

Abstract
C-terminal Binding Protein (CtBP) is a transcriptional co-regulator that downregulates the expression of many tumor-suppressor genes. Utilizing a crystal structure of CtBP with its substrate 4-methylthio-2-oxobutyric acid (MTOB) and NAD+ as a guide, we have designed, synthesized, and tested a series of small molecule inhibitors of CtBP. From our first round of compounds, we identified 2-(hydroxyimino)-3-phenylpropanoic acid as a potent CtBP inhibitor (IC50 = 0.24 μM). A structure-activity relationship study of this compound further identified the 4-chloro- (IC50 = 0.18 μM) and 3-chloro- (IC50 = 0.17 μM) analogues as additional potent CtBP inhibitors. Evaluation of the hydroxyimine analogues in a short-term cell growth/viability assay showed that the 4-chloro- and 3-chloro- analogues are 2-fold and 4-fold more potent, respectively, than the MTOB control. A functional cellular assay using a CtBP-specific transcriptional readout revealed that the 4-chloro- and 3-chloro-hydroxyimine analogues were able to block CtBP transcriptional repression activity. This data suggests that substrate-competitive inhibition of CtBP dehydrogenase activity is a potential mechanism to reactivate tumor-suppressor gene expression as a therapeutic strategy for cancer.
Keywords: C-Terminal Binding Protein, CtBP, MTOB, hydroxyimine, oxime transcriptional co-repressor, dehydrogenase, tumor suppressor gene
Graphical abstract
To create your abstract, type over the instructions in the template box below.
Fonts or abstract dimensions should not be changed or altered.
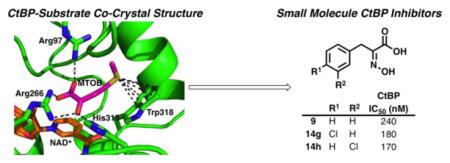
1. Introduction
C-terminal binding protein (CtBP) is a transcriptional co-regulator known to repress tumor suppressor genes and activate proto-oncogenes, and is overexpressed in a majority of colon,1 breast,2 and ovarian3 cancers. The endogenous function of CtBP is to regulate gene expression by interacting with DNA-binding transcription factors and to recruit repressor or activator complexes to targeted promoters. These transcription complexes, which include histone deacetylases,4 histone methyltransferases,5 and other chromatin-modifying proteins,6 modify the target gene epigenetically to silence or activate its expression.
Multiple tumor suppressor genes are repressed by CtBP including E-cadherin,7 PTEN,8 the apoptosis genes Bik, Noxa, Puma, and PERP,9 and the breast cancer susceptibility gene Brca1.2 Repression of these tumor suppressor genes by CtBP promotes multiple pro-oncogenic activities, including epithelial-mesenchymal transition (EMT),7 cell migration and invasion,8, 10, 11 and cell survival via proapoptotic pathways.7, 12 Activated oncogenes and cancer relevant genes include TIAM1 and the MDR1 gene, which promote metastasis and chemotherapeutic drug efflux and resistance, respectively.13 Genomic studies14 have shown that: 1) CtBP expression induces mesenchymal and stem cell-like features, 2) multiple CtBP repression gene targets are selectively downregulated in aggressive breast cancer subtypes, 3) differential expression of CtBP-targeted genes predicts poor clinical outcome in breast cancer patients, and 4) elevated levels of CtBP in patient tumors predict shorter median survival.
CtBP is only activated as a transcriptional co-regulator under conditions of hypoxia or glycolysis where NADH levels are elevated,15 as might be found in hypoxic tumors or those undergoing aerobic glycolysis (the Warburg effect). Under conditions with a low concentration of NADH, CtBP remains in an inactive, monomeric form. Elevation of NADH levels results in dimerization of two CtBP subunits into the transcriptionally active form of the protein.16-19 In this manner, CtBP acts as an intracellular metabolic sensor of NADH concentration.
Inhibition of CtBP’s transcriptional co-regulatory activity has been shown to reverse the neoplastic phenotype. Depletion of CtBP using siRNA restored expression of mRNA from repressed target genes, synthesis of the gene product by translation, and protein function.2 A cyclic peptide (cyclo-SGWTVVRMY) has been reported that inhibits CtBP dimerization in vitro and in cells by binding to an allosteric site.20 This peptide reduced mitotic fidelity, proliferation, and colony forming potential in cancer cells. A small molecule that disrupts CtBP association with a transcriptional partner has also been reported.21
In addition to acting as a transcriptional co-regulator, CtBP contains a catalytically active dehydrogenase domain that binds NADH and a putative substrate, 4-methylthio-2-oxobutyric acid (MTOB, 1, Figure 1a).22 The dehydrogenase active site catalyzes the reduction of the ketone in MTOB (1) to the alcohol in the product 4-methylthio-2-hydroxy-butyric acid (MTHB, 2), converting NADH to NAD+ in the process (Figure 1a).
Figure 1.

a. Enzymatic reaction catalyzed by CtBP. b. Crystal structure of CtBP1 with MTOB (1) and NAD+ (PBDID: 4LCE).
Pharmacological inhibition of CtBP’s dehydrogenase activity also reverses the cancer phenotype and induces apoptosis. High concentrations of MTOB substrate (1) inhibits the dehydrogenase activity of recombinant CtBP in vitro (IC50 = 300 μM), disrupts transcriptional repression/activation, promotes the expression of the BH3 protein Bik in p53−/− HCT-116 colon cells, and is specifically cytotoxic to both wild-type and p53−/− HCT-116 cells but not MEF cells.1 Further studies have shown that MTOB (1) disrupts transcriptional repression in breast cancer cell lines14 and suppressed cell survival in an ovarian cell line with CtBP overexpression.23
Here we report the design, synthesis, and biological evaluation of small molecule inhibitors of CtBP’s dehydrogenase activity. From our first round of compounds, we identified 2-(hydroxyimino)-3-phenylpropanoic acid (9) as a potent CtBP inhibitor (IC50 = 0.24 μM). A structure-activity relationship study of this compound further identified the 4-chloro- and 3-chloro-analogues as additional potent CtBP inhibitors which not only inhibit the dehydrogenase activity in vitro but also restore the transcription of Bik , a CtBP target gene, in cells.
2. Results and Discussion
2.1. First-Generation Substrate-Competitive CtBP Inhibitors
The crystal structure of MTOB (1) in complex with CtBP and NAD+ has been reported (Figure 1b).24 The α-ketoacid functional group in MTOB (1) shows clear interactions with the CtBP catalytic triad (Arg97, Arg266, and His315). The sulfur atom in MTOB (1) is positioned ~4 Å from Trp318 and forms a sulfur–π-interaction with the indole ring.
Using the CtBP1/MTOB (1)/NAD+ crystal structure as a starting point, we designed, synthesized, and evaluated small molecules that would inhibit the dehydrogenase activity of CtBP. Starting from MTOB (1, IC50 = 300 μM), the first modification was to replace the thioether with a phenyl ring in order to improve the π-interactions with Trp318 of CtBP (Scheme 1). Phenylpyruvic acid (3) was tested for its ability to inhibit the reduction of MTOB (1) to MTHB (2) by CtBP in an NADH consumption assay. Phenylpyruvic acid (3) was found to be a ~3-fold better inhibitor than MTOB with an IC50 of 116 μM (see Table S1 in the Supplementary Material).
Scheme 1.
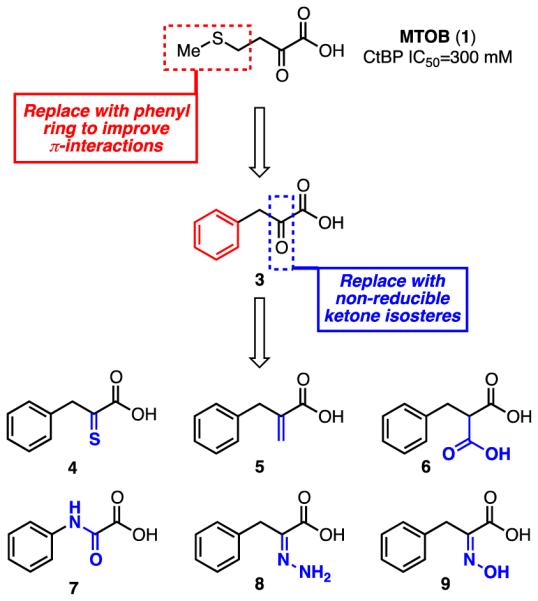
Design of CtBP Inhibitors from MTOB (1).
While phenylpyruvic acid (3) does inhibit the dehydrogenase activity of CtBP, it also acts as a substrate for the enzyme. To stop this chemical reduction of phenylpyruvic acid (3) and improve inhibition of CtBP, we next designed compounds based on the phenylpyruvic acid structure that contained isosteres of the α–ketone that could not be reduced by NADH (Scheme 1). These compounds included α–thioketone 4, acrylic acid 5, malonic acid 6, amide 7, hydrazone 8, and hydroxyimine 9. All of these compounds were either synthesized or purchased and tested for their ability to inhibit CtBP. While most of these compounds were less active than phenylpyruvic acid itself (Table S1), hydroxyimine 9 inhibited CtBP with an IC50 of 0.24 μM, a ~480-fold improvement. Hydroxyimine 9 was therefore chosen for further structure-activity relationship studies. In these studies, none of the compounds with a bioisosteric replacement for the α–ketone (4-9) acted as a substrate for CtBP.
In parallel with the ketone isosteres, we also prepared a series of deconstruction analogues of 3 to determine which structural features were necessary for inhibition. These deconstruction analogues included: pyruvate 10, where the phenyl ring has been removed; hydrocinnamic acid 11, where the ketone has been removed; 1-phenylpropan-2-one 12, where the carboxylic acid has been removed; and phenylglyoxylic acid 13, where the methylene spacer between the phenyl ring and the α-ketoacid has been removed. These compounds were also either synthesized or purchased and tested for their ability to inhibit CtBP. All of these compounds were less active than phenylpyruvic acid (Table S1).
2.2. Structure-Activity Relationship Studies on the hydroxyimine scaffold
With an understanding of the minimum structural elements needed to inhibit CtBP, we designed a large set of analogues based on the best inhibitor from our first set of compounds, hydroxyimine 9. For this set, we chose to focus on exploring the structure-activity relationship of the phenyl ring. Our set of analogues included electronically and sterically diverse substituents; all were evaluated computationally by docking them into the site identified in the CtBP–9 crystal structure25 to prioritize synthesis and evaluation. Docking scores calculated with the HINT scoring function26 (Table 1), which has been shown to correlate with free energy of binding, identified thirteen compounds that we selected for synthesis and evaluation as inhibitors of CtBP (14a-m, Figure 2).
Table 1.
Inhibition of Recombinant CtBP and Inhibition of Cell Growth of HCT-116 p53−/− Colon Cancer Cells by Cell Growth/Viability Assay for Hydroxyimine Analogues.
| Compound | Substituent | HINT Score | CtBP IC50 (μM)a | Cellular IC50 (mM)a | C Log P |
|---|---|---|---|---|---|
| 9 | H | 963 | 0.24 (0.21, 0.27) | 4.12 (2.96, 5.73) | 1.36 |
| 14a | 4-Me | 946 | 0.32 (0.29, 0.37) | 3.28 (2.51, 4.28) | 1.86 |
| 14b | 3-Me | 756 | 0.48 (0.43, 0.54) | 3.26 (2.71, 3.93) | 1.86 |
| 14c | 2-Me | 1078 | 8.73 (6.19, 12.29 | 0.23 (0.16, 0.35) | 1.81 |
| 14d | 4-OMe | 925 | 2.16 (1.19, 3.90) | 1.93 (1.65, 2.25) | 1.28 |
| 14e | 3-OMe | 919 | 0.88 (0.81, 0.97) | 5.60 (3.56, 8.84) | 1.28 |
| 14f | 2-OMe | 858 | >100 | 1.24 (1.02, 1.51) | 1.28 |
| 14g | 4-Cl | 861 | 0.18 (0.16, 0.20) | 1.74 (1.47, 2.06) | 2.07 |
| 14h | 3-Cl | 844 | 0.17 (0.15, 0.19) | 0.85 (0.76, 0.96) | 2.07 |
| 14i | 2-Cl | 974 | 7.65 (5.93, 9.86) | 2.37 (1.83, 3.08) | 2.07 |
| 14j | 4-OH | 767 | 7.34 (5.26, 10.25) | >10 | 0.69 |
| 14k | 3-OH | 1003 | 0.72 (0.67, 0.78) | >10 | 0.69 |
| 14l | 4-F | n.d.b | 0.30 (0.27, 0.33) | 3.97 (3.52, 4.49) | 1.50 |
| 14m | 4-CN | 574 | 0.90 (0.82, 0.98) | 1.10 (0.81, 1.49) | 0.79 |
| MTOB (1) | – | n.d.b | n.d.b | 4.0c | − 0.34 |
Data represents the averages with 95% confidence intervals shown in parentheses of three independent experiments.
n.d. = not determined.
From reference 1.
Figure 2.
Analogues of 9 designed to explore the structure-activity relationship.
To synthesize the hydroxyimine analogues, we chose hydantoin chemistry as a general route that would offer the flexibility needed to introduce a variety of substituents on the phenyl ring (Scheme 3). Condensation of substituted benzaldehydes (15a-b, d-g, i-m) with hydantoin under basic conditions afforded the 5-benzylideneimidazolidine-2,4-diones (16a-b, d-g, i-m), which were then hydrolyzed to afford a variety of substituted phenylpyruvic acid analogues, which upon characterization were shown to exist as the enol tautomers (17a-b, d-g, i-m), as shown in Scheme 3. These phenylpyruvic acid analogues were then condensed with hydroxylamine to form the hydroxyimine analogues (14a-b, d-g, i-m). We found this synthetic route to be highly tolerant of substitutions on the starting benzaldehyde and were able to synthesize eleven of the thirteen analogues using this methodology. For the two compounds (14c and 14h) where condensation of the benzaldehyde with the hydantoin failed to afford the 5-benzylideneimidazolidine-2,4-dione, we used the alternative methodology shown in Scheme 4. This alternative method utilized a condensation of benzaldehydes 15c and 15h with 1,4-diacetylpiperazine-2,5-dione to afford the 1-acetyl-3-benzylidenepiperazine-2,5-diones 18c and 18h. These intermediates were then hydrolyzed to afford the phenylpyruvic acid analogues 17c and 17h, which were also found to exist as the enol tautomers, followed by condensation with hydroxylamine to form the hydroxyimine analogues 14c and 14h.
Scheme 3.

Synthesis of analogues 14a-b, d-g, and i-m. Reagents and Conditions: (a) Hydantoin, Na2CO3 (sat aq), ethanolamine, EtOH/H2O (1:1), 120 °C, 5-10 h. (b) i. 20% NaOH (aq), 100 °C, 3h; 12N HCl, 54-88% for 2 steps. (c) NH2OH·HCl, NaOH, H2O, rt, 12h, 54-91%.
Scheme 4.

Synthesis of analogues 14cand 14h. Reagents and Conditions: (a) 1,4-diacetylpiperazine-2,5-dione, NEt3, DMF, rt, 12 h. (b) 6N HCl, reflux, 4 h; 58% (17c), 56% (17h). (c) NH2OH·HCl, NaOH, H2O, rt, 12h; 92% (17c), 66% (17h).
Hydroxyimine analogues 14a-m were tested for their ability to inhibit the functional dehydrogenase activity of CtBP in a NADH consumption assay. Relative to the parent hydroxyimine 9, we observed that ortho-substitutions on the ring were detrimental to inhibitory activity [14c (2-Me), 14f (2-OMe), 14i (2-Cl), Table 1]. Para- and meta-substitution by electron-donating groups [14d (4-OMe), 14e (3-OMe), 14j (4-OH),14k (3-OH)] resulted in a large decrease in inhibitory activity, while para- and meta-substitution by the electronically neutral methyl group [14a (4-Me), 14b (3-Me)] resulted in only a small decrease in inhibitory activity. However, para- and meta-substitution by an electron-withdrawing chlorine group [14g (4-Cl), 14h (3-Cl)] resulted in analogues that were more active as inhibitors than parent hydroxyimine 9. Interestingly, this effect seems specific to chlorine, as para-substitutions with other electron-withdrawing groups [14l (4-F), 14m (4-CN)] resulted in no increase in inhibition (14l) or a decrease in inhibitory activity (14m). The three best inhibitors, hydroxyimines 9, 14g, and 14h, were tested for off-target inhibition of lactate dehydrogenase (LDH), a metabolic dehydrogenase with a catalytic mechanism similar to CtBP. All three hydroxyimines were >170-fold selective for CtBP2 over LDH [9: LDH IC50 = 42.5 ± 1.1 μM; 14g: LDH IC50 = 181.9 ± 12.9 μM; and 14h: LDH IC50 = 94.9 ± 7.5 μM].
To determine whether inhibition of the dehydrogenase activity of CtBP by hydroxyimine 9 and analogues 14a-m is effective at inhibiting cancer cell growth, we carried out a short-term cell growth/viability assay (Table 1). HCT-116 p53−/− colorectal cancer cells were treated with the compounds for 72 hours and cell growth and viability determined by MTT assay. The cellular IC50’s for this set of compounds ranged from 0.23 to >10 mM. A complex structure-activity relationship was noted between in vitro enzyme inhibition and in vivo cell inhibitory activity, as demonstrated by the plot in Figure 3. A number of compounds appeared to have cytotoxic effects out of proportion to their in vitro activity, consistent with off-target or non-specific toxicity (14c, 14d, 14f, 14i), while others had lower than expected cellular inhibitory effects consistent with instability or poor intracellular penetration (14e, 14k). One subset of the compounds exhibited a relatively consistent correlation of in vitro and in vivo inhibitory activity (14a,b,g,h,l), with 14g and 14h demonstrating the strongest in vitro and in vivo inhibitory activity. However, the cellular IC50’s for these compounds (0.85 to ~4 mM) were much higher than expected given their potent inhibition of recombinant CtBP2 (0.17 to 0.48 μM) in the in vitro enzyme inhibition assay.
Figure 3.
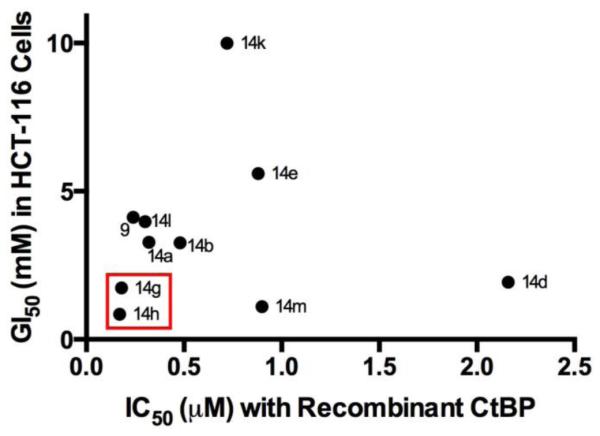
Plot of IC50 with recombinant enzyme (from NADH assay) vs. GI50 from cell growth/viability assay for hydroxyimine analogues. The most potent compounds (14g and 14h) are highlighted by the red box. Analogues 14c, 14f, 14i, and 14j have been omitted for clarity.
One potential explanation for the poor correlation between the enzyme inhibition and cell inhibitory activity is that the hydroxyimine compounds are not stable to extended exposure to aqueous conditions and are hydrolyzed to the keto-acids. To evaluate the stability of the hydroxyimines, we incubated compounds 9, 14g, and 14h in phosphate buffer (25 mM, pH 7.2) for 72 hours and monitored for degradation by HPLC (See Figure S1 A-C in the Supplementary Material). All three compounds were stable under these conditions for the entire 72 hours and no hydrolysis products were observed.
Previous studies have shown that inhibition of CtBP de-repression the transcription of the pro-apoptotic gene Bik.1 To determine whether the hydroxyimines that inhibit recombinant CtBP2 in vitro have a functional CtBP inhibition effect in a cellular environment, we performed an assay to measure restoration of Bik promoter transcription using a luciferase reporter system. In the absence of inhibitors, CtBP2 binds to the Bik promoter and prevents transcription of the firefly luciferase reporter gene cloned downstream of the Bik promoter. Functional inhibition of CtBP2 results in de-repression of the Bik promoter and an increase in the firefly luciferase signal. The hydroxyimine CtBP2 inhibitors 9, 14g, 14h, 14f, 14k, and 1 were evaluated in this assay (Figure 4). The hydroxyimines that were the best inhibitors of recombinant CtBP2 (9, 14g, 14h) also showed a strong increase in Bik transcription (9: 1.2-fold increase at 2 mM, 1.8-fold increase at 4 mM; 14g: 1.32-fold increase at 1 mM, 2.91-fold increase at 2 mM; 14h: 1.41-fold increase at 1 mM, 2.98-fold increase at 2 mM). For compounds 14g and 14h, the increase in Bik transcription could not be determined at 4 mM due to cellular cytotoxicity at this concentration. High concentrations of the substrate MTOB (1) resulted in little increase in Bik transcription in this assay (≤1.15-fold increase at both 2 and 4 mM). Hydroxyimines 14f (CtBP2 IC50 > 100 μM; cellular IC50 = 1.24 mM) and 14k (CtBP2 IC50 = 0.72 μM; cellular IC50 > 10 mM) were also evaluated as control compounds and resulted in only modest increase in Bik transcription (14f: 1.45-fold increase at 2 mM; 14k: 1.34-fold increase at 2 mM, 1.60-fold increase at 4 mM). The ~3-fold increase in Bik transcription by treatment with 14g and 14h (at 2 mM) demonstrates that the inhibition of cellular growth by these compounds most likely arises from on-target inhibition of CtBP and not off-target effects.9
Figure 4.

Change in Bik promoter expression in HCT-116 p53−/− cells upon treatment with CtBP inhibitors. * P<0.05 compared to 1, 9, 14f, and 14h. ** P<0.05 compared to 1, 9, 14f, and 14g.
In parallel with this study to explore the structure-activity relationship of hydroxyimine 9, our multidisciplinary team co-crystallized 9 with CtBP and NADH (Figure 5). This crystal structure and associated biophysical data have been reported separately.25 More detailed computational docking with this crystal structure model was performed to support the SAR studies. While no clear trend in docking scores vs. protein or cellular inhibition IC50’s was found, the docked poses of designed analogues at the CtBP active site revealed factors important for binding and inhibition (Figure 6). Most importantly, the phenyl rings of the ligands fit well into a hydrophobic region primarily formed by Trp318, Tyr76 and Met327. Compounds 14a and 14g form stronger hydrophobic and aryl-X interactions in the pocket, compared to 14j, where its polar (OH) substitution results in unfavorable hydrophobic-polar interactions. These results will inform our design of next-generation analogues.
Figure 5.
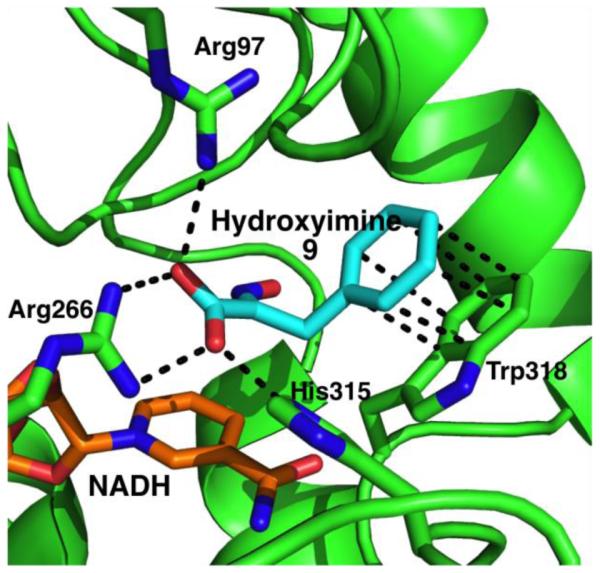
Crystal structure of CtBP1 with hydroxyimine 9 and NADH (PBDID: 4U6Q).
Figure 6.

Docked poses of 9 (white), 14a (cyan), 14g (magenta) and 14j (yellow) within CtBP active site.
2.3. Discussion
There is compelling evidence in the literature that targeting CtBP could be a highly tumor-selective and effective strategy for treating cancer. CtBP is activated as a transcriptional co-regulator only under conditions that are relatively unique to tumor cells (hypoxia and/or high NADH concentration). Disrupting the transcriptional co-regulatory function of CtBP in a tumorigenic environment has wide ranging effects on multiple cellular mechanisms, as was seen in the genomic study by Gardner and coworkers,14 and can restore the natural, endogenous function of tumor suppressor genes.
While genetic inhibition of CtBP (with siRNA) and pharmacological inhibition with peptides are effective tools for studying the role of CtBP in cancer biology, small molecule inhibitors of CtBP are needed for further clinical drug development. Here we report the first small molecule inhibitors of CtBP dehydrogenase activity, and have optimized inhibition to a submicromolar level.
While 14g and 14h are effective inhibitors of recombinant CtBP dehydrogenase activity, high concentrations are still required to inhibit growth in cells and increase Bik promoter activity in the luciferase assay. Earlier work with the parent compound MTOB, however, suggests that in long term colony formation assays, GI50 values are usually 10-fold lower than those seen in short term assays, and these studies are ongoing with the hydroxyimine derivatives.
Even taking the possible 10-fold lower GI50 of CtBP inhibitors in long-term assays into account, the cellular inhibition for the current series of CtBP inhibitors remain 2-3 orders of magnitude higher than the in vitro IC50’s. A possible rationale for this effect is that in cancer cells, CtBP has already formed transcriptional co-regulator complexes with NADH, transcription factors, and chromatin modifiers, and is not available for effective inhibitor binding. Binding of the inhibitor to CtBP either must break up the transcription complex or wait until the complex dissociates naturally in order to bind. This mechanistic model is consistent with our data that the compounds effectively bind to and inhibit the dimeric form of CtBP generated in the in vitro assay. Alternatively, the disparity between in vitro IC50 data and cell-based assays may be related to the incompletely understood relationship between enzymatic activity and transcriptional function of CtBP. We are currently working to investigate this mechanistic model and develop analogues with improved activity in the cellular assays. We are also using these new CtBP inhibitors as tools for studying the role of CtBP in cell survival and migration, as well as the impact of restoring tumor suppressor gene expression and inhibiting oncogenes, such as TIAM1, in tumor cells. These ongoing investigations by our collaborative team will be reported in due course.
3. Conclusion
In conclusion, we have developed two compounds (14g and 14h) that are substrate-competitive inhibitors of the dehydrogenase enzymatic activity of CtBP in vitro, inhibit growth in HCT-116 p53−/− colorectal cancer cells that express activated CtBP, and cause a ~3-fold increase in the transcription of the pro-apoptotic gene Bik in a luciferase reporter assay. These compounds serve as proof-of concept that substrate-competitive inhibition of CtBP dehydrogenase activity is a potential mechanism to reactivate tumor-suppressor gene expression.
4. Experimental Section
4.1. Synthesis
4.1.1. General Chemical Methods
Reagents/chemicals, catalysts, solvents were purchased from Sigma-Aldrich, Fisher and Alfa-Aesar. The following compounds were purchased from commercial sources and tested for CtBP inhibition without further purification: 4-methylthio-2-oxobutyric acid (MTOB (1), Sigma-Aldrich), phenyl pyruvic acid (3, Sigma-Aldrich), 2-benzylacrylic acid (5, TCI America), benzylmalonic acid (6, Sigma Aldrich), anilino(oxo)acetic acid (7, Sigma-Aldrich), pyruvate (10, Sigma-Aldrich), hydrocinnamic acid (11, Sigma Aldrich), and phenylglyoxylic acid (13, Sigma-Aldrich). Analytical Thin Layer Chromatography (TLC) was performed using silica gel GHLF plates (Analtech Inc.). Flash chromatography was performed on Teledyne Isco CombiFlash® Rf instrument using RediSep Rf Normal-phase Flash Columns (4-gm, 12-gm, 24-gm or 40-gm). 1H NMR and 13C NMR experiments were recorded on Bruker 400MHz NMR instrument in deuterated solvents - chloroform (CDCl3), acetone (acetone-d6), dimethyl sulfoxide (DMSO-d6) or methanol (CD3OD). All chemical shifts are reported in parts per million (ppm) with reference to chloroform, acetone, DMSO and methanol residual peaks at 7.26, 2.05, 2.50 and 3.31 respectively (1H NMR spectra); 77.16, 29.84, 39.52 and 49.00 respectively (13C NMR spectra). The data is reported as: chemical shifts (ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), coupling constant(s) (Hz) and integral values.
4.1.2. First Generation and Deconstruction Analogues
3-Phenyl-2-thioxopropanoic acid (4)
Rhodanine (1.25 g, 9.4 mmol), anhydrous sodium acetate (2.16 g, 26.4 mmol), and benzaldehyde (1.0 g, 9.4 mmol) were added to a vial followed by glacial acetic acid (17.8 mL, 0.53 M). The reaction was heated to 115 °C for three hours, upon which time the reaction had turned orange and developed a precipitate. The reaction mixture was cooled to room temperature and poured into ice water, which caused additional precipitate to form. Upon warming to room temperature, the precipitate was filtered and then dried in an oven for 36 hours to afford a dry powder (1.66 g, 80% yield). This intermediate was dissolved in water (10 mL) and 6N sodium hydroxide (10 mL) and heated to 95 °C for 1 h. The reaction mixture was cooled to room temperature, water (10 mL) was added, and 6N HCl was added to crash out a white precipitate. This precipitate was filtered and purified by flash chromatography (silica gel, 5% MeOH/DCM) to afford the product (0.86 g, 63% yield). 1H NMR (400 MHz, (DMSO-d6) δ 7.74 (s, 1H), 7.68 (d, J = 7.4 Hz, 2H), 7.48 (t, 2H), 7.42-7.36 (m, 1H). HRMS C9H7O2S [M−H]− Expected: 179.0167, Found: 179.0164.
2-hydrazono-3-phenylpropanoic acid (8)
Phenylpyruvic acid (0.1 g, 0.61 mmol) was dissolved in ethanol (0.4 ml). Hydrazine (0.02 ml, 0.61 mmol) was added and the reaction mixture is stirred overnight at room temperature. Sodium carbonate (0.03 g, 0.31 mmol) was added and the reaction mixture was stirred for 2 h at room temperature. The precipitated solid was filtered and dried to give the title compound in 62% yield (0.07 g, 0.38 mmol). 1H NMR (400 MHz, DMSO-d6) δ 7.26 (d, J = 7.5 Hz, 2H), 7.19 (t, J = 7.7 Hz, 2H), 7.10 (t, J = 7.5 Hz, 1H), 6.26 (s, 2H), 3.78 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ 168.60, 146.52, 138.01, 128.68, 127.88, 125.37, 30.21. HRMS C9H10N2O2 [M+Na]− Expected: 201.0634, Found: 201.0622.
2-(hydroxyimino)-3-phenylpropanoic acid (9)
Phenyl pyruvic acid (3.0 g, 18.28 mmol) was dissolved in a solution of NaOH (2.2 g, 54.83 mmol) in water (1 ml). Hydroxylamine hydrochloride (1.9 g, 27.41 mmol) was added to the reaction and stirred overnight at room temperature. 1N HCl was added to the reaction, the precipitated product was filtered and dried. The crude product was purified using flash chromatography (silica gel, 5% MeOH/DCM). Yield: 76% (2.50 g, 13.95 mmol). 1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 7.27 (m, 2H), 7.19 (m, 3H), 3.82 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ 165.14, 150.13, 136.68, 128.52, 128.32, 126.13, 29.85. HRMS C9H9NO3 [M−H]− Expected: 178.0509, Found: 178.0494.
1-phenylpropan-2-one (12)
Phenylacetic acid (0.5 g, 3.7 mmol) was dissolved in DCM (9.2 ml). To this, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (0.8 g, 4.4 mmol), 4-methylmorpholine (0.4 g, 3.7 mmol), N,O-dimethylhydroxylamine hydrochloride (0.4 g, 4.0 mmol) were added. The reaction mixture was stirred at room temperature for 4 hours. It was concentrated and diethyl ether was added. The organic layer was washed with brine, dried over Na2SO4 and evaporated. Purification of the residue on silica gel (5:1 hexanes/EtOAc) afforded the product N-methoxy-N-methyl-2-phenylacetamide in 87% yield. 0.6 g (3.2 mmol) of this product was dissolved in ether (10.6 ml) under nitrogen at 0 °C. Methylmagnesium bromide solution (3.4 ml, 4.8 mmol, 1.4 M in THF:toluene 1:3) was added over 30 minutes. The reaction was stirred at 0 °C for 1 hour and at room temperature for 30 minutes. It was quenched at 0 °C with the slow addition of 1 M HCl. The reaction was extracted with ether. The organic layer was washed with brine, dried over Na2SO4 and evaporated. Purification of the residue on silica gel (5:1 hexanes/EtOAc) afforded the product 1-phenylpropan-2-one in 89% yield (0.38 g, 2.83 mmol). 1H NMR (400 MHz, MeOD) δ 7.21-7.33 (m, 5H), 3.75 (s, 2H), 2.15 (s, 3H); 13C NMR (125 MHz, MeOD) δ 209.24, 136.00, 130.58, 129.67, 127.97, 51.37, 29.27. HRMS C9H10O [M−H]− Expected: 133.0659, Found: 132.0813.
4.1.3. Hydroxyimine Analogues for Structure - Activity Relationship Study
4.1.3.1. Keto - Acid Intermediates
General Procedure 1
Hydantoin (15.0 mmol) was dissolved in water (15.0 ml) at 70 °C. The solution was adjusted to pH 7 using saturated sodium bicarbonate solution. Ethanolamine (1.4 ml) was added and the temperature was raised to 90 °C. The corresponding aldehyde (15.0 mmol) in ethanol (15.0 ml) was added drop wise to the reaction mixture. The reaction was refluxed at 120 °C for 5-10 hours. After being cooled to room temperature, the product was filtered, washed with alcohol/water (1:5) and dried to give the corresponding benzylhydantoin intermediate. This corresponding benzylhydantoin (8.98 mmol) was dissolved in 20% aqueous NaOH solution (28.4 ml) and refluxed at 100 °C for 3 hours. After being cooled to room temperature, 12N HCl (11.8 ml) was added. Sodium bicarbonate was added to bring the pH to 7. The reaction mixture was extracted with ether until the ether layer was clear. This layer was discarded. To the aqueous layer, 12N HCl (7.1 ml) was added. It was extracted with ether until no more acid was obtained. The ether layer was dried to give the crude keto-acid. It was recrystallized in water to give pure product, which was judged by proton NMR to exist as the enol tautomer.
2-Hydroxy-3-(p-tolyl)acrylic acid (17a)
Procedure 1 was followed using 4-methylbenzaldehyde. Yield 54% (1.0 g, 5.61 mmol). 1H NMR (400 MHz, MeOD) δ 7.64-7.66 (d, J = 8.1 Hz, 2H), 7.12-7.14 (d, J = 8.1 Hz, 2H), 6.46 (s, 1H), 2.32 (s, 3H); 13C NMR (125 MHz, MeOD) δ 168.46, 141.49, 138.44, 133.49, 130.72, 129.87, 111.79, 21.28. HRMS C10H10O3 [M−H]− Expected: 177.0557, Found: 177.0562.
2-Hydroxy-3-(m-tolyl)acrylic acid (17b)
Procedure 1 was followed using 3-methylbenzaldehyde. Yield 57% (0.3 g, 1.68 mmol). 1H NMR (400 MHz, MeOD) δ 7.57 (d, J = 4.1 Hz, 1H), 7.55 (s, 1H), 7.18-7.22 (t, J = 7.5 Hz, 1H), 7.03-7.05 (d, J = 7.9 Hz, 1H), 6.45 (s, 1H), 2.32 (s, 3H); 13C NMR (125 MHz, MeOD) δ 170.0, 143.64, 140.38, 137.85, 132.89, 130.72, 129.55, 113.34, 23.07. HRMS C10H10O3 [M−H]− Expected: 177.0557, Found: 177.0565.
2-Hydroxy-3-(4-methoxyphenyl)acrylic acid (17d)
Procedure 1 was followed using 4-methoxybenzaldehyde. Yield 88% (1.8 g, 9.27 mmol). 1H NMR (400 MHz, MeOD) δ 7.71-7.73 (d, J = 8.8 Hz, 2H), 6.88-6.90 (d, J = 8.9 Hz, 2H), 6.46 (s, 1H), 3.80 (s, 3H); 13C NMR (125 MHz, MeOD) δ 168.61, 160.57, 140.55, 132.24, 129.08, 114.76, 111.83, 55.69. HRMS C10H10O4 [M−H]− Expected: 193.0506, Found: 193.0511.
2-Hydroxy-3-(3-methoxyphenyl)acrylic acid (17e)
Procedure 1 was followed using 3-methoxylbenzaldehyde. Yield 56% (0.5 g, 2.57 mmol). 1H NMR (400 MHz, MeOD) δ 7.44 (t, J = 2 Hz, 1H), 7.20-7.29 (m, 2H), 6.79-6.82 (m, 1H), 6.46 (s, 1H), 3.79 (s, 3H); 13C NMR (125 MHz, MeOD) δ 168.32, 161.04, 142.44, 137.66, 130.11, 123.52, 115.80, 114.39, 111.51, 55.62. HRMS C10H10O4 [M−H]− Expected: 193.0506, Found: 193.0495.
2-Hydroxy-3-(2-methoxyphenyl)acrylic acid (17f)
Procedure 1 was followed using 2-methoxybenzaldehyde. Yield 56% (0.23 g, 1.29 mmol). 1H NMR (400 MHz, MeOD) δ 8.20-8.22 (m, 1H), 7.19-7.23 (m, 1H), 6.90-6.95 (m, 3H), 3.86 (s, 3H); 13C NMR (125 MHz, MeOD) δ 168.64, 158.20, 141.79, 131.72, 129.78, 124.98, 121.41, 111.46, 105.03, 56.11. HRMS C10H10O4 [M−H]− Expected: 193.0506, Found: 193.0496.
3-(4-Chlorophenyl)-2-hydroxyacrylic acid (17g)
Procedure 1 was followed using 4-chlorobenzaldehyde. Yield 62% (1.1 g, 5.54 mmol). 1H NMR (400 MHz, MeOD) δ 7.74-7.76 (d, J = 8.8 Hz, 2H), 7.30-7.32 (d, J = 8.5 Hz, 2H), 6.45 (s, 1H); 13C NMR (125 MHz, MeOD) δ 168.05, 142.95, 135.22, 133.81, 132.06, 129.35, 110.01. HRMS C9H7O3Cl [M−H]− Expected: 197.0011, Found: 197.0016.
3-(2-Chlorophenyl)-2-hydroxyacrylic acid (17i)
Procedure 1 was followed using 2-chlorobenzaldehyde. Yield 56% (0.3 g, 1.51 mmol). 1H NMR (400 MHz, MeOD) δ 8.32-8.35 (d, J = 7.9 Hz, 1H), 7.38-7.40 (d, J = 8.1 Hz, 1H), 7.26-7.30 (t, J = 7.6 Hz, 1H), 7.19-7.21 (t, J = 7.8 Hz, 1H), 6.89 (s, 1H); 13C NMR (125 MHz, MeOD) δ 167.97, 143.77, 134.42, 134.04, 132.20, 130.31, 129.41, 127.73, 105.93. HRMS C9H7O3Cl [M−H]− Expected: 197.0011, Found: 197.0010.
2-Hydroxy-3-(4-hydroxyphenyl)acrylic acid (17j)
Procedure 1 was followed using 4-hydroxybenzaldehyde. Yield 74% (1.3 g, 7.22 mmol). 1H NMR (400 MHz, MeOD) δ 7.62-7.64 (d, J = 8.6 Hz, 2H), 6.75-6.77 (d, J = 8.8 Hz, 2H), 6.45 (s, 1H); 13C NMR (125 MHz, MeOD) δ 168.74, 158.19, 139.97, 132.41, 127.94, 116.15, 112.35. HRMS C9H8O4 [M−H]− Expected: 179.0350, Found: 179.0354.
2-Hydroxy-3-(3-hydroxyphenyl)acrylic acid (17k)
Procedure 1 was followed using 3-hydroxybenzaldehyde. Yield 88% (0.7 g, 3.89 mmol). 1H NMR (400 MHz, (DMSO-d6) δ 7.29 (s, 1H), 7.07-7.14 (m, 2H), 6.63-6.66 (m, 1H), 6.29 (s, 1H); 13C NMR (125 MHz, (DMSO-d6) δ 166.28, 157.11, 141.56, 135.96, 129.02, 120.48, 115.78, 114.47, 109.76. HRMS C9H8O4 [M−H]− Expected: 179.0350, Found: 179.0348.
3-(4-Fluorophenyl)-2-hydroxyacrylic acid (17l)
Procedure 1 was followed using 4-fluorobenzaldehyde. Yield 71% (0.5 g, 2.74 mmol). 1H NMR (400 MHz, MeOD) δ 7.78-7.81 (m, 2H), 7.02-7.07 (t, J = 8.9 Hz, 2H), 6.47 (s, 1H); 13C NMR (125 MHz, MeOD) δ 168.26, 164.52, 162.07, 142.02, 132.57, 132.65, 115.88, 116.10, 110.42. HRMS C9H7O3F [M−H]− Expected: 181.0306, Found: 181.0306.
3-(4-Cyanophenyl)-2-hydroxyacrylic acid (17m)
Procedure 1 was followed using 4-cyanobenzaldehyde. Yield 70% (0.05 g, 0.26 mmol). 1H NMR (400 MHz, (acetone-d6) δ 8.62 (d, J = 1.7 Hz, 1H), 8.01 (d, J = 8.6 Hz, 2H), 7.76 (d, J = 8.6 Hz, 2H), 6.60 (s, 1H); 13C NMR (125 MHz, (acetone-d6) δ 167.49, 145.32, 141.54, 133.02, 132.69, 132.65, 131.03, 120.00, 110.87, 108.92. HRMS C10H7NO3 [M−H]− Expected: 188.0353, Found: 188.0346.
General Procedure 2
1,4-diacetylpiperazine-2,5-dione27 (0.5 mmol) was dissolved in DMF (1 mL). Triethylamine (0.5 mmol) was added followed by corresponding aldehyde (0.5 mmol). The reaction mixture was stirred for 12 hours at room temperature. It was extracted with DCM, washed with NH4Cl solution. The organic layer was dried over Na2SO4 and evaporated. Purification of the residue on silica gel afforded the product (benzylidene)-piperazine-2,5-dione intermediate. This intermediate (0.1 mmol) was refluxed in 6N HCl (4.0 ml) for 4 hours. The reaction was cooled to room temperature, extracted with ether, dried over Na2SO4 and evaporated to give the crude keto-acid, which was recrystallized in water to give pure product and judged by proton NMR to exist as the enol tautomer.
2-Hydroxy-3-(o-tolyl)acrylic acid (17c)
Procedure 2 was followed using 2-methylbenzaldehyde. Yield 58% (0.08 g, 0.45 mmol). 1H NMR (400 MHz, (acetone-d6) δ 8.18 (d, J = 8.2 Hz, 1H), 7.85 (s, 1H), 7.11-7.23 (m, 4H), 6.74 (s, 1H), 2.38 (s, 3H); 13C NMR (125 MHz, (acetone-d6) δ 167.10, 141.15, 137.11, 133.96, 130.84, 130.64, 128.34, 126.63, 107.60, 20.13. HRMS C10H10O3 [M−H]− Expected: 177.0557, Found: 177.0549.
3-(3-Chlorophenyl)-2-hydroxyacrylic acid (17h)
Procedure 2 was followed using 3-chlorobenzaldehyde. Yield 56% (0.12 g, 0.60 mmol). 1H NMR (400 MHz, (acetone-d6) δ 7.96 (t, J = 1.96 Hz, 1H), 7.76 (dt, J = 7.8 Hz, 1H), 7.25-7.41 (m, 3H), 6.54 (s, 1H), 4.28 (s, 0.6H), 4.05 (d, J = 6.2 Hz, 0.5H); 13C NMR (125 MHz, (acetone-d6) δ 166.54, 137.85, 134.59, 130.74, 130.67, 129.80, 128.83, 127.99, 109.31. HRMS C9H7O3Cl [M−H]− Expected: 197.0011, Found: 197.0005.
4.1.3.2. Hydroxyimine Analogues
General Procedure 3
The corresponding keto-acid (0.50 mmol) was dissolved in a solution of NaOH (1.51 mmol) in water (1 ml). Hydroxylamine hydrochloride (0.76 mmol) was added to the reaction and stirred overnight at room temperature. 1N HCl was added to the reaction, the precipitated product was filtered and dried. The crude product was purified using flash chromatography (silica gel, 5% MeOH/DCM) as necessary.
2-(hydroxyimino)-3-p-tolylpropanoic acid (14a)
Title compound was prepared following general procedure 3. Yield 55% (0.03 g, 0.16 mmol). 1H NMR (400 MHz, MeOD) δ 7.05-7.14 (m, 4H), 3.86 (s, 2H), 2.27 (s, 3H); 13C NMR (125 MHz, MeOD) δ 166.90, 152.35, 136.96, 134.84, 129.96, 129.89, 30.51, 21.03. HRMS C10H11NO3 [M−H]− Expected: 192.0666, Found: 192.0673.
2-(hydroxyimino)-3-m-tolylpropanoic acid (14b)
Title compound was prepared following general procedure 3. Yield 74% (0.04 g, 0.21 mmol). 1H NMR (400 MHz, MeOD) δ 7.03-7.13 (m, 3H), 6.98 (d, J = 7.31 Hz, 1H), 3.87 (s, 2H), 2.28 (s, 3H); 13C NMR (125 MHz, MeOD) δ 166.88, 152.22, 139.03, 137.84, 130.61, 129.26, 128.02, 127.02, 30.85, 21.42. HRMS C10H11NO3 [M−H]− Expected: 192.0666, Found: 192.0656.
2-(hydroxyimino)-3-o-tolylpropanoic acid (14c)
Title compound was prepared following general procedure 3. Yield 92% (0.12 g, 0.62 mmol). 1H NMR (400 MHz, (acetone-d6) δ 7.01-7.10 (m, 4H), 3.88 (s, 2H), 2.35 (s, 3H); 13C NMR (125 MHz, (acetone-d6) δ 167.77, 153.63, 137.10, 136.37, 130.67, 129.10, 126.82, 126.60, 28.37, 19.93. HRMS C10H11NO3 [M-H]− Expected: 192.0666, Found: 192.0652.
2-(hydroxyimino)-3-(4-methoxyphenyl)propanoic acid (14d)
Title compound was prepared following general procedure 3. Yield 71% (0.08 g, 0.36 mmol). 1H NMR (400 MHz, MeOD) δ 7.18 (d, J = 8.6 Hz, 2H), 6.79 (d, J = 8.8 Hz, 2H), 3.86 (s, 2H), 3.74 (s, 3H); 13C NMR (125 MHz, MeOD) δ 167.48, 159.71, 153.20, 131.07, 130.09, 114.79, 55.67, 30.16. HRMS C10H11NO4 [M-H]− Expected: 208.0615, Found: 208.0601.
2-(hydroxyimino)-3-(3-methoxyphenyl)propanoic acid (14e)
Title compound was prepared following general procedure 3. Yield 72% (0.08 g, 0.37 mmol). 1H NMR (400 MHz, MeOD) δ 7.13-7.17 (t, J = 8.1 Hz, 1H), 6.83 (m, 2H), 6.74 (dd, J = 8.3 Hz, 1H), 3.89 (s, 2H), 3.75 (s, 3H); 13C NMR (125 MHz, MeOD) δ 166.87, 161.21, 152.07, 139.41, 130.29, 122.35, 115.73, 112.86, 55.56, 30.95. HRMS C10H11NO4 [M-H]− Expected: 208.0615, Found: 208.0612.
2-(hydroxyimino)-3-(2-methoxyphenyl)propanoic acid (14f)
Title compound was prepared following general procedure 3. Yield 56% (0.07 g, 0.33 mmol). 1H NMR (400 MHz, (acetone-d6) δ 7.17 (t, J = 7.8 Hz, 1H), 7.00 (d, J = 7.6 Hz, 1H), 6.93 (d, J = 8.5 Hz, 1H), 6.83 (t, J = 7.6 Hz, 1H), 3.90 (s, 2H), 3.82 (s, 3H); 13C NMR (125 MHz, (acetone-d6) δ 165.17, 158.28, 151.76, 129.60, 28.34, 125.57, 121.06, 111.32, 55.72, 25.53. HRMS C10H11NO4 [M-H]− Expected: 208.0615, Found: 208.0607.
3-(4-chlorophenyl)-2-(hydroxyimino)propanoic acid (14g)
Title compound was prepared following general procedure 3. Yield 56% (0.06 g, 0.28 mmol). 1H NMR (400 MHz, DMSO-d6) δ 12.34 (br s, 1H), 7.33 (d, J = 8.5 Hz, 2H), 7.20 (d, J = 8.5 Hz, 2H), 3.79 (s, 2 H); 13C NMR (125 MHz, MeOD) δ 166.71, 151.68, 136.88, 133.20, 131.63, 129.40, 30. 39. HRMS C9H8NO3Cl [M-H]− Expected: 212.0120, Found: 212.0102.
3-(3-chlorophenyl)-2-(hydroxyimino)propanoic acid (14h)
Title compound was prepared following general procedure 3. Yield 66% (0.15 g, 0.70 mmol). 1H NMR (400 MHz, DMSO-d6) δ 7.24-7.34 (m, J = 8.8 Hz, 3H), 7.17 (d, J = 7.3 Hz, 1H), 3.82 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ 165.16, 139.26, 132.81, 130.14, 128.32, 127.32, 126.14, 29.64. HRMS C9H8O3NCl [M-H]− Expected: 212.0120, Found: 212.0108.
3-(2-chlorophenyl)-2-(hydroxyimino)propanoic acid (14i)
Title compound was prepared following general procedure 3. Yield 54% (0.02 g, 0.07 mmol). 1H NMR (400 MHz, DMSO-d6) δ 7.41-7.44 (m, 1H), 7.21-7.28 (m, 2H), 7.03 (m, 1H), 3.88 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ 164.97, 149.01, 134.08, 132.83, 129.22, 129.11, 127.96, 127.11, 27.93. HRMS C9H8O3NCl [M-H]− Expected: 212.0120, Found: 212.0134.
2-(hydroxyimino)-3-(4-hydroxyphenyl)propanoic acid (14j)
Title compound was prepared following general procedure 3. Yield 91% (0.10 g, 0.51 mmol). 1H NMR (400 MHz, MeOD) δ 7.08 (d, J = 8.5 Hz, 2H), 6.66 (d, J = 8.5 Hz, 2H), 3.80 (s, 2H); 13C NMR (125 MHz, MeOD) δ 166.93, 156.93, 152.63, 131.06, 128.70, 116.13, 30.03. HRMS C9H9NO4 [M-H]− Expected: 194.0459, Found: 194.0459.
2-(hydroxyimino)-3-(3-hydroxyphenyl)propanoic acid (14k)
Title compound was prepared following general procedure 3. Yield 65% (0.07 g, 0.36 mmol). 1H NMR (400 MHz, DMSO-d6) δ 7.04 (t, J = 8.0 Hz, 1H), 6.55-6.62 (m, 3H), 3.72 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ 165.18, 157.22, 137.89, 129.09, 119.24, 115.44, 113.07, 48.52, 29.74. HRMS C9H9NO4 [M-H]− Expected: 194.0459, Found: 194.0442.
3-(4-fluorophenyl)-2-(hydroxyimino)propanoic acid (14l)
Title compound was prepared following general procedure 3. Yield 74% (0.08 g, 0.41 mmol). 1H NMR (400 MHz, DMSO-d6) δ 7.20-7.24 (m, 2H), 7.10 (t, J = 8.8 Hz, 2H), 3.79 (s, 2H); 13C NMR (125 MHz, MeOD) δ 165.06, 161.94, 159.54, 150.03, 132.82, 132.79, 130.38, 130.30, 115.12, 114.91, 29.06. HRMS C9H8NO3F [M-H]− Expected: 196.0415, Found: 196.0398.
3-(4-cyanophenyl)-2-(hydroxyimino)propanoic acid (14m)
Title compound was prepared following general procedure 3. Yield 64% (0.09 g, 0.44 mol). 1H NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 7.6 Hz, 2H), 7.37 (d, J = 8.1 Hz, 2H), 3.89 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ 164.93, 149.11, 142.74, 132.26, 129.52, 118.77, 109.06, 30.20. HRMS C10H8N2O3 [M-H]− Expected: 203.0462, Found: 203.0452.
4.2. Biological Assays
4.2.1. Protein Production and Purification of CtBP2
Truncated CtBP2 (amino acids 31-384), which lacks both the N- and C-terminal domains, was expressed as a His6-tagged protein in BL21-CodonPlus®(DE3)-RIL competent cells (Stratagene). Protein expression was induced in cultures with 200 μM IPTG for 4 hours and cells were homogenized and incubated in the presence of NiNTA beads (Thermo Scientific). CtBP2(31-384) was eluted with 300 mM imidazole after several washes and further purified by overnight dialysis and passage through a 6-8,000 kDa MWCO filter (Fisher Scientific). Purity was assessed by Coomassie stain (Invitrogen).
4.2.2. Inhibition of Dehydrogenase Activity of Recombinant CtBP (NADH inhibition assay)22
Purified CtBP2 in 50% glycerol was added to 150 μM NADH, 48 μM MTOB and various concentrations of inhibitor in buffer containing 25 mM HEPES, pH 7.1, 25 mM potassium chloride, and 1 mM DTT. The final concentration of CtBP2 was 40 μg/mL (986 nM) per reaction. Inhibitors dissolved in DMSO constituted 1% of the total volume. For compounds 4-13, inhibitor concentrations ranging from 300 μM to 10 μM and a no-drug (DMSO) control were tested. For 9and analogues 14a-m, inhibitor concentrations of 5 μM, 2.5 μM, 1 μM, 500nM, 100nM, 50nM, 10nM, and 0 nM (DMSO control) were used. Three triplicate runs were performed for each inhibitor.
Reaction components were added to 96-well UV-Star Microplates (Greiner Bio-One) and upon addition of CtBP, reactions were mixed vigorously and immediately read by a Synergy H1 microplate reader (BioTek). Absorbance was recorded at A=340nm every 30 seconds for 15 minutes at 25 °C to measure CtBP2 dehydrogenase function (NADH, but not NAD+ absorbs light at 340nm). Change in absorbance was plotted after 15 minutes and IC50 concentrations were determined using Prism (Graphpad, Version 5.04).
4.2.3. Inhibition of Dehydrogenase Activity of Lactate Dehydrogenase (LDH)
Lactate dehydrogenase (Sigma-Aldrich L1254, 0.5 U/mL, in 50% glycerol) was added to 300 μM NADH, 200 μM sodium pyruvate, and various concentrations of inhibitor in 100 mM phosphate buffer (pH 7.2). Inhibitors dissolved in DMSO constituted 1.25% of the total reaction volume (80 μL). Inhibitor concentrations of 192, 96, 48, 24, 12, 6, and 3 μM and 0 μM (DMSO control) were used. Three triplicate runs were performed for each inhibitor. Sodium oxamate was used as a control for LDH inhibition and was found to have an IC50 of 45.8 ±1.9 μM.
Reaction components were added to 96-well Microplates (Greiner Bio-One), mixed, and immediately read on a FlexStation 3 microplate reader (Molecular Devices). Absorbance was recorded at A=340nm every 10 seconds for 30 minutes at 28 °C to measure LDH dehydrogenase function (NADH, but not NAD+ absorbs light at 340nm). Change in absorbance was plotted and IC50 concentrations were determined using Prism (Graphpad).
4.2.4. Inhibition of Cell Growth (MTT Assay)
HCT-116 p53−/− colorectal cancer cell (~1,000 cells) in 100 μL media were plated in a 96 well plate. After a 24-hour incubation, inhibitors in DMSO were further diluted in NaHCO3 (0.8% diluted DMSO final volume/well) and added to plates at 4, 2, 1, and 0.5mM concentrations. 72 hours after addition of inhibitor, 20 μL of MTT solution (Alfa Aesar) was added to each well and cells were incubated a further 4 hours. Media was then aspirated and the MTT metabolic product formazan was resuspended in 200 μL DMSO. Optical density was measured at 560 nm (subtracting background at 670 nm) using a microplate reader and IC50 concentrations were determined using Prism (Graphpad, Version 5.04).
4.2.5. Restoration of Bik Transcription (Luciferase Assay)
HCT-116 p53−/− colorectal cancer cell (800 cells per well) were plated in a 96-well plate. After 24 hours, cells were transfected with pCDNA3-V5-CtBP2, the transcription factor pCDNA3-V5-KLF8, the firefly luciferase vector pGL3-Bik (promoter region - 1710 to +203), and control luciferase plasmid expressing Renilla luciferase, pRL-TK, using Lipofectamine 2000 (Thermo Fisher). Following a further 24-hour incubation, vehicle (DMSO) or inhibitors (9, 14g, 14h, 14f, 14k, and 1) were added to their final concentrations (1, 2, or 4 mM) and the expression of luciferase reporter genes was determined using the Dual Luciferase assay (Promega). For analysis, Firefly expression was normalized to Renilla control, and values were reported as a proportion of the luciferase expression of the vehicle (DMSO) control. Statistical analyses were determined by One-way ANOVA using a Tukey’s post-test using Graphpad Prism software for 2 mM drug concentrations. Data points display the average of three independent experiments, and error bars represent standard deviation.
4.3. Stability of Hydroxyimines to Aqueous Hydrolysis
Compounds 9, 14g, and 14h were diluted in 25 mM phosphate buffer (pH 7.2) to a final concentration of 4 mM. Compounds were incubated for the time indicated and then analyzed by HPLC (Agilent 1200). Aliquots (20 uL) were injected on a C18 column at a flow rate of 1.0 mL/min and eluted over 12 minutes (Linear Gradient: 95% H2O/5% ACN to 5% H2O/95% ACN). Compound elution was detected at 254 nm on a diode-array detector. The appropriate keto-acid (3, 17g, or 17h) were used as controls to monitor for hydrolysis of the oxime to the keto-acid.
4.4. HINT Scoring
Analogues of hydroxyimine 9 were modeled with SYBYL-X 2.1 (Tripos Inc.); Gasteiger–Hückel charges were assigned and models were energy minimized with the Tripos forcefield (10,000 iterations, termination gradient of 0.01 kcal/mol-Å). The crystal structure25 of CtBP1/9/NAD+ was used as the target for docking studies using GOLD v5.2,28 with the binding defined as all the residues within 10 Å of the bound ligand 9. Since the ligands are analogs of 9, it is reasonable to expect that they will adopt binding modes very similar to that of the parent compound. Thus, the binding pose of 9 defined a scaffold match constraint, i.e., the common substructure forced the corresponding atoms of ligands to lie in the exact, or very close, position within the binding site. Default GOLD genetic algorithm parameters were used and a total of 50 solutions per compound were generated. The generated conformations were re-ranked using the free-energy based HINT force-field,29 that quantifies all non-bonded interactions in a biological environment, including hydrogen-bonding, electrostatic interactions, hydrophobic interactions as well as desolvation energy. The ligand in its most energetically favorable binding conformation and the CtBP1/NAD+ complex were subjected to minimization (Powell, 2500 iterations, termination gradient 0.01 kcal/mol-Å), to remove steric clashes and optimize the protein-ligand interactions within the active site. HINT scores for the thus optimized complex were then recalculated.
Supplementary Material
Scheme 2.
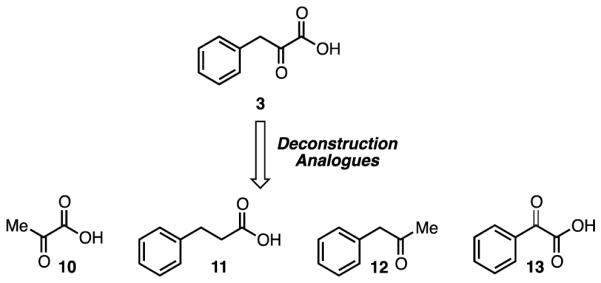
Design of Deconstruction Analogues of Phenylpyruvic Acid (3).
Acknowledgments
The authors thank Professor Richard G. Moran and the Massey Cancer Center Developmental Therapeutics group for valuable feedback and suggestions. This research was supported by a Pilot Project Grant from the VCU Massey Cancer Center (to KCE and SRG) and VCU Massey Cancer Center Shared Resource Cores supported in part with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
These authors contributed equally to this work.
Supplementary Material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.bmc.xxxx.xx. These data include Table S1, Figure S2 A-C, and NMR spectra for compounds synthesized.
References and notes
- 1.Straza MW, Paliwal S, Kovi RC, Rajeshkumar B, Trenh P, Parker D, Whalen GF, Lyle S, Schiffer CA, Grossman SR. Cell Cycle. 2010;9:3764. doi: 10.4161/cc.9.18.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deng Y, Deng H, Liu J, Han G, Malkoski S, Liu B, Zhao R, Wang X-J, Zhang Q. Mol. Carcinog. 2012;51:500. doi: 10.1002/mc.20813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barroilhet L, Yang J, Hasselblatt K, Paranal RM, Ng SK, Rauh-Hain JA, Welch WR, Bradner JE, Berkowitz RS, Ng SW. Oncogene. 2013;32:3896. doi: 10.1038/onc.2012.380. [DOI] [PubMed] [Google Scholar]
- 4.Subramanian T, Chinnadurai G. FEBS Lett. 2003;540:255. doi: 10.1016/s0014-5793(03)00275-8. [DOI] [PubMed] [Google Scholar]
- 5.Shi Y, Sawada J.-i., Sui G, Affar EB, Whetstine JR, Lan F, Ogawa H, Po-Shan Luke M, Nakatani Y, Shi Y. Nature. 2003;422:735. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 6.Chinnadurai G. Int. J. Biochem. Cell Biol. 2007;39:1593. doi: 10.1016/j.biocel.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 7.Grooteclaes M, Deveraux Q, Hildebrand J, Zhang Q, Goodman RH, Frisch SM. Proc. Natl. Acad. Sci. U. S. A. 2003;100:4568. doi: 10.1073/pnas.0830998100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paliwal S, Kovi RC, Nath B, Chen Y-W, Lewis BC, Grossman SR. Cancer Res. 2007;67:9322. doi: 10.1158/0008-5472.CAN-07-1743. [DOI] [PubMed] [Google Scholar]
- 9.Kovi RC, Paliwal S, Pande S, Grossman SR. Cell Death Differ. 2010;17:513. doi: 10.1038/cdd.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Q, Wang S-Y, Nottke AC, Rocheleau JV, Piston DW, Goodman RH. Proc. Natl. Acad. Sci. U. S. A. 2006;103:9029. doi: 10.1073/pnas.0603269103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y-W, Paliwal S, Draheim K, Grossman SR, Lewis BC. Cancer Res. 2008;68:476. doi: 10.1158/0008-5472.CAN-07-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paliwal S, Pande S, Kovi RC, Sharpless NE, Bardeesy N, Grossman SR. Mol. Cell. Biol. 2006;26:2360. doi: 10.1128/MCB.26.6.2360-2372.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin W, Scotto KW, Hait WN, Yang J-M. Biochem. Pharmacol. 2007;74:851. doi: 10.1016/j.bcp.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di L-J, Byun JS, Wong MM, Wakano C, Taylor T, Bilke S, Baek S, Hunter K, Yang H, Lee M, Zvosec C, Khramtsova G, Cheng F, Perou CM, Ryan Miller C, Raab R, Olopade OI, Gardner K. Nat. Commun. 2013;4:1449. doi: 10.1038/ncomms2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fjeld CC, Birdsong WT, Goodman RH. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9202. doi: 10.1073/pnas.1633591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mani-Telang P, Sutrias-Grau M, Williams G, Arnosti DN. FEBS Lett. 2007;581:5241. doi: 10.1016/j.febslet.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q, Piston DW. Science. 2002;295:1895. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- 18.Kumar V, Carlson JE, Ohgi KA, Edwards TA, Rose DW, Escalante CR, Rosenfeld MG, Aggarwal AK. Mol. Cell. 2002;10:857. doi: 10.1016/s1097-2765(02)00650-0. [DOI] [PubMed] [Google Scholar]
- 19.Thio SSC, Bonventre JV, Hsu SI-H. Nucleic Acids Res. 2004;32:1836. doi: 10.1093/nar/gkh344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birts CN, Nijjar SK, Mardle CA, Hoakwie F, Duriez PJ, Blaydes JP, Tavassoli A. Chem. Sci. 2013;4:3046. doi: 10.1039/c3sc50481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blevins MA, Kouznetsova J, Krueger AB, King R, Griner LM, Hu X, Southall N, Marugan JJ, Zhang Q, Ferrer M, Zhao R. J. Biomol. Screening. 2015;20:663. doi: 10.1177/1087057114561400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Achouri Y, Noël G, Van Schaftingen E. Biochem. Biophys. Res. Commun. 2007;352:903. doi: 10.1016/j.bbrc.2006.11.111. [DOI] [PubMed] [Google Scholar]
- 23.May T, Yang J, Shoni M, Liu S, He H, Gali R, Ng S-K, Crum C, Berkowitz RS, Ng S-W. Neoplasia. 2013;15:600. doi: 10.1593/neo.121674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilbert BJ, Grossman SR, Schiffer CA, Royer WE., Jr FEBS Lett. 2014;588:1743. doi: 10.1016/j.febslet.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hilbert BJ, Morris BL, Ellis KC, Paulsen JL, Schiffer CA, Grossman SR, Royer WE. ACS Chem. Biol. 2015;10:1118. doi: 10.1021/cb500820b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarkar A, Kellogg GE. Curr. Top. Med. Chem. 2010;10:67. doi: 10.2174/156802610790232233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balducci D, Conway PA, Sapuppo G, Müller-Bunz H, Paradisi F. Tetrahedron. 2012;68:7374. [Google Scholar]
- 28.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins: Struct., Funct., Bioinf. 2003;52:609. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 29.Kellogg GE, Abraham DJ. Eur. J. Med. Chem. 2000;35:651. doi: 10.1016/s0223-5234(00)00167-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.