

Abstract
Few systematic assessments of developmental forms of hydrocephalus exist. We reviewed MRIs and clinical records of patients with infancy-onset hydrocephalus. Among 411 infants, 236 had hydrocephalus with no recognizable extrinsic cause. These children were assigned to one of five subtypes and compared on the basis of clinical characteristics, developmental and surgical outcomes. At an average age of 5.3 years, 72% of children were walking independently and 87% could eat by mouth. 18% had epilepsy. Distinct patterns of associated malformations and syndromes were observed within each subtype. On average, children with aqueductal obstruction, cysts and encephaloceles had worse clinical outcomes than those with other forms of developmental hydrocephalus. 53% of surgically-treated patients experienced at least one shunt failure, but hydrocephalus associated with posterior fossa crowding required fewer shunt revisions. We conclude that each subtype of developmental hydrocephalus is associated with distinct clinical characteristics, syndromology, and outcomes, suggesting differences in underlying mechanisms.
Keywords: hydrocephalus, aqueductal stenosis, myelomenigocele, encephaloceles
Hydrocephalus, characterized by progressive accumulation of cerebrospinal fluid (CSF) within the ventricular system of the brain, affects approximately one in 1,000 births1. Hydrocephalus is a well-established consequence of acquired events such as intraventricular hemorrhage, but also occurs without a clear extrinsic cause, especially in infants. Our understanding of the causes of non-extrinsic forms of hydrocephalus, which we refer to as developmental hydrocephalus, is particularly limited.
When hydrocephalus develops during infancy, it has significant clinical implications. Early-onset hydrocephalus conveys a high risk of neurodevelopmental impairment, but because developmental subtypes of hydrocephalus are often not defined, the extent to which outcome depends upon subtype is unclear. Similarly, the extent to which surgical outcome (in particular, the rate of shunt failure) differs by subtype is also unclear. As a result, most available information about the both the causes and the clinical consequences of developmental hydrocephalus is generic, rather than subtype-specific.
Using existing medical records, we investigated the clinical characteristics of a large cohort of infants with hydrocephalus, with particular emphasis on hydrocephalus without an extrinsic cause. We sought to better define the major clinical-radiographic subtypes of developmental hydrocephalus and their relative frequency, as well as the additional physical malformations and syndromes seen within each group, which could hold clues to underlying functional mechanisms. We then compared subtypes on the basis of concrete and quantifiable markers of developmental and surgical outcome.
METHODS
Patient identification
With the intention of performing a retrospective review, and with the approval of the Seattle Children’s Hospital Institutional Review Board, we searched the hospital’s imaging database using the terms “hydrocephalus,” “ventriculomegaly,” and “aqueductal stenosis” for MRIs that had been performed in children 12 months of age or less.
Patient inclusion and exclusion criteria
We used the definition of hydrocephalus proposed by the International Hydrocephalus Working Group: “an active [and progressive] distension of the ventricular system…resulting from inadequate passage of CSF from its point of production… to its point of absorption…2” Accordingly, we included children with ventricular distension, regardless of whether they had clinical signs of increased intracranial pressure. We excluded children with hydranencephaly, children in whom excessive CSF was purely extraaxial, and children with hydrocephalus ex vacuo, unless progressive ventricular distension was also evident.
Confirmation of hydrocephalus and review of anatomy
Two authors (HMT, GEI) assessed scans to confirm ventricular dilatation. When we observed only equivocal ventricular dilatation, we looked for evidence of progression on follow-up imaging studies, or accelerated head growth on growth charts. To quantify ventricular dilatation, we calculated an Evans index using standard methods3. We also assessed brain anatomy in detail.
Clinical information
We reviewed the medical records of all identified children. In addition to basic demographic characteristics, date of birth and date of death or last follow-up, we recorded associated medical conditions, results of diagnostic testing, surgical history and cause of death (if known, and if applicable). Time of onset of hydrocephalus was defined as the age at which it was first confirmed on imaging. We recorded whether children of any age were able to eat safely by mouth and whether they required physical or speech therapy. We also recorded whether children had epilepsy. We recorded whether children 2 years of age and older were able to walk independently. We recorded the type and timing of all hydrocephalus-related surgical procedures, and whether there was a history of shunt infection or mechanical shunt failure.
Classification of subtypes
We sought to delineate subtypes of developmental hydrocephalus on the basis of major radiographic findings, particularly the apparent point of CSF obstruction. Since infants with developmental forms of hydrocephalus may have multiple points of CSF obstruction, not all of which can be easily defined on MRI2, we classified them as having apparent obstruction at the level of the aqueduct, at the level of the posterior fossa, or neither. We also incorporated readily apparent imaging findings such as intracranial cysts and encephaloceles, as well as the distinctive Chiari II malformation seen in association with myelomeningoceles.
Statistical analysis
To compare differences in outcome variables across subtypes of hydrocephalus, we performed Chi-square, Fisher’s exact, anova and non-parametric mean tests. All analyses were performed using Stata12 software (Stata Statistical Software: Release 12. College Station, TX: StataCorp LP).
RESULTS
We identified 424 infants who were diagnosed with or treated for hydrocephalus between 2002 and 2012, 411 of whom had sufficiently detailed records to allow assessment of etiology (Supplementary Table 1). Of these infants, 155 (37.7%) had hydrocephalus attributable to a known extrinsic event, including intraventricular (N=96) or intraparenchymal (N=10) hemorrhage, neoplasm (N=20), infection (N=16), and trauma (N=8). Another 20 had clinical or imaging signs that implied a cryptic extrinsic cause, including chorioretinal scarring (suggesting intrauterine infection), or apparent hemosiderin on MRI (suggesting intrauterine hemorrhage). The remaining 236 patients had hydrocephalus without any evident extrinsic cause, so were classified as having developmental hydrocephalus.
Subtypes of developmental hydrocephalus
232 of 236 children could be placed in one of five clinical-radiographic categories (Supplementary Table 2): hydrocephalus associated with myelomeningocele (MMH) (Figure 1), (N=78); hydrocephalus associated with apparent aqueductal obstruction (AQ) (Figure 2), (N=59); hydrocephalus associated with posterior fossa crowding (PFC) (Figure 3), (N=25); hydrocephalus associated with cysts or cephaloceles (Figure 4) (N=40); and communicating hydrocephalus (Figure 5), with no radiographic evidence of obstruction (N=31). Four children could not be categorized: two with Vein of Galen malformations and one with a dural AV fistula, none of whom had detailed pre-surgical imaging to allow the point of obstruction to be determined, and one with neurocutaneous melanosis, with multiple subarachnoid adhesions. These children were included in the overall analyses, but not within subgroup analyses.
Figure 1. MM-associated hydrocephalus. A–D: Classic Chiari II malformation.
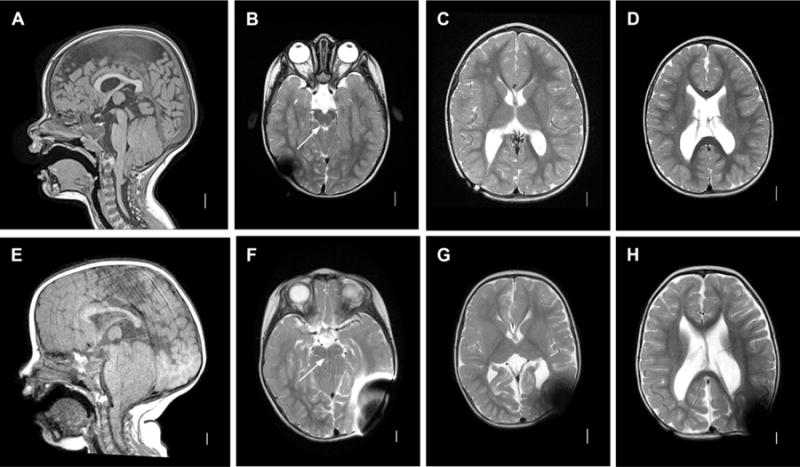
Sagittal T1 MRI image (A) demonstrating classic features of a Chiari II malformation including elongated pons and downwardly displaced medulla, tectal beaking, small posterior fossa with vertically oriented tentorium, with cerebellar tonsillar ectopia below the foramen magnum line. Axial l T2 images (B–D). Note patency of aqueduct in B (arrow). E–H: Chiari II with aqueductal compression. Sagittal T1 image (A) demonstrating similar anatomic configuration as A, but with more prominent posterior fossa crowding and aqueductal compression. Axial T2 images (F–H). Note absence of patent aqueduct in F (arrow).
Figure 2. Aqueductal obstruction. A–D: Aqueductal obstruction associated with L1CAM mutation.
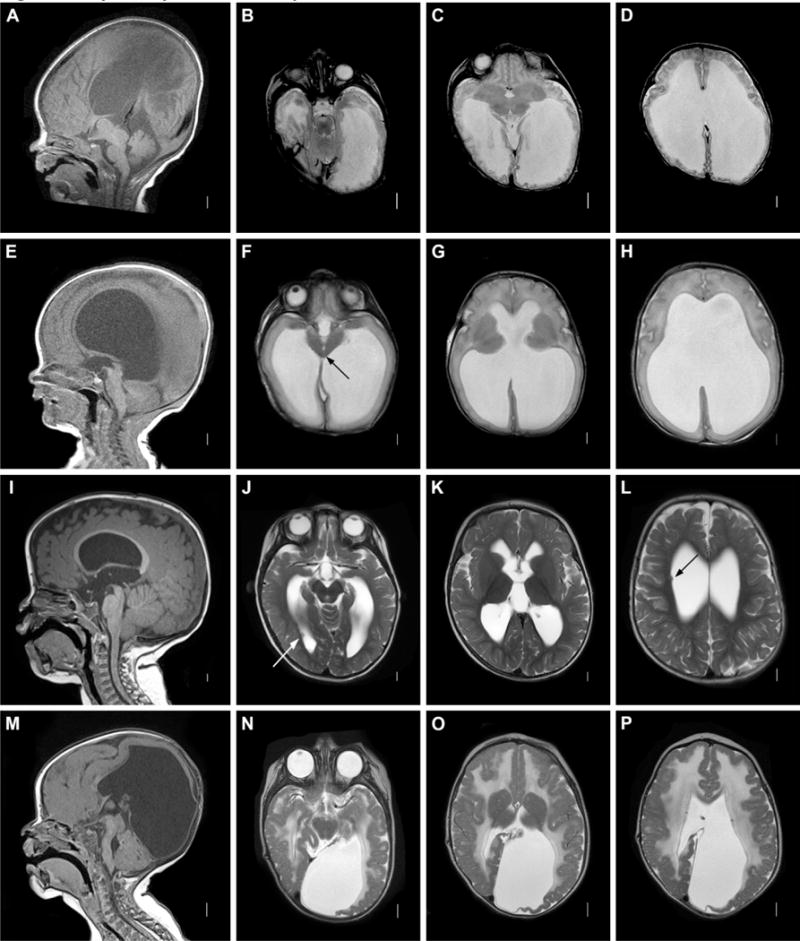
Sagittal T1 MRI image (A) showing complete aqueductal occlusion (arrow) and small cerebellum. Axial T2 images (B–D) demonstrating extensive dilation of lateral ventricles. E–H: Mesencephalosynapsis. Sagittal T1 images (A) demonstrating inferior aqueductal occlusion with funneling (arrow). Axial T2 images (F–H) showing fused inferior colliculi (arrow) and severe ventricular dilatation. I–L: Periventricular nodular heterotopia with aqueductal nodule. Sagittal T1 image (A) showing obstructive nodule within aqueduct (arrow). Axial T2 images (J–L) demonstrating moderate ventricular dilatation and periventricular nodular heterotopia (arrows.) M–P: Muscle-Eye-Brain disease. Sagittal T1 image (M) showing enlarged tectum with complete aqueductal obstruction (arrow), hypoplastic and kinked brainstem, and cerebellar dysplasia with cysts. Axial T2 images (N–P) showing cobblestone cortex and abnormal white matter.
Figure 3. Posterior fossa crowding. A–D: Chiari I malformation.
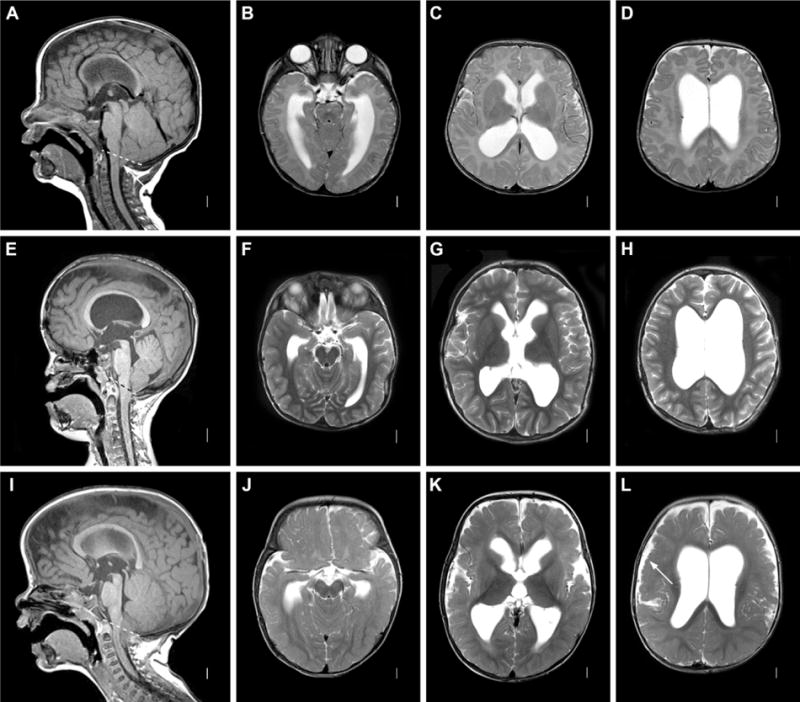
Axial T1 MRI image (A) showing open aqueduct and relatively large-appearing cerebellum, with herniation of tonsils below foramen magnum (dashed line). Axial T2 images (B–D) showing moderate dilatation of lateral ventricles. E–H: Pfeiffer Syndrome with multi-suture synostosis. Sagittal T2 images (E) showing midface retrusion, widely patent aqueduct and small, crowded posterior fossa with tonsillar herniation below the foramen magnum (dashed line) in patient with a confirmed FGFR2 mutation. Axial T2 images (F–H) showing moderate ventricular dilatation. I–L: MPPH syndrome. Sagittal T1 image (I) showing widely patent aqueduct with tonsillar herniation through foramen magnum (dashed line). Axial T2 image showing moderate ventricular dilation (J–L) and extensive bilateral perisylvian polymicrogyria (L, arrow).
Figure 4. Cysts and cephaloceles. A–D: 3rd ventricular cyst.
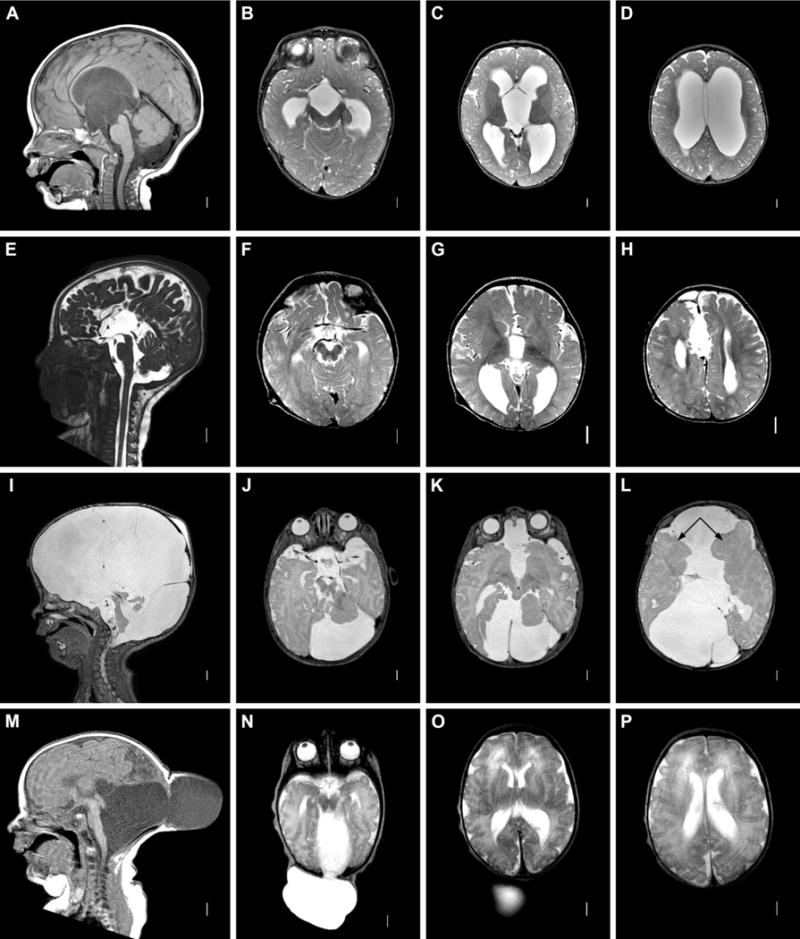
Sagittal T1 (A) and axial T2 (B-d) MRI images demonstrating small, obstructive cyst with lack of additional brain malformations or cortical dysplasia. E–H: intrahemispheric cyst. Sagittal CISS and axial T2 images showing interhemispheric cyst with absent corpus callosum. I–L: complex cystic malformation. Sagittal (I) and axial (J–L) T2 images demonstrating brainstem hypoplasia and extensive infolded, dysplastic cerebral hemispheres (L, arrows). M–P: Encephalocele. Sagittal T1 images (M) showing cephalocele, with fluid collection in continuity with posterior fossa. Axial T2 images (N–P) demonstrating relatively normal-appearing cerebral hemispheres.
Figure 5. Communicating hydrocephalus. A–D: mild idiopathic communicating hydrocephalus.
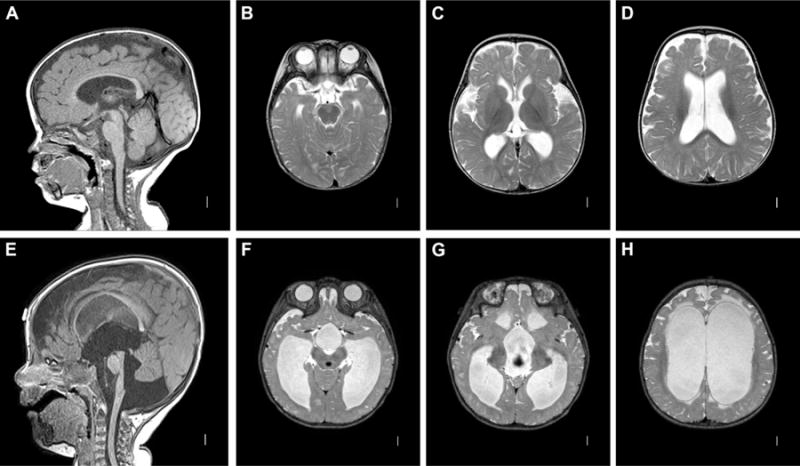
Sagittal T1 MRI image (AS) showing open aqueduct and absence of posterior fossa crowding. Axial T2 images (B–D) showing rounded, mildly dilated ventricles with generous extraaxial space. E–H: severe idiopathic communicating hydrocephalus. Sagittal T1 image (E) showing enlarged aqueduct, 4th ventricle, and excess fluid within the posterior fossa. Axial T2 images (E–H) demonstrate marked ventriculomegaly with transependymal flow.
Additional brain malformations (Supplementary Table 3)
All but two children with MM-associated hydrocephalus had classic Chiari II malformations. The other two had the brainstem features of a Chiari II, but with a partially absent cerebellum, a feature presumed to be the result of a prenatal disruption4. Among children with AQ, 14 had additional midline brainstem and cerebellar malformations. Three had nodular aqueductal obstruction, and two had the characteristic features of muscle-eye-brain disease. PFC was associated with Chiari I malformations in 11 children, four of whom had megalencephaly.
Among children in the cysts and cephaloceles group, agenesis of the corpus callosum (ACC) was seen in seven, always in conjunction with midline cysts. Cortical dysplasia, sometimes extensive, was seen in eight children. Only two children in this category had classic Dandy-Walker malformations. Among children with communicating hydrocephalus, two had midline malformations, including ACC and absent septum pellucidum; otherwise, brain malformations were uncommon.
Additional physical malformations and identifiable clinical syndromes
Additional physical anomalies were identified in 48 of 232 children (21%), 27 of whom had been diagnosed with specific syndromes (Table 2). No specific syndromes were present in patients with MMH, and additional physical malformations were rare. Among children with AQ, eight of 59 (14%) had a defined syndrome, including six with Hydrocephalus with Stenosis of the Aqueduct of Sylvius (HSAS) associated with L1CAM mutations (sometimes referred to as Bickers-Adams syndrome). Only one additional physical anomaly was seen.
Table 2.
Clinical syndromes and additional anomalies by category
| Clinical syndrome/Additional anomalies | Genetic cause (N/N tested) |
|---|---|
| MM-associated | |
| Defined syndrome: 0/78 (0%) | |
| Additional physical anomalies: 3/78 (4%) | |
| MM with structural cardiac (1), atypical thoracic MM with vert seg defects, absent L kidney, absent R testis, structural cardiac (1), atypical thoracic MM with multiple vert seg defects (1) |
|
|
| |
| Proximal obstruction | |
| Defined syndrome: 8/59 (14%) | |
| Aqueductal stenosis without additional findings (47), including HSAS. | L1CAM (6/8) |
| Muscle-eye-brain (2) | POMGNT1 (2/2) |
| Additional physical anomalies without defined syndrome: 1/51 (2%) | |
| Unilateral anophthalmia (1) | |
|
| |
| Distal obstruction | |
| Defined syndrome: 15/25 (60%) | |
| Crouzon (4) | FGFR2 (1/1) |
| Pfeiffer (3) | FGFR2 (3/3) |
| Carpenter (1) | Not tested |
| Achondroplasia (3) | FGFR3 (1/1) |
| Thanatophoric dysplasia (1) | FGFR3 (1/1) |
| Spondyloepiphyseal dysplasia (1) | Not tested |
| Undefined skeletal dysplasia | FGFR3 (0/1) |
| MPPH (1) | PiK3CA/AKT3 pathway genes not tested |
| Additional physical anomalies without defined syndrome 2/10 (20%) | |
| Additional anomalies: unilateral microphthalmia (with megalencephaly), upper cervical fusion anomaly | |
|
| |
| Cysts and cephaloceles | |
| Defined syndrome: 3/39 (8%) | |
| Chudley-McCullough (1) | GPSM2 (1/1) |
| Oro-facial-digital type 1 (1) | OFD1 not tested |
| Opitz G/BBB (1) | MID1 not tested |
| Additional physical anomalies without defined syndrome: 7/36 (19%) | |
| Cysts in multiple organ systems and polysyndactyly (1), multicystic kidneys (1), ambiguous genitalia and polydactyly (1), syndactyly and limb reduction with skin appendages (1), structural renal with vert seg defects and interrupted aortic arch (1), vert seg defects and cleft palate (1), structural renal (1), TEF (1) | (OFD1 0/1,other genes not known/not tested) |
|
| |
| Communicating | |
| Defined syndrome: 2/31 (6%) | |
| Cardio-facio-cutaneous with pulmonic stenosis (1) | BRAF (1/1) |
| Gorlin (1) | PTCH1 not tested |
| Additional physical anomalies without defined syndrome: 4/29 (14%) | |
| CDH (3), structural cardiac (1) | (genes not known) |
MM: myelomeningocele, HSAS: Hydrocephalus with Stenosis of the Aqueduct of Sylvius (associated with L1CAM mutations), TEF: tracheoesophageal fistula, MPPH: megalencephaly, polydactyly, polymicrogyria and hydrocephalus syndrome.
Only three of 39 children (6%) with cysts and cephaloceles had an identifiable syndrome, but seven (19%) had additional malformations, most conspicuously renal cysts and digit abnormalities.
Among children with communicating hydrocephalus, only two of 31 (6%) had a defined syndrome, but five had major physical anomalies, including two structural cardiac defects and three congenital diaphragmatic hernias. Remarkably, 15 of 25 children (60%) with PFC had defined disorders, most frequently multi-suture craniosynostosis or skeletal dysplasia syndromes. Additional physical malformations were seen in two of the ten remaining children (20%).
Developmental and surgical outcome
After assessing basic characteristics of children with hydrocephalus (Table 3), we compared children on the basis of objective clinical criteria. At an average age of 5.3 years, 87% could eat by mouth, and 72% of children over two years of age (86% of children without MM) were walking independently. Seventy percent required physical therapy (56% of those without MM), 41% were receiving speech therapy, and 18% had epilepsy. We noted statistically significant differences in the need for physical therapy, speech therapy, and the presence of epilepsy across subtypes.
Table 3.
Clinical outcome of children with developmental hydrocephalus
| All hydrocephalus (n=236) | MM (n= 78) | Proximal obstruction (n=60) | Distal obstruction (n=25) | Cysts and celes (n=38) | Communicating (n=31) | P value for heterogeneity | |
|---|---|---|---|---|---|---|---|
|
| |||||||
|
NGT- or GT-fed N (%) |
29 (12. 8) | 6 (7.9) | 10 (17.0) | 5 (20.8) | 4 (11.1) | 4 (12.9) | 0.40 |
|
| |||||||
| Mobility (age ≥2 years) | |||||||
| Walking independently | 114 (65.9) | 19 (31.1) | 32 (74.4) | 19 (95.0) | 24 (85.7) | 16 (94.1) | 0.28 (excluding MM) |
| Crutches or walker | 14 (8.1) | 121 (19.7) | 2 (4.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Wheelchair | 45 (26.0) | 301 (49.2) | 9 (20.9) | 1 (5.0) | 4 (14.3) | 1 (5.8) | |
|
| |||||||
|
Physical therapy N (%) |
126 (70.0) | 631 (92.7) | 28 (68.3) | 10 (47.6) | 17 (68.0) | 8 (32.0) | 0.02 (excluding MM) |
|
| |||||||
|
Speech therapy N (%) |
71 (40.5) | 19 (29.2) | 23 (59.0) | 12 (54.6) | 10 (58.3) | 7 (28.0) | 0.02 |
|
| |||||||
|
Epilepsy N (%) |
40 (17.3) | 5 (6.5) | 20 (33.3) | 2 (8.0) | 11 (29.0) | 2 (6.5) | <0.001 |
|
| |||||||
|
Deceased N (%) |
14 (6.0) | 1 (1.3) | 7 (11.5) | 2 (8.0) | 2 (5.3) | 2 (6.5) | 0.16 |
Note: figures in bold reflect p values less than 0.005
May primarily reflect disability from myelomenigocele.
Surgery for hydrocephalus was performed in 72% of children, with the highest proportion in MMH (87%) and the lowest in communicating hydrocephalus (19%) (Table 4). Among patients who underwent VP shunt placement, 53% experienced at least one shunt failure, a result that did not differ statistically by subtype. However, the 10-year shunt failure rate demonstrated highly statistically significant difference across subtypes, a result driven by the lower revision rates seen in children with PFC.
Table 4.
Surgical outcome of children with developmental hydrocephalus
| All hydrocephalus (n=236) | MM (n= 78) | Proximal obstruction (n=60) | Distal obstruction (n=25) | Cysts and celes (n=38) | Communicating (n=31) | P value for heterogeneity | |
|---|---|---|---|---|---|---|---|
|
Any surgery1 N (%) |
167 (72.0) | 68 (87.2) | 47 (78.3) | 13 (52.0)2 | 33 (86.8) | 6 (19.4) | <0.001 |
|
Total surgeries2 Mean +/−SD (min, max) |
2.3 ± 1.6 (1, 9) | 2.3 ± 1.7 (1,9) | 2.3 ± 1.5 (1,6) | 1.3 ± 0.8 (1,3) | 2.8 ± 1.8 (1,7) | 1.8 ± 1.2 (1,3) | 0.04 |
| VP shunt N (%) | 162 (68.6) | 67 (85.9) | 47 (78.3) | 11 (44.0) | 28 (73.7) | 6 (19.4) | <0.001 |
|
Any shunt failure3 N (%) |
85 (52.8) | 39 (58.2) | 26 (55.3) | 2 (18.2) | 15 (55.6) | 2 (33.3) | 0.13 |
|
Failure rate3 (number of failures per 10 child-years)3 |
2.0 ± 3.9 | 2.0 ± 3.6 | 1.8 ± 3.6 | 0.4 ± 0.9 | 3.0 ± 5.5 | 1.4 ± 2.8 | <0.001 |
Shunt, ETV, or cyst fenestration.
Among children who underwent surgery.
Among children with VP shunt.
DISCUSSION
We investigated a large series of infants with hydrocephalus and found that 58% had no obvious extrinsic cause of their condition. Almost all children could be placed into one of five subtypes based on key clinical and radiographic features. The additional malformations and syndromes seen within subtypes suggest distinct underlying mechanisms, a notion further underscored by differences in basic clinical characteristics, developmental and surgical outcomes.
Hydrocephalus associated with myelomeningocele
The MM-associated Chiari II malformation is characterized by several anatomic features that combine to cause apparent aqueductal and posterior fossa crowding5, which may contribute to the earlier onset and greater ventricular dilatation seen in this group of children compared to those with posterior fossa crowding alone. Though these children had mobility problems as a result of their myelomeningoceles, the proportion of children requiring speech therapy, who had epilepsy and who were deceased was relatively low.
Mechanistically, the Chiari II malformation is usually viewed as a consequence of chronic intrauterine CSF leakage6, 7, a notion supported by animal models7–9 and by the results of clinical trials in utero repair of myelomeningocele, which show improvement in hydrocephalus10. Of note, mutations in planar cell polarity genes play a role in the pathogenesis of some neural tube defects in humans11–13, while mutations in other planar cell polarity genes give rise to hydrocephalus independent of MM in mice14, which is postulated to be the result of impaired development and function of ependymal cilia15. The hydrocephalus that accompanies MM may therefore be both a consequence of mechanical obstruction and, in a subset of patients, genetically-based differences in CSF flow.
Aqueductal obstruction
Hydrocephalus associated with aqueductal obstruction was early in onset and associated with the greatest severity of ventricular dilation. Not surprisingly, this group of children had the worst developmental outcomes of any group, though the need for multiple surgeries and the total number of surgical procedures undergone by each patient was similar to most other subtypes.
Of the eight children with aqueductal obstruction tested, six had mutations in L1CAM, which plays key roles in neuronal migration and axon guidance16. Two children had muscle-eye-brain disease caused by mutations in POMGNT1, which also leads to aberrant migration of neurons and likely contributes to an obstructive brainstem malformation17, 18. Notably, 15 children with aqueductal obstruction had additional mid-hindbrain malformations, most often mesencephalosynapsis with or without rhombencephalosynapsis. These malformations are thought to be genetically based, though the genes involved are not known.
Though we excluded patients with known IVH or infection, some children in this group could have aqueductal obstruction as the result of an unrecognized extrinsic event. Evidence of obstructive microhemorrhage in a structurally normal aqueduct is sometimes evident only upon autopsy19. IVH has also been shown to induce nodules of neural progenitor cells within the ventricular system20. This mechanism could potentially explain the nodular obstruction seen in three children.
Posterior fossa crowding
PFC, with or without hydrocephalus, is often described in conjunction with alterations of skull shape21–24. Skeletal dysplasias and multi-suture synostosis syndromes were seen in over half the patients in this category, with mutations in FGFR genes found in all but one of those children who underwent testing. The bony changes associated with FGFR-related syndromes are well recognized. However, mutations in FGFR-mediated signaling pathways can also cause excessive growth of the brain itself25–28. This provides a link between FGFR-associated hydrocephalus and megalencephaly-associated hydrocephalus, which was present in six children in this category.
The mechanism leading to this subtype of hydrocephalus may be a progressive mismatch between skull size and brain size, which is underscored by the relatively late onset of hydrocephalus seen in this group of children. Clinical outcomes were similar to the group as a whole, though fewer children had epilepsy. Notably, children in this category who underwent hydrocephalus-related surgery were much less likely to experience shunt failure, possibly because many also underwent skull surgery, possibly rendering these children less shunt-dependent though improvedment of CSF flow dynamics.
Cysts and cephaloceles
Cysts and cephaloceles are known causes of hydrocephalus29, 30, but the pathogenesis of these malformations is poorly understood. Simple cysts have been attributed to accidental entrapment of CSF within a split layer of arachnoid31. However, more complex cystic malformations can have a genetic basis, with numerous syndromes described in the literature, including oro-facial-digital syndrome32, Chudley-McCullough syndrome33 and Aicardi syndrome34. This subtype was associated with the highest proportion of children with epilepsy, likely reflecting the inclusion of complex cystic malformations and encephaloceles with associated cortical dysplasia.
We suspect that several molecular mechanisms underlie cyst- and cephalocele-associated hydrocephalus, which is supported by the spectrum of MRI findings seen in this group of children. Only three patients had a defined syndrome, but additional physical anomalies were more common in this subtype than in any other. Several of the malformations seen in association with complex cystic malformations and encephaloceles, including renal cysts and poly- or syndactyly, hint at defective ciliary signaling35.
Communicating hydrocephalus
Hydrocephalus without apparent obstruction is a known consequence of IVH and infection, presumably due to inflammation in the subarachnoid space. The pathophysiology of idiopathic communicating hydrocephalus is less clear, with cryptic hemorrhage36, immaturity of the arachnoid granulations37, 38, excessive skull growth39, lymphatic dysplasia40 and elevated venous outflow resistance41–43 all invoked as possible causes. A genetic underpinning of idiopathic communicating hydrocephalus has long been suspected, based on the observation that a substantial minority of affected children have close family members with macrocephaly38, 44, 45. In our series, this subtype had a much higher proportion of males than others, which suggests an X-linked contribution.
The highly variable age of onset and severity seen among children with communicating hydrocephalus suggests that multiple functional mechanisms may be operating. Notably, five patients in this group had malformations associated with increased vascular pressure, confirming that high venous outflow resistance may be important in this form of hydrocephalus.
Limitations of this study
This study provides detailed anatomic and clinical information on a large cohort of children; however, it is limited to those who underwent MRI scans. Those who underwent only CT or ultrasound were not included, which could bias the results towards more severely affected children. This study is also limited by its retrospective and observational nature. Children with multiple medical needs are likely to have frequent medical appointments with detailed documentation available for review; children who are more mildly affected may be more easily lost to follow-up. Therefore, this study may be biased towards more severely affected children, with a resulting overestimation of the proportion with disabilities. In contrast, this study may underestimate the frequency of outcomes such as epilepsy and shunt failure; these outcomes accrue over time and would be expected to increase if the cohort were followed longer.
CONCLUSION
Among 411 infants with hydrocephalus, 60% had no recognizable extrinsic cause of their condition. All but four of these infants could be placed in one of five categories based on key clinical and radiographic features. The clinical characteristics, patterns of additional malformations and syndromes, as well as statistically significant differences in developmental and surgical outcome observed across subtypes suggest distinct underlying mechanisms. We suspect that these mechanisms will be better elucidated, and subtypes further refined, with advances in imaging and discovery of new genes. This in turn will allow for more nuanced counselling of affected families and more clinically relevant comparisons of outcome and response to treatment.
Supplementary Material
Table 1.
Basic characteristics of 236 children with developmental hydrocephalus
| All hydrocephalus (n=236) | MM (n= 78) | Proximal obstruction (n=60) | Distal obstruction (n=25) | Cysts and celes (n=38) | Communicating (n=31) | |
|---|---|---|---|---|---|---|
|
| ||||||
|
Male N (%) |
131 (55.3) | 43 (55.1) | 14 (56.0) | 14 (56.0) | 18 (47.4) | 22 (71.0) |
|
| ||||||
| Age at diagnosis N (%) | ||||||
| Prenatal and <1w | 166 (72.2) | 78 (100.0) | 51 (86.4) | 2 (8.0) | 24 (64.9) | 11 (35.4) |
| 1w-6m | 37 (16.1) | 0 (0.0) | 7 (11.9) | 9 (36.0) | 9 (24.3) | 12 (38.7) |
| >6m-12mo | 27 (11.7) | 0 (0.0) | 1 (1.7) | 14 (56.0) | 4 (10.8) | 8 (25.8) |
|
| ||||||
|
Age at last visit (yrs) mean +/− SD (min, max) |
4.6 +/− 3.1 (0.0, 13.1) | 4.9 +/− 3.1 (0.1, 11.7) | 4.9 +/− 3.2 (0.0, 11.8) | 5.2 +/− 3.2 (0.3, 11.3) | 4.7 +/− 3.1 0.4, 13.1) | 3.0 +/− 2.1 (0.1, 10.0) |
|
| ||||||
|
Evans index median (min, max) |
0.44 (0.26, 0.88) | 0.41 (0.30, 0.76) | 0.56 (0.34, 0.88) | 0.41 (0.31, 0.71) | 0.43 (0.31, 0.59) | 0.37 (0.26, 0.80) |
Acknowledgments
This work was performed at Seattle Children’s Hospital and the University of Washington. An earlier version was presented at the Child Neurology Society annual meeting.
Funding (Financial Disclosure). Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number T32NS051171, as well as the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number KL2TR000421 and the Child Neurology Foundation through a Shields Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions. Hannah Tully designed the study, collected and analyzed the data and drafted the manuscript; Gisele Ishak reviewed MRIs; Tessa Rue and Jennifer Dempsey performed statistical analysis of the data; Sam Browd, Kathy Millen, Dan Doherty and Bill Dobyns provided conceptual framework for delineating subtypes of hydrocephalus as well as clinical outcomes.
Declaration of Conflicting Interests. No conflicts of interest have been identified by any of the authors or their collaborators.
Ethical Approval. This study was reviewed by the Human Subjects Protection Program of the Seattle Children’s Research Institute and was granted approval number 14299.
References
- 1.Jeng S, Gupta N, Wrensch M, et al. Prevalence of congenital hydrocephalus in California, 1991–2000. Pediatr Neurol. 2011;45(2):67–71. doi: 10.1016/j.pediatrneurol.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Rekate HL. A contemporary definition and classification of hydrocephalus. Semin Pediatr Neurol. 2009;16(1):9–15. doi: 10.1016/j.spen.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 3.van der Knaap MS, Bakker CJ, Faber JA, et al. Comparison of skull circumference and linear measurements with CSF volume MR measurements in hydrocephalus. J Comput Assist Tomogr. 1992;16(5):737–43. doi: 10.1097/00004728-199209000-00013. [DOI] [PubMed] [Google Scholar]
- 4.Sener RN. Cerebellar agenesis versus vanishing cerebellum in Chiari II malformation. Comput Med Imaging Graph. 1995;19(6):491–4. doi: 10.1016/0895-6111(96)00002-x. [DOI] [PubMed] [Google Scholar]
- 5.Geerdink N, van der Vliet T, Rotteveel JJ, et al. Essential features of Chiari II malformation in MR imaging: an interobserver reliability study–part 1. Childs Nerv Syst. 2012;28(7):977–85. doi: 10.1007/s00381-012-1761-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McLone DG, Knepper PA. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15(1):1–12. doi: 10.1159/000120432. [DOI] [PubMed] [Google Scholar]
- 7.Sweeney KJ, Caird J, Sattar MT, et al. Spinal level of myelomeningocele lesion as a contributing factor in posterior fossa volume, intracranial cerebellar volume, and cerebellar ectopia. J Neurosurg Pediatr. 2013;11(2):154–9. doi: 10.3171/2012.10.PEDS12177. [DOI] [PubMed] [Google Scholar]
- 8.Encinas Hernandez JL, Soto C, Garcia-Cabezas MA, et al. Brain malformations in the sheep model of myelomeningocele are similar to those found in human disease: preliminary report. Pediatr Surg Int. 2008;24(12):1335–40. doi: 10.1007/s00383-008-2276-8. [DOI] [PubMed] [Google Scholar]
- 9.Encinas JL, Garcia-Cabezas MA, Barkovich J, et al. Maldevelopment of the cerebral cortex in the surgically induced model of myelomeningocele: implications for fetal neurosurgery. J Pediatr Surg. 2011;46(4):713–22. doi: 10.1016/j.jpedsurg.2010.11.028. [DOI] [PubMed] [Google Scholar]
- 10.Adzick NS, Thom EA, Spong CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. The New England journal of medicine. 2011;364(11):993–1004. doi: 10.1056/NEJMoa1014379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seo JH, Zilber Y, Babayeva S, et al. Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans. Hum Mol Genet. 2011;20(22):4324–33. doi: 10.1093/hmg/ddr359. [DOI] [PubMed] [Google Scholar]
- 12.Allache R, De Marco P, Merello E, et al. Role of the planar cell polarity gene CELSR1 in neural tube defects and caudal agenesis. Birth Defects Res A Clin Mol Teratol. 2012;94(3):176–81. doi: 10.1002/bdra.23002. [DOI] [PubMed] [Google Scholar]
- 13.Bartsch O, Kirmes I, Thiede A, et al. Novel VANGL1 Gene Mutations in 144 Slovakian, Romanian and German Patients with Neural Tube Defects. Mol Syndromol. 2012;3(2):76–81. doi: 10.1159/000339668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tissir F, Qu Y, Montcouquiol M, et al. Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat Neurosci. 2010;13(6):700–7. doi: 10.1038/nn.2555. [DOI] [PubMed] [Google Scholar]
- 15.Sotak BN, Gleeson JG. Can’t get there from here: cilia and hydrocephalus. Nat Med. 2012;18(12):1742–3. doi: 10.1038/nm.3011. [DOI] [PubMed] [Google Scholar]
- 16.Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10(1):19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- 17.Dobyns WB, Pagon RA, Armstrong D, et al. Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet. 1989;32(2):195–210. doi: 10.1002/ajmg.1320320213. [DOI] [PubMed] [Google Scholar]
- 18.Miller G, Ladda RL, Towfighi J. Cerebro-ocular dysplasia–muscular dystrophy (Walker Warburg) syndrome. Findings in 20-week-old fetus. Acta Neuropathol. 1991;82(3):234–8. doi: 10.1007/BF00294451. [DOI] [PubMed] [Google Scholar]
- 19.Lategan B, Chodirker BN, Del Bigio MR. Fetal hydrocephalus caused by cryptic intraventricular hemorrhage. Brain Pathol. 2010;20(2):391–8. doi: 10.1111/j.1750-3639.2009.00293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yung YC, Mutoh T, Lin ME, et al. Lysophosphatidic acid signaling may initiate fetal hydrocephalus. Sci Transl Med. 2011;3(99):99ra87. doi: 10.1126/scitranslmed.3002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furtado SV, Thakre DJ, Venkatesh PK, et al. Morphometric analysis of foramen magnum dimensions and intracranial volume in pediatric Chiari I malformation. Acta Neurochir (Wien) 2010;152(2):221–7. doi: 10.1007/s00701-009-0480-5. discussion 7. [DOI] [PubMed] [Google Scholar]
- 22.Aydin S, Hanimoglu H, Tanriverdi T, et al. Chiari type I malformations in adults: a morphometric analysis of the posterior cranial fossa. Surg Neurol. 2005;64(3):237–41. doi: 10.1016/j.surneu.2005.02.021. discussion 41. [DOI] [PubMed] [Google Scholar]
- 23.Trigylidas T, Baronia B, Vassilyadi M, Ventureyra EC. Posterior fossa dimension and volume estimates in pediatric patients with Chiari I malformations. Childs Nerv Syst. 2008;24(3):329–36. doi: 10.1007/s00381-007-0432-4. [DOI] [PubMed] [Google Scholar]
- 24.Massimi L, Caldarelli M, Frassanito P, Di Rocco C. Natural history of Chiari type I malformation in children. Neurol Sci. 2011;32(Suppl 3):S275–7. doi: 10.1007/s10072-011-0684-3. [DOI] [PubMed] [Google Scholar]
- 25.Hevner RF. The cerebral cortex malformation in thanatophoric dysplasia: neuropathology and pathogenesis. Acta Neuropathol. 2005;110(3):208–21. doi: 10.1007/s00401-005-1059-8. [DOI] [PubMed] [Google Scholar]
- 26.Thomson RE, Kind PC, Graham NA, et al. Fgf receptor 3 activation promotes selective growth and expansion of occipitotemporal cortex. Neural Dev. 2009;4:4. doi: 10.1186/1749-8104-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khonsari RH, Delezoide AL, Kang W, et al. Central nervous system malformations and deformations in FGFR2-related craniosynostosis. American journal of medical genetics Part A. 2012;158A(11):2797–806. doi: 10.1002/ajmg.a.35598. [DOI] [PubMed] [Google Scholar]
- 28.Hill CA, Martinez-Abadias N, Motch SM, et al. Postnatal brain and skull growth in an Apert syndrome mouse model. American journal of medical genetics Part A. 2013;161A(4):745–57. doi: 10.1002/ajmg.a.35805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moritake K, Nagai H, Miyazaki T, et al. Nationwide survey of the etiology and associated conditions of prenatally and postnatally diagnosed congenital hydrocephalus in Japan. Neurol Med Chir (Tokyo) 2007;47(10):448–52. doi: 10.2176/nmc.47.448. discussion 52. [DOI] [PubMed] [Google Scholar]
- 30.Massimi L, Paternoster G, Fasano T, Di Rocco C. On the changing epidemiology of hydrocephalus. Childs Nerv Syst. 2009;25(7):795–800. doi: 10.1007/s00381-009-0844-4. [DOI] [PubMed] [Google Scholar]
- 31.Westermaier T, Schweitzer T, Ernestus RI. Arachnoid cysts. Adv Exp Med Biol. 2012;724:37–50. doi: 10.1007/978-1-4614-0653-2_3. [DOI] [PubMed] [Google Scholar]
- 32.Bisschoff IJ, Zeschnigk C, Horn D, et al. Novel mutations including deletions of the entire OFD1 gene in 30 families with type 1 orofaciodigital syndrome: a study of the extensive clinical variability. Hum Mutat. 2013;34(1):237–47. doi: 10.1002/humu.22224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doherty D, Chudley AE, Coghlan G, et al. GPSM2 mutations cause the brain malformations and hearing loss in Chudley-McCullough syndrome. Am J Hum Genet. 2012;90(6):1088–93. doi: 10.1016/j.ajhg.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aicardi J. Aicardi syndrome. Brain Dev. 2005;27(3):164–71. doi: 10.1016/j.braindev.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 35.Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011;26(7):1039–56. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Govaert P, Oostra A, Matthys D, Vanhaesebrouck P, Leroy J. How idiopathic is idiopathic external hydrocephalus? Dev Med Child Neurol. 1991;33(3):274–6. doi: 10.1111/j.1469-8749.1991.tb05121.x. [DOI] [PubMed] [Google Scholar]
- 37.Barlow CF. CSF dynamics in hydrocephalus–with special attention to external hydrocephalus. Brain Dev. 1984;6(2):119–27. doi: 10.1016/s0387-7604(84)80060-1. [DOI] [PubMed] [Google Scholar]
- 38.Zahl SM, Egge A, Helseth E, Wester K. Benign external hydrocephalus: a review, with emphasis on management. Neurosurg Rev. 2011;34(4):417–32. doi: 10.1007/s10143-011-0327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nogueira GJ, Zaglul HF. Hypodense extracerebral images on computed tomography in children. “External hydrocephalus”: a misnomer? Childs Nerv Syst. 1991;7(6):336–41. doi: 10.1007/BF00304833. [DOI] [PubMed] [Google Scholar]
- 40.Clericuzio C, Roberts A, Kucherlapati R, et al. Communicating hydrocephalus in Noonan syndrome: A consequence of lymphatic dysplasia? Proc Greenwood Gen Ctr. 2008;27(81) [Google Scholar]
- 41.Rosman NP, Shands KN. Hydrocephalus caused by increased intracranial venous pressure: a clinicopathological study. Ann Neurol. 1978;3(5):445–50. doi: 10.1002/ana.410030516. [DOI] [PubMed] [Google Scholar]
- 42.McLaughlin JF, Loeser JD, Roberts TS. Acquired hydrocephalus associated with superior vena cava syndrome in infants. Childs Nerv Syst. 1997;13(2):59–63. doi: 10.1007/s003810050042. [DOI] [PubMed] [Google Scholar]
- 43.Karmazyn B, Dagan O, Vidne BA, et al. Neuroimaging findings in neonates and infants from superior vena cava obstruction after cardiac operation. Pediatr Radiol. 2002;32(11):806–10. doi: 10.1007/s00247-002-0770-z. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez LA, Maytal J, Shinnar S. Idiopathic external hydrocephalus: natural history and relationship to benign familial macrocephaly. Pediatrics. 1986;77(6):901–7. [PubMed] [Google Scholar]
- 45.Castro-Gago M, Pérez-Gómez C, Novo-Rodríguez M, et al. Benign idiopathic external hydrocephalus (benign subdural collection) in 39 children: its natural history and relation to familial macrocephaly. Rev Neurol. 2005;40(9):513–17. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.