

Abstract
Fibrodysplasia Ossificans Progressiva (FOP) is a rare and as yet untreatable, genetic disorder of progressive extraskeletal ossification, is the most disabling form of heterotopic ossification (HO) in humans and causes skeletal deformities, movement impairment and premature death. Most FOP patients carry an activating mutation in a BMP type I receptor gene, ACVR1R206H, that promotes ectopic chondrogenesis and osteogenesis and in turn HO. We showed previously that the retinoic acid receptor γ (RARγ) agonist Palovarotene effectively inhibited HO in injury-induced and genetic mouse models of the disease. Here we report that the drug additionally prevents spontaneous HO, using a novel conditional-on knock-in mouse line carrying the human ACVR1R206H mutation for classic FOP. In addition, Palovarotene restored long bone growth, maintained growth plate function, and protected growing mutant neonates when given to lactating mothers. Importantly, Palovarotene maintained joint, limb and body motion, providing clear evidence for its encompassing therapeutic potential as a treatment for FOP.
Keywords: Fibrodysplasia ossificans progressiva (FOP), retinoic acid receptor (RAR), Palovarotene, heterotopic ossification, ACVR1
Introduction
Fibrodysplasia Ossificans Progressiva (FOP; MIM 135100) is an extremely disabling genetic disorder in which excess bone tissue forms episodically and postnatally at multiple ectopic sites around the skeletal bones and joints and within soft connective tissues such as tendons, ligaments, and skeletal muscles, leading to progressive joint ankylosis, skeletal deformities, growth impairment, breathing difficulty, and premature death(1). Analyses of surgical retrieval specimens showed that the extra-skeletal bone tissue that forms in FOP patients – a process described as heterotopic ossification (HO) – is invariably endochondral in nature(2). As such, its formation involves recruitment and proliferation of progenitor mesenchymal cells, their differentiation into chondrocytes, maturation and hypertrophy of cartilage, and transition to endochondral bone and mature heterotopic ossification. An anatomical site where HO initiates is recognized by FOP patients as a local ‘flare-up’ and is characterized by swelling, pain, erythema, and stiffness preceding overt bone formation(3). Such symptoms indicate local inflammation, and early stage lesions are often characterized by the presence and accumulation of innate immune cells, including mast cells that are thought to have an important role in inducing and initiating the HO formation process(4). As a consequence, the current standard of care for FOP patients includes systemic treatment with corticosteroids within 24 hours of the onset of a flare-up, with treatment continued for several days to reduce inflammation and pain(5). However, corticosteroids, or any other current drug treatments, are unable to reliably prevent HO(6).
Patients with a classic clinical presentation of FOP carry a specific germline heterozygous gain-of-function mutation – R206H – in the bone morphogenetic protein (BMP) type I receptor ACVR1 (also known as ALK2)(7). BMP signaling plays a major role in skeletogenesis and promotes chondrogenesis(8, 9). ACVR1/ALK2 acts as a BMP receptor and a constitutively active Alk2 mouse mutant induces ectopic endochondral ossification(10, 11). Importantly, the activating ACVR1R206H mutation promotes excess chondrogenic differentiation of mesenchymal progenitor cells recruited to the flare-up site, eliciting a skeletogenic response that leads to formation of heterotopic bone through an endochondral process(3, 12).
Previous findings showed that endogenous retinoid signaling is normally attenuated during chondrogenesis, that this attenuation is required for chondrogenic differentiation(13, 14), and that exogenous retinoid agonists can block chondrogenesis effectively and rapidly(15). We therefore targeted the RA signaling pathway to inhibit the obligate chondrogenic phase of HO in FOP and in turn reduce or prevent HO(16, 17). Synthetic retinoid agonists, each selective for different nuclear retinoic acid receptors (RARα or RARγ)(18, 19) were tested in mouse models of subcutaneous and intramuscular injury-induced HO as well as mice bearing an inducible and constitutive active Acvr1Q207D transgene(20), the only model of genetically-driven HO available at the time, but one that is not fully representative of the human FOP genotype(21). We found that agonists for RARα and RARγ were indeed successful in inhibiting chondrogenesis and preventing HO, but the RARγ agonists were far more effective(16, 17). This is likely due to the fact that RARγ is highly and selectively expressed in chondrogenic cells and chondrocytes compared to the other RAR members (22, 23). One of the RARγ class drugs was Palovarotene, a highly-specific RARγ agonist that was previously tested in a two-year Phase 2 clinical trial for another condition(24).
Although the previous studies were quite promising, many questions remained unanswered and most importantly whether Palovarotene would be as effective in preventing HO triggered by the human ACVR1R206H mutation as it did for Acvr1Q207D and whether this treatment could ameliorate overall skeletal function and restore skeletal growth. The Acvr1Q207D mutation is a constitutively activating Acvr1 mutation that induces robust downstream BMP pathway activation in the absence of ligand; this mutation does not occur in FOP patients. In contrast, the Acvr1R206H mutation is the most common mutation in FOP, occurring in >97% of FOP patients; it is a mildly-activating mutation in the absence of exogenous ligand, but is also very highly ligand responsive. The data in the present study, using a novel conditional-on knock-in ACVR1[R206H]FlEx mouse model that faithfully phenocopies classic FOP, provide strong and clear support for Palovarotene as an effective treatment to prevent FOP heterotopic ossification and protect skeletal function.
Materials and Methods
Mice
A conditional-on knock-in mouse model Acvr1[R206H]FlEx was developed to encode the common R206H mutant allele in FOP following recombination by Cre(25). For doxycycline-inducible global allele expression, Acvr1[R206H]FlEx/+ mice were mated with mice double transgenic for R26-rtTA and tetO-Cre (heterozygous Gt(ROSA)26Sortm1(rtTA*M2)Jae and hemizygous Tg(tetO-Cre)1Jaw; Jackson Laboratories) to generate Acvr1[R206H]FlEx/+;Gt(ROSA)26Sortm1(rtTA*M2)Jae; Tg(tetO-Cre)1Jaw mice (which we refer to as Acvr1cR206H/+). For expression in Prrx1+ progenitor cells, Acvr1[R206H]FlEx/+ mice were mated with heterozygous B6.Cg-Tg(Prrx1-Cre)1Cjt (Jackson Laboratories) to generate Acvr1[R206H]FlEx/+;Prrx1-Cre+/− mice (Prrx1-R206H). Acvr1+/+ and Acvr1[R206H]FlEx/+(without Cre) littermates were used as controls. To confirm the distribution of Prrx1-expressing cells in mouse limbs, Prrx1-Cre mice were crossed with Gt(ROSA)26Sortm4(ACTB-tdTomato-EGFP)Luo reporter mice (mT/mG; Jackson Laboratories). To detect PTHrP expression in growth plates, Prrx1-R206H and control mice in a heterozygous PTHrP-lacZ reporter background(26) (a gift from Arthur Broadus, Yale) were used. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at University of Pennsylvania.
Injury-induced heterotopic ossification in Acvr1R206H/+;R26-rtTA;tetO-Cre mice and treatment with Palovarotene
One-month-old Acvr1cR206H/+ mice were provided doxycycline chow (Harlan Laboratories, Madison, WI) for 3 days to induce mutant gene expression globally. Mouse quadriceps muscles were injured by injection with 50 μl of 10 μM cardiotoxin (Sigma). Beginning on the day of injury, Palovarotene or vehicle (1:4 DMSO in corn oil) was administered daily for 14 days by oral gavage (100μg/mouse from day 1–3 and 15μg/mouse from day 4–14) using a 20-gauge gavage needle (Fine Science Tools). Palovarotene (R667, Atomax Chemicals, China) solution in DMSO was stored at −20°C under argon and diluted (1:4) with corn oil for administration.
Induction of HO in Prrx1-R206H mice and treatment with Palovarotene
Heterotopic ossification in Prrx1-R206H mice occurs spontaneously in the absence of injury. Palovarotene treatment of Prrx1-R206H and control mice was administered orally to lactating mice (50μg/mouse/day) beginning on day of delivery and continuing for 15 days. Treatment to pups was continued by oral gavage (20μg/pup) on alternate days from P16 to P30.
Imaging analyses
Heterotopic ossification was detected and quantified by micro-computed tomography (μCT) of paraformaldehyde (PFA)-fixed whole mouse specimens (eXplore Locus SP μCT Specimen Scanner; GE Healthcare). Volumetric data were acquired using the following parameters: 80 kVp and 80 μA X-ray tube voltage and current, 250 μm aluminum filter, 1.7 s integration time, 400 views at 0.5° increments, 2×2 detector bin mode, 4 averages, 1 hr scan time. Image data were reconstructed at a resolution of 40.5 μm isotropic voxels using a Feldkamp cone beam algorithm. Reconstructed 3D data were analyzed and volumes rendered using OsiriX software (www.osirix-viewer.com). High-resolution, cross-sectional images of hindlimbs were obtained using a VivaCT 40 μCT (Scanco Medical AG, Brüttisellen, Switzerland) at a source voltage of 55kV, a source current of 145 μA, and an isotropic voxel size of 10.5 μm. Reconstructed 3-D images and Scanco μCT software were used for measuring lengths of femurs and tibia.
Histology and immunohistochemistry
Fixed tissues (4% PFA) were decalcified using Immunocal™ (Decal Chemical Corporation, Tallman, NY) for 3 days, embedded in paraffin, and sectioned serially at 7 μm. Deparaffinized sections were stained with hematoxylin/eosin (Sigma) or Alcian blue/hematoxylin/Orange G for cartilage and bone. Mast cells were detected by modified CEM staining (American MasterTech, KTCEM).
For immunohistochemical staining, deparaffinized sections were treated for antigen retrieval with 10 mM Na citrate buffer (pH 6.0) or 1% hyaluronidase at 37°C for 60 min. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide solution. Sections were blocked (Background Buster; American MasterTech), and incubated with primary antibody overnight at 4°C, followed by appropriate HRP linked secondary antibody and DAB detection (SuperPicTure™ Polymer, Invitrogen). Primary antibodies used were for: Collagen II (Abcam, ab21291), Sox9(Abcam, ab26414), PTHrP (Abcam, ab93121), phosphorylated-Smad1/5/8 (Cell Signaling, 9511S),. Hematoxylin (Vector Labs) was the counterstain. For β-galactosidase detection, fixed tissues were processed with a LacZ staining kit (Invivogen, rep-lz-t), decalcified in EDTA (pH6.5), transferred to 30% sucrose (Fisher Scientific) and embedded in OTC (American MasterTech) before serial sectioning at 14 μm.
To detect proliferating cells in vivo, mice were injected intraperitoneally with 5-bromo-2′-deoxyuridine (BrdU) solution (Thermo Fisher Scientific) at 10 μl/g mouse(27). Tissues were harvested after 3 or 36 hr, fixed in 4% PFA, processed as above, and detected by BrdU immunohistochemical staining (Thermo Fisher Scientific).
Microscopy imaging and quantification
Histological images were captured using Eclipse 90i microscope (Nikon) and processed using NIS-Elements microscope imaging software (Nikon). Measurements of the lengths of growth plate zones were performed using the NIS-Elements microscope imaging software and data obtained was analyzed statistically. Stained sections were analyzed by independent investigators without bias and with random blinding methods.
Whole mount skeletal staining
For skeletal staining, skin and internal organs were removed from neonates and adult mice and skeletons were processed for differential staining using Alcian blue and Alizarin red as described(28). Cleared and stained skeleton images were captured and lengths of long bones were measured using a Leica DFC 450c instrument and software.
Statistical analysis
Data obtained were analyzed statistically using Graph Pad Prism software (t-test and two-way Anova); values are expressed as the mean ± SEM in bar graphs. All data are from a minimum of three independent experiments.
Results
Palovarotene inhibits heterotopic ossification (HO) in a mouse model of FOP
To create a mouse model of FOP, we used a knock-in mouse line in which exon 5 containing the human R206H mutation replaces a wild-type exon 5 within the Acvr1 locus upon Cre recombinase activity (Supplemental Fig. 1; see Methods). In a first set of studies, 1-month-old heterozygous Acvr1[R206H]FlEx/+;Gt(ROSA)26Sortm1(rtTA*M2)Jae;Tg(tetO-Cre)1Jaw mice (briefly referred to as Acvr1cR206H/+) were treated with doxycycline to globally induce Cre recombinase and concomitant recombination of the Acvr1[R206H]FlEx/+ allele leading to expression of Acvr1R206H in the tissues where Acvr1 is normally expressed. Subsequently, the mice were injected with cardiotoxin into the quadriceps muscle of one leg to provoke local inflammation, muscle damage and HO(17). Starting from day 1 of injury, half of the Acvr1cR206H/+ mice were treated with Palovarotene by daily gavage for 14 days and the other half received vehicle as control. Analysis by μCT and 3D image reconstruction at day 14 showed that large HO tissue masses had formed in the targeted leg of Acvr1cR206H/+ mutant mice receiving vehicle (Fig. 1A, arrow, left panel), but HO formation was greatly diminished in Palovarotene-treated companions (Fig. 1A, arrow, right panel) by over 80% based on bone volume/total volume quantification (Fig. 1B). Histochemical analysis confirmed that newly formed cartilaginous tissue and endochondral bone were present in untreated mutants (Fig. 1C, circled area), but not in treated Acvr1cR206H/+ mutant mice (Fig. 1C, right panel). This analysis revealed also that untreated mutants showed a strong local fibroproliferative response and numerous mast cells within the muscle tissues at and around the site of incipient HO formation (Fig. 1D, F; left panels), but both responses were greatly reduced in Palovarotene-treated animals (Fig. 1D, F; right panels and Supplemental Fig. 2) by 60 to 80% (Fig. 1E, G).
Fig. 1.
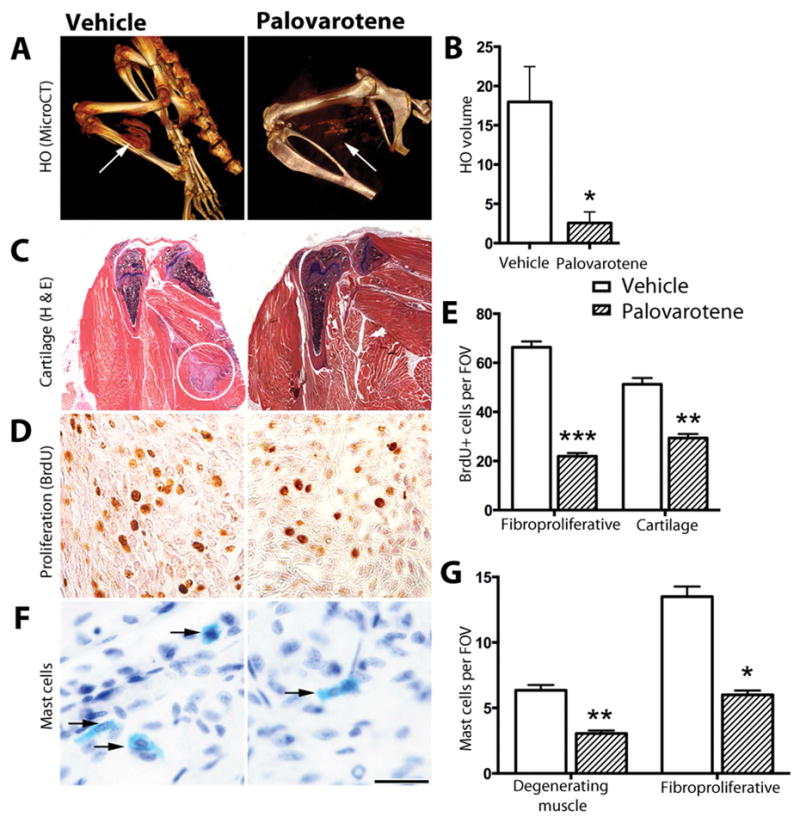
Injury-induced heterotopic ossification in Acvr1cR206H/+ mutant mice is inhibited by the RARγ agonist Palovarotene. (A) Acvr1cR206H/+;R26-rtTA;tetO-cre mice treated with doxycycline to induce global expression of Acvr1cR206H/+ were subjected to hindlimb muscle injury with cardiotoxin and examined by μCT imaging 14 days after treatment with Palovarotene (right panel) or vehicle (left panel) to detect heterotopic ossification (HO; arrows) (vehicle, n= 4; Palovarotene, n=6). (B) Heterotopic bone (arrows in A) was quantified as total mineralized tissue volume (vehicle, n= 4; Palovarotene, n=6). (C) Tissue sections of hindlimbs were stained with hematoxylin and eosin. Areas of cartilage and bone were detected in untreated (left panel, circle) but not in Palovarotene-treated samples (right panel) (n=3 mice per group). (D) Proliferating cells were detected by BrdU incorporation at the site of lesion formation in untreated (left panel) and treated (right panel) samples, and (E) quantified in the early stage fibroproliferative region following injury and in ectopic cartilage at later stages. Positive cells in 3 fields of view (FOV) in each of 4 sections were counted (n=3 mice per group). (F) Activated mast cells were detected by histochemical staining (bright blue; indicated by arrows) in early stage lesions (untreated, left panel and treated, right panel), and (G) quantified in FOV of degenerating muscle and fibroproliferative regions (n=3 mice per group). Scale bar (D and F), 25 μm. In B, E, G, hatched bars represent data from Palovarotene-treated mice and white bars from vehicle-treated. Data shown are mean±SEM; statistics compared Palovarotene-treated vs. vehicle-treated using an unpaired t-test; ***p<0.001, **p<0.01, *p<0.05.
A severe functional consequence of HO is that it can impair or completely block skeletal movement and joint function(29). Indeed, video of live, untreated ACVR1cR206H/+ mutant mice at ~14 days after HO induction by injury showed that movement of HO-affected legs was overtly impaired, but in Palovarotene-treated mice the cardiotoxin-injured legs appeared to move and function well and similarly to the contralateral uninjured leg (Supplemental Video 1).
Prenatal expression of Acvr1R206H causes limb skeletal malformations and HO
FOP patients are characterized by a great toe (first digit) malformation that is present at birth and reflects aberrant embryonic skeletal development(29). Thus, we investigated whether prenatal expression of Acvr1R206H would affect skeletal development, growth and morphogenesis. Since unrestricted endogenous activation of Acvr1R206H during early embryonic development leads to perinatal lethality(30), we limited expression to a population of skeletal progenitor cells (Prrx1+) in order to investigate effects on local skeletal development. Acvr1[R206H]FlEx/+ mice were mated with Prrx1-Cre mice to generate mutant embryos expressing Acvr1R206H throughout the lateral plate mesoderm-derived limb mesenchyme(31). Acvr1[R206H]FlEx/+: Prrx1-Cre mice (which will be referred to as Prrx1-R206H) were born at a normal Mendelian frequency and were not grossly different from companion control littermates (Acvr1+/+ and Acvr1[R206H]FlEx/+ without Cre) (Fig. 2A). However, when mice were examined after birth, the first digits of the hindlimbs were observed to be malformed as clearly revealed by radiographic imaging (Fig. 2C, arrows, right panel). While no obvious HO was present within the limbs themselves at these early ages (Fig. 2B), spontaneous HO became extensive by 1 month of age in mutants (Fig. 2D, E, arrows, right panels) and, and was detected first in the hindlimbs at ~P7 and then in the forelimbs beginning at ~P14. HO in both hind and forelimbs progressively increased over time (Supplemental Fig. 3) but was absent in control littermates. These phenotypes were fully penetrant, and the sites of HO formation were consistent with the locations of Prrx1+ lineage cells. The appearance of HO in this Prrx1-R206H mouse model was also consistent with the appearance of HO in patients with FOP. Patients do, however, develop HO in other areas of their body, including the back and neck, indicating that HO in FOP can involve cell types belonging to lineages other than those descending from Prrx1-expressing cells.
Fig. 2.

Acvr1cR206H expression in Prrx1+ skeletal progenitor cells induces malformation of hindlimb first digits and heterotopic ossification. (A) At birth (P1), Prrx1-R206H mice were shorter compared to control mice (n=12 mice per group). (B) Radiography of control and mutant mice at P1; no heterotopic ossification was detected (n=3 mice per group). (C) Radiography of hindlimbs at two weeks (P14) showed FOP-like malformation of the first digits (arrows) in mutant mice (n=3 mice per group). (D, E) At 4 weeks of age, μCT imaging showed spontaneous (non-injury-induced) heterotopic ossification (arrows) in forelimbs (D, right panel) and hindlimbs (E, right panel) of mutant mice. Control mice are Acvr1cR206H/+ without Prrx1 activation of cre (n=3 mice per group).
Long bone elongation was significantly impaired in Prrx1-R206H mice (Fig. 3A). Lengths of humerus, radius, femur and tibia were reduced in 5 day-old mutants (Fig. 3B), and growth retardation persisted at 1 month (Supplemental Fig. 4). Of note, lengths of bones did not appear to be proportionally reduced. Clinical observations have suggested that disproportionate limb lengths may occur in some FOP patients, however this remains to be verified in detailed studies. Histological analysis showed that the epiphyseal area was not grossly altered in the mutants, retaining a largely normal shape and size, however there was a significant reduction in the height of the growth plate hypertrophic zone (Fig. 3C, right panel; and Fig. 3D), a region of the growth plate primarily responsible for long bone elongation(32, 33).
Fig. 3.
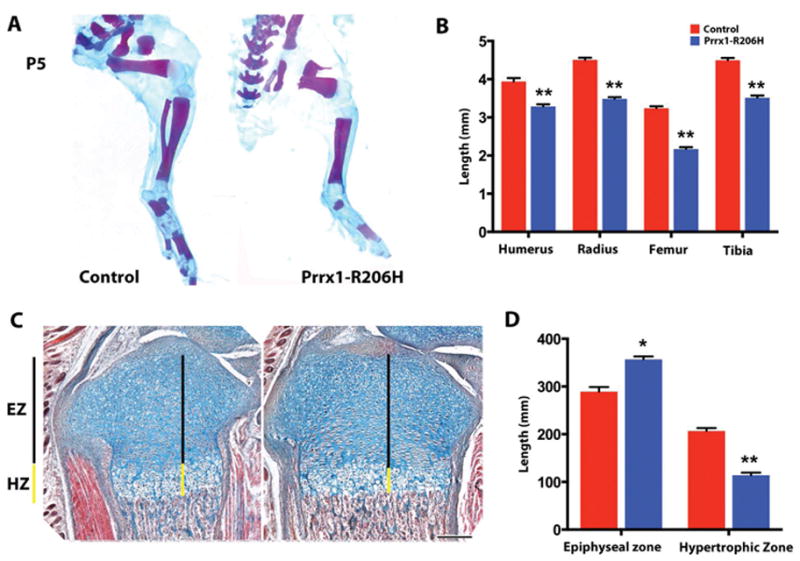
Long bone elongation is impaired by expression of Acvr1cR206H in Prrx1+ cells. (A) Hindlimb skeletons from 5-day-old (P5) control and mutant mice were stained to detect cartilage (Alcian blue) and bone (Alizarin red) (n=3 mice per group). (B) Lengths of bones in forelimbs (humerus, radius) and hindlimbs (femur, tibia) were quantified in control (red bars) and mutant (blue bars) mice (n=3 mice per group). (C, D) Proximal region of P7 tibias from control (C, right panel) and mutant (C, left panel) mice were stained with Alcian blue, Orange G, and eosin. Epiphyseal zones (EZ; black line) and hypertrophic zones (HZ; yellow line) are indicated in (C) and quantified in (D) (n=3 mice per group). Scale bar, 200 μm. Data shown are mean±SEM; statistics compared control vs. mutant using an unpaired t-test; **p<0.01, *p<0.05.
To further characterize the phenotype of the growth plate, longitudinal sections of tibias from day 14 mutant and control mice were processed for cartilage marker analysis by immunohistochemistry. Overall growth plate homeostasis and functioning were found to be defective in mutants as indicated by: (i) increased levels of phosphorylated SMAD1/5/8 and Sox9 in mutant hypertrophic zone (Fig. 4A, B, right panels), both of which are normally expressed at barely detectable levels in that area of the elongating growth plate(34, 35) (Fig. 4A, B, left panels); and (ii) higher levels of collagen II and PTHrP throughout the mutant growth plates (Fig. 4C, D, right panels) compared to the more restricted patterns in controls (Fig. 4C, D, left panels). To verify cells transcribing PTHrP, we used PTHrP-lacZ mice(26) to create reporter;Prrx1-R206H mice. We found that PTHrP expression was expanded in mutants (Fig. 4E, right panel), correlating well with the expanded distribution of PTHrP protein (Fig. 4D), while reporter activity was present more narrowly in control growth plates (Fig. 4E, left panel). Together, dysregulated PTHrP expression and high levels of phosphorylated SMAD1/5/8 and Sox9 may have overwhelmed the growth plate, disturbing hypertrophic differentiation of chondrocytes and delaying the cartilage to bone transition in the growth-plates of Acvr1R206H/+ mutant mice.
Fig. 4.

Prrx1-Acvr1cR206H/+ expression alters growth plate homeostasis. Proximal tibial growth plate sections from 14 day old mutant (Prrx1-Acvr1cR206H/+) mice (left panels) show altered distributions of growth plate markers and regulatory proteins compared to controls (Acvr1cR206H/+) (right panels) as revealed by immunohistochemical staining for: (A) pSmad 1/5/8; (B) Sox 9; (C) Collagen II; and (D) PTHrP. (E) Cells expressing PTHrP were identified in control and mutant mice in a PTHrP-LacZ background by staining for β-galactosidase. Upper zones (black line) and hypertrophic zones (yellow line) are indicated. Scale bar, 100 μm. (n=3 mice per group, similar data was observed at P1, and P7 (data not shown)).
In addition to hypertrophic zone elongation, physiologic activity of the growth plate is also influenced by chondrocyte proliferation, a process that is restricted to the distal end of the growth plate where commitment of proliferating chondrocytes to mature and differentiate occurs. To determine whether this process was altered in mutants, 2-week-old Prrx1-R206H mice and control littermates were injected with the cell proliferation marker BrdU, sacrificed at 3 or 36 hours after injection, and processed for immunohistochemical detection of BrdU-labeled chondrocytes in long bone growth plates. At the 3-hour time point, both control and mutant long bones displayed labeled chondrocytes in the upper portion of their growth plates that includes the proliferative and prehypertrophic zones (Fig. 5A). Interestingly, mutant growth plates contained a consistently higher number of labeled chondrocytes (Fig. 5B, upper zone), indicating higher cell proliferation rates compared to controls. At 36 hours, substantial numbers of BrdU+ cells in control growth plates have transitioned to the hypertrophic zone (Fig. 5C, left panel); however, mutant growth plates retained a considerable number of labeled cells in the upper zone and only a few in the hypertrophic zone (Fig. 5C, right panel; and Fig. 5D), indicating that mutant cells did not advance through the growth plate zones at normal rates.
Fig. 5.
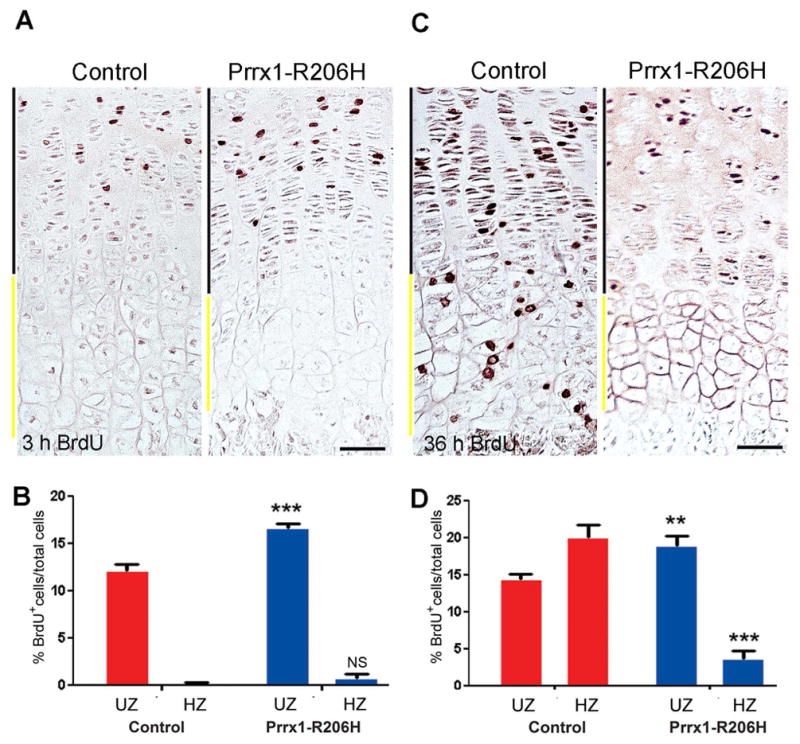
Chondrocyte proliferation and progression are altered in Prrx1-R206H growth plates. (A) Proximal tibial growth plates of control (left panel) and mutant (right panel) 14-day-old mice were labeled for 3 hours with BrdU and then processed for BrdU immunostaining to identify proliferating cells. Sections were counterstained with haemotoxylin. Upper zones (UZ; black line) and hypertrophic zones (HZ; yellow line) are indicated. Scale bar, 50 um. (B) In each of 3 sections per mouse (n=3 mice per group), BrdU positive cells in UZ and HZ regions were counted. The percentage of BrdU+ cells was determined relative to the total number of cells with haemotyoxylin-stained nuclei in each zone. (C) After 36-hour of BrdU labeling the fate of labeled cells in control (left panel) and mutant (right panel) over time was determined. Upper zones (UZ; black line) and hypertrophic zones (HZ; yellow line) are indicated. Scale bar, 50 um. (D) In each of 3 sections per mouse (n=3 mice per group), BrdU positive cells in UZ and HZ regions were counted and percentage of BrdU+ per total cells calculated. In B and D, red bars indicate data from control and blue bars from mutant mice. Data shown are mean±SEM; in each graph, statistics compared control vs. mutant in UZ or HZ using an unpaired t-test; **p<0.01, ***p<0.001, NS=no significant difference.
Palovarotene reduces spontaneous HO and improves skeletal malformations and growth in Acvr1R206H/+ mutants
To determine whether Palovarotene could prevent or alleviate the severe neonatal and postnatal skeletal defects in Prrx1-R206H mice, treatment was initiated from a neonatal stage. Palovarotene was administered to nursing transgenic females (that had been mated to Prrx1-Cre males) by daily gavage with the expectation that the drug would be passed to the pups through breastfeeding. Companion nursing females received vehicle. Litters included both mutants and control littermates, and drug or vehicle administration to the nursing mothers was continued until day 14. Thereafter, the drug (or vehicle) was administered directly to the young mice by oral gavage on alternate days until 1 month of age. Whole body skeletal examination by μCT showed that compared to controls (Fig. 6A, first panel), extensive HO had developed in the limbs of mutants, in particular their hindlimbs (Fig. 6A, third panel, arrows). Treatment of control mice with Palovarotene caused reduction in skeletal growth (Fig. 6A, second panel; and Fig. 6B), as previously observed in response to other retinoid agonists(36, 37), but no other major obvious phenotype was noted. Palovarotene-treated mutants showed markedly reduced HO that was nearly absent in their forelimbs and much reduced in hindlimbs (Fig. 6A, fourth panel). Of particular note, long bone length was considerably protected in Palovarotene-treated mutants (Fig. 6B) in contrast to the negative effects of Palovarotene on control long bone growth (Fig. 6B). Furthermore, while vehicle-treated mutants had severe movement impediment and difficulties, drug-treated mutants were mobile and appeared to function well (Supplemental Video 2). This was consistent with the beneficial effects of Palovarotene on mobility of injury-induced older mutants shown above (Supplemental Video 1). When Palovarotene treatment was delayed until day 14 and given only to the young mice, it was not as effective at inhibiting HO and maintaining mobility (not shown), suggesting importance of early treatment.
Fig. 6.

Palovarotene preserves long bone growth and growth plate organization in Prrx1-R206H mice. (A) Control and mutant mice were treated with Palovarotene (drug) or vehicle from birth (P1) to 30 days of age and then examined by μCT analysis to visualize skeletons and heterotopic bone (arrows) (n= 6 mice per group). The observed spinal curvatures are an artifact of sample position in μCT tubes, not the result of defects in the spine. (B) Quantification of femur and tibia lengths by μCT imaging (n=6 mice per group). (C) Proximal tibial growth plates were stained with Alcian blue, Orange G, and eosin. Note that Alcian blue staining cartilage matrix is markedly reduced in drug-treated controls (second panel from right) but is preserved in drug-treated mutants (leftmost panel) (n=4 mice per group). Scale bar, 100 μm. (D) Quantification of upper zones and hypertrophic zones from histological analysis. In B and D, red bars indicate data from control and blue bars from mutant mice; hatched bars represent data from palovarotene-treated mice. Data shown are mean±SEM; statistics compared control vs. control+drug or mutant vs. mutant+drug using a two-way Anova; **p<0.01, *p<0.05. Note that (in panel D), lengths of hypertrophic zones in mutants treated with drug (blue hatched bar) are protected and not significantly (NS) different from untreated controls (red bar).
Control mice receiving Palovarotene from birth were not only stunted (Fig. 6A) but exhibited also a significant reduction in safranin-O positive cartilage matrix in their growth plates (Fig. 6C, second panel) compared to untreated controls (Fig. 6C, first panel), likely the result of decreased matrix production or enhanced catabolism(38, 39). In sharp contrast, Palovarotene treatment not only protected long bone growth of the mutant mice (Fig. 6B), but also preserved the organization of their growth plates and matrix deposition (Fig. 6C, third panel; and Fig. 6D). These data suggest that interactions between Palovarotene and Acvr1R206H compensated for the negative effects of each on skeletal growth and phenotype, and that the effects of, and responses to, drug treatment differ in control and mutant cells and tissues.
Discussion
This study demonstrates that the highly specific RARγ agonist Palovarotene is a potent inhibitor of both trauma-induced and spontaneous HO elicited by the human ACVR1R206H FOP mutation in a novel mouse model of classic FOP. We further show that Palovarotene can inhibit HO that was triggered by post-natal tissue trauma or provoked systemically from an early age in the absence of overt trauma. Our data additionally demonstrate that Palovarotene can counteract the broader ravages of HO, including deleterious, often debilitating, effects on skeletal growth and mobility. Previous studies conducted through a phase 2 clinical trial for emphysema showed that Palovarotene is well tolerated by patients with relatively few and minor side effects(24). Those studies and the data here provide further support for Palovarotene as a safe and effective drug for the treatment and prevention of HO in FOP patients and potentially also for non-FOP patients who can form endochondral HO following severe injury or invasive surgeries(40, 41).
Acvr1R206H knock-in mice mimic the defects seen in FOP patients(29, 30), including skeletal growth retardation which is likely a result of (i) the extensive HO within muscles and connective tissues surrounding the skeletal elements that indirectly hampers skeletal growth and (ii) direct effects of the Acvr1R206H mutation and enhanced BMP signaling on growth plate physiology. Normally, BMP signaling - mainly via Bmpr1a and 1b - is active in the proliferative and prehypertrophic zones where it promotes and sustains Sox9 expression and chondrocyte proliferation, and is markedly reduced in the hypertrophic zone in order to facilitate transition to bone(34, 42). Previous in vitro data indicate that the ACVR1 receptor plays a key role in early steps of chondrocyte differentiation and commitment(12). Acvr1 was recently shown to be expressed in growth plates, and its conditional ablation in cartilage was found to cause growth plate dysfunction and reduced chondrocyte proliferation(43). Consistently, we observe greater proliferation in the Acvr1R206H growth plates, with failure or significant delay in hypertrophic chondrocyte differentiation. As a result, the overall effects of enhanced activation of Acvr1R206H led to compromised bone growth, supporting the notion that the appropriate magnitude and distribution of BMP receptor signaling are critical for normal longitudinal bone growth(9). Because Palovarotene inhibits BMP signaling(16, 17), it may restore skeletal growth in mutant mice by reducing BMP signaling to re-establish more physiologic signaling levels. Importantly, patients with FOP have multiple, variable and often severe skeletal anomalies affecting long bone growth predominantly in the lower limbs, and the skeletal effects noted in the mouse model emphasize the necessity for a more robust clinical characterization of this often overlooked feature in patients(44, 45). Such studies are ongoing.
In this study, we observed that Palovarotene counteracted multiple soft tissue and skeletal pathologies in Acvr1R206H/+ mutant mice. In their damaged muscle tissue, Palovarotene treatment was associated with fewer mast cells and reductions in fibroproliferation, cartilage formation, and the amount of HO. Within the skeletons of mutant mice, Palovarotene treatment led to restoration of proliferative and hypertrophic zones of growth plates and improvement of compromised bone growth. The articular malformations in the great toe, the heterotopic endochondral ossification and the severe skeletal dysplasia observed in our knock-in mouse model of classic FOP confirm the widespread effects of the Acvr1R206H mutation on a broad population of cartilage cells and precursor cells in the skeleton and soft connective tissues. Furthermore, in conjunction with our previous studies showing a role of retinoid signaling in hypertrophic cartilage to bone transition(22, 39), the abrogation of heterotopic ossification and reversal of skeletal defects by Palovarotene provide robust in vivo support for the idea that a fine balance between BMP and retinoid signaling, and in particular between Acvr1 and RARγ, is normally critical for growth plate function and skeletal growth, as occurs in other biological processes(46). For example, one role for endogenous retinoid signaling in hypertrophic cartilage would be to reduce BMP signaling and facilitate transition to bone. Thus, interactions between retinoid and BMP signaling are likely to be context dependent and/or receptor isoform specific, and to be greatly modulated by altered activity of mutant BMP receptors. Further characterization of Acvr1 and RARγ interactions will be useful not only for understanding the action and mechanisms of such key regulators, but also for identifying additional druggable targets and even more effective treatment regimens.
HO is highly damaging to the well being of FOP patients because it progressively interferes with, and limits, multiple body functions including walking, bending, breathing, mastication, and swallowing. Since Palovarotene inhibits the chondrogenic stage of heterotopic endochondral ossification, to be effective Palovarotene treatment would be needed at each flare-up to reduce or possibly prevent each new round of HO, possibly starting from a young age. Once formed, HO is permanent in FOP patients; the ectopic bone cannot be removed by surgery because the resulting tissue damage triggers additional episodes of HO(29). Given the potency of Palovarotene to prevent HO, this drug has the potential to suppress HO initiation following surgery in FOP patients, a therapeutic feat if indeed possible. Notably, the complete recovery of growth plate structure and function that we observe in Palovarotene-treated mutants supports the possibility that drug treatment of skeletally-immature patients might enable suppression of HO during childhood while restoring skeletal growth, an attainment that originally seemed counter-intuitive with an RARγ agonist.
The goal of the present study was to test whether Palovarotene can block HO elicited by the human Acvr1 R206H mutation and whether this treatment could also correct skeletal defects caused by this mutation in FOP patients. The data presented demonstrate that Palovarotene is in fact able to do so and, quite significantly, appears to be able to act on neonates via the mother’s milk. While the cellular mechanisms through which Palovarotene and Acvr1 R206H operate remain to be elucidated in further detail, these findings are a major step in establishing an effective and preventative treatment for FOP, possibly administrable from infancy.
Supplementary Material
Conditional-on knock-in mouse containing the FOPAcvr1R206H allele.
Cell proliferation is reduced in Palovarotene-treated mutant mice following injury.
Radiographs of Prrx1-R206H mice at 4 and 8 weeks of age.
Long bone elongation defects persist in 30-day-old Prrx1-R206H mice.
Palovarotene-treated Prrx1-R206H mice have improved mobility.
Palovarotene-treated ACVR1-R206H mice maintain leg movement.
Acknowledgments
We thank A. Broadus (Yale) for the PTHrP-LacZ reporter mice, the Penn Center for Musculoskeletal Diseases (NIH P30) core facilities, and A. Wright, R. Caron, W. Tseng, J. Richa, G. Ramaswamy, A. Culbert for advice and suggestions. This work was supported by the International Fibrodysplasia Ossificans Progressiva Association (IFOPA), the Center for Research in FOP and Related Disorders, the Ian Cali Endowment for FOP Research, the Whitney Weldon Endowment for FOP Research, the Isaac and Rose Nassau Professorship of Orthopaedic Molecular Medicine (FSK), the Cali-Weldon Professorship of FOP Research (EMS), the Penn Center for Musculoskeletal Disorders, contract no. W81XWH-07-1-0212 from the Department of the Army, United States Army Medical Research Acquisition Activity (MP) and US National Institutes of Health R01 grants AR056837 (MI and MP) and R01-AR41916 (FSK and EMS).
SC, KU, MC, DZ performed experiments. All authors analyzed data and/or participated in experimental design. A.E. developed the conditional Acvr1 mouse model. MP and EMS wrote the manuscript with contributions by FSK, MI and AE. All authors contributed and approved of the final version of the manuscript. EMS, MP, and MI directed the project and take responsibility for the integrity of the data.
Footnotes
Disclosures
MI and MP are consultants of Clementia Pharmaceuticals. ANE is an employee of Regeneron Pharmaceuticals, Inc. and holds stock in the company. The other authors have no disclosures related to this work.
References
- 1.Shore EM, Kaplan FS. Inherited human diseases of heterotopic bone formation. Nat Rev Rheumatol. 2010;6:518–27. doi: 10.1038/nrrheum.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplan FS, Tabas JA, Gannon FH, Finkel G, Hahn GV, Zasloff MA. The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. J Bone Joint Surg Am. 1993;75:220–30. doi: 10.2106/00004623-199302000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Shore E, Kaplan FS. Role of altered signal transduction in heterotopic ossification and fibrodysplasia ossificans progressiva. Curr Osteoporos Rep. 2011;9:83–8. doi: 10.1007/s11914-011-0046-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gannon FH, Glaser DL, Caron R, Thompson LDR, Shore E, Kaplan FS. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum Pathol. 2001;32:842–8. doi: 10.1053/hupa.2001.26464. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan FS, Pignolo RJ, Shore EM. Medical management of fibrodysplasia ossificans progressiva: Current treatment considerations. Clinical Proceedings International Clinical Consortium for FOP [Internet] 2011;4:1–100. [Google Scholar]
- 6.Kaplan FS, Chakkalakal SA, Shore EM. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Disease Models & Mechanisms. 2012;5(6):756–62. doi: 10.1242/dmm.010280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shore E, Xu M, Feldman GJ, Fenstermacher DA, Brown MA, et al. Consortium TFIR. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genet. 2006;38:525–7. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 8.Haas AR, Tuan RS. Chondrogenic differentiation of murine C3HT1/2 multipotential mesenchymal cells: II. Simulation by bone morphogenetic protein-2 requires modulation of N-cadherin expression and function. Differentiation. 1999;64:77–89. doi: 10.1046/j.1432-0436.1999.6420077.x. [DOI] [PubMed] [Google Scholar]
- 9.Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA. 2005;102:5062–7. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koefoed M, Ito H, Gromov K, Reynolds DG, Awad HA, Rubery PT, et al. Biological effects of rAAV-caAlk2 coating on structural allograft healing. Molecular Therapy. 2005;12(2):212–8. doi: 10.1016/j.ymthe.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 11.Zhang DH, Schwarz EM, Rosier RN, Zuscik MJ, Puzas JE, O’Keefe RJ. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. Journal of Bone and Mineral Research. 2003;18(9):1593–604. doi: 10.1359/jbmr.2003.18.9.1593. [DOI] [PubMed] [Google Scholar]
- 12.Culbert AL, Chakkalakal SA, Theosmy EG, Brennan TA, Kaplan FS, Shore EM. Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem Cells. 2014;32(5):1289–300. doi: 10.1002/stem.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weston AD, Chandraratna RAS, Torchia J, Underhill TM. Requirement for RAR-mediated gene repression in skeletal progenitor differentiation. J Cell Biol. 2002;158:39–51. doi: 10.1083/jcb.200112029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weston AD, Rosen V, Chandraratna RAS, Underhill TM. Regulation of skeletal progenitor differentiation by the BMP and retinoid signaling pathways. J Cell Biol. 2000;148:679–90. doi: 10.1083/jcb.148.4.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pacifici M, Cossu G, Molinaro M, Tato’ F. Vitamin A inhibits chondrogenesis but not myogenesis. Exp Cell Res. 1980;129:469–74. doi: 10.1016/0014-4827(80)90517-0. [DOI] [PubMed] [Google Scholar]
- 16.Shimono K, Morrison TN, Tung W-E, Chandraratna RAS, Williams JA, Iwamoto M, et al. Inhibition of ectopic bone formation by a selective retinoic acid receptor α-agonist: a new therapy for heterotopic ossification? J Orthop Res. 2010;28:271–7. doi: 10.1002/jor.20985. [DOI] [PubMed] [Google Scholar]
- 17.Shimono K, Tung W-E, Macolino C, Chi A, Didizian JH, Mundy C, et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nature Med. 2011;17:454–60. doi: 10.1038/nm.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernard BA, Bernardon J-M, Deleschluse C, Martin B, Lenoir M-C, Maignan J, et al. Identification of synthetic retinoids for selectivity for human nuclear retinoic acid receptor γ. Biochem Biophys Res Commun. 1992;186:977–83. doi: 10.1016/0006-291x(92)90842-9. [DOI] [PubMed] [Google Scholar]
- 19.Klein ES, Pino ME, Johnson AT, Davies PJA, Napgal S, Scott SM, et al. Identification and functional separation of retinoic acid receptor neutral antagonists and inverse agonists. J Biol Chem. 1996;271:22692–6. doi: 10.1074/jbc.271.37.22692. [DOI] [PubMed] [Google Scholar]
- 20.Fukuda T, Scott G, Komatsu Y, Araya R, Kawano M, Ray MK, et al. Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis. 2006;44:159–67. doi: 10.1002/dvg.20201. [DOI] [PubMed] [Google Scholar]
- 21.Haupt J, Deichsel A, Stange K, Ast C, Bocciardi R, Ravazzolo R, et al. ACVR1 p.Q207E causes classic fibrodysplasia ossificans progressiva and is functionally distinct from the engineered constitutively active ACVR1 p.Q207D variant. Human Molecular Genetics. 2014;23(20):5364–77. doi: 10.1093/hmg/ddu255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koyama E, Golden EB, Kirsch T, Adams SL, Chandraratna RAS, Michaille JJ, et al. Retinoid signaling is required for chondrocyte maturation and endochondral bone formation during limb skeletogenesis. Developmental Biology. 1999;208(2):375–91. doi: 10.1006/dbio.1999.9207. [DOI] [PubMed] [Google Scholar]
- 23.Weston AD, Hoffman LM, Underhill TM. Revisiting the role of retinoid signaling in skeletal development. Birth Defects Research (Part C) 2003;69:156–73. doi: 10.1002/bdrc.10010. [DOI] [PubMed] [Google Scholar]
- 24.Hind M, Stinchcombe S. Palovarotene, a novel retinoic acid receptor gamma agonist for the treatment of emphysema. Curr Opin Invest Drugs. 2009;10:1243–50. [PubMed] [Google Scholar]
- 25.Hatsell SJ, Idone V, Wolken DMA, Huang L, Kim HJ, Wang L, et al. ACVR1 R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Science Translational Medicine. 2015;7(303):303ra137. doi: 10.1126/scitranslmed.aac4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen XS, Macica CM, Dreyer BE, Hammond VE, Hens JR, Philbrick WM, et al. Initial characterization of PTH-related protein gene-driven lacZ expression in the mouse. Journal of Bone and Mineral Research. 2006;21(1):113–23. doi: 10.1359/JBMR.051005. [DOI] [PubMed] [Google Scholar]
- 27.Dy P, Wang WH, Bhattaram P, Wang QQ, Wang L, Ballock RT, et al. Sox9 Directs Hypertrophic Maturation and Blocks Osteoblast Differentiation of Growth Plate Chondrocytes. Developmental Cell. 2012;22(3):597–609. doi: 10.1016/j.devcel.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ovchinnikov D. Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harbor Protocols. 2009;3 doi: 10.1101/pdb.prot5170. pdb.prot5170. [DOI] [PubMed] [Google Scholar]
- 29.Kaplan FS, Le Merre M, Glaser DL, Pignolo RJ, Goldsby R, Kitterman JA, et al. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191–205. doi: 10.1016/j.berh.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakkalakal SA, Zhang DY, Culbert AL, Convente MR, Caron RJ, Wright AC, et al. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. Journal of Bone and Mineral Research. 2012;27(8):1746–56. doi: 10.1002/jbmr.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33(2):77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 32.Breur GJ, VanEnkevort BA, Farnum CE, Wilsman NJ. Linear relationship between the volume of hypertrophic chondrocytes and the rate of longitudinal bone growth in growth plates. J Ortho Res. 1991;9:348–59. doi: 10.1002/jor.1100090306. [DOI] [PubMed] [Google Scholar]
- 33.Hunziker EB. Mechanism of longitudinal bone growth and its regulation by growth plate chondrocytes. Microsc Res Tech. 1994;28:505–19. doi: 10.1002/jemt.1070280606. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi T, Lyons KM, McMahon AP, Kronenberg HM. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc Natl Acad Sci USA. 2005;102:18023–7. doi: 10.1073/pnas.0503617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lefebvre V, Smits P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Research, PtC. 2005;75:200–12. doi: 10.1002/bdrc.20048. [DOI] [PubMed] [Google Scholar]
- 36.Standeven AM, Davies PJA, Chandraratna RAS, Mader DR, Johnson AT, Thomazy VA. Retinoid-induced epiphyseal closure in guinea pigs. Fundam Appl Toxicol. 1996;34:91–8. doi: 10.1006/faat.1996.0179. [DOI] [PubMed] [Google Scholar]
- 37.Wolbach SB. Vitamin-A deficiency and excess in relation to skeletal growth. J Bone Joint Surg. 1947;29:201–33. [PubMed] [Google Scholar]
- 38.Williams JA, Kane M, Okabe T, Enomoto-Iwamoto M, Napoli JL, Pacifici M, et al. Endogenous retinoids in mammalian growth plate cartilage - analysis and roles in matrix homeostasis and turnover. Journal of Biological Chemistry. 2010;285(47):36674–81. doi: 10.1074/jbc.M110.151878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams JA, Kondo N, Okabe T, Takeshita N, Pilchak DM, Koyama E, et al. Retinoic acid receptors are required for skeletal growth, matrix homeostasis and growth plate function in postnatal mouse. Developmental Biology. 2009;328(2):315–27. doi: 10.1016/j.ydbio.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forsberg JA, Pepek JM, Wagner S, Wilson K, Flint J, Andersen RC, et al. Heterotopic ossification in high-energy wartime extremity injuries: prevalence and risk factors. J Bone Joint Surg. 2009;91:1084–91. doi: 10.2106/JBJS.H.00792. [DOI] [PubMed] [Google Scholar]
- 41.Vanden Booshe L, Vanderstraeten G. Heterotopic ossification: a review. J Rehabil Med. 2005;37:129–36. doi: 10.1080/16501970510027628. [DOI] [PubMed] [Google Scholar]
- 42.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 43.Rigueur D, Brugger S, Anbarchian T, Kim JK, Lee YJ, Lyons KM. The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J Bone Min Res. 2015 doi: 10.1002/jbmr.2385. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaplan FS, Groppe JC, Seemann P, Pignolo RJ, Shore EM. Fibrodysplasia ossificans progressive: developmental implications of a novel metamorphogene. In: Bronner F, Farah-Carson MC, Roach HI, editors. Bone and Development. London: Springer-Verlag; 2010. pp. 233–49. [Google Scholar]
- 45.Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, et al. Classic and Atypical FOP Phenotypes are Caused by Mutations in the BMP Type I Receptor ACVR1. Human Mutation. 2009;30:379–90. doi: 10.1002/humu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cunningham TJ, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nature Reviews Molecular Cell Biology. 2015;16(2):110–23. doi: 10.1038/nrm3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Conditional-on knock-in mouse containing the FOPAcvr1R206H allele.
Cell proliferation is reduced in Palovarotene-treated mutant mice following injury.
Radiographs of Prrx1-R206H mice at 4 and 8 weeks of age.
Long bone elongation defects persist in 30-day-old Prrx1-R206H mice.
Palovarotene-treated Prrx1-R206H mice have improved mobility.
Palovarotene-treated ACVR1-R206H mice maintain leg movement.