

Abstract
Acid‐sensing ion channels (ASICs) and the epithelial Na+ channel (ENaC) are both members of the ENaC/degenerin family of amiloride‐sensitive Na+ channels. ASICs act as proton sensors in the nervous system where they contribute, besides other roles, to fear behaviour, learning and pain sensation. ENaC mediates Na+ reabsorption across epithelia of the distal kidney and colon and of the airways. ENaC is a clinically used drug target in the context of hypertension and cystic fibrosis, while ASIC is an interesting potential target. Following a brief introduction, here we will review selected aspects of ASIC and ENaC function. We discuss the origin and nature of pH changes in the brain and the involvement of ASICs in synaptic signalling. We expose how in the peripheral nervous system, ASICs cover together with other ion channels a wide pH range as proton sensors. We introduce the mechanisms of aldosterone‐dependent ENaC regulation and the evidence for an aldosterone‐independent control of ENaC activity, such as regulation by dietary K+. We then provide an overview of the regulation of ENaC by proteases, a topic of increasing interest over the past few years. In spite of the profound differences in the physiological and pathological roles of ASICs and ENaC, these channels share many basic functional and structural properties. It is likely that further research will identify physiological contexts in which ASICs and ENaC have similar or overlapping roles.
Abbreviations
- AQP
aquaporin
- ASDN
aldosterone‐sensitive distal nephron
- ASIC
acid‐sensing ion channel
- BASIC
bile acid‐activated ion channel
- BK
big calcium‐activated K+ channel
- CA
carbonic anhydrase
- CAP‐1, ‐2, or‐3
channel activating proteases
- Cav
voltage‐gated Ca2+ channel
- CCD
cortical collecting duct
- CD
collecting duct
- PHA‐1
pseudohypoaldosteronism type 1
- CFTR
cystic fibrosis transmembrane conductance regulator
- CNT
connecting tubule
- CF
cystic fibrosis
- CLCN/Kb
voltage‐sensitive chloride channel Kb
- DCT
distal convoluted tubule
- DRG
dorsal root ganglion
- ENaC
amiloride‐sensitive epithelial sodium channel
- EPSP
excitatory post‐synaptic potential
- FaNaC
FMRFa‐activated Na+ channel
- FMRFa
Phe‐Met‐Arg‐Phe‐amide
- GMQ
2‐guanidine‐4‐methylquinazoline
- GPI
glycosylphosphatidyl‐inositol
- HAI
hepatocyte growth factor activator inhibitor
- HCN
hyperpolarization‐activated cyclic nucleotide‐gated channel
- IA
A‐type current of rapid inactivating K+ channels
- iGluR
ionotropic glutamate receptor
- IK
K+ current
- INa
Na+ current
- Ih
current produced by HCN channels
- Imax
maximal current amplitude
- Kir
inward rectifier K+ channel
- Kv
voltage‐gated K+ channel
- MR
mineralocorticoid receptor
- Nav
voltage‐gated Na+ channel
- NBC
Na+, ‐HCO3‐ cotransporter
- NCC
Na+-Cl− cotransporter
- NHE
Na+-H+ exchanger
- OSR1/SPAK
Ste20‐related protein kinases
- P2X
purinergic receptor
- PcTx1
Psalmotoxin 1
- pH50
pH of half‐maximal activation
- pHe
extracellular pH
- pH50Inh./Act.
pH of half-maximal inhibition/activation
- PMCA
plasma membrane Ca2+-ATPase
- PNS
peripheral nervous system
- PPK
pickpocket
- ROMK
renal outer medullary potassium channel
- SPLUNC1
the short palate, lung, and nasal epithelial clone 1
- TASK
two‐pore domain K+ channel
- TRAAK
TWIK‐related arachidonic acid‐stimulated K+ channel
- TREK
TWIK‐related K+ channel
- TRPM
transient receptor potential cation channel, subfamily M
- TRPV
transient receptor potential cation channel subfamily V
- TWIK
tandem of P domains in a weak inwardly rectifying K+ channel
- V‐ATPase
vacuolar‐type H+-ATPase
Tables of Links
| LIGANDS | |
|---|---|
| A‐317567 | GMQ |
| Amiloride | Nafamostat |
| APETx2 | P552‐02 |
| Agmatine | Phenamil |
| Aldosterone | Psalmotoxin 1 |
| Arcaine | Triamterene |
| Benzamil | Vasopressin |
| α‐CGRP (calcitonine gene‐related peptide α) |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,d,eAlexander et al., 2015a, 2015c, 2015b, 2015d, 2015e).
Introduction
The amiloride‐sensitive epithelial sodium channel (ENaC)/degenerin (DEG) superfamily of ion channels includes, besides ENaC and acid‐sensing ion channels (ASICs), the DEGs that are part of mechanotransduction complexes in C. elegans (Arnadottir et al., 2011), the peptide‐gated channel Phe‐Met‐Arg‐Phe‐amide (FMRFa)‐activated Na+ channel (FaNaC) of snails (Lingueglia et al., 2006), the mammalian bile acid‐sensitive ion channel (BASIC) (Wiemuth et al., 2014) and Drosophila ENaC/DEG channels such as pickpocket, ripped pocket and others (Adams et al., 1998) (Figure 1A). Amino acid sequence identity between different ENaC/DEG subfamilies is 15–20%.
Figure 1.
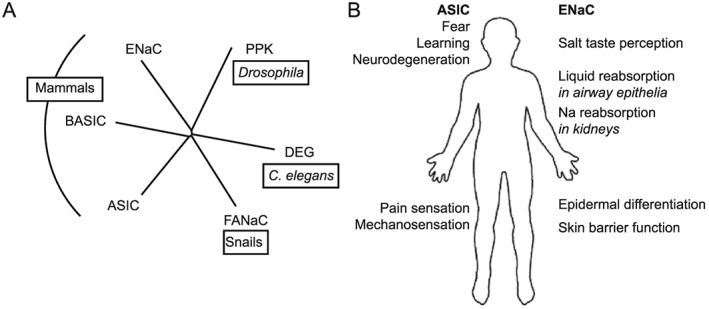
Relations and roles of ENaC and ASICs. (A) Phylogenetic tree of the ENaC/degenerin (DEG) family, showing besides ASIC and ENaC the subfamilies pickpocket (PPK), degenerin, the FMRFa‐activated channel FaNaC and the BASIC (also known as hINaC or BLINaC). (B) Illustration of the different physiological and pathological roles of ASICs and ENaC.
ASICs and FaNaC are expressed in the nervous system, DEGs are present in touch‐sensitive neurons, BASIC shows highest expression in the brain, liver and intestine, and ENaC is found at highest levels in tight epithelia, while members of the Drosophila ENaC/DEG are probably expressed in many different tissues. FaNaC is an excitatory ion channel of the nervous system of snails, DEGs are critical for C. elegans touch sensation, and members of the Drosophila ENaC/DEG may also be involved in touch sensation, among other roles. BASIC is activated by bile acids; however, its physiological role is currently not known. ASICs are involved in fear behaviours, learning and memory functions, and pain sensation (Figure 1B). They contribute to neurodegeneration after ischaemic stroke (reviewed in (Wemmie et al., 2013; Kellenberger and Schild, 2015). There is also evidence for an involvement of ASICs in mechanosensation (Chen and Wong, 2013). ENaC plays a well‐established role in Na+ reabsorption in the distal nephron, distal colon and airway epithelia. In addition, it is involved in salt taste perception, epidermal differentiation and skin barrier function (Figure 1B).
Crystal structures of chicken ASIC1, which in some studies was co‐crystallized with the ASIC toxins psalmotoxin 1 (PcTx1) or MIT‐toxin, showed a channel made up of three subunits (Jasti et al., 2007; Gonzales et al., 2009; Baconguis and Gouaux, 2012; Dawson et al., 2012; Baconguis et al., 2014). The shape of each subunit was compared with that of a hand holding a small ball, and accordingly, the different extracellular domains were labelled palm, knuckle, finger, thumb and β‐ball (Figure 2A). The palm domain is the extracellular continuation of the transmembrane segments and forms a β‐strand‐rich scaffold of the extracellular channel part. The knuckle and β‐ball are located on top and along the upper half of the palm, respectively (Figure 2A and B). The finger and thumb are oriented towards the outside of the protein. Details of the crystal structures and their differences have been recently discussed (Grunder and Augustinowski, 2012; Kellenberger and Grutter, 2015; Kellenberger and Schild, 2015). Structure–function studies indicate that the ENaC and ASIC ectodomains play important roles in controlling the opening of the channel pore (reviewed in Kellenberger and Schild, 2015). The sequence homology suggests that all ENaC/DEG members share the same subunit topology. Models of ENaC subunits have been constructed based on the ASIC crystal structures. The highest homology of the ectodomain between ASICs and ENaC is found in the palm and the β‐ball (Kashlan and Kleyman, 2011; Kashlan et al., 2011). The predicted secondary structures of most other ENaC domains match the ASIC structure moderately well except for the finger that has the lowest homology and contains a ~80 amino acid insertion in ENaC (Figure 2C).
Figure 2.
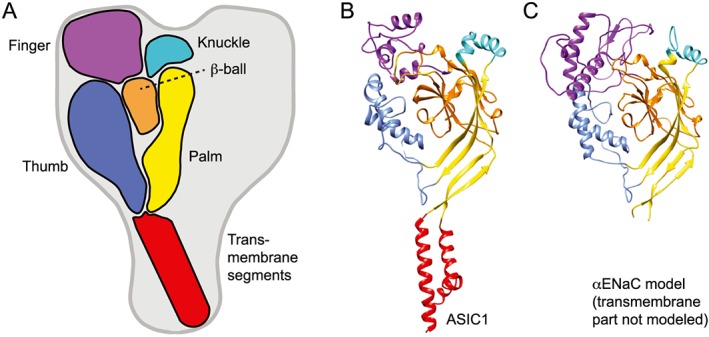
ASIC and ENaC subunit organization. (A) Schematic view of one subunit in the context of the trimeric ASIC, highlighting the different domains finger (purple), knuckle (turquoise), β‐ball (orange), palm (yellow), thumb (blue) and transmembrane domains (red). (B) Structure of an ASIC1 subunit based on the crystal structure obtained from chicken ASIC1 binding Mit‐Tx (Baconguis et al., 2014). The domains are coloured as in (A). (C) Model of the extracellular part of αENaC (Kashlan and Kleyman, 2011). Colouring as in (A); the transmembrane part was not modeled.
Stoichiometry predictions of ENaC and ASIC that were based on functional and biochemical data indicated that these channels are tetramers (Firsov et al., 1998; Kosari et al., 1998; Anantharam and Palmer, 2007; van Bemmelen et al., 2015). In contrast, all crystal structures describe ASIC as a trimer. In a recent study, ASIC1a and ASIC2a containing fluorescently labelled subunits were expressed in Xenopus oocytes, and the number of bleaching steps of plasma membrane‐resident channels was counted to determine the subunit stoichiometry of these functional channels at the cell surface (Bartoi et al., 2014). This analysis indicated that functional ASICs at the cell surface are trimers. Similarly, an earlier study using fluorescence ratio measurements had concluded that ENaC is a trimer (Staruschenko et al., 2005). In conclusion, the available data strongly support a trimeric structure of ASICs. Most likely, the subunit stoichiometry is conserved within the ENaC/DEG family.
Acid‐sensing ion channels
Basic information on ASICs
Physiological and pathological roles of ASICs
ASIC1a, ‐2a and ‐2b are widely expressed in the CNS. ASIC4, for which no channel activity has yet been demonstrated, shows a more dispersed expression in the CNS (Lin et al., 2015); all ASIC subunits except ASIC4 are found in the adult peripheral nervous system (PNS; reviewed in Wemmie et al., 2013; Kellenberger and Schild, 2015). Because ASICs are Na+‐selective ion channels, their activation is expected to induce a neuronal depolarization. Indeed, activation of ASICs in neurons of the CNS and PNS induces membrane depolarization and generation of action potentials (Figure 3A) (Deval et al., 2003; Vukicevic and Kellenberger, 2004; Poirot et al., 2006). ASIC1a shows, in addition to its Na+ permeability, a small permeability for Ca2 + that is probably important for some of its roles (Waldmann et al., 1997b; Bassler et al., 2001; Boillat et al., 2014).
Figure 3.
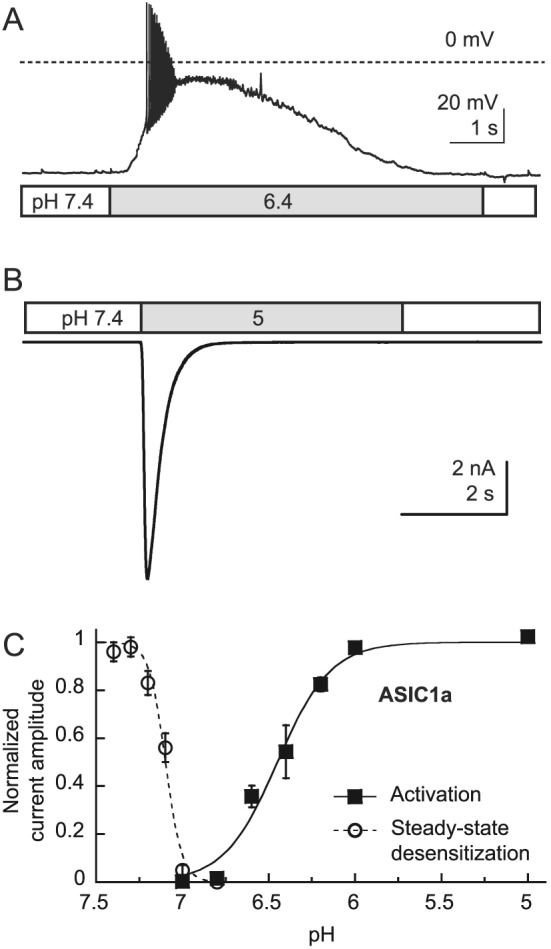
Functional properties of ASIC. (A) Action potential induction by extracellular acidification to pH 6.4, mediated by ASICs, measured by whole‐cell current‐clamp from a mouse hippocampal neuron. (B) A pH 5‐induced current recorded in whole‐cell voltage clamp to −60 mV from a Chinese hamster ovary cell stably transfected with ASIC1a. (C) The pH‐dependence of steady‐state desensitization and of activation of ASIC1a. In steady‐state desensitization experiments, cells were exposed for 55 s to the indicated conditioning pH, and a stimulation pH 6 solution was applied for 5 s to open the not yet desensitized channels. The normalized current response at pH 6 is plotted as a function of the conditioning pH. For ASIC1a activation, the cells were perfused by a pH 7.4 solution, and once per minute, this solution was changed to one of acidic pH to open the channels. The normalized current is plotted as a function of the stimulation pH.
Synaptic signalling involves an acidification of the synaptic cleft, which can activate ASICs. Disruption of the ASIC1a‐encoding gene in mice eliminated most of the ASIC currents of CNS neurons, impaired long‐term potentiation (LTP) in the hippocampus and induced a mild deficit in spatial learning (Wemmie et al., 2002). Recent studies have added important new information on the role of ASICs in synaptic functions (see below). ASIC1a is highly expressed in the amygdala, and there is strong evidence that ASIC1a of the amygdala contributes to fear behaviour (Wemmie et al., 2013). Disruption of ASIC1a or inhibition of ASIC1a activity in the brain by PcTx1‐containing venom reduced the infarct volume in an experimental stroke model by >50%, strongly suggesting that ASIC1a activation contributes to neurodegeneration in this situation (Xiong et al., 2004). Further studies showed that disruption of ASIC1a had a protective effect in several neurodegenerative diseases, including multiple sclerosis, Huntington's and Parkinson's disease (reviewed in Wemmie et al., 2013). The extracellular pH (pHe) is lowered in inflammation and ischaemia, which both involve pain. There is also strong evidence for a role of sensory neuron ASICs in pain sensation. ASICs belong to the same ion channel family as the C. elegans DEGs that form the channel parts of mechanotransduction complexes. Several ASIC isoforms are expressed in mechanosensory structures and may have similar functions as DEGs. ASIC knockout mice show defects in mechanosensation in many different tissues, indicating that ASIC mechanosensation is involved in touch and pain sensation, baroreceptor function, blood volume control, digestive functions and possibly hearing (reviewed in Chen and Wong, 2013; Omerbasic et al., 2015).
Currently, ASIC inhibition is not used clinically. However, pharmacological inhibition of ASICs is expected to be beneficial in several human disorders. ASIC inhibitors may be used as anxiolytic and analgesic drugs, and to limit neurodegeneration after ischaemic stroke. Several ASIC inhibitors are currently in preclinical trials and clinical phase I trials, mostly in the context of pain (https://clinicaltrials.gov).
ASIC function and regulation
Exposure of ASICs to an acidic pHe leads to rapid channel opening, followed by a slower entry into a non‐conducting desensitized state. This results in a transient current (Figure 3B). In some ASIC subtypes, such as ASIC3 and some heteromeric ASICs, desensitization is not complete, and a small sustained current persists after the initial peak (Lingueglia et al., 1997; Waldmann et al., 1997a; Yagi et al., 2006). Desensitization can also occur without apparent channel opening (termed steady‐state desensitization in this case) during moderate lowering of the pH and can limit the availability of ASICs for opening. The pH dependence of these two processes, channel activation and steady‐state desensitization, is illustrated by the example of ASIC1a in Figure 3C. The steady‐state desensitization occurs at pH < 7.4, with a mid‐point, termed pH of half‐maximal desensitization of ~7.15. ASIC1a opening occurs at pH < 7 and is characterized by a pH of half‐maximal activation (pH50) of ~6.5. These parameters determine the open probability of ASICs under given pH conditions. As shown in Table 1, ASIC1a and ASIC3 are the most sensitive ASICs and are activated by acidification to pH values only slightly below pH 7. In contrast, ASIC2a needs much more acidic pH (<5.5) for activation. ASIC1a is the ASIC isoform that is most sensitive to desensitization. It shows partial desensitization already at pH slightly below 7.4 (Figure 3C and Table 1).
Table 1.
Biophysical properties of ASICs
| pH50 activation | pHD50 steady‐state desensitization | Desensitization time constant (s) | Most important sites of expression | |
|---|---|---|---|---|
| ASIC1a | 6.2–6.6 | ~7.2 | ~0.4 | CNS, PNS |
| ASIC1b | 5.9–6.3 | ~6.7 | ~0.9 | PNS |
| ASIC2a | 4.0–4.9 | ~5.6 | ~1.4 | CNS, PNS |
| ASIC3 | 6.4–6.7 | ~7.1 | ~0.3 | PNS |
The desensitization time constant is indicated for stimulation pH close to the pH50. These data are from different articles and reviews (Hesselager et al., 2004; Poirot et al., 2004; Blanchard and Kellenberger, 2011; Alijevic and Kellenberger, 2012; Wemmie et al., 2013). ASIC4, not indicated in the table, has not been shown so far to form functional channels and is mainly expressed in the CNS (Lin et al., 2015).
For several ASICs it has been shown that with fast solution changes the opening time constant at a pH that fully activates the channels is of the order of ~10 ms (Bassler et al., 2001). The kinetics of current desensitization depend on the subunit composition, as shown in Table 1. Besides protons, there are only very few ASIC activators known so far. The small synthetic molecule 2‐guanidine‐4‐methylquinazoline (GMQ) activates ASIC3 at pH 7.4 and inhibits other ASIC subtypes by changing the pH dependence of activation and of steady‐state desensitization (EC50 ~ 1 mM; Yu et al., 2010; Alijevic and Kellenberger, 2012). Interestingly, endogenous arginine metabolites that contain a guanidinium group, as does GMQ, were found to have similar effects as GMQ (EC50 > 3 mM; Li et al., 2011). A recent study showed activation of ASIC3 at pH 7.4 by administration of the two lipids lysophosphatidylcholine (EC50 ~ 4.3 μM) and arachidonic acid (used at 10 μM), which are both present in inflammatory exudates (Marra et al., 2016). The authors showed that this activation is due to a shift in the pH‐dependence of activation. Administration of this combination of lipids induced pain that was prevented by ASIC3 inhibitors in rats and reduced in ASIC3 knockout mice.
ASIC activity is regulated by many different modulators, such as divalent and polyvalent cations, neuropeptides, arachidonic acid, protein kinases and proteases, as summarized in Table 2. Many of these regulatory mechanisms are likely to be active under physiological conditions. Divalent and polyvalent cations such as Ca2 +, Mg2 + and spermine appear to compete with protons for binding sites [with apparent affinities of the order of millimolar concentrations (Babini et al., 2002)], since an increase in their concentration shifts the ASIC pH‐dependence towards more acidic pH values. The peptide FMRFa activates the ENaC/DEG family member FaNaC (Lingueglia et al., 1995). FMRFa and related mammalian neuropeptides slow the desensitization kinetics of ASIC1 and ASIC3 and induce a sustained current, with EC50 values that are for most peptides in the order of 10–50 μM (Askwith et al., 2000; Vick and Askwith, 2015). Arachidonic acid, whose tissue concentration is increased in ischaemia and inflammation, was shown to potentiate ASIC currents in the CNS and the PNS at concentrations of 5–10 μM by mechanisms that include an alkaline shift in the pH‐dependence of activation (Allen and Attwell, 2002; Smith et al., 2007; Deval et al., 2008). Trypsin (at ≥2 μg·mL−1) was shown to shift the ASIC1a pH‐dependence of activation and steady‐state desensitization to more acidic values by a cleavage in the ectodomain. This led to reduced acidification‐induced ASIC currents and neuronal signalling at a physiological conditioning pH 7.4 and to increased activity if the conditioning pH was slightly reduced (Vukicevic and Kellenberger, 2004; Vukicevic et al., 2006). The serine protease tissue kallikrein, at a concentration of 3 μM, was also shown to cleave and regulate ASIC1a (Su et al., 2011). Finally, ASIC function is modulated by interaction with other proteins, as discussed in Wemmie et al. (2006) and updated in Kellenberger and Schild (2015).
Table 2.
ASIC modulators
| Modulator class | Important example | Effect | Site of action | Reference |
|---|---|---|---|---|
| Divalent cations | Ca2 +, Mg2 +, Ba2 + | Acidic shift of pH dependence | Not known | (Babini et al., 2002; Immke and McCleskey, 2003) |
| Divalent cations | Ca2+ | Pore block | Acidic residues in pore entry of ASIC1a | (Paukert et al., 2004) |
| Neuropeptides | Dynorphin, FMRFa | Acidic shift of pH dependence of steady‐state desensitization, slowing of desensitization and induction of sustained current | Not known | (Askwith et al., 2000; Vick and Askwith, 2015) |
| Proteases | Tissue kallikrein, trypsin | Acidic shift of pH dependence | Cleavage in finger domain | (Poirot et al., 2004; Vukicevic et al., 2006; Su et al., 2011) |
| Protein kinases | PKA, PKC | Changes expression and function | Intracellular | Rev. in (Kellenberger and Schild, 2015; Wemmie et al., 2013) |
| Other | Arachidonic acid | Increase of peak current amplitude | Not known | (Allen and Attwell, 2002; Smith et al., 2007) |
| Other | Nitric oxide | Increase of peak current amplitude | Extracellular | (Cadiou et al., 2007) |
ASIC pharmacology
As mentioned above, ASIC inhibition is currently not used clinically. The present compounds except for amiloride, which is clinically used as ENaC inhibitor (IC50 = 100–200 nM), have been characterized in cell systems and in part also in animal models. An interesting recent review of ASIC pharmacology is provided by Baron and Lingueglia (2015). Amiloride has a low potency (EC50 of 10–100 μM) and selectivity on ASIC peak currents and does not inhibit the sustained ASIC currents. Amiloride binds into the pore of ENaC and ASICs (Schild et al., 1997; Adams et al., 1999; Alijevic and Kellenberger, 2012). The site of action of other small molecule inhibitors on ASICs is not known. Amiloride derivatives modified at the five position of the pyrazine ring by hydrophobic groups increased the potency for ASIC3 inhibition by up to 100‐fold (Kuduk et al., 2009). Nafamostat mesylate, an anti‐inflammatory agent and protease inhibitor, contains a guanidinium moiety as do amiloride and GMQ and was shown to inhibit ASIC currents, including the sustained current of ASIC3, with IC50 values of 2–70 μM (Ugawa et al., 2007). The chemically unrelated compound A‐317 567 inhibits peak and sustained currents of neuronal and recombinant ASICs with IC50 values between 2 and 30 μM (Dube et al., 2005). The development of A‐317 567 derivatives yielded substances with a higher affinity for ASICs, which, however, lost some of their selectivity (IC50 on ASIC3 of 400–500 nM, and for other neurotransmitter receptors of <10 μM) (Kuduk et al., 2010).
Inflammation increases ASIC mRNA expression, and it was shown that several non‐steroidal anti‐inflammatory drugs at doses close to those used in clinics prevent or suppress this RNA overexpression (Voilley et al., 2001). These drugs also inhibit ASIC currents; however, with potencies that are orders of magnitude lower than that on cyclooxygenases (Voilley et al., 2001). Several antiprotozoal diarylamidines inhibit ASICs with IC50 values of 0.3–38 μM (Chen et al., 2010). A recent screening of a fragment library followed by optimization led to ASIC3‐inhibiting 2‐aminopyridine derivatives with an IC50 of ~3 μM (Wolkenberg et al., 2011).
Venom toxins acting on ASICs have been used to elucidate some of the physiological and pathological roles of ASICs (Wemmie et al., 2013). In addition, complexes of ASIC1 with toxins were used to determine the crystal structure of ASIC1 in the likely open conformation. The most important ASIC toxins are the gating modifiers PcTx1 of the spider Psalmopoeus cambridgei and the Mambalgins of the black mamba, the ASIC3 inhibitor APETx2 of the sea anemone Anthopleura elegantissima and the activating Mit‐toxin of the Texas coral snake that generates a sustained ASIC opening at pH 7.4 (reviewed in Baron et al., 2013). MIT‐toxin and Mambalgins target several ASIC subtypes, while PcTx1 is selective for ASIC1a homomers and ASIC1a/2b heteromers, and APETx2 for ASIC3‐containing channels. IC50 and EC50 values of these toxins range from nanomolar to micromolar concentrations. Some of them have a high affinity for selected targets (IC50 of PcTx1 for ASIC1a: 0.4–13 nM, EC50 of Mit‐toxin for ASIC1a: 9 nM; Baron et al., 2013) and may be used in binding studies after labelling, as shown for PcTx1 (Salinas et al., 2006). The ASIC toxins have so far not been shown to target other channels besides ASICs, with the exception of APETx2, which also inhibits some voltage‐gated Na+ channel isoforms (IC50 in the range of nanomolar to low micromolar concentrations) (Blanchard et al., 2012; Peigneur et al., 2012). PcTx1 inhibits mammalian ASIC1a by an alkaline shift in the pH‐dependence of steady‐state desensitization (leading to complete desensitization at pH 7.4), while Mambalgin inhibition is due to an acidic shift in the pH‐dependence of activation. The mechanisms of action of the other ASIC toxins are currently not known. Co‐crystallization showed that PcTx1 binds to the acidic pocket of ASIC1 and that the much larger Mit‐toxin binds to the wrist, palm and thumb domains, without however reaching into the acidic pocket (Baconguis and Gouaux, 2012; Dawson et al., 2012; Baconguis et al., 2014). Site‐directed mutagenesis indicated that Mambalgins also bind to the acidic pocket (Salinas et al., 2014; Schroeder et al., 2014).
pH changes in the brain and role of ASICs in synaptic signalling in the CNS
Physiological extracellular pH changes during neuronal activity
The resting pHe value in the interstitial fluid of brain tissues can vary between 7.15 and 7.33 depending on the brain area and is generally about 0.1–0.2 pH units more acidic than the blood pH (Mutch and Hansen, 1984; Syková and Svoboda, 1990). Because membrane depolarization, ion transport and metabolic activity affect the pHe, changes in pHe are commonly observed during neuronal activity and in changed metabolic states, such as ischaemia (Kraig et al., 1983; Siesjö et al., 1985; Krishtal et al., 1987; Dmitriev and Mangel, 2001; Chesler, 2003). Information about the variations of pHe in the CNS of mammals comes mainly from studies with anaesthetised rodents and in vitro preparations in which the pHe was recorded using optical techniques or pH‐sensitive microelectrodes (Chesler, 2003). Upon repetitive electrical stimulation, the neuronal activity of the mammalian CNS produces a general pattern of pHe changes containing three phases (Chesler, 1990; Chesler, 2003; Makani and Chesler, 2010). An initial short (<200 ms) transient alkaline shift of typically 0.01–0.2 pH units, occurring within tens of milliseconds after the beginning of the stimulus, is followed by a long‐lasting, slowly developing acidosis that reaches a maximal acidic shift of 0.1–0.25 pH units 20–60 s after the beginning of the repetitive stimulation. In the third phase, the pH returns to the initial value. The brain pH is buffered by the bicarbonate‐carbon dioxide buffer. The buffering capacity of the interstitial fluid depends therefore on the bicarbonate and CO2 concentration and codetermines the magnitude of pHe changes (Du et al., 2014; Highstein et al., 2014). The initial transient alkalinization mentioned above has been reported in the hippocampus by several studies and occurs in the same time frame as do excitatory postsynaptic currents. It is therefore fast enough to modulate fast synaptic transmission (Krishtal et al., 1987; Chen and Chesler, 1992; Gottfried and Chesler, 1996; Chen and Chesler, 2015). The alkaline shift is reduced by pharmacological blockade of various ion channels involved in neurotransmission, pointing to a direct association between the alkalinization and neuronal activity (Smith and Chesler, 1999). Figure 4 illustrates the different mechanisms leading to pH changes in the synapse. Inhibition of the plasma membrane Ca2 +‐ATPase prevented the transient alkaline shift in hippocampal neurons effectively, suggesting that the alkaline shift is generated by the activity of this transporter that exchanges internal Ca2 + for external H+ (Kreitzer et al., 2007; Makani and Chesler, 2010). The initial alkaline shift is small or even inexistent in some brain regions (Yamamoto et al., 1992; Venton et al., 2003). Several observations indicate that the acidic change that follows the initial alkalinization is mostly due to the activity of glial cells (Jendelová and Syková, 1991; Chesler, 2003). It was suggested that the Na+‐HCO3 − cotransporter (NBC) contributes to the lowering of the pHe by the glia. Activation of NBC leads to transport of Na+ and HCO3 − into the glial cell and thereby to extracellular acidification (Deitmer, 2002) (Figure 4).
Figure 4.
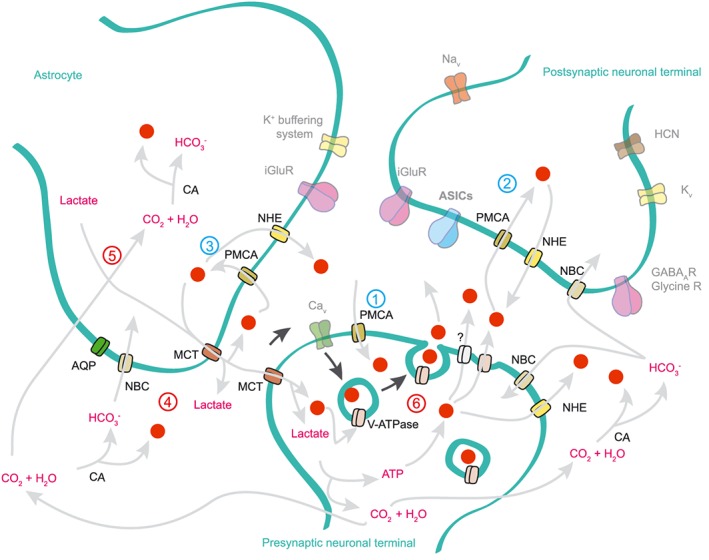
Extracellular pH changes during neurotransmission in the CNS. Illustration based on experiments performed in hippocampal, photoreceptor, amygdala and calyx synapses, showing a synapse between a pre‐ and postsynaptic neuronal terminal and an astrocyte. For simplification, only the transport of protons and of HCO3 − is shown in the figure (grey arrows; protons are red dots). The black arrows indicate the activation of synaptic vesicle release by calcium entry. The numbers 1–3 (coloured in blue) represent potential mechanisms for the initial alkaline shift recorded in hippocampal synapses: neurotransmission stimulates calcium signalling in the perisynaptic cells. As a consequence, the mechanisms of calcium buffering system are stimulated. One of these mechanisms involves the PMCA, which pumps the accumulated intracellular calcium ions out of the cell in exchange for external protons, leading to extracellular alkalinization. The numbers 4–6 (coloured in red) represent potential mechanisms underlying the acidic shift. The glial NBC causes a gradual extracellular acidification (4). Intense neurotransmission increases astrocytic energy demand, resulting in lactate and CO2 production (5). For clarity, only the lactate shuttle into presynaptic terminals is shown. Lactate is transported outside the astrocyte by the H+‐coupled monocarboxylate transporter (MCT) and CO2 can freely diffuse, also leading to extracellular acidification at the synaptic cleft. Fast acidifications at the synaptic cleft may also occur as a result of synaptic vesicle release and also by non‐vesicular release involving an undefined transporter (6).
Protons and ASICs, a neurotransmitter‐receptor pair in the CNS?
In this section, we present the experimental evidence for a role of protons as a neurotransmitter. We discuss then how ASICs contribute to synaptic signalling and LTP, thereby affecting learning and fear sensation. Finally, we review different observations, suggesting that ASICs can strongly influence glutamate receptor function.
Indirect evidence for synaptic cleft acidification during electrical signalling has been provided by studies that measured the inhibition of presynaptic pH‐sensitive voltage‐gated Ca2 + channel (Cav) currents and estimated the acidification in the synaptic cleft to 0.2–0.6 pH units (DeVries, 2001; Palmer et al., 2003; Vessey et al., 2005). Recent studies with fluorescent pH indicators showed that presynaptic stimulation led to acidification of the synaptic cleft in synapses formed by vestibular hair cells and the calyx nerve terminal of the turtle Trachemys scripta elegans (Highstein et al., 2014) and in synapses of mouse lateral amygdala (Du et al., 2014). These pH patterns differed from the alkalinization–acidification observed in earlier studies, probably due to different locations of the studied synapses or due to the configuration of the measurements. Several mechanisms may contribute to the acidification of the synaptic cleft. The synaptic vesicles containing neurotransmitters that are continuously replenished with protons by the H+‐ATPase pumps are an important source of protons in synapses (Beyenbach and Wieczorek, 2006; Vavassori and Mayer, 2014). There is evidence that the pH of cholinergic, glutamatergic and GABAergic synaptic vesicles can reach values as acidic as pH ~5.5 (Michaelson and Angel, 1980; Fuldner and Stadler, 1982; Miesenbock et al., 1998; DeVries, 2001; Palmer et al., 2003; Dietrich and Morad, 2010). The acidification of the synaptic cleft observed in the mouse lateral amygdala had only a small amplitude (<0.1 pH unit), and a time constant of 0.5–1 s, thus much slower than the release of synaptic vesicles (Du et al., 2014). Highstein et al. also observed an acidification time course of ~0.5 s (Highstein et al., 2014). The slow kinetics may point to acid–base transporters as the source of this proton release into the synaptic cleft. Consistent with this hypothesis, transient synaptic cleft acidification in cultured rat cerebellar granule cells during GABAergic transmission was found to be at least in part due to the activity of the Na+/H+ exchanger (Dietrich and Morad, 2010).
The activity of many neuronal ion channels is pH‐dependent (Table 3), suggesting that the pHe changes during neuronal activity modulate ion channel function. In general, alkaline pHe favours inward currents, thus enhancing excitability, while acidic pHe depresses excitability in many circumstances (Chesler, 2003) and can be considered as negative feedback because it is caused by neuronal stimulation. ASICs in contrast are activated by extracellular acidification. Administration of specific ASIC1a antagonists or disruption of the ASIC1a gene eliminates the majority of the acid‐induced currents in CNS neurons (Wemmie et al., 2013; Wu et al., 2013). This demonstrates that the ASIC1a homomers and ASIC1a‐containing heteromers are the principal sensors of rapid extracellular acidification in the brain. ASIC1a, ‐2a and ‐2b are widely expressed in the CNS (reiewed in Wemmie et al., 2013; Kellenberger and Schild, 2015). Localization by immunohistochemistry studies, evidence for the interaction with the postsynaptic proteins PICK1 and AKAP150, and co‐localization with PSD‐95 in spines together indicate that ASIC1a has a somatodendritic distribution (Zha, 2013) and is well situated for the detection of rapid synaptic pH changes. Several recent studies expressed light‐activated proton pumps in neurons or astrocytes. Light‐induced activation of these pumps led to extracellular acidification and ASIC activation (Li et al., 2014; Zeng et al., 2015; Ferenczi et al., 2016).
Table 3.
Extracellular pH‐sensitive ion channels expressed in CNS and PNS neurons
| Inhibition by pHe (pH50Inh.) | Ion channels | Mechanisms / Comments |
|---|---|---|
| pH 8.6 (PNS) | (1) TASK2 | Decrease of K+ pore occupancy and open probability |
| pH 7.4–pH 7.3 (CNS, PNS) | (2) TREK1, (3) Kv1.4, (4) TASK1, (5) TRAAK, (6) KCNQ2/3 | (2) C‐type inactivation enhanced, (4) open probability reduced, (6) Imax reduced, depolarizing shift of the activation |
| pH 7.3–pH 7 (CNS, PNS) | (7) NMDAR, (8) GABAAR, (9) voltage‐gated calcium channels (Cav), (10) glycine receptors | (7) Reduced burst duration, decreased opening frequency, (8), (10) subunit‐ and agonist‐dependent, (9) Imax reduced, depolarizing shift of the activation |
| Mixed pH effects (CNS, PNS) | (11) TWIK1 | From pH 7.5 to pH 6.5: current enhanced. Below pH 6.5: current inhibited. |
| pH 6.7–pH 6.5 (CNS, PNS) | (12) Kir2.3, (13) TASK3, (14) TRPM2, | (12), (13) conductance and open probability reduced |
| pH 6.3–pH 6.2 (CNS, PNS) | (15) AMPAR, (16) Ca2 +‐sensing non‐selective cation channels, (17) Kv1.5 | (15) enhancing desensitization, (17) facilitation of a non‐conducting state |
| pH 6–pH 4.6 (CNS, PNS) | (18) Kainate receptors, (19) Kv1.3, (20) IA, IK, (21) INa, (22) Nav1.2 | (18) Subunit dependent, (20) depolarizing shift of the inactivation, (19), (21) Imax reduced, (21), (22) depolarizing shift of the activation |
| Stimulation by pHe (pH50Act.) | Ion channels | Mechanisms / Comments |
|---|---|---|
| pH 7.3 (CNS, PNS) | (23) TREK2, (24) P2X2 | (24) Sensitization to activation by ATP |
| pH 6.5–pH 6 (CNS, PNS) | (25) ASIC1a, (26) ASIC1a/2b, (27) ASIC3, (28) δβγENaC | – |
| pH 5.8–pH 3 (CNS, PNS) | (29) ASIC1a/2a, (30) ASIC2a, (31) ASIC2a/2b | – |
| Mixed pH effects (CNS, PNS) | (32) BK and (33) Kv1.3 associated with ASICs | ~50% inhibition of BK and Kv1.3 currents at pH 7.4. Removal of this inhibition at acidic pH. |
| Mixed pH effects (CNS, PNS) | (34) P2X3 | The ATP concentration‐dependence of the current is shifted to higher concentrations and its Imax is increased |
| (35), (36)~pH 5.4 for proton activation (35) pH 7–pH 6 for proton sensitization (CNS, PNS) | (35) TRPV1, (36) TRPV4 | (35), (36) Direct activation by strong acidosis (< pH 6). (35) Sensitization to other stimuli such as capsaicin and temperature by mild acidosis (pH 7–6) |
| pH 6.5‐pH 4 (CNS, PNS) | (37) nAChRs α3/β4 (38) HCN, Ih | (37) Increase of the agonist‐induced current and changed kinetics. Agonist‐ and subunit‐ dependent. (38) Activation of Ih current |
The ion channels are classified based on their pH sensitivity. Table citations: (1), (2), (4), (5), (13), (23): (Lesage and Barhanin, 2011; Ehling et al., 2015) (3): (Claydon et al., 2000). (4), (13): (Plant, 2012). (6): (Prole et al., 2003). (7), (15), (18): (Traynelis and Cull‐Candy, 1990; Traynelis and Cull‐Candy, 1991). (8) (Zhai et al., 1998; Wilkins et al., 2005). (9), (20), (21) (Tombaugh and Somjen, 1996). (10) (Chen et al., 2004a). (11) (12) (Zhu et al., 1999). (14) (Starkus et al., 2010). (15) (Lei et al., 2001). (16) (Chu et al., 2003). (17) (Kehl et al., 2002). (18) (Mott et al., 2003). (19) (Somodi et al., 2004; Petroff et al., 2008). (21) (Daumas and Andersen, 1993). (22) (Vilin et al., 2012). (24) (King et al., 1997). (25), (30) (Alijevic and Kellenberger, 2012) (26) (Sherwood et al., 2011). (27) (Waldmann et al., 1997a). (28) (Ji and Benos, 2004; Yamamura et al., 2004). (29) (Bartoi et al., 2014). (31) (Ugawa et al., 2003). (32), (33) (Petroff et al., 2008). (34) (Gerevich et al., 2007. (35) Tominaga et al., 1998). (36), (Suzuki et al., 2003). (37) (Abdrakhmanova et al., 2002; Abdrakhmanova et al., 2004). (38) (Cichy et al., 2015). BK, big calcium‐activated K+ channel; HCN, hyperpolarization‐activated cyclic nucleotide‐gated channel; IA, A‐type current of rapid inactivating K+ channels; Ih, current produced by HCN channels; IK, delayed rectifier K+ current; Imax, maximal current amplitude; Kir, inward rectifier K+ channel; Kv, voltage‐gated K+ channel; nAChR, nicotinic acetylcholine receptor; Nav, voltage‐gated Na+ channel; TASK, two‐pore domain K+ channel; TRAAK, TWIK‐related arachidonic acid‐stimulated K+ channel; TREK, TWIK‐related K+ channel; TRPM, transient receptor potential cation channel, subfamily M; TRPV, transient receptor potential cation channel subfamily V; TWIK, tandem of P‐domain in a weak inwardly rectifying K+ channel.
In the lateral amygdala, presynaptic stimulation activated postsynaptic ASIC currents. Perfusion of glutamate receptor blockers inhibited 95% of the amplitude of the observed excitatory postsynaptic currents. The remaining 5% of the excitatory postsynaptic current amplitude were mediated by ASICs, because this current was absent in the presence of amiloride or if the ASIC1a gene was deleted (Du et al., 2014). A similar situation with a contribution of ASICs to 5% of the excitatory postsynaptic current amplitude was also found in nucleus accumbens (Kreple et al., 2014). High‐frequency stimulation induced LTP of EPSPs in the hippocampus of wild type but not ASIC1a(−/−) mice (Wemmie et al., 2002). The ASIC1a(−/−) mice displayed mildly defective spatial learning and eyeblink conditioning, consistent with decreased LTP. A recent study confirmed these initial observations by showing that pharmacological blockade of ASIC1a in hippocampal synapses impaired LTP (Quintana et al., 2015). In a different model of ASIC1a knockout mice, however, in which ASIC1a was deleted at early embryonic stages in contrast to the classical knockout used in the study by Wemmie et al., normal LTP at CA3‐CA1 synapses was observed (Wu et al., 2013). The origin of this discrepancy may involve roles of ASIC in synapse development, or heterogeneity of ASIC expression in synaptic connections in the hippocampus. A study that used a multi‐electrode array system to record the synaptic plasticity within different neuronal populations in brain slices of the CA1 hippocampal area from WT and ASIC1a KO mice confirmed the involvement of ASIC1a in LTP at many synapses but showed also that at some synapses LTP induction was independent of ASICs (Liu et al., 2016). The same study also showed that long‐term depression, another form of synaptic plasticity, does not require ASIC1a at these synapses.
ASIC1a(−/−) mice have reduced innate fear and show deficits in conditioned fear behaviour (Wemmie et al., 2013). The fear‐related behaviour is in many cases correlated with CO2 and acid sensing and was restored in ASIC1a(−/−) mice by injection of a viral vector in the basolateral nuclei of the amygdala that restored ASIC1a expression locally (Coryell et al., 2009; Ziemann et al., 2009). Two recent studies showed that ASIC1a is critical for LTP at the synapses of the fear circuitry between the cortex and the basolateral nuclei of the amygdala (Du et al., 2014; Chiang et al., 2015). The study by Chiang et al. investigated LTP at various synapses of amygdala neurons and found that the extent of LTP at different synapses correlated with the ASIC current density in postsynaptic neurons. Cell type‐specific deletion of ASIC1a showed that ASIC‐dependent LTP is required at several amygdala synapses for fear learning. ASIC4 does not form functional channels but is known to down‐regulate the expression of other ASIC subunits (Donier et al., 2008). ASIC1a expression is therefore expected to be up‐regulated in ASIC4−/− mice, and it was indeed shown that ASIC4 knockout mice have an increased freezing response (Lin et al., 2015). A recent study with rats suggests a species difference with regard to the role of ASICs in the fear circuitry. This study showed that ASIC1a activation reduces anxiety in rats, by enhancing inhibition in the basolateral amygdala (Pidoplichko et al., 2014). The reason for this opposite role of ASICs in fear behaviour of mice and rats is currently not understood, and the rat data rely so far on one single study. Given the complexity of the expression and role of ASIC1a in the mouse fear circuitry (Chiang et al., 2015), it seems plausible that the ASIC expression pattern in the fear circuitry may be different between mice and rats. The role of ASICs in fear expressions of humans is currently not known.
Several studies have shown that ASICs interact functionally with glutamate receptors in synaptic signalling and that a functional ASIC is required for LTP, as discussed above (Wemmie et al., 2002; Du et al., 2014; Kreple et al., 2014; Quintana et al., 2015; Liu et al., 2016). The initial LTP study in hippocampus suggested that activation of postsynaptic ASICs removes the Mg2 + block of NMDA receptors, because LTP was only disrupted in ASIC1a(−/−) mice in physiological extracellular Mg2 + concentrations, but was normal at low Mg2 + concentrations (Wemmie et al., 2002). This does not, however, explain the more recent observations in the amygdala, the nucleus accumbens and in hippocampal cultures after oxygen–glucose deprivation. In the amygdala, the absence of ASIC1a decreased the EPSC amplitude only slightly, but markedly impaired the LTP (Du et al., 2014). Similarly, the presence or absence of ASIC1a strongly influenced glutamate receptor function in the nucleus accumbens (Kreple et al., 2014). The mechanism of this functional interaction of ASIC1a with glutamate receptors is not understood. An earlier study had shown that during ischaemia, NMDA receptor activity leads to phosphorylation of ASIC1a by CaMKII that enhances ASIC currents and leads to ischaemic cell death (Gao et al., 2005). There are also indications that the presence of ASICs can influence the density of dendritic spines and the glutamate receptor composition (Zha et al., 2006; Kreple et al., 2014). Quintana et al. have shown that apart from influencing NMDA receptors, ASIC1a can induce a special form of LTP in the ischaemic hippocampus via AMPA receptors (Quintana et al., 2015). The AMPA receptors show a high degree of post‐ischaemic plasticity that contributes to the excitotoxicity in the CA1 region by two mechanisms, anoxic LTP during the first hours, and an increased expression of Ca2 +‐permeable AMPA receptor types several hours later (Pellegrini‐Giampietro et al., 1992; Hsu and Huang, 1997). After an oxygen–glucose deprivation, anoxic LTP was observed in organotypic hippocampal slice cultures of WT mice, but was absent in slice cultures of ASIC1a(−/−) mice or after pharmacological blockade of ASIC1a (Quintana et al., 2015). Inhibition of ASIC1a or of Ca2 +‐permeable AMPA receptors was sufficient to protect neurons of the CA1 area, illustrating the important role of ASICs in neurodegeneration in this context.
In summary, pH changes occur in the CNS during neuronal and metabolic activity. The synaptic cleft is acidified upon presynaptic stimulation, leading to the activation of postsynaptic ASICs. In spite of their small contribution to the postsynaptic currents, ASICs play a critical role in synaptic signalling.
ASICs and other ion channels sense the extracellular pH in the PNS
Nociceptive fibres conduct signals from the periphery to the CNS that are induced by a variety of potential tissue‐damaging stimuli such as heat, pressure and chemicals. Many different substances that can modulate this signalling are released during tissue damage and inflammation. Although protons are the smallest modulators, they have dramatic effects on diverse properties of sensory neurons. Many different types of acid sensors are expressed by primary sensory neurons and especially by nociceptors, illustrating the vital importance of acid–base sensing and regulation. Tissue acidification occurs, for example, during ischaemia, inflammation, cancer pain and in muscle during exercise. It has been proposed that rapid localized pH changes may occur in the environment of peripheral nerves (Martin and Jain, 1994). In this section, we review firstly the roles of ASICs in pain sensation. We then describe the ASIC currents in PNS neurons. We discuss the apparent paradox that ASICs sense slowly developing and long‐lasting pH changes although their activity is mostly transient. Finally, we present observations showing that ASICs are not the only pH sensors in the PNS and that they cover together with other ion channels proton sensing over a wide pH range.
Role of ASICs in pain sensation
A large body of experimental data underlines the importance of sensory neuron ASICs in acid‐induced nociception (Deval and Lingueglia, 2015; Sluka and Gregory, 2015). Studies with human volunteers showed that the pain due to iontophoresis or injection of acid solutions in the skin was inhibited by amiloride and that it followed the pH‐dependence of ASIC1a and ASIC3 (Jones et al., 2004; Ugawa et al., 2002). Deletion of ASIC3 in mice reduced several forms of inflammation‐ and chronic acidification‐related pain (Sluka et al., 2009). Intrathecal administration of ASIC3‐specific siRNA to rats diminished pain behaviours induced by inflammation or wounding (Deval et al., 2008; Deval et al., 2011). The role of ASIC3 in pain sensation was further confirmed when it was shown that local injection of the ASIC3 activator GMQ induced pain behaviours that depended on the presence of ASIC3 (Yu et al., 2010). The use of toxins showed that ASIC1 channels of the PNS are also involved in pain sensation. Injection of the ASIC‐activating MIT‐toxin of the Texas coral snake in the mouse paw induced pain that depended on the presence of ASIC1a (Bohlen et al., 2011). Peripheral injection of Mambalgin, which inhibits several ASIC subtypes, exerted an analgesic action due to its inhibition of ASIC1b (Diochot et al., 2012). ASICs are also expressed in pain‐processing areas of the CNS. Administration of PcTx1 or Mambalgin to the CNS inhibited pain behaviours, indicating a role of central ASICs in pain sensation (Mazzuca et al., 2007; Diochot et al., 2012), in addition to the more obvious role of ASICs as pH sensors in the PNS. Two mouse models in which ASIC currents were suppressed – expression of a dominant negative ASIC3 mutant and a triple ASIC1a/ASIC2/ASIC3 knockout – showed increased pain behaviour, indicating that some aspects of the role of ASICs in pain sensation are not yet understood (Mogil et al., 2005; Kang et al., 2012).
The neuropeptides CGRP and substance P contribute to inflammation and are secreted from sensory nerve terminals in response to tissue acidification. CGRP induces vasodilatation and substance P promotes increased vascular permeability leading to plasma extravasation during neurogenic inflammation (Walker et al., 2010; Hoyer and Bartfai, 2012). Because the neuropeptide secretion is Ca2 +‐dependent, TRPV1 and ASICs, which induce Ca2 + entry directly or via depolarization‐induced activation of Cavs, may mediate acidification‐induced neuropeptide secretion. Experiments using specific pharmacological inhibitors or gene knockout identified TRPV1 but not ASICs as the pH sensors for the acid‐induced neuropeptide secretion from sensory neurons (Fischer et al., 2003; Pan et al., 2010; Weller et al., 2011; Boillat et al., 2014). It is likely that the transient ASIC current is too short to induce neuropeptide release and that a sustained current, as the one mediated by TRPV1, is required.
Peng et al. showed recently that disruption of ASIC3 in mice reduced itch and pain in response to local co‐injection of acid and a pruritogen (Peng et al., 2015). A similar effect was obtained when GMQ was injected alone, suggesting that persistent ASIC3 activity may be sufficient to induce itch. Interestingly, it has previously been shown that ASIC3 is potentiated by another pruritogen, 5‐HT (Wang et al., 2013). However, whether the pruritogenic effect of 5‐HT is mediated by ASIC3 has not yet been explored.
Expression pattern and cellular functions of ASICs in the PNS
Expression of the different ASIC subunits has been shown in primary afferent neurons innervating the skin, eye, ear, taste buds, heart, gut, skeletal muscle and joints (reviewed in Wemmie et al., 2013; Kellenberger and Schild, 2015). ASIC function has been measured in small‐ and large‐diameter neurons of the trigeminal, nodose and dorsal root ganglia of mice and rats (Benson et al., 2002; Poirot et al., 2006; Boillat et al., 2014; Deval and Lingueglia, 2015). ASIC‐like currents have also been measured from human dorsal root ganglion (DRG) neurons (Baumann et al., 2004). Sensory neurons express homo‐ and heterotrimeric ASICs, producing current subtypes with different pH dependence. In rat DRG neurons, pH50 values ranging from 6.6 to <4.5 were measured (Poirot et al., 2006). The presence of ASIC2 in heteromeric ASICs tends to decrease the pH sensitivity of the channel (Table 3). Rat DRG neurons express heteromeric ASICs of different compositions or homomeric ASIC1a or ASIC3. The proportion of these current subtypes varied between studies (Poirot et al., 2006; Deval et al., 2008). Rat DRG neurons express a high proportion of ASIC1 and ASIC3, with fewer ASIC2a and ‐2b (Poirot et al., 2006; Deval et al., 2008; Boillat et al., 2014). This is similar to the ASIC expression in humans (Delaunay et al., 2012) but different from mouse DRGs that show a predominant expression of ASIC2a and ‐2b (Drew et al., 2004; Hughes et al., 2007).
Because ASIC currents are mostly transient due to rapid desensitization, they last only a few seconds, even if an acidic extracellular environment persists. In addition, at a pH slightly below the physiological 7.4, some ASIC subtypes desensitize without apparently opening and are not available for subsequent activation in the case of further acidification. From these observed properties, it is expected that ASICs are best adapted to sense rapid changes in pH, but will not report a persistent changed pH. If a pH change occurs slowly, as is the case in ischaemia and inflammation, it may even desensitize ASICs rather than activating them. Two different mechanisms may allow the ASICs to sense pH in such conditions, (i) the sustained current of some ASICs and (ii) modulation of ASIC function by diverse endogenous mediators. Several studies detected a sustained fraction of ASIC currents in DRG neurons and in cells expressing ASIC3 (Poirot et al., 2006; Yagi et al., 2006; Deval et al., 2011). Native ASIC currents in DRG neurons are positively modulated by several inflammatory mediators. 5‐HT, ATP, lactic acid, arachidonic acid and hypertonicity are able to enhance the proton‐induced ASIC3 current, in several cases by changing its pH‐dependence (Light et al., 2008; Baron and Lingueglia, 2015). Also, the synthetic molecule GMQ and related endogenous polyamines such as agmatine and arcaine (EC50 ≥ 1 mM) have recently been shown to activate a sustained current in ASIC3 at physiological pHe (Yu et al., 2010; Li et al., 2011).
Extracellular pH modulation of ion channels in nociceptors
In the following paragraph, we present the different pH sensors in PNS neurons, organized according to their pH sensitivity. Detailed information is provided in Table 3. The varied expression pattern of ASICs and other pH‐sensitive ion channels suggests that pH changes may affect neuronal excitability to different extents in the various subpopulations of nociceptors (Figure 5).
Figure 5.
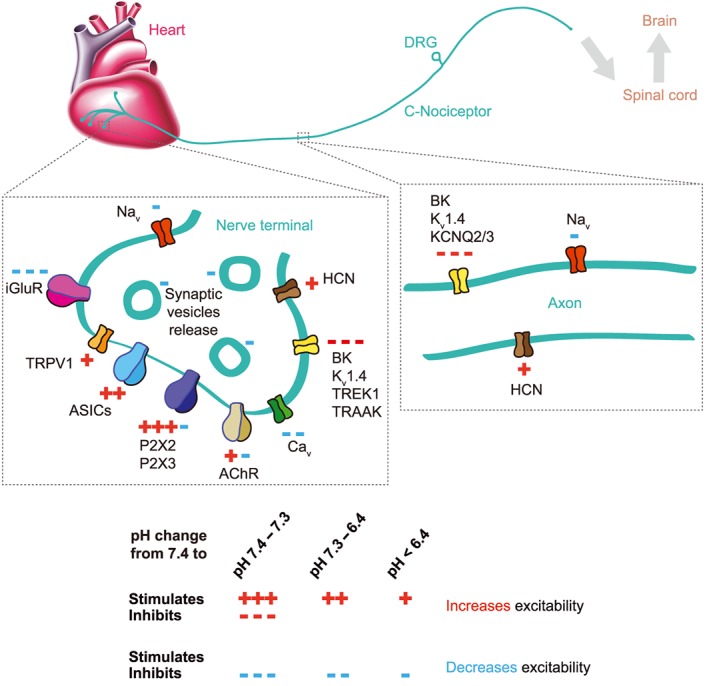
Extracellular pH‐sensitive ion channels in the PNS. Illustration based on experiments performed in small‐diameter DRG neurons innervating peripheral organs. The scheme indicates the pathway of a signal from the sensory nerve terminals in an organ (in the example the heart) to the spinal cord and the brain and focuses on two specific locations, the peripheral nerve terminal (left) and the signalling along the sensory nerve axon (right). Only the ion channels that are modulated by pHe on the nerve terminal and along the axon are shown, indicated by the symbols ‘+’ (stimulation by lowering of pHe) and ‘−‘ (inhibition by lowering of pHe). Red symbols indicate that this modulation increases excitability, and blue symbols indicate that it decreases excitability. The number of symbols correlates with the pH‐dependence of the regulation (Table 3) as indicated at the bottom of the figure.
Fluctuations of the resting pH (pH 7.4–7.3): members of the two‐pore K+ (K2P) channel family are constitutively open at resting membrane potentials. Their activity codetermines the hyperpolarized resting membrane potential of primary afferent neurons (Lesage and Lazdunski, 2000; Kang and Kim, 2006). Most K2P channels are inhibited by extracellular protons with a pH50 of ~7.4. A slight lowering of the pH will therefore shift the resting membrane potential towards more positive values (Lesage and Barhanin, 2011) (Table 3).
Mild extracellular acidosis (acidification to pH 7.2–6.4) inhibits different classes of K+ channels. Studies with human and rat DRG neurons provided evidence for an acidification‐induced depolarization that is mediated by K2P channels in this pH range (Baumann et al., 2004; Cooper et al., 2004). For several K2P channels, it was shown that their loss of function increased the sensation of pain (reviewed in Mathie and Veale, 2015). Acidification activates several types of ASICs in DRG neurons (Dirajlal et al., 2003; Baumann et al., 2004; Poirot et al., 2006). In this pH range, the native ASIC currents are mediated by different ASIC heteromers containing ASIC1a and/or ASIC3 or by ASIC1a or ASIC3 homomers.
Mild extracellular acidosis inhibits efficiently glutamatergic (Traynelis and Cull‐Candy, 1990; Traynelis and Cull‐Candy, 1991) and GABAergic transmission (Zhai et al., 1998; Valeyev et al., 1999), thus neutralizing the excitatory and inhibitory tone on neurotransmission. Voltage‐gated Ca2 + channels are inhibited by extracellular acidification by mechanisms involving pore block and a shift in voltage‐dependence (Hille, 1992).
Severe extracellular acidosis (acidification to pH ≤ 6.4) activates the whole repertoire of ASIC subtypes, including heteromers with a strong ASIC2 contribution. ASIC3 and several ASIC heteromers develop a sustained current at severe acidosis below pH 4.5. Thus, severe acidosis provides persistent ASIC‐mediated depolarizing inputs. Patch‐clamp and Ca2 + imaging experiments with rat DRG neurons showed that pH 6 induces an ASIC current in 50–70% of DRG neurons (Poirot et al., 2006; Deval et al., 2008; Boillat et al., 2014). 40–80% of the putative nociceptors investigated in these studies showed TRPV1‐like activity. TRPV1 is a polymodal ion channel activated by capsaicin and a number of other stimuli including noxious heat (>42°C) endogenous arachidonic acid derivatives, ethanol, camphor and protons (Caterina et al., 1997; Mickle et al., 2015). TRPV1‐mediated sustained currents due to acidification have a pH50 of ~5.4. Therefore, TRPV1 activation by protons alone requires severely acidic conditions (pH ≤ 6). TRPV1 is, however, also modulated by protons and has a substantially higher pH sensitivity for modulation (pH50 ~ 7–6). Mild acidosis can therefore lower the temperature threshold of TRPV1 activation, leading to TRPV1 activation at normal body temperature (Tominaga et al., 1998). Behavioural studies demonstrated an important role of TRPV1 in nociception associated with thermal/mechanical hypersensitivity, with models of inflammation and with different chronic pain conditions (Caterina et al., 2000; Honore et al., 2005; Keeble et al., 2005; Barton et al., 2006). Approximately 20% of measured DRG neurons presented pH‐sensitive inward currents that were not mediated by ASICs or TRPV1, indicating that they were mediated by other ion channels, for example, by the inhibition of K2P channels that would appear like an inward current. Upon acidification to pH 6, ASICs mediated larger currents and charge movements than TRPV1, due to their different pH sensitivity (Poirot et al., 2006; Blanchard and Kellenberger, 2011).
P2X receptors are trimeric cation channels that are activated by extracellular ATP. P2X2 and P2X3 are highly expressed in nociceptors (Chen et al., 1995; Lewis et al., 1995). Disrupting P2X3 in mice decreased nociceptive behaviour (Cockayne et al., 2000; Souslova et al., 2000). P2X receptor activity is modulated by pH changes in sensory neurons. Extracellular acidification increases the P2X2 currents and inhibits P2X3 currents at low and increases them at high ATP concentrations (Gerevich et al., 2007; King et al., 1997). Simultaneous ATP release and extracellular lactic acidification occur in skeletal muscle under exercise. Interestingly, there is evidence for a physical interaction between P2X receptors and ASICs, and it was shown that ATP‐induced P2X2 receptor activation potentiates the acid‐induced ASIC3 current by twofold. It is therefore likely that ATP and acidosis can converge to increase nociceptor excitability (Birdsong et al., 2010).
Amiloride‐sensitive epithelial sodium channel
Function and regulation of ENaC
ENaC is composed of three different subunits, namely α, β and γENaC, encoded by three different genes (SCNN1A, SCNN1B and SCNN1G, respectively; see for review Kellenberger and Schild, 2015). In the kidney, ENaC is present in the aldosterone‐sensitive distal nephron (ASDN), that is composed of the distal convoluted tubule (DCT), connecting tubule (CNT) and collecting duct (CD) segments (Figure 6) (Rossier et al., 2013), to allow fine‐tuning of whole body Na+ homeostasis. Expression of ENaC is regulated transcriptionally but equally by post‐translational modifications, which thereby determine its synthesis, trafficking and activity at the cell membrane (Rossier, 2014). This regulation occurs on the ENaC subunits themselves (Loffing et al., 2001) and/or on ENaC‐regulating proteins such as the serum‐ and glucocorticoid‐regulated kinase 1 (Lang and Shumilina, 2013). Aldosterone modulates ENaC activity by transcription‐dependent and ‐independent mechanisms (Thomas et al., 2007). Due to space constraints in this review, we refer to the extensive literature on ENaC regulation (Kellenberger and Schild, 2015; Rossier et al., 2015; Verouti et al., 2015).
Figure 6.
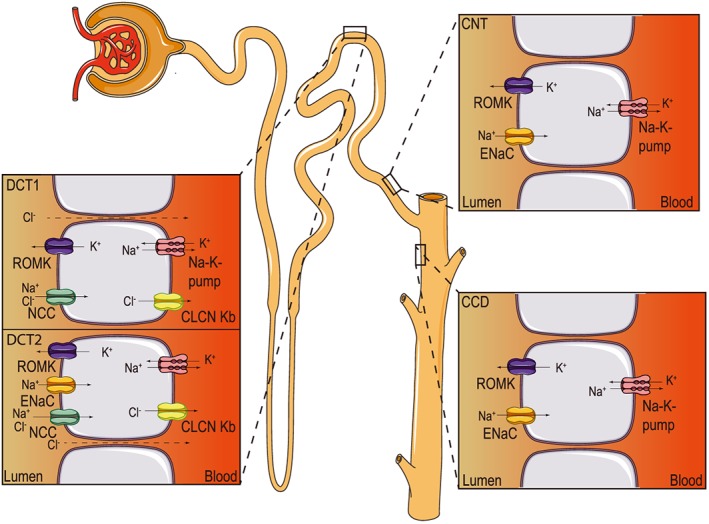
ENaC expression along the aldosterone‐sensitive distal nephron (ASDN). Schematic view of a nephron, with detailed views of the ion transport mechanisms in cells of the DCT, CNT and CCD. CLCN Kb, voltage‐sensitive chloride channel Kb; ROMK, renal outer medullary potassium channel.
ENaC function in different tissues
Role of ENaC in classical and non‐classical tissues and organs
The syndrome pseudohypoaldosteronism type 1 (PHA‐1) was first described by Cheek and Perry in 1958 (Cheek and Perry, 1958). Patients suffering from PHA‐1 present the following symptoms: renal salt wasting, hypovolaemia and hypotension, hyperkalaemia, metabolic acidosis, high plasma levels of renin and aldosterone, and respiratory illness. PHA‐1 is caused by either mutations in mineralocorticoid receptors (MRs) (autosomal dominant PHA‐1; Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 177 735, http://omim.org/) or by loss‐of‐function mutations in ENaC subunits (autosomal recessive PHA‐1; OMIM number 264 350) (Furgeson and Linas, 2010). The genetic dissection of ENaC along the mouse nephron highly suggests that the early ASDN (DCT and/or CNT, Figure 6) is crucial for Na+ and K+ balance. Whereas animals with ENaC deficiency in the CD survive well even under salt restriction or K+ loading (Rubera et al., 2003), ENaC deficiency in the CNT and CD (Christensen et al., 2010), or reduced ENaC expression solely in the CNT (Poulsen et al., 2015) alone is sufficient to induce a mild salt‐losing phenotype. However, only the deletion of ENaC subunit expression in mice along the whole nephron mimics the severe human adult PHA‐1 (Perrier et al., 2015). An additional relevant role of ENaC in the cortical collecting duct (CCD) can currently not be excluded. Table 4 provides an overview of studies with ENaC mouse models. The constitutive αENaC knockout mice develop respiratory distress syndrome and die soon after birth, thus revealing an important role of the αENaC subunit in lung liquid clearance at birth (Hummler et al., 1996). The γENaC subunit seems to transiently facilitate neonatal lung liquid clearance at birth, suggesting equally important roles for the other ENaC subunits (Barker et al., 1998). Increased airway epithelial Na+ absorption produces cystic fibrosis (CF)‐like lung disease in transgenic mice (Mall et al., 2004), unveiling an important role of ENaC in the regulation of the airway surface liquid volume (Mall et al., 2010). In the colon, αENaC‐deficiency causes Na+ loss and aldosterone resistance, that is normally compensated by the renin‐angiotensin‐aldosterone system in the kidney (Malsure et al., 2014); thus, these mice are able to maintain their Na+ and K+ homeostasis.
Table 4.
ENaC animal models
| Mouse model | Tissue/organ | Phenotype | Reference |
|---|---|---|---|
| αENaC KO (constitutive) | Kidney and lung | Neonatal death, lung fluid clearance failure, hyperkalemia and sodium loss | (Hummler et al., 1996) |
| Increased number of angiotensin II subtype 1 receptors in renal tissues and lowered blood pressure during the angiotensin II receptor blockade | (Wang et al., 2001) | ||
| Skin | Decreased transcription of the alpha1‐subunit of Na+,K+‐ATPase | (Blot‐Chabaud et al., 2001) | |
| Increased transepidermal water loss, higher skin surface pH, disturbed stratum corneum lipid composition and lamellar body secretion | (Charles et al., 2008) | ||
| Impaired directionality of galvanotaxis in keratinocytes | (Yang et al., 2013) | ||
| Altered epidermal differentiation | (Mauro et al., 2002) | ||
| Embryo | Inability to rescue the lethal embryonic phenotype in double αENaC and HAI‐1 KOs | (Szabo et al., 2012) | |
| Ear | Normal mechano‐electrical transducer apparatus | (Rusch and Hummler, 1999) | |
| βENaC KO (constitutive) | Kidney | Hyperkalemia, neonatal death and type 1 pseudohypoaldosteronism | (McDonald et al., 1999) |
| γENaC KO (constitutive) | Kidney | Hyperkalemia, neonatal death and type 1 pseudohypoaldosteronism | (Barker et al., 1998) |
| CNT‐specific αENaC KO (Hoxb7, constitutive) | Kidney | Normal sodium and potassium balance | (Rubera et al., 2003) |
| Normal ascites development | (Mordasini et al., 2015) | ||
| Abolished benzamil‐sensitive component of Cl− absorption | (Pech et al., 2012) | ||
| Protection from lithium‐induced nephrogenic diabetes insipidus | (Christensen et al., 2011) | ||
| Presence of electroneutral, amiloride‐resistant, thiazide‐sensitive, transepithelial NaCl absorption in CNT | (Leviel et al., 2010) | ||
| Rosiglitazone‐induced fluid retention | (Vallon et al., 2009) | ||
| Normal urinary acidification following furosemide alone and in combination with hydrochlorothiazide treatment | (Kovacikova et al., 2006) | ||
| CNT‐specific αENaC KO (AQP2, constitutive) | Kidney | Higher urinary sodium excretion and hyperkalemia under Na+‐deficient diet | (Christensen et al., 2010) |
| Attenuation of body weight and water increase following RGZ (rosiglitazone) treatment | (Fu et al., 2015) | ||
| nephron‐specific αENaC KO (inducible) | – | Hyperkalemia and body weight loss under regular salt diet | (Perrier et al., 2015) |
| CNT‐specific αENaC KO (constitutive) | – | Type 1 pseudohypoaldosteronism symptoms during high dietary K+ loading | (Poulsen et al., 2015) |
| β and γENaC KOs (constitutive) | Sensory neurons | Normal mechanosensory behaviour | (Raouf et al., 2012) |
| airway‐specific overexpression βENaC‐transgene (constitutive) | Lung | Increased airway Na+ absorption, airway mucus obstruction and chronic airway inflammation | (Zhou et al., 2011) |
| Unaltered development of airway cartilage | (Bonora et al., 2011) | ||
| Therapeutic effects of α1‐antitrypsin on Pseudomonas aeruginosa infection | (Nichols et al., 2015) | ||
| Defective regulation of airway surface liquid volume and ENaC‐mediated Na+ absorption | (Mall et al., 2010) | ||
| αENaC KO (constitutive) | Tongue | Complete loss of salt attraction and sodium taste response | (Chandrashekar et al., 2010) |
| βENaC ‐Liddle knock‐in (constitutive) | Kidney | High blood pressure, metabolic alkalosis, hypokalemia with cardiac and renal hypertrophy under high salt intake | (Pradervand et al., 1999b) |
| Constitutive hyperactivity of ENaC in cortical connecting ducts | (Pradervand et al., 2003) | ||
| Maintained mineralocorticoid regulation of ENaC | (Dahlmann et al., 2003) | ||
| Increased vasopressin‐stimulated CFTR Cl− currents in CCD cells | (Chang et al., 2005) | ||
| Lung | No effect of hypoxia on amiloride‐sensitive alveolar fluid clearance | (Gille et al., 2014) | |
| Increased alveolar fluid clearance and reduced severity of hydrostatic pulmonary oedema | (Randrianarison et al., 2007) | ||
| Pendrin gene ablation in Liddle homozygous does not eliminate nitric oxide‐sensitive net Cl− flux and transepithelial potential difference | (Pech et al., 2013) | ||
| Intact regulation of airway surface liquid volume and ENaC‐mediated Na+ absorption | (Mall et al., 2010) | ||
| Intestine | Increased aldosterone responsiveness of ENaC in colon | (Bertog et al., 2008) | |
| Heart | Decreased action potential duration, intracellular Ca2 + transient amplitude and contraction | (Perrier et al., 2005) | |
| βENaC hypomorphic mice (constitutive) | Kidney | Weight loss, hyperkalemia and decreased blood pressure on low salt diet | (Pradervand et al., 1999a) |
| Lung | Impaired lung fluid clearance | (Randrianarison et al., 2008) | |
| αENaC(−/−)Tg (CMV‐αENaC) (constitutive) | Lung, kidney and intestine | Rescued perinatal lethal pulmonary phenotype and partially restored Na+ transport in renal, colonic, and pulmonary epithelia | (Hummler et al., 1997) |
| Lung | Reduced Na+ transport rate probably insufficient for airway fluid clearance and favouring pulmonary oedema | (Olivier et al., 2002) | |
| Predisposition to pulmonary oedema and delayed resolution | (Egli et al., 2004) | ||
| Heart | Absence of cardiac remodelling and fibrosis under a normal‐salt diet | (Wang et al., 2004) | |
| increased action potential duration, intracellular Ca2 + transient amplitude and contraction | (Perrier et al., 2005) | ||
| Colon‐specific αENaC KO (constitutive) | Intestine | Sodium loss and aldosterone resistance | (Malsure et al., 2014) |
Apart from its classical role as a membrane constituent of many salt‐reabsorbing epithelia that facilitates Na+ movement across the tight epithelial in the distal nephron, distal colon, the ducts of salivary and sweat glands, and the lung, the expression of ENaC subunits is also reported in tissues and/or organs that seem not to be implicated in whole body Na+ homeostasis, such as the skin, tongue, eye and blood vessels (reviewed in Rossier et al., 2013; Kellenberger and Schild, 2015). The analysis of genetically engineered mouse models started to reveal the role of ENaC in these tissues/organs, unveiling non‐classical roles, namely in epidermal differentiation, barrier function, lipid synthesis and secretion, and also migration of keratinocytes (Charles et al., 2008; Yang et al., 2013), and salt perception [tongue; (Chandrashekar et al., 2010), Table 4]. In the human eye, all ENaC subunits (α, β, γ and δ) are expressed within the cornea, ciliary body, iris and retina. β and γENaC subunits present distinct localizations (Krueger et al., 2012). The β subunit is present in basal regions of the limbal epithelium, while the γ subunit is present throughout all layers of the corneal epithelium but not in the basal regions of the limbal epithelium. Measurements of electrical potential difference confirmed functional ENaC‐mediated Na+‐transport, which is probably involved in maintaining hydration of the ocular surface (Yu et al., 2012). ENaC is also expressed in endothelial and vascular smooth muscle cells, where its increased surface expression can lead to membrane stiffening and reduced release of nitric oxide (Jeggle et al., 2013). However, the contribution to hypertension of ENaC expressed in these cells still remains to be determined (Kusche‐Vihrog et al., 2014).
ENaC and hypertension
According to the World Health Organization, hypertension or increased blood pressure strongly correlates with risk of cardiovascular events, stroke and kidney disease and thus represents the leading cause of death worldwide (Santulli, 2013). It was predicted that the number of hypertensive patients will reach 1.56 billion in 2025 (Kearney et al., 2005) and will thus affect one third of adults in most developing and developed communities. Clinical practice guidelines have been written to provide a straightforward approach to manage hypertension (Weber and Anlauf, 2014). Ninety‐five percent of adults with high blood pressure have primary or essential hypertension with unknown cause, but it is widely accepted that genetic and environmental factors affect blood pressure. According to the American Society of Hypertension and the International Society of Hypertension, the treatment aims to control blood pressure and to deal with all risk factors for cardiovascular diseases, like e.g. overweight or stress (Weber and Anlauf, 2014). Lifestyle interventions have been identified to reduce blood pressure, for example, weight loss, salt reduction, exercise and reduced alcohol consumption (Levenson et al., 2002). Blood pressure is known to depend on salt balance, although there is a heterogeneity in salt‐induced blood pressure responses in both normotensive and hypertensive populations (Weinberger, 1996). Recent evidence indicates that in addition to sodium, chloride may also independently contribute to this salt response (McCallum et al., 2015). Lowering the salt intake decreases blood pressure (Vollmer et al., 2001; Bray et al., 2004) and prevents hypertension (The Trials of Hypertension Prevention Collaborative Research Group, 1997). In addition, increased K+ intake improved blood pressure (Obel, 1989), although the combination of lower Na+ and higher K+ intake did not further decrease blood pressure (Chalmers et al., 1986). These findings highly suggest that food consumption may be primordial in the prevention and/or the treatment of hypertension, and the ‘Dietary Approaches to Stop Hypertension’ reports that a diet composed of fruits, vegetables and low‐fat dairy products rich in Ca2 +, Mg2 + and K+ exerts antihypertensive effects (Zemel, 1997).
The most used pharmacological treatments against hypertension are the β‐blockers, the diuretics, the calcium blockers and the angiotensin‐converting enzyme inhibitors such as enalapril or ramipril (Rahimi et al., 2015). To avoid diuretic‐induced hypokalaemia, K+‐sparing diuretics such as the ENaC inhibitor amiloride are used (see for review Saha et al., 2005; Weber and Anlauf, 2014). Treatment with inhibitors of ENaC resulted in substantial improvement in blood pressure, highly suggesting that increase in Na+ transport by ENaC may be a common and requisite component of salt‐dependent forms of hypertension (Pratt, 2005). Thus, the combination of two or more drugs that includes ENaC inhibitors was proposed (Vidt and Borazanian, 2003; Pratt, 2005). ENaC may play an even more central role in Na+ retention in the generation of hypertension than previously thought (Pratt, 2005). Thiazide diuretics are currently among the most prescribed anti‐hypertension drugs. They are often combined with other antihypertensive drugs (Weber, 2014). Next generation diuretics may block synergistically the Na+Cl− cotransporter (NCC) and ENaC, and possibly in addition the Cl−/HCO3 −‐exchanger pendrin in patients with fluid overloads such as congestive heart failure, nephrotic syndrome, diuretic resistance or generalized oedema. They may also block one or more pathways known to up‐regulate ENaC activity (reviewed in Rossier, 2014; Verouti et al., 2015).
ENaC and cystic fibrosis
CF is one of the most common hereditary life‐threatening diseases. It is characterized by mutations in the gene coding for the CF transmembrane conductance regulator (CFTR) (Brennan and Schrijver, 2016). The most frequent mutation, the in‐frame phenylalanine 508 deletion (∆F508) is responsible for 70% of CF mutations worldwide and has existed since the Palaeolithic period (Morral et al., 1994). The respiratory failure and finally the death of patients suffering from CF are due to airway obstruction, inflammation and chronic bacterial infections. CFTR allows hydration and thus mucus clearance of the airway surface of the epithelia by secreting chloride to the lumen of the airway epithelia (Boucher, 2007). In the case of CFTR mutations, the mutated receptor cannot reach the plasma membrane due to a trafficking defect, and as a consequence, the airway epithelium gets dehydrated because of the inability of CFTR to secrete chloride (Denning et al., 1992; Kartner et al., 1992). Although medical treatment strategies for CF have evolved rapidly over the past years, there is still no cure today. Among the new approaches, miRNAs allow the regulation of post‐transcriptional gene expression. miRNAs can act either by repressing or by up‐regulating genes of interest (Ramachandran et al., 2012; Valinezhad Orang et al., 2014). miRNA approaches have been recently proposed as a promising therapeutic treatment of CF that could up‐regulate the expression and the number of CFTR proteins that reach the cell surface, as demonstrated in primary human airway epithelia (Ramachandran et al., 2012; Sonneville et al., 2015).
The depth of surface liquid of airways is regulated by Cl− secretion through CFTR and by Na+ absorption through ENaC. Reduced CFTR expression at the cell surface leads to ENaC up‐regulation, resulting in an increase of Na+ reabsorption and, consequently, to a dehydration of the airway surface (Clunes and Boucher, 2007). Interestingly, it was shown that a mouse model overexpressing the β subunit of the epithelial Na+ channel (ENaC) in the lower airways shows typical features of CF, namely reduction of the periciliary liquid weight in bronchi and tracheae, depletion of the airway surface volume, abnormal mucus transport and reduction of the clearance of bacteria, with a mortality of ~50% (Mall et al., 2004). For 30 years, inhalation of amiloride has been known to inhibit Na+ reabsorption in CF patients. ENaC inhibition improves mucociliary clearance and thereby retards the lung infection (Kohler et al., 1986). Amiloride therapy may reduce morbidity and mortality if it is given early in life before the onset of the lung disease (Zhou et al., 2008). As amiloride has a short duration of action on airway surfaces, second and third generation amiloride analogues, such as benzamil and phenamil, were generated and tested to improve both the affinity to ENaC and decrease the reversibility of the ENaC inhibition (Hirsh et al., 2006). Benzamil presents the highest potency and is rapidly absorbed from the mucosal surface to the cytosol. However, in vivo pharmacodynamic experiments in sheep have shown that amiloride and benzamil have the same efficiency (Hirsh et al., 2004). More recently, Parion compound N‐(3,5‐diamino‐6‐chloropyrazine‐2‐carbonyl)‐N‐4‐[4‐(2,3‐dihydroxypropoxy)phenyl]butyl‐guanine methanesulfonate (552–02) was tested in CF bronchial epithelial cells and in sheep for mucociliary clearance after aerosol dosing. Compared with amiloride, 552–02 induced greater airway surface liquid expansion in bronchial epithelial cells. In addition, 552–02 increased the mucociliary clearance in sheep at the time of administration and 4 to 6 h later (Hirsh et al., 2008). In summary, the development of new ENaC blockers, such as 552–02, with a higher potency, higher selectivity and durability, is of clinical importance for the treatment of CF lung disease.
Aldosterone‐dependent and ‐independent regulation of ENaC
Aldosterone‐dependent ENaC regulation
Several hormones regulate ENaC activity (e.g. aldosterone, vasopressin, angiotensin, insulin; reviewed by Verouti et al., 2015). Among them, the mineralocorticoid aldosterone acts on the principal cells in the ASDN (Figure 6). Aldosterone is secreted by the adrenal gland in response to a small increase in K+ concentration in blood, or decrease in vascular volume, which activates the renin‐angiotensin‐aldosterone system, thereby maintaining the extracellular Na+ concentration and the blood pressure within a physiological range (Bollag, 2014). In addition to angiotensin II, potassium itself also serves as an independent mediator of aldosterone secretion from the adrenal gland (Williams, 2005). The genomic response to aldosterone in target tissues has been extensively analysed and occurs 4–24 h following an aldosterone stimulus. In this phase, aldosterone is translocated into the nucleus, where it binds to the MR and stimulates further synthesis of ‘aldosterone‐induced proteins’ like ion transporting proteins, such as the α subunit of ENaC and the α1 subunit of the Na+‐K+‐ATPase (Pearce et al., 2015). The rapid effects of aldosterone on target tissues are far less well understood and are still controversial, because they are coupled to MR or to a yet unidentified membrane‐associated aldosterone receptor (Dooley et al., 2012). These non‐genomic effects occur within minutes to 2 h after the aldosterone stimulus (Bollag, 2014) and do not require synthesis of ion transporter proteins (Le Moellic et al., 2004). Aldosterone may activate the ERK1/2 MAPK cascade independently of MR (Rossol‐Haseroth et al., 2004). Furthermore, activation of the PKC/PKD signalling pathway through the c‐Src‐dependent transactivation of the epidermal growth factor receptor may also contribute to early ENaC trafficking in response to aldosterone (Dooley et al., 2012). Thus, some of the early responses may potentiate the genomic effects of aldosterone through, for example, the phosphorylation of channels and/or transporters.
Aldosterone‐independent ENaC regulation
ENaC activity is preserved in MR knockout mice (Berger et al., 1998), demonstrating that aldosterone is not the only modulator of ENaC. Furthermore, aldosterone synthase(−/−) mice survive even under low salt diet, demonstrating again an aldosterone‐independent compensatory mechanism (Makhanova et al., 2006). Interestingly, a study of Nesterov and colleagues clearly demonstrated by measuring ENaC activity in cut open tubules of aldosterone synthase(−/−) mice that aldosterone regulates ENaC only in the distal part of the nephron (CNT/CCD), whereas in the DCT2/CNT, ENaC regulation is aldosterone‐independent (Nesterov et al., 2012). Other hormones such as insulin, vasopressin and angiotensin II regulate ENaC activity as well (Verouti et al., 2015). Indeed, in adrenalectomized mice, vasopressin is up‐regulated, and ENaC function is not compromised (Mironova et al., 2012). Furthermore, more recently, angiotensin II has been proposed to up‐regulate γENaC in the DCT in AS(−/−) mice treated with high K+ diet (2% K+), thereby regulating Na+ and K+ homeostasis (Todkar et al., 2015) by increasing the electrochemical driving force for K+ excretion through renal K+ channels (van der Lubbe et al., 2013a). This phenomenon is aldosterone‐independent and involves a decrease in the upstream NCC activity (Sorensen et al., 2013; van der Lubbe et al., 2013a, 2013b). Several recent in vivo studies confirm that upon hyperkalaemia, the distal Na+ delivery is increased, by decreasing the NCC activity, while upon hypovolaemia, both NCC and ENaC are up‐regulated to improve Na+ absorption (Rengarajan et al., 2014; van der Lubbe et al., 2013a, 2013b; Vitzthum et al., 2014). Thus, angiotensin II acts on the switch from K+ secretion upon hyperkalaemia to Na+ reabsorption under hypovolaemia that is mediated via several kinases (Arroyo et al., 2011; Mamenko et al., 2013). This so‐called aldosterone paradox phenomenon (Arroyo and Gamba, 2012) determines NCC and ENaC as key players for Na+ and K+ balance within the DCT. As expected for an aldosterone‐dependent ENaC regulation, treating WT mice with high Na+ and high K+ diet leads to an increase in plasma aldosterone and an increase in the βENaC subunit and serum‐ and glucocorticoid‐regulated kinase 1 protein expression (Vitzthum et al., 2014). However, even in presence of the MR antagonist spironolactone or the ENaC blocker amiloride, blood pressure stays increased (Vitzthum et al., 2014). This strongly suggests the existence of other, currently unknown regulatory mechanisms in a Na+‐repleted state. Several other aspects of ENaC regulation are still unresolved, for example, the molecular mechanisms underlying the aldosterone‐dependent and ‐independent regulation of ENaC, the differential distribution of ENaC channels along the nephron or the possibility of a crosstalk between NCC and ENaC within the DCT2.
Is ENaC regulated by potassium diet?
Up to now, evidence for ENaC regulation through K+ is poor. In the case of hyperkalaemia, angiotensin II is known to improve the delivery of Na+ to the distal part of the nephron to increase ENaC‐mediated electrochemical secretion of K+ through renal K+ channels. As discussed above, this phenomenon is aldosterone‐independent and leads to a decrease in the upstream NCC activity. To date, more is known about the regulation of NCC activity upon K+ loading than on ENaC regulation in this context. Low K+ diet is a strong stimulus to increase NCC activity (Sorensen et al., 2013; van der Lubbe et al., 2013b) by an aldosterone‐independent mechanism (Vallon et al., 2009; Castaneda‐Bueno et al., 2014). Acute oral K+ loading resulted in rapid NCC dephosphorylation and inactivation of NCC (Sorensen et al., 2013). In contrast, a high Na+ diet mediates a decrease in NCC phosphorylation by the oxidative‐stress‐responsive kinase 1/Ste20‐related proline alanine rich kinase in an aldosterone‐dependent manner, thereby increasing NCC activity (Chiga et al., 2008). ENaC activity within the DCT2/CNT segments is however independent of aldosterone (Nesterov et al., 2012; Todkar et al., 2015), and the mechanism of ENaC regulation in the DCT2/CNT in this context is currently unknown. Furthermore, the effect of solely a low K+ diet on ENaC has not been investigated so far. The recent study by Perrier et al. (2015) clearly demonstrates that in nephron‐specific αENaC knockout mice, hyperkalaemia becomes the determining factor in regulating NCC activity, regardless of Na+ loss, and thus remains the predominant and life‐threatening feature to be avoided.
Regulation of ENaC by proteases
In addition to the hormonal regulation through, for example, vasopressin (Ecelbarger et al., 2001) and insulin (Marunaka et al., 1992), ENaC is regulated by changes in both extracellular and intracellular sodium. An increase in extracellular Na+ reduces ENaC activity (Fuchs et al., 1977), and this Na+ self‐inhibition can be abolished by treatment with extracellular proteases (Chraibi and Horisberger, 2002). The first evidence of ENaC regulation by proteases was reported in 1983 when Garty and Edelman applied high concentrations of trypsin (1 mg·mL−1) to toad urinary bladder and recorded a decrease in the amiloride‐sensitive Na+ current (INa) probably as a result of proteolytic digestion of the ENaC channel (Garty and Edelman, 1983). When lower concentrations of trypsin or chymotrypsin (1–5 μg·mL−1) were applied, ENaC activity expressed in the Xenopus oocytes was strongly stimulated by an increase in channel open probability (Chraibi et al., 1998). Since then, trypsin and chymotrypsin are commonly used as experimental tools to achieve maximal ENaC activation. In 1997, Vallet and colleagues screened a Xenopus A6 cell complementary DNA library to detect proteins involved in the control of ENaC activity and isolated the first membrane‐bound serine protease whose co‐expression with ENaC induced a threefold increase in the ENaC‐mediated INa amplitude (Vallet et al., 1997). Consequently, this serine protease was termed channel‐activating protease‐1 (CAP1). It is encoded by the Prss8 gene and is orthologous to human prostasin containing a glycosylphosphatidyl‐inositol (GPI) domain as a plasma membrane anchor (Vallet et al., 1997). The mouse counterpart was cloned 2 years later from a CCD cell line derived from mouse kidney (Vuagniaux et al., 2000). Measurements of this cell line revealed that the ENaC‐mediated INa was less sensitive (only ~50% of control) to the serine protease inhibitor aprotinin (Vuagniaux et al., 2000). This suggested that ENaC activation depends on more than one protease and led to the discovery of two additional membrane‐bound serine proteases that increased ENaC currents 6‐ to 10‐fold. Accordingly, these type II‐oriented membrane‐bound serine proteases were called CAP2 (also known as Tmprss4) and CAP3 (also known as matriptase) (Vuagniaux et al., 2000). Interestingly, CAP3 has been shown to decrease an acid‐activated ASIC current as measured by two‐electrode voltage clamp in Xenopus oocytes (Clark et al., 2010). Since then, several other proteases were added to the list of ENaC regulatory molecules, all increasing ENaC currents. TMPRSS3 is mutated in deafness (Guipponi et al., 2002), furin (Hughey et al., 2004), neutrophil elastase (Caldwell et al., 2005), kallikrein (Picard et al., 2008; Patel et al., 2012), plasmin (Svenningsen et al., 2009; Buhl et al., 2014), metalloprotease meprin β subunit (Garcia‐Caballero et al., 2011), cathepsin‐S (Haerteis et al., 2012), urokinase plasminogen activator (Chen et al., 2014; Ji et al., 2015), alkaline phosphatase from Pseudomonas aeruginosa (Butterworth et al., 2012; Butterworth et al., 2014) and trypsin IV (Haerteis et al., 2014) are all proteases able to activate ENaC in in vitro settings. So far, only CAP1, kallikrein and the urokinase plasminogen activator have been confirmed as physiological regulators of ENaC activity by in vivo studies using genetically modified or spontaneous mutant mice (Picard et al., 2008; Planes et al., 2009; Frateschi et al., 2012; Chen et al., 2014; Malsure et al., 2014), and it was shown that the serine protease CAP2 is not involved in ENaC regulation in vivo in kidney and colon (Keppner et al., 2015). The mechanism by which these proteases activate the channel is still largely debated. Several researchers claim and provide evidence for a direct activation mediated by proteolytic cleavage of ENaC that would lead to the removal of inhibitory domains and/or changes in the three‐dimensional structure of the channel, resulting in its activation (Shi et al., 2013). Indeed, this hypothesis is quite tempting, as most of these enzymes are membrane‐bound proteins facing the extracellular side of the plasma membrane, or soluble molecules and thus potentially located in close proximity with ENaC. Although cleavage products of the different ENaC subunits have been observed under various conditions, functional evidence is still missing. In this regard, CAP1 is peculiar and its action on ENaC still controversial. A CAP1 cleavage site (RKRK) was identified in the γ subunit of ENaC downstream of the proposed consensus cleavage site for furin. On the one hand, the mutation of either the CAP1 cleavage site or the furin cleavage site abolished the activation of ENaC by CAP1 in Xenopus oocytes and prevented the appearance of a smaller fragment of the γ subunit (Bruns et al., 2007; Carattino et al., 2008). On the other hand, mutation of the CAP1 catalytic triad that should render the protease catalytically inactive did not prevent full ENaC activation and moreover induced γ subunit cleavage equally well as did its wild‐type counterpart (Andreasen et al., 2006; Bruns et al., 2007; Carattino et al., 2014). Other studies indicated that there is no consistent correlation between γ subunit cleavage and ENaC activation (Fejes‐Toth et al., 2008; Harris et al., 2008). In addition, although CAP1 is indeed a positive regulator of ENaC in vivo, because CAP1 loss of function or absence significantly decreases ENaC basal activity (Planes et al., 2009; Frateschi et al., 2012; Malsure et al., 2014), the cleaved fragment originating from γENaC was still observed in the colon or in the lung of tissue‐specific CAP1 knockouts (Planes et al., 2009; Malsure et al., 2014). Interestingly, both the wild‐type and the catalytically inactive mutant CAP1 are able to elicit skin pathology when overexpressed in mice (Frateschi et al., 2011; Crisante et al., 2014). This indicates that as for ENaC activation, the catalytic triad of CAP1 is not essential for provoking pathophysiological effects. The presence of cleaved fragments originating from ENaC subunits may nevertheless be physiologically significant, even if they most likely do not derive from direct cleavage by CAP1. Very recently, Zachar et al. reported that, in the human kidney, γENaC is subjected to proteolytic processing, yielding fragments compatible with furin cleavage and that proteinuria is accompanied by cleavage at the putative CAP1 site of γENaC (Zachar et al., 2015). Table 5 highlights the proteases involved in the regulation of ENaC with particular emphasis on their physiological implications.
Table 5.
Proteases modulating ENaC activity in vitro and in vivo
| Protease | Type | ENaC regulation in vitro | ENaC cleavage | ENaC regulation in vivo |
|---|---|---|---|---|
| CAP1/Prss8, also known as prostasin | Membrane‐bound serine protease, GPI‐anchored | Catalytically independent and increased ENaC activity in X. oocytes and mCCD cells (Vallet et al., 1997; Vuagniaux et al., 2000; Andreasen et al., 2006). | Cleavage of γENaC in transfected MDCK cells through catalytically active and inactive CAP1 (Bruns et al., 2007). Probable involvement of a second protease (Carattino et al., 2014). | 40% decrease in ENaC‐mediated Na+ currents in alveolar epithelial cells; unaltered ENaC cleavage in lung‐specific CAP1 KO mice (Planes et al., 2009). Reduced ΔPDAmil but normal ENaC processing in intestine‐specific CAP1 KO (Malsure et al., 2014). |
| CAP2/Tmprss4 | Membrane‐bound serine protease, type II | Catalytically dependent and increased ENaC current in X. oocytes (Vuagniaux et al., 2000). | Cleavage of all three ENaC subunits in X. oocytes (Garcia‐Caballero et al., 2008). Cleavage only in γENaC (Passero et al., 2012). | Normal ΔPDAmil and ENaC processing in constitutive CAP2 KO (Keppner et al., 2015). |
| CAP3/matriptase | Membrane‐bound serine protease, type II | Catalytically‐dependent and increased ENaC current in X. oocytes (Vuagniaux et al., 2000). | Mediates cleavage of ENaC at basic residues near the γENaC furin site (Kota et al., 2012). | Not determined. |
| TMPRSS3 | Membrane‐bound serine protease, type II | Increased ENaC current in X. oocytes. TMPRSS3 mutants fail to activate ENaC (Guipponi et al., 2002). | Not determined. | Not determined. |
| Furin | Intracellular (subtilisin‐like proprotein convertase family) | Activated ENaC through suppression of Na+ self‐inhibition (Sheng et al., 2006). | Cleaved α and γENaC in X. oocytes (Hughey et al., 2003; Hughey et al., 2004). | Not determined. |
| Neutrophil elastase | Soluble serine protease | Increased ENaC activity in cultured human bronchial epithelia. Increased open probability of near‐silent channels in NIH‐3 T3 cells (Caldwell et al., 2005). | Neutrophil elastase‐dependent activation of γENaC (Caldwell et al., 2005; Harris et al., 2007). | Not determined. |
| Kallikrein | Soluble serine proteinase | Activated ENaC in X. oocytes (Patel et al., 2012). | ENaC activation and cleavage abolished in mouse ENaC mutants (Patel et al., 2012). | Blunted urinary Na+ excretion after amiloride injection and decreased colonic ΔPDAmil (Picard et al., 2008), and absence of the cleaved (70‐kDa) γENaC in kallikrein KO mice (Picard et al., 2008). |
| Plasmin | Soluble serine proteinase | Activated ENaC in M‐1 cells and X. oocytes (Svenningsen et al., 2009). | Released γENaC peptide (Passero et al., 2008; Svenningsen et al., 2009). | Not determined. |
| Metalloprotease meprin β subunit | Cell surface or secreted meprin metalloproteinase | Increased ENaC in X. oocytes and epithelial cells (Garcia‐Caballero et al., 2011). | Cleavage of α and γENaC subunits (Garcia‐Caballero et al., 2011). | Not determined. |
| Cathepsin‐S | Lysosomal cysteine protease | Activated ENaC in X. oocytes and M‐1 cells (Haerteis et al., 2012). | γENaC cleavage (Haerteis et al., 2012). | Not determined. |
| Urokinase plasminogen activator | Membrane‐bound serine protease, GPI‐anchored | Catalytically dependent activation of ENaC in X. oocytes (Ji et al., 2015). | γ but not αENaC cleavage in X. oocytes (Ji et al., 2015). | Decreased ENaC activity in primary tracheal epithelial cells from urokinase plasminogen activator KO (Chen et al., 2014). |
| Alkaline phosphatase from Pseudomonas aeruginosa | Phosphomonoesterase (periplasmic space in Gram‐negative bacteria) | Activated ENaC in immortalized and primary human bronchial epithelial cells from both CF and non‐CF patients (Butterworth et al., 2012; Butterworth et al., 2014). | Cleaved γENaC (Butterworth et al., 2012). | Not determined. |
| Trypsin IV and I | – | Increased ENaC activity by trypsin IV in human airway epithelial cells (Haerteis et al., 2014). | ENaC cleavage and activation by trypsin IV (but not by trypsin I) (Haerteis et al., 2014). | Not determined. |
GPI, glycosylphosphatidyl‐inositol
Protease inhibitors achieve another level of regulation of ENaC activity. Using a proteomic approach, Garcia‐Caballero et al. identified the short palate, lung and nasal epithelial clone 1 as an ENaC inhibitor highly expressed in the airways, colon and kidney (Garcia‐Caballero et al., 2009), with the inhibitory domain located at the N terminus (Hobbs et al., 2013). α1‐Antitrypsin, an anti‐serine protease found in human plasma and lung epithelial fluid (Lazrak et al., 2009), inhibits amiloride‐sensitive Na+ transport in Xenopus oocytes and alveolar fluid clearance in mice (Lazrak et al., 2009). A role for SerpinB1, a highly efficient inhibitor of neutrophil serine proteases, was described in the CF airways, suggesting a possible link to ENaC regulation (Cooley et al., 2011). Although a putative impact of the pulmonary protease inhibitors secretory leukocyte protease inhibitor I and trappin‐2 on ENaC and Na+ transport in the airways remains unknown, their antibacterial and anti‐fungal properties towards common micro‐organisms in CF patients have been reported (Sallenave, 2010; Zani et al., 2011). Similarly, loss of either of the two Kunitz‐type transmembrane serine protease inhibitors, hepatocyte growth factor activator inhibitor (HAI)‐1 or ‐2, expressed by tubular epithelium in kidney (Yamauchi et al., 2004), is associated with embryonic lethality in mice (Mitchell et al., 2001; Tanaka et al., 2005). All developmental defects in HAI‐1‐ and HAI‐2‐deficient embryos are prevented by simultaneous CAP1 deficiency, but not by the absence of ENaC, suggesting that ENaC activity is not involved (Szabo et al., 2012). The protease activity of CAP1 can be inhibited by the serine protease inhibitor protease nexin‐1 (also known as SerpinE2) (Chen et al., 2004b; Crisante et al., 2014). Nexin‐1 is known to be an endogenous inhibitor of α‐thrombin, plasmin and plasminogen activators, and Wakida et al. showed an inhibitory effect of nexin‐1 on CAP1‐induced ENaC currents and proposed that nexin‐1 could exert a natriuretic effect by inhibiting CAP1 activity (Wakida et al., 2006). We also recently reported an inhibitory effect of nexin‐1 on both wild‐type and catalytically inactive mutant CAP1 in vitro and in an in vivo model, indicating that the catalytic site of CAP1 is dispensable for nexin‐1 inhibition (Crisante et al., 2014), and demonstrating a novel inhibitory interaction between CAP1 and nexin‐1. This opens the search for specific CAP1 antagonists that are independent of its catalytic activity.
Altogether, proteases (in particular serine proteases) and their inhibitors are important regulators of ENaC function. Although progress has been made on the mechanism of such regulations, the precise signalling pathways remain to be disclosed, which necessitates the usage of in vivo models to define their physiological significance.
Outlook
As discussed above, all ENaC/DEG family members share the same subunit topology and form Na+ channels that are inhibited by amiloride (Kellenberger and Schild, 2015). More recently, it was found that ENaC and ASICs are both regulated by proteases and also that ENaC is pH‐dependent (Table 3). The tissue expression and physiological roles are, however, very different between ASICs and ENaC. Whereas ENaC is well known for its role in mediating Na+ entry in epithelial cells, especially in the kidney, lung and colon, ASICs are predominantly found in the CNS and the PNS. The question arises whether in tissues expressing both ASIC and ENaC channels, overlapping functions can be identified. No function has so far been found for ENaC in the ear and in DRG neurons, in contrast to ASICs (Rusch and Hummler, 1999; Raouf et al., 2012). There is currently evidence for roles of both ENaC and ASICs in mechanosensation in arterial baroreceptors (Drummond et al., 2001; Lu et al., 2009). ENaC β and γ subunits are also expressed in vascular smooth muscle cells where they contribute to pressure‐induced constriction. Similarly, ASICs in vascular smooth muscle cells probably regulate vascular tone and chemotactic migration (Drummond et al., 2008). Other overlapping functions of ENaC and ASICs may still be discovered. Interestingly, two recent studies indicate that overexpression of ASICs together with ENaC may lead to the formation of hybrid ENaC/ASIC channels (Kapoor et al., 2011; Jeggle et al., 2015).
What will be the next challenges and advancements in ENaC and ASIC research? For ASICs, it will be important to identify and develop specific and potent small molecule inhibitors, with two aims: firstly, to confirm the physiological and pathological roles of ASICs; and secondly, to take advantage of ASIC pharmacology to treat human diseases. The structural information together with complementary approaches will lead to a better understanding of the molecular mechanisms of ASIC function, allowing rational drug development. For the ENaC field, the distribution and function of ENaC have still to be unveiled in classical and non‐classical tissues and organs and these may reveal as yet unknown functions of ENaC, for example, a defect in lipid synthesis and processing in skin (Charles et al., 2008). Conditional and inducible gene targeting of single ENaC subunits in adult mice may help to shed some light on these important issues.
Author contributions
The authors contributed equally to the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
Work in the author's laboratories was funded by Swiss national science foundation grants 31003A_163347 (to E.H.) and 31003A_153419 (to S.K.), and by the Swiss National Center of Competence in Research, kidney.ch, Leducq Foundation, and the networking support by the COST Action ADMIRE BM 1301 to E.H. NC‐IUPHAR receives financial support from the Wellcome Trust.
Boscardin, E. , Alijevic, O. , Hummler, E. , Frateschi, S. , and Kellenberger, S. (2016) The function and regulation of acid‐sensing ion channels (ASICs) and the epithelial Na+ channel (ENaC): IUPHAR Review 19. British Journal of Pharmacology, 173: 2671–2701. doi: 10.1111/bph.13533.
This article was contributed by the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC‐IUPHAR).
References
- Abdrakhmanova G, Cleemann L, Lindstrom J, Morad M (2004). Differential modulation of beta2 and beta4 subunits of human neuronal nicotinic acetylcholine receptors by acidification. Mol Pharmacol 66: 347–355. [DOI] [PubMed] [Google Scholar]
- Abdrakhmanova G, Dorfman J, Xiao Y, Morad M (2002). Protons enhance the gating kinetics of the alpha3/beta4 neuronal nicotinic acetylcholine receptor by increasing its apparent affinity to agonists. Mol Pharmacol 61: 369–378. [DOI] [PubMed] [Google Scholar]
- Adams CM, Anderson MG, Motto DG, Price MP, Johnson WA, Welsh MJ (1998). Ripped pocket and pickpocket, novel drosophila deg/enac subunits expressed in early development and in mechanosensory neurons. J Cell Biol 140: 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams CM, Snyder PM, Welsh MJ (1999). Paradoxical stimulation of a DEG/ENaC channel by amiloride. J Biol Chem 274: 15500–15504. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: voltage‐gated ion channels. Brit J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: enzymes. Brit J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: other ion channels. Brit J Pharmacol 172: 5942–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015d). THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: transporters. Brit J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015e). THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: ligand‐gated ion channels. Brit J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alijevic O, Kellenberger S (2012). Subtype‐specific modulation of acid‐sensing ion channel (ASIC) function by 2‐guanidine‐4‐methylquinazoline. J Biol Chem 287: 36059–36070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Attwell D (2002). Modulation of ASIC channels in rat cerebellar Purkinje neurons by ischaemia‐related signals. J Physiol‐London 543: 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam A, Palmer LG (2007). Determination of epithelial Na+ channel subunit stoichiometry from single‐channel conductances. J Gen Physiol 130: 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen D, Vuagniaux G, Fowler‐Jaeger N, Hummler E, Rossier BC (2006). Activation of epithelial sodium channels by mouse channel activating proteases (mCAP) expressed in xenopus oocytes requires catalytic activity of mCAP3 and mCAP2 but not mCAP1. J Am Soc Nephrol 17: 968–976. [DOI] [PubMed] [Google Scholar]
- Arnadottir J, O'Hagan R, Chen Y, Goodman MB, Chalfie M (2011). The DEG/ENaC protein MEC‐10 regulates the transduction channel complex in Caenorhabditis elegans touch receptor neurons. J Neurosci 31: 12695–12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo JP, Gamba G (2012). Advances in WNK signaling of salt and potassium metabolism: clinical implications. Am J Nephrol 35: 379–386. [DOI] [PubMed] [Google Scholar]
- Arroyo JP, Ronzaud C, Lagnaz D, Staub O, Gamba G (2011). Aldosterone paradox: differential regulation of ion transport in distal nephron. Physiology 26: 115–123. [DOI] [PubMed] [Google Scholar]
- Askwith CC, Cheng C, Ikuma M, Benson C, Price MP, Welsh MJ (2000). Neuropeptide FF and FMRFamide potentiate acid‐evoked currents from sensory neurons and proton‐gated DEG/ENaC channels. Neuron 26: 133–141. [DOI] [PubMed] [Google Scholar]
- Babini E, Paukert M, Geisler HS, Grunder S (2002). Alternative splicing and interaction with di‐ and polyvalent cations control the dynamic range of acid‐sensing ion channel 1 (ASIC1). J Biol Chem 277: 41597–41603. [DOI] [PubMed] [Google Scholar]
- Baconguis I, Bohlen CJ, Goehring A, Julius D, Gouaux E (2014). X‐ray structure of acid‐sensing ion channel 1‐snake toxin complex reveals open state of a Na+‐selective channel. Cell 156: 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baconguis I, Gouaux E (2012). Structural plasticity and dynamic selectivity of acid‐sensing ion channel‐spider toxin complexes. Nature 489: 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker PM, Nguyen MS, Gatzy JT, Grubb B, Norman H, Hummler E et al. (1998). Role of gammaENaC subunit in lung liquid clearance and electrolyte balance in newborn mice. Insights into perinatal adaptation and pseudohypoaldosteronism. J Clin Invest 102: 1634–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron A, Diochot S, Salinas M, Deval E, Noel J, Lingueglia E (2013). Venom toxins in the exploration of molecular, physiological and pathophysiological functions of acid‐sensing ion channels. Toxicon 75: 187–204. [DOI] [PubMed] [Google Scholar]
- Baron A, Lingueglia E (2015). Pharmacology of acid‐sensing ion Channels – physiological and therapeutical perspectives. Neuropharmacology 94: 19–35. [DOI] [PubMed] [Google Scholar]
- Bartoi T, Augustinowski K, Polleichtner G, Grunder S, Ulbrich MH (2014). Acid‐sensing ion channel (ASIC) 1a/2a heteromers have a flexible 2:1/1:2 stoichiometry. Proc Natl Acad Sci U S A 111: 8281–8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NJ, McQueen DS, Thomson D, Gauldie SD, Wilson AW, Salter DM et al. (2006). Attenuation of experimental arthritis in TRPV1R knockout mice. Exp Mol Pathol 81: 166–170. [DOI] [PubMed] [Google Scholar]
- Bassler EL, Ngo‐Anh TJ, Geisler HS, Ruppersberg JP, Grunder S (2001). Molecular and functional characterization of acid‐sensing ion channel (ASIC) 1b. J Biol Chem 276: 33782–33787. [DOI] [PubMed] [Google Scholar]
- Baumann TK, Chaudhary P, Martenson ME (2004). Background potassium channel block and TRPV1 activation contribute to proton depolarization of sensory neurons from humans with neuropathic pain. Eur J Neurosci 19: 1343–1351. [DOI] [PubMed] [Google Scholar]
- Benson CJ, Xie JH, Wemmie JA, Price MP, Henss JM, Welsh MJ et al. (2002). Heteromultimers of DEG/ENaC subunits form H+‐gated channels in mouse sensory neurons. Proc Natl Acad Sci U S A 99: 2338–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H et al. (1998). Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A 95: 9424–9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertog M, Cuffe JE, Pradervand S, Hummler E, Hartner A, Porst M et al. (2008). Aldosterone responsiveness of the epithelial sodium channel (ENaC) in colon is increased in a mouse model for Liddle's syndrome. J Physiol 586: 459–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyenbach KW, Wieczorek H (2006). The V‐type H+ ATPase: molecular structure and function, physiological roles and regulation. J Exp Biol 209: 577–589. [DOI] [PubMed] [Google Scholar]
- Birdsong WT, Fierro L, Williams FG, Spelta V, Naves LA, Knowles M et al. (2010). Sensing muscle ischemia: coincident detection of acid and ATP via interplay of two ion channels. Neuron 68: 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard MG, Kellenberger S (2011). Effect of a temperature increase in the non‐noxious range on proton‐evoked ASIC and TRPV1 activity. Pflugers Arch 461: 123–139. [DOI] [PubMed] [Google Scholar]
- Blanchard MG, Rash LD, Kellenberger S (2012). Inhibition of voltage‐gated Na+ currents in sensory neurones by the sea anemone toxin APETx2. Br J Pharmacol 165: 2167–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot‐Chabaud M, Djelidi S, Courtois‐Coutry N, Fay M, Cluzeaud F, Hummler E et al. (2001). Coordinate control of Na,K‐atpase mRNA expression by aldosterone, vasopressin and cell sodium delivery in the cortical collecting duct. Cell Mol Biol 47: 247–253. [PubMed] [Google Scholar]
- Bohlen CJ, Chesler AT, Sharif‐Naeini R, Medzihradszky KF, Zhou S, King D et al. (2011). A heteromeric Texas coral snake toxin targets acid‐sensing ion channels to produce pain. Nature 479: 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillat A, Alijevic O, Kellenberger S (2014). Calcium entry via TRPV1 but not ASICs induces neuropeptide release from sensory neurons. Mol Cell Neurosci 61: 13–22. [DOI] [PubMed] [Google Scholar]
- Bollag WB (2014). Regulation of aldosterone synthesis and secretion. Compr Physiol 4: 1017–1055. [DOI] [PubMed] [Google Scholar]
- Bonora M, Riffault L, Marie S, Mall M, Clement A, Tabary O (2011). Morphological analysis of the trachea and pattern of breathing in betaENaC‐Tg mice. Respir Physiol Neurobiol 178: 346–348. [DOI] [PubMed] [Google Scholar]
- Boucher RC (2007). Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med 58: 157–170. [DOI] [PubMed] [Google Scholar]
- Bray GA, Vollmer WM, Sacks FM, Obarzanek E, Svetkey LP, Appel LJ et al. (2004). A further subgroup analysis of the effects of the DASH diet and three dietary sodium levels on blood pressure: results of the DASH‐Sodium Trial. Am J Cardiol 94: 222–227. [DOI] [PubMed] [Google Scholar]
- Brennan ML, Schrijver I (2016). Cystic Fibrosis: A Review of Associated Phenotypes, Use of Molecular Diagnostic Approaches, Genetic Characteristics, Progress, and Dilemmas. J Mol Diagn 18: 3–14. [DOI] [PubMed] [Google Scholar]
- Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM et al. (2007). Epithelial Na+ channels are fully activated by furin‐ and prostasin‐dependent release of an inhibitory peptide from the gamma‐subunit. J Biol Chem 282: 6153–6160. [DOI] [PubMed] [Google Scholar]
- Buhl KB, Oxlund CS, Friis UG, Svenningsen P, Bistrup C, Jacobsen IA et al. (2014). Plasmin in urine from patients with type 2 diabetes and treatment‐resistant hypertension activates ENaC in vitro. J Hypertens 32: 1672–1677. [DOI] [PubMed] [Google Scholar]
- Butterworth MB, Zhang L, Heidrich EM, Myerburg MM, Thibodeau PH (2012). Activation of the epithelial sodium channel (ENaC) by the alkaline protease from Pseudomonas aeruginosa. J Biol Chem 287: 32556–32565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth MB, Zhang L, Liu X, Shanks RM, Thibodeau PH (2014). Modulation of the epithelial sodium channel (ENaC) by bacterial metalloproteases and protease inhibitors. PLoS One 9: e100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadiou H, Studer M, Jones NG, Smith ES, Ballard A, McMahon SB et al. (2007). Modulation of acid‐sensing ion channel activity by nitric oxide. J Neurosci 27: 13251–13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell RA, Boucher RC, Stutts MJ (2005). Neutrophil elastase activates near‐silent epithelial Na+ channels and increases airway epithelial Na + transport. Am J Physiol Lung Cell Mol Physiol : L813–L819. [DOI] [PubMed] [Google Scholar]
- Carattino MD, Hughey RP, Kleyman TR (2008). Proteolytic processing of the epithelial sodium channel {gamma} subunit has a dominant role in channel activation. J Biol Chem 283: 25290–25295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carattino MD, Mueller GM, Palmer LG, Frindt G, Rued AC, Hughey RP et al. (2014). Prostasin interacts with the epithelial Na+ channel and facilitates cleavage of the gamma‐subunit by a second protease. Am J Physiol Renal Physiol 307: F1080–F1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda‐Bueno M, Cervantes‐Perez LG, Rojas‐Vega L, Arroyo‐Garza I, Vazquez N, Moreno E et al. (2014). Modulation of NCC activity by low and high K(+) intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol 306: F1507–F1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen‐Zeitz KR et al. (2000). Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288: 306–313. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997). The capsaicin receptor: a heat‐activated ion channel in the pain pathway. Nature 389: 816–824. [DOI] [PubMed] [Google Scholar]
- Chalmers J, Morgan T, Doyle A, Dickson B, Hopper J, Mathews J et al. (1986). Australian National Health and Medical Research Council dietary salt study in mild hypertension. J Hypertens Suppl 4: S629–S637. [PubMed] [Google Scholar]
- Chandrashekar J, Kuhn C, Oka Y, Yarmolinsky DA, Hummler E, Ryba NJ et al. (2010). The cells and peripheral representation of sodium taste in mice. Nature 464: 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CT, Bens M, Hummler E, Boulkroun S, Schild L, Teulon J et al. (2005). Vasopressin‐stimulated CFTR Cl‐ currents are increased in the renal collecting duct cells of a mouse model of Liddle's syndrome. J Physiol 562: 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles RP, Guitard M, Leyvraz C, Breiden B, Haftek M, Haftek‐Terreau Z et al. (2008). Postnatal requirement of the epithelial sodium channel for maintenance of epidermal barrier function. J Biol Chem 283: 2622–2630. [DOI] [PubMed] [Google Scholar]
- Cheek DB, Perry JW (1958). A salt wasting syndrome in infancy. Arch Dis Child 33: 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Akopian AN, Sivilotti L, Colquhoun D, Burnstock G, Wood JN (1995). A P2X purinoceptor expressed by a subset of sensory neurons. Nature 377: 428–431. [DOI] [PubMed] [Google Scholar]
- Chen CC, Wong CW (2013). Neurosensory mechanotransduction through acid‐sensing ion channels. J Cell Mol Med 17: 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H‐Y, Chesler M (2015). Autocrine boost of NMDAR current in hippocampal CA1 pyramidal neurons by a PMCA‐dependent, perisynaptic, extracellular pH shift. J Neurosci 35: 873–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Chesler M (1992). Modulation of extracellular pH by glutamate and GABA in rat hippocampal slices. J Neurophysiol 67: 29–36. [DOI] [PubMed] [Google Scholar]
- Chen LM, Zhang X, Chai KX (2004b). Regulation of prostasin expression and function in the prostate. Prostate 59: 1–12. [DOI] [PubMed] [Google Scholar]
- Chen X, Qiu L, Li M, Durrnagel S, Orser BA, Xiong ZG et al. (2010). Diarylamidines: high potency inhibitors of acid‐sensing ion channels. Neuropharmacology 58: 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Dillon GH, Huang R (2004a). Molecular determinants of proton modulation of glycine receptors. J Biol Chem 279: 876–883. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhao R, Zhao M, Liang X, Bhattarai D, Dhiman R et al. (2014). Regulation of epithelial sodium channels in urokinase plasminogen activator deficiency. Am J Physiol Lung Cell Mol Physiol 307: L609–L617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M (1990). The regulation and modulation of pH in the nervous system. Prog Neurobiol 34: 401–427. [DOI] [PubMed] [Google Scholar]
- Chesler M (2003). Regulation and modulation of pH in the brain. Physiol Rev 83: 1183–1221. [DOI] [PubMed] [Google Scholar]
- Chiang PH, Chien TC, Chen CC, Yanagawa Y, Lien CC (2015). ASIC‐dependent LTP at multiple glutamatergic synapses in amygdala network is required for fear memory. Sci Rep 5: 10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S et al. (2008). Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 74: 1403–1409. [DOI] [PubMed] [Google Scholar]
- Chraibi A, Horisberger JD (2002). Na self inhibition of human epithelial Na channel: temperature dependence and effect of extracellular proteases. J Gen Physiol 120: 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chraibi A, Vallet V, Firsov D, Hess SK, Horisberger JD (1998). Protease modulation of the activity of the epithelial sodium channel expressed in Xenopus oocytes. J Gen Physiol 111: 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen BM, Perrier R, Wang Q, Zuber AM, Maillard M, Mordasini D et al. (2010). Sodium and potassium balance depends on alphaENaC expression in connecting tubule. J Am Soc Nephrol 21: 1942–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen BM, Zuber AM, Loffing J, Stehle JC, Deen PM, Rossier BC et al. (2011). alphaENaC‐mediated lithium absorption promotes nephrogenic diabetes insipidus. J Am Soc Nephrol 22: 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X‐P, Zhu X‐M, Wei W‐L, Li G‐H, Simon RP, MacDonald JF et al. (2003). Acidosis decreases low Ca(2+)‐induced neuronal excitation by inhibiting the activity of calcium‐sensing cation channels in cultured mouse hippocampal neurons. J Physiol (Lond) 550: 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichy A, Ackels T, Tsitoura C, Kahan A, Gronloh N, Sochtig M et al. (2015). Extracellular pH regulates excitability of vomeronasal sensory neurons. J Neurosci 35: 4025–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EB, Jovov B, Rooj AK, Fuller CM, Benos DJ (2010). Proteolytic cleavage of human acid‐sensing ion channel 1 by the serine protease matriptase. J Biol Chem 285: 27130–27143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claydon TW, Boyett MR, Sivaprasadarao A, Ishii K, Owen JM, O'Beirne HA et al. (2000). Inhibition of the K+ channel kv1.4 by acidosis: protonation of an extracellular histidine slows the recovery from N‐type inactivation. J Physiol (Lond) 526 (Pt 2): 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clunes MT, Boucher RC (2007). Cystic fibrosis: the mechanisms of pathogenesis of an inherited lung disorder. Drug Discov Today Dis Mech 4: 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockayne DA, Hamilton SG, Zhu QM, Dunn PM, Zhong Y, Novakovic S et al. (2000). Urinary bladder hyporeflexia and reduced pain‐related behaviour in P2X(3)‐deficient mice. Nature 407: 1011–1015. [DOI] [PubMed] [Google Scholar]
- Cooley J, Sontag MK, Accurso FJ, Remold‐O'Donnell E (2011). SerpinB1 in cystic fibrosis airway fluids: quantity, molecular form and mechanism of elastase inhibition. Eur Respir J 37: 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper BY, Johnson RD, Rau KK (2004). Characterization and function of TWIK‐related acid sensing K+ channels in a rat nociceptive cell. Neuroscience 129: 209–224. [DOI] [PubMed] [Google Scholar]
- Coryell MW, Wunsch AM, Haenfler JM, Allen JE, Schnizler M, Ziemann AE et al. (2009). Acid‐sensing ion channel‐1a in the amygdala, a novel therapeutic target in depression‐related behavior. J Neurosci 29: 5381–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisante G, Battista L, Iwaszkiewicz J, Nesca V, Merillat AM, Sergi C et al. (2014). The CAP1/Prss8 catalytic triad is not involved in PAR2 activation and protease nexin‐1 (PN‐1) inhibition. FASEB J 28: 4792–4805. [DOI] [PubMed] [Google Scholar]
- Dahlmann A, Pradervand S, Hummler E, Rossier BC, Frindt G, Palmer LG (2003). Mineralocorticoid regulation of epithelial Na + channels is maintained in a mouse model of Liddle's syndrome. Am J Physiol Renal Physiol 285: F310–F318. [DOI] [PubMed] [Google Scholar]
- Daumas P, Andersen OS (1993). Proton block of rat brain sodium channels. Evidence for two proton binding sites and multiple occupancy. J Gen Physiol 101: 27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson RJ, Benz J, Stohler P, Tetaz T, Joseph C, Huber S et al. (2012). Structure of the acid‐sensing ion channel 1 in complex with the gating modifier Psalmotoxin 1. Nat Commun 3: 936 doi: 10.1038/ncomms1917. [DOI] [PubMed] [Google Scholar]
- Deitmer JW (2002). A role for CO(2) and bicarbonate transporters in metabolic exchanges in the brain. J Neurochem 80: 721–726. [DOI] [PubMed] [Google Scholar]
- Delaunay A, Gasull X, Salinas M, Noel J, Friend V, Lingueglia E et al. (2012). Human ASIC3 channel dynamically adapts its activity to sense the extracellular pH in both acidic and alkaline directions. Proc Natl Acad Sci U S A 109: 13124–13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning GM, Ostedgaard LS, Welsh MJ (1992). Abnormal localization of cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia. J Cell Biol 118: 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deval E, Baron A, Lingueglia E, Mazarguil H, Zajac JM, Lazdunski M (2003). Effects of neuropeptide SF and related peptides on acid sensing ion channel 3 and sensory neuron excitability. Neuropharmacology 44: 662–671. [DOI] [PubMed] [Google Scholar]
- Deval E, Lingueglia E (2015). Acid‐sensing ion channels and nociception in the peripheral and central nervous systems. Neuropharmacology 94: 49–57. [DOI] [PubMed] [Google Scholar]
- Deval E, Noel J, Gasull X, Delaunay A, Alloui A, Friend V et al. (2011). Acid‐sensing ion channels in postoperative pain. J Neurosci 31: 6059–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deval E, Noel J, Lay N, Alloui A, Diochot S, Friend V et al. (2008). ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J 27: 3047–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries SH (2001). Exocytosed protons feedback to suppress the Ca2 + current in mammalian cone photoreceptors. Neuron 32: 1107–1117. [DOI] [PubMed] [Google Scholar]
- Dietrich CJ, Morad M (2010). Synaptic acidification enhances GABAA signaling. J Neurosci 30: 16044–16052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diochot S, Baron A, Salinas M, Douguet D, Scarzello S, Dabert‐Gay AS et al. (2012). Black mamba venom peptides target acid‐sensing ion channels to abolish pain. Nature 490: 552–555. [DOI] [PubMed] [Google Scholar]
- Dirajlal S, Pauers LE, Stucky CL (2003). Differential response properties of IB4‐positive and ‐negative unmyelinated sensory neurons to protons and capsaicin. J Neurophysiol 89: 513–524. [DOI] [PubMed] [Google Scholar]
- Dmitriev AV, Mangel SC (2001). Circadian clock regulation of pH in the rabbit retina. J Neurosci 21: 2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donier E, Rugiero F, Jacob C, Wood JN (2008). Regulation of ASIC activity by ASIC4‐‐new insights into ASIC channel function revealed by a yeast two‐hybrid assay. Eur J Neurosci 28: 74–86. [DOI] [PubMed] [Google Scholar]
- Dooley R, Harvey BJ, Thomas W (2012). Non‐genomic actions of aldosterone: from receptors and signals to membrane targets. Mol Cell Endocrinol 350: 223–234. [DOI] [PubMed] [Google Scholar]
- Drew LJ, Rohrer DK, Price MP, Blaver KE, Cockayne DA, Cesare P et al. (2004). Acid‐sensing ion channels ASIC2 and ASIC3 do not contribute to mechanically activated currents in mammalian sensory neurones. J Physiol (Lond) 556: 691–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond HA, Jernigan NL, Grifoni SC (2008). Sensing tension: epithelial sodium channel/acid‐sensing ion channel proteins in cardiovascular homeostasis. Hypertension 51: 1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond HA, Welsh MJ, Abboud FM (2001). ENaC subunits are molecular components of the arterial baroreceptor complex. Ann N Y Acad Sci 940: 42–47. [DOI] [PubMed] [Google Scholar]
- Du J, Reznikov LR, Price MP, Zha XM, Lu Y, Moninger TO et al. (2014). Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proc Natl Acad Sci U S A 111: 8961–8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube GR, Lehto SG, Breese NM, Baker SJ, Wang X, Matulenko MA et al. (2005). Electrophysiological and in vivo characterization of A‐317567, a novel blocker of acid sensing ion channels. Pain 117: 88–96. [DOI] [PubMed] [Google Scholar]
- Ecelbarger CA, Kim GH, Wade JB, Knepper MA (2001). Regulation of the abundance of renal sodium transporters and channels by vasopressin. Exp Neurol 171: 227–234. [DOI] [PubMed] [Google Scholar]
- Egli M, Duplain H, Lepori M, Cook S, Nicod P, Hummler E et al. (2004). Defective respiratory amiloride‐sensitive sodium transport predisposes to pulmonary oedema and delays its resolution in mice. J Physiol 560: 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehling P, Cerina M, Budde T, Meuth SG, Bittner S (2015). The CNS under pathophysiologic attack–examining the role of K2p channels. Pflugers Arch 467: 959–972. [DOI] [PubMed] [Google Scholar]
- Fejes‐Toth G, Frindt G, Naray‐Fejes‐Toth A, Palmer LG (2008). Epithelial Na+ channel activation and processing in mice lacking SGK1. Am J Physiol Renal Physiol 294: F1298–F1305. [DOI] [PubMed] [Google Scholar]
- Ferenczi EA, Vierock J, Atsuta‐Tsunoda K, Tsunoda SP, Ramakrishnan C, Gorini C et al. (2016). Optogenetic approaches addressing extracellular modulation of neural excitability. Sci Rep 6: 23947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firsov D, Gautschi I, Merillat AM, Rossier BC, Schild L (1998). The heterotetrameric architecture of the epithelial sodium channel (ENaC). EMBO J 17: 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MJ, Reeh PW, Sauer SK (2003). Proton‐induced calcitonin gene‐related peptide release from rat sciatic nerve axons, in vitro, involving TRPV1. Eur J Neurosci 18: 803–810. [DOI] [PubMed] [Google Scholar]
- Frateschi S, Camerer E, Crisante G, Rieser S, Membrez M, Charles RP et al. (2011). PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat Commun 2: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frateschi S, Keppner A, Malsure S, Iwaszkiewicz J, Sergi C, Merillat AM et al. (2012). Mutations of the serine protease CAP1/Prss8 lead to reduced embryonic viability, skin defects, and decreased ENaC activity. Am J Pathol 181: 605–615. [DOI] [PubMed] [Google Scholar]
- Fu Y, Gerasimova M, Batz F, Kuczkowski A, Alam Y, Sanders PW et al. (2015). PPARgamma agonist‐induced fluid retention depends on alphaENaC expression in connecting tubules. Nephron 129: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs W, Larsen EH, Lindemann B (1977). Current–voltage curve of sodium channels and concentration dependence of sodium permeability in frog skin. J Physiol 267: 137–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuldner HH, Stadler H (1982). 31P‐NMR analysis of synaptic vesicles. Status of ATP and internal pH. Eur J Biochem 121: 519–524. [DOI] [PubMed] [Google Scholar]
- Furgeson SB, Linas S (2010). Mechanisms of type I and type II pseudohypoaldosteronism. J Am Soc Nephrol 21: 1842–1845. [DOI] [PubMed] [Google Scholar]
- Gao J, Duan B, Wang DG, Deng XH, Zhang GY, Xu L et al. (2005). Coupling between NMDA receptor and acid‐sensing ion channel contributes to ischemic neuronal death. Neuron 48: 635–646. [DOI] [PubMed] [Google Scholar]
- Garcia‐Caballero A, Dang Y, He H, Stutts MJ (2008). ENaC proteolytic regulation by channel‐activating protease 2. J Gen Physiol 132: 521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Caballero A, Ishmael SS, Dang Y, Gillie D, Bond JS, Milgram SL et al. (2011). Activation of the epithelial sodium channel by the metalloprotease meprin beta subunit. Channels (Austin) 5: 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Caballero A, Rasmussen JE, Gaillard E, Watson MJ, Olsen JC, Donaldson SH et al. (2009). SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc Natl Acad Sci U S A 106: 11412–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garty H, Edelman IS (1983). Amiloride‐sensitive trypsinization of apical sodium channels. Analysis of hormonal regulation of sodium transport in toad bladder. J Gen Physiol 81: 785–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerevich Z, Zadori ZS, Koles L, Kopp L, Milius D, Wirkner K et al. (2007). Dual effect of acid pH on purinergic P2X3 receptors depends on the histidine 206 residue. J Biol Chem 282: 33949–33957. [DOI] [PubMed] [Google Scholar]
- Gille T, Randrianarison‐Pellan N, Goolaerts A, Dard N, Uzunhan Y, Ferrary E et al. (2014). Hypoxia‐induced inhibition of epithelial Na(+) channels in the lung. Role of Nedd4‐2 and the ubiquitin‐proteasome pathway. Am J Respir Cell Mol Biol 50: 526–537. [DOI] [PubMed] [Google Scholar]
- Gonzales EB, Kawate T, Gouaux E (2009). Pore architecture and ion sites in acid‐sensing ion channels and P2X receptors. Nature 460: 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried JA, Chesler M (1996). Temporal resolution of activity‐dependent pH shifts in rat hippocampal slices. J Neurophysiol 76: 2804–2807. [DOI] [PubMed] [Google Scholar]
- Grunder S, Augustinowski K (2012). Toxin binding reveals two open state structures for one acid‐sensing ion channel. Channels (Austin) 6: 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guipponi M, Vuagniaux G, Wattenhofer M, Shibuya K, Vazquez M, Dougherty L et al. (2002). The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet 11: 2829–2836. [DOI] [PubMed] [Google Scholar]
- Haerteis S, Krappitz A, Krappitz M, Murphy JE, Bertog M, Krueger B et al. (2014). Proteolytic activation of the human epithelial sodium channel by trypsin IV and trypsin I involves distinct cleavage sites. J Biol Chem 289: 19067–19078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerteis S, Krappitz M, Bertog M, Krappitz A, Baraznenok V, Henderson I et al. (2012). Proteolytic activation of the epithelial sodium channel (ENaC) by the cysteine protease cathepsin‐S. Pflugers Arch 464: 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M, Firsov D, Vuagniaux G, Stutts MJ, Rossier BC (2007). A novel neutrophil elastase inhibitor prevents elastase activation and surface cleavage of the epithelial sodium channel expressed in Xenopus laevis oocytes. J Biol Chem 282: 58–64. [DOI] [PubMed] [Google Scholar]
- Harris M, Garcia‐Caballero A, Stutts MJ, Firsov D, Rossier BC (2008). Preferential assembly of epithelial sodium channel (ENaC) subunits in Xenopus oocytes: role of furin‐mediated endogenous proteolysis. J Biol Chem 283: 7455–7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselager M, Timmermann DB, Ahring PK (2004). pH dependency and desensitization kinetics of heterologously expressed combinations of acid‐sensing ion channel subunits. J Biol Chem 279: 11006–11015. [DOI] [PubMed] [Google Scholar]
- Highstein SM, Holstein GR, Mann MA, Rabbitt RD (2014). Evidence that protons act as neurotransmitters at vestibular hair cell‐calyx afferent synapses. Proc Natl Acad Sci U S A 111: 5421–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (1992). Ionic Channels of Excitable Membranes, 2nd edn. Sinauer Associates, Inc: Sunderland, MA. [Google Scholar]
- Hirsh AJ, Molino BF, Zhang J, Astakhova N, Geiss WB, Sargent BJ et al. (2006). Design, synthesis, and structure–activity relationships of novel 2‐substituted pyrazinoylguanidine epithelial sodium channel blockers: drugs for cystic fibrosis and chronic bronchitis. J Med Chem 49: 4098–4115. [DOI] [PubMed] [Google Scholar]
- Hirsh AJ, Sabater JR, Zamurs A, Smith RT, Paradiso AM, Hopkins S et al. (2004). Evaluation of second generation amiloride analogs as therapy for cystic fibrosis lung disease. J Pharmacol Exp Ther 311: 929–938. [DOI] [PubMed] [Google Scholar]
- Hirsh AJ, Zhang J, Zamurs A, Fleegle J, Thelin WR, Caldwell RA et al. (2008). Pharmacological properties of N‐(3,5‐diamino‐6‐chloropyrazine‐2‐carbonyl)‐N′‐4‐[4‐(2,3‐dihydroxypropoxy)phenyl] butyl‐guanidine methanesulfonate (552‐02), a novel epithelial sodium channel blocker with potential clinical efficacy for cystic fibrosis lung disease. J Pharmacol Exp Ther 325: 77–88. [DOI] [PubMed] [Google Scholar]
- Hobbs CA, Blanchard MG, Alijevic O, Tan CD, Kellenberger S, Bencharit S et al. (2013). Identification of the SPLUNC1 ENaC‐inhibitory domain yields novel strategies to treat sodium hyperabsorption in cystic fibrosis airway epithelial cultures. Am J Physiol Lung Cell Mol Physiol 305: L990–L1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, Wismer CT, Mikusa J, Zhu CZ, Zhong C, Gauvin DM et al. (2005). A‐425619 [1‐isoquinolin‐5‐yl‐3‐(4‐trifluoromethyl‐benzyl)‐urea], a novel transient receptor potential type V1 receptor antagonist, relieves pathophysiological pain associated with inflammation and tissue injury in rats. J Pharmacol Exp Ther 314: 410–421. [DOI] [PubMed] [Google Scholar]
- Hoyer D, Bartfai T (2012). Neuropeptides and neuropeptide receptors: drug targets, and peptide and non‐peptide ligands: a tribute to Prof. Dieter Seebach. Chem Biodivers 9: 2367–2387. [DOI] [PubMed] [Google Scholar]
- Hsu KS, Huang CC (1997). Characterization of the anoxia‐induced long‐term synaptic potentiation in area CA1 of the rat hippocampus. Br J Pharmacol 122: 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PA, Brierley SM, Young RL, Blackshaw LA (2007). Localization and comparative analysis of acid‐sensing ion channel (ASIC1, 2, and 3) mRNA expression in mouse colonic sensory neurons within thoracolumbar dorsal root ganglia. J Comp Neurol 500: 863–875. [DOI] [PubMed] [Google Scholar]
- Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD et al. (2004). Epithelial sodium channels are activated by furin‐dependent proteolysis. J Biol Chem 279: 18111–18114. [DOI] [PubMed] [Google Scholar]
- Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL et al. (2003). Maturation of the epithelial Na+ channel involves proteolytic processing of the alpha‐ and gamma‐subunits. J Biol Chem 278: 37073–37082. [DOI] [PubMed] [Google Scholar]
- Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A et al. (1996). Early death due to defective neonatal lung liquid clearance in alpha‐ENaC‐deficient mice. Nat Genet 12: 325–328. [DOI] [PubMed] [Google Scholar]
- Hummler E, Barker P, Talbot C, Wang Q, Verdumo C, Grubb B et al. (1997). A mouse model for the renal salt‐wasting syndrome pseudohypoaldosteronism. Proc Natl Acad Sci U S A 94: 11710–11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW (2003). Protons open acid‐sensing ion channels by catalyzing relief of Ca2 + blockade. Neuron 37: 75–84. [DOI] [PubMed] [Google Scholar]
- Jasti J, Furukawa H, Gonzales EB, Gouaux E (2007). Structure of acid‐sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449: 316–323. [DOI] [PubMed] [Google Scholar]
- Jeggle P, Callies C, Tarjus A, Fassot C, Fels J, Oberleithner H et al. (2013). Epithelial sodium channel stiffens the vascular endothelium in vitro and in Liddle mice. Hypertension 61: 1053–1059. [DOI] [PubMed] [Google Scholar]
- Jeggle P, Smith ES, Stewart AP, Haerteis S, Korbmacher C, Edwardson JM (2015). Atomic force microscopy imaging reveals the formation of ASIC/ENaC cross‐clade ion channels. Biochem Biophys Res Commun 464: 38–44. [DOI] [PubMed] [Google Scholar]
- Jendelová P, Syková E (1991). Role of glia in K+ and pH homeostasis in the neonatal rat spinal cord. Glia 4: 56–63. [DOI] [PubMed] [Google Scholar]
- Ji HL, Benos DJ (2004). Degenerin sites mediate proton activation of dbg‐epithelial sodium channel. J Biol Chem 279: 26939–26947. [DOI] [PubMed] [Google Scholar]
- Ji HL, Zhao R, Komissarov AA, Chang Y, Liu Y, Matthay MA (2015). Proteolytic regulation of epithelial sodium channels by urokinase plasminogen activator: cutting edge and cleavage sites. J Biol Chem 290: 5241–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NG, Slater R, Cadiou H, McNaughton P, McMahon SB (2004). Acid‐induced pain and its modulation in humans. J Neurosci 24: 10974–10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Kim D (2006). TREK‐2 (K2P10.1) and TRESK (K2P18.1) are major background K+ channels in dorsal root ganglion neurons. Am J Physiol Cell Physiol 291: C138–C146. [DOI] [PubMed] [Google Scholar]
- Kang S, Jang JH, Price MP, Gautam M, Benson CJ, Gong H et al. (2012). Simultaneous disruption of mouse ASIC1a, ASIC2 and ASIC3 genes enhances cutaneous mechanosensitivity. PLoS One 7: e35225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor N, Lee W, Clark E, Bartoszewski R, McNicholas CM, Latham CB et al. (2011). Interaction of ASIC1 and ENaC subunits in human glioma cells and rat astrocytes. Am J Physiol Cell Physiol 300: C1246–C1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartner N, Augustinas O, Jensen TJ, Naismith AL, Riordan JR (1992). Mislocalization of delta F508 CFTR in cystic fibrosis sweat gland. Nat Genet 1: 321–327. [DOI] [PubMed] [Google Scholar]
- Kashlan OB, Adelman JL, Okumura S, Blobner BM, Zuzek Z, Hughey RP et al. (2011). Constraint‐based, homology model of the extracellular domain of the epithelial Na+ channel alpha subunit reveals a mechanism of channel activation by proteases. J Biol Chem 286: 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashlan OB, Kleyman TR (2011). ENaC structure and function in the wake of a resolved structure of a family member. Am J Physiol Renal Physiol 301: F684–F696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J (2005). Global burden of hypertension: analysis of worldwide data. Lancet 365: 217–223. [DOI] [PubMed] [Google Scholar]
- Keeble J, Russell F, Curtis B, Starr A, Pinter E, Brain SD (2005). Involvement of transient receptor potential vanilloid 1 in the vascular and hyperalgesic components of joint inflammation. Arthritis Rheum 52: 3248–3256. [DOI] [PubMed] [Google Scholar]
- Kehl SJ, Eduljee C, Kwan DCH, Zhang S, Fedida D (2002). Molecular determinants of the inhibition of human Kv1.5 potassium currents by external protons and Zn(2+). J Physiol (Lond) 541: 9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, Grutter T (2015). Architectural and functional similarities between trimeric ATP‐gated P2X receptors and acid‐sensing ion channels. J Mol Biol 427: 54–66. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, Schild L (2015). International Union of Basic and Clinical Pharmacology. XCI. Structure, function, and pharmacology of acid‐sensing ion channels and the epithelial Na+ channel. Pharmacol Rev 67: 1–35. [DOI] [PubMed] [Google Scholar]
- Keppner A, Andreasen D, Merillat AM, Bapst J, Ansermet C, Wang Q et al. (2015). Epithelial sodium channel‐mediated sodium transport is not dependent on the membrane‐bound serine protease CAP2/Tmprss4. PLoS One 10 .e0135224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BF, Wildman SS, Ziganshina LE, Pintor J, Burnstock G (1997). Effects of extracellular pH on agonism and antagonism at a recombinant P2X2 receptor. Br J Pharmacol 121: 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler D, App E, Schmitz‐Schumann M, Wurtemberger G, Matthys H (1986). Inhalation of amiloride improves the mucociliary and the cough clearance in patients with cystic fibroses. Eur J Respir Dis Suppl 146: 319–326. [PubMed] [Google Scholar]
- Kosari F, Sheng S, Li J, Mak DO, Foskett JK, Kleyman TR (1998). Subunit stoichiometry of the epithelial sodium channel. J Biol Chem 273: 13469–13474. [DOI] [PubMed] [Google Scholar]
- Kota P, Garcia‐Caballero A, Dang H, Gentzsch M, Stutts MJ, Dokholyan NV (2012). Energetic and structural basis for activation of the epithelial sodium channel by matriptase. Biochemistry 51: 3460–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova J, Winter C, Loffing‐Cueni D, Loffing J, Finberg KE, Lifton RP et al. (2006). The connecting tubule is the main site of the furosemide‐induced urinary acidification by the vacuolar H+‐ATPase. Kidney Int 70: 1706–1716. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Ferreira‐Filho CR, Nicholson C (1983). Alkaline and acid transients in cerebellar microenvironment. J Neurophysiol 49: 831–850. [DOI] [PubMed] [Google Scholar]
- Kreitzer MA, Collis LP, Molina AJA, Smith PJS, Malchow RP (2007). Modulation of extracellular proton fluxes from retinal horizontal cells of the catfish by depolarization and glutamate. J Gen Physiol 130: 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreple CJ, Lu Y, Taugher RJ, Schwager‐Gutman AL, Du J, Stump M et al. (2014). Acid‐sensing ion channels contribute to synaptic transmission and inhibit cocaine‐evoked plasticity. Nat Neurosci 17: 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishtal OA, Osipchuk YV, Shelest TN, Smirnoff SV (1987). Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Res 436: 352–356. [DOI] [PubMed] [Google Scholar]
- Krueger B, Schlotzer‐Schrehardt U, Haerteis S, Zenkel M, Chankiewitz VE, Amann KU et al. (2012). Four subunits (αβγΔ) of the epithelial sodium channel (ENaC) are expressed in the human eye in various locations. Invest Ophthalmol Vis Sci 53: 596–604. [DOI] [PubMed] [Google Scholar]
- Kuduk SD, Di Marco CN, Bodmer‐Narkevitch V, Cook SP, Cato MJ, Jovanovska A et al. (2010). Synthesis, structure–activity relationship, and pharmacological profile of analogs of the ASIC‐3 inhibitor A‐317567. ACS Chem Neurosci 1: 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuduk SD, Di Marco CN, Chang RK, Dipardo RM, Cook SP, Cato MJ et al. (2009). Amiloride derived inhibitors of acid‐sensing ion channel‐3 (ASIC3). Bioorg Med Chem Lett 19: 2514–2518. [DOI] [PubMed] [Google Scholar]
- Kusche‐Vihrog K, Tarjus A, Fels J, Jaisser F (2014). The epithelial Na+ channel: a new player in the vasculature. Curr Opin Nephrol Hypertens 23: 143–148. [DOI] [PubMed] [Google Scholar]
- Lang F, Shumilina E (2013). Regulation of ion channels by the serum‐ and glucocorticoid‐inducible kinase SGK1. FASEB J 27: 3–12. [DOI] [PubMed] [Google Scholar]
- Lazrak A, Nita I, Subramaniyam D, Wei S, Song W, Ji HL et al. (2009). Alpha(1)‐antitrypsin inhibits epithelial Na+ transport in vitro and in vivo. Am J Respir Cell Mol Biol 41: 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Moellic C, Ouvrard‐Pascaud A, Capurro C, Cluzeaud F, Fay M, Jaisser F et al. (2004). Early nongenomic events in aldosterone action in renal collecting duct cells: PKCalpha activation, mineralocorticoid receptor phosphorylation, and cross‐talk with the genomic response. J Am Soc Nephrol 15: 1145–1160. [PubMed] [Google Scholar]
- Lei S, Orser BA, Thatcher GR, Reynolds JN, MacDonald JF (2001). Positive allosteric modulators of AMPA receptors reduce proton‐induced receptor desensitization in rat hippocampal neurons. J Neurophysiol 85: 2030–2038. [DOI] [PubMed] [Google Scholar]
- Lesage F, Barhanin J (2011). Molecular Physiology of pH‐Sensitive Background K2P Channels. Physiology 26: 424–437. [DOI] [PubMed] [Google Scholar]
- Lesage F, Lazdunski M (2000). Molecular and functional properties of two‐pore‐domain potassium channels. Am J Physiol Renal Physiol 279: F793–F801. [DOI] [PubMed] [Google Scholar]
- Levenson JW, Skerrett PJ, Gaziano JM (2002). Reducing the global burden of cardiovascular disease: the role of risk factors. Prev Cardiol 5: 188–199. [DOI] [PubMed] [Google Scholar]
- Leviel F, Hubner CA, Houillier P, Morla L, El Moghrabi S, Brideau G et al. (2010). The Na+‐dependent chloride‐bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis C, Neidhart S, Holy C, North RA, Buell G, Surprenant A (1995). Coexpression of P2X2 and P2X3 receptor subunits can account for ATP‐gated currents in sensory neurons. Nature 377: 432–435. [DOI] [PubMed] [Google Scholar]
- Li T, Yang Y, Canessa CM (2014). A method for activation of endogenous acid‐sensing ion channel 1a (ASIC1a) in the nervous system with high spatial and temporal precision. J Biol Chem 289: 15441–15448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WG, Yu Y, Zhang ZD, Cao H, Xu TL (2011). ASIC3 channels integrate agmatine and multiple inflammatory signals through the nonproton ligand sensing domain. Mol Pain 6: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light AR, Hughen RW, Zhang J, Rainier J, Liu Z, Lee J (2008). Dorsal root ganglion neurons innervating skeletal muscle respond to physiological combinations of protons, ATP, and lactate mediated by ASIC, P2X, and TRPV1. J Neurophysiol 100: 1184–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S‐H, Chien Y‐C, Chiang W‐W, Liu Y‐Z, Lien C‐C, Chen C‐C (2015). Genetic mapping of ASIC4 and contrasting phenotype to ASIC1a in modulating innate fear and anxiety. Eur J Neurosci 41: 1553–1568. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, Champigny G, Lazdunski M, Barbry P (1995). Cloning of the amiloride‐sensitive FMRFamide peptide‐gated sodium channel. Nature 378: 730–733. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, de Weille JR, Bassilana F, Heurteaux C, Sakai H, Waldmann R et al. (1997). A modulatory subunit of acid sensing ion channels in brain and dorsal root ganglion cells. J Biol Chem 272: 29778–29783. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, Deval E, Lazdunski M (2006). FMRFamide‐gated sodium channel and ASIC channels: a new class of ionotropic receptors for FMRFamide and related peptides. Peptides 27: 1138–1152. [DOI] [PubMed] [Google Scholar]
- Liu MG, Li HS, Li WG, Wu YJ, Deng SN, Huang C et al. (2016). Acid‐sensing ion channel 1a contributes to hippocampal LTP inducibility through multiple mechanisms. Sci Rep 6: 23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffing J, Zecevic M, Feraille E, Kaissling B, Asher C, Rossier BC et al. (2001). Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Renal Physiol 280: F675–F682. [DOI] [PubMed] [Google Scholar]
- Lu Y, Ma X, Sabharwal R, Snitsarev V, Morgan D, Rahmouni K et al. (2009). The ion channel ASIC2 is required for baroreceptor and autonomic control of the circulation. Neuron 64: 885–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makani S, Chesler M (2010). Rapid rise of extracellular pH evoked by neural activity is generated by the plasma membrane calcium ATPase. J Neurophysiol 103: 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhanova N, Sequeira‐Lopez ML, Gomez RA, Kim HS, Smithies O (2006). Disturbed homeostasis in sodium‐restricted mice heterozygous and homozygous for aldosterone synthase gene disruption. Hypertension 48: 1151–1159. [DOI] [PubMed] [Google Scholar]
- Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC (2004). Increased airway epithelial Na+ absorption produces cystic fibrosis‐like lung disease in mice. Nat Med 10: 487–493. [DOI] [PubMed] [Google Scholar]
- Mall MA, Button B, Johannesson B, Zhou Z, Livraghi A, Caldwell RA et al. (2010). Airway surface liquid volume regulation determines different airway phenotypes in liddle compared with betaENaC‐overexpressing mice. J Biol Chem 285: 26945–26955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malsure S, Wang Q, Charles RP, Sergi C, Perrier R, Christensen BM et al. (2014). Colon‐specific deletion of epithelial sodium channel causes sodium loss and aldosterone resistance. J Am Soc Nephrol 25: 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamenko M, Zaika O, Prieto MC, Jensen VB, Doris PA, Navar LG et al. (2013). Chronic angiotensin II infusion drives extensive aldosterone‐independent epithelial Na+ channel activation. Hypertension 62: 1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra S, Ferru‐Clement R, Breuil V, Delaunay A, Christin M, Friend V et al. (2016). Non‐acidic activation of pain‐related acid‐sensing ion channel 3 by lipids. EMBO J 35: 414–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GR, Jain RK (1994). Noninvasive measurement of interstitial pH profiles in normal and neoplastic tissue using fluorescence ratio imaging microscopy. Cancer Res 54: 5670–5674. [PubMed] [Google Scholar]
- Marunaka Y, Hagiwara N, Tohda H (1992). Insulin activates single amiloride‐blockable Na channels in a distal nephron cell line (A6). Am J Physiol 263: F392–F400. [DOI] [PubMed] [Google Scholar]
- Mathie A, Veale EL (2015). Two‐pore domain potassium channels: potential therapeutic targets for the treatment of pain. Pflugers Arch 467: 931–943. [DOI] [PubMed] [Google Scholar]
- Mauro T, Guitard M, Behne M, Oda Y, Crumrine D, Komuves L et al. (2002). The ENaC channel is required for normal epidermal differentiation. J Invest Dermatol 118: 589–594. [DOI] [PubMed] [Google Scholar]
- Mazzuca M, Heurteaux C, Alloui A, Diochot S, Baron A, Voilley N et al. (2007). A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat Neurosci 10: 943–945. [DOI] [PubMed] [Google Scholar]
- McCallum L, Lip S, Padmanabhan S (2015). The hidden hand of chloride in hypertension. Pflug Arch Eur J Phy 467: 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald FJ, Yang B, Hrstka RF, Drummond HA, Tarr DE, McCray PB Jr et al. (1999). Disruption of the beta subunit of the epithelial Na+ channel in mice: hyperkalemia and neonatal death associated with a pseudohypoaldosteronism phenotype. Proc Natl Acad Sci U S A 96: 1727–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson DM, Angel I (1980). Determination of delta pH in cholinergic synaptic vesicles: its effect on storage and release of acetylcholine. Life Sci 27: 39–44. [DOI] [PubMed] [Google Scholar]
- Mickle AD, Shepherd AJ, Mohapatra DP (2015). Sensory TRP channels: the key transducers of nociception and pain. Prog Mol Biol Transl Sci 131: 73–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE (1998). Visualizing secretion and synaptic transmission with pH‐ sensitive green fluorescent proteins. Nature 394: 192–195. [DOI] [PubMed] [Google Scholar]
- Mironova E, Bugaj V, Roos KP, Kohan DE, Stockand JD (2012). Aldosterone‐independent regulation of the epithelial Na+ channel (ENaC) by vasopressin in adrenalectomized mice. Proc Natl Acad Sci U S A 109: 10095–10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell KJ, Pinson KI, Kelly OG, Brennan J, Zupicich J, Scherz P et al. (2001). Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat Genet 28: 241–249. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Breese NM, Witty M‐F, Ritchie J, Rainville M‐L, Ase A et al. (2005). Transgenic expression of a dominant‐negative ASIC3 subunit leads to increased sensitivity to mechanical and inflammatory stimuli. J Neurosci 25: 9893–9901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordasini D, Loffing‐Cueni D, Loffing J, Beatrice R, Maillard MP, Hummler E et al. (2015). ENaC activity in collecting ducts modulates NCC in cirrhotic mice. Pflugers Arch 467: 2529–2539. [DOI] [PubMed] [Google Scholar]
- Morral N, Bertranpetit J, Estivill X, Nunes V, Casals T, Gimenez J et al. (1994). The origin of the major cystic fibrosis mutation (delta F508) in European populations. Nat Genet 7: 169–175. [DOI] [PubMed] [Google Scholar]
- Mott DD, Washburn MS, Zhang S, Dingledine RJ (2003). Subunit‐dependent modulation of kainate receptors by extracellular protons and polyamines. J Neurosci 23: 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch WA, Hansen AJ (1984). Extracellular pH changes during spreading depression and cerebral ischemia: mechanisms of brain pH regulation. J Cereb Blood Flow Metab 4: 17–27. [DOI] [PubMed] [Google Scholar]
- Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C (2012). Aldosterone‐dependent and ‐independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol 303: F1289–F1299. [DOI] [PubMed] [Google Scholar]
- Nichols DP, Jiang D, Happoldt C, Berman R, Chu HW (2015). Therapeutic effects of alpha1‐antitrypsin on psedumonas aeruginosa infection in ENaC transgenic mice. PLoS One 10 .e0141232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obel AO (1989). Placebo‐controlled trial of potassium supplements in black patients with mild essential hypertension. J Cardiovasc Pharmacol 14: 294–296. [DOI] [PubMed] [Google Scholar]
- Olivier R, Scherrer U, Horisberger JD, Rossier BC, Hummler E (2002). Selected contribution: limiting Na(+) transport rate in airway epithelia from alpha‐ENaC transgenic mice: a model for pulmonary edema. J Appl Physiol 93: 1881–1887. [DOI] [PubMed] [Google Scholar]
- Omerbasic D, Schuhmacher LN, Bernal Sierra YA, Smith ES, Lewin GR (2015). ASICs and mammalian mechanoreceptor function. Neuropharmacology 94: 80–86. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Hull C, Vigh J, von Gersdorff H (2003). Synaptic cleft acidification and modulation of short‐term depression by exocytosed protons in retinal bipolar cells. J Neurosci 23: 11332–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan XQ, Gonzalez JA, Chang S, Chacko S, Wein AJ, Malykhina AP (2010). Experimental colitis triggers the release of substance P and calcitonin gene‐related peptide in the urinary bladder via TRPV1 signaling pathways. Exp Neurol 225: 262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passero CJ, Mueller GM, Myerburg MM, Carattino MD, Hughey RP, Kleyman TR (2012). TMPRSS4‐dependent activation of the epithelial sodium channel requires cleavage of the gamma‐subunit distal to the furin cleavage site. Am J Physiol Renal Physiol 302: F1–F8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passero CJ, Mueller GM, Rondon‐Berrios H, Tofovic SP, Hughey RP, Kleyman TR (2008). Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J Biol Chem 283: 36586–36591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, Chao J, Palmer LG (2012). Tissue kallikrein activation of the epithelial Na channel. Am J Physiol Renal Physiol 303: F540–F550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paukert M, Babini E, Pusch M, Grunder S (2004). Identification of the Ca2 + blocking site of acid‐sensing ion channel (ASIC) 1: implications for channel gating. J Gen Physiol 124: 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PMT, Kohan DE (2015). Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 10: 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pech V, Thumova M, Dikalov SI, Hummler E, Rossier BC, Harrison DG et al. (2013). Nitric oxide reduces Cl‐ absorption in the mouse cortical collecting duct through an ENaC‐dependent mechanism. Am J Physiol Renal Physiol 304: F1390–F1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pech V, Thumova M, Kim YH, Agazatian D, Hummler E, Rossier BC et al. (2012). ENaC inhibition stimulates Cl− secretion in the mouse cortical collecting duct through an NKCC1‐dependent mechanism. Am J Physiol Renal Physiol 303: F45–F55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peigneur S, Beress L, Moller C, Mari F, Forssmann WG, Tytgat J (2012). A natural point mutation changes both target selectivity and mechanism of action of sea anemone toxins. FASEB J 26: 5141–5151. [DOI] [PubMed] [Google Scholar]
- Pellegrini‐Giampietro DE, Zukin RS, Bennett MV, Cho S, Pulsinelli WA (1992). Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc Natl Acad Sci U S A 89: 10499–10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Li WG, Huang C, Jiang YM, Wang X, Zhu MX et al. (2015). ASIC3 mediates itch sensation in response to coincident stimulation by acid and nonproton ligand. Cell Rep 13: 387–398. [DOI] [PubMed] [Google Scholar]
- Perrier R, Boscardin E, Malsure S, Sergi C, Maillard MP, Loffing J et al. (2015). Severe salt‐losing syndrome and hyperkalemia induced by adult nephron‐specific knockout of the epithelial sodium channel alpha‐subunit. J Am Soc Nephrol. Epub ahead of print: pii: ASN.2015020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier R, Richard S, Sainte‐Marie Y, Rossier BC, Jaisser F, Hummler E et al. (2005). A direct relationship between plasma aldosterone and cardiac L‐type Ca2+ current in mice. J Physiol 569: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroff EY, Price MP, Snitsarev V, Gong H, Korovkina V, Abboud FM et al. (2008). Acid‐sensing ion channels interact with and inhibit BK K+ channels. Proc Natl Acad Sci U S A 105: 3140–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard N, Eladari D, El Moghrabi S, Planes C, Bourgeois S, Houillier P et al. (2008). Defective ENaC processing and function in tissue kallikrein‐deficient mice. J Biol Chem 283: 4602–4611. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, Aroniadou‐Anderjaska V, Prager EM, Figueiredo TH, Almeida‐Suhett CP, Miller SL et al. (2014). ASIC1a activation enhances inhibition in the basolateral amygdala and reduces anxiety. J Neurosci 34: 3130–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planes C, Randrianarison NH, Charles RP, Frateschi S, Cluzeaud F, Vuagniaux G et al. (2009). ENaC‐mediated alveolar fluid clearance and lung fluid balance depend on the channel‐activating protease 1. EMBO Mol Med 2: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD (2012). A Role for K2P channels in the operation of somatosensory nociceptors. Front Mol Neurosci 5: 21. doi: 10.3389/fnmol.2012.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirot O, Berta T, Decosterd I, Kellenberger S (2006). Distinct ASIC currents are expressed in rat putative nociceptors and are modulated by nerve injury. J Physiol 576: 215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirot O, Vukicevic M, Boesch A, Kellenberger S (2004). Selective regulation of acid‐sensing ion channel 1 by serine proteases. J Biol Chem 279: 38448–38457. [DOI] [PubMed] [Google Scholar]
- Poulsen SB, Praetorius J, Damkier HH, Miller L, Nelson RD, Hummler E et al. (2015). Reducing alphaENaC expression in kidney connecting tubule induces pseudohypoaldosteronism type 1 symptoms during K+ loading. Am J Physiol Renal Physiol 310: F300–F310. [DOI] [PubMed] [Google Scholar]
- Pradervand S, Barker PM, Wang Q, Ernst SA, Beermann F, Grubb BR et al. (1999a). Salt restriction induces pseudohypoaldosteronism type 1 in mice expressing low levels of the beta‐subunit of the amiloride‐sensitive epithelial sodium channel. Proc Natl Acad Sci U S A 96: 1732–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradervand S, Vandewalle A, Bens M, Gautschi I, Loffing J, Hummler E et al. (2003). Dysfunction of the epithelial sodium channel expressed in the kidney of a mouse model for Liddle syndrome. J Am Soc Nephrol 14: 2219–2228. [DOI] [PubMed] [Google Scholar]
- Pradervand S, Wang Q, Burnier M, Beermann F, Horisberger JD, Hummler E et al. (1999b). A mouse model for Liddle's syndrome. J Am Soc Nephrol 10: 2527–2533. [DOI] [PubMed] [Google Scholar]
- Pratt JH (2005). Central role for ENaC in development of hypertension. J Am Soc Nephrol 16: 3154–3159. [DOI] [PubMed] [Google Scholar]
- Prole DL, Lima PA, Marrion NV (2003). Mechanisms underlying modulation of neuronal KCNQ2/KCNQ3 potassium channels by extracellular protons. J Gen Physiol 122: 775–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana P, Soto D, Poirot O, Zonouzi M, Kellenberger S, Muller D et al. (2015). Acid‐sensing ion channel 1a drives AMPA receptor plasticity following ischaemia and acidosis in hippocampal CA1 neurons. J Physiol 593: 4373–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi K, Emdin CA, MacMahon S (2015). The epidemiology of blood pressure and its worldwide management. Circ Res 116: 925–936. [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Karp PH, Jiang P, Ostedgaard LS, Walz AE, Fisher JT et al. (2012). A microRNA network regulates expression and biosynthesis of wild‐type and DeltaF508 mutant cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci U S A 109: 13362–13367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randrianarison N, Clerici C, Ferreira C, Fontayne A, Pradervand S, Fowler‐Jaeger N et al. (2008). Low expression of the beta‐ENaC subunit impairs lung fluid clearance in the mouse. Am J Physiol Lung Cell Mol Physiol 294: L409–L416. [DOI] [PubMed] [Google Scholar]
- Randrianarison N, Escoubet B, Ferreira C, Fontayne A, Fowler‐Jaeger N, Clerici C et al. (2007). beta‐Liddle mutation of the epithelial sodium channel increases alveolar fluid clearance and reduces the severity of hydrostatic pulmonary oedema in mice. J Physiol 582: 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raouf R, Rugiero F, Kiesewetter H, Hatch R, Hummler E, Nassar MA et al. (2012). Sodium channels and mammalian sensory mechanotransduction. Mol Pain 8: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan S, Lee DH, Oh YT, Delpire E, Youn JH, McDonough AA (2014). Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am J Physiol Renal Physiol 306: F1059–F1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossier BC (2014). Epithelial sodium channel (ENaC) and the control of blood pressure. Curr Opin Pharmacol 15C: 33–46. [DOI] [PubMed] [Google Scholar]
- Rossier BC, Baker ME, Studer RA (2015). Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev 95: 297–340. [DOI] [PubMed] [Google Scholar]
- Rossier BC, Staub O, Hummler E (2013). Genetic dissection of sodium and potassium transport along the aldosterone‐sensitive distal nephron: importance in the control of blood pressure and hypertension. FEBS Lett 587: 1929–1941. [DOI] [PubMed] [Google Scholar]
- Rossol‐Haseroth K, Zhou Q, Braun S, Boldyreff B, Falkenstein E, Wehling M et al. (2004). Mineralocorticoid receptor antagonists do not block rapid ERK activation by aldosterone. Biochem Biophys Res Commun 318: 281–288. [DOI] [PubMed] [Google Scholar]
- Rubera I, Loffing J, Palmer LG, Frindt G, Fowler‐Jaeger N, Sauter D et al. (2003). Collecting duct‐specific gene inactivation of alphaENaC in the mouse kidney does not impair sodium and potassium balance. J Clin Invest 112: 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch A, Hummler E (1999). Mechano‐electrical transduction in mice lacking the alpha‐subunit of the epithelial sodium channel. Hear Res 131: 170–176. [DOI] [PubMed] [Google Scholar]
- Saha C, Eckert GJ, Ambrosius WT, Chun TY, Wagner MA, Zhao Q et al. (2005). Improvement in blood pressure with inhibition of the epithelial sodium channel in blacks with hypertension. Hypertension 46: 481–487. [DOI] [PubMed] [Google Scholar]
- Salinas M, Besson T, Delettre Q, Diochot S, Boulakirba S, Douguet D et al. (2014). Binding site and inhibitory mechanism of the mambalgin‐2 pain‐relieving peptide on acid‐sensing ion channel 1a. J Biol Chem 289: 13363–13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas M, Rash LD, Baron A, Lambeau G, Escoubas P, Lazdunski M (2006). The receptor site of the spider toxin PcTx1 on the proton‐gated cation channel ASIC1a. J Physiol 570: 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallenave JM (2010). Secretory leukocyte protease inhibitor and elafin/trappin‐2: versatile mucosal antimicrobials and regulators of immunity. Am J Respir Cell Mol Biol 42: 635–643. [DOI] [PubMed] [Google Scholar]
- Santulli G (2013). Epidemiology of cardiovascular disease in the 21st century: updated numbers and updated facts. J Cardiorespir Dis 1: 1–2. [Google Scholar]
- Schild L, Schneeberger E, Gautschi I, Firsov D (1997). Identification of amino acid residues in the alpha, beta, and gamma subunits of the epithelial sodium channel (ENaC) involved in amiloride block and ion permeation. J Gen Physiol 109: 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder CI, Rash LD, Vila‐Farres X, Rosengren KJ, Mobli M, King GF et al. (2014). Chemical synthesis, 3D structure, and ASIC binding site of the toxin mambalgin‐2. Angew Chem Int Ed Engl 53: 1017–1020. [DOI] [PubMed] [Google Scholar]
- Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR (2006). Furin cleavage activates the epithelial Na+ channel by relieving Na+ self‐inhibition. Am J Physiol Renal Physiol 290: F1488–F1496. [DOI] [PubMed] [Google Scholar]
- Sherwood TW, Lee KG, Gormley MG, Askwith CC (2011). Heteromeric acid‐sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis‐induced neuronal death. J Neurosci 31: 9723–9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Carattino MD, Hughey RP, Kleyman TR (2013). ENaC regulation by proteases and shear stress. Curr Mol Pharmacol 6: 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjö BK, von Hanwehr R, Nergelius G, Nevander G, Ingvar M (1985). Extra‐ and intracellular pH in the brain during seizures and in the recovery period following the arrest of seizure activity. J Cereb Blood Flow Metab 5: 47–57. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Gregory NS (2015). The dichotomized role for acid sensing ion channels in musculoskeletal pain and inflammation. Neuropharmacology 94: 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka KA, Winter OC, Wemmie JA (2009). Acid‐sensing ion channels: a new target for pain and CNS diseases. Curr Opin Drug Discov Devel 12: 693–704. [PMC free article] [PubMed] [Google Scholar]
- Smith ES, Cadiou H, McNaughton PA (2007). Arachidonic acid potentiates acid‐sensing ion channels in rat sensory neurons by a direct action. Neuroscience 145: 686–698. [DOI] [PubMed] [Google Scholar]
- Smith SE, Chesler M (1999). Effect of divalent cations on AMPA‐evoked extracellular alkaline shifts in rat hippocampal slices. J Neurophysiol 82: 1902–1908. [DOI] [PubMed] [Google Scholar]
- Somodi S, Varga Z, Hajdu P, Starkus JG, Levy DI, Gaspar R et al. (2004). pH‐dependent modulation of Kv1.3 inactivation: role of His399. Am J Physiol Cell Physiol 287: C1067–C1076. [DOI] [PubMed] [Google Scholar]
- Sonneville F, Ruffin M, Guillot L, Rousselet N, Le Rouzic P, Corvol H et al. (2015). New insights about miRNAs in cystic fibrosis. Am J Pathol 185: 897–908. [DOI] [PubMed] [Google Scholar]
- Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G et al. (2013). Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824. [DOI] [PubMed] [Google Scholar]
- Souslova V, Cesare P, Ding Y, Akopian AN, Stanfa L, Suzuki R et al. (2000). Warm‐coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature 407: 1015–1017. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus JG, Fleig A, Penner R (2010). The calcium‐permeable non‐selective cation channel TRPM2 is modulated by cellular acidification. J Physiol (Lond) 588: 1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A, Adams E, Booth RE, Stockand JD (2005). Epithelial Na + Channel Subunit Stoichiometry. Biophys J 88: 3966–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Tang Y, Liu L, Zhou H, Dong Q (2011). Regulation of acid‐sensing ion channel 1a function by tissue kallikrein may be through channel cleavage. Neurosci Lett 490: 46–51. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Mizuno A, Kodaira K, Imai M (2003). Impaired pressure sensation in mice lacking TRPV4. J Biol Chem 278: 22664–22668. [DOI] [PubMed] [Google Scholar]
- Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B et al. (2009). Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol 20: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syková E, Svoboda J (1990). Extracellular alkaline‐acid‐alkaline transients in the rat spinal cord evoked by peripheral stimulation. Brain Res 512: 181–189. [DOI] [PubMed] [Google Scholar]
- Szabo R, Uzzun Sales K, Kosa P, Shylo NA, Godiksen S, Hansen KK et al. (2012). Reduced prostasin (CAP1/PRSS8) activity eliminates HAI‐1 and HAI‐2 deficiency‐associated developmental defects by preventing matriptase activation. PLoS Genet 8 .e1002937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Nagaike K, Takeda N, Itoh H, Kohama K, Fukushima T et al. (2005). Hepatocyte growth factor activator inhibitor type 1 (HAI‐1) is required for branching morphogenesis in the chorioallantoic placenta. Mol Cell Biol 25: 5687–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas W, McEneaney V, Harvey BJ (2007). Rapid responses to steroid hormones in the kidney. Nephron Physiol 107: 1–9. [DOI] [PubMed] [Google Scholar]
- Todkar A, Picard N, Loffing‐Cueni D, Sorensen MV, Mihailova M, Nesterov V et al. (2015). Mechanisms of renal control of potassium homeostasis in complete aldosterone deficiency. J Am Soc Nephrol 26: 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Somjen GG (1996). Effects of extracellular pH on voltage‐gated Na+, K+ and Ca2 + currents in isolated rat CA1 neurons. J Physiol (Lond) 493 (Pt 3): 719–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K et al. (1998). The cloned capsaicin receptor integrates multiple pain‐producing stimuli. Neuron 21: 531–543. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Cull‐Candy SG (1990). Proton inhibition of N‐methyl‐D‐aspartate receptors in cerebellar neurons. Nature 345: 347–350. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Cull‐Candy SG (1991). Pharmacological properties and H+ sensitivity of excitatory amino acid receptor channels in rat cerebellar granule neurones. J Physiol (Lond) 433: 727–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugawa S, Ishida Y, Ueda T, Inoue K, Nagao M, Shimada S (2007). Nafamostat mesilate reversibly blocks acid‐sensing ion channel currents. Biochem Biophys Res Commun 363: 203–208. [DOI] [PubMed] [Google Scholar]
- Ugawa S, Ueda T, Ishida Y, Nishigaki M, Shibata Y, Shimada S (2002). Amiloride‐blockable acid‐sensing ion channels are leading acid sensors expressed in human nociceptors. J Clin Invest 110: 1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugawa S, Yamamoto T, Ueda T, Ishida Y, Inagaki A, Nishigaki M et al. (2003). Amiloride‐insensitive currents of the acid‐sensing ion channel‐ 2a (ASIC2a)/ASIC2b heteromeric sour‐taste receptor channel. J Neurosci 23: 3616–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeyev AY, Hackman JC, Holohean AM, Wood PM, Katz JL, Davidoff RA (1999). GABA‐induced Cl− current in cultured embryonic human dorsal root ganglion neurons. J Neurophysiol 82: 1–9. [DOI] [PubMed] [Google Scholar]
- Valinezhad Orang A, Safaralizadeh R, Kazemzadeh‐Bavili M (2014). Mechanisms of miRNA‐Mediated Gene Regulation from Common Downregulation to mRNA‐Specific Upregulation. Int J Genomics 2014: 970607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC (1997). An epithelial serine protease activates the amiloride‐sensitive sodium channel. Nature 389: 607–610. [DOI] [PubMed] [Google Scholar]
- Vallon V, Hummler E, Rieg T, Pochynyuk O, Bugaj V, Schroth J et al. (2009). Thiazolidinedione‐induced fluid retention is independent of collecting duct alphaENaC activity. J Am Soc Nephrol 20: 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bemmelen MX, Huser D, Gautschi I, Schild L (2015). The human acid‐sensing ion channel ASIC1a: evidence for a homotetrameric assembly state at the cell surface. PLoS One 10 .e0135191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AH et al. (2013a). K+‐induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+‐Cl− cotransporter. Am J Physiol Renal Physiol 305: F1177–F1188. [DOI] [PubMed] [Google Scholar]
- van der Lubbe N, Zietse R, Hoorn EJ (2013b). Effects of angiotensin II on kinase‐mediated sodium and potassium transport in the distal nephron. Curr Opin Nephrol Hypertens 22: 120–126. [DOI] [PubMed] [Google Scholar]
- Vavassori S, Mayer A (2014). A new life for an old pump: V‐ATPase and neurotransmitter release. J Cell Biol 205: 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venton BJ, Michael DJ, Wightman RM (2003). Correlation of local changes in extracellular oxygen and pH that accompany dopaminergic terminal activity in the rat caudate‐putamen. J Neurochem 84: 373–381. [DOI] [PubMed] [Google Scholar]
- Verouti SN, Boscardin E, Hummler E, Frateschi S (2015). Regulation of blood pressure and renal function by NCC and ENaC: lessons from genetically engineered mice. Curr Opin Pharmacol 21: 60–72. [DOI] [PubMed] [Google Scholar]
- Vessey JP, Stratis AK, Daniels BA, Da Silva N, Jonz MG, Lalonde MR et al. (2005). Proton‐mediated feedback inhibition of presynaptic calcium channels at the cone photoreceptor synapse. J Neurosci 25: 4108–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vick JS, Askwith CC (2015). ASICs and neuropeptides. Neuropharmacology 94: 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidt DG, Borazanian RA (2003). Treat high blood pressure sooner: tougher, simpler JNC 7 guidelines. Cleve Clin J Med 70: 721–728. [DOI] [PubMed] [Google Scholar]
- Vilin YY, Peters CH, Ruben PC (2012). Acidosis differentially modulates inactivation in na(v)1.2, na(v)1.4, and na(v)1.5 channels. Front Pharmacol 3: 109. doi: 10.3389/fphar.2012.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitzthum H, Seniuk A, Schulte LH, Muller ML, Hetz H, Ehmke H (2014). Functional coupling of renal K+ and Na+ handling causes high blood pressure in Na+ replete mice. J Physiol 592: 1139–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voilley N, de Weille J, Mamet J, Lazdunski M (2001). Nonsteroid anti‐inflammatory drugs inhibit both the activity and the inflammation‐induced expression of acid‐sensing ion channels in nociceptors. J Neurosci 21: 8026–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer WM, Sacks FM, Ard J, Appel LJ, Bray GA, Simons‐Morton DG et al. (2001). Effects of diet and sodium intake on blood pressure: subgroup analysis of the DASH‐sodium trial. Ann Intern Med 135: 1019–1028. [DOI] [PubMed] [Google Scholar]
- Vuagniaux G, Vallet V, Jaeger NF, Pfister C, Bens M, Farman N et al. (2000). Activation of the amiloride‐sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J Am Soc Nephrol 11: 828–834. [DOI] [PubMed] [Google Scholar]
- Vukicevic M, Kellenberger S (2004). Modulatory effects of acid‐sensing ion channels on action potential generation in hippocampal neurons. Am J Physiol Cell Physiol 287: C682–C690. [DOI] [PubMed] [Google Scholar]
- Vukicevic M, Weder G, Boillat A, Boesch A, Kellenberger S (2006). Trypsin cleaves acid‐sensing ion channel 1a in a domain that is critical for channel gating. J Biol Chem 281: 714–722. [DOI] [PubMed] [Google Scholar]
- Wakida N, Kitamura K, Tuyen DG, Maekawa A, Miyoshi T, Adachi M et al. (2006). Inhibition of prostasin‐induced ENaC activities by PN‐1 and regulation of PN‐1 expression by TGF‐beta1 and aldosterone. Kidney Int 70: 1432–1438. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Bassilana F, Deweille J, Champigny G, Heurteaux C, Lazdunski M (1997a). Molecular cloning of a non‐inactivating proton‐gated Na+ channel specific for sensory neurons. J Biol Chem 272: 20975–20978. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M (1997b). A proton‐gated cation channel involved in acid‐sensing. Nature 386: 173–177. [DOI] [PubMed] [Google Scholar]
- Walker CS, Conner AC, Poyner DR, Hay DL (2010). Regulation of signal transduction by calcitonin gene‐related peptide receptors. Trends Pharmacol Sci 31: 476–483. [DOI] [PubMed] [Google Scholar]
- Wang Q, Clement S, Gabbiani G, Horisberger JD, Burnier M, Rossier BC et al. (2004). Chronic hyperaldosteronism in a transgenic mouse model fails to induce cardiac remodeling and fibrosis under a normal‐salt diet. Am J Physiol Renal Physiol 286: F1178–F1184. [DOI] [PubMed] [Google Scholar]
- Wang Q, Hummler E, Maillard M, Nussberger J, Rossier BC, Brunner HR et al. (2001). Compensatory up‐regulation of angiotensin II subtype 1 receptors in alpha ENaC knockout heterozygous mice. Kidney Int 59: 2216–2221. [DOI] [PubMed] [Google Scholar]
- Wang X, Li WG, Yu Y, Xiao X, Cheng J, Zeng WZ et al. (2013). Serotonin facilitates peripheral pain sensitivity in a manner that depends on the nonproton ligand sensing domain of ASIC3 channel. J Neurosci 33: 4265–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Anlauf M (2014). Treatment resistant hypertension–investigation and conservative management. Dtsch Arztebl Int 111: 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MA (2014). The evolving clinical management of hypertension. J Clin Hypertens (Greenwich) 16: 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger MH (1996). Salt sensitivity of blood pressure in humans. Hypertension 27: 481–490. [DOI] [PubMed] [Google Scholar]
- Weller K, Reeh PW, Sauer SK (2011). TRPV1, TRPA1, and CB1 in the isolated vagus nerve‐axonal chemosensitivity and control of neuropeptide release. Neuropeptides 45: 391–400. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Chen J, Askwith CC, Hruska‐Hageman AM, Price MP, Nolan BC et al. (2002). The acid‐activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34: 463–477. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Price MP, Welsh MJ (2006). Acid‐sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci 29: 578–586. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Taugher RJ, Kreple CJ (2013). Acid‐sensing ion channels in pain and disease. Nat Rev Neurosci 14: 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Trials of Hypertension Prevention Collaborative Research Group . (1997). Effects of weight loss and sodium reduction intervention on blood pressure and hypertension incidence in overweight people with high‐normal blood pressure. The Trials of Hypertension Prevention, phase II. Arch Intern Med 157: 657–667. [PubMed] [Google Scholar]
- Wiemuth D, Assmann M, Grunder S (2014). The bile acid‐sensitive ion channel (BASIC), the ignored cousin of ASICs and ENaC. Channels (Austin) 8: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins ME, Hosie AM, Smart TG (2005). Proton modulation of recombinant GABA(A) receptors: influence of GABA concentration and the beta subunit TM2‐TM3 domain. J Physiol 567: 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GH (2005). Aldosterone biosynthesis, regulation, and classical mechanism of action. Heart Fail Rev 10: 7–13. [DOI] [PubMed] [Google Scholar]
- Wolkenberg SE, Zhao Z, Mulhearn JJ, Harrison ST, Sanders JM, Cato MJ et al. (2011). High concentration electrophysiology‐based fragment screen: discovery of novel acid‐sensing ion channel 3 (ASIC3) inhibitors. Bioorg Med Chem Lett 21: 2646–2649. [DOI] [PubMed] [Google Scholar]
- Wu P‐Y, Huang Y‐Y, Chen C‐C, Hsu T‐T, Lin Y‐C, Weng J‐Y et al. (2013). Acid‐sensing ion channel‐1a is not required for normal hippocampal LTP and spatial memory. J Neurosci 33: 1828–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL et al. (2004). Neuroprotection in ischemia: blocking calcium‐permeable acid‐ sensing ion channels. Cell 118: 687–698. [DOI] [PubMed] [Google Scholar]
- Yagi J, Wenk HN, Naves LA, McCleskey EW (2006). Sustained currents through ASIC3 ion channels at the modest pH changes that occur during myocardial ischemia. Circ Res 99: 501–509. [DOI] [PubMed] [Google Scholar]
- Yamamoto F, Borgula GA, Steinberg RH (1992). Effects of light and darkness on pH outside rod photoreceptors in the cat retina. Exp Eye Res 54: 685–697. [DOI] [PubMed] [Google Scholar]
- Yamamura H, Ugawa S, Ueda T, Nagao M, Shimada S (2004). Protons activate the delta‐subunit of the epithelial Na + channel in humans. J Biol Chem 279: 12529–12534. [DOI] [PubMed] [Google Scholar]
- Yamauchi M, Kataoka H, Itoh H, Seguchi T, Hasui Y, Osada Y (2004). Hepatocyte growth factor activator inhibitor types 1 and 2 are expressed by tubular epithelium in kidney and down‐regulated in renal cell carcinoma. J Urol 171: 890–896. [DOI] [PubMed] [Google Scholar]
- Yang HY, Charles RP, Hummler E, Baines DL, Isseroff RR (2013). The epithelial sodium channel mediates the directionality of galvanotaxis in human keratinocytes. J Cell Sci 126: 1942–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Thelin WR, Rogers TD, Stutts MJ, Randell SH, Grubb BR et al. (2012). Regional differences in rat conjunctival ion transport activities. Am J Physiol Cell Physiol 303: C767–C780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Chen Z, Li WG, Cao H, Feng EG, Yu F et al. (2010). A nonproton ligand sensor in the acid‐sensing ion channel. Neuron 68: 61–72. [DOI] [PubMed] [Google Scholar]
- Zachar RM, Skjodt K, Marcussen N, Walter S, Toft A, Nielsen MR et al. (2015). The epithelial sodium channel gamma‐subunit is processed proteolytically in human kidney. J Am Soc Nephrol 26: 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zani ML, Tanga A, Saidi A, Serrano H, Dallet‐Choisy S, Baranger K et al. (2011). SLPI and trappin‐2 as therapeutic agents to target airway serine proteases in inflammatory lung diseases: current and future directions. Biochem Soc Trans 39: 1441–1446. [DOI] [PubMed] [Google Scholar]
- Zemel MB (1997). Dietary pattern and hypertension: the DASH study. Dietary approaches to stop hypertension. Nutr Rev 55: 303–305. [DOI] [PubMed] [Google Scholar]
- Zeng WZ, Liu DS, Liu L, She L, Wu LJ, Xu TL (2015). Activation of acid‐sensing ion channels by localized proton transient reveals their role in proton signaling. Sci Rep 5: 14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XM (2013). Acid‐sensing ion channels: trafficking and synaptic function. Mol Brain 6: 1. doi: 10.1186/1756‐6606‐6‐1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XM, Wemmie JA, Green SH, Welsh MJ (2006). Acid‐sensing ion channel 1a is a postsynaptic proton receptor that affects the density of dendritic spines. Proc Natl Acad Sci U S A 103: 16556–16561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai J, Peoples RW, Li C (1998). Proton inhibition of GABA‐activated current in rat primary sensory neurons. Pflugers Arch 435: 539–545. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Duerr J, Johannesson B, Schubert SC, Treis D, Harm M et al. (2011). The ENaC‐overexpressing mouse as a model of cystic fibrosis lung disease. J Cyst Fibros 10 (Suppl 2): S172–S182. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Treis D, Schubert SC, Harm M, Schatterny J, Hirtz S et al. (2008). Preventive but not late amiloride therapy reduces morbidity and mortality of lung disease in betaENaC‐overexpressing mice. Am J Respir Crit Care Med 178: 1245–1256. [DOI] [PubMed] [Google Scholar]
- Zhu G, Chanchevalap S, Cui N, Jiang C (1999). Effects of intra‐ and extracellular acidifications on single channel Kir2.3 currents. J Physiol (Lond) 516 (Pt 3): 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemann AE, Allen JE, Dahdaleh NS, Drebot II, Coryell MW, Wunsch AM et al. (2009). The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell 139: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]