

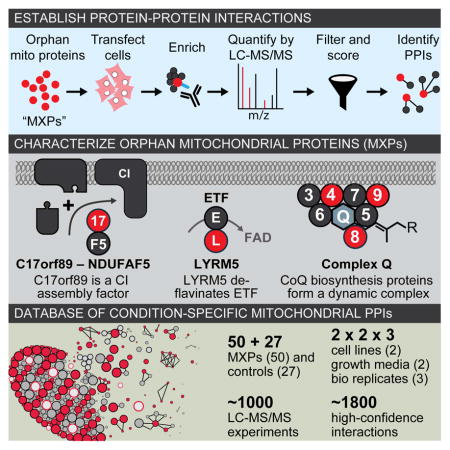
SUMMARY
Mitochondria are essential for numerous cellular processes, yet hundreds of their proteins lack robust functional annotation. To reveal new functions for these proteins (termed MXPs) we assessed condition-specific protein-protein interactions for 50 select MXPs using affinity enrichment mass spectrometry. Our data connect MXPs to diverse mitochondrial processes, including multiple aspects of respiratory chain function. Building upon these observations, we validated C17orf89 as a complex I (CI) assembly factor. Disruption of C17orf89 markedly reduced CI activity, and its depletion is found in an unresolved case of CI deficiency. We likewise discovered that LYRM5 interacts with and deflavinates the electron transferring flavoprotein that shuttles electrons to coenzyme Q (CoQ). Finally, we identified a dynamic human CoQ biosynthetic complex involving multiple MXPs whose topology we map using purified components. Collectively, our data lend new mechanistic insight into respiratory chain-related activities and prioritize hundreds of additional interactions for further exploration of mitochondrial protein function.
eTOC Blurb
Mitochondria are essential organelles, yet hundreds of their proteins lack robust functional characterization. Floyd et al. define interaction partners for 50 such proteins, providing hypotheses about their roles in mitochondria. In particular, their work lends new mechanistic insight into respiratory chain activities related to complex I, the electron transferring flavoprotein, and coenzyme Q.
INTRODUCTION
Mitochondria are centers of metabolism for nearly all eukaryotic cells. Once considered to be mere sites of ATP generation, it is now appreciated that mitochondria participate in a wide range of essential functions related to cellular metabolism, signaling, and programmed cell death (Pagliarini and Rutter, 2013). Consistently, large-scale proteomics- and computation-based efforts during the past decade have revealed that the mitochondrial proteome is much more extensive than once thought, and dysfunction of these organelles is now associated with hundreds of inborn errors of metabolism and common diseases (Koopman et al., 2012; Nunnari and Suomalainen, 2012).
Despite our advanced cataloging of the mammalian mitochondrial proteome, functional characterization of these proteins is far from complete (Calvo et al., 2016; Pagliarini et al., 2008). This gap in knowledge has limited our understanding of basic mitochondrial biology and has obscured the nature and cause of many mitochondrial diseases. For instance, many patients with biochemically-established mitochondrial disease lack mutations in known mitochondrial disease genes, implying the existence of unidentified proteins whose proper function is necessary for the affected process (Calvo et al., 2010; Haack et al., 2011). Alternatively, other diseases arise from mutations in mitochondrial proteins with no known function, making it difficult to interrogate the molecular mechanism of the disease.
Protein-protein interactions (PPIs) can provide powerful insight into protein function. Recent advancements in affinity enrichment mass spectrometry (AE-MS) (Hein et al., 2015; Hosp et al., 2015; Huttlin et al., 2015; Keilhauer et al., 2015) have improved the ability to accurately detect PPIs, enabling researchers to overcome the systematic bias against poorly characterized proteins inherent in many large-scale “interactome” analyses (Sahni et al., 2015). With AE-MS, a protein of interest (i.e., the bait) is enriched from a sample; it, along with any co-enriching proteins, is then analyzed by mass spectrometry (MS). In this process, the majority of captured proteins are typically not meaningful interactors of the bait. To sort the so-called wheat from the chaff, the AE-MS approach combines the analyses of multiple baits with a robust quantitative MS platform and a scoring algorithm to differentiate between informative interactions and nonspecific background co-enrichment.
In this study, we began by curating a list of proteins from the mitochondrial proteome that lack significant functional annotation, which we call mitochondrial uncharacterized (x) proteins (MXPs). Using stringent criteria, we conservatively estimate there to be 228 MXPs in humans, including more than 25 that have been associated with human diseases. We then designed a robust AE-MS strategy to define an extensive cell type- and condition-specific interactome of 50 select MXPs, enabling us to propose new functions and disease associations for these uncharacterized proteins.
Informed by our large-scale AE-MS analyses, we conducted extensive in vitro biochemistry and cell biology experiments that validated roles for MXPs in various aspects of the mitochondrial respiratory chain. We establish C17orf89 as a complex I (CI) assembly factor, whose silencing markedly impairs CI activity, and whose depletion is found in an unresolved case of isolated CI deficiency. We also establish LYRM5 as a “deflavinase” that directly regulates the electron transferring flavoprotein (ETF), and identify a dynamic human coenzyme Q biosynthetic complex that includes multiple MXPs. We propose functions for a variety of other MXPs and make our data freely available to the community for accelerate further annotation of mitochondrial proteins.
RESULTS
At least twenty percent of the mitochondrial proteome lacks functional annotation
Previous analyses have suggested that much of the mitochondrial proteome is uncharacterized (Pagliarini et al., 2008; Pagliarini and Rutter, 2013). To capture an up-to-date assessment of mitochondrial proteins and their functions, we combined the recently updated MitoCarta 2.0 list (Calvo et al., 2016) with additional literature sources to generate a MitoCarta+ list of 1,166 human proteins with validated mitochondrial localization (Table S1). Next, we annotated these proteins based on the integration of online databases and analysis of the current literature (see Experimental Procedures). Our data indicate that at least 228 mitochondrial proteins have no known, or poorly established, biochemical function, and that an additional 26 proteins with dual localization to the mitochondrion and another cellular compartment do not have clear roles within the mitochondrion. These MXPs constitute approximately 20% of the mammalian mitochondrial proteome, and include many proteins associated with human disease (Table S1, and see Supplemental Experimental Procedures).
Overall experimental strategy
To begin characterizing the functions of MXPs, we elected to establish MXP-specific interactions via affinity enrichment-mass spectrometry (AE-MS)—an approach proven to be capable of efficiently connecting uncharacterized proteins to known pathways (Figure 1A). We prioritized an initial set of 50 MXPs (Table S1) based on disease-relevance, evolutionary conservation, and confirmed localization to mitochondria when possessing a C-terminal FLAG-tag (Figure 1B, S1). We supplemented these bait proteins with 27 mitochondrial proteins of known function (Table S1), and a variant of green fluorescent protein harboring an N-terminal mitochondrial localization sequence (MLS-GFP-FLAG). We performed our interaction analyses in two cell lines (HEK293 and HepG2) grown in both glucose- and galactose-based media conditions. Galactose is known to increase oxygen consumption and dependence on mitochondrial respiratory chain function, which we observed in both lines (Figure 1C). We collected three replicates (i.e., distinct cell transfections) of each condition for a total of 12 analyses per bait protein.
Figure 1. AE-MS methodology.

(A) Schematic workflow of the AE-MS method.
(B) Localization of all FLAG-tagged constructs was established based on MLS-GFP and anti-FLAG fluorescence microscopy (see Figure S1). Venn diagrams report the percentage of the MitoCarta+ list (Table S1) associated with each category.
(C) Galactose induces mitochondrial respiration. Ratio of rates of oxygen consumption (OCR) and extracellular acidification (ECAR) in HEK293 (left) and HepG2 (right) cells grown in 10 mM glucose (Glu) or galactose (Gal) for 24 hours prior to assay. OCR/ECAR is proportional to mitochondrial vs. glycolytic flux (* indicates t-test p-value < 0.05). Error bars indicate ± SEM.
Following anti-FLAG immunoaffinity enrichment, protein eluent was analyzed using nanoflow liquid chromatography coupled to high resolution mass spectrometry (quadrupole linear ion trap-Orbitrap hybrid, nLC-MS/MS). Analysis of these 78 unique baits required 936 nLC-MS/MS experiments that generated 20 million MS/MS spectra, identified 5 million unique peptides, and resulted in the discovery of 10,000 unique proteins. On average, each nLC-MS/MS experiment detected ~ 1,000 unique proteins; however, each bait likely has far fewer bona fide interactions. To help distinguish genuine interactions from background, we utilized CompPASS, a known and validated algorithmic approach to highlighting high-confidence interactions and removing non-specific binders (Sowa et al., 2009), modified to incorporate label-free quantitation data.
AE-MS analyses identify hundreds of new mitochondrial interactions
To assess the performance of our approach and to determine an appropriate cutoff score, we focused on our positive-control bait proteins that have known binding partners. We curated a list of literature-established PPIs involving our positive-control baits and compared their CompPASS scores to those for all mitochondrial preys (Table S2). We selected a stringent cutoff score that achieved 93% sensitivity for known PPIs while simultaneously eliminating 95% of all observed, likely background, interactions (Figure 2A).
Figure 2. Overall analyses of our AE-MS approach.

(A) CompPASS scoring accuracy. CompPASS scores were calculated for all bait-prey interactions, including those involving only mitochondrial prey (gray), and those determined a priori to be high-confidence PPIs based on the literature (black). At each score threshold, the percent of remaining PPI per bin was calculated. Scores to the right of the vertical gray bar exceeded the threshold set for this study and are counted as high-confidence interactions.
(B) Quantitative scoring enriches for high-confidence interactors. Top: Schematic of the results of CompPASS score filtering. Bottom left: Heat map where white indicates a prey was not observed, and shades of gray indicate quantified abundance. Prey proteins are in rows, and FLAG-tagged baits are in columns. Data are averages from 6 replicates per cell line (3 glucose, 3 galactose). Bottom right: Heat map showing scores above CompPASS threshold. Preys and baits are organized in the same order as in left heat map. Black indicates a score above the threshold.
(C) Venn diagram of high-confidence PPIs from each cell line. Mitochondrial interactions above threshold in HEK293 (blue) and HepG2 (green) cells are indicated (see Figure S2).
(D) Venn diagram of high-confidence PPIs from each carbon source. Mitochondrial interactions above threshold in glucose (orange) and galactose (purple) cells are indicated.
(E) Histogram of the fold change abundances of PPIs between carbon sources. Select dynamic PPIs involving proteins from the coenzyme Q biosynthetic pathway (see Figure S7) are indicated.
Applying this threshold to the rest of our data, and filtering for proteins in our MitoCarta+ list, we identified 1,829 interactions from a total observed set of 109,817 (Figure 2B, Table S3). Consistent with previous efforts (Jager et al., 2012), we found that using more than one cell type is an effective means for identifying and prioritizing interactions, as only 32% of interactions are shared between the cell lines when limited to our stringent cutoff score (Figure 2C). Changing the carbon source (i.e., glucose vs. galactose) had a more modest effect with 61% of interactions shared between conditions at our threshold cutoff (Figure 2D and S2); however, the abundances of hundreds of these shared interactions are modulated by these changing conditions (Figure 2E), suggesting that mitochondrial metabolism may be linked to the regulation of protein-protein interactions. Thus, our experimental design expanded our search space, enabled the identification of meaningful interactions by excluding nonspecific background, and detected interactions affected by nutrient state.
C17orf89 is a CI assembly factor
We designed our study with an awareness that many known mitochondrial pathways and processes are “missing” enzymes or functional components that could be completed by our MXPs. For example, 45–60% of biochemically-validated cases of CI (Calvo et al., 2010; Haack et al., 2011), CII (Jain-Ghai et al., 2013), and CIII deficiency (Fernandez-Vizarra and Zeviani, 2015) lack molecular diagnoses. As such, we prioritized MXPs that interacted with respiratory chain components for functional investigations.
A particularly noteworthy MXP in this category was C17orf89, a 7 kDa protein that interacted with the known CI assembly factor (CIAF) NDUFAF5 (Figure 3A). Out of 1415 observed preys for C17orf89, 31 mitochondrial proteins were above our cutoff score, and only four interactions were observed in both cell lines. Of these, the highest-scoring interaction was between C17orf89 and NDUFAF5. Reciprocally, only three mitochondrial preys interacted with NDUFAF5 in both cell lines, one of which was C17orf89. We validated this C17orf89-NDUFAF5 interaction by showing that immunoprecipitation of FLAG-tagged C17orf89 from HEK-293 cells captured endogenous NDUFAF5 (Figure 3B).
Figure 3. C17orf89 is required for complex I assembly.

(A) Schematic of top-scoring C17orf89 interactions. Arrows originate from bait proteins and point to high-confidence interactors.
(B) Immunoblot of immunoprecipitated FLAG-tagged C17orf89, LYRM5, and MLS-GFP with anti-FLAG (red) or anti-NDUFAF5 (green).
(C) Activity of complex I in C17orf89 knock down (kd) and control (c) HEK293 cell lines and control lines. Error bars indicate ± SD (see also Figure S3).
(D) Measurement of oxygen consumption rate (OCR) for the same kd or c lines as in (C) using a Seahorse Extracellular Flux Analyzer (FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone). Error bars indicate ± SEM.
(E) Complex I activity in kd and c cell lines after transfection with GFP-FLAG (negative control) or C17orf89-FLAG (rescue). Error bars indicate ± SD.
To test the hypothesis that C17orf89 is necessary for CI activity, we used RNAi to knock down (KD) C17orf89 expression in HEK-293 cells via lentiviral shRNA constructs. Indeed, we found that silencing of C17orf89 had a dramatic effect on CI activity (Figure 3C) and subunit levels (Figure S3A), with no consistent change in the abundance of subunits of other respiratory chain complexes. We also observed a slight decrease in the abundance of a subunit of CIV (Figure S3A) and of CIV activity (Figure S3B), consistent with a recent study suggesting that loss of NDUFAF5 also perturbs CIV (Saada et al., 2012). We further observed a dramatic reduction in the oxygen consumption rates of live C17orf89 KD cells, both basally and upon stimulation with the uncoupler FCCP (Figure 3D), without change to the extracellular acidification rate (ECAR, a measure of glycolytic activity) (Figure S3C). Notably, transfection of C17orf89-FLAG, but not GFP-FLAG, into KD cell lines was able to rescue much of the lost CI activity (Figure 3E).
To further assess the functional relationship between C17orf89 and NDUFAF5, we established siRNA-mediated HEK-293 KD of C17orf89, NDUFAF4, NDUFAF5, NDUFAF6, and the CI subunit NDUFS3 (Figures S4A–E) and analyzed each using mass spectrometry. Silencing of the target genes resulted in an overall decrease in CI subunits; however, the pattern of subunit changes for the C17orf89 kd cells was most similar to that of NDUFAF5 (Figure S4F), further strengthening the C17orf89–NDUFAF5 functional relationship. Strikingly, silencing of C17orf89, but not other CIAFs or NDUFS3, resulted in a drastic loss of NDUFAF5 protein (Figure 4A), suggesting that the role of C17orf89 in CI assembly likely involves the direct stabilization of NDUFAF5.
Figure 4. C17orf89 stabilizes NDUFAF5 and is depleted in a case of CI deficiency.

(A) Immunoblots of mitochondrial proteins in cells treated with siRNA for CI and CIAF genes. C17orf89 knockdown results in loss of CI subunits and a marked depletion of NDUFAF5 (red box). See also Figure S4.
(B) RNAseq analysis of C17orf89 expression in 96 cell lines from patients with respiratory chain dysfunction (RPKM, Reads Per Kilobase of transcript per Million mapped reads). Arrow indicates a line with severe loss of C17orf89 expression.
(C) Respiratory chain complex analyses of the patient line indicated in (B), revealing an isolated CI deficiency (P, patient; C, control). Error bars represent mean +/− SD, n=25.
(D) Proposed model of C17orf89–NDUFAF5 complex function in CI assembly.
Our implication of C17orf89 as a new CI assembly factor motivated us to investigate the possibility that its disruption might contribute to unresolved patient cases of isolated CI deficiency. We sequenced C17orf89 in 125 such cases but did not identify likely pathogenic variants. However, RNAseq analysis on 96 cell lines from mitochondrial disease patients that had previously been analyzed by whole exome sequencing revealed one case that exhibited an 80% reduction of C17orf89 reads (Figure 4B). This patient, now 26 years of age, was born to unrelated parents and presented in childhood with acute encephalopathy, seizures, mild spasticity and neuroradiological features consistent with a diagnosis of Leigh syndrome (bilateral high signal intensities in the basal ganglia and brainstem); a diagnostic muscle biopsy at the age of 14 revealed evidence of an isolated CI defect (<40% control activity; Figure 4C). Respiratory chain activities were normal in the sequenced cultured skin fibroblasts (Figure S4G), as is the case for ~50% of patients exhibiting a respiratory chain defect detected in skeletal muscle, liver, or heart (Kirby et al., 2007), thereby precluding our ability to rescue the defect by reintroduction of wild type C17orf89 cDNA. Nonetheless, based on its key role in CI function and its interaction with NDUFAF5 (Figure 4D), we propose to rename C17orf89 as NDUFAF8, and suggest that it is candidate gene for human CI deficiency.
LYRM5 binds and deflavinates the electron transferring flavoprotein
A second MXP that our AE-MS analyses connected to the respiratory chain is LYRM5, an 11 kDa member of the mitochondrial family of LYR motif-containing (LYRM) proteins, which also include MXPs LYRM1, LYRM2, and LYRM9. LYRM proteins were first identified as supernumerary subunits of CI (NDUFA6 and NDUFB9), and have recently been found to act as assembly factors for CII (SDHAF1/LYRM8, SDHAF3/ACN9) (Atkinson et al., 2011; Ghezzi et al., 2009; Sánchez et al., 2013), CV (FMC1) (Lefebvre-Legendre et al., 2001), and iron-sulfur cluster biosynthetic enzymes (ISD11/LYRM4) (Adam et al., 2006; Shan et al., 2007). Our data captures many of these expected LYRM interactions (Figure 5A). Interestingly, all seven LYRM proteins in our study were found to interact with NDUFAB1, a poorly characterized CI subunit that also serves as an acyl carrier protein (Angerer et al., 2014) (Figure 5A).
Figure 5. LYRM5 forms a complex with ETF.

(A) Schematic of top-scoring LYRM PPIs. LYRM5-ETF interactions are shaded.
(B) Validation of the interaction between LYRM5 and both ETFA and ETFB. C-terminally FLAG-tagged GFP, IVD, NDUFA4, and LYRM5 were immunoprecipitated (IP) from HEK cells and immunoblotted (IB) with anti-ETFA and anti-ETFB (upper) or anti-FLAG (lower). LYRM5 enriched for both ETF proteins more efficiently than IVD, a known ETF interactor.
(C) IP of LYRM5-FLAG or MLS-GFP-FLAG from HEK293 cells analyzed by Blue Native-PAGE analysis and IB. The same membrane was blotted for ETFB (left) and then FLAG (right) (see also Figure S5).
(D) Recombinant N-terminally His-tagged LYRM5 and untagged ETFA/B were co-expressed in E. coli. Purification of LYRM5 by metal affinity chromatography led to the co-purification of ETFA/B.
(E) LYRM5 and ETF form a stable complex. Size exclusion chromatography of LYRM5 alone (red), ETF alone (green), or the co-purified LYRM5-ETF complex (blue) noted in (D).
Unlike the other LYRM proteins, LYRM5 interacted robustly with ETFA and ETFB, which comprise the electron transfer flavoprotein (ETF) (Figure 5A). ETF partners with ETF dehydrogenase to shuttle electrons to CoQ. Notably, LYRM5-FLAG immunoprecipitation enriched for endogenous ETFA and ETFB to a much greater extend than does IVD—a known ETF substrate (Kim and Miura, 2004) (Figure 5B, S5). Furthermore, the LYRM5 IP elution generated a single band containing LYRM5, ETFA, and ETFB on a BN-PAGE immunoblot (Figure 5C). To test whether the LYRM5-ETF interaction is direct, we purified recombinant ETF and LYRM5 from E. coli (Figure 5D) and demonstrated that they form a stable complex by size-exclusion chromatography (Figure 5E). LYRM5 alone eluted as a broad peak around 44kDa, indicating that it may exist as a tetramer.
Surprisingly, the purified LYRM5-ETF complex lacked the characteristic yellow color of flavoproteins, suggesting that LYRM5 either prevents the incorporation of flavin adenine dinucleotide (FAD) into ETF, or that it is capable of removing this cofactor. To test the ability of LYRM5 to “deflavinate” the ETF complex, we mixed LYRM5 and ETF at increasing ratios and measured ETF activity. The addition of LYRM5 led to a linear reduction of ETF activity up to a ratio of 4:1, at which point ETF lost all activity (Figure 6A). This decrease in activity was concomitant with a proportional release of FAD from the ETF complex, as observed by fluorescence emission spectroscopy (Figure 6B). The interaction of FAD with ETF protein residues can be seen by the 420 nm and 460 nm peaks on an absorbance spectrum. These peaks disappear upon addition of LYRM5 (Figure 6C), lending further evidence of its direct deflavination activity. Collectively, these experiments support a direct, specific interaction between LYRM5 and ETF that results in deflavination (Figure 6D)—a unique and unexpected activity that perhaps suggests a non-electron transferring role for ETF (see discussion).
Figure 6. LYRM5 deflavinates ETF.

(A) ETF activity in the presence of increasing amounts of LYRM5 (± SD from triplicate measurements).
(B) FAD release from ETF upon incubation with varying amounts of LYRM5, measured by fluorescence emission spectroscopy.
(C) Visible spectra of ETF in the presence of LYRM5. The flavin visible spectrum shows two shoulder peaks at 420nm and 460nm due to the interaction between FAD and ETF protein residues that are lost upon addition of LYRM5. Spectrum a, b, and c are for LYRM5:ETF ratios of 0, 2, and 4, respectively. For clarity, spectra a and b have been shifted by +0.04 and 0.02 OD units, respectively.
(D) Proposed model of the functional interaction between LYRM5 and ETF. Binding of four molar equivalents of LYRM5 (red dot) to ETF (green trace of PDB ID 1EFV) leads to the loss of FAD from ETF (F).
CoQ is synthesized by a dynamic biosynthetic complex
Both CI and ETF operate by shuttling electrons to CoQ—a requisite gateway for electron transport along the mitochondrial respiratory chain. CoQ is synthesized within the mitochondrion in a process that involves at least 13 human proteins, six of which we consider MXPs, as they lack well established biochemical roles in the pathway (Figure S6A). Across a series of isolated studies, seven S. cerevisiae CoQ proteins, Coq3p-9p, have been found to physically interact and to potentially form a biosynthetic complex (He et al., 2014; Marbois et al., 2005). We and others have speculated that a CoQ complex might exist in other species; however, species-specific aspects of CoQ biosynthesis exist and, to date, there are only four known human CoQ-related PPIs (Ashraf et al., 2013; Lohman et al., 2014; Nguyen et al., 2014).
Our AE-MS data have now established direct evidence of a highly interconnected mammalian CoQ biosynthetic complex, which we call complex Q (Figure 7A). Intriguingly, the abundances of CoQ pathway PPIs involving COQ8A/ADCK3 increased in galactose-treated cells, and those with COQ8B/ADCK4 reciprocally decreased (Figure 7B)—a phenomenon that accompanied a marked elevation of CoQ levels in HepG2 cells (Figure 7C) (Note that ADCK3 and ADCK4 are now referred to as COQ8A and COQ8B, respectively—see Stefely, et al. in this issue). We consider COQ8A and COQ8B to be MXPs due to their lack of clear biochemical function; however, consistent with these results, we recently revealed that COQ8A is essential for the stability of CoQ proteins in a mouse model of Coq8a deficiency (Stefely et al., this issue of Mol Cell), and COQ8B has been shown to interact with COQ6 (Ashraf et al., 2013). These data suggest an important connection between CoQ production and interactions among CoQ biosynthetic complex members, and may indicate reciprocal regulation of the activities of the paralogous COQ8A and COQ8B.
Figure 7. Interaction analysis reveals the dynamic human CoQ-related complex.

(A) All CoQ-related proteins used as baits and/or observed as preys in this study are shown as white nodes. Interactions above our score threshold are shown for HEK cells (gray arrows) or HepG2 cells (green arrows).
(B) CoQ-related interactions involving COQ8A or COQ8B as bait were assessed for the effect of cellular metabolic status. Interactions more abundant in galactose or glucose media are shown in red or blue, respectively, while those with no clear media effect are in white.
(C) Relative abundance of CoQ10 in HEK293 and HepG2 cells after treatment with 10 mM glucose or galactose for 24 hours (*** indicates t-test p-value < 0.001, error bars indicate the 95% confidence interval).
(D-E) Representative results of in vitro protein translation and purification of each core CoQ complex protein individually (D) or in pairs (E). Proteins were run on SDS-PAGE and detected by Coomassie stain. See Figure S6 for all interactions observed.
(F) Schematic of COQ protein network direct interactions established in vitro. All robust interactions are represented as edges between the COQ protein nodes.
(G) In vitro co-purification of all six core COQ complex proteins.
(H) Three-dimensional model of predicted COQ complex structure based on in vitro interaction data. Colors are as in Figure 3F.
We next aimed to reconstruct the CoQ interactions using purified recombinant proteins. This is a powerful means to validate our observed AE-MS interactions, ascertain the direct interactions between the CoQ proteins, and begin mapping the topology of complex Q. Our in vitro analyses using a cell-free protein translation and purification system revealed that while nearly all COQ proteins were unstable or insoluble when produced alone, many coexpression pairs between COQ3-7 and COQ9 were stabilized by direct interaction (Figure 7D, E, S6B). Strikingly, using this same method, we were then able to rebuild a CoQ complex containing six of the core members in vitro (Figure 7G, H). Combined with the binary interactions noted above, these data allow us to propose the subunit topology of the complex (Figure 7F, H). While it is likely that other CoQ-related proteins interact with the complex directly or indirectly and are important for its stability in vivo, such as COQ8A and COQ8B, this work suggests that these six core CoQ-related proteins are sufficient to form a complex. These data lend clarity to an evolutionarily conserved complex Q and provide a platform for the future interrogation of the roles of each complex subunit, including the MXPs COQ4 and COQ9.
Associating MXPs with other established pathways
Our analyses above focused on MXPs related to CI and CoQ in order to build upon our recent work in these areas (Khadria et al., 2014; Lohman et al., 2014; Stefely et al., 2015). However, our data reveals many other connections between MXPs and diverse mitochondrial processes. For instance, C15orf48 also interacts with multiple subunits of CI and CIV (Figure S7), suggesting that it regulates the activity of one or both of these complexes, or perhaps supercomplex formation. C2orf47 interacts with AFG3L2 and SPG7, the two members of the human m-AAA protease complex that is responsible for the maturation of several membrane-associated proteins and for mitochondrial protein quality processes (Ehses et al., 2009) (Figure S7). As a final example, we observed that DHRS4—a poorly characterized member of the short-chain dehydrogenases/reductases (SDR) family (Persson and Kallberg, 2013)—interacts with other members of the SDR family, including DHRS4L2 and CBR4, as well as with SIRT3 and EHHADH (Figure S7). DHRS4 is reported to have dual-localization to peroxisomes and mitochondria (Matsunaga et al., 2008; Pagliarini et al., 2008), and is dynamically phosphorylated and acetylated (Grimsrud et al., 2012; Still et al., 2013); however, no direct function is known. These interactions suggest that DHRS4 might have an important unappreciated role in coordinating lipid metabolism between these organelles. Our other top-scoring interactions, and many others that fall just below our stringent cutoff, can likewise enable new hypotheses about MXP function (Table S2).
DISCUSSION
Insights into mitochondrial respiratory chain function
Our identification of C17orf89/NDUFAF8 as a complex I assembly factor (CIAF) pinpoints a new disease gene candidate for unresolved cases of isolated CI deficiency. Given the presence of twin CX9C domains in its primary structure, C17orf89/NDUFAF8 is likely a member of the coiled-coil-helix-coiled-coil-helix (CHCH) domain family of proteins (Modjtahedi et al., 2016). Many CHCH proteins are mitochondrial, and several are involved in respiratory chain functions, including four CI subunits (Modjtahedi et al., 2016). Our data revealed a robust interaction between C17orf89/NDUFAF8 and NDUFAF5—a putative methyltransferase that is essential for early stages of CI assembly (Pagliarini et al., 2008; Sugiana et al., 2008), but for which neither a substrate nor a direct role in CI maturation have been elucidated. Interestingly, several similar sub-complexes of CIAFs exist, including the mitochondrial CI assembly (MCIA) complex (Guarani et al., 2014), and the NDUFAF3:NDUFAF4 complex (Saada et al., 2009). Given that C17orf89/NDUFAF8 interacts with and stabilizes NDUFAF5, we suggest that C17orf89 may facilitate the methyltransferase activity of NDUFAF5, or even be its substrate. Furthermore, as we identified a patient with CI deficiency whose molecular diagnosis eluded whole genome sequencing but who is deficient in C17orf89/NDUFAF8 transcript, we both prioritize this as a disease gene and highlight the importance of complementary methods for the diagnosis of genetic diseases.
CI is one of several sources of electrons entering the CoQ pool for transport along the respiratory chain. A second source is ETF, which interacts with and accepts electrons from various mitochondrial dehydrogenases before passing them to ETFDH (Roberts et al., 1996). ETF harbors causal mutations in glutaric acidemia type 2 (GA2) (Vockley and Whiteman, 2002), and other cases of GA2 remain unresolved (Schiff et al., 2006). Given this, we were particularly interested in our observed interaction of ETF with LYRM5.
We discovered that the interaction between LYRM5 and ETF results in an efficient—and surprising—removal of FAD from the ETF holoenzyme. Much work needs to be done to understand the in vivo utility of this process, but a few possibilities stand out. First, the FAD of ETF could become damaged and need to be replaced. Second, removal of FAD may facilitate the destabilization of ETF and allow for proteolysis and recycling of the flavin cofactor. Third, if in a given metabolic state ETF is not interacting with dehydrogenases (i.e., its electron transfer partners), its FAD would be free to interact with water or other matrix metabolites and generate reactive oxygen species (Rodrigues and Gomes, 2012). In this scenario, LYRM5 could act to shut down ETF activity without the need to completely turn over the enzyme. Finally, it is possible that ETF possesses a distinct, non-electron transferring function—a possibility we most favor. Consistent with this hypothesis is our observation that LYRM5, like other LYRM proteins (Figure 5A), interacts with the CI subunit NDUFAB1—the mitochondrial acyl carrier protein (ACP) (Angerer, 2015; Angerer et al., 2014). It is possible that LYRM5 serves as an adaptor to bring ETF to CI via NDUFAB1 to perform an unidentified function. Given our functional discoveries, we propose renaming C17orf89 and LYRM5 as NDUF Assembly Factor 8 (NDUFAF8) and ETF Regulatory Factor 1 (ETFRF1), respectively.
Insights into CoQ biosynthesis
CoQ is a requisite gateway in the respiratory chain that accepts electrons from many sources, including CI and ETF (via ETFDH). Although it was discovered 60 years ago, multiple aspects of eukaryotic CoQ biosynthesis remain unexplained, including roles for MXPs COQ8A, COQ8B, COQ4, and COQ9 (Figure S6A).
Two notable features emerged from our work with the CoQ machinery. First, it is abundantly clear that the mammalian CoQ proteins physically interact to form what appears to be a biosynthetic complex—complex Q. Saccharomyces cerevisiae is also known to have a partially-defined complex, but it is not known how this complex facilitates CoQ biosynthesis. Our work here provides new insights into the binary interactions that likely enable the formation of complex Q in both species. Second, our work implicates MXPs ADCK3 and ADCK4 (aka COQ8A and COQ8B, respectively) in complex Q, and show that their interactions with the complex are both dynamic and reciprocal. COQ8A and COQ8B are paralogs that are each likely orthologs of yeast Coq8p, and we have speculated that they possess redundant functions in a condition- or tissue-specific manner. We recently have shown that COQ8A adopts an atypical protein kinase-like fold (Stefely et al., 2015) and, building off of the work here, have now determined that its function is essential for maintaining complex Q stability (Stefely et al., this issue of Mol Cell).
Overall, we have extended the annotation of the mammalian mitochondrial proteome by identifying robust PPIs involving MXPs and by validating roles for these proteins in well-characterized mitochondrial pathways. Other prominent PPIs from our study now await further biological investigation, and many more that fell below our stringent cutoff score will nonetheless serve as important clues for the functional annotation of MXPs moving forward (Table S3). As such, our compendium of state-specific MXP associations will continue to serve as a powerful resource for the characterization of mitochondrial proteins, thereby advancing our understanding of basic mitochondrial biology and its associated pathophysiology.
EXPERIMENTAL PROCEDURES
Mitochondrial proteome compilation
Entrez GeneIDs of the mouse MitoCarta (Pagliarini et al., 2008) were converted from mouse to human using HomoloGene and reciprocal BLASTP searches. Data from the mitochondrial matrix (Rhee et al., 2013), and inter-membrane space (Hung et al., 2014) proteomic studies, and other high-quality studies, were then integrated with the human MitoCarta 2.0 (Calvo et al., 2016) to generate our MitoCarta+ list of mitochondrial proteins.
Generation of tagged constructs
Mitochondrial open reading frames were obtained from The Broad Institute and DNASU (Seiler et al., 2014) and cloned into a pcDNA3.1 mammalian expression vector with a C-terminal FLAG tag.
Mammalian cell culture
HEK293 or HepG2 cells were transiently transfected with pcDNA3.1 gene-FLAG plasmids using linear polyethylenimine (PEI, PolySciences) and Opti-MEM (LifeTechnologies). After 48 hours, cells were washed with PBS and media was replaced with DMEM containing either 10 mM glucose or 10 mM galactose. After 24 hours, cells were washed with and harvested into phosphate-buffered saline (PBS), snap frozen in liquid nitrogen, and stored at −80°C.
Affinity enrichment
Cell pellets were lysed in 200 μL buffer (see supplemental experimental procedures). After vortexing on ice, insoluble materials were pelleted (16,000 g, 10 min, 4°C). Equal masses of cell supernatant were mixed with 30 μL pre-washed anti-FLAG magnetic beads (Sigma M8823) for 2–3 h at 4°C with end-over-end agitation. Following incubation, beads were washed four times and proteins were eluted in 70 μL elution buffer containing 0.2 mg/mL FLAG-peptide for 30 min at room temperature with constant agitation.
LC-MS/MS analysis
All experiments were performed using a NanoAcquity UPLC system (Waters, Milford, MA) coupled to an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Reverse-phase columns were made in-house by packing a fused silica capillary with 3.5 μm diameter, 130 Å pore size Bridged Ethylene Hybrid C18 particles (Waters) to a final length of 30 cm. The column was heated to 55°C for all experiments. Precursor trypsin digested peptide cations were generated from the eluent through the utilization of a nanoESI source. Mass spectrometry instrument methods consisted of MS1 survey scans that were used to guide fifteen subsequent data-dependent MS/MS scans. Raw data can be found on Chorus (https://chorusproject.org) under project ID 1043.
Data Analysis
Data was processed using the MaxQuant software suite (Cox and Mann, 2008; Cox et al., 2011). Searches were performed against a target-decoy database using the default settings for high-resolution mass spectra. Results were filtered to 1% FDR at the unique peptide level and grouped into proteins within MaxQuant. Proteins were quantified across all replicates within each bait set using MaxLFQ (Cox et al., 2014).
Generation of C17orf89 knockdown cells
Viral particles were produced in HEK293 cells by transient transfection with PEI of lentiviral shRNA construct, psPAX2, and pMD2.G packaging plasmids. HEK293 cells were transduced in 6-well plates and were selected in culture medium supplemented with 2 μg/ml puromycin for at least two weeks. For siRNA knockdowns, HEK293 cells were transfected with 10 nM RNA for each target or the Non-Targeting siRNA based on the manufacturer’s protocol. After 2 days, the cells were passaged, and the next day were transfected again with 10 nM siRNA, and after another 2 days were collected for real-time qPCR, immunoblot, and mass spectrometry-based proteomic analyses.
CI activity and C17orf89 sequencing and expression analysis
Patients with biochemical evidence of isolated complex I deficiency identified via diagnostic work-up for suspected mitochondrial disease were screened for disruptive variants in C17orf89 by Sanger sequencing (primers available on request). C17orf89 transcript abundance in control and patient lines was measured as described previously (Haack et al., 2015).
Purification and functional analyses of LYRM5 and ETF
LYRM5 was cloned into a pET28a vector and purified as an N-terminally His-tagged protein by immobilized metal affinity chromatography. Size exclusion chromatography was performed using a Pharmacia 300×10mm Superdex 200 column. ETF activity +/− LYRM5 was measured by monitoring DCPIP reduction at 600 nm. For assay conditions, 40μM DCPIP, 20uM C8-CoA, and 0.1~0.2μM ETF were mixed in 20mM Tris-HCl buffer, pH 8.0. The assay reactions were initiated by adding 0.2μM MCAD and the UV absorbance decrease at 600nm was followed for 3 minutes. FAD release was measured by first mixing 10 μM ETF with varying amounts of LYRM5 and incubated on ice for 2 hours. The samples were then diluted 5 times in 20 mM Tris-HCl buffer (pH 8.0) and the fluorescence emission spectra were taken at room temperature (λex = 436nm). Visible spectra measurements of 10 μM ETF was measured in the presence of 0, 20μM and 40μM LYRM5 in 20mM Tris-HCl buffer, pH 8.0.
Blue Native-PAGE
Samples were mixed with NativePAGE sample buffer to 1x final concentration, and were loaded onto NativePAGE gels alongside NativeMARK standard. Gels were run for a total of two hours and subsequently subjected to western analysis.
Coenzyme Q Quantification
Tissue culture cells were lysed by vortexing with glass beads and spiked with an internal standard (CoQ6). Lipids were extracted with CHCl3/MeOH (1:1, v/v) and analyzed by LC-MS/MS.
Cell-free expression and purification
Purified plasmid DNA was used as individual transcription templates with SP6 RNase polymerase. Transcription and translation methods are as previously described (Makino et al., 2013).
Supplementary Material
HIGHLIGHTS.
PPI mapping of 50 uncharacterized mitochondrial proteins reveals new functions
C17orf89 is a novel CI assembly factor depleted in a case of CI deficiency
LYRM5 interacts with and deflavinates the electron transferring flavoprotein
Proteins involved in coenzyme Q biosynthesis form a dynamic ‘complex Q’
Acknowledgments
We thank David Aceti, John Primm, Brian Fox, and John Markley for technical and managerial assistance, and Clay Williams for mass spectrometry technical support. This work was supported by a Searle Scholars Award and NIH grants U01GM94622, R01DK098672, R01GM112057, and R01GM115591 (to D.J.P.), R35GM118110 (to J.J.C.), R01GM029076 (to J.-J.P.K), T32DK007665 and T32GM008692 MSTP (to B.J.F.), T15LM007359 (to. C.E.M.), T32HL007899 (to E.M.W.), F30AG043282 (to J.A.S.), and T32GM07215 (to K.M.W.), and also by a Wisconsin Distinguished Graduate Fellowship (to B.J.F.), and an NSF Graduate Research Fellowship (to M.T.V.). RWT is funded by a Wellcome Trust Strategic Award (096919/Z/11/Z), MRC Centre for Neuromuscular Diseases (G0601943), UK NHS Highly Specialised “Rare Mitochondrial Disorders of Adults and Children” Service and The Lily Foundation. C.L.A. is the recipient of a National Institute for Health Research (NIHR) doctoral fellowship (NIHR-HCS-D12-03-04). H.P. was supported by the German Bundesministerium für Bildung und Forschung (BMBF) through the German Network for mitochondrial disorders (mitoNET, 01GM1113C) and the E-Rare project GENOMIT (01GM1603), the EC FP7-PEOPLE-ITN Mitochondrial European Educational Training Project (GA #317433) and EU Horizon2020 Collaborative Research Project SOUND (633974).
Footnotes
AUTHOR CONTRIBUTIONS
B.J.F. and D.J.P. conceived of the project and its design. B.J.F, C.E.M., E.M.W., M.T.V., C.X., E.T.B., H.C., L.S.K., C.L.A., K.A.G., B.K.D., A.U., J.A.S., S.L.B., K.M.W., C.E.W., J.W.R., R.W.T., H.P., J.J.K., J.J.C., D.J.P., performed experiments and data analysis. R.L.W., A.J., M.S.W., J.W.R., R.W.T., H.P., J.J.K., J.J.C., D.J.P., provided key experimental resources and/or aided in experimental design. B.J.F. and D.J.P. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam AC, Bornhövd C, Prokisch H, Neupert W, Hell K. The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. The EMBO Journal. 2006;25:174–183. doi: 10.1038/sj.emboj.7600905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer H. Eukaryotic LYR Proteins Interact with Mitochondrial Protein Complexes. Biology (Basel) 2015;4:133–150. doi: 10.3390/biology4010133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer H, Radermacher M, Mankowska M, Steger M, Zwicker K, Heide H, Wittig I, Brandt U, Zickermann V. The LYR protein subunit NB4M/NDUFA6 of mitochondrial complex I anchors an acyl carrier protein and is essential for catalytic activity. Proc Natl Acad Sci U S A. 2014;111:5207–5212. doi: 10.1073/pnas.1322438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, Fang H, Song X, Cattran DC, Avila-Casado C, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. The Journal of clinical investigation. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson A, Smith P, Fox JL, Cui TZ, Khalimonchuk O, Winge DR. The LYR protein Mzm1 functions in the insertion of the Rieske Fe/S protein in yeast mitochondria. Molecular and Cellular Biology. 2011;31:3988–3996. doi: 10.1128/MCB.05673-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo S, Tucker EJ, Compton AG, Kirby DM, Crawford G, Burtt NP, Rivas M, Guiducci C, Bruno DL, Goldberger OA, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nature Genetics. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. Journal of proteome research. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. The Journal of cell biology. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Zeviani M. Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front Genet. 2015;6:134. doi: 10.3389/fgene.2015.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, Lochmüller H, D'Adamo P, Gasparini P, Strom TM, Prokisch H, et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nature Genetics. 2009;41:654–656. doi: 10.1038/ng.378. [DOI] [PubMed] [Google Scholar]
- Grimsrud PA, Carson JJ, Hebert AS, Hubler SL, Niemi NM, Bailey DJ, Jochem A, Stapleton DS, Keller MP, Westphall MS, et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 2012;16:672–683. doi: 10.1016/j.cmet.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarani V, Paulo J, Zhai B, Huttlin EL, Gygi SP, Harper JW. TIMMDC1/C3orf1 functions as a membrane-embedded mitochondrial complex I assembly factor through association with the MCIA complex. Mol Cell Biol. 2014;34:847–861. doi: 10.1128/MCB.01551-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Madignier F, Herzer M, Lamantea E, Danhauser K, Invernizzi F, Koch J, Freitag M, Drost R, Hillier I, et al. Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. Journal of medical genetics. 2011;49 doi: 10.1136/jmedgenet-2011-100577. jmedgenet-2011-100577-100589. [DOI] [PubMed] [Google Scholar]
- Haack TB, Staufner C, Kopke MG, Straub BK, Kolker S, Thiel C, Freisinger P, Baric I, McKiernan PJ, Dikow N, et al. Biallelic Mutations in NBAS Cause Recurrent Acute Liver Failure with Onset in Infancy. Am J Hum Genet. 2015;97:163–169. doi: 10.1016/j.ajhg.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CH, Xie LX, Allan CM, Tran UC, Clarke CF. Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochimica et biophysica acta. 2014;1841:630–644. doi: 10.1016/j.bbalip.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell. 2015;163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- Hosp F, Scheltema RA, Eberl HC, Kulak NA, Keilhauer EC, Mayr K, Mann M. A Double-Barrel Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) System to Quantify 96 Interactomes per Day. Mol Cell Proteomics. 2015;14:2030–2041. doi: 10.1074/mcp.O115.049460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung V, Zou P, Rhee H-W, Udeshi ND, Cracan V, Svinkina T, Carr SA, Mootha VK, Ting AY. Proteomic Mapping of the Human Mitochondrial Intermembrane Space in Live Cells via Ratiometric APEX Tagging. Molecular cell. 2014 doi: 10.1016/j.molcel.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al. Global landscape of HIV-human protein complexes. Nature. 2012;481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain-Ghai S, Cameron JM, Al Maawali A, Blaser S, MacKay N, Robinson B, Raiman J. Complex II deficiency--a case report and review of the literature. Am J Med Genet A. 2013;161A:285–294. doi: 10.1002/ajmg.a.35714. [DOI] [PubMed] [Google Scholar]
- Keilhauer EC, Hein MY, Mann M. Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS) Mol Cell Proteomics. 2015;14:120–135. doi: 10.1074/mcp.M114.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadria AS, Mueller BK, Stefely JA, Tan CH, Pagliarini DJ, Senes A. A Gly-zipper motif mediates homodimerization of the transmembrane domain of the mitochondrial kinase ADCK3. J Am Chem Soc. 2014;136:14068–14077. doi: 10.1021/ja505017f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Miura R. Acyl-CoA dehydrogenases and acyl-CoA oxidases. Structural basis for mechanistic similarities and differences. Eur J Biochem. 2004;271:483–493. doi: 10.1046/j.1432-1033.2003.03948.x. [DOI] [PubMed] [Google Scholar]
- Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Methods Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Willems PHGM, Smeitink JAM. Monogenic Mitochondrial Disorders. New England Journal of Medicine. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- Lefebvre-Legendre L, Vaillier J, Benabdelhak H, Velours J, Slonimski PP, di Rago JP. Identification of a nuclear gene (FMC1) required for the assembly/stability of yeast mitochondrial F(1)-ATPase in heat stress conditions. The Journal of biological chemistry. 2001;276:6789–6796. doi: 10.1074/jbc.M009557200. [DOI] [PubMed] [Google Scholar]
- Lohman DC, Forouhar F, Beebe ET, Stefely MS, Minogue CE, Ulbrich A, Stefely JA, Sukumar S, Luna-Sanchez M, Jochem A, et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc Natl Acad Sci U S A. 2014;111:E4697–4705. doi: 10.1073/pnas.1413128111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S-i, Beebe ET, Markley JL, Fox BG. Cell-Free Protein Synthesis for Functional and Structural Studies. Totowa, NJ: Humana Press; 2013. pp. 161–178. [DOI] [PubMed] [Google Scholar]
- Marbois B, Gin P, Faull KF, Poon WW, Lee PT, Strahan J, Shepherd JN, Clarke CF. Coq3 and Coq4 define a polypeptide complex in yeast mitochondria for the biosynthesis of coenzyme Q. J Biol Chem. 2005;280:20231–20238. doi: 10.1074/jbc.M501315200. [DOI] [PubMed] [Google Scholar]
- Matsunaga T, Endo S, Maeda S, Ishikura S, Tajima K, Tanaka N, Nakamura KT, Imamura Y, Hara A. Characterization of human DHRS4: an inducible short-chain dehydrogenase/reductase enzyme with 3beta-hydroxysteroid dehydrogenase activity. Arch Biochem Biophys. 2008;477:339–347. doi: 10.1016/j.abb.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Modjtahedi N, Tokatlidis K, Dessen P, Kroemer G. Mitochondrial Proteins Containing Coiled-Coil-Helix-Coiled-Coil-Helix (CHCH) Domains in Health and Disease. Trends Biochem Sci. 2016;41:245–260. doi: 10.1016/j.tibs.2015.12.004. [DOI] [PubMed] [Google Scholar]
- Nguyen TPT, Casarin A, Desbats MA, Doimo M, Trevisson E, Santos-Ocaña C, Navas P, Clarke CF, Salviati L. Molecular characterization of the human COQ5 C-methyltransferase in coenzyme Q10 biosynthesis. Biochimica et biophysica acta. 2014;1841:1628–1638. doi: 10.1016/j.bbalip.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Rutter J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013;27:2615–2627. doi: 10.1101/gad.229724.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson B, Kallberg Y. Classification and nomenclature of the superfamily of short-chain dehydrogenases/reductases (SDRs) Chemico-biological interactions. 2013;202:111–115. doi: 10.1016/j.cbi.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science (New York, NY) 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DL, Frerman FE, Kim JJ. Three-dimensional structure of human electron transfer flavoprotein to 2.1-A resolution. Proceedings of the National Academy of Sciences. 1996;93:14355–14360. doi: 10.1073/pnas.93.25.14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues JV, Gomes CM. Mechanism of superoxide and hydrogen peroxide generation by human electron-transfer flavoprotein and pathological variants. Free Radic Biol Med. 2012;53:12–19. doi: 10.1016/j.freeradbiomed.2012.04.016. [DOI] [PubMed] [Google Scholar]
- Saada A, Edvardson S, Shaag A, Chung WK, Segel R, Miller C, Jalas C, Elpeleg O. Combined OXPHOS complex I and IV defect, due to mutated complex I assembly factor C20ORF7. J Inherit Metab Dis. 2012;35:125–131. doi: 10.1007/s10545-011-9348-y. [DOI] [PubMed] [Google Scholar]
- Saada A, Vogel RO, Hoefs SJ, van den Brand MA, Wessels HJ, Willems PH, Venselaar H, Shaag A, Barghuti F, Reish O, et al. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease. American journal of human genetics. 2009;84:718–727. doi: 10.1016/j.ajhg.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, Peng J, Weile J, Karras GI, Wang Y, et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. 2015;161:647–660. doi: 10.1016/j.cell.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez E, Lobo T, Fox JL, Zeviani M, Winge DR, Fernandez-Vizarra E. LYRM7/MZM1L is a UQCRFS1 chaperone involved in the last steps of mitochondrial Complex III assembly in human cells. Biochimica et biophysica acta. 2013;1827:285–293. doi: 10.1016/j.bbabio.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff M, Froissart R, Olsen RK, Acquaviva C, Vianey-Saban C. Electron transfer flavoprotein deficiency: functional and molecular aspects. Mol Genet Metab. 2006;88:153–158. doi: 10.1016/j.ymgme.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Seiler CY, Park JG, Sharma A, Hunter P, Surapaneni P, Sedillo C, Field J, Algar R, Price A, Steel J, et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Research. 2014;42:D1253–1260. doi: 10.1093/nar/gkt1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, Napoli E, Cortopassi G. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Human molecular genetics. 2007;16:929–941. doi: 10.1093/hmg/ddm038. [DOI] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefely JA, Reidenbach AG, Ulbrich A, Oruganty K, Floyd BJ, Jochem A, Saunders JM, Johnson IE, Minogue CE, Wrobel RL, et al. Mitochondrial ADCK3 employs an atypical protein kinase-like fold to enable coenzyme Q biosynthesis. Mol Cell. 2015;57:83–94. doi: 10.1016/j.molcel.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still AJ, Floyd BJ, Hebert AS, Bingman CA, Carson JJ, Gunderson DR, Dolan BK, Grimsrud PA, Dittenhafer-Reed KE, Stapleton DS, et al. Quantification of mitochondrial acetylation dynamics highlights prominent sites of metabolic regulation. J Biol Chem. 2013;288:26209–26219. doi: 10.1074/jbc.M113.483396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiana C, Pagliarini DJ, McKenzie M, Kirby DM, Salemi R, Abu-Amero KK, Dahl HH, Hutchison WM, Vascotto KA, Smith SM, et al. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am J Hum Genet. 2008;83:468–478. doi: 10.1016/j.ajhg.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vockley J, Whiteman DA. Defects of mitochondrial beta-oxidation: a growing group of disorders. Neuromuscular disorders : NMD. 2002;12:235–246. doi: 10.1016/s0960-8966(01)00308-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.