

Abstract
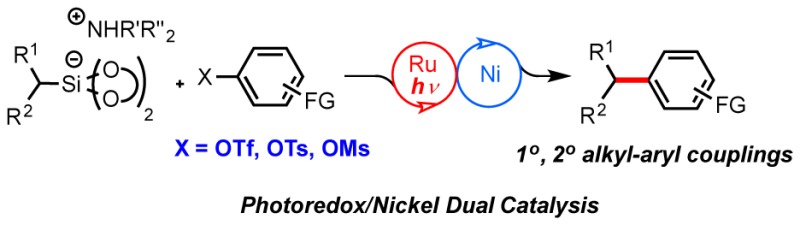
Photoredox/nickel dual catalysis via single electron transmetalation allows coupling of Csp3–Csp2 hybridized centers under mild conditions. A procedure for the coupling of electron-deficient aryl triflates, -tosylates, and -mesylates with alkylbis(catecholato)silicates is presented. This method represents the first example of the use of phenol derivatives as electrophilic coupling partners in photoredox/nickel dual catalysis.
Transition-metal-catalyzed cross-couplings have become the foundation by which synthetic chemists generate C–C and C–heteroatom bonds.1 Despite the tremendous impact of such methods, C–C bond-forming reactions using transition metal catalysts are often restricted to the construction of Csp2–Csp2 bonds, with extension to Csp3–Csp2 cross-couplings proving challenging.2 Recent developments, however, have enabled Csp3–Csp2 bonds to be forged with ease via photoredox/nickel dual catalysis.3 In particular, we and others have recently highlighted the success of alkylbis(catecholato)silicates as efficient radical precursors in dual catalysis.4 Alkylbis(catecholato)silicates undergo facile SET oxidation via reductive quenching of a variety of photocatalysts that results in C–Si bond homolysis to produce Csp3-centered radicals.5 Because of their low oxidation potentials, the use of [Ru(bpy)3](PF6)2 as a photocatalyst, which is both commercially available or simply made in-house, is feasible.6
To date, Csp3–Csp2 cross-couplings via photoredox/nickel dual catalysis have been carried out using only aryl/alkenyl halide starting materials.3a,3b,4,7 However, phenol derivatives, or “pseudo-halides”, can often be used as replacements for aryl halides in traditional transition-metal-catalyzed cross-couplings.8 Despite their use in these reactions, the ability to cross-couple aryl sulfonate esters with alkyl nucleophiles is limited, analogous to their halide counterparts.9 Because they are derived from different feedstocks than aryl halides, aryl triflates, -tosylates, and -mesylates allow access to cross-coupled substructures that may be complementary to those derived from the aryl halides alone. In addition, the ease of synthesis of aryl sulfonates from the corresponding phenols contrasts with that of the corresponding halides, which requires halogenation of arenes or aryl diazonium salts.
In an effort to expand the scope of alkylsilicate photoredox/Ni dual catalytic cross-coupling with suitable electrophiles, the compatibility of aryl sulfonates was assessed. Because they possess a similar propensity toward oxidative addition as that of the corresponding aryl iodides and bromides,10 aryl triflates and other phenol derivatives can be thought of as suitable replacements in the proposed catalytic cycle (Figure 1).11
Figure 1.
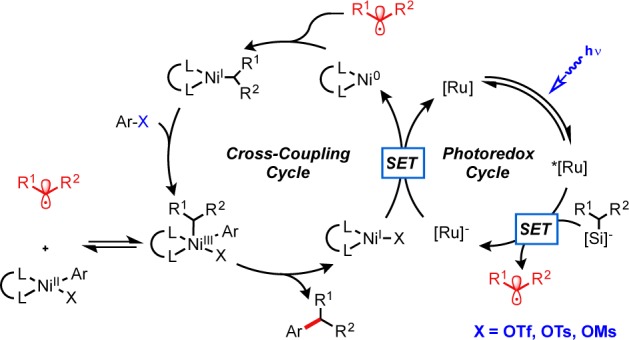
Proposed catalytic cycle for photoredox/Ni dual catalysis with alkylsilicates and phenol derivatives.
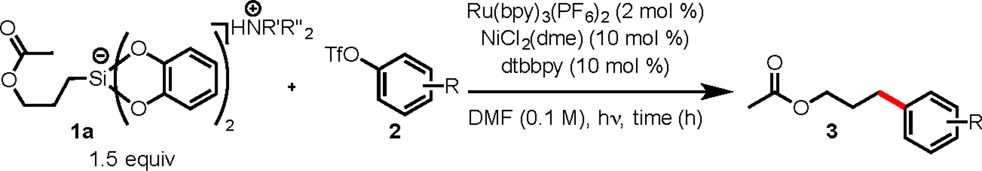
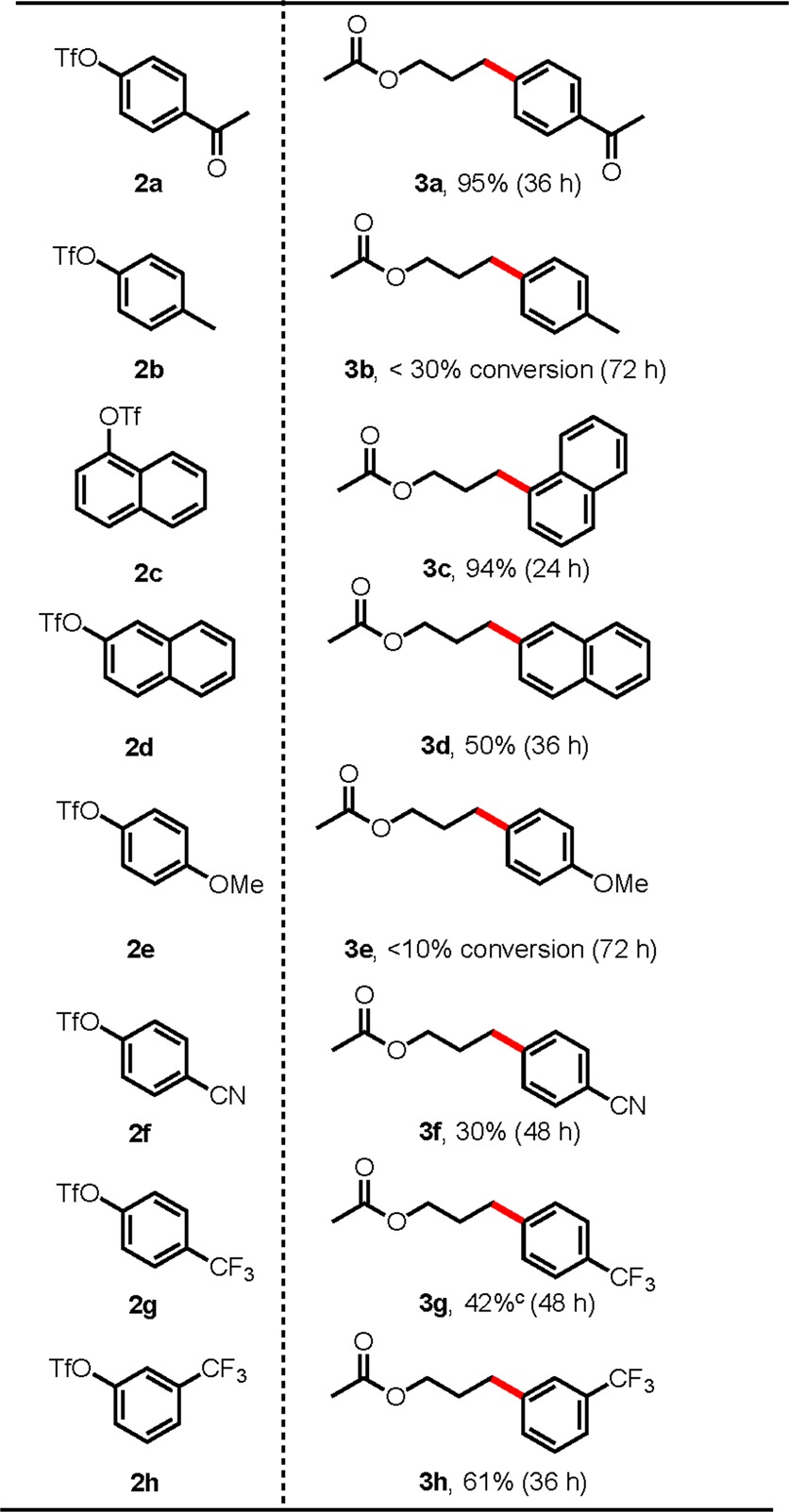
To assess the viability of aryl sulfonates in photoredox/Ni dual catalysis, a preliminary screening revealed an 87% conversion of 4-acetylphenyl triflate (2a) when the reaction was conducted in the presence of 1a under our previously established conditions {2 mol % [Ru(bpy)3](PF6)2, 5 mol % NiCl2(dme), 5 mol % dtbbpy in DMF (0.1 M), 26 W compact fluorescence lamp (CFL) or blue LEDs} after 24 h.4b,4c,12 Further optimization of this reaction allowed full consumption of triflate 2a when increasing NiCl2(dme) and dtbbpy loading to 10 mol % (see Supporting Information), which gave 3a in 95% yield on a 0.5 mmol scale after 36 h.13 Using these conditions, the full scope of this reaction was explored using 1a as the silicate coupling partner (Table 1). In addition to triflate 2a, silicate 1a coupled successfully to both 1- and 2-naphthyl triflates in 94% (3c) and 50% (3d) yields, respectively. Minimal conversion was observed in the case of 3e, possessing an electron-donating methoxy group at the para position of the arene. Additionally, although this reaction has been shown to work well with electron-deficient aryl triflates, low yields were obtained when attempting to couple silicate 1a with cyano- and trifluoromethyl-containing aryl triflates, 2f and 2g, respectively. Although unreacted starting material remained in the reaction of 2f, complete conversion of triflate was observed in the case of 2g, in which the reaction yielded a mixture of cross-coupled product 3g as well as aryl dimer from the triflate starting material. It should be noted that dimerization was not observed in reactions other than those containing trifluoromethyl functional groups. Interestingly, this reaction proved viable for meta-substituted aryl triflate derivative 2h, providing cross-coupled product 3h in good yield.
Table 1. Scope of Aryl Triflates in Photoredox/Ni Cross-Couplinga,b.
R′ = R″ = Et or R′ = H, R″ = i-Pr.
Conversion determined by HPLC.
Inseparable aryl dimer present in 1H and 13C NMRs.
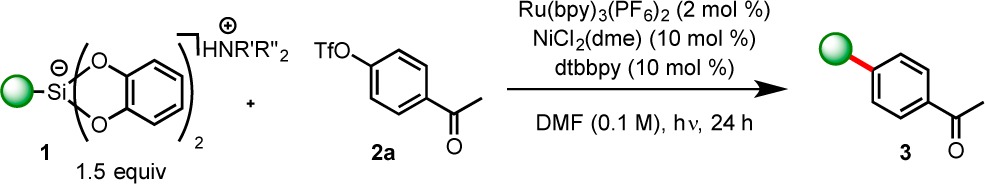
The scope of alkylbis(catecholato)silicates in this reaction was next examined (Table 2).14 Moderate to excellent yields were obtained for a variety of primary and secondary alkylsilicates when reacted with triflate 2a. Secondary silicates 1b and 1c were amenable to cross-coupling, generating the expected products in 63% and 92% yields, respectively. Additionally, simple primary silicates were amenable to cross-coupling. Hexyl (1e) and isobutyl (1f) silicate derivatives thus provided cross-coupled products in 74% and 78% yields, respectively. Silicate 1f performed well on gram scale, generating cross-coupled product in 82% yield using 5 mol % NiCl2(dme) and dtbbpy. Furthermore, the conditions of this reaction successfully allowed alkyl chains containing both alkenyl and methoxy functional groups (3n and 3o, respectively), as well as a seven-membered ring lactam/urea (1i) to be installed with ease. Importantly, the reaction could be carried out in the presence of oxygen, and thus no degassing of solvents was necessary.
Table 2. Scope of Alkylbis(catecholato)silicates in Photoredox/Ni Cross-Couplinga.
R′ = R″ = Et or R′ = H, R″ = i-Pr.
NMR yield.
Reaction provided comparable yield when no measures were taken to remove oxygen.
Reaction with 3m run on gram scale using 5 mol % [NiCl2(dme)] and dtbbpy with white LEDs.
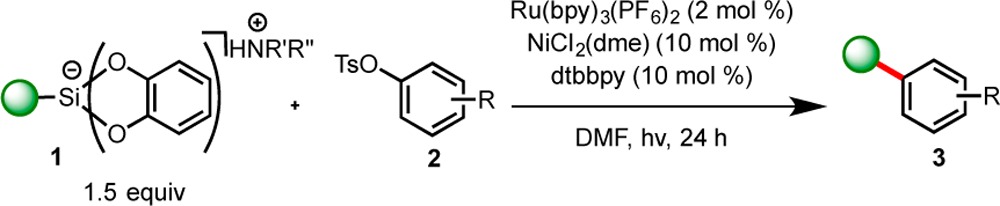
Although aryl triflates are often prone to hydrolysis under basic conditions, aryl tosylates and -mesylates exhibit better stability and are easier to handle and store. Given the observed reactivity utilizing aryl triflates as electrophilic coupling partners in photoredox/nickel dual catalysis in the presence of alkylbis(catecholato)silicates, the scope of this reaction using aryl tosylates was explored (Table 3). In the event, acetyl-containing aryl tosylate 2i showed similar reactivity to its analogous triflate derivative, furnishing 3a in 80% yield. In addition, 1-naphthyl tosylate 2j proved a sufficient coupling partner with secondary silicate 1b. Despite the success of meta-substituted triflate derivative 2h; however, tosylate 2k did not prove to be an adequate coupling partner.
Table 3. Cross-Coupling of Aryl Tosylates with Alkylbis(catecholato)silicates in Photoredox/Ni Cross-Couplinga.
R′ = R″ = Et or R′ = H, R″ = i-Pr.
Conversion determined by HPLC.
Inseparable cyclohexyl dimer impurity present in 1H and 13C NMRs
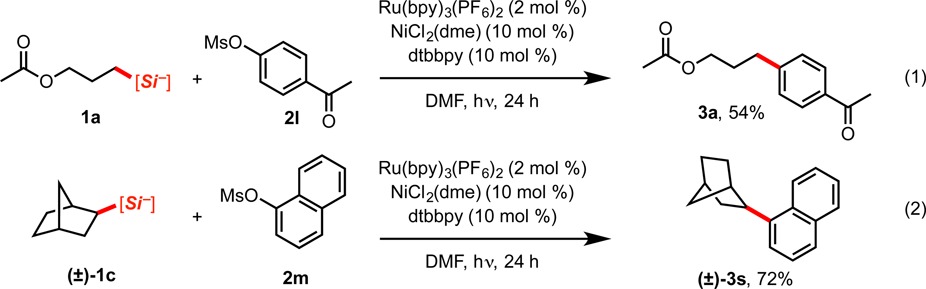
Along with triflates and tosylates, mesylates were found to be competent electrophilic coupling partners in this transformation. Coupling products 3a and 3s were produced in 54% and 72% yields, respectively, using aryl methansulfonates 2l and 2m (eqs 1 and 2). Unfortunately, sulfamate and carbamate derivatives proved to be unsuccessful coupling partners under this set of conditions.
 |
1 |
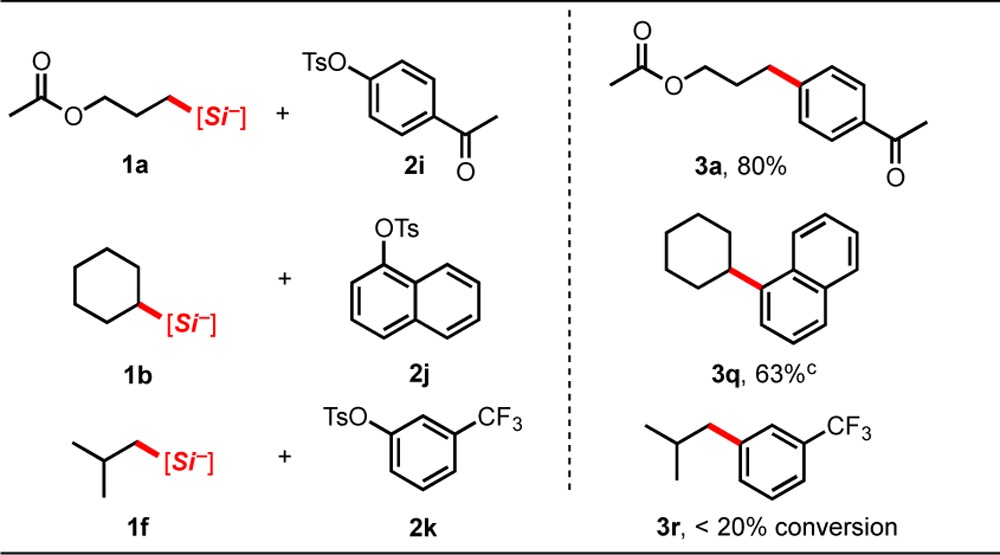
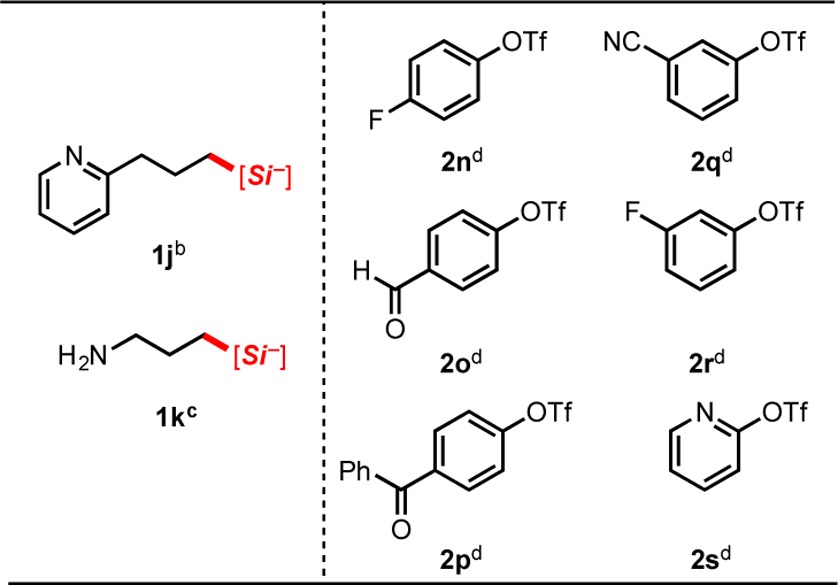
In addition to the substrates described above, other alkylsilicates and aryl triflates were attempted in the cross-coupling (Table 4). In general, we observed reduced yields using electron-rich substrates, likely because of the additive effects of this class of substrate with the diminished rate of oxidative addition into aryl sulfonate esters compared to their analogous halide counterparts. Despite the previous success of nitrogen-containing silicates with aryl- and alkenyl halides,4b,4c1j and 1k proved to be poor nucleophilic coupling partners for this transformation when reacted with 2a and 2c. In addition, triflates 2n–2s proved to be unsuccessful when reacted with silicate 1a, generating little to no cross-coupled product after 48 h.
Table 4. Unsuccessful Cross-Coupling Partners in Photoredox/Ni Cross-Couplinga.
Reacted using 2 mol % [Ru(bpy)3](PF6), 10 mol % [NiCl2(dme)] and 10 mol % dttbpy in DMF (0.1 M).
Reacted with 2a.
Reacted with 2c.
Reacted with silicate 1a.
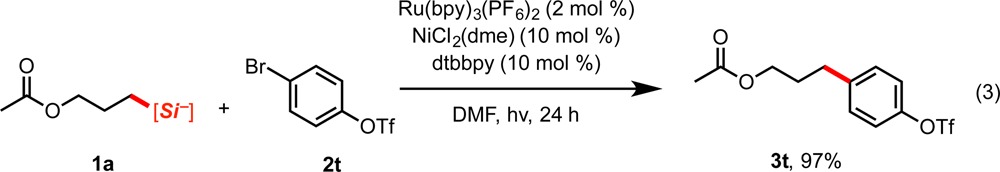
In view of the previously reported relative reactivity of aryl halides in this reaction,4b the competition between aryl triflates and -bromides in the cross-coupling was next assessed. When subjecting 2t to the conditions of the cross-coupling reaction in the presence of silicate 1a (eq 3), the reaction occurred solely at the halide, leaving the triflate virtually unreacted (3t). This suggests that the faster rate of oxidative addition of the aryl halide onto Ni(I) is critical in determining reactivity. Reactivity solely at the halide could allow selective photoredox/Ni-catalyzed cross-coupling at the bromide followed by subsequent reactivity at the triflate via photoredox/Ni dual catalysis or by means of traditional Pd- or Ni-catalyzed cross-coupling.
 |
3 |
In summary, electron-deficient aryl sulfonates can be readily cross-coupled with ammonium alkylbis(catecholato)silicates via photoredox/nickel dual catalysis—processes that proceed via single electron transmetalation under mild conditions. The ease of SET oxidation of alkylbis(catecholato)silicates to produce Csp3 centered radicals permits the use of a readily available photocatalyst. The compatibility of phenol derivatives toward oxidative addition of nickel centers allows the expansion of aryl–alkyl couplings via photoredox-nickel dual catalysis to include sulfonate ester based electrophiles. Electron-deficient aryl triflates, -tosylates, and -mesylates have been shown to be successful electrophilic coupling partners in the described cross-coupling reaction. This method represents the first example of the use of phenol derivatives as electrophilic coupling partners in photoredox/nickel dual catalysis.
Experimental Section
General Procedure for Photoredox Cross-Coupling
3-(4-Acetylphenyl)propyl Acetate (3a).15
To a 2 dram, clear glass vial equipped with a Teflon-coated magnetic stir bar were added 4,4′-di-tert-butyl-2,2′-bipyridine (13.4 mg, 0.050 mmol) and [NiCl2(dme)] (11 mg, 0.050 mmol). The vial was sealed and evacuated three times via an inlet needle and purged with argon. Once purged, 1.5 mL of THF was introduced. The resulting suspension was heated briefly with a heat gun until the nickel and ligand were fully solubilized, yielding a pale green solution. Solvents were then evaporated in vacuo to give a fine coating of the ligated nickel complex. Once dry, a phenol derivative (134 mg, 0.5 mmol, 1.0 equiv) (liquid phenol derivatives were added with solvent), alkylsilicate (335 mg, 0.75 mmol, 1.5 equiv), and [Ru(bpy)3]2PF6 (8.6 mg, 0.01 mmol) were added in succession. Under an inert atmosphere, DMF (5 mL) was introduced. The cap was sealed with Parafilm, and the solution was irradiated in front of a 26 W CFL bulb or blue LEDs. The temperature of the reaction was maintained at approximately 27 °C via a fan. The solution was stirred vigorously while being irradiated. The reaction progress was monitored by HPLC or GC/MS. Once judged to be complete, the solution was transferred to a separatory funnel and diluted with deionized H2O (∼20 mL) and Et2O (∼20 mL). The layers were separated, and the aqueous layer was extracted with Et2O (3 × ∼20 mL). The combined organic layers were washed with 1 M NaOH (∼30 mL), 1 M HCl (∼30 mL), and brine (∼50 mL). The organic layer was dried (MgSO4), and the solvent was removed in vacuo by rotary evaporation. Further purification was accomplished via column chromatography, eluting with hexane/EtOAc to give the desired compound as a light yellow oil (102 mg, 95%). 1H NMR (CDCl3, 500 MHz): δ 7.89 (d, J = 8.3 Hz, 2H), 7.28 (d, J = 8.3 Hz, 2H), 4.09 (t, J = 6.5 Hz, 2H), 2.75 (t, J = 7.6 Hz, 2H), 2.59 (s, 3H), 2.05 (s, 3H), 2.02–1.93 (m, 2H). 13C {1H} NMR (CDCl3, 125 MHz): δ 197.8, 171.2, 147.1, 135.4, 128.7, 128.7, 63.7, 32.4, 29.9, 26.7, 21.0.
3-(Naphthalen-1-yl)propyl Acetate (3c).4b
Obtained as a colorless oil (104 mg, 94%). 1H NMR (CDCl3, 500 MHz): δ 8.02 (d, J = 8.1 Hz, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.72 (d, J = 7.9 Hz, 1H), 7.55–7.44 (m, 2H), 7.39 (t, J = 7.3 Hz, 1H), 7.32 (d, J = 7.0 Hz, 1H), 4.16 (t, J = 6.5 Hz, 2H), 3.15 (t, J = 7.7 Hz, 2H), 2.15–2.03 (m, 5H). 13C {1H} NMR (CDCl3, 125 MHz): δ 171.2, 137.3, 134.0, 131.9, 128.9, 127.0, 126.1, 126.0, 125.6, 125.6, 123.7, 64.2, 29.6, 29.4, 21.1.
3-(Naphthalen-2-yl)propyl Acetate (3d).16
Obtained as a colorless oil (57 mg, 50%). 1H NMR (CDCl3, 500 MHz): δ 7.83–7.74 (m, 3H), 7.62 (s, 1H), 7.48–7.38 (m, 2H), 7.32 (dd, J = 8.4, 1.8 Hz, 1H), 4.13 (t, J = 6.6 Hz, 2H), 2.85 (t, J = 7.6 Hz, 2H), 2.09–2.01 (m, 5H). 13C {1H} NMR (CDCl3, 125 MHz): δ 171.2, 138.8, 133.7, 132.2, 128.1, 127.7, 127.5, 127.2, 126.6, 126.1, 125.3, 64.0, 32.5, 30.2, 21.1.
3-(4-Cyanophenyl)propyl Acetate (3f)
Obtained as a colorless oil (31 mg, 30%). 1H NMR (CDCl3, 500 MHz): δ 7.58 (d, J = 7.3 Hz, 2H), 7.27 (d, J = 7.7 Hz, 2H), 4.09 (t, J = 6.2 Hz, 2H), 2.75 (t, J = 7.7 Hz, 2H), 2.04 (s, 3H), 2.04–1.93 (m, 2H). 13C {1H} NMR (CDCl3, 125 MHz): δ 171.3, 146.9, 132.4, 129.3, 119.0, 110.2, 63.5, 32.5, 29.8, 21.0. IR (neat): ν = 2957, 2227, 1734, 1608, 1505, 1453, 1415, 1387, 1366, 1234, 1178, 1038, 915, 878, 846, 815, 733, 633, 606, 559 cm–1. HRMS (ESI): m/z calcd for C12H13NO2Na [M + Na]+ 226.0844, found 226.0834.
3-(4-(Trifluoromethyl)phenyl)propyl Acetate (3g).4e
Obtained as a colorless oil (61 mg, 42%). 1H NMR (CDCl3, 500 MHz): δ 7.56 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 7.9 Hz, 2H), 4.10 (t, J = 6.5 Hz, 2H), 2.76 (t, J = 7.8 Hz, 2H), 2.06 (s, 3H), 2.03–1.95 (m, 2H). 13C {1H} NMR (CDCl3, 125 MHz): δ 171.2, 145.4, 128.8, 128.6 (q, J = 32.3 Hz), 125.5 (q, J = 3.8 Hz), 122.3 (q, J = 271.9 Hz), 63.7, 32.2, 30.0, 21.1.
3-(3-(Trifluoromethyl)phenyl)propyl Acetate (3h)
Obtained as a colorless oil (103 mg, 84%). 1H NMR (CDCl3, 500 MHz): δ 7.49–7.34 (m, 4H), 4.10 (t, J = 6.5 Hz, 2H), 2.75 (t, J = 7.5 Hz, 2H), 2.05 (s, 3H), 2.03–1.93 (m, 2H). 13C {1H} NMR (CDCl3, 125 MHz): δ 171.2, 142.2, 131.9, 129.0, 130.9 (q, J = 32.0 Hz), 125.2 (q, J = 3.7 Hz), 124.3 (q, J = 272.2 Hz), 123.1 (q, J = 3.9 Hz), 63.7, 32.2, 30.1, 21.0. 19F NMR (CDCl3, 470 MHz): −62.6. IR (neat): ν = 1737, 1450, 1388, 1367, 1328, 1236, 1199, 1161, 1120, 1072, 1039, 1002, 951, 904, 799, 733, 702, 661, 631, 606 cm–1. HRMS (ESI): m/z calcd for C12H13F3O2Na [M + Na]+ 269.0765, found 269.0754.
1-(4-Cyclohexylphenyl)ethanone (3i).17
Obtained as a white solid (63 mg, 63%); mp, 57–58 °C. 1H NMR (CDCl3, 500 MHz): δ 7.89 (d, J = 8.3 Hz, 2H), 7.29 (d, J = 8.2 Hz, 2H), 2.58 (m, 4H), 1.92–1.81 (m, 4H), 1.80–1.72 (m, 1H), 1.50–1.34 (m, 4H), 1.33–1.18 (m, 1H). 13C {1H} NMR (CDCl3, 125 MHz): δ 197.9, 153.8, 135.2, 128.6, 127.1, 44.8, 34.2, 26.8, 26.6, 26.1.
(±)-1-(4-(Bicyclo[2.2.1]heptan-2-yl)phenyl)ethanone (3j)
Obtained as a yellow oil (92 mg, 92%). 1H NMR (CDCl3, 500 MHz): δ 7.87 (d, J = 8.5 Hz, 2H), 7.30 (d, J = 8.3 Hz, 2H), 2.83–2.75 (m, 1H), 2.57 (s, 3H), 2.42–2.34 (m, 2H), 1.84–1.70 (m, 1H), 1.69–1.48 (m, 3H), 1.41–1.34 (m, 1H), 1.33–1.18 (m, 3H). 13C {1H} NMR (CDCl3, 125 MHz): δ 197.9, 153.5, 134.8, 128.5, 127.3, 47.6, 42.8, 39.2, 37.0, 36.3, 30.7, 28.9, 26.6. IR (neat): ν = 2949, 2869, 1679, 1604, 1568, 1454, 1411, 1357, 1308, 1267, 1212, 1185, 1139, 1014, 955, 848, 822, 605, 591, 570 cm–1. HRMS (ESI): m/z calcd for C15H19O [M + H]+ 215.1436, found 215.1436.
1-(4-Hexylphenyl)ethanone (3l).18
Obtained as a colorless oil (76 mg, 74%). 1H NMR (CDCl3, 500 MHz): δ 7.88 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 2.69–2.62 (t, J = 7.7 Hz, 2H), 2.58 (s, 3H), 1.67–1.58 (m, 2H), 1.38–1.24 (m, 6H), 0.92–0.82 (m, 3H). 13C {1H} NMR (CDCl3, 125 MHz): δ 197.9, 148.9, 135.0, 128.7, 128.5, 36.1, 31.8, 31.2, 29.0, 26.6, 22.7, 14.2.
1-(4-Isobutylphenyl)ethanone (3m).19
Obtained as a colorless oil (69 mg, 78%). 1H NMR (CDCl3, 500 MHz): δ 7.87 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 8.3 Hz, 2H), 2.58 (s, 3H), 2.53 (d, J = 7.2 Hz, 2H), 1.90 (m, 1H), 0.91 (d, J = 6.6 Hz, 6H). 13C {1H} NMR (CDCl3, 125 MHz): δ 198.0, 147.7, 135.1, 129.4, 128.4, 45.5, 30.2, 26.6, 22.4.
1-(4-(2-(Cyclohex-3-en-1-yl)ethyl)phenyl)ethanone (3n)
Obtained as a light yellow oil (62 mg, 55%). 1H NMR (CDCl3, 500 MHz): δ 7.88 (d, J = 8.1 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H), 5.71–5.62 (m, 2H), 2.71 (t, J = 7.8 Hz, 2H), 2.58 (s, 3H), 2.20–1.99 (m, 3H), 1.83–1.52 (m, 5H), 1.34–1.21 (m, 1H). 13C {1H} NMR (CDCl3, 125 MHz): δ 197.9, 148.9, 135.1, 128.7, 128.6, 127.2, 126.4, 38.2, 33.4, 33.2, 31.9, 28.9, 26.6, 25.2. IR (neat): ν = 3021, 2912, 2836, 1680, 1605, 1433, 1412, 1356, 1302, 1265, 1181, 1017, 954, 871, 847, 818, 689, 653, 596, 581 cm–1. HRMS (ESI): m/z calcd for C16H21O [M + H]+ 229.1592, found 229.1590.
1-(4-(3-Methoxypropyl)phenyl)ethanone (3o)
Obtained as a colorless oil (72 mg, 75%). 1H NMR (CDCl3, 500 MHz): δ 7.89 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 3.38 (t, J = 6.3 Hz, 2H), 3.34 (s, 3H), 2.75 (t, J = 7.8 Hz, 2H), 2.58 (s, 3H), 1.95–1.85 (m, 2H). 13C {1H} NMR (CDCl3, 125 MHz): δ 197.9, 147.9, 135.2, 128.8, 128.6, 71.7, 58.7, 32.4, 31.0, 26.6. IR (neat): ν = 2926, 2867, 1679, 1606, 1430, 1412, 1386, 1357, 1266, 1207, 1181, 1115, 1073, 1017, 955, 886, 845, 814, 597, 584 cm–1. HRMS (ESI): m/z calcd for C12H17O2 [M + H]+ 193.1229, found 193.1231.
N-(3-(4-Acetylphenyl)propyl)-2-oxoazepane-1-carboxamide (3p)
Obtained as a colorless oil (86 mg, 57%). 1H NMR (CDCl3, 500 MHz): δ 9.35 (br s, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.29 (d, J = 8.2 Hz, 2H), 4.03–3.93 (m, 2H), 3.32 (q, J = 5.7 Hz, 2H), 2.77–2.66 (m, 4H), 2.58 (s, 3H), 1.96–1.86 (m, 2H), 1.81–1.67 (m, 6H). 13C {1H} NMR (CDCl3, 125 MHz): δ 197.9, 179.6, 155.0, 147.4, 135.2, 128.7, 128.7, 43.9, 40.0, 39.9, 33.3, 30.8, 29.2, 28.4, 26.7, 23.6. IR (neat): ν = 1695, 1680, 1651, 1606, 1526, 1453, 1435, 1397, 1358, 1333, 1267, 1213, 1179, 1163, 1082, 969, 844, 729, 645, 595 cm–1. HRMS (ESI): m/z calcd for C18H25N2O3 [M + H]+ 317.1865, found 317.1877.
1-Cyclohexylnaphthalene (3q).20
Obtained as a yellow semisolid (69 mg, 63%). 1H NMR (CDCl3, 500 MHz): 8.11 (d, J = 8.5, 1H), 7.84 (d, J = 7.8, 1H), 7.68 (d, J = 8.0, 1H), 7.52–7.35 (m, 4H), 3.34–3.30 (m, 1H), 2.10–1.80 (m, 5H), 1.75–1.49 (m, 4H), 1.40–1.28 (m, 1H). 13C {1H} NMR (CDCl3, 125 MHz): δ 143.9, 134.0, 131.5, 129.0, 126.3, 125.8, 125.7, 125.3, 123.3, 122.4, 39.4, 34.3, 27.4, 26.7.
(±)-1-Bicyclo[2.2.1]heptan-2-yl)naphthalene (3s)
Obtained as a colorless oil (80 mg, 72%). 1H NMR (CDCl3, 500 MHz): δ 8.09 (d, J = 8.3 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 7.5 Hz, 1H), 7.53–7.43 (m, 2H), 7.43–7.34 (m, 2H), 3.43–3.32 (m, 1H), 2.59 (d, J = 3.5 Hz, 1H), 2.38 (s, 1H), 2.02–1.94 (m, 1 H), 1.79–1.58 (m, 4H), 1.57–1.22 (m, 3H). 13C {1H} NMR (CDCl3, 125 MHz): δ 143.2, 134.2, 132.1, 128.9, 126.1, 125.6, 125.5, 125.4, 124.4, 121.7, 43.4, 41.7, 39.7, 37.1, 36.7, 30.6, 29.4. IR (neat): ν = 2948, 2868, 1739, 1596, 1509, 1452, 1396, 1372, 1311, 1297, 1239, 1139, 1045, 950, 907, 793, 775, 731, 648, 560 cm–1. HRMS (ESI): m/z calcd for C17H18 [M+] 222.1409, found 222.1414.
3-(4-(((Trifluoromethyl)sulfonyl)oxy)phenyl)propyl Acetate (3t)
Obtained as a yellow oil (167 mg, 97%). 1H NMR (CDCl3, 500 MHz): δ 7.26 (d, J = 8.6 Hz, 2H), 7.19 (d, J = 8.6 Hz, 2H), 4.09 (t, J = 6.4 Hz, 2H), 2.72 (t, J = 7.6 Hz, 2H), 2.05 (s, 3H), 2.00–1.92 (m, 2H). 13C {1H} NMR (CDCl3, 125 MHz): δ 171.2, 148.0, 141.9, 130.2, 121.4, 118.8 (q, J = 319.5 Hz), 63.6, 31.7, 30.1, 21.0. 19F NMR (CDCl3, 470 MHz): −72.9. IR (neat): ν = 1737, 1501, 1419, 1367, 1246, 1205, 1179, 1135, 1039, 1017, 884, 846, 813, 734, 638, 606, 568, 535, 517, 498 cm–1. HRMS (ESI): m/z calcd for C12H13F3O5SNa [M + Na]+ 349.0333, found 349.0334.
Acknowledgments
We thank Dr. Matthieu Jouffroy and Kingson Lin (University of Pennsylvania) for the preparation of alkylsilicates. We thank NIGMS (RO1 GM113878) for financial support of this research. We thank Evonik for their generous donation of trialkoxysilanes for the synthesis of alkylbis(catecholato)silicates.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b00800.
Additional experimental details and spectral data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Metal-Catalyzed Cross-Coupling Reactions; de Meijere A., Diederich F., Eds.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Hartwig J. F.Organotransition Metal Chemistry: From Bonding to Catalysis, 3rd ed.; University Science: Sausalito, CA, 2010. [Google Scholar]
- a Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Osawa M.; Nagai H.; Akita M. Dalton Trans. 2007, 827. 10.1039/b618007h. [DOI] [PubMed] [Google Scholar]; d Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 18566. 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ye Y.; Sanford M. S. J. Am. Chem. Soc. 2012, 134, 9034. 10.1021/ja301553c. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Rueping M.; Koenigs R. M.; Poscharny K.; Fabry D. C.; Leonori D.; Vila C. Chem. - Eur. J. 2012, 18, 5170. 10.1002/chem.201200050. [DOI] [PubMed] [Google Scholar]; g Sahoo B.; Hopkinson M. N.; Glorius F. J. Am. Chem. Soc. 2013, 135, 5505. 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]; h Shu X. Z.; Zhang M.; He Y.; Frei H.; Toste F. D. J. Am. Chem. Soc. 2014, 136, 5844. 10.1021/ja500716j. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Tasker S. Z.; Jamison T. F. J. Am. Chem. Soc. 2015, 137, 9531. 10.1021/jacs.5b05597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Corce V.; Chamoreau L.-M.; Derat E.; Goddard J.-P.; Ollivier C.; Fensterbank L. Angew. Chem., Int. Ed. 2015, 54, 11414. 10.1002/anie.201504963. [DOI] [PubMed] [Google Scholar]; b Jouffroy M.; Primer D. N.; Molander G. A. J. Am. Chem. Soc. 2016, 138, 475. 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Patel N. R.; Kelly C. K.; Jouffroy M.; Molander G. A. Org. Lett. 2016, 18, 764. 10.1021/acs.orglett.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jouffroy M.; Kelly C. K.; Molander G. A. Org. Lett. 2016, 18, 876. 10.1021/acs.orglett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Leveque C.; Chenneberg L.; Corce V.; Goddard J.-P.; Ollivier C.; Fensterbank L. Org. Chem. Front. 2016, 3, 462. 10.1039/C6QO00014B. [DOI] [PubMed] [Google Scholar]
- Nishigaichi Y.; Suzuki A.; Takuwa A. Tetrahedron Lett. 2007, 48, 211. 10.1016/j.tetlet.2006.11.057. [DOI] [Google Scholar]
- For the redox potential of [Ru(bpy)3](PF6)2, see:Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Karakaya I.; Primer D. N.; Molander G. A. Org. Lett. 2015, 17, 3294. 10.1021/acs.orglett.5b01463. [DOI] [PMC free article] [PubMed] [Google Scholar]; c El Khatib M.; Serafim R. A. M.; Molander G. A. Angew. Chem., Int. Ed. 2016, 55, 254. 10.1002/anie.201506147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews, see:; a Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.; Garg N. K.; Percec V. Chem. Rev. 2011, 111, 1346. 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mesganaw T.; Garg N. K. Org. Process Res. Dev. 2013, 17, 29. 10.1021/op300236f. [DOI] [Google Scholar]; c Tasker S. Z.; Standley E. A.; Jamison T. F. Nature 2014, 509, 299. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For select examples, see:; a Furstner A.; Leitner A. Angew. Chem., Int. Ed. 2002, 41, 609.. [DOI] [Google Scholar]; b Joshi-Pangu A.; Wang C.-Y.; Biscoe M. R. J. Am. Chem. Soc. 2011, 133, 8478. 10.1021/ja202769t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Agrawal T.; Cook S. P. Org. Lett. 2013, 15, 96. 10.1021/ol303130j. [DOI] [PubMed] [Google Scholar]; d Chen X.; Quan Z.-J.; Wang X.-C. Appl. Organomet. Chem. 2015, 29, 296. 10.1002/aoc.3289. [DOI] [Google Scholar]; e Doucet H. Eur. J. Org. Chem. 2008, 2008, 2013. 10.1002/ejoc.200700984. [DOI] [Google Scholar]
- a Jutand A.; Mosleh A. Organometallics 1995, 14, 1810. 10.1021/om00004a038. [DOI] [Google Scholar]; b Jutand A.; Mosleh A. J. Org. Chem. 1997, 62, 261. 10.1021/jo961464b. [DOI] [PubMed] [Google Scholar]; c Alcazar-Roman L. M.; Hartwig J. F. Organometallics 2002, 21, 491. 10.1021/om0108088. [DOI] [Google Scholar]
- For a detailed computational study of photoredox/nickel dual catalysis utilizing aryl halides, see:Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despite extensive studies using alkyltrifluoroborates as radical precursors in photoredox/nickel dual catalysis, the use of such starting materials has yet to prove fruitful for coupling to aryl sulfonate esters.
- Control studies showed that light, a Ru photocatalyst, and Ni as well as ligand catalysts were essential for reaction success; without these components, trace conversion to a cross-coupled product was observed.
- Note that both diisopropylammonium and triethylammonium counterions on alkylbis(catecholato)silicates show no difference in reactivity or yield.
- Li X.; Wang W.; Zhang C. Adv. Synth. Catal. 2009, 351, 2342. 10.1002/adsc.200900428. [DOI] [Google Scholar]
- Kim D. W.; Hong D. J.; Seo J. W.; Kim H. S.; Kim H. K.; Song C. E.; Chi D. Y. J. Org. Chem. 2004, 69, 3186. 10.1021/jo035563i. [DOI] [PubMed] [Google Scholar]
- Guo X.; Li C. Org. Lett. 2011, 13, 4977. 10.1021/ol202081c. [DOI] [PubMed] [Google Scholar]
- Shen Z.; Goh K. K. K.; Yang Y.; Lai Y.; Wong C. H. A.; Cheong H.; Loh T. Angew. Chem., Int. Ed. 2011, 50, 511. 10.1002/anie.201005798. [DOI] [PubMed] [Google Scholar]
- Bellale E. V.; Bhalerao D. S.; Akamanchi K. G. J. Org. Chem. 2008, 73, 9473. 10.1021/jo801580g. [DOI] [PubMed] [Google Scholar]
- Czaplik W. M.; Mayer M.; von Wangelin A. J. Angew. Chem., Int. Ed. 2009, 48, 607. 10.1002/anie.200804434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.