HMW substrate acquisition (Dupont et al., 2012). An OTU affiliated with the SAR92 clade (Figure 4) assimilated more protein than any other OTU (Figure 4, BD shift; 0.036 g ml−1), suggesting that this clade may be important for protein recycling in areas where the SAR92 clade reaches high abundance, such as coastal waters (Stingl et al., 2007). Flavobacterial cells have a preference for HMW-DON over LMW-DON (Cottrell and Kirchman, 2000; Kirchman, 2002), but in our open ocean study, a lower percentage of free-living Flavobacteria assimilated protein compared with other groups (Table 1), which is also low compared with the percentage of Flavobacteria assimilating protein in coastal waters (Cottrell and Kirchman, 2000).
Our study also links protein utilization to several diverse and uncultivated marine bacterial taxa that previously had not been implicated in dissolved protein cycling. In the free-living size fraction, OTUs affiliated with marine Actinobacteria utilized proteins (Table 1). Within the Actinobacteria, the majority of OTUs utilizing dissolved protein were affiliated with the enigmatic actinobacterial Sva0996 clade (Supplementary Table S1), a group that has been detected in both sediment (Ravenschlag et al., 1999) and water column (Bano and Hollibaugh, 2002) habitats. To our knowledge, this is the first demonstration of the substrate assimilation ability of these uncultivated actinobacteria.
We found that some taxa previously shown to respond to HMW-DOM are especially active in utilizing proteins on particles. The prevalent assimilation of protein by particle-attached Flavobacteria clades observed here (41% Table 1,Figure 5c) is consistent with their common adhesion to particles (Fernandez-Gomez et al., 2012). Particle-attached bacteria can have much higher protein degradation rates compared with free-living bacteria (Taylor, 1995), and thus particle-attached Flavobacteria may have been ‘primed' for protein assimilation. Our results indicate that in addition to degrading particulate matter, particle-attached Flavobacteria also act as a sink of HMW-DOM through the utilization of dissolved proteins. An OTU affiliated with flavobacterial genus Fluviicola was the most abundant (7.4% of total reads) taxon assimilating protein in the >3 μm fraction, and a total of six Fluviicola-affiliated OTUs assimilated protein in this fraction (Supplementary Table S1). This strongly suggests that some members of the Fluviicola, represented by the freshwater strain Fluviicola taffensis RW262 (O'Sullivan et al., 2005), are important degraders of proteins, and potentially particulate organic nirtrogen, in marine ecosystems.
We also found diverse, uncultivated Planctomycetes clades assimilating protein in the particle-attached size fraction (Table 1, Supplementary Table S1). This is consistent with their detection in particle-associated size fractions in previous studies (DeLong et al., 1993; Crump et al., 1999). A high percentage (10%) of the Planctomycetes OTUs in the >0.8 μm fractions assimilated protein compared with most other microbial groups, and did not assimilate protein in the free-living fraction (Table 1). Utilization of M. pusilla proteins by planctomycetes is consistent with their previous detection following phytoplankton blooms (Morris et al., 2006) and metagenomic evidence indicating that metabolism of marine planctomycetes is linked to phytoplankton-derived substrates, such as the diatom cell wall protein silaffin (Zeigler Allen et al., 2012). The success of planctomycete cells on particles may be due to their unique holdfast structures and stalks, which allow them to attach to surfaces (Fuerst, 1995). Most Planctomycetes OTUs that assimilated protein were affiliated with the CL500–3 clade (Supplementary Table S1), an as-yet uncultured subgroup found in oligotrophic lakes (Urbach et al., 2001). Another Planctomycete-affiliated OTU that assimilated protein in the >3 μm fraction was affiliated with the uncultivated Urania-1B-19 sediment group (Supplementary Table S1), reported from deep sea sediment of the Mediterranean (Heijs et al., 2008).
The discovery of protein utilization by a diversity of uncultivated Verrucomicrobia taxa in all size fractions (Supplementary Table S1) suggests that Verrucomicrobia are important for DON cycling both on and off of particles. The relatively high proportion of Verrucomicrobia taxa assimilating protein in the free-living (51%) and >3 μm fractions (46%) indicates that they have a relatively strong preference for dissolved proteins compared with most other groups detected (Table 1). In the free-living fraction, Verrucomicrobia protein utilization was dominated by representatives of the Puniceicoccaceae, previously shown to assimilate exudates from Synechococcus in euphotic zone waters (Nelson and Carlson, 2012). This supports biogeographic and genomic evidence suggesting that certain marine Verrucomicrobia species have a preference for phytoplankton-derived HMW-DOM (Herleman et al., 2013). Verrucomicrobia have also been identified as degraders of polysaccharides in the Arctic (Cardman et al., 2014) and given their ubiquity in the ocean and presence on POM (Freitas et al., 2014), chemoheterotrophic activity of Verrucomicrobia taxa should contribute markedly to DOM and POM turnover. OTUs affiliated with Roseibacillus were the dominant verrucomicrobial group assimilating protein in the particle-associated fraction (Supplementary Table S1). The few cultivated Roseibacillus species do not exhibit N-acetyl-β-glucosaminidase, trypsin or chymotrypsin activity (Yoon et al., 2008), enzymes which degrade HMW-DON such as chitin and protein. Thus, it seems likely that some particle-attached Roseibacillus cells might instead acquire protein C and N by transporting the oligopeptides released through protein hydrolysis from extracellular enzymes manufactured by other bacteria.
Protein assimilation was identified in OM27 clade organisms (Supplementary Table S1), which are most closely related to the predatory deltaproteobacterial genus Bdellovibrio (Fuchs et al., 2005) and have a geographically wide distribution (for example, Rappe et al., 1997; Fuchs et al., 2005). The OM27 clade had the highest percentage of OTUs (21%) assimilating protein compared to all other groups in the 0.8–3 μm fraction suggesting their activity was relatively high in this size range (Table 1). This may indicate the formation and activity of bdelloplasts, structures that are formed as predatory Bdellovibrio divide inside the periplasmic space causing the prey cell size to increase (Abram et al., 1974). This cross-feeding phenomenon of predatory bacteria has been observed in previous DNA-SIP studies, whereby Bdellovibrio populations assimilated C after preying on methanotrophs incubated with 13C methane (Morris et al., 2002). We hypothesize that OM27 clade organisms correspond to an uncultivated group of predatory bacteria because (i) the closest phylogenetic relation of this clade is to the predatory Bdellovibrio genus (Fuchs et al., 2005) and (ii) 1–2 μm-sized bdelloplasts (Fenton et al., 2010) should be enriched in the 0.8–3 μm size range, which is the size range where OM27 group-specific protein assimilation (21%) was higher than all other groups (Table 1). We conclude that OM27 clade organisms acquired the label through predation on bacteria that had assimilated the dissolved protein substrate (that is, cross-feeding), and were then captured as swollen bdelloplasts in the 0.8–3 μm fraction.
We confirmed protein utilization by uncultivated taxa affiliated with marine Euryarchaeota (Figure 5a). Our study provides the first direct link between MGII cells and protein recycling, as well as identification of the specific MGII taxa utilizing dissolved proteins. Compared with the other major microbial groups, a relatively high percentage (52%) of free-living MGII OTUs assimilated protein, suggesting that they have a strong affinity for this substrate (Table 1). The physiology of planktonic MGII is hypothesized to be based on protein metabolism (Reysenbach and Flores, 2008; Iverson et al., 2012), and heterotrophic euryarchaea in sediments also appear to utilize detrital proteins (Lloyd et al., 2013; Orsi et al., 2013). The strong affinity for protein substrates by marine euryarchaea shown here (Table 1) provides direct evidence supporting these hypotheses. Assimilation of protein by MGII (Figure 6a) was markedly larger compared with thaumarchaea (Figure 6b), which according to our criteria did not assimilate protein.
We originally hypothesized that MGII would also be active in protein assimilation on particles, because of an enrichment of MGII genes involved in HWM-DOM degradation in particle-associated metagenomes (Orsi et al., 2015). Although we did not observe any MGII OTUs assimilating protein in the >3 μm fraction (Table 1), qPCR analysis with MGII-specific primers did show protein assimilation by MGII in this size fraction (Figure 6a). Thus, MGII OTUs assimilating protein in the >3 μm fraction, and potentially other size fractions, were missed using the ‘universal' tag-sequencing PCR primers.
The SAR11 clade (Giovannoni et al., 1990), which includes Pelagibacter (Rappe et al., 2002), readily assimilates DFAA (Malmstrom et al., 2004; Nikrad et al., 2012) and was the most abundant free-living taxon (Figure 2). However, no SAR11 OTUs assimilated protein in our incubations. This led us to investigate the question: Do some groups acquire proteins via a HMW transmembrane transport mechanism that is absent in SAR11 cells? TonB-dependent receptor (TBDR) proteins transport HMW compounds that exceed the typical range of normal porins (<600 daltons) by catalyzing high affinity transport of Ni-, Cu-, Fe-chelates, proteins, siderophores and polysaccharides across the outer membrane (Schauer et al., 2008), and are absent in all Pelagibacter genomes (n=19) available in IMG (as of March 2015). TBDR are important for microbial competition in the ocean, as cells with TBDR were observed to outcompete other microbes lacking TBDR in the presence of labile HMW-DOM released after a phytoplankton bloom in the North Sea (Teeling et al., 2012) and were also overexpressed in marine communities following HMW-DOM additions (McCarren et al., 2010). TBDR have been identified as important components of algal glycan utilization in Gramella forsetti, a widespread marine flavobacterium pivotal to remineralization of complex organic matter (Kabisch et al., 2014). However, it is unknown whether TBDR also assist in protein acquisition by marine microbes.
Two observations suggest that TBDR may facilitate protein uptake by certain marine microbes. First, a positive correlation (Spearman correlation: r=0.55, P=0.02) exists between the number of OTUs per group assimilating protein and the percent of genomes in that group that encode TBDR proteins (Figure 7). Second, the gross majority of lineages that assimilated protein encode TBDR transmembrane transporters (Supplementary Table S1), whereas SAR11-affiliated OTUs that lack these transporters did not. The dominance of SAR11 taxa in the free-living fraction (Figure 2) combined with their lack of substrate assimilation suggests that hydrolyzed amino acids from the labeled protein substrate were not readily available for assimilation during the 24 h incubation. Given the correlation between groups containing TBDR and the number of OTUs per group assimilating protein (Figure 7), it seems likely that some TBDR encoding marine microbes may be able to transport larger oligopeptides (>600 daltons) quickly, before they are hydrolyzed into smaller amino acids. As suggested for polysaccharides (Teeling et al., 2012), this would help them compete against cells lacking TBDR during periods of increased protein availability (for example, during blooms). This hypothesis is consistent with experimental demonstration of TBDR transport of large proteins (>600 daltons) such as serum transferrin and hemoglobin across cell membranes (Noinaj et al., 2010), suggesting that this is a mechanism of protein acquisition in the ocean as well.
Figure 7.

Relationship between the number of OTUs assimilating protein and the percentage of genomes per group that encode TBDR. Searches for TBDR in genomes of each group were performed using the control term search function in IMG (img.jgi.doe.gov) against all finished and draft genomes in each group (as of March 2015), using ‘tonb' as a keyword. The correlation was performed on all data points (including the number of Flavobacteria OTUs in >3 μm outlier); the y axis break was inserted to reduce plotting space. See Supplementary Table S1 for a complete summary of values used in this plot and the number of genomes per group used to calculate percent values on the x axis.
Despite uptake of the labeled proteins by abundant, diverse lineages, it is not likely that cells acquired the label after remineralized enriched protein C and N passed through an inorganic dissolved phase (as DIC or NO3−). The δ13C of DIC at the end of the experiment was only enriched 3‰ (~1 atm%) relative to the controls. Such a low level of enrichment would be insufficient to produce a detectable labeling of the DNA meeting our threshold of 0.012 g ml−1. Similarly, the highest final δ15NNOx in the experimental incubations was 17‰ (< 0.4 atm%), the uptake of which would not produce a detectable shift in DNA density because 25 atm% is the minimal 15N enrichment in DNA needed to detect a nitrogen-based assimilation signal with DNA-SIP (Buckley et al., 2007b). For similar reasons, it is unlikely that 15N was acquired via 15NH4+ regenerated from the labeled proteins. The maximum shift in DNA BD possible from an N source with 50 atm% enrichment is 0.01 g ml−1 (Buckley et al., 2007b). Thus, OTUs being labeled solely through 15NH4+ assimilation is highly unlikely because the starting 15N enrichment of our protein extract was 42 atm% and we used a DNA BD threshold of 0.012 g ml−1 for defining assimilation.
Protein remineralization to DIC in the in situ array and SIP incubation (Figure 1c) indicates that the amended proteins were not only assimilated, but also used as an energy source. The profile of protein C remineralization rates (Figure 1c) is similar in shape to previously reported hemoglobin remineralization rates in the Southern California Bight, which were also found to peak in the subsurface chlorophyll maximum (Hollibaugh and Azam, 1983). We acknowledge that our calculated C remineralization rates (0.002–0.35 μmol l−1 per day) based on the assumption of 16 atm% labeling of the added proteins may be overestimates, but are similar in magnitude to previous studies (Keil and Kirchman, 1993; Nagata et al., 2003). Calculations using the calculated value of the M. pusilla cells of 95 atm% yields rates that are much lower (0.001–0.10 μmol l−1 per day), but a profile that is similar in shape.
Conclusion
Our study demonstrates a direct link between phytoplankton-derived dissolved protein and several dominant, yet uncultivated, members of the bacterial and archaeal community and significantly expands the known microbial diversity responsible for mediating protein turnover. Substrate assimilation was observed after a relatively short incubation time (24 h), indicating that the high-throughput DNA-SIP method used here is sufficiently sensitive in detecting substrate assimilation. Correlative evidence suggests that TBDR receptors are a potential mechanism used by certain marine microbes for protein utilization, a finding that warrants further investigation. Although protein assimilation was widespread within certain taxonomic groups (Verrucomicrobia and MGII archaea), in others, fewer sub-clades were found to be active in protein uptake (for example, α-Proteobacteria, SAR86, and Actinobacteria). Some taxa found in particle-attached fractions were particularly active in protein assimilation (Flavobacteria, Verrucomicrobia and Planctomycetes), while their free-living counterparts were not. Microbial utilization of protein in particle-associated fractions indicates that some particle-attached microbes are active in the degradation of dissolved protein. Particle-attached bacteria are typically thought to act as sources of DOM through hydrolysis of particulate HMW organic matter (Cho and Azam, 1988). However, the particle-attached bacteria and Euryarchaeaota, utilizing dissolved HMW-DON shown here demonstrate that a diversity of particle-attached microbial communities also act as a sink of important DON substrates over relatively short timescales.
Acknowledgments
We thank the captain and crew of the R/V Western Flyer, J. Timothy Pennington, Marguerite Blum, Valeria Jimenez, Christopher Wahl, Noriko Okamoto, Jarred Swalwell and Francisco Chavez for logistical assistance prior to and during the cruise. We also thank Michael Morando and Elizabeth Kujawinski for advice and conversations on SIP methodology, and Mak Saito and Dawn Moran for advice on for protein extraction. We thank Amy Apprill for sharing the modified, barcoded PCR primers, Alexandra Welch for assistance with qPCR, and Paul Carini and Tristan Horner for discussions. We thank John Hobbie for discussions on protein remineralization rate kinetics, and three anonymous reviewers for their helpful comments. Support for this work was provided by GBMF3307 to PJK, TAR, AZW and AES, United States National Science Foundation award DBI-1318455 to AES, and the David and Lucile Packard Foundation (to AZW). This is UMCES contribution number 5169.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Abram D, Castro e Melo J, Chou D. (1974). Penetration of Bdellovibrio bacteriovorus into host cells. J Bacteriol 118: 663–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderkamp AC, Sintes E, Herndl GJ. (2006). Abundance and activity of major groups of prokaryotic plankton in the coastal North Sea during spring and summer. Aquatic Microbial Ecology 45: 237–246. [Google Scholar]
- Aluwihare LI, Meador TB. (2009) Chemical composition of marine dissolved organic nitrogen. In: Capone DG, Bronk DA, Mulholland MR, Carpenter EJ (eds), Nitrogen in the Marine Environment. Academic Press: San Diego, CA, USA, pp 95–133. [Google Scholar]
- Aluwihare LI, Repeta DJ, Pantoja S, Johnson CG. (2005). Two chemically distinct pools of organic nitrogen accumulate in the ocean. Science 308: 1007–1010. [DOI] [PubMed] [Google Scholar]
- Apprill A, McNally S, Parsons R, Weber L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquatic Microbial Ecology 75: 129–137. [Google Scholar]
- Bano N, Hollibaugh JT. (2002). Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean. Appl Environ Microbiol 68: 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billen G, Fontigny A. (1987). Dynamics of a Phaeocystis dominated spring bloom in Belgian coastal waters. II. Bacterioplankton dynamics. Mar Ecol Prog Ser 37: 249–257. [Google Scholar]
- Bronk DA. (2002) Dynamics of DON. In: Hansell DA, Carlson CA (eds), Biogeochemistry of Marine Dissolved Organic matter. Academic Press: San diego, CA, USA, pp 153–247. [Google Scholar]
- Bronk DA, Steinberg DK. (2009) Nitrogen regeneration. In: Capone DG, Bronk DA, Mulholland MR, Carpenter EJ (eds), Nitrogen in the Marine Environment. Academic Pres: San Diego, CA, USA, pp 385–449. [Google Scholar]
- Buckley DH, Huangyutitham V, Hsu SF, Nelson TA. (2007. a). Stable isotope probing with 15N2 reveals novel noncultivated diazotrophs in soil. Appl Environ Microbiol 73: 3196–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DH, Huangyutitham V, Hsu SF, Nelson TA. (2007. b). Stable isotope probing with 15N achieved by disentangling the effects of genome G+C content and isotope enrichment on DNA density. Appl Environ Microbiol 73: 3189–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6: 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardman Z, Arnosti C, Durbin A, Ziervogel K, Cox C, Steen AD et al. (2014). Verrucomicrobia are candidates for polysaccharide-degrading bacterioplankton in an arctic fjord of Svalbard. Appl Environ Microbiol 80: 3749–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CA, Ducklow H. (1995). Dissolved organic carbon in the upper ocean of the central equatorial Pacific Ocean, 1992: Daily and finescale vertical variations. Deep Sea Res II 42: 639–656. [Google Scholar]
- Cho BC, Azam F. (1988). Major role of bacteria in biogeochemical fluxes in the ocean's interior. Nature 332: 441–443. [Google Scholar]
- Collins CA, Pennington JT, Castro CG, Rago TA, Chavez FP. (2003). The California Current system off Monterey, California: physical and biological coupling. Deep Sea Res II 50: 2389–2404. [Google Scholar]
- Cottrell MT, Kirchman DL. (2000). Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacteria cluster consuming low- and high- molecular weight dissolved organic matter. Appl Environ Microbiol 66: 1692–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump BC, Armbrust V, Baross JA. (1999). Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia River, its estuary, and the adjacent ocean. App Environ Microbiol 65: 3192–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvelier ML, Allen AE, Monier A, McCrow JP, Messie M et al. (2010). Targeted metagenomics and ecology of globally important uncultured eukaryotic phytoplankton. Proc Nat Acad Sci USA 107: 14679–14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong EF, Franks DG, Alldredge AL. (1993). Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol Oceanogr 38: 924–934. [Google Scholar]
- Dugdale RC, Goering JJ. (1967). Uptake of new and regenerated forms of nitrogen in primary productivity. Limnol Oceanogr 12: 196–206. [Google Scholar]
- Dunford EA, Neufeld JD. (2010). DNA stable-isotope probing (DNA-SIP). J Vis Exp; e-pub ahead of print 2 August 2010; doi:10.3791/2027. [DOI] [PMC free article] [PubMed]
- Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R et al. (2012). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. [DOI] [PubMed] [Google Scholar]
- Fenton AK, Lambert C, Wagstaff PC, Sockett RE. (2010). Manipulating each MreB of Bdellovibrio bacteriovorus gives diverse morphological and predatory phenotypes. J Bacteriol 192: 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gomez B, Richter M, Schuler M, Pinhassi J, Acinas SG, Gonzalez JM et al. (2012). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J 7: 1026–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogg GE. (1971). The extracellular products of algae in freshwater. Arch Hydrobiol 5: 1–25. [Google Scholar]
- Freitas S, Hatosy S, Fuhrman JA, Huse SM, Welch DB, Sogin ML et al. (2014). Global distribution and diversity of marine Verrucomicrobia. ISME J 6: 1499–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigaard NU, Martinez A, Mincer TJ, DeLong EF. (2006). Proteorhodopsin lateral gene transfer between marine planktonic Bacteria and Archaea. Nature 439: 847–850. [DOI] [PubMed] [Google Scholar]
- Fuchs BM, Woebken D, Zubkov MV, Burkill P, Amann R. (2005). Molecular identification of picoplankton populations in contrasting waters of the Arabian Sea. Aquatic Microbial Ecology 39: 145–157. [Google Scholar]
- Fuerst JA. (1995). The planctomycetes: emerging models for microbial ecology, evolution and cell biology. Microbiology 141: 1493–1506. [DOI] [PubMed] [Google Scholar]
- Fuhrman JA. (1999). Marine viruses and their biogeochemical and ecological effects. Nature 399: 541–548. [DOI] [PubMed] [Google Scholar]
- Giovannoni SJ, Britschgi TB, Moyer CL, Field KG. (1990). Genetic diversity in Sargasso Sea bacterioplankton. Nature 345: 60–63. [DOI] [PubMed] [Google Scholar]
- Guillard RRL, Hargraves PE. (1993). Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia 32: 234–236. [Google Scholar]
- Heijs SK, Laverman AM, Forney LJ, Hardoim PR, van Elsas JD. (2008). Comparison of deep-sea sediment microbial communities in the Eastern Mediterranean. FEMS Microbiol Ecol 64: 362–377. [DOI] [PubMed] [Google Scholar]
- Herleman DP, Lundin D, Labrenz M, Jürgens K, Zheng Z, Aspeborg H et al. (2013). Metagenomic de novo assembly of an aquatic representative of the verrucomicrobial class Spartobacteria. MBio 4: e00569–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbie JE, Crawford CC, Webb KL. (1968). Amino acid flux in an estuary. Science 159: 1463–1464. [DOI] [PubMed] [Google Scholar]
- Hollibaugh JT, Azam F. (1983). Microbial degradation of proteins in seawater. Limnol Oceanogr 28: 1104–1116. [Google Scholar]
- Holmes RM, Aminot A, Kerouel R, Hooker BA, Petersen BJ. (1999). A simple and precise method for measuring ammonium in marine and freshwater ecosystems. Can J Fish Aquat Sci 56: 1801–1808. [Google Scholar]
- Hungate BA, Mau RL, Schwartz E, Caporaso JG, Dijkstra P, van Gestel N et al. (2015). Quantitative microbial ecology through stable isotope probing. Appl Environ Microbiol 81: 7570–7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. (2012). Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335: 587–590. [DOI] [PubMed] [Google Scholar]
- Kabisch A, Otto A, König S, Becher D, Albrecht D, Schüler M et al. (2014). Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes 'Gramella forsetti' KT0803. IMSE J 8: 1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl DM, Björkman KM, Dore JE, Fujieki L, Hebel DV, Houlihan T et al. (2001). Ecological nitrogen-to-phosphorus stoichiometry at station ALOHA. Deep Sea Res II 48: 1529–1566. [Google Scholar]
- Keil RG, Kirchman DL. (1993). Dissolved combined amino acids: chemical form and utilization by marine bacteria. Limnol Oceanogr 38: 1256–1270. [Google Scholar]
- Keil RG, Kirchman DL. (1994). Abiotic transformation of labile protein to refractory protein in sea water. Marine Chemistry 45: 187–196. [Google Scholar]
- Keil RG, Tsamakis E, Hedges JI. (2000) Early diagenesis of particulate amino acids in marine systems. In: Goodfriend GA, Fogel MJ, Collins ML, Macko SA, Wehmiller JF (eds), Perspectives in Amino Acid and Protein Geochemistry. Oxford University Press: New York, pp 69–82. [Google Scholar]
- Kerouel R, Aminot A. (1997). Fluorometric determination of ammonia in sea and estuarine waters by direct segmented flow analysis. Marine Chemistry 57: 265–275. [Google Scholar]
- Kirchman DL. (2000) Uptake and regeneration of inorganic nutrients by marine heterotrophic bacteria. In: Kirchman DL. (ed) Microbial Ecology of the Oceans. Wiley-Liss: New York, pp 261–288. [Google Scholar]
- Kirchman DL. (2002). The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39: 91–100. [DOI] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79: 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova M, Lee C. (2002). Dissolved free and combined amino acids in nearshore seawater, sea surface microlayers and foams: Influence of extracellular hydrolysis. Aquatic Science 64: 1–17. [Google Scholar]
- Lee C, Wakeham SG, Hedges JI. (2000). Composition and flux of particulate amino acids and chloropigments in equatorial Pacific seawater and sediments. Deep Sea Res I 47: 1535–1568. [Google Scholar]
- Lipschultz F. (2008). Isotope tracer methods for studies of the marine nitrogen cycle. In: Capone DG, Bronk DA, Mulholland MR, Carpenter EJ (eds), Nitrogen in the Marine Environment, 2nd Edition, Academic Press: Burlington, MA, USA, pp 1345–1384.
- Liu Z, Liu S, Liu J, Gardner WS. (2013). Differences in peptide decomposition rates and pathways between hypoxic and oxic coastal environments. Marine Chemistry 157: 67–77. [Google Scholar]
- Lloyd KG, Schreiber L, Petersen DG, Kjeldsen KU, Lever MA, Steen AD et al. (2013). Predominant archaea in marine sediments degrade detrital proteins. Nature 496: 215–218. [DOI] [PubMed] [Google Scholar]
- Loh AN, Bauer JE, Druffel ER. (2004). Variable ageing and storage of dissolved organic components in the open ocean. Nature 430: 877–881. [DOI] [PubMed] [Google Scholar]
- Malmstrom RR, Cottrell MT, Elifantz H, Kirchman DL. (2005). Biomass production and assimilation of dissolved organic matter by SAR11 bacteria in the Northwest Atlantic Ocean. Appl Environ Microbiol 71: 2979–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmstrom RR, Kiene RP, Cottrell MT, Kirchman DL. (2004). Contribution of SAR11 bacteria to dissolved dimethylsulfoniopropionate and amino acid uptake in the North Atlantic ocean. Appl Environ Microbiol 70: 4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayali X, Stewart B, Mabery S, Weber PKTemporal succession in carbon incorporation from macromolecules by particle-attached bacteria in marine microcosms. Environ Microbiol Rep; e-pub ahead of print 3 November 2015; doi:10.1111/1758-2229.12352. [DOI] [PubMed]
- McCarren J, Becker JW, Repeta DJ, Shi Y, Young CR, Malmstrom RR et al. (2010). Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci USA 107: 16420–16427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy M, Benner R, Lee C, Fogel MJ. (2007). Amino acid nitrogen isotopic fractionation patterns as indicators of heterotrophy in plankton, particulate, and dissolved organic matter. Geochimica et Cosmochimica Acta 71: 4727–4744. [Google Scholar]
- McCarthy M, Pratum T, Hedges JI, Benner R. (1997). Chemical composition of dissolved organic nitrogen in the ocean. Nature 390: 150–153. [Google Scholar]
- McCarthy MD, Benner R, Lee C, Hedges JI, Fogel ML. (2004). Amino acid carbon isotopic fractionation patterns in oceanic dissolved organic matter: an unaltered photoautotrophic source for dissolved organic nitrogen in the ocean? Marine Chemistry 92: 123–124. [Google Scholar]
- McCarthy MD, Hedges JI, Benner R. (1998). Major bacterial contribution to marine dissolved organic nitrogen. Science 281: 231–234. [DOI] [PubMed] [Google Scholar]
- Meador TB, Aluwihare LI, Mahaffey C. (2007). Isotopic heterogeneity and cycling of organic nitrogen in the oligotrophic ocean. Limnol Oceanogr 430: 877–881. [Google Scholar]
- Morando M, Capone DG in review. Intraclade heterogeneity in nitrogen utilization by marine prokaryotes revealed using stable isotope probing coupled with tag sequencing (Tag-SIP). [DOI] [PMC free article] [PubMed]
- Morris RM, Longnecker K, Giovannoni SJ. (2006). Pirellula and OM43 are among the dominant lineages identified in an Oregon coast diatom bloom. Environ Microbiol 8: 1361–1370. [DOI] [PubMed] [Google Scholar]
- Morris SA, Radajewski S, Willison TW, Murrell JC. (2002). Identification of the functionally active methanotroph population in a peat soil microcosm by stable-isotope probing. Appl Environ Microbiol 68: 1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata T, Fukuda R, Koike I, Kogure K, Kirchman DL. (1998). Degradation by bacteria of membrane and soluble protein in seawater. Aquatic Microbial Ecology 14: 29–37. [Google Scholar]
- Nagata T, Meon B, Kirchman DL. (2003). Microbial degradation of peptidoglycan in seawater. Limnol Oceanogr 48: 745–754. [Google Scholar]
- Nelson CE, Carlson CA. (2012). Tracking differential incorporation of dissolved organic carbon types among diverse lineages of Sargasso Sea bacterioplankton. Environ Microbiol 14: 1500–1516. [DOI] [PubMed] [Google Scholar]
- Neufeld JD, Vohra J, Dumont MG, Lueders T, Manefield M, Friedrich MW et al. (2007). DNA stable-isotope probing. Nat Protoc 2: 860–866. [DOI] [PubMed] [Google Scholar]
- Nguyen RT, Harvey RH. (1997). Protein and amino acid cycling during phytoplankton decomposition in oxic and anoxic waters. Organic Geochemistry 27: 115–128. [Google Scholar]
- Nikrad MP, Cottrell MT, Kirchman DL. (2012). Abundance and single-cell activity of heterotrophic bacterial groups in the western Arctic Ocean in summer and winter. Appl Environ Microbiol 78: 2402–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikrad MP, Cottrell MT, Kirchman DL. (2014). Uptake of dissolved organic carbon by gammaproteobacterial subgroups in coastal waters of the West Antarctic Peninsula. Appl Environ Microbiol 80: 3362–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N, Guillier M, Barnard TJ, Buchanan SK. (2010). TonB-dependent transporters: regulation, structure, and function. Ann Rev Microbiol 64: 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan LA, Rinna J, Humphreys G, Weightman AJ, Fry JC. (2005). Fluviicola taffensis gen. nov., sp. nov., a novel freshwater bacterium of the family Cryomorphaceae in the phylum 'Bacteroidetes'. Int J Syst Evol Microbiol 55: 2189–2194. [DOI] [PubMed] [Google Scholar]
- Orsi WD, Edgcomb VP, Christman GD, Biddle JF. (2013). Gene expression in the deep biosphere. Nature 499: 205–208. [DOI] [PubMed] [Google Scholar]
- Orsi WD, Smith JM, Wilcox HM, Swalwell JE, Carini P, Worden AZ et al. (2015). Ecophysiology of uncultivated marine euryarchaea is linked to particulate organic matter. ISME J 9: 1747–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington JT, Castro CG, Collins CA, Evans WWI, Friederich GE, Michisaki RP et al. (2010). The Northern and Central California Coastal Upwelling System. In: Liu K-K (ed), Carbon and Nutrient Fluxes in Continental Margins. Springer-Verlag: Berlin Heidelberg, pp 29–44.
- Pennington TJ, Chavez FP. (2000). Seasonal fluctuations of temperature, salinity, nitrate, chlorophyll, and primary production at station H3/M1 over 1989-1996 in Monterey Bar, California. Deep Sea Res II 47: 947–973. [Google Scholar]
- Pernthaler A, Preston CM, Pernthaler J, Delong EF, Amann R. (2002). Comparison of fluorescently labeled oligonucleotide and polynucleotide probes for the detection of pelagic marine bacteria and archaea. Appl Environ Microbiol 68: 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappe MS, Connon SA, Vergin KL, Giovannoni SJ. (2002). Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418: 630–633. [DOI] [PubMed] [Google Scholar]
- Rappe MS, Kemp PF, Giovannoni SJ. (1997). Phylogenetic diversity of marine coastal picoplankton 16S rRNA genes clones from the continental shelf off Cape Hatteras, North Carolina. Limnol Oceanogr 42: 811–826. [Google Scholar]
- Ravenschlag K, Sahm K, Pernthaler J, Amann R. (1999). High bacterial diversity in permanently cold marine sediments. Appl Environ Microbiol 65: 3982–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reysenbach AL, Flores GE. (2008). Electron microscopy encounters with unusual thermophiles helps direct genomic analysis of Aciduliprofundum boonei. Geobiology 6: 331–336. [DOI] [PubMed] [Google Scholar]
- Rinta-Kanto JM, Sun S, Sharma S, Kiene RP, Moran MA. (2012). Bacterial community transcription patterns during a marine phytoplankton bloom. Environ Microbiol 14: 228–239. [DOI] [PubMed] [Google Scholar]
- Santoro AE, Casciotti KL, Francis CA. (2010). Activity, abundance and diversity of nitrifying archaea and bacteria in the central California Current. Environ Microbiol 12: 1989–2006. [DOI] [PubMed] [Google Scholar]
- Santoro AE, Dupont CL, Richter RA, Craig MT, Carini P, McIlvin MR et al. (2015). Genomic and proteomic characterization of ‘Candidatus Nitrosopelagicus brevis:' An ammonia-oxidizing archaeon from the open ocean. Proc Natl Acad Sci U S A 112: 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer K, Rodionov DA, de Reuse H. (2008). New substrates for TonB-dependent transport: do we only see the 'tip of the iceberg'? Trends Biochem Sci 33: 330–338. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AK, Becker JW, Ottesen EA, Bryant JA, Duhamel S, Karl DM et al. (2014). Distinct dissolved organic matter sources induce rapid transcriptional responses in coexisting populations of Prochlorococcus, Pelagibacter and the OM60 clade. Environ Microbiol 16: 2815–2830. [DOI] [PubMed] [Google Scholar]
- Sigman DM, Casciotti KL, Andreani M, Barford C, Galanter M, Bohlke JK. (2001). A bacterial method for the nitrogen isotopic analysis of nitrate in seawater and freshwater. Anal Chem 73: 4145–4153. [DOI] [PubMed] [Google Scholar]
- Sizemore RK, Stevenson LH. (1974). Environmental factors associated with proteolytic activity of estuarine bacteria. Life Sci 15: 1425–1432. [DOI] [PubMed] [Google Scholar]
- Smith DC, Simon M, Alldredge AL, Azam F. (1982). Intense hydrolytic enzyme activity on marine aggregates and implications for rapid particle dissolution. Nature 359: 139–142. [Google Scholar]
- Smith JM, Chavez FP, Francis CA. (2014). Ammonium uptake by phytoplankton regulates nitrification in the sunlit ocean. PLoS One 9: e108173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl DA, Amann R. (1991) Development and application of nucleic acid probes. In: Stackebrandt E, Goodfellow M (eds), Nucleic Acid Techniques in Bacterial Systematics. John Wiley & Sons, Ltd.: Chichester, England, pp 205–248. [Google Scholar]
- Stingl U, Desiderio RA, Cho JC, Vergin KL, Giovannoni SJ. (2007). The SAR92 clade: an abundant coastal clade of culturable marine bacteria possessing proteorhodopsin. App Environ Microbiol 73: 2290–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue E, Nishiyama S, Kamo M, Tsugita A. (1995). Bacterial membranes: possible source of a major dissolved protein in seawater. Geochimica et Cosmochimica Acta 59: 2643–2648. [Google Scholar]
- Taylor GT. (1995). Microbial degradation of sorbed and dissolved protein in seawater. Limnol Oceanogr 40: 875–885. [Google Scholar]
- Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336: 608–611. [DOI] [PubMed] [Google Scholar]
- Thomsen HA, Buck KR. (1998). Nanoflagellates of the central California waters: taxonomy, biogeography and abundance of primitive, green flagellates (Pedinophyceae, Prasinophyceae). Deep Sea Res II 45: 1687–1707. [Google Scholar]
- Tiera E, Reinthaler T, Pernthaler J, Herndl GJ. (2004). Combining catalyzed reporter deposition-fluorescence in situ hybridization and microautoradiography to detect substrate utilization by bacteria and archaea in the deep ocean. Appl Environ Microbiol 70: 4411–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach E, Vergin KL, Young L, Morse A, Larson GL, Giovannoni SJ. (2001). Unusual bacterioplankton community structure in ultra-oligotrophic Crater Lake. Limnol Oceanogr 46: 557–572. [Google Scholar]
- Worden AZ, Follows MJ, Giovannoni SJ, Wilken S, Zimmerman AE, Keeling PJ. (2015). Rethinking the marine carbon cycle: factoring in the multifarious lifestyles of microbes. Science 347: 1257594. [DOI] [PubMed] [Google Scholar]
- Yoon J, Matsuo Y, Adachi K, Nozawa M, Matsuda S, Kasai H et al. (2008). Description of Persicirhabdus sediminis gen. nov., sp. nov., Roseibacillus ishigakijimensis gen. nov., sp. nov., Roseibacillus ponti sp. nov., Roseibacillus persicicus sp. nov., Luteolibacter pohnpeiensis gen. nov., sp. nov. and Luteolibacter algae sp. nov., six marine members of the phylum ‘Verrucomicrobia', and emended descriptions of the class Verrucomicrobiae, the order Verrucomicrobiales and the family Verrucomicrobiaceae. Int J Sys Evol Microbiol 58: 998–1007. [DOI] [PubMed] [Google Scholar]
- Zeigler Allen L, Allen EE, Badger JH, McCrow JP, Paulsen IT, Elbourne LD et al. (2012). Influence of nutrients and currents on the genomic composition of microbes across an upwelling mosaic. ISME J 6: 1403–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, Xie W, Martin-Cuadrado AB, Rodriguez-Valera F. (2015). Marine Group II Archaea, potentially important players in the global carbon cycle. Front Microbiol 6: 1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.