

Abstract

Iridium- and ruthenium-free approaches to protected allylic amines and alkyl nitriles under photoredox conditions are reported. An inexpensive organic dye, eosin Y, catalyzes coupling of Boc-protected potassium α-aminomethyltrifluoroborates with a variety of substituted alkenyl sulfones through an α-aminomethyl radical addition–elimination pathway. Allylic and homoallylic amines were formed in moderate yields with high E/Z selectivity. The mechanistic approach was extended using tosyl cyanide as a radical trap, enabling the conversion of alkyltrifluoroborates to nitriles via a Fukuzumi acridinium-catalyzed process.
Visible-light-mediated organic transformations are an area of interest because there is an increased desire to develop sustainable reaction conditions and to access novel radical intermediates and pathways.1 In particular, visible light is increasingly appreciated as an abundant, renewable, and clean energy source for chemical reactions. However, implementing visible light in organic synthesis is limited by the fact that most organic molecules do not absorb visible light and, thus, are unreactive under photochemical conditions. Photoredox catalysis is an attractive approach to activating organic molecules by translating visible light energy via single-electron transfer (SET)2 or energy transfer. Most commonly, ruthenium and iridium photocatalysts are employed because of their advantageous properties, such as relatively long excited state lifetimes and favorable redox potentials. However, ruthenium and iridium are rare and expensive. Environmentally sustainable and inexpensive organic dyes are attractive replacements of traditional photoredox catalysts, although methods employing these organic catalysts are underdeveloped.3 Herein, two methods featuring single-electron oxidation of potassium alkyltrifluoroborates by organic photoredox catalysts are reported.
Alkyl radicals are widely known to add to unsaturated systems, such as acrylates and styrenes, generating α-carbonyl or benzylic radicals, respectively. These stabilized radicals may be captured in polymerization or 1,4 addition reactions. Alternatively, β-leaving groups have been implemented in radical addition–elimination reactions to forge C(sp2)–C(sp3) bonds.4 Recently, designed β-elimination processes were utilized by MacMillan et al. in a photoredox approach to allylic amines via Ir-catalyzed α-alkenylation of α-amino acids.5 Therein, electron-rich α-aminomethyl radicals—generated from single-electron oxidation of cesium carboxylates—reacted with electron-poor alkenyl sulfones through a radical addition–elimination pathway (Scheme 1). Although this method is attractive in its use of readily available α-amino acids, it is limited by the use of an expensive iridium photoredox catalyst. To overcome this limitation, Boc-protected potassium α-aminomethyltrifluoroborates were explored as alternative radical precursors. Potassium alkyltrifluoroborate salts are bench-stable, redox-active solids.6 They have been employed in various radical processes, including 1,4 additions by Akita et al. and dual catalytic cross-couplings by Molander and co-workers.7 Importantly, potassium alkyltrifluoroborates have oxidation potentials lower than those of their corresponding cesium carboxylates, enabling the removal of iridium in favor of mild and inexpensive organic dyes, such as eosin Y. Thus, the oxidation potential of potassium α-pyrrolidinyltrifluoroborate (Ered1/2 = +0.78 V vs SCE) falls within the redox window of eosin Y (Ered1/2 = +0.83 V),8 whereas cesium α-pyrrolidinyl carboxylate (Ered1/2 = +0.95 V vs SCE)5 lies outside of eosin Y’s oxidation window.
Scheme 1. Reaction of α-Amino Radicals with Alkenyl Sulfones under Photoredox Conditions.
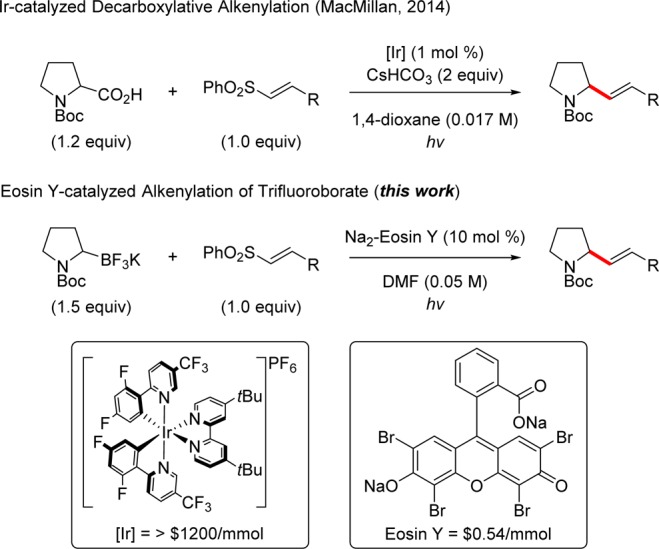
Studies began by exploring the reaction of potassium α-pyrrolidinyltrifluoroborate (1) and phenyl trans-styryl sulfone (2a) in the presence of eosin Y (Scheme 2). After an initial product hit, conditions were optimized to provide the desired product in 73% yield (>98:2 E/Z selectivity). Notably, eosin Y was a more effective catalyst than alternative fluorescein derivatives, including eosin B, rose bengal, and ethyl eosin. Furthermore, the more oxidizing 9-mesityl-10-methylacridinium perchlorate (MesAcr+) catalyst (Ered1/2 = +2.06 V vs SCE)9 results in poor E/Z selectivity. Control reactions were performed to show that photocatalyst and light were essential for reactivity. The addition of 2,6-lutidine, which is a common additive in photoredox reactions of alkyltrifluoroborates,7d does not significantly alter the yield. Interestingly, switching light sources from 26 W CFL to a green LED (λ = 530 nm) did not influence conversion or selectivity.
Scheme 2. Control Studies for α-Alkenylation of Potassium Alkyltrifluoroborate.
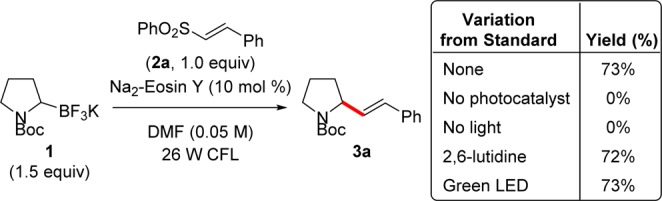
With satisfactory conditions in hand, the reaction scope was explored, beginning with the sulfonyl partner. Attractively, aryl-substituted sulfones are stable, crystalline solids. Alkenyl sulfones were readily synthesized by several methods, including iodosulfonation–dehydroiodination of styrene derivatives,10 olefination of α-sulfonylphosphonates,11 or Heck reaction of phenyl vinyl sulfone with aryl halides.12 The reaction conditions developed allowed a wide range of aryl subunits to be incorporated, including electron-deficient (3c–f) and electron-rich arenes (3g and 3h), providing the desired alkenyl pyrrolidines in acceptable yields (Table 1). Halide substitution, including that in fluoride 3i and bromides 3j and 3k, withstood the reaction conditions and may serve as handles for further diversification. In addition, an electron-deficient pyridine cleanly provided the desired product, 3l, rather than Minisci-type products. Unfortunately, nitroarenes (e.g., 3n), which are known to undergo reduction by excited eosin Y, were not tolerated.13 Notably, stabilization of the radical addition intermediate is essential. Unsubstituted (3o) or alkyl-substituted (3p) products were not formed, likely stemming from the thermodynamically disfavored formation of the primary or tertiary radical addition intermediates from secondary α-amino radicals. Furthermore, 1,4 addition products were not observed owing to the absence of a quenching pathway—either H atom abstraction or reduction–protonation—under the standard conditions. As a result, the reaction conditions provided styrene 3q and acrylate 3r in synthetically useful yields via alkyl radical addition to allylic sulfones, which form analogous β-sulfonyl radical intermediates (Scheme 3).
Table 1. Scope of Reaction with Alkenyl and Allylic Sulfonesa.
All reactions were conducted on 0.5 mmol scale. Reaction times were 48–72 h. E/Z selectivity determined by 1H NMR.
Scheme 3. Radical Addition and β-Fragmentation of Sulfonyl Leaving Group.
Unfortunately, the eosin Y-catalyzed alkenylation is largely limited to secondary α-aminomethyltrifluoroborates. Attempts to extend the method to primary α-aminomethyl (Ered1/2 = 0.9 V), primary or secondary α-alkoxy (Ered1/2 = 0.9 V), and benzylic (Ered1/2 = 1.2 V) trifluoroborates provided products in low or trace yield.
Based on the reactivity of potassium alkyltrifluoroborates with alkenyl and allylic sulfones, tosyl cyanide (TsCN) was envisioned as an alternative radical acceptor. Renaud et al. has reported a hyponitrite-mediated deboronative cyanation of boronate esters, generated in situ via hydroboration of alkenes, with TsCN.14 The reaction required several additions of a radical initiator. It was thought that employing TsCN in a photoredox manifold with alkyltrifluoroborates would render the reaction truly catalytic in organic radical initiator and benefit further from utilization of a bench-stable organoboron source. Furthermore, the more oxidizing MesAcr+ catalyst—which provided inadequate selectivity in alkenylations because of the presence of electron-rich alkene-containing products—could oxidize both stabilized and unstabilized trifluoroborates, providing access to a larger range of alkyl nitriles. The trend in radical stability was anticipated to favor formation of substituted alkyl nitriles that are not easily synthesized via traditional SN2 reactions. When we began our studies, photoredox cyanations were largely limited by substrate scope (e.g., N-aryl tetrahydroisoquinolines)15 or unfavorable conditions (e.g., iridium/NaCN/AcOH system).16 However, a deboronative cyanation of potassium alkyltrifluoroborates with TsCN was recently reported.17 The latter reaction utilized a ruthenium photocatalyst, excess of a hypervalent iodine oxidant, and TFA.17 Therefore, a precious metal and stoichiometric, oxidant-free photoredox cyanation remained desirable.
Minor optimization to reaction parameters provided suitable conditions for nitrile formation without the addition of external oxidants or acids (Table 2). A series of γ-, β-, and α-amino nitriles (6a, 6b, and 6c) were synthesized. This could provide a route to amino acids from simple alkenes via a hydroboration/cyanation/hydrolysis sequence rather than accessing acids from oxidation/reduction manipulation (e.g., oxidation of aldehydes or ketones). Although protection of cyanohydrins is often problematic because deprotonation to the alkoxide results in decyanation,18 returning the aldehyde starting material, the MesAcr+-catalyzed cyanation of an α-alkoxytrifluoroborate provided protected cyanohydrin 6e in good yield. Primary nitriles may be synthesized through the Kolbe reaction, but the isolation of nitrile 6f shows that unactivated alkyl radicals are formed and react under these highly oxidizing conditions. Unfortunately, the standard conditions provide β-cyano ketones in reduced yield with the α,β-unsaturated ketone as a side product. In addition, aryltrifluoroborates may be oxidized by MesAcr+,7a but protodeboronation rather than cyanation is observed.
Table 2. Cyanation of Potassium Alkyltrifluoroboratesa.
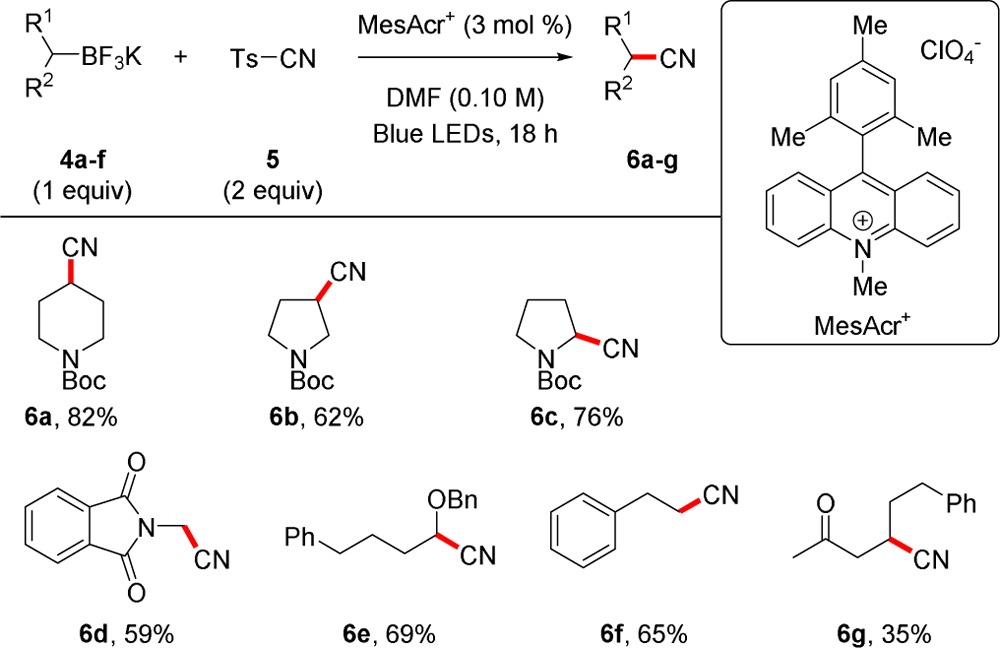
All reactions were performed on 0.35 mmol scale.
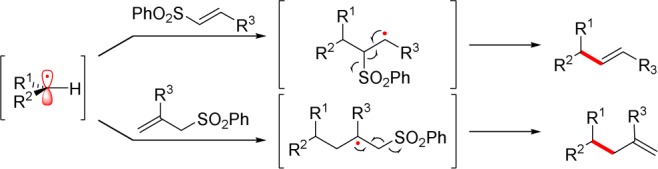
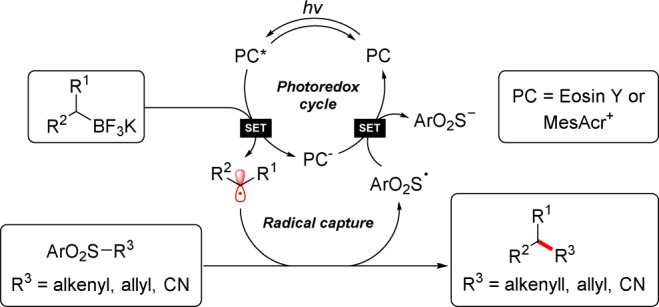
Mechanistically, photoredox alkenylation, allylation, and cyanation are thought to proceed through similar pathways (Scheme 4). Visible light can promote the ground state photoredox catalyst (PC), either eosin Y or MesAcr+, to a singlet excited state that may undergo intersystem crossing to a relatively long-lived triplet state (PC*). This excited state may oxidize an appropriate potassium alkyltrifluoroborate, forming an alkyl radical. In the presence of a sulfonyl partner, the carbon-centered radical may undergo radical addition–elimination, forging a new C–C bond and producing an equivalent of a more thermodynamically favored sulfonyl radical. For turnover of the catalytic cycle, the sulfonyl radical (Ered1/2 = +0.50 V)19 can accept an electron from the reduced photoredox catalyst (PC–) (for eosin Y,3eEred1/2 = −1.14 V; for MesAcr+,20Ered1/2 = −0.49 V vs SCE). Thus, the sulfonyl partner acts as radical acceptor and oxidant.
Scheme 4. Plausible Reaction Mechanism.
In summary, inexpensive organic dyes have been employed as sustainable alternatives to iridium photoredox catalysts in potassium alkyltrifluoroborate transformations. Eosin Y is an effective catalyst for α-alkenylation of a pyrrolidine-derived trifluoroborate with high stereoselectivity (E/Z selectivity). Furthermore, Fukuzumi’s acridinium catalyst facilitates the formation of alkyl nitriles from various alkyltrifluoroborates, including stabilized α-aminomethyl precursors and unactivated primary substrates. These processes largely rely upon the ability of sulfonyl radical traps as leaving groups and oxidants for catalytic turnover.
Experimental Section
General Considerations
All reactions were carried out under an inert atmosphere of argon. Photoredox reactions were irradiated with a 26 W CFL or a blue LED strip (460–480 nm). Temperatures were controlled using an external fan. Column chromatography was performed by Combiflash using RediSep Rf gold normal-phase silica columns. Melting points (°C) are uncorrected. 1H (500 MHz) and 13C (126 MHz) NMR chemical shifts are reported relative to internal TMS. Data are presented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, br = broad), coupling constant J (Hz), and integration. Thin-layer chromatography (TLC) was performed on TLC silica gel plates (0.25 mm) precoated with a fluorescent indicator. Visualization of the TLC plates was effected with ultraviolet light. HRMS spectra (ESI-TOF) were recorded in CH2Cl2 or acetonitrile.
General Procedure for Eosin Y-Catalyzed Alkenylation and Allylation
Sulfone (0.50 mmol, 1.0 equiv), potassium alkyltrifluoroborate (208 mg, 0.75 mmol, 1.5 equiv), and eosin Y disodium salt (35 mg, 0.05 mmol, 10 mol %) were added to a reaction vial. The vial was placed under argon and filled with degassed, anhydrous DMF (10 mL, 0.05 M). The vial was placed between two 26 W CFLs. Consumption of sulfone was monitored by TLC or HPLC. Upon completion (typically 40–72 h), the reaction was transferred to a separatory funnel with Et2O (30 mL) and H2O (40 mL). The aqueous layer was washed twice with Et2O (30 mL). The combined organic layers were washed with brine (40 mL), dried (MgSO4), and concentrated by rotary evaporation. The crude product was purified by flash column chromatography with hexanes/EtOAc.
tert-Butyl (E)-2-Styrylpyrrolidine-1-carboxylate (3a)
The general procedure provided the titled compound as a white solid (99.6 mg, 73% yield, >98:2 E/Z): mp = 96–98 °C; 1H NMR (500 MHz, CDCl3) δ 7.33 (m, 4H), 7.21 (t, J = 7.4 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 6.18–5.99 (br s, 1H), 4.60–4.32 (br m, 1H), 3.53–3.30 (br s, 2H), 2.15–1.99 (br s, 1H), 1.99–1.70 (m, 3H), 1.43 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.8, 137.2, 130.9, 129.6, 128.6, 127.4, 126.4, 79.3, 59.1, 46.4, 32.8, 28.7, 23.2; IR (neat) ν = 2973, 2875, 1687, 1493, 1477, 1388, 1363, 1253, 1160, 1107, 960, 917, 895, 877, 860, 771, 747, 692, 507 cm–1; MS (ESI) m/z calcd for C17H23NO2Na ([M + Na]+) 296.1626, found 296.1615.
tert-Butyl (E)-2-(4-Methyl)styrylpyrrolidine-1-carboxylate (3b)
The general procedure provided the titled compound as a colorless oil (94.8 mg, 66% yield, >98:2 E/Z): 1H NMR (500 MHz, CDCl3) δ 7.24 (d, J = 8.0 Hz, 2H), 7.11 (d, J = 7.6 Hz, 2H), 6.36 (d, J = 15.4 Hz, 1H), 6.11–5.95 (br s, 1H), 4.56–4.30 (br m, 1H), 3.53–3.32 (br s, 2H), 2.33 (s, 3H), 2.14–1.99 (br s, 1H), 1.99–1.67 (m, 3H), 1.42 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.8, 137.1, 134.4, 129.9, 129.5, 129.3, 126.3, 79.3, 59.2, 46.4, 32.7, 28.7, 23.2, 21.3; IR (neat) ν = 2972, 2927, 2874, 1688, 1513, 1477, 1454, 1388, 1363, 1253, 1161, 1107, 1040, 962, 917, 896, 859, 800, 770, 510 cm–1; MS (ESI) m/z calcd for C18H25NO2 (M+) 287.1885, found 287.1882.
tert-Butyl (E)-2-(3-Trifluoromethyl)styrylpyrrolidine-1-carboxylate (3c)
The general procedure provided the titled compound as a colorless oil (127.9 mg, 75% yield, 98:2 E/Z): 1H NMR (500 MHz, CDCl3) δ 7.61–7.34 (m, 4H), 6.42 (d, J = 14.5 Hz, 1H), 6.28–6.08 (br s, 1H), 4.63–4.28 (br m, 1H), 3.58–3.27 (br s, 2H), 2.19–1.98 (br s, 1H), 1.98–1.63 (m, 3H), 1.41 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.7, 140.7, 138.0, 133.8, 129.1, 128.2, 126.5, 125.6, 124.4 (q, 1JC–F = 272.2 Hz), 123.9, 79.5, 59.0, 46.5, 32.6, 28.6, 23.2; 19F NMR (470.8 MHz, CDCl3) δ −62.5, −62. Characterization data matched that reported in the literature.5
tert-Butyl (E)-2-(4-Cyano)styrylpyrrolidine-1-carboxylate (3d)
The general procedure provided the titled compound as a colorless oil (114.8 mg, 77% yield, >98:2 E/Z): 1H NMR (500 MHz, CDCl3) δ 7.57 (d, J = 7.8 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 6.40 (d, J = 14.9 Hz, 1H), 6.31–6.14 (br s, 1H), 4.58–4.32 (br m, 1H), 3.53–3.30 (br s, 2H), 2.18–2.00 (br s, 1H), 1.97–1.62 (m, 3H), 1.40 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.7, 141.7, 135.2, 132.5, 128.0, 126.9, 119.1, 110.7, 100.1, 79.5, 59.0, 46.5, 32.6, 28.6, 23.2; IR (neat) ν = 2974, 2225, 1686, 1603, 1504, 1477, 1454, 1389, 1364, 1301, 1254, 1161, 1111, 968, 918, 896, 860, 811, 772, 547 cm–1; MS (ESI) m/z calcd for C18H22N2O2Na ([M + Na]+) 321.1579, found 321.1579.
tert-Butyl (E)-2-(4-Acetyl)styrylpyrrolidine-1-carboxylate (3e)
The general procedure provided the titled compound as a colorless oil (116.6 mg, 74% yield, 98:2 E/Z): 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 8.0 Hz, 2H), 7.40 (d, J = 8.4, 2H), 6.42 (d, J = 14.8 Hz, 1H), 6.30–6.15 (br s, 1H), 4.58–4.32 (br m, 1H), 3.50–3.33 (br s, 2H), 2.56 (s, 3H), 2.17–2.02 (br s, 1H), 1.98–1.74 (m, 3H), 1.39 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 197.6, 154.7, 141.9, 136.0, 134.0, 128.9, 128.6, 126.4, 79.4, 59.1, 46.5, 28.6, 26.7, 23.2; IR (neat) ν = 2973, 2875, 1678, 1601, 1478, 1454, 1389, 1362, 1302, 1266, 1160, 1106, 956, 918, 896, 859, 810, 773, 730, 593 cm–1; MS (ESI) m/z calcd for C19H25NO3 ([M + Na]+) 338.1732, found 338.1737.
tert-Butyl (E)-2-(4-Methoxycarbonyl)styrylpyrrolidine-1-carboxylate (3f)
The general procedure provided the titled compound as an off-white semisolid (125.8 mg, 76% yield, 97:3 E/Z); 1H NMR (500 MHz, CDCl3) δ 7.96 (d, J = 7.4 Hz, 2H), 7.39 (d, J = 7.9 Hz, 2H), 6.42 (d, J = 13.0 Hz, 1H), 6.30–6.13 (br s, 1H), 4.58–4.33 (br m, 1H), 3.89 (s, 3H), 3.51–3.34 (br s, 2H), 2.16–2.01 (br s, 1H), 2.00–1.69 (m, 3H), 1.40 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 167.0, 154.7, 141.7, 137.2, 133.8, 130.0, 128.7, 126.3, 79.4, 59.1, 52.1, 46.5, 32.7, 28.6, 23.2; IR (neat) ν = 2974, 2877, 1719, 1690, 1606, 1568, 1478, 1434, 1391, 1365, 1278, 1164, 1108, 1017, 968, 918, 897, 867, 764, 698 cm–1; MS (ESI) m/z calcd for C19H25NO4Na ([M + Na]+) 354.1681, found 354.1678.
tert-Butyl (E)-2-(4-Methoxy)styrylpyrrolidine-1-carboxylate (3g)
The general procedure provided the titled compound as a colorless oil (103.1 mg, 68% yield, >98:2 E/Z): 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 8.7, 2H), 6.83 (d, J = 7.2 Hz, 2H), 6.33 (d, J = 15.6 Hz, 1H), 6.02–5.87 (br s, 1H), 4.54–4.29 (br m, 1H), 3.79 (m, 3H), 3.50–3.31 (br s, 2H), 2.16–1.98 (br s, 1H), 1.98–1.66 (m, 3H), 1.42 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 159.1, 154.8, 130.0, 129.0, 128.7, 127.5, 114.1, 79.2, 59.2, 55.4, 46.4, 32.8, 28.7, 23.2; IR (neat) ν = 2972, 1686, 1607, 1510, 1477, 1455, 1389, 1363, 1300, 1244, 1161, 1105, 1033, 961, 917, 842, 806, 765, 731, 524 cm–1; MS (ESI) m/z calcd for C18H25NO3Na ([M + Na]+) 326.1744, found 326.1732.
tert-Butyl (E)-2-(2-(Benzo[d][1,3]dioxol-5-yl)vinyl)pyrrolidine-1-carboxylate (3h)
The general procedure provided the titled compound as a colorless oil (88.8 mg, 56% yield, >98:2 E/Z): 1H NMR (500 MHz, CDCl3) δ 6.89 (s, 1H), 6.75 (m, 2H), 6.30 (d, J = 15.3 Hz, 1H), 6.00–5.83 (br m, 3H), 4.54–4.28 (br m, 1H), 3.51–3.32 (br s, 2H), 2.14–2.00 (br s, 1H), 2.00–1.69 (m, 3H), 1.42 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 148.1, 147.1, 131.7, 129.2, 120.9, 108.4, 105.8, 101.1, 79.3, 59.1, 46.4, 32.8, 28.7, 23.4; IR (neat) ν = 2973, 2878, 1685, 1502, 1489, 1444, 1390, 1364, 1248, 1189, 1161, 1099, 1037, 959, 931, 886, 860, 799, 773, 732 cm–1; MS (ESI) m/z calcd for C18H23NO4Na ([M + Na]+) 340.1525, found 340.1518.
tert-Butyl (E)-2-(4-Fluoro)styrylpyrrolidine-1-carboxylate (3i)
The general procedure provided the titled compound as a colorless oil (91.7 mg, 63% yield, 96:4 E/Z): 1H NMR (500 MHz, CDCl3) δ 7.30 (dd, J = 8.4, 5.4 Hz, 2H), 6.99 (dd, J = 8.7, 6.9 Hz, 2H), 6.35 (d, J = 14.7 Hz, 1H), 6.07–5.93 (br s, 1H), 4.54–4.28 (br m, 1H), 3.51–3.35 (br s, 2H), 2.14–2.01 (br s, 1H), 2.01–1.75 (m, 3H), 1.42 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 162.3 (d, 1JC–F = 246.1 Hz), 154.7, 133.3, 130.8, 128.4, 127.8, 115.6, 79.3, 59.1, 46.4, 32.7, 28.6, 23.2; 19F NMR (470.8 MHz, CDCl3) δ −115.0, −115.3; IR (neat) ν = 2974, 2876, 1687, 1601, 1508, 1478, 1454, 1390, 1364, 1226, 1158, 1110, 1093, 963, 897, 852, 810, 790, 771, 517 cm–1; MS (ESI) m/z calcd for C17H22FNO2Na ([M + Na]+) 314.1544, found 314.1540.
tert-Butyl (E)-2-(4-Bromo)styrylpyrrolidine-1-carboxylate (3j)
The general procedure provided the titled compound as a white semisolid (107.0 mg, 61% yield, >98:2 E/Z): 1H NMR (500 MHz, CDCl3) δ 7.40 (d, J = 8.1 Hz, 2H), 7.20 (d, J = 8.2 Hz, 2H), 6.32 (d, J = 15.2 Hz, 1H), 6.16–6.00 (br s, 1H), 4.57–4.28 (br m, 1H), 3.52–3.32 (br s, 2H), 2.14–2.01 (br s, 1H), 1.97–1.65 (m, 3H), 1.40 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.7, 136.2, 131.7, 129.1, 128.4, 127.9, 121.1, 79.4, 59.1, 46.5, 32.6, 28.6, 23.3; IR (neat) ν = 2973, 2875, 1687, 1487, 1454, 1389, 1364, 1253, 1161, 1109, 1072, 1008, 962, 917, 896, 854, 802, 773, 736, 504 cm–1; MS (ESI) m/z calcd for C17H22BrNO2Na ([M + Na]+) 374.0732, found 374.0727.
tert-Butyl (E)-2-(3-Bromo)styrylpyrrolidine-1-carboxylate (3k)
The general procedure provided the titled compound as a white solid (101.8 mg, 58% yield, >98:2 E/Z): mp = 53–55 °C; 1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 7.17 (d, J = 7.9 Hz, 1H), 6.31 (d, J = 16.2 Hz, 1H), 6.17–6.02 (br s, 1H), 4.56–4.31 (br m, 1H), 3.51–3.28 (br s, 2H), 2.18–1.99 (br s, 1H), 1.99–1.64 (m, 3H), 1.41 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.7, 139.4, 132.6, 130.2, 129.6, 129.2, 128.2, 127.9, 125.1, 79.4, 59.0, 46.5, 32.7, 28.7, 23.1; IR (neat) ν = 2973, 2930, 1687, 1590, 1560, 1475, 1454, 1388, 1364, 1250, 1161, 1108, 1071, 994, 959, 906, 876, 860, 773, 684 cm–1; MS (ESI) m/z calcd for C17H22BrNO2Na ([M + Na]+) 374.0732, found 374.0730.
tert-Butyl (E)-2-(2-(Pyridin-2-yl)vinyl)pyrrolidine-1-carboxylate (3l)
The general procedure provided the titled compound as a colorless oil (97.3 mg, 71% yield, >98:2 E/Z): 1H NMR (500 MHz, CDCl3) δ 8.49 (d, J = 4.9 Hz, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.18 (d, J = 8.2 Hz, 1H), 7.06 (t, J = 6.0 Hz, 1H), 6.58 (d, J = 15.6 Hz, 1H), 6.52–6.38 (br m, 1H), 4.58–4.35 (br m, 1H), 3.53–3.18 (br s, 2H), 2.12–1.97 (br s, 1H), 1.98–1.72 (m, 3H), 1.37 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 155.5, 154.7, 149.6, 136.6, 135.4, 129.3, 122.0, 121.7, 79.3, 58.8, 58.4, 46.7, 46.3, 32.4, 31.6, 28.6, 23.8, 23.1; IR (neat) ν = 2973, 2875, 1686, 1585, 1564, 1470, 1430, 1388, 1363, 1254, 1150, 1109, 1089, 990, 968, 918, 900, 860, 766, 745 cm–1; MS (ESI) m/z calcd for C16H23N2O2 ([M + H]+) 275.1760, found 275.1761.
tert-Butyl 2-(2,2-Diphenylvinyl)pyrrolidine-1-carboxylate (3m)
The general procedure provided the titled compound as a white solid (108.3 mg, 62% yield): mp = 87–90 °C; 1H NMR (500 MHz, CDCl3) δ 7.44–7.07 (m, 10H), 6.02 (d, J = 8.7 Hz, 1H), 4.35–4.20 (br s, 1H), 3.65–3.26 (br m, 2H), 2.19–2.04 (br s, 1H), 2.00–1.66 (m, 3H), 1.40 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.8, 142.5, 140.6, 139.7, 132.3, 130.2, 128.3, 128.2, 127.5, 127.3, 127.3, 79.3, 56.5, 47.0, 35.0, 28.8, 24.0; IR (neat) ν = 2974, 1687, 1492, 1477, 1444, 1389, 1363, 1250, 1162, 1108, 1074, 1030, 910, 877, 860, 762, 731, 698, 645, 626 cm–1; MS (ESI) m/z calcd for C23H27NO2 (M+) 349.2042, found 349.2040.
tert-Butyl 2-(2-Phenylallyl)pyrrolidine-1-carboxylate (3q)
The general procedure provided the titled compound as a colorless oil (83.3 mg, 58% yield): 1H NMR (500 MHz, CDCl3) δ 7.58–7.41 (m, 2H), 7.35–7.27 (m, 3H), 5.35 (s, 1H), 5.07 (s, 1H), 4.00–3.79 (br m, 1H), 3.46–3.03 (m, 3H), 2.35–2.23 (br m, 1H), 1.89–1.62 (m, 4H), 1.55–1.42 (br m, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.7, 146.1, 128.4, 127.7, 126.3, 125.0, 115.0, 77.4, 55.8, 46.4, 40.0, 28.8, 28.7, 22.7; IR (neat) ν = 2978, 2875, 1689, 1478, 1449, 1394, 1365, 1252, 1171, 1108, 899, 777, 705, 537 cm–1; MS (ESI) m/z calcd for C18H25NO2 (M+) 287.1885, found 287.1889.
tert-Butyl 2-(2-(Methoxycarbonyl)allyl)pyrrolidine-1-carboxylate (3r)
The general procedure provided the titled compound as a colorless oil (65.9 mg, 49% yield): 1H NMR (500 MHz, CDCl3) δ 6.21 (s, 1H), 5.61–5.51 (br m, 1H), 4.08–3.96 (br s, 1H), 3.76 (s, 3H), 3.45–3.27 (br m, 2H), 2.78–2.61 (m, 1H), 2.46–2.27 (m, 1H), 1.91–1.77 (m, 3H), 1.73–1.64 (br s, 1H), 1.45 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 167.9, 154.7, 138.0, 127.6, 79.4, 56.6, 52.0, 46.0, 36.8, 30.2, 28.6, 22.8; IR (neat) ν = 2973, 1722, 1691, 1630, 1478, 1439, 1392, 1365, 1274, 1253, 1217, 1166, 1144, 1115, 997, 948, 903, 864, 817, 772 cm–1; MS (ESI) m/z calcd for C14H23NO4Na ([M + Na]+) 292.1525, found 292.1522.
General Procedure for Cyanation of Potassium Alkyltrifluoroborates
Potassium alkyltrifluoroborate (0.35 mmol, 1 equiv), TsCN (127.4 mg, 0.70 mmol, 2 equiv), and 9-mesityl-10-methylacridinium perchlorate (4.3 mg, 0.011 mmol, 3 mol %) were added to a reaction vial. The reaction vial was placed under argon. Dried and degassed DMF (3.5 mL, 0.10 M) was added to the vial. The reaction was placed in a beaker that was wrapped with a blue LED strip. A fan was placed above the beaker to maintain a temperature of 30–35 °C. Upon completion, the reaction was transferred to a separatory funnel with Et2O (15 mL) and H2O (20 mL). The aqueous layer was washed with Et2O (15 mL, 3 times), and the combined organic layers were washed with H2O (20 mL) and brine (20 mL). The organic layer was dried (MgSO4) and concentrated by rotary evaporation. The crude product was isolated by flash column chromatography with hexanes/EtOAc.
tert-Butyl 4-Cyanopiperidine-1-carboxylate (6a):
Isolated as a white solid (60.3 mg, 82% yield); mp = 46–47 °C; 1H NMR (500 MHz, CDCl3) δ 3.67–3.60 (m, 2H), 3.35–3.28 (m, 2H), 2.82–2.74 (m, 1H), 1.91–1.72 (m, 4 H), 1.44 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.5, 121.2, 80.2, 41.9, 28.6, 28.5, 26.4. Characterization data matched that reported in the literature.21
tert-Butyl 3-Cyanopyrrolidine-1-carboxylate (6b):
Isolated as a colorless oil (42.6 mg, 62% yield); 1H NMR (500 MHz, CDCl3) δ 3.72–3.63 (br s, 1H), 3.63–3.48 (br s, 2H), 3.48–3.37 (br s, 1H), 3.12–3.04 (m, 1H), 2.30–2.14 (m, 2H), 1.46 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 154.0, 120.1, 80.4, 49.0, 44.7, 30.4, 29.5, 28.6, 27.8. Characterization data matched that reported in the literature.22
tert-Butyl 2-Cyanopyrrolidine-1-carboxylate (6c):
Isolated as a colorless oil (52.2 mg, 76% yield); 1H NMR (500 MHz, CDCl3) δ 4.60–4.40 (m, 1H), 3.57–3.25 (m, 2H), 2.30–1.93 (m, 4H), 1.51–1.47 (m, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 153.1, 119.3, 81.6, 47.3, 45.9, 31.8, 28.5, 24.0. Characterization data matched that reported in the literature.23
2-(1,3-Dioxoisoindolin-2-yl)acetonitrile (6d):
Isolated as a white solid (38.4 mg, 59% yield): mp = 121–123 °C; 1H NMR (500 MHz, CDCl3) δ 7.96–7.91 (m, 2H), 7.83–7.79 (m, 2H), 4.58 (s, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 166.1, 135.0, 131.6, 124.3, 113.7, 25.3. Characterization data matched that reported in the literature.24
2-(Benzyloxy)-5-phenylpentanenitrile (6e):
Isolated as a colorless oil (64.0 mg, 69% yield); 1H NMR (500 MHz, CDCl3) δ 7.38–7.14 (m, 10H), 4.84 (d, J = 11.6 Hz, 1H), 4.51 (d, J = 11.6 Hz, 1H), 4.14 (t, J = 6.0 Hz, 1H), 2.64 (s, 2H), 1.92–1.82 (m, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 141.3, 136.0, 128.8, 128.6, 128.6, 128.5, 128.4, 126.2, 118.4, 72.4, 67.5, 35.2, 33.1, 26.5. FT-IR (neat): ν = 3063, 3029, 2927, 2866, 1735, 1603, 1496, 1454, 1334, 1208, 1101, 1028, 911, 747, 698, 495 cm–1. HRMS (ESI): m/z calc. for C18H19NO (M+) 265.1467, found 265.1468.
3-Phenylpropanenitrile (6f):
Isolated as a colorless oil (29.8 mg, 65% yield); 1H NMR (500 MHz, CDCl3) δ 7.29 (m, 5H), 2.96 (t, J = 7.0 Hz, 2H), 2.62 (t, J = 6.8 Hz, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 138.2, 129.0, 128.4, 127.4, 119.3, 31.7, 19.5. Characterization data matched that reported in the literature.25
4-Oxo-2-phenethylpentanenitrile (6g):
Isolated as a colorless oil (25.6 mg, 35% yield); 1H NMR (500 MHz, CDCl3) δ 7.31–7.19 (m, 5H), 3.07–2.99 (m, 1H), 2.95–2.85 (m, 2H), 2.80–2.65 (m, 2H), 2.18 (s, 3H), 1.95–1.83 (m, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 203.6, 139.9, 128.8, 128.5, 126.7, 121.4, 45.4, 33.5, 33.4, 30.1, 25.7. Characterization data matched that reported in the literature.26
Acknowledgments
This research was generously supported by the National Institute of General Medical Sciences (R01 GM 113878) and the National Science Foundation (CHE 1362841). We are grateful to Frontier Scientific for the gift of several potassium alkyltrifluoroborates. We thank Pfizer for the 9-mesityl-10-methylacridinium perchlorate photocatalyst. We are grateful to the Higher Education Commission (HEC), Pakistan, for a scholarship (PIN No. 112-24510-2PS1-388) to Komal Rizwan. We thank Dr. Rakesh Kohli (University of Pennsylvania) for acquisition of HRMS spectra.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b01207.
1H, 13C, and 19NMR spectra for reported compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Narayanam J. M. R.; Stephenson C. R. Chem. Soc. Rev. 2011, 40, 102. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]
- a Reckenthaler M.; Griesbeck A. G. Adv. Synth. Catal. 2013, 355, 2727. 10.1002/adsc.201300751. [DOI] [Google Scholar]; b Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Koike T.; Akita M. Inorg. Chem. Front. 2014, 1, 562. 10.1039/C4QI00053F. [DOI] [Google Scholar]
- a Nicewicz D. A.; Nguyen T. M. ACS Catal. 2014, 4, 355. 10.1021/cs400956a. [DOI] [Google Scholar]; b Ravelli D.; Fagnoni M. ChemCatChem 2012, 4, 169. 10.1002/cctc.201100363. [DOI] [Google Scholar]; c Sun C.-L.; Shi Z.-J. Chem. Rev. 2014, 114, 9219. 10.1021/cr400274j. [DOI] [PubMed] [Google Scholar]; d Hari D. P.; Konig B. Chem. Commun. 2014, 50, 6688. 10.1039/C4CC00751D. [DOI] [PubMed] [Google Scholar]; e Ravelli D.; Fagnoni M.; Albini A. Chem. Soc. Rev. 2013, 42, 97. 10.1039/C2CS35250H. [DOI] [PubMed] [Google Scholar]
- a Russell G. A.; Ngoviwatchai P.; Tashtoush H.; Pla-Dalmau A.; Khanna R. K. J. Am. Chem. Soc. 1988, 110, 3530. 10.1021/ja00219a030. [DOI] [Google Scholar]; b Xiang J.; Jiang W.; Gong J.; Fuchs P. L. J. Am. Chem. Soc. 1997, 119, 4123. 10.1021/ja963636s. [DOI] [Google Scholar]; c Amaoka Y.; Nagatomo M.; Watanabe M.; Tao K.; Kamijo S.; Inoue M. Chem. Sci. 2014, 5, 4339. 10.1039/C4SC01631A. [DOI] [Google Scholar]; d Baralle A.; Fensterbank L.; Goddard J.; Ollivier C. Chem. - Eur. J. 2013, 19, 10809. 10.1002/chem.201301449. [DOI] [PubMed] [Google Scholar]; e Corce V.; Chamoreau L.-M.; Derat E.; Goddard J.-P.; Ollivier C.; Fensterbank L. Angew. Chem., Int. Ed. 2015, 54, 11414. 10.1002/anie.201504963. [DOI] [PubMed] [Google Scholar]
- Noble A.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 11602. 10.1021/ja506094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander G. A. J. Org. Chem. 2015, 80, 7837. 10.1021/acs.joc.5b00981. [DOI] [PubMed] [Google Scholar]
- a Yasu Y.; Koike T.; Akita M. Adv. Synth. Catal. 2012, 354, 3414. 10.1002/adsc.201200588. [DOI] [Google Scholar]; b Miyazawa K.; Yasu Y.; Koike T.; Akita M. Chem. Commun. 2013, 49, 7249. 10.1039/c3cc42695e. [DOI] [PubMed] [Google Scholar]; c Chinzei T.; Miyazawa K.; Yasu Y.; Koike T.; Akita M. RSC Adv. 2015, 5, 21297. 10.1039/C5RA01826A. [DOI] [Google Scholar]; d Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A. U.; Slanina T.; Yao C.; Konig B. ACS Catal. 2016, 6, 369. 10.1021/acscatal.5b02410. [DOI] [Google Scholar]
- Benniston A. C.; Harriman A.; Li P.; Rostron J. P.; van Ramesdonk H. J.; Groeneveld M. M.; Zhang H.; Verhoeven J. W. J. Am. Chem. Soc. 2005, 127, 16054. 10.1021/ja052967e. [DOI] [PubMed] [Google Scholar]
- Sawangphon T.; Katrun P.; Chaisiwamongkhol K.; Pohmakotr M.; Reutrakul V.; Jaipetch T.; Soorukram D.; Kuhakarn C. Synth. Commun. 2013, 43, 1692. 10.1080/00397911.2012.663448. [DOI] [Google Scholar]
- a Le W. J.; Oh D. Y. Synth. Commun. 1989, 19, 2209. 10.1080/00397918908052616. [DOI] [Google Scholar]; b Enders D.; Nguyen D. Synthesis 2000, 2000, 2092. 10.1055/s-2000-8732. [DOI] [Google Scholar]
- Geoghegan K.; Kelleher S.; Evans P. J. Org. Chem. 2011, 76, 2187. 10.1021/jo200023r. [DOI] [PubMed] [Google Scholar]
- Yang X.-J.; Chen B.; Zheng L.-Q.; Wu L.-W.; Tung C.-H. Green Chem. 2014, 16, 1082. 10.1039/C3GC42042F. [DOI] [Google Scholar]
- Schaffner A.-P.; Darmency V.; Renaud P. Angew. Chem., Int. Ed. 2006, 45, 5847. 10.1002/anie.200601206. [DOI] [PubMed] [Google Scholar]
- Hari D. P.; Konig B. Org. Lett. 2011, 13, 3852. 10.1021/ol201376v. [DOI] [PubMed] [Google Scholar]
- Rueping M.; Zhu S.; Koenigs R. M. Chem. Commun. 2011, 47, 12709. 10.1039/c1cc15643h. [DOI] [PubMed] [Google Scholar]
- Dai J.-J.; Zhang W.-M.; Shu Y.-J.; Sun Y.-Y.; Xu J.; Feng Y.-S.; Xu H.-J. Chem. Commun. 2016, 52, 6793. 10.1039/C6CC01530A. [DOI] [PubMed] [Google Scholar]
- Kraus G. A.; Dneprovskaia E. Tetrahedron Lett. 2000, 41, 21. 10.1016/S0040-4039(99)02017-1. [DOI] [Google Scholar]
- Persson B. Acta Chem. Scand. 1977, 31B, 88. 10.3891/acta.chem.scand.31b-0088. [DOI] [Google Scholar]
- Fukuzumi S.; Kotani H.; Ohkubo K.; Ogo S.; Tkachenko N. V.; Lemmetyinen H. J. Am. Chem. Soc. 2004, 126, 1600. 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Romaire J. P.; Newhouse T. R. J. Am. Chem. Soc. 2015, 137, 5875. 10.1021/jacs.5b02243. [DOI] [PubMed] [Google Scholar]
- Kim Y. J.; Kaiser D. A.; Pollard T. D.; Ichikawa Y. Bioorg. Med. Chem. Lett. 2000, 10, 2417. 10.1016/S0960-894X(00)00486-8. [DOI] [PubMed] [Google Scholar]
- Kamijo S.; Hoshikawa T.; Inoue M. Org. Lett. 2011, 13, 5928. 10.1021/ol202659e. [DOI] [PubMed] [Google Scholar]
- Getz J. J.; Prankerd R. J.; Sloan K. B. J. Org. Chem. 1993, 58, 4913. 10.1021/jo00070a029. [DOI] [Google Scholar]
- Kelly C. B.; Lambert K. M.; Mercadante M. A.; Ovian J. M.; Bailey W. F.; Leadbeater N. E. Angew. Chem., Int. Ed. 2015, 54, 4241. 10.1002/anie.201412256. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2008, 130, 6072. 10.1021/ja801201r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.