

Abstract
Over 70% of diffuse intrinsic pediatric gliomas, an aggressive brainstem tumor, harbor heterozygous mutations that create a K27M amino acid substitution (methionine replaces lysine 27) in the tail of histone H3.3. The role of the H3.3K27M mutation in tumorigenesis not fully understood. Here, we use a human embryonic stem cell system to model this tumor. We show that H3.3K27M expression synergizes with p53 loss and PDGFRA activation in neural progenitor cells derived from human embryonic stem cells, resulting in neoplastic transformation. Genome-wide analyses indicate a resetting of the transformed precursors to a developmentally more primitive stem cell state, with evidence of major modifications of histone marks at several master regulator genes. Drug screening assays identified a compound targeting the protein menin as an inhibitor of tumor cell growth in vitro and in mice.
Diffuse intrinsic pontine gliomas (DIPGs) are rare, highly aggressive brainstem tumors primarily affecting children. Over 70% of DIPGs carry somatic mutations in the H3F3A gene encoding histone H3.3, leading to a p.Lys27Met amino acid substitution (methionine replaces lysine 27) (1–3). Tumors positive for the mutation are associated with poorer prognosis and diminished survival. Comprehensive whole-genome analyses have shown that the H3.3K27M mutation identifies a distinct subgroup of DIPGs that has substantial overlap with p53 mutations and platelet-derived growth factor receptor, α polypeptide (PDGFRA) amplification (60% and 40%, respectively) (4, 5). These genetic studies have paved the way for investigations of the pathogenesis and treatment of this rapidly fatal tumor. However, tissue access remains a substantial challenge because of the infiltrative nature and sensitive location of the tumor in the brainstem. Key features of K27M-mutated DIPGs are the restricted developmental window during which they emerge [mean age at diagnosis is 8 years (5)] and their specific midline location, which implicate a developmentally early and anatomically specific cell of origin.
We reasoned that human pluripotent stem cells (hPSCs) (6) might be a valuable model for studying DIPG. These cells provide an attractive platform for functional analysis of oncogenic mutations in a genetically defined human background. In addition, neural differentiation protocols allow the derivation of relevant developmentally early neural stem cells that are often inaccessible; thus, tumorigenesis can be studied in the proper cell context. We first derived early neural progenitor cells (NPCs) from human embryonic stem (hES) cells (H9, WA-09), using the dual Smad inhibition protocol (7). We then cotransduced the cells with lentiviruses encoding (i) a constitutively active form of the PDGFRA in which valine replaces aspartic acid 842 (D842V); (ii), a small hairpin RNA (shRNA) against p53 tagged with red fluorescent protein (RFP); and (iii) a hemagglutinin (HA)–tagged wild-type (WT) or K27M mutant form of histone H3.3 (Fig. 1A). These oncogenes were selected on the basis of their high frequency of expression and/or mutations in K27M-mutated DIPGs (5, 8). After transduction and double selection under puromycin and G418, the cells maintained NPC-like morphology and expression of two NPC marker genes, Nestin and SOX2 (Fig. 1B). Consistent with previous reports (9–11), the expression of H3.3K27M led to a reduction in histone H3K27 trimethylation (H3K27me3), as shown by immunohistochemistry and Western blotting (Fig. 1, B and C). Expression of H3.3K27M alone increased cell proliferation (Ki-67 of ~ 27% versus15 to 17%) and total cell number, in comparison to WT H3.3 or mock (empty vector) conditions (Fig. 1D and fig. S1A). Overexpression of constitutively active PDGFRA (D842V) and knockdown of p53 (hereafter referred to as P5) also increased the proliferation of NPCs. The combination of H3.3K27M and P5 was even more effective in increasing the proliferative capacity of the P5 cells, up to a Ki-67 index of >30%. This result was confirmed by using a second independent shRNA against p53 (sh-p53) (fig. S1, B to D). The proliferative effect on neural precursors is specific to H3.3K27M and is not seen in the G34R/V mutations of H3.3 (glycine 34 is replaced by arginine or valine), which are mostly reported in supratentorial glioblastomas (Fig. 1E). It is also highly specific to the cell context, because H3.3K27M expression in undifferentiated hES cells or in differentiated somatic cells—such as hES-derived astrocytes, primary human astrocytes, or MRC-5 human lung fibroblast cells—did not affect proliferation rates and, in some cases, induced senescence (Fig. 1F and fig. S2). Expression of Olig2, reported in DIPGs (4), was increased in both the H3.3K27M and the P5 condition (fig. S3).
Fig. 1. The impact of H3.3 K27M mutation on neural precursors.
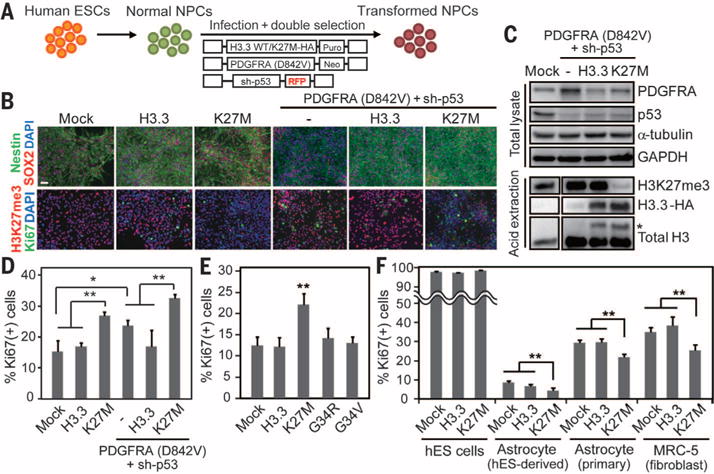
(A) hES-derived NPCs were transformed using a combination of lentiviruses encoding H3.3 WT or H3.3K27M, plus PDGFRA and sh-p53. (B) Immunohistochemistry of NPCs transduced with different vector combinations demonstrates maintenance of SOX2 and Nestin expression. DAPI, 4′,6′-diamidino-2-phenylindole. Expression of H3.3K27M (K27M) is associated with significantly reduced H3K27 trimethylation mark (H3K27me3). Ki-67 immunopositivity depicts proliferation and is most elevated in the K27M, PDGFRA, and sh-p53 conditions. Scale bar, 50 μm. (C) Western blotting of transduced NPCs. Total lysates and acid-extracted histones were separated by SDS-polyacrylamide gel electrophoresis and blotted with the indicated antibodies. Asterisk (*) beside higher bands indicates HA-tagged H3.3 transgene. (D) Quantification of Ki-67 immunostaining in transduced NPCs. (E) Proliferation index of NPCs transduced with various H3.3 mutations. (F) Impact of K27M mutation on the proliferation index of various cell lines. Error bars in panels (D) to (F) indicate means ± SD (n = 4 to 5 in each). *P < 0.05; **P < 0.01.
We next explored whether the transduced NPCs had acquired features of neoplastic cells. Under low-density culture conditions, all cell groups showed poor survival, with the exception of the P5+H3.3K27M (P5K) cells, which formed robust colonies that grew to confluence, consistent with a neoplastic phenotype (Fig. 2A and fig. S4A). The combination of PDGFRA, sh-p53, and H3.3K27M had a synergistic effect on cell survival. After growth factor withdrawal, the apoptotic cell fraction was higher in the H3.3K27M-expressing cells, in comparison with normal and WT H3.3-expressing NPCs (fig. S4B); this suggests that the proliferative effect imparted by the histone mutation was balanced with an increased apoptotic rate, a phenomenon often associated with premalignant states (12). The introduction of p53 knockdown in the P5K condition abrogated the apoptotic response, and possibly promoting genomic instability as the cells continued to proliferate. The P5 condition also conferred greater efficiency in neurosphere formation, in all groups, including the P5K combination, whereas cells expressing only H3.3K27M were similar to unperturbed NPCs (fig. S4C). These results support a synergistic effect of H3.3K27M and P5 in the oncogenic transformation of NPCs, consistent with the high frequency of comutation of H3.3K27M and mutated p53 in human DIPGs (1, 4, 5).
Fig. 2. Synergistic effect of K27M and oncogenes in inducing transformation and tumorigenicity of NPCs.
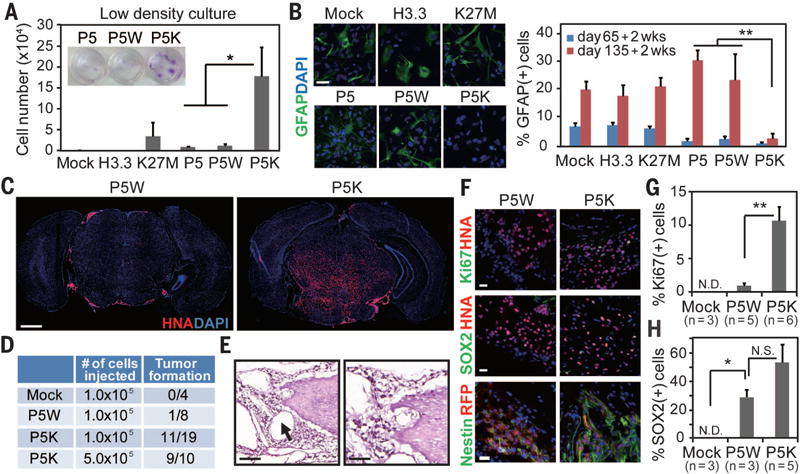
(A) Transduced NPCs were grown at very low density for 4 weeks, and the number of viable cells was counted by trypan blue staining. Crystal violet staining is shown in the inset. P5, P5W, and P5K indicate PDGFRA (D842V) + sh-p53, P5 + WT histone H3.3, and P5 + K27M mutant, respectively. Bars indicate means ± SD (n = 4). (B) Immunohistochemistry and quantification for the glial marker (GFAP) under the differentiation condition. Bars indicate means ± SD (n = 4 to 5). Scale bar, 20 μm. (C) Transcranial injection of P5K cells into the brainstem of mice led to tumor formation, whereas P5W cells gave rise to isolated cell clusters. Low-magnification immunofluorescence images of representative sections labeled for human nuclear antigen (HNA) and counterstained with DAPI. Scale bar, 1 mm. (D) Summary of transplantation and tumor formation. (E) Light microscopy of hematoxylin and eosin–stained representative section taken from the pons of a mouse bearing P5K tumor demonstrates encasement of the basilar artery (arrow) by tumor cells, microcystic change, and infiltration in the pons. Scale bar, 100 μm (left panel), 50 μm (right panel). (F to H) Immunophenotyping of the xenografts. Immunofluorescence images and quantification for HNA, Ki67 (F and G), SOX2 (F and H) and human-specific Nestin (F). Error bars in panels (G) and (H) indicate means ± SEM (n = 3 to 6). Scale bar, 20 μm. *P < 0.05; **P < 0.01; NS, not significant; ND, not detected.
We also analyzed transduced NPCs for radiation response and migratory capacity. After irradiation, cells transduced with the combination of H3.3K27M and P5 maintained a high proliferation rate despite substantial DNA damage (fig. S4, D to G), a hallmark of cancer cells (13). Expression of H3.3K27M also increased cell migration (fig. S4H) and invasion (fig. S4I) in in vitro assays. High migration capacity of tumor cells is a major feature of DIPGs that renders the tumors unsuitable for surgical eradication. There was a near-complete differentiation block in the astrocytic lineage in the P5K cells, despite an extended culture period in vitro [normal NPCs acquire a capacity for robust differentiation into astrocytes (glial competence) only after several weeks in vitro] (Fig. 2B). Note that neither the P5 nor the H3.3K27M condition alone inhibited differentiation, but the combination blocked differentiation into astrocytes and to some extent differentiation into oligodendrocytes (fig. S5).
To evaluate tumor formation in vivo, we injected transduced NPCs into the brainstem (pons) of immunocompromised mice. Massive brain tumor formation occurred in the P5K mice, but not in the P5W or mock-injected mice (Fig. 2, C and D). Immunohistochemistry demonstrated human cells infiltrating the pons, with encasement of the basilar artery, microcystic changes, and subarachnoid and subventricular spread, as is often seen in patients with DIPG (fig. S6) (14). Tumor growth was relatively slow, with symptoms and/or substantial tumor volume appearing 3 months after injection of 500,000 cells or 5 months after injection of 100,000 cells. Phenotypic analysis of the tumor cells confirmed expression of the H3.3K27M and sh-p53 constructs (HA and RFP tags in 78% and 76.4% of all human cells, respectively) (fig. S7, A and B), as well as markers of immature NPCs (Nestin, Sox2, and Olig2) and high proliferation rates in the P5K group compared with P5W cells or normal NPCs (10.4% versus 0.7% and 0%, respectively) (Fig. 2, F to H, and fig. S7C). GFAP (glial fibrillary acidic protein) colabeled with tumor cells that had down-regulated H3K27M expression (fig. S7D). Pathologically, the tumors resembled lower-grade DIPGs and not full-blown glioblastomas (GBMs), as determined by the absence of necrosis and microvascular proliferation. We speculate that longer in vivo growth periods may be required for the accumulation of mutations leading to a more malignant phenotype. Animals bearing P5W cells showed evidence of a few cell clusters and minimal infiltration in the pons, whereas mock cell injections were associated with minimal cell survival (Fig. 2C).
We next examined gene expression profiles of P5K cells maintained in vitro or sorted by fluorescence-activated cell sorting from tumor-bearing animals. These profiles were compared with those of patient tumors, including DIPGs with H3K27M or H3G34R/V mutations and adult GBMs without histone mutations (Fig. 3A and fig. S8A). Upon unsupervised hierarchical clustering, the P5K cells from in vitro or in vivo sources clustered closer to the H3K27M group (P < 4.36 × 10−8 and P < 2.26 × 10−4) than the G34R/V or GBMs without histone mutation (fig. S8B). We next analyzed differentially expressed genes among the different NPC groups (Fig. 3B, fig. S8C, and tables S1 and S2). We noted an enrichment in the K27M-expressing cells of a subset of transcripts known to be expressed in neuroepithelial cells at a very early developmental stage—i.e., the neural plate, which precedes the emergence of NPCs. To validate this finding, we derived from public datasets (GEO accession no. GSE9921) (15) gene sets that are uniquely expressed at the neural rosette (early neural plate) stage and the neural precursor stage (L-NSCs), as well as gene sets that are shared between rosette and undifferentiated ES cells. A gene set enrichment analysis revealed a significant correlation between H3.3K27M differentially regulated genes and genes enriched in rosette cells (R-NPCs), but not in normal NPCs (L-NPCs) (fig. S8D). We also found a highly significant intersection of genes that were up-regulated in H3.3K27M cells with those in the rosette or rosette-human ES groups. In contrast, most of the genes that were differentially down-regulated in the H3.3K27M cells were shared with those in the NPC group (fig. S8E). Reverse transcription with quantitative polymerase chain reaction (RT-qPCR) validated the increased expression of LIN28B, PLAG1, and PLAGL1 in hES cells and rosettes and substantial downregulation of all three genes in normal NPCs. Expression of H3.3K27M in NPCs up-regulated expression levels of the same genes (Fig. 3C). Note that analysis of patient samples revealed that these genes were expressed at higher levels in DIPGs with the H3K27M mutation than in DIPGs with the G34R/V mutation or without either mutation in histone H3.3 (1) (Fig. 3D and fig. S9). Knockdown of LIN28B or PLAG1 led to a significant decrease in cell number and proliferation in P5K cells (fig. S10). These data suggest that expression of mutant H3.3K27M leads to a developmental resetting of neural precursors to a more primitive stem cell state, which in combination with growth factor signaling, results in the acquisition or consolidation of oncogenic features.
Fig. 3. Genomewide analysis of the K27M transformed NPCs.
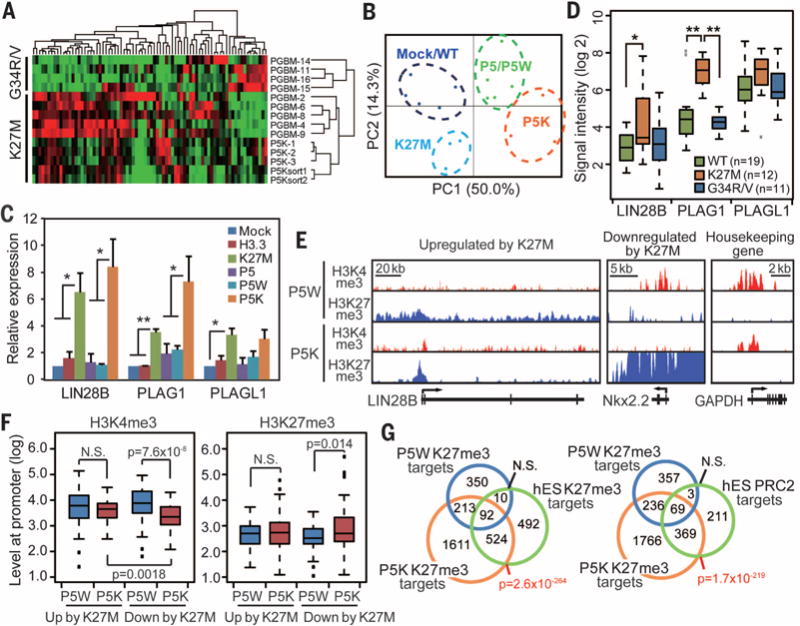
(A) Hierarchical clustering of microarray data from P5K cells, xenograft-derived P5K cells, and patient tumor samples bearing the K27M or G34R/V mutations (GSE34824) obtained from (1). (B) Principal component analysis (PCA) of microarray data obtained from NPCs transduced with the different H3.3 and oncogene combinations. (C) RT-qPCR demonstrates increased expression levels of LIN28B, PLAG1, and PLAGL1 in the P5K cells. Bars indicate means ± SEM (n = 4 to 6). (D) Boxplot shows the expression levels of the genes in patient tumor samples. (E to G) The chromatin landscape of transformed cells. P5K cells showed a global decrease in H3K27me3, but enrichment at the gene body or promoter regions of select genes as shown in representative profiles [(E) H3K4me3 in red and H3K27me3 in blue]. (F) Levels of H3K4me3 and H3K27me3 marks at the promoter region (+1 kilobase to ~500 base pairs) of the genes differentially regulated by K27M. P values were calculated by the Wilcoxon rank-sum test. (G) Venn diagrams comparing genes that are marked by H3K27me3 or bound by PRC2. *P < 0.05; **P < 0.01; NS, not significant.
We next investigated whether the transcriptional changes in the K27M groups were associated with chromatin modifications. We performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) analysis to map active H3K4me3 and repressive H3K27me3 marks in the P5W and P5K cells. Consistent with previous reports (11, 16), H3K27me3 peaks undergo genomic redistribution in the P5K condition (Fig. 3E). Notably, the genes up-regulated by K27M had significantly lower levels of H3K27me3 at their gene-body regions in P5K cells in comparison with P5W cells (P < 3.3 × 10−5) (Fig. 3E and fig. S11, A to D). Concomitantly, H3K4me3 peaks remained stable at promoter regions, which implied that K27M may release these genes from the poised state. On the other hand, the genes down-regulated by K27M gained H3K27me3 and lost H3K4me3 marks in P5K cells at their promoter regions (Fig. 3, E and F; fig. S11C; and table S3). Of these, oligodendrocyte differentiation genes Nkx2.2 and MBP were highly marked by H3K27me3, and their expression levels were decreased in the NPCs expressing K27M alone or the P5K combination (Fig. 3E and fig. S11, B and E). Master genes that initiate acquisition of astrocyte cell fate are not fully elucidated in humans and could not be analyzed. We further analyzed a set of genes that gain H3K27me3 marks in P5K and P5W cells by comparing them with reported H3K27me3 or PRC2 target genes in undifferentiated hES cells (17). There was a highly significant intersection of P5K-specific H3K27me3-target genes with H3K27me3-target genes, as well as PRC2-target genes in ES cells (P < 2.6 × 10−264 and P < 1.7 × 10−219) (Fig. 3G and table S4), whereas P5W-specific H3K27me3-target genes have no significant intersection. These data support our hypothesis that the expression of K27M leads to a resetting of neural precursors to a more primitive stem cell state.
To explore whether the hES-based tumor model can be used for drug discovery, we performed a chemical screen using a commercially available small-molecule library of compounds that target epigenetic regulators (n = 80), such as BET bromodomain inhibitors, deacetylase and demethylase inhibitors, including selective JMJD3 inhibitors (table S5 and the methods section of supplementary materials). Green fluorescent protein (GFP)–labeled normal NPCs and RFP-labeled P5K cells were mixed and seeded onto 96-well plates. Cells were then treated with each compound at eight different concentrations for 6 days, and GFP and RFP fluorescence was quantified by a plate reader (Fig. 4A). The top hit was the menin inhibitor MI-2 (18), which reduced survival of P5K cells at submicromolar concentrations [median inhibitory concentration (IC50) = 155 nM] but had no effect on normal NPCs (Fig. 4B, fig S12A, and table S5). The effect of MI-2 on cell survival was independent of the P5 combination and was restricted to cells with the H3K27M mutation (Fig. 4, C and D) (fig. S12, B to D). shRNA-mediated silencing of the gene encoding menin (MEN1) resulted in decreased proliferation of P5K but not P5W cells (Fig. 4E and fig. S12, E to H). It also restored astrocytic differentiation in P5K cells (fig. S12, I and J), which suggests that a common mechanism underlies the major transformation features, i.e., proliferation and impaired differentiation. Menin is expressed in undifferentiated hES cells and rosettes, but its levels decrease in normal NPCs and astrocytes; however, expression of the P5K combination in NPCs increases menin transcription sixfold (fig S13). Pontine injections of P5K cells transduced with shRNA against menin resulted in significantly reduced tumor formation in mice (fig. S14A). We also tested the impact of systemic treatment with MI-2 on tumor growth in vivo. To this end, we injected P5K cells transduced with a luciferase vector in the brainstem of a large group of mice (n = 26), monitored for tumor development via bioluminescence imaging (BLI), then started treatment with intraperitoneal injections of MI-2. Control mice received tumor cell injections in the brain but were treated with dimethyl sulfoxide (DMSO) intraperitoneally. After 4 weeks of drug treatment, the MI-2 group demonstrated significantly smaller tumors by BLI (P = 0.026) (Fig. 4F and fig. S14B) in comparison with the DMSO group. We also live-sorted P5K cells from mouse tumors and tested their response to MI-2. The cells exhibited a decrease in cell survival and proliferation and increased apoptosis, similar to P5K cells that were never injected in mice (fig. S15). P5K cells live-sorted from mouse tumors also exhibited a decrease in cell survival when LIN28B or PLAGL1 are silenced (fig. S15H). Finally, we tested MI-2 on cell cultures derived from a patient sample. Cells from a human DIPG positive for the H3.3K27M mutation showed a robust antiproliferative response (Fig. 4G and fig. S16). Menin plays a tumor-suppressor role in endocrine cancers but is highly oncogenic in mixed lineage–associated leukemias (19, 20). It is a member of the trithorax family histone methyltransferase complex but also interacts with a wide range of proteins and is thought to be involved in transcriptional regulation. Our data suggest that it may be a potential therapeutic target for pediatric gliomas harboring the H3.3K27M mutation.
Fig. 4. Chemical screen of the transformed NPCs.
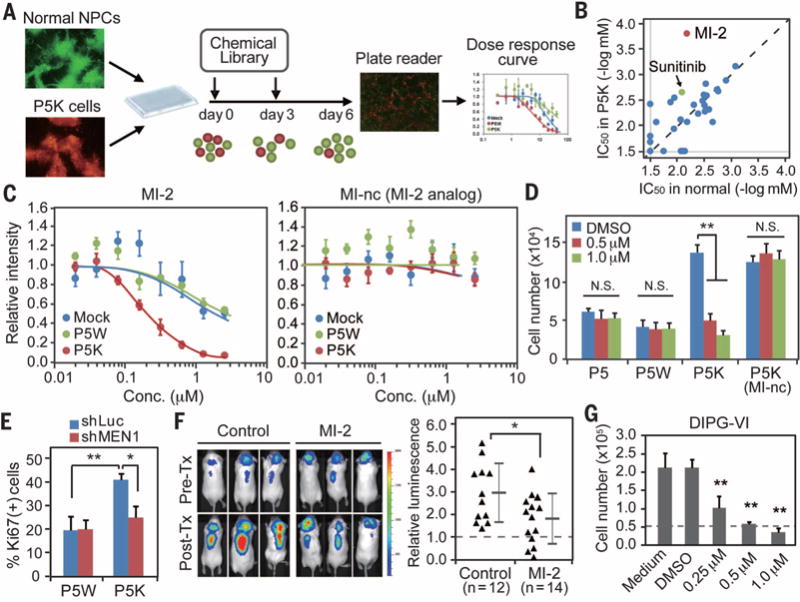
(A) Schematic representation of the screening strategy. A mixture of GFP-labeled normal NPCs and RFP-labeled P5K cells in a 1:3 ratio were plated into 96-well plates, and each compound in the library was added in a twofold serial dilution for a total of eight doses. (B) IC50 was calculated for each compound after 6 days in vitro by using a fluorescence plate reader. (C) Representative dose-response curves in normal NPCs (mock, blue), P5W (green), and P5K (red) cells treated with MI-2 or MI-nc (MI-2 analog) show selectivity of MI-2, a menin inhibitor (n = 4). (D) Viability assay demonstrates a significant effect of MI-2 on P5K cells, with no impact on normal or P5Wcells (n = 4). (E) Silencing of menin via shRNA also decreased the proliferation of P5K cells. (F) Administration of MI-2 suppressed in vivo growth of P5K cells. Intracranial growth of luciferase-labeled P5K cells was measured by quantitative in vivo bioluminescence imaging. Values indicate fold change of luminescence before and after the treatment. (G) MI-2 treatment suppressed the proliferation of a patient DIPG-derived cell line. Cells were treated with MI-2 for 7 days, and the number of viable cells was counted by trypan blue staining (n = 4). Error bars in panels (C) to (G) indicate means ± SD. *P < 0.05; **P < 0.01; NS, not significant.
In summary, using hES cells as a platform to model diffuse intrinsic gliomas, we demonstrate a driver role of the H3.3K27M mutation in the appropriate cell context and developmental window; the model also showed that the altered chromatin landscape induced by H3K27M facilitates the reacquisition of an earlier developmental program with subsequent activation of factors crucial to reprogramming and oncogenesis, such as the micro-RNA binding protein LIN28B (21). A chemical screen identified the menin pathway as a contributor to tumor maintenance, which provides a potential opportunity for therapeutic intervention.
Supplementary Material
Acknowledgments
K.F. was supported by grants from the Brain Tumor Center and the Center for Stem Cell Biology at Memorial Sloan Kettering Cancer Center; C.D.A. is supported by The Rockefeller University. Funding in part from The Geoffrey Beene Cancer Center, The Starr Cancer Consortium and the New York State Stem Cell Board (NYSTEM). Microarray data generated in this manuscript are deposited in GEO (GSE55541). We thank J. Huse, Sloan Kettering, for the pathology assessment of the mouse tumors, and M. Monje, Stanford University, for the human DIPG line, which is available from her under a material transfer agreement. V.T. and K.F. are coinventors on a patent application filed by Memorial Sloan Kettering Cancer Center related to the use of the MI-2 drug in tumors and the ES-based modeling system.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/346/6216/1529/suppl/DC1
Materials and Methods
References (22–30)
REFERENCES AND NOTES
- 1.Schwartzentruber J, et al. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 2.Wu G, et al. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, et al. Nat Genet. 2013;45:602–612. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sturm D, et al. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 5.Khuong-Quang DA, et al. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabar V, Studer L. Nat Rev Genet. 2014;15:82–92. doi: 10.1038/nrg3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chambers SM, et al. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paugh BS, et al. Cancer Res. 2013;73:6219–6229. doi: 10.1158/0008-5472.CAN-13-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis PW, et al. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan KM, et al. Genes Dev. 2013;27:985–990. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bender S, et al. Cancer Cell. 2013;24:660–672. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Halazonetis TD, Gorgoulis VG, Bartek J. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan D, Weinberg RA. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 14.Sethi R, et al. J Neurooncol. 2011;102:121–127. [Google Scholar]
- 15.Elkabetz Y, et al. Genes Dev. 2008;22:152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis PW, Allis CD. Cell Cycle. 2013;12:3241–3242. doi: 10.4161/cc.26356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-Porath I, et al. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grembecka J, et al. Nat Chem Biol. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matkar S, Thiel A, Hua X. Trends Biochem Sci. 2013;38:394–402. doi: 10.1016/j.tibs.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yokoyama A, et al. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 21.Viswanathan SR, Daley GQ. Cell. 2010;140:445–449. doi: 10.1016/j.cell.2010.02.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.