

Structured Abstract
Background
The high sequence and structural homology among the hsp90 paralogs – Hsp90α, Hsp90β, Grp94, and Trap-1 – has made the development of paralog-specific inhibitors a challenging proposition.
Objective
This review surveys the state of developments in structural analysis, compound screening, and structure-based design that have been brought to bear on this problem.
Results
First generation compounds that selectively bind to Hsp90, Grp94, or Trap-1 have been identified.
Conclusion
With the proof of principle firmly established, the prospects for further progress are bright.
Keywords: Hsp90, Grp94, Trap-1, paralog-selective inhibitor, structure-based design, screening
Introduction
It is now widely appreciated that the hsp90 family of molecular chaperones occupies a key place in the maintenance of the cellular proteome. Client proteins that are dependent on the hsp90s for maturation currently number over 500 and are wildly diverse in sequence, structure, cellular location, and function [1]. Examples include regulatory and checkpoint kinases, growth factors and their receptors, angiogenesis promoters, transcription factors, metalloproteinases, and telomerases. It has not been lost on the research and pharmaceutical communities that targeting this client pool by inhibiting the chaperone that matures them represents the equivalent of a ‘one stop’ therapeutic extravaganza for a host of interventions, including cancer, neurodegenerative diseases, and viral and fungal infections [2–8].
There are four Hsp90 paralogs in higher eukaryotes. Hsp90α and Hsp90β, the inducible and constituitive forms, respectively [9], are found in the cytoplasm. Grp94 is the endoplasmic reticulum paralog [10], while Trap-1 is localized to the mitochondria [11, 12]. (In this review, lowercase hsp90 will refer to the set of 4 cellular paralogs, while uppercase Hsp90 refers to the cytoplasmic paralogs. Residue numbering refers to the human homologs of Grp94, Trap1, and Hsp90α unless otherwise indicated.) The hsp90s are dimeric and their architecture consists of 3 major domains whose overall folds are conserved (Figure 1) [13–15]. The C-terminal domain is the site of the obligatory dimerization interface [16, 17], the middle domain has been implicated in client protein binding [18–23], and the N-terminal domain contains the ATP binding site [24, 25]. As befits the structural homology, the sequence homology of the paralogs is also high, with overall pairwise identities of 50–60%. Despite the high structural and sequence homology among the four paralogs, however, no paralog or paralog domain has yet been shown to substitute for that of another paralog [26]. The client protein pools for each paralog are also distinct.
Figure 1.
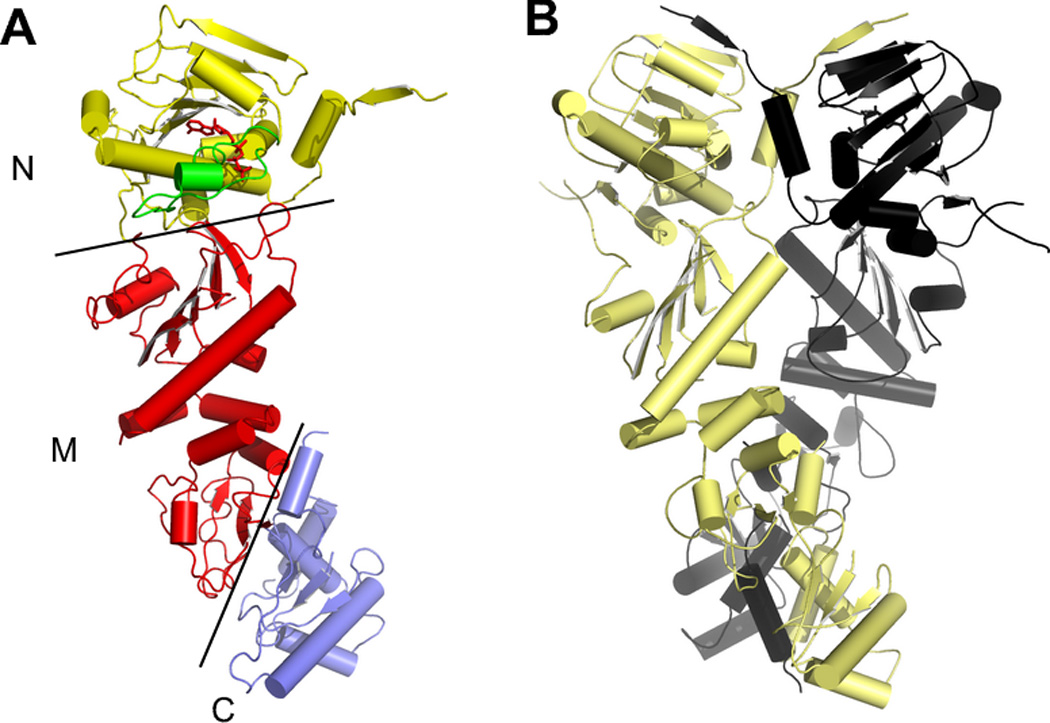
The structure of full-length yeast Hsp90 (PDB 2cg9). A) One subunit of the dimer, showing the N-terminal, Middle, and C-terminal domains. B) The structure of the intact Hsp90 dimer, showing the dimerization of both the N and C domains. The co-chaperone p23, which was crystallized with the Hsp90, has been omitted for clarity.
Our understanding of the biology of hsp90 chaperones has been closely tied to the discovery of highly selective inhibitors. Prior to the discovery of Geldanamycin, an ansamycin antibiotic derived from bacteria (see Figure 2 for compounds described in this review), relatively little was known about Hsp90 other than the fact that it comprised over 2 percent of the cellular polypeptide mass and that it was associated with unliganded steroid receptors and a small set of cellular kinases [27–30]. Given the very low levels of these proteins in the cell, and the high abundance of Hsp90, this obvious mismatch between the amount of Hsp90 and its only known function constituted a significant mystery. Geldanamycin, which was originally misidentified as a novel cellular kinase inhibitor [31], was subsequently shown to have no direct interaction with any kinase but instead to bind with high specificity to Hsp90 [32–34]. The impact of this discovery is hard to overstate. Indeed, the use of this selective inhibitor alone led directly to the identification of hundreds of client proteins whose function was dependent upon Hsp90 [1, 35]. This provided convincing proof that Hsp90 was in fact a broadly acting molecular chaperone.
Figure 2.

Hsp90 family inhibitor compounds discussed in this review.
Molecular chaperones harness the energy of ATP to alter the conformational state of a client protein. For Hsp90, a key step in this mechanism is the binding of ATP to the N-terminal domain [24]. ATP binding drives tertiary and quaternary structural rearrangements in the chaperone that potentiate client protein association (Figure 3) [36, 37]. Within the N-terminal domain, ATP binding causes the ‘lid’, which consists of helices 1, 4 and 5, to close over the bound nucleotide and configure a second dimerization interface [13, 38]. Concomitantly, the gamma phosphate of the bound ATP forms hydrogen bonds to Arg399 from the Hsp90 middle domain, a tethering interaction that requires a precise alignment of the N and M domains about the rotationally flexible junction that connects them. This juxtaposes the two N-terminal domain dimer interfaces such that a client-binding “double dimer” conformation of the chaperone is achieved. Hydrolysis of the bound ATP reverses the conformational rearrangements and is associated with the release of the bound client and cofactor proteins. Underscoring importance of ATP binding for these mechanistic steps to occur, point mutants that preclude ATP binding result in chaperones that are completely defective in vivo [36, 37].
Figure 3.

The ATP cycle of hsp90s.
All of the small molecule inhibitors of Hsp90 that are currently under clinical investigation occupy the ATP binding pocket of the N-terminal domain and are competitive inhibitors of ATP binding [39]. As such, they block chaperone action by preventing the conformational rearrangements that lead to chaperone activity. Although the ATP hydrolysis cycle of hsp90s requires contributions from all three hsp90 domains, the structural basis for inhibitor affinity can be understood from studying the N-terminal domain in isolation [25, 40, 41]. In large part this is due to the fact that all of the conformational rearrangements that lead to the active state of the chaperone, including lid closure and N-terminal domain dimerization, occur subsequent to ATP binding. Inhibitor binding thus reduces to a problem of competing with ATP for the binding pocket, which is located entirely within the N-terminal domain. This circumstance has proven to be experimentally fortuitous because the N-terminal domains of hsp90s have been generally amenable to crystallization. Thus, while the structure of an inhibited complex of any intact hsp90 chaperone has yet to be reported, over 300 crystal structures of N-terminal domain:ligand complexes have been determined. Interestingly, while structure determinations of hsp90:ligand complexes have employed the N-terminal domain, the major biochemical assay for measuring inhibitor binding is a fluorescence polarization displacement assay that utilizes the intact hsp90 chaperone for maximal signal to noise [42]. The fact that the structure determination and assay techniques, which have been optimized using different chaperone constructs, are nonetheless experimentally congruent accounts for much of the continuing progress of the hsp90 inhibitor development field.
The success of Geldanamycin in identifying the client pool of Hsp90, and the subsequent realization that inhibition of Hsp90 had the potential to be therapeutically useful, has led to an explosion of efforts to develop high affinity inhibitor molecules that bind to the N-terminal domain. Compounds based on no fewer than 19 different scaffolds that target the ATP binding pocket are currently undergoing clinical trials [39].
Despite the success in identifying novel scaffolds for Hsp90 inhibition, two significant challenges remain. First, the current generation of inhibitors now in clinical trials target all four paralogs. These ‘pan-hsp90’ inhibitors are of limited use, however, in deconvoluting the biological role of any individual paralog. If inhibitors that targeted a single paralog could be developed, it is clear from the experience of Geldanamycin that our understanding of the role of each chaperone in the cell would be significantly advanced. Second, as might be expected from inhibitory strategies that target a broad range of client proteins indiscriminately, enthusiasm for the clinical utility of the current set of hsp90 inhibitors has been tempered by the observation of adverse side effects associated with treatment [39, 43]. These include hepatotoxicity, hypoatremia, hypoglycemia, fatigue, diarrhea, general toxicities associated with DMSO formulations, and the upregulation of compensatory chaperone pathways such as Hsp70. Because it is axiomatic that the first route to minimizing side effects is by increased selectivity in targeting, a significant challenge is to develop compounds that target just a single hsp90 paralog.
While the idea of targeting individual hsp90 paralogs using selective inhibitors is attractive in principle, in practice the high sequence and structural homology of the individual members of the hsp90 family would appear to make them poor candidates for this approach. Within their N-terminal domains, the four cellular paralogs exhibit sequence identities of 50% or more (Figure 4). Even worse, the amino acids that line the ATP/ligand binding pocket are over 70% identical, with 21 out of 29 residues completely conserved, and the remaining 8 are highly conserved. Despite these daunting prospects, however, paralog selective inhibitors have been developed. The key to these advances, as will be discussed in the following sections, has been the identification and exploitation of three pockets, termed Site 1, Site 2, and Site 3, that form a halo of potential selectivity immediately adjacent to the ATP binding cavity (Figure 6D). These pockets form pairwise compound binding sites with the central ATP binding cavity serving as the common partner. The ability of a ligand to successfully access and stably bind these compound sites in large part accounts for selective paralog binding.
Figure 4.

Alignment of human Hsp90α, Hsp90β, Grp94, and Trap-1 N-terminal domains. Identical residues are shaded black, homologies are shaded grey. Residues comprising Sites 1, 2, and 3 are indicated by numbered squares above the residues. The core ATP binding pocket is indicated by squares labeled with the letter C.
Figure 6.
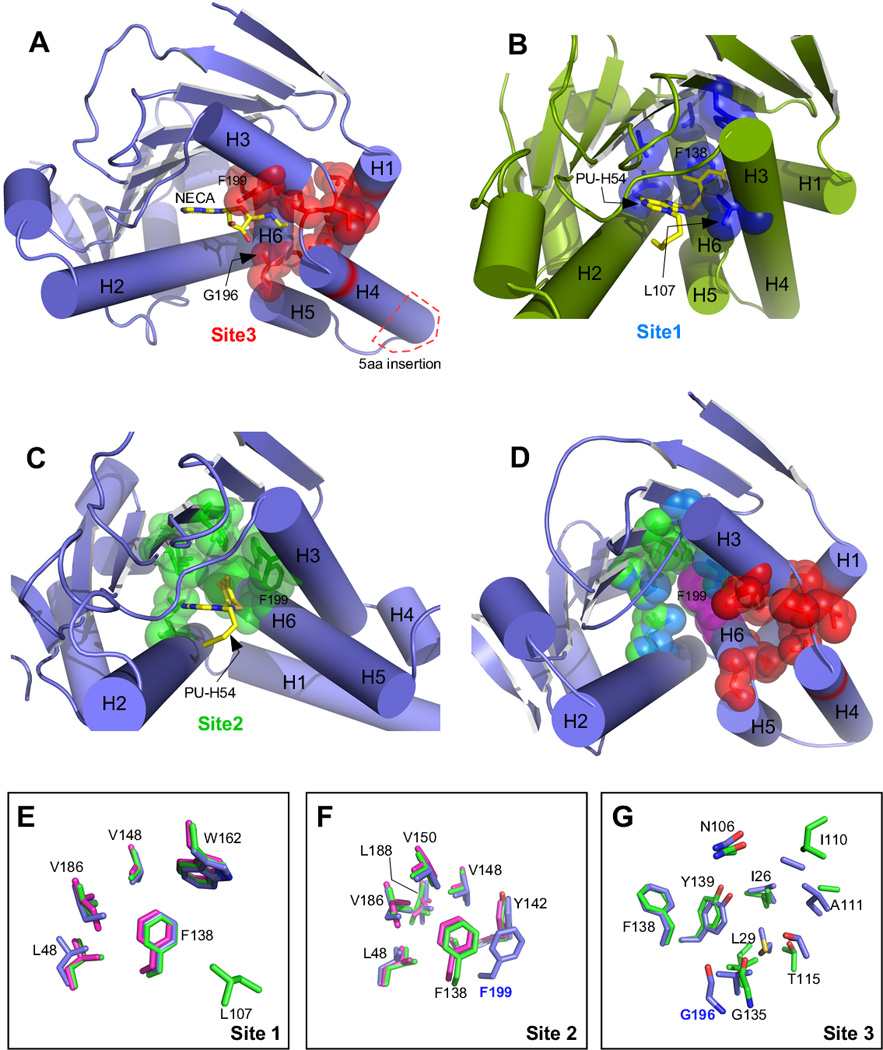
N-terminal domain structures showing ligand binding sites and overlay of binding site residues from individual paralogs. A) Grp94 in complex with NECA (PDB 1qy5). Site 3 residues are colored red, and the location of the Grp94 5 amino acid insertion is shown in the red box. B) Hsp90α in complex with PU-H54 (PDB 3o0i). Site 1 residues are colored blue. C) Grp94 in complex with PU-H54 (PDB 3o2f). Site 2 residues are colored green. D) Composite of Sites 1, 2, and 3 on Grp94 (PDB 1qy5). Phe199, which is common to all 3 sites is colored magenta. E) Overlay of Site 1 residues from hHsp90:PU-H54 (PDB 3o0i, green), Grp94:SNX0723 (PDB 4nh9, blue), and Trap-1:PU-H71 (PDB 4z1f, magenta). Numbering is for hHsp90; the equivalent of L107 is disordered in the Grp94 and Trap-1 structures. F) Overlay of Site 2 residues from hHsp90:PU-H54 (PDB 3o0i), Grp94:PU-H54 (PDB 3o2f), and Trap-1:PU-H71 (PDB 4z1f). Coloring is as in (E). Numbering is for hHsp90 except for F199 which is from Grp94. G) Overlay of Site 3 residues from hHsp90:apo (PDB 1yes, green) and Grp94:NECA (PDB 1qy5, blue). Trap-1 is not shown due to lack of a suitable model for comparison. Numbering is for hHsp90 except for G196 which is from Grp94.
NECA – proof of principle and identification of Site 3
Despite the challenges posed by their high sequence and structural similarity, there were hints that selective hsp90 paralog inhibition might be achievable. NECA (N-ethyl-carboximido adenosine) is a well characterized agonist of adenosine receptors [44, 45]. Biochemical purification of NECA binding proteins from placental extracts, however, identified Grp94 as the other major NECA binding protein in cells [46, 47]. Hsp90 was not recovered from these purifications, a fact that was considered unremarkable at the time these results were reported in the early 1990s. A decade later, however, armed with an increasing appreciation for the role of hsp90s in the cell, these studies were revisited and showed that NECA indeed did bind to purified Grp94 with a dissociation constant of 200 nM while its binding to purified Hsp90 was not detectable [48]. NECA was thus shown to be the first paralog-selective hsp90 inhibitor.
The structure of the N-terminal domain of Grp94 in complex with NECA (PBD code 1qy5) explained the basis for this discrimination [49]. In Grp94, the N-ethyl carboximido moiety of NECA was inserted into a previously unrecognized pocket off the main adenosine binding pocket (for schematic, see Figure 5A). This pocket, now termed Site 3 (Figure 6A,D), was bounded by Val82, Met85, Asn162, Ile166, Ala167, Thr171, Gly196, Val197, Phe199, and Tyr200. In Hsp90, the equivalent Site 3 residues are nearly identical (Ile26, Leu29, Asn106, Ile110, Ala111, Thr115, Gly135, Val136, Phe138, and Tyr139; homologous residues given in italics). Comparison of the structures of Hsp90 and Grp94:NECA showed that the N-ethyl moiety was effectively blocked from entry into Site 3 in Hsp90 by a threatened van der Waals clash between the terminal methyl group of the N-ethyl moiety and the backbone carbonyl oxygen of Hsp90 Gly135 (Figure 6G). In Grp94, this clash was avoided because the equivalent residue, Gly196 was displaced from the entrance to Site 3 by 3.6 Å compared to its Hsp90 counterpart, and thus allowed entry of the N-ethyl group into Site 3 of Grp94.
Figure 5.
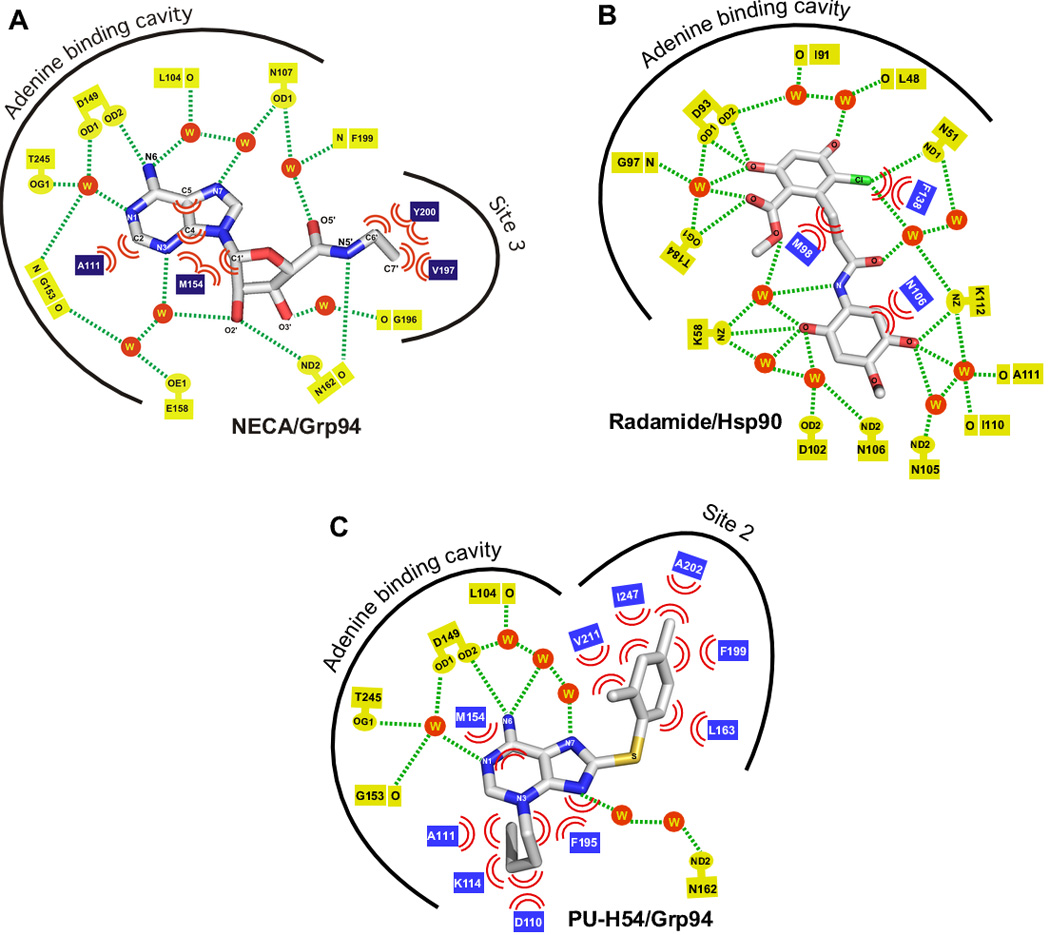
Schematic illustrations of the interaction of Grp94 or Hsp90 with NECA, Radamide, and PU-H54. Hydrogen bond interactions are denoted by dashed lines, van der Waals contacts are shown as pairs of arcs, and waters are shown as spheres. A) Grp94:NECA (PDB 1qy5). B) yeast Hsp90:Radamide (PDB 2fxs). Residues are numbered according to their human Hsp90α equivalents. C) Grp94:PU-H54 (PDB 3o2f).
Given the near-complete conservation of the Site 3 residues between Grp94 and Hsp90, what caused the repositioning of Gly196 in Grp94 to allow NECA binding? Gly196, like its counterpart Gly135 in Hsp90, is part of the helix 1,4,5 lid subdomain. In Grp94 the lid contains a unique 5 amino acid insertion between helices 4 and 5 (Figure 4, 6A). Rather than simply expand the turn between the two antiparallel aligned helices, however, the additional residues are incorporated into helix 4, lengthening it relative to its Hsp90 counterpart. Because the N-terminal end of helix 4 abuts the ATP binding pocket, this lengthening has several structural consequences. First, it causes a small movement of the entire lid unit away from the ATP binding pocket, relative to the lid of Hsp90. Second, the movement of the lid in Grp94 repositions Gly196, which is at the N-terminal end of helix 5, moving it away from the pocket cavity. Third, it repositions the backbone and sidechains of Grp94 residues Ala167 and Lys168, which are at the base of helix 4, displacing them away from the pocket. Together, these differences between Grp94 and Hsp90, which, notably, are found outside of the ATP binding pocket, lead to the expansion of Site 3 in Grp94. These experiments showed that structural rearrangements, dictated by an insertion that was remote from the conserved binding pocket, were sufficient to remodel the pocket and allow binding of a ligand to one hsp90 paralog but not another.
The structure of Grp94:NECA showed that the N-ethyl moiety of NECA does not fully occupy the volume of Site 3 in Grp94. Based on this observation, N-propyl, N-hydroxyethyl, and N-aminoethyl variants of NECA were synthesized and structures of Grp94 in complex with these ligands were determined (PDB codes 2gqp, 2hch, 2hg1). While the longer 5’ substituents further fill the Site 3 cavity, as predicted, these compounds did not exhibit higher affinity binding to Grp94 than the parent compound, NECA [50]. The fold selectivity of the modified NECA compounds was not reported. N-propyl carboximido adenosine, like NECA, is also an agonist of adenosine receptors [51, 52]. Thus, although NECA binds preferentially to Grp94 and not Hsp90, the prospects for developing Site 3 targeted therapeutics based on the NECA scaffold are likely to be remote due to the strong potential off target activation of the adenosine receptor pathway.
Resorcinilic scaffolds targeting Site 3 in Grp94
While targeting Site 3 in Grp94 appeared to be a possible route for developing Grp94-selective inhibitors, the question remained whether Site 3 in Grp94 could be accessed by inhibitors that were not based on the NECA scaffold. It was also not clear whether access to Site 3 in Hsp90 would remain blocked to other classes of inhibitors. The prospects for selectively targeting Site 3 in Grp94 got an unexpected boost from an analysis of the binding of Radamide to Hsp90 and Grp94 [50]. Radamide is a Radicicol-Geldanamycin chimera that joins the resorcinol moiety derived from the high affinity pan-hsp90 inhibitor Radicicol to the quinone derived from Geldanamycin via an amide linkage [53]. Previous structures of Hsp90 in complex with diaryl resorcinylic inhbitors had shown that the resorcinylic hydroxyl makes a direct hydrogen bond to the conserved Hsp90 Asp93 of the ATP binding pocket (Asp149 in Grp94), thereby positioning the resorcinol group in the site normally occupied by the adenine ring of bound ATP [54–57]. Binding data indicated that Radamide bound both Grp94 and Hsp90 with approximately the same sub-micromolar affinity [50].
The crystal structures of Hsp90:Radamide and Grp94:Radamide (PDB codes 2fxs and 2gfd, respectively) supported the idea that access to Site 3 was selective for Grp94 [50]. In the Grp94:Radamide crystal structure, two poses of the quinone substituent were observed. These poses differed in the orientation of the quinone about the amide linkage. Pose 1 exhibited a cis-amide orientation that placed the quinone ring parallel to the plane of the resorcinol ring, while the second pose exhibited a bent trans-amide conformation that resulted in the plane of the quinone ring lying perpendicular to the plane of the resorcinol ring. Despite being far bulkier than the N-ethyl moiety of NECA, the Radamide quinone occupies Site 3 in Grp94 in both the cis- and the trans- poses. In the trans-amide pose the quinone blocks the mouth of Site 3, while in the cis-amide pose the quinone inserts further into Site 3 and makes van der Waals interactions with the side chains of Ile166 and Tyr200, and the backbone of Asn162.
The Grp94:Radamide structure also highlighted a novel plasticity of the Grp94 binding pocket. Modeling showed that in both Radamide poses the bulkier quinone moiety could not be accommodated into Site 3 of Grp94 as observed in the Grp94:NECA structure because the side chain of Thr171 of Grp94 makes a potential steric clash with the exocyclic o-methyl substituent of the quinone. Remarkably, however, this steric clash was avoided by the partial unwinding of helix 4 from its N-terminal end. The resulting remodeled helix 4 displaces Thr171 in Grp94:Radamide by 4.5 Å from its Grp94:NECA position, relieving the potential clash with the quinone. The Grp94:Radamide structure thus showed that 1) Site 3 in Grp94 could be accessed by non-NECA compounds, 2) Site 3 in Grp94 could expand to accommodate larger substituents, and 3) that amide linkage of Radamide was flexible.
Radamide also binds to Hsp90, but in contrast to Grp94 the quinone was not found to occupy Site 3. Instead, in Hsp90 the flexible amide linkage of Radamide adopts a third conformation: a linear, trans-amide pose that rotates the quinone moiety away from Site 3 and towards the solvent exposed mouth of the ATP binding pocket. This results in an extensive set of direct and water mediated hydrogen bonds between the polar groups of the quinone and Lys58, Asp102, Asn105, Asn106, Ile110, Ala111 and Lys112 of Hsp90 (Figure 5B). These residues are not part of Site 3. The extensive hydrogen bonding between the quinone and the mouth of the ATP binding pocket in Hsp90 thus appears to compensate for the loss of the interactions between the quinone and Site 3 in Grp94. This compensation likely accounts for the nearly identical binding constants for Radamide to Hsp90 and Grp94. Importantly, however, the Hsp90:Radamide structure showed that Site 3 was still inaccessible in Hsp90.
Second generation resorcinillic inhibitors
Based on the above studies, it was proposed that compounds disfavoring the linear trans-amide pose seen in the Hsp90-bound conformation of Radamide would prevent binding to Hsp90 while still allowing binding to Grp94 [58]. This required restricting the conformational freedom of the resorcinylic substituent to fix it in the cis-amide binding pose in order to target it to Site 3. Replacing the polar quinone with a non-polar benzyl group was also proposed to disfavor the hydrogen bonding interactions observed between the Radamide quinone and the mouth of the Hsp90 binding pocket. The result of this design process produced BnIm, which contained the resorcinyl group of Radamide to target the compound to the adenine binding site of the ATP binding pocket, linked to a non-polar phenyl group via an imidazole linker. While there is no structural data available to confirm whether BnIm binds to Grp94 with the benzyl ring inserted into Site 3 as predicted, Toll-like receptor surface expression, IGF-II secretion, and Grp94 antibody 9G10 recognition were all suppressed upon treatment with BnIm, confirming interaction with Grp94.
Whether Bnlm is a true paralog-selective inhibitor is unclear. The affinity of Bnlm binding to Grp94 was determined to be 1.2 µM in a fluorescent displacement assay, but the affinity of BnIm for purified Hsp90 was not reported. Indirect evidence for the Grp94 selectivity of BnIm includes the failure to exhibit cytotoxicity in MCF7, SKBr3, and HEK293 cells – all hallmarks of Hsp90 inhibition – even at 100 µM treatment. The levels of Hsp90 client proteins Raf and Akt in HEK293 cells were also not affected by BnIm treatment until 100 µM concentrations were applied. Despite these suggestions of specific targeting to Grp94, an independent analysis [59] found that the binding of BnIm to purified Grp94 and Hsp90 was only 10-fold selective for Grp94 (1.2 µM vs. 14 µM). Further indications of modest activity for BnIm include a failure to induce BiP expression upon treatment with < 75 µM BnIm, and failure to reduce HER2 levels in SKBr3 cells, a previously determined effect of bona fide Grp94 inhibition. Hsp70 induction, a hallmark of Hsp90 inhibition, was also observed in BnIm-treated SKBr3 cells. In light of these inconsistencies, further development of the BnIm scaffold would likely benefit greatly from structural data that identifies the precise location of the resorcinylic substituent and confirms the targeting of Site 3. Rational designs based on this structural data may lead to higher affinity inhibitors with greater intrinsic selectivity for Grp94.
Purine-based Grp94 selective inhibitors and the identification of Site 2
The 8-aryl purine (PU) derivatives have seen extensive development as hsp90 inhibitors over the past decade [60, 61]. PU compounds consist of a purine moiety substituted with an aryl group that is connected to the C8 by a carbon or sulfanyl linker. A short aliphatic tail is attached to the purine at the N9 or N3 position. Crystal structures of PU compounds in complex with Hsp90 showed that the purine moiety targets the site occupied by the adenine of ATP, while the N9 or N3 tail extends outward from the plane of the purine ring towards helix 2 of the N-terminal domain, and down towards the solvent exposed mouth of the ATP binding pocket [62, 63]. The 8-aryl substituent occupies a large hydrophobic cavity above the plane of the purine ring. This cavity, termed Site 1 (Figure 6B, D), is lined by the side chains of Leu48, Leu107, Phe138, Val148, Trp162, and Val186. The observed placement of the 8-aryl group into Site 1 was a surprise because in the apo form of Hsp90, Site 1 is occluded by the side chain of Leu107. Modeling shows that the 8-aryl group would clash with this sidechain. Upon PU binding to Hsp90, however, Site 1 becomes accessible to the 8-aryl moiety because of a rearrangement of the terminal end of helix 3 between residues 101–114, which causes Leu107 to move out of the way.
The Hsp90 Site 1 residues are completely conserved in Grp94 (Leu104, Leu163, Phe199, Val209, Trp223, Ile247) (Figure 6E). Because of this, the prospects for developing selective inhibitors based on the 8-aryl purine scaffold would appear to be limited. Surprisingly, however, high throughput screening of a set of 230 8-aryl PU compounds for binding to purified recombinant Grp94 and Hsp90 revealed a subset of 14 compounds that exhibited 10- to >1000-fold greater affinity for Grp94 compared to Hsp90 [64]. The crystal structures of one of these Grp94-selective compounds, PU-H54, in complex with both Grp94 and Hsp90 (PDB codes 3o2f, 3o0i), revealed the basis for this selectivity [64]. As expected, in complex with Hsp90, PU-H54 binds in a manner analogous to all other Hsp90:PU complexes. The purine ring sits in the adenine binding pocket, and the 8-aryl group is inserted into Site 1, displacing Leu107 and remodeling residues 101–114 in helix 3.
In contrast, the structure of Grp94:PU-H54 showed that rearrangements occur in the lid region, resulting in a substantially altered ATP-binding pocket. While the purine moiety still sits in the adenine binding site, as in the Hsp90:PU-54 complex, in Grp94 the 8-aryl group adopts a dramatically different conformation. Instead of bending ‘forward’ and occupying Site 1, the 8-aryl group in Grp94-bound PU-H54 undergoes an 80 degree rotation about the dihedral angle made by the C8-S bond of the sulfanyl linker. Concomitant with this ‘backwards-bent’ dihedral angle, Phe199 of Grp94 swings away from the binding pocket by 4 Å to expose a deep hydrophobic cleft. This cleft, which is called Site 2 (Figure 6C,D), is lined by the side chains of Leu104, Leu163, Phe199, Ala202, Phe203 Val209, Val211, Ile247, and Leu249 (for schematic, see Figure 5C). The hydrophobic 8-aryl group of PU-H54 is inserted into Site 2 and makes stabilizing contacts with several of the cleft residues. Interestingly, Site 2, which is part of the hydrophobic core of the N-terminal domain, is almost completely conserved between Grp94 and Hsp90 (Figure 6F) (Hsp90: Leu48, Leu107, Phe138, Ala141, Tyr142, Val148, Val150, Val186, and Leu188; conserved but not identical residues are in italics). Unlike in Grp94, however, access to Site 2 in Hsp90 is blocked by the side chain of Phe138, the equivalent of Grp94 Phe199.
The origins of the swing movement of Phe199 in Grp94 that exposes Site 2 for 8-aryl group occupancy are not yet fully understood. Movement of Phe199 is predicated, however, on a series of correlated motions of nearby side chains and secondary structural elements that are driven by the need to avoid steric clashes. These motions include, in order, the rotation of Tyr200 away from the new position of Phe199, the rotation of helix 1 to avoid clashes with the new position of Tyr200, and the partial closing of the lid to avoid clashes with the new position of helix 1. Because the remodeling of the Grp94 lid is the ultimate root of the movement of Phe199, it seems reasonable to conclude that the ability of the Grp94 lid to adopt different conformations in response to ligand perturbations of the ATP binding pocket is ultimately responsible for the ability of ligands to access Site 2 in Grp94, and not in Hsp90. These Grp94 lid movements, in turn, likely reflect the destabilizing effects of the 5 amino acid insertion found in the Grp94 lid, and which are absent in Hsp90.
Extensive structure-activity analysis has been carried out on Grp94-selective PU ligands [59]. The results of these studies indicated that 3’,5’- or 2’,4’- chlorine or bromine substituents on the 8-aryl moiety, which inserts into Site 2, yielded the highest affinity binding to Grp94. Polar, methyl or larger substituents on the 8-aryl ring significantly weakened the binding, reflecting the strongly hydrophobic nature of Site 2 and the narrow confines into which the 8-aryl moiety is inserted. Interestingly, the small, hydrophobic 8-aryl substitutents that are preferred for Grp94 binding are the same ones that were found to be the most unfavorable for PU ligands that bind to Site 1 in Hsp90 [65]. This reflects the less hydrophobic character and larger volume of Site 1 compared to Site 2 [64]. Selectivity of PU ligands for Grp94 over Hsp90 is thus due to the combined effects of 8-aryl substituents that favor insertion into Site 2 of Grp94 and simultaneously disfavor interactions with Site 1 in Hsp90.
The ability of the PU ligand to adopt the backward-bent pose that allows for the insertion of the 8-aryl group into Site 2 of Grp94 is a key parameter for selective binding. SAR of the linker about which 8-aryl moiety rotates indicates a 2 to 4-fold preference for C8-S-aryl linkers over C8-C-aryl and C8-O-aryl linkers, and a 20-fold preference over the more highly constrained mono- and di-oxygenated sulfoxide linkers [59]. Energetics calculations show, however, that while certain ligands prefer the backward bent conformation, the intrinsic difference in energy between the forward and backward bent conformations is not enough to account for the observed selectivity. Interestingly, however, when the C1’ position of the N9 substituent was modified, this sterically disfavors the forward bent conformation. The binding of these compounds to Hsp90 could not be detected.
As a demonstration of the power of paralog selective inhibition, a lead compound based on the Grp94-selective PU analysis, PU-WS13, has been shown to be selective for Grp94 in numerous cell-based assays and has provided key insights into the role of Grp94 in stabilizing HER2 in breast cancer cells [64].
Turning the tables on Grp94 pocket flexibility - Benzamadine tetrahydroindolones point the way to Hsp90-selective inhibitors
The first attempts to develop paralog-selective inhibitors focused on Grp94, in part because the plasticity of the region around the ATP binding pocket allowed for moieties on Grp94-selective ligands to target Sites 2 and 3, which were not accessible in the more rigid Hsp90 N-terminal domain. As early as 2008, however, there were indications that some inhibitors in fact bound better to Hsp90 than Grp94. NVP-AUY922 gave a reported IC50 of 21 nM to Hsp90, but bound to Grp94 and Trap-1 25-fold and 4-fold weaker, respectively [66]. SNX2112 had an IC50 of 30 nM to Hsp90, but bound 140- and 30-fold weaker to Grp94 and Trap-1 [67]. NVP-BEP800 had an IC50 of 58 nM to Hsp90 but bound Grp94 and Trap-1 70- and 95-fold weaker [68]. Finally, a more recent optimization of the purine-based EC144 showed a Ki of 0.2 nM to Hsp90 and 300- or 1200-fold weaker binding to Grp94 and Trap-1, respectively [69]. The common feature of these Hsp90-preferring compounds is the presence of a substituent moiety that occupies Site 1 of the ATP binding pocket. As discussed previously, Site 1 is ordinarily blocked in Hsp90 by residues 104–111. These residues link helices 3 and 4 and adopt a closed or open loop configuration in apo- or Geldanamycin-bound Hsp90, respectively. When ligands targeting Site 1 bind, however, residues 104–111 are remodeled into an alpha helical configuration, which exposes the Site 1 pocket. In this configuration, residues Leu107, Ile110, and Ala111 of Hsp90 form one side of the Site 1 and make stabilizing interactions with the bound ligand and shield it from exposure to solvent.
The crystal structure of Grp94:SNX0723 (PDB code 4nh9) (a benzamadine tetrahydroindolone which is similar to SNX2112 and exhibits 125-fold weaker binding to Grp94 than to Hsp90) showed that while the tetrahydroindolone moiety also occupies Site 1 in Grp94, the equivalent of Hsp90 residues 104–111 in Grp94 (160–167) were disordered and failed to make the stabilizing and shielding interactions with the SNX ligand as they did in Hsp90 (PDB code 4nh8) (Figure 6E) [70]. It was proposed that the loss of these interactions accounts for the ~100-fold lower affinity observed when these Site 1 utilizing ligands bind to Grp94. In support of this hypothesis, it was observed that when the 3-methyl group of the SNX0723 tetrahydroindolone, which is in close proximity to Leu107 (Grp94 Leu163), was removed, the fold selectivity for Hsp90 dropped from 140-fold to 40-fold. Thus, it appears that where previously the increased plasticity of the Grp94 pocket allowed for the selective binding of ligands that the more rigid Hsp90 could not accommodate, the Grp94 flexibility could now be exploited to favor the reverse selectivity.
Extensive SAR analysis of SNX0723 and its derivatives was carried out with the twin goals of improving the fold-selectivity of binding to Hsp90 and increasing potency in a Huntington’s disease Htt degradation model [3]. Two logs of selectivity for Hsp90 over Grp94 were already provided by targeting the tetrahydroindolone moiety into Site 1. To further improve the selective binding, the authors turned their attention to the benzamide moiety, which occupies the position of the adenine base in the ATP binding pocket. Selectively targeting this pocket is a particularly challenging prospect. The ATP binding pockets of Hsp90 and Grp94 are structurally conserved and conformationally rigid. To date all of the protein elements that have been exploited to achieve paralog selective ligand binding have been in the sites that adjoin the ATP pocket.
Despite these challenges, however, a careful analysis of the Hsp90:SNX0723 and Grp94:SNX0723 structures pointed to a possible exploitable difference in the binding of benzamides to the ATP pocket [3]. In the back of the pocket, residue 186 of Hsp90 is a valine. The equivalent residues in Grp94 and Trap-1 are Ile247 and Ile253. From the Grp94 and Hsp90 structures, the extra delta methyl of the larger Ile247 in Grp94 projects into the ATP cavity by ~1 Å more than the gamma methyl of Hsp90 Val186. In the substituted benzamides, substituents ortho to the amide, which in SNX0723 is a fluorine, point directly at Val186 or Ile247 and make a tight fit. It was hypothesized that optimization of the benzylic substituent at this position could maximize the fit with Val186 in Hsp90 but sterically clash with the extra methyl of Ile247 in Grp94 and Ile253 in Trap-1. To test this, the ortho fluorine in SNX0723 was replaced with a benzolactam. This modification increased the bulk of the benzamide moiety compared to the fluorine on SNX0723 and rigidified the ortho substituent by cyclization with the primary amine. Remarkably, the resulting compound showed a 10-fold improvement in selectivity for Hsp90 compared to the parent compound, confirming the validity of targeting the ATP pocket. The addition of a cyclopentyl group ortho to the lactam, resulting in Compound 31 improved the binding to Hsp90 by an order of magnitude to 5 nM, and the fold selectivity for Hsp90 over Grp94 and Trap-1 by another order of magnitude as well. The successful development of Hsp90-selective ligands thus exploited both the conformational flexibility of Grp94 (and presumably Trap-1) in the vicinity of Site 1 to lose potentially stabilizing interactions, combined with the potential for a steric clash deep within the nearly conserved ATP binding pocket itself.
Recently, a second highly selective benzamide compound, TAS-116, was reported that exhibits Ki’s of 34 nM and 21 nM to Hsp90α and Hsp90β, respectively, and >50 µM binding to Grp94 and Trap-1 [71]. Although no structure of a complex between TAS-116 and Hsp90 has been reported, the mode of ligand binding can be anticipated based on a comparison with the SNX compounds described above. The benzamide moiety likely occupies the ATP binding site, while the heavily substituted pyridinyl group likely occupies Site 1. Should this be confirmed structurally, this would further establish the principle that moieties targeting Site 1 impart selective binding advantage for Hsp90α and β.
Hsp90 α/β Selectivity – looking outside the ATP pocket
Hsp90β is generally considered to be the constituitive paralog in the cell [9], while Hsp90α is the inducible form that is frequently overexpressed under conditions of malignant transformation [72]. Given the centrality of Hsp90’s function for the well being of normal cells in the body, targeting just the cancer-associated species is an attractive prospect. Unfortunately, as subtle as the differences between the ATP binding pockets of Hsp90, Grp94, and Trap-1 may be, they loom large compared to the differences between the pockets of Hsp90α and Hsp90β. All of the amino acids in the ATP binding pocket and its vicinity are conserved between the α and β paralogs, and the few crystal structures of the Hsp90β N-terminal domain (PDB codes 1uym, 3pry) show it to be virtually superimposable with that of Hsp90α. Nevertheless, the same screen that identified the Grp94 selective PU compounds also identified six other weakly binding PU compounds that exhibited 3- to 5-fold selectivity for Hsp90α over Hsp90β [64]. These compounds, examples of which include PU-11trans and PU-29F, bind to Hsp90α with affinities in the 5–20 µM range. They consist of a purine ring connected to a 3’,4’,5’ trimethoxy- 8-aryl group via a C8-C-aryl linker. The N9 substituent is large and non-polar. A similar compound that is not Hsp90 α/β selective, PU-7, differs from PU-11trans only in the N9 substituent. In PU-11trans this is a trans-butyl moiety, while in PU-7 the N9 substituent is a cis-pentanyl moiety. In the absence of a structural comparison of Hsp90α and β in complex with an α/β selective compound, it is difficult to explain the origins of the observed ligand selectivity. Assuming that a comparison of the structures of two N-terminal domain ligand complexes shows no apparent differences, however, it is tempting to speculate that differences in the binding of the α/β selective compounds may instead be due to structural rearrangements that take place outside of the N-terminal domains. Hsp90s are known to undergo dramatic quaternary changes in response to nucleotide binding [73], and it is possible that the energetics of these transitions are not the same in the α and β paralogs. A detailed thermodynamic analysis of the binding of the α/β selective compounds to the two paralogs may be needed to shed light on this mechanism of selectivity.
Trap-1 inhibitors – exploiting organelle targeting
There are considerably fewer structures of the mitochondrial paralog Trap-1 compared to Hsp90 and Grp94. Nonetheless, the recent determination of the structure of the N+Middle domain construct of human Trap-1 in complex with the pan-hsp90 PU inhibitors PU-H71 and BIIB-021 (PDB codes 4zif, 4z1g) [74] confirms the important themes discovered in the course of developing Hsp90-selective inhibitors described above. In particular, the crystal structures show that the Trap-1 lid and adjoining helix 3-helix 4 connector are disordered in these complexes (Figure 6E). This confirms the prediction that substitutents targeting Site 1 in Grp94 and Trap-1 would lose the stabilizing benefit of these interactions [3].
It is not yet been confirmed by a crystal structure whether Sites 2 and 3 in Trap-1 are accessible to substituent moieties on bound ligands. The fact that some Grp94-specific PU ligands also show weak to moderate binding to Trap-1 [64], however, suggests that Site 2 may indeed be utilized in this paralog in a manner analogous to that of Grp94. Exposure of Site 2 ultimately derives from the altered lid energetics brought about the 5 amino acid insertion between helices 4 and 5 of the lid in Grp94. A comparison of the equivalent region in Trap-1 (Figure 4) shows that Trap-1 occupies a middle ground between Hsp90 and Grp94. On the one hand, Trap-1 has a 2 amino acid insertion (Ala191, Glu192) between helices 4 and 5 of the lid, but on the other hand, where the sequence of helix 4 was not conserved between Grp94 and Hsp90, helix 4 of Trap-1 is highly similar to that of Hsp90. This partial Grp94-like character likely accounts for the weak affinity for Trap-1 shown by the Grp94-selective PU inhibitors, as well as for the disorder of the helix 3-helix 4 connector seen when ligand substituents target Site 1. Given these observations, the prospects for the design of inhibitors that are selective for Trap-1 alone are not hopeful. Any attempt to target Site 2, and possibly Site 3 in Trap-1 in order to avoid binding to Hsp90 will likely encounter significant cross-affinity with Grp94.
Trap-1 is exclusively localized in the mitochondria. Selective inhibition may also thus be achieved by preferentially targeting what are otherwise pan-hsp90 inhibitors to this organelle. This was tested in the case of PU-H71, where the N9 alkyl group was replaced with a triphenylphosphonium (TPP) conjugated alkyl group, also at the N9 position [74]. TPP modification is an established mechanism of mitochondrial targeting [75]. The resulting compound, SMTIN-P01, showed a 100-fold accumulation in the mitochondria, compared to the surrounding media. SMTIN-P01 also induced mitochondrial membrane depolarization in HeLa cells, suggesting that the TPP targeting was effective. The crystal structure of SMTIN-P01 in complex with Trap-1 (PDB code 4z1h) showed that the modified ligand bound in the same manner as the parent compound PU-H71, and inhibited the ATPase activity of Trap-1 to a similar degree as well. Unlike PU-H71, however, SMTIN-P01 did not induce depletion of the Hsp90 client proteins Chk1 and Akt, nor did it upregulate the Hsp70 response, suggesting that the effects on cytosolic Hsp90 have been minimized by the mitochondrial targeting. These results thus highlight the potential for selective inhibition of individual hsp90 paralogs by targeting their cellular compartments. It will be interesting to see if mitochondrial targeting of compounds exhibiting selective binding to Trap-1 and Grp94 yield better overall selectivity in cell based assays.
Summary and Future prospects
Given the challenges posed by the high sequence and structural homology of the hsp90 family, it is remarkable that selective inhibitors for these paralogs have been identified. What was at first a daunting prospect has now given way to an appreciation of the idiosyncratic structural nuances that rendered the development of such inhibitors possible. From the current progress, a few key lessons have emerged. First, exploiting the conformational flexibility of Grp94 is the key to targeting this paralog. Grp94-selective ligands all contain moieties that target Sites 2 or 3 of the N-terminal domain. These sites are present in all of the paralogs, but access to them is sterically blocked in Hsp90, dramatically lowering the affinity of ligands whose low energy pose targets these sites. The flexibility of the Grp94 lid, derived ultimately from the destabilizing effect of the 5 amino acid insertion between helices 4 and 5, allows for the movement of the backbone and sidechains to expose these sites. Second, Hsp90-selective ligands target Site 1 of the N-terminal domain. The same plasticity that exposes Sites 2 and 3 in Grp94 renders one side of the Site 1 cavity unstable when ligand moieties inhabit this pocket. In the absence of other stabilizing interactions, ligands that target Site 1 exhibit ~100-fold higher affinity for Hsp90 than Grp94 or Trap-1 due to the loss of stabilizing interactions between the ligand and Site 1. Site 1 is not destabilized in Hsp90, and as a result can make productive interactions with the inserted ligand moieties. Third, subtle differences in the ATP binding pockets of Hsp90, Grp94, and Trap-1 can be exploited for selective ligand binding. Val186 in Hsp90 and its Grp94 equivalent, Ile247 differ by one methyl group. The delta methyl in Ile247, however, projects 1 Å deeper into the ATP binding pocket than the gamma methyl of Val186. Bulkier ligands that reach further into the back of the ATP binding pocket can exploit this 1 Å difference such that preference for Hsp90 over Grp94 or Trap can be achieved. This was shown to be particularly powerful in improving the selectivity of Hsp90-favoring ligands when combined with the targeting of Site 1. Finally, it is important to acknowledge that both screening and structure-based design have played key roles in the development of the first set of paralog selective inhibitors. Screening using ligand affinity as a metric first identified NECA, the PU compounds, and the benzamadine tetrahydroindolones as having selective affinity for individual paralogs. Understanding their mode of selectivity, however, required high resolution structures of the representative protein:ligand complexes due to the uncertainties of modeling the ligands into a conformationally plastic binding pocket. In the absence of co-crystal structures with the bound ligands, modeling the binding of the selective ligands would have resulted in identical sets of binding interactions. Under these circumstances, the path to further development and the confidence to pursue it would have been unclear.
The proof of principle that hsp90 paralog selective inhibitors can be developed has now been established. Two broad avenues lie ahead. The first is to improve the overall affinity and pharmacokinetic and pharmacodynamics properties of the current crop of inhbitors. The Hsp90-selective Benzamadine tetrahydroindolones show low nanomolar affinity for Hsp90 and >1000-fold selectivity, but the Grp94-selective PU compounds have only high nanomolar affinities and ~100-fold selectivities. Ligands that could access Site 2 but are sterically restricted from accessing Site 1 as an alternative mode of binding would likely improve the selecitivty of these Grp94-targeted compounds. Alternatively, ligands that target Site 3 in Grp94 have yet to be fully explored. The future prospects for the NECA scaffold are limited due to the cross-reactivity with the adenosine receptor family, but scaffolds that replace the adenine-ribose combination of NECA with either a resorcinol or benzamadine may prove to be more promising.
The second avenue for future development is in the exploitation of the selective ligands themselves. The study of the biology of the individual paralogs is still in its infancy. High affinity inhibitors with several logs of paralog selectivity will be key to identifying the authentic clients and pathways regulated by these chaperones. No paralog-selective inhibitors are as yet in clinical trials, but, given the progress to date, the therapeutic prospects for these selective inhibitors are indeed wide open.
Acknowledgments
I thank Dr. N. Que for help with the figures and for a critical reading of the manuscript. DG is supported by grants from the NIH (R01CA095130 and P01CA186866) and from the Richard W. and Mae Stone Goode Foundation.
Footnotes
Conflict of Interest
The author declares no conflicts of interest.
References
- 1.Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS One. 2011;6(10):e26044. doi: 10.1371/journal.pone.0026044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baldo B, Weiss A, Parker CN, Bibel M, Paganetti P, Kaupmann K. A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (Hsp90) client protein. The Journal of biological chemistry. 2012;287(2):1406–1414. doi: 10.1074/jbc.M111.294801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ernst JT, Neubert T, Liu M, Sperry S, Zuccola H, Turnbull A, Fleck B, Kargo W, Woody L, Chiang P, Tran D, Chen W, Snyder P, Alcacio T, Nezami A, Reynolds J, Alvi K, Goulet L, Stamos D. Identification of novel HSP90alpha/beta isoform selective inhibitors using structure-based drug design. demonstration of potential utility in treating CNS disorders such as Huntington's disease. J Med Chem. 2014;57(8):3382–3400. doi: 10.1021/jm500042s. [DOI] [PubMed] [Google Scholar]
- 4.Geller R, Taguwa S, Frydman J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim Biophys Acta. 2012;1823(3):698–706. doi: 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pallavi R, Roy N, Nageshan RK, Talukdar P, Pavithra SR, Reddy R, Venketesh S, Kumar R, Gupta AK, Singh RK, Yadav SC, Tatu U. Heat shock protein 90 as a drug target against protozoan infections: biochemical characterization of HSP90 from Plasmodium falciparum and Trypanosoma evansi and evaluation of its inhibitor as a candidate drug. The Journal of biological chemistry. 2010;285(49):37964–37975. doi: 10.1074/jbc.M110.155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy N, Nageshan RK, Ranade S, Tatu U. Heat shock protein 90 from neglected protozoan parasites. Biochim Biophys Acta. 2012;1823(3):707–711. doi: 10.1016/j.bbamcr.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Shahinas D, Liang M, Datti A, Pillai DR. A repurposing strategy identifies novel synergistic inhibitors of Plasmodium falciparum heat shock protein 90. J Med Chem. 2010;53(9):3552–3557. doi: 10.1021/jm901796s. [DOI] [PubMed] [Google Scholar]
- 8.Whitesell L, Lin NU. HSP90 as a platform for the assembly of more effective cancer chemotherapy. Biochim Biophys Acta. 2012;1823(3):756–766. doi: 10.1016/j.bbamcr.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Meng X, Jerome V, Devin J, Baulieu EE, Catelli MG. Cloning of chicken hsp90 beta: the only vertebrate hsp90 insensitive to heat shock. Biochem Biophys Res Commun. 1993;190(2):630–636. doi: 10.1006/bbrc.1993.1095. [DOI] [PubMed] [Google Scholar]
- 10.Koch G, Smith M, Macer D, Webster P, Mortara R. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. Journal of cell science. 1986;86:217–232. doi: 10.1242/jcs.86.1.217. [DOI] [PubMed] [Google Scholar]
- 11.Felts SJ, Owen BA, Nguyen P, Trepel J, Donner DB, Toft DO. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. The Journal of biological chemistry. 2000;275(5):3305–3312. doi: 10.1074/jbc.275.5.3305. [DOI] [PubMed] [Google Scholar]
- 12.Song HY, Dunbar JD, Zhang YX, Guo D, Donner DB. Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. The Journal of biological chemistry. 1995;270(8):3574–3581. [PubMed] [Google Scholar]
- 13.Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440(7087):1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Molecular cell. 2007;28(1):41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavery LA, Partridge JR, Ramelot TA, Elnatan D, Kennedy MA, Agard DA. Structural asymmetry in the closed state of mitochondrial Hsp90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Molecular cell. 2014;53(2):330–343. doi: 10.1016/j.molcel.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris SF, Shiau AK, Agard DA. The Crystal Structure of the Carboxy-Terminal Dimerization Domain of htpG, the Escherichia coli Hsp90, Reveals a Potential Substrate Binding Site. Structure. 2004;12:1087–1097. doi: 10.1016/j.str.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Nemoto T, Ohara-Nemoto Y, Ota M, Takagi T, Yokoyama K. Mechanism of dimer formation of the 90-kDa heat-shock protein. Eur J Biochem. 1995;233(1):1–8. doi: 10.1111/j.1432-1033.1995.001_1.x. [DOI] [PubMed] [Google Scholar]
- 18.Fontana J, Fulton D, Chen Y, Fairchild TA, McCabe TJ, Fujita N, Tsuruo T, Sessa WC. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circulation research. 2002;90(8):866–873. doi: 10.1161/01.res.0000016837.26733.be. [DOI] [PubMed] [Google Scholar]
- 19.Meyer P, Prodromou C, Hu B, Vaughan C, Roe SM, Panaretou B, Piper PW, Pearl LH. Structural and functional analysis of the middle segment of hsp90. Implications for ATP hydrolysis and client protein and cochaperone interactions. Molecular cell. 2003;11(3):647–658. doi: 10.1016/s1097-2765(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 20.Meyer P, Prodromou C, Liao C, Hu B, Mark Roe S, Vaughan CK, Vlasic I, Panaretou B, Piper PW, Pearl LH. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. The EMBO journal. 2004;23(3):511–519. doi: 10.1038/sj.emboj.7600060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(20):10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Street TO, Lavery LA, Agard DA. Substrate binding drives large-scale conformational changes in the Hsp90 molecular chaperone. Molecular cell. 2011;42(1):96–105. doi: 10.1016/j.molcel.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaughan CK, Gohlke U, Sobott F, Good VM, Ali MM, Prodromou C, Robinson CV, Saibil HR, Pearl LH. Structure of an hsp90-cdc37-cdk4 complex. Molecular cell. 2006;23(5):697–707. doi: 10.1016/j.molcel.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90(1):65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 25.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89(2):239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 26.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nature cell biology. 2001;3(10):891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 27.Brugge JS, Erikson E, Erikson RL. The specific interaction of the Rous sarcoma virus transforming protein, pp60src, with two cellular proteins. Cell. 1981;25(2):363–372. doi: 10.1016/0092-8674(81)90055-6. [DOI] [PubMed] [Google Scholar]
- 28.Dalman FC, Bresnick EH, Patel PD, Perdew GH, Watson SJ, Jr, Pratt WB. Direct evidence that the glucocorticoid receptor binds to hsp90 at or near the termination of receptor translation in vitro. The Journal of biological chemistry. 1989;264(33):19815–19821. [PubMed] [Google Scholar]
- 29.Oppermann H, Levinson W, Bishop JM. A cellular protein that associates with the transforming protein of Rous sarcoma virus is also a heat-shock protein. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(2):1067–1071. doi: 10.1073/pnas.78.2.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature. 1990;348(6297):166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- 31.Uehara Y, Murakami Y, Mizuno S, Kawai S. Inhibition of transforming activity of tyrosine kinase oncogenes by herbimycin A. Virology. 1988;164(1):294–298. doi: 10.1016/0042-6822(88)90649-6. [DOI] [PubMed] [Google Scholar]
- 32.Whitesell L, Cook P. Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Molecular endocrinology (Baltimore, Md. 1996;10(6):705–712. doi: 10.1210/mend.10.6.8776730. [DOI] [PubMed] [Google Scholar]
- 33.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(18):8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitesell L, Shifrin SD, Schwab G, Neckers LM. Benzoquinonoid ansamycins possess selective tumoricidal activity unrelated to src kinase inhibition. Cancer Res. 1992;52(7):1721–1728. [PubMed] [Google Scholar]
- 35.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Experimental biology and medicine (Maywood, N.J. 2003;228(2):111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 36.Obermann WM, Sondermann H, Russo AA, Pavletich NP, Hartl FU. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. The Journal of cell biology. 1998;143(4):901–910. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panaretou B, Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. The EMBO journal. 1998;17(16):4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prodromou C, Panaretou B, Chohan S, Siligardi G, O'Brien R, Ladbury JE, Roe SM, Piper PW, Pearl LH. The ATPase cycle of Hsp90 drives a molecular 'clamp' via transient dimerization of the N-terminal domains. The EMBO journal. 2000;19(16):4383–4392. doi: 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823(3):742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42(2):260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 41.Zubriene A, Gutkowska M, Matuliene J, Chaleckis R, Michailoviene V, Voroncova A, Venclovas C, Zylicz A, Zylicz M, Matulis D. Thermodynamics of radicicol binding to human Hsp90 alpha and beta isoforms. Biophysical chemistry. 2010;152(1–3):153–163. doi: 10.1016/j.bpc.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Kim J, Felts S, Llauger L, He H, Huezo H, Rosen N, Chiosis G. Development of a fluorescence polarization assay for the molecular chaperone Hsp90. J Biomol Screen. 2004;9(5):375–381. doi: 10.1177/1087057104265995. [DOI] [PubMed] [Google Scholar]
- 43.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bruns RF, Lu GH, Pugsley TA. Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol Pharmacol. 1986;29(4):331–346. [PubMed] [Google Scholar]
- 45.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474(7352):521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hutchison KA, Fox IH. Purification and characterization of the adenosine A2-like binding site from human placental membrane. The Journal of biological chemistry. 1989;264(33):19898–19903. [PubMed] [Google Scholar]
- 47.Hutchison KA, Nevins B, Perini F, Fox IH. Soluble and membrane-associated human low-affinity adenosine binding protein (adenotin): properties and homology with mammalian and avian stress proteins. Biochemistry. 1990;29(21):5138–5144. doi: 10.1021/bi00473a020. [DOI] [PubMed] [Google Scholar]
- 48.Rosser MF, Nicchitta CV. Ligand interactions in the adenosine nucleotide-binding domain of the Hsp90 chaperone, GRP94. I. Evidence for allosteric regulation of ligand binding. The Journal of biological chemistry. 2000;275(30):22798–22805. doi: 10.1074/jbc.M001477200. [DOI] [PubMed] [Google Scholar]
- 49.Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. The Journal of biological chemistry. 2003;278(48):48330–48338. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- 50.Immormino RM, Metzger LEt, Reardon PN, Dollins DE, Blagg BS, Gewirth DT. Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: implications for paralog-specific drug design. Journal of molecular biology. 2009;388(5):1033–1042. doi: 10.1016/j.jmb.2009.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Zwart M, Kourounakis A, Kooijman H, Spek AL, Link R, von Frijtag Drabbe Kunzel JK, AP IJ. 5'-N-substituted carboxamidoadenosines as agonists for adenosine receptors. J Med Chem. 1999;42(8):1384–1392. doi: 10.1021/jm9804984. [DOI] [PubMed] [Google Scholar]
- 52.Tosh DK, Phan K, Gao ZG, Gakh AA, Xu F, Deflorian F, Abagyan R, Stevens RC, Jacobson KA, Katritch V. Optimization of adenosine 5'-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment screening. J Med Chem. 2012;55(9):4297–4308. doi: 10.1021/jm300095s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clevenger RC, Blagg BS. Design, synthesis, and evaluation of a radicicol and geldanamycin chimera, radamide. Org Lett. 2004;6(24):4459–4462. doi: 10.1021/ol048266o. [DOI] [PubMed] [Google Scholar]
- 54.Brough PA, Barril X, Beswick M, Dymock BW, Drysdale MJ, Wright L, Grant K, Massey A, Surgenor A, Workman P. 3-(5-Chloro-2,4-dihydroxyphenyl)-pyrazole-4-carboxamides as inhibitors of the Hsp90 molecular chaperone. Bioorganic & medicinal chemistry letters. 2005;15(23):5197–5201. doi: 10.1016/j.bmcl.2005.08.091. [DOI] [PubMed] [Google Scholar]
- 55.Cheung KM, Matthews TP, James K, Rowlands MG, Boxall KJ, Sharp SY, Maloney A, Roe SM, Prodromou C, Pearl LH, Aherne GW, McDonald E, Workman P. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorganic & medicinal chemistry letters. 2005;15(14):3338–3343. doi: 10.1016/j.bmcl.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 56.Dymock BW, Barril X, Brough PA, Cansfield JE, Massey A, McDonald E, Hubbard RE, Surgenor A, Roughley SD, Webb P, Workman P, Wright L, Drysdale MJ. Novel, potent small-molecule inhibitors of the molecular chaperone Hsp90 discovered through structure-based design. J Med Chem. 2005;48(13):4212–4215. doi: 10.1021/jm050355z. [DOI] [PubMed] [Google Scholar]
- 57.Kreusch A, Han S, Brinker A, Zhou V, Choi HS, He Y, Lesley SA, Caldwell J, Gu XJ. Crystal structures of human HSP90alpha-complexed with dihydroxyphenylpyrazoles. Bioorganic & medicinal chemistry letters. 2005;15(5):1475–1478. doi: 10.1016/j.bmcl.2004.12.087. [DOI] [PubMed] [Google Scholar]
- 58.Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O, Shinogle HE, Moore DS, Argon Y, Nicchitta CV, Blagg BS. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134(23):9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patel HJ, Patel PD, Ochiana SO, Yan P, Sun W, Patel MR, Shah SK, Tramentozzi E, Brooks J, Bolaender A, Shrestha L, Stephani R, Finotti P, Leifer C, Li Z, Gewirth DT, Taldone T, Chiosis G. Structure-activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum Hsp90 paralog Grp94. J Med Chem. 2015;58(9):3922–3943. doi: 10.1021/acs.jmedchem.5b00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiosis G. Targeting chaperones in transformed systems--a focus on Hsp90 and cancer. Expert opinion on therapeutic targets. 2006;10(1):37–50. doi: 10.1517/14728222.10.1.37. [DOI] [PubMed] [Google Scholar]
- 61.Taldone T, Chiosis G. Purine-scaffold Hsp90 inhibitors. Current topics in medicinal chemistry. 2009;9(15):1436–1446. doi: 10.2174/156802609789895737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Immormino RM, Kang Y, Chiosis G, Gewirth DT. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. J Med Chem. 2006;49(16):4953–4960. doi: 10.1021/jm060297x. [DOI] [PubMed] [Google Scholar]
- 63.Wright L, Barril X, Dymock B, Sheridan L, Surgenor A, Beswick M, Drysdale M, Collier A, Massey A, Davies N, Fink A, Fromont C, Aherne W, Boxall K, Sharp S, Workman P, Hubbard RE. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem Biol. 2004;11(6):775–785. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 64.Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA, Gewirth DT, Chiosis G. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013 doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Llauger L, He H, Kim J, Aguirre J, Rosen N, Peters U, Davies P, Chiosis G. Evaluation of 8-arylsulfanyl, 8-arylsulfoxyl, and 8-arylsulfonyl adenine derivatives as inhibitors of the heat shock protein 90. J Med Chem. 2005;48(8):2892–2905. doi: 10.1021/jm049012b. [DOI] [PubMed] [Google Scholar]
- 66.Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K, Prodromou C, Pearl L, James K, Matthews TP, Cheung KM, Kalusa A, Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B, Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood M, Wright L, Workman P. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68(8):2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 67.Chandarlapaty S, Sawai A, Ye Q, Scott A, Silinski M, Huang K, Fadden P, Partdrige J, Hall S, Steed P, Norton L, Rosen N, Solit DB. SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase-dependent cancers. Clin Cancer Res. 2008;14(1):240–248. doi: 10.1158/1078-0432.CCR-07-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Massey AJ, Schoepfer J, Brough PA, Brueggen J, Chene P, Drysdale MJ, Pfaar U, Radimerski T, Ruetz S, Schweitzer A, Wood M, Garcia-Echeverria C, Jensen MR. Preclinical antitumor activity of the orally available heat shock protein 90 inhibitor NVP-BEP800. Molecular cancer therapeutics. 2010;9(4):906–919. doi: 10.1158/1535-7163.MCT-10-0055. [DOI] [PubMed] [Google Scholar]
- 69.Shi J, Van de Water R, Hong K, Lamer RB, Weichert KW, Sandoval CM, Kasibhatla SR, Boehm MF, Chao J, Lundgren K, Timple N, Lough R, Ibanez G, Boykin C, Burrows FJ, Kehry MR, Yun TJ, Harning EK, Ambrose C, Thompson J, Bixler SA, Dunah A, Snodgrass-Belt P, Arndt J, Enyedy IJ, Li P, Hong VS, McKenzie A, Biamonte MA. EC144 is a potent inhibitor of the heat shock protein 90. J Med Chem. 2012;55(17):7786–7795. doi: 10.1021/jm300810x. [DOI] [PubMed] [Google Scholar]
- 70.Ernst JT, Liu M, Zuccola H, Neubert T, Beaumont K, Turnbull A, Kallel A, Vought B, Stamos D. Correlation between chemotype-dependent binding conformations of HSP90alpha/beta and isoform selectivity-Implications for the structure-based design of HSP90alpha/beta selective inhibitors for treating neurodegenerative diseases. Bioorganic & medicinal chemistry letters. 2014;24(1):204–208. doi: 10.1016/j.bmcl.2013.11.036. [DOI] [PubMed] [Google Scholar]
- 71.Ohkubo S, Kodama Y, Muraoka H, Hitotsumachi H, Yoshimura C, Kitade M, Hashimoto A, Ito K, Gomori A, Takahashi K, Shibata Y, Kanoh A, Yonekura K. TAS-116, a highly selective inhibitor of heat shock protein 90alpha and beta, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Molecular cancer therapeutics. 2015;14(1):14–22. doi: 10.1158/1535-7163.MCT-14-0219. [DOI] [PubMed] [Google Scholar]
- 72.Yufu Y, Nishimura J, Nawata H. High constitutive expression of heat shock protein 90 alpha in human acute leukemia cells. Leukemia research. 1992;16(6–7):597–605. doi: 10.1016/0145-2126(92)90008-u. [DOI] [PubMed] [Google Scholar]
- 73.Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta. 2012;1823(3):624–635. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 74.Lee C, Park HK, Jeong H, Lim J, Lee AJ, Cheon KY, Kim CS, Thomas AP, Bae B, Kim ND, Kim SH, Suh PG, Ryu JH, Kang BH. Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. J Am Chem Soc. 2015;137(13):4358–4367. doi: 10.1021/ja511893n. [DOI] [PubMed] [Google Scholar]
- 75.Armstrong JS. Mitochondrial medicine: pharmacological targeting of mitochondria in disease. Br J Pharmacol. 2007;151(8):1154–1165. doi: 10.1038/sj.bjp.0707288. [DOI] [PMC free article] [PubMed] [Google Scholar]