

Significance
The molecular chaperone heat-shock protein 90 (Hsp90) is a key member of the cellular proteostasis network, and as such helps to protect cells against proteotoxic stress. Cancer cells have up-regulated members of this network, including Hsp90, to promote their survival and growth. Several Hsp90 inhibitors have undergone clinical trials, but these drugs, which bind to a shared nucleotide pocket in the N-terminal domain, do not differentiate between the four Hsp90 family members [Hsp90α, Hsp90β, GRP94 (glucose-regulated protein 94 kDa), and TRAP1 (tumor necrosis receptor-associated protein 1)]. In this report, we identify a pharmacophore contained within the natural product gambogic acid that binds uniquely to a site in Hsp90β, thus identifying this compound as a prototype of a new class of isoform-specific Hsp90 inhibitors.
Keywords: molecular chaperone, heat-shock protein 90, isoform-specific inhibitor, molecular docking, caged xanthone
Abstract
Because of their importance in maintaining protein homeostasis, molecular chaperones, including heat-shock protein 90 (Hsp90), represent attractive drug targets. Although a number of Hsp90 inhibitors are in preclinical/clinical development, none strongly differentiate between constitutively expressed Hsp90β and stress-induced Hsp90α, the two cytosolic paralogs of this molecular chaperone. Thus, the importance of inhibiting one or the other paralog in different disease states remains unknown. We show that the natural product, gambogic acid (GBA), binds selectively to a site in the middle domain of Hsp90β, identifying GBA as an Hsp90β-specific Hsp90 inhibitor. Furthermore, using computational and medicinal chemistry, we identified a GBA analog, referred to as DAP-19, which binds potently and selectively to Hsp90β. Because of its unprecedented selectivity for Hsp90β among all Hsp90 paralogs, GBA thus provides a new chemical tool to study the unique biological role of this abundantly expressed molecular chaperone in health and disease.
Heat shock protein 90 (Hsp90) is a molecular chaperone essential for eukaryotic cell viability and protein homeostasis (proteostasis). Within the cell, Hsp90 plays a critical role in helping to fold, assemble, and maintain the proper 3D structures of a variety of proteins, referred to as clients (1, 2). In cancer cells, these client proteins are involved in many oncogenic processes such as aberrant cell proliferation, metastasis, and angiogenesis (3). Moreover, cancer cells that experience various damaging effects due to microenvironmental stresses such as hypoxia and nutrient deficiency up-regulate Hsp90 to enable survival (4, 5). Consequently, Hsp90 is the subject of numerous drug discovery efforts in cancer and other diseases characterized by disordered proteostasis (6).
By virtue of its function as a master regulator of protein folding, Hsp90 is one of the most highly conserved and abundantly expressed proteins across all species. In eukaryotic cells, under nonstress conditions, Hsp90 accounts for 1 to 2% of total protein load, but under cellular stress these levels can increase to 4 to 6%. There are two major cytoplasmic isoforms of Hsp90: the stress-inducible Hsp90α and the constitutively expressed Hsp90β. These isoforms share 85% sequence identity and display significant functional redundancy, although each paralog has also developed distinct cellular functions during evolution (7, 8). Unfortunately, current pharmacologic approaches do not allow interrogation of isoform-specific function, because currently available agents are not able to clearly distinguish between Hsp90α and Hsp90β (9). Other organelle-specific Hsp90 paralogs include glucose-regulated protein 94 kDa (GRP94) in the endoplasmic reticulum and tumor necrosis receptor-associated protein 1 (TRAP1) in mitochondria (10, 11). All Hsp90 family members couple ATP binding and hydrolysis to a cycle of conformational rearrangements that drive chaperone activity (12). Hsp90α and Hsp90β require the physical interaction of a group of cohort proteins, referred to as cochaperones, to aid in client delivery, ATP hydrolysis, and conformational cycling (13–15). When the chaperone cycle is blocked by Hsp90 inhibitors, client proteins are unable to fully mature, and instead become ubiquitinated and degraded within the proteasome (16, 17).
At physiological conditions, each Hsp90 paralog exists as a homodimer composed of three highly conserved and structurally distinct domains: the N-terminal domain (NTD) with an adenosine triphosphate (ATP)-binding pocket, which regulates N-domain conformational dynamics; the middle domain (MD), which is involved in client recognition/binding and is necessary for ATPase competence; and the C-terminal domain (CTD), which ensures Hsp90 dimerization and also contains binding sites for various cochaperones including Hop, FKBP52, Cyp40, and CHIP (18). Upon ATP binding, the two NTDs dimerize and initiate a conformational cycle that drives client protein folding with the assistance of cochaperones including Aha1, p23, and Cdc37 (19–21).
The conserved ATP-binding pocket of all Hsp90 paralogs has been the target of most drug discovery efforts and is the binding site for various clinically evaluated inhibitors such as 17-allylaminogeldanamycin (17-AAG), ganetespib (STA-9090), and SNX-2112 (22–24). In addition, the CTD domain of Hsp90 contains a second drug-binding site (e.g., for novobiocin and other C-terminal Hsp90 inhibitors) (25–27). However, despite robust preclinical data, clinical studies with these inhibitors have reported only modest single-agent activity, highlighting the need to identify additional druggable sites on Hsp90, as well as new small molecules with improved paralog selectivity (28, 29). Although recent drug discovery efforts have moved in this direction (30), to date the only paralog-specific inhibitors identified are for GRP94 (31–33) as well as an inhibitor with a moderate preference for Hsp90α (34). At present, there are no selective Hsp90β inhibitors, making it difficult to explore the distinct cellular roles for this constitutively expressed Hsp90 protein without resorting to genetic approaches.
The natural product gambogic acid (GBA), a polyprenylated xanthone derived from the resin of Garcinia hanburyi trees (35), is a promising anticancer agent currently in phase II clinical trials in China in patients with non–small-cell lung, colon, and renal cancers (36). GBA potently inhibits cancer cell proliferation in vitro and in mouse xenograft models (37–41). Although GBA is reported to have multiple effects in cancer cells (42, 43), recent studies have ascribed some of GBA’s antitumor activity to its binding to Hsp90 (44, 45). In this report, we further define the interaction of GBA with Hsp90. Unexpectedly, our findings identify GBA as an Hsp90β-specific inhibitor. Using a series of Hsp90 deletion mutants and molecular docking of GBA to the Hsp90 MD, we have uncovered a previously unrecognized druggable binding site distinct from the NTD ATP pocket- and CTD novobiocin-binding sites. Thus, our findings provide access to bioprobes able to pharmacologically dissect the isoform-specific functions of Hsp90α and Hsp90β. In addition, they demonstrate that GBA represents a lead with which to pursue new drug discovery efforts exploiting a novel mechanism of Hsp90 inhibition.
Results
GBA Preferentially Binds to the Hsp90β Isoform.
The chemical structures of GBA and biotinylated GBA (Bio-GBA) are shown in Fig. 1A. Using streptavidin bead pulldowns from cell lysates, we first examined Bio-GBA binding to FLAG-tagged Hsp90α, Hsp90β, and TRAP1 proteins that were transiently expressed in HEK293 cells. We found that Bio-GBA was only able to bind to FLAG-Hsp90β, whereas STA-7346 (a biotinylated derivative of the NTD ATP pocket-binding inhibitor STA-9090) binds strongly to all three isoforms (Fig. 1B). Next, we examined Bio-GBA’s ability to bind to purified Hsp90 proteins. Consistent with the transient transfection data, GBA binds markedly less to Hsp90α and TRAP1 proteins than it does to Hsp90β (Fig. 1C). To investigate the binding preferences of endogenous Hsp90 proteins, we used lysate from HEK293 and SKBR3 cell lines to compare Bio-GBA’s ability to bind Hsp90 paralogs. Again, GBA does not detectably bind to endogenous Hsp90α and GRP94; the only detectable binding was to Hsp90β. Conversely, STA-7346 could not distinguish between paralogs and binds all three (Fig. 1 D and E). To compare the relative affinities of GBA to Hsp90α and Hsp90β, we set up a competition assay in which lysates were incubated with increasing concentrations of unlabeled GBA before pulldown of FLAG and HA-tagged Hsp90 proteins with Bio-GBA. We found that unlabeled GBA was able to compete with Hsp90β binding to Bio-GBA in a dose-dependent manner, whereas Hsp90α displayed a low level of binding not affected by increasing concentrations of unlabeled GBA (Fig. 1F). A plate-based (luminescence-based mammalian interactome; LUMIER) drug-binding assay confirmed the isoform specificity of Bio-GBA, which interacted with Hsp90β 10 times more strongly than it did with Hsp90α (Fig. 1G).
Fig. 1.

Gambogic acid preferentially binds to Hsp90β. (A) Chemical structures of gambogic acid and GBA-biotin. (B–E) Hsp90 from cell lysate or purified protein was isolated with biotinylated GBA and streptavidin beads (abbreviated as Strep beads in all figures). STA-7346, the biotinylated version of the NTD-targeted inhibitor ganetespib, was used for comparison. AP, affinity purification; WB, Western blot. (B) FLAG-Hsp90α, FLAG-Hsp90β, and FLAG-TRAP1 were transfected into HEK293 cells. Only Hsp90β bound to Bio-GBA, whereas STA-7346 interacted strongly with all three isoforms. (C) Purified proteins (10 μg; Hsp90α, Hsp90β, and TRAP1) were incubated with Bio-GBA and STA-7346. Bio-GBA bound strongly to Hsp90β, only weakly to Hsp90α, and not at all to TRAP1. (D and E) Binding preferences of endogenous Hsp90α, Hsp90β, and GRP94 were evaluated in lysate from HEK293 (D) and SKBR3 (E) cells. Bio-GBA bound only to Hsp90β, whereas STA-7346 bound to all isoforms tested. (F) HEK293 cells were transfected with 3×F(LAG)-Hsp90α, 3×F-Hsp90β, HA-Hsp90α, or HA-Hsp90β. The following day, cells were lysed and lysates were treated with increasing concentrations of unlabeled GBA. After incubation for 30 min, Hsp90 protein was isolated with Bio-GBA and streptavidin beads. Compared with 3×F-Hsp90α and HA-Hsp90α, 3×F-Hsp90β and HA-Hsp90β bound strongly to Bio-GBA and binding was competitively and dose-dependently inhibited by pretreatment of cell lysate with unlabeled GBA. (G) LUMIER analysis confirmed that Bio-GBA binds to 3×F-Hsp90β at 10 times the binding strength of 3×F-Hsp90α [using lysates of HEK293 cells transfected with Hsp90 and control (EYFP) vector expression plasmids]. Error bars represent SDs. *P < 0.05.
Gambogic Acid Promotes Degradation of Hsp90-Dependent Clients and Demonstrates a Unique Client and Cochaperone Binding Profile.
To explore the cellular consequences of GBA-mediated inhibition of Hsp90β, we assessed depletion of selected endogenous Hsp90 clients. We treated SKBR3 cells with 0 to 10 μM GBA for 6 h (DMSO was used as a negative control) and measured the levels of the Hsp90-dependent kinases ErbB2, phospho-Akt, Akt, and Cdk4 and the Hsp90-dependent nuclear receptor glucocorticoid receptor (GR). α-Tubulin was used as a loading control. GBA promoted the loss of these Hsp90-dependent clients in a concentration-dependent manner (Fig. 2A). Importantly, and in contrast to ATP-competitive NTD-targeted inhibitors, GBA treatment did not cause a significant cotemporal induction of either of the cytoprotective chaperones Hsp70 and Hsp27 (Fig. 2A). Next, we asked whether Bio-GBA could isolate client–Hsp90 protein complexes, as most currently identified Hsp90 inhibitors are unable to do so. GBA was able to bind Hsp90 associated with several endogenous clients in HEK293, SKBR3, and 22Rv1 cells, including ErbB2, Cdk4, Raf-1, GR, and androgen receptor (AR), a second Hsp90-dependent nuclear receptor (Fig. 2B).
Fig. 2.

Gambogic acid promotes degradation of Hsp90-dependent clients and demonstrates a unique client and cochaperone binding profile. (A) SKBR3 cells were treated with GBA for 6 h at increasing concentrations. GBA induced potent, dose-dependent degradation of several endogenous Hsp90 client proteins, although not significantly impacting expression of either of the cytoprotective chaperones Hsp70 and Hsp27. (B and C) Hsp90β–client and Hsp90β–cochaperone complexes were isolated from HEK293, SKBR3, and 22Rv1 cell lysates with Bio-GBA and streptavidin beads. (B) Bio-GBA is able to pull down Hsp90 complexed to several endogenous client proteins, unlike most classic NTD-targeted Hsp90 inhibitors. (C) Bio-GBA recognizes Hsp90 in complex with multiple cochaperones recognizing distinct Hsp90 conformational states.
Cochaperones bind to distinct conformations of Hsp90 and deliver clients or facilitate structural changes in the chaperone to inhibit or accelerate Hsp90 ATPase activity. Consequently, cochaperones are important regulators of Hsp90 dynamics, and their binding identifies distinct conformational states of the chaperone. We examined which cochaperone–Hsp90 complexes are accessible to GBA in HEK293 cells. Bio-GBA was able to pull down Hsp90 associated with the cochaperones Cdc37, Aha1, FKBP52, and Cyp40, while excluding Hop and p23 (Fig. 2C). These data suggest that GBA, unlike most N-terminal ATP pocket-binding inhibitors, recognizes a range of ATP-dependent and ATP-independent Hsp90 conformations.
To confirm that copulldown of clients and cochaperones was indirect and dependent on GBA binding to Hsp90β, we transfected HEK293 cells with either control or Hsp90β-specific siRNA for 72 h and then blotted cell lysates for the degree of Hsp90β knockdown and client/cochaperone levels. At the same time, we affinity-purified Hsp90β complexes with Bio-GBA and blotted for selected copurifying cochaperones and clients (Fig. S1). The data show clearly that neither client nor cochaperone levels in lysates were affected by Hsp90β siRNA (whereas Hsp90β expression was reduced). In contrast, clients and cochaperones in a Bio-GBA pulldown from lysates of Hsp90β siRNA-treated cells were clearly reduced, concomitant with a reduction in the amount of affinity-purified Hsp90β.
Fig. S1.

GBA does not bind directly to Hsp90 clients and cochaperones. HEK293 cells were transfected with control siRNA or Hsp90β siRNA (20 nM). Cell lysates were prepared after 72 h and incubated with biotinylated GBA to isolate Hsp90–client/cochaperone complexes. “Input” lanes reflect lysate concentrations of Hsp90β and each client or cochaperone. Biotinylated GBA pulldown lanes are shown (Left). Although cochaperone and client expression was not affected by silencing Hsp90β, copulldown of these proteins was reduced, consistent with the degree of reduction in expression of Hsp90β following siRNA silencing.
GBA Binds at a Site Distinct from N-Terminal and C-Terminal Hsp90 Inhibitors.
Current Hsp90 inhibitors target nucleotide-binding sites in either the NTD or CTD (46, 47). Because these regions are highly conserved throughout the four Hsp90 paralogs, these agents do not selectively target one paralog over another (48–50). The apparent selectivity of GBA for Hsp90β suggested that its binding site was unique. To determine whether GBA bound to the NTD ATP pocket, we pretreated HEK293 lysate with STA-9090, SNX-2112, or 17AAG (each at 10 μM). None of these drugs had any effect on the amount of Hsp90β subsequently pulled down by Bio-GBA (Fig. 3A). To confirm these findings, we demonstrated that pretreatment with unlabeled GBA (10 μM) had no effect on the ability of STA-7346 or SNX-2112 to pull down Hsp90β. As a positive control, we showed that N-terminal inhibitors were able to compete with each other. To test whether C-terminal inhibitors are competitive with GBA, we pretreated cell lysate with the novobiocin derivative KU32 (10 μM) (51), which also did not affect Bio-GBA binding to Hsp90β (Fig. 3A). Finally, we used the LUMIER assay to provide quantitative support for these observations (Fig. 3B).
Fig. 3.

Gambogic acid binds at a site distinct from NTD and CTD Hsp90 inhibitors. (A) HEK293 cells were lysed and lysates were treated with various Hsp90 inhibitors (10 μM). Bio-GBA and streptavidin beads were then added to isolate Hsp90β. Only unlabeled GBA was able to compete with Bio-GBA for binding. Conversely, HEK293 lysate was also incubated with free GBA and pulled down with STA-7346– or SNX-2112–conjugated drug beads. GBA did not block binding of these NTD-targeted inhibitors. As a positive control, STA-9090 (ganetespib) was added to lysate followed by Hsp90 pulldown with SNX-2112–conjugated beads and competitive binding inhibition was observed. Similar competitive binding inhibition was seen when SNX-2112 was added to lysate followed by Hsp90 pulldown with STA-7346. (B) LUMIER analysis of drug competition confirmed that no NTD- or CTD-targeted inhibitors compete with GBA for binding to Hsp90β. Error bars represent SDs. *P < 0.05.
Domain Dissection of Hsp90β Reveals a Druggable Site in the Hsp90β MD.
To identify the GBA-binding site on Hsp90β, we constructed a series of recombinant C-terminal 3×F(LAG)-Hsp90β truncation mutants. These constructs were transfected into HEK293 cells, and we subsequently subjected cell lysates to Bio-GBA and streptavidin beads. We found that the first 432 residues of Hsp90β are needed to confer GBA binding, eliminating any requirement of the CTD that begins at residue 602 in Hsp90β (Fig. 4 and Fig. S2). In addition, because GBA does not bind to the NTD alone, these data suggest that the MD of Hsp90β is the site of GBA binding. Specifically, it appears that residues between amino acids 368 and 453 are critical for binding. In support of this model, Bio-GBA binds to NTD-deleted Hsp90β (Fig. 4 and Fig. S2). In contrast, STA-7346, which binds within the N-terminal ATP pocket, requires only the Hsp90β NTD for binding. However, when this region is deleted in the 268–642 Hsp90β mutant, STA-7346, unlike Bio-GBA, is no longer able to bind (Fig. 4 and Fig. S2). Taken together, these findings are consistent with GBA recognizing a druggable site within the MD of Hsp90β that provides paralog specificity.
Fig. 4.

Domain dissection of Hsp90β reveals a druggable site in the MD. Various 3×F-Hsp90β truncation mutants were made and transfected into HEK293 cells. STA-7346 was used as a representative NTD-targeted inhibitor and bound to all fragments that included the NTD. Bio-GBA was only able to bind to Hsp90β fragments that contained at least the first 432 residues. In contrast to STA-7346, GBA binding did not require the NTD. See Fig. S2 for the raw data supporting this figure.
Fig. S2.

Biotinylated GBA and STA-7346 (biotinylated N-terminal domain inhibitor) binding to Hsp90β truncation mutants. HEK293 cells were transfected with various 3×F-Hsp90β truncation mutants. After cell lysis, biotinylated GBA and STA-7346 were added to isolate 3×F-Hsp90β. (A–C) C-terminal truncations extending into the MD beyond amino acid 432 in Hsp90β are not recognized by biotinylated GBA, whereas STA-7346 bound all C-terminal truncation mutants retaining the NTD. (D) Densitometric quantification of selected data obtained in C, normalized for input. IP, immunoprecipitation. (E) N-terminally truncated Hsp90β (C268–648) retains binding to GBA but not to STA-7346.
Identification of the GBA-Binding Pocket in Hsp90β Using Computational Modeling and Site-Specific Mutagenesis.
Based on the above domain dissection data, we performed blind docking of GBA over the entire MD of Hsp90β and identified a single potential binding pocket located around residues 350 to 436 (Fig. 5A). This pocket contains a deep hydrophobic cavity formed by residues I353, L369, I370, and S365 of Hsp90β that is able to accommodate the hydrophobic side chain located at the C2 position of GBA. Moreover, the hydrogen bonds formed with residues E372, N436, N375, and N351 play an important role in stabilizing the interaction of GBA with Hsp90β (Fig. 5 A and B). In contrast, blind docking of GBA over the equivalent site of Hsp90α (residues 359 to 444) revealed no suitable binding pocket able to accommodate GBA (Fig. 5C). Specifically, the structurally related hydrophobic cavity on Hsp90α is occluded and cannot accommodate the C2 hydrophobic side chain of GBA; further, the related protein surface of Hsp90α is spatially crowded and cannot bind the rigid framework of GBA (Fig. 5 B and C).
Fig. 5.

Molecular docking model of GBA bound to Hsp90β. (A) Molecular modeling suggests that GBA (shown in blue stick representation) binds to a pocket within a region composed of residues 350 to 436 in Hsp90β (shown in pale cyan). Hydrogen bonds formed between GBA and the pocket are shown as red dashed lines. Oxygens are shown in red and nitrogens are shown in blue. (B and C) Surface electrostatic potential map of Hsp90β (residues 350 to 436) and Hsp90α (residues 359 to 444). Red, blue, and white colors correspond to negatively charged, positively charged, and neutral areas, respectively. (D) LUMIER analysis showed decreased GBA binding to 3×F-Hsp90β S365N and 3×F-Hsp90β 369SSA (the triple mutant L369S, I370S, E372A) compared with wild-type 3×F-Hsp90β. Error bars represent SDs. *P < 0.05 relative to wild-type. (E) HEK293 cells were transfected with HA-Hsp90β, HA-S365N, or HA-369SSA. The next day, cells were lysed and increasing amounts of Bio-GBA were used to pull down Hsp90β from cells expressing equivalent amounts of wild-type HA-Hsp90β or the mutants HA-S365N and HA-369SSA. The binding-pocket mutants displayed reduced ability to bind to Bio-GBA, consistent with the data obtained by LUMIER.
To experimentally test the computational predictions of the GBA-binding pocket on Hsp90β, we mutated key residues within the binding region and measured the effect of mutation on Bio-GBA binding. Based on surface topography and sequence similarity between Hsp90α (PDB ID code 3Q6M) and Hsp90β (PDB ID code 3PRY), we created two mutants in 3×F-Hsp90β: the single mutant S365N and the triple mutant L369S, I370S, E372A, denoted as 369SSA. In the S365N single mutation, the serine 365 residue found at the top of the hydrophobic cavity of Hsp90β was replaced by asparagine, reminiscent of the N373 residue that is found at the similar position in Hsp90α. The 369SSA triple mutation was designed to evaluate the effect of the hydrophobic wall at the left side of the GBA-binding pocket. Thus, hydrophobic residues L369 and I370 were mutated to serine, a small uncharged residue. Moreover, the E372 glutamic acid, predicted to form a hydrogen bond with GBA, was replaced by alanine, an amino acid that has no ability to form hydrogen bonds. As envisioned, both mutant proteins demonstrated significantly reduced binding to Bio-GBA (Fig. 5 D and E). The S365N mutation introduces steric hindrance at the top of the hydrophobic cavity and reduces hydrophobicity, as shown by comparing the molecular surfaces of wild-type Hsp90β and the S365N mutant (Fig. S3 A and B). The 369SSA triple mutation results in markedly more impaired GBA binding. This is consistent with the significant changes that occur in both surface topography and electrostatic potential of the GBA-binding pocket of this mutant (Fig. S3C). The triple mutation simultaneously reduces the hydrophobicity contributed by the leucine and isoleucine side chains, eliminates the negative charge contributed by glutamic acid, and eliminates the hydrogen bond formed between E372 and the hydroxyl group of Bio-GBA. To confirm that the triple mutant retains structural integrity, we used HA beads to immunoprecipitate HA-tagged wild-type and triple-mutant Hsp90β transiently expressed in HEK293 cells. Blotting for associating cochaperones Hop (interacts with the Hsp90 open conformation), Cdc37 (interacts with the open conformation), and p23 (interacts with the ATP-bound closed conformation) detected no differences in cochaperone association with wild-type and triple-mutant Hsp90β (Fig. S3D). Similarly, equivalent amounts of both HA-tagged Hsp90β proteins were pulled down from HEK293 lysate with the biotinylated, ATP-competitive, NTD-targeted inhibitor STA-7346 (Fig. S3E). Taken together, these data confirm that triple mutation of Hsp90β residues L369, I370, and E372 does not compromise the chaperone’s structural integrity.
Fig. S3.

Surface electrostatic potential map of the GBA-binding site (residues 350 to 436) in Hsp90β and the impact of binding-site mutation on the interaction of cochaperones. (A) Wild-type human Hsp90β. (B) S375N mutant. (C) Triple mutant L369S, I370S, E372A. Red color corresponds to negatively charged areas, blue color corresponds to positively charged areas, and white color corresponds to neutral areas. The negatively charged area around L369 is because of the D367 (not shown). (D) Plasmids expressing HA-Hsp90β or HA-Hsp90β 369SSA (triple mutant) were transfected into HEK293 cells and then isolated with anti-HA beads. (E) HA-Hsp90β and HA-Hsp90β 369SSA were transfected into HEK293 cells (4 μg plasmid per 10-cm dish) and allowed to express overnight. After 24 h, transfected proteins were immunoprecipitated with HA beads (D) or affinity-purified with STA-7346 and streptavidin beads (E). In D, three cochaperones associated to an equivalent degree with wild-type and mutated Hsp90β. In E, the biotinylated N-terminal inhibitor STA-7346 affinity-purified both wild-type and mutant Hsp90β with equal efficiency. Taken together, the data in D and E confirm that Hsp90β 369SSA is structurally intact.
Chemical Modification of GBA Allows for Increased or Decreased Binding to Hsp90β.
Based on the binding mode of GBA to Hsp90β, both the C2 hydrophobic motif and C29 carboxylic acid group significantly contribute to GBA’s ability to bind to Hsp90β. Consequently, modifications were made to the backbone of GBA to confirm our virtual docking model and to point the way forward to developing more effective/specific Hsp90β inhibitors (Fig. 6A). Along these lines, we synthesized MAD28 (52–54), which maintains the central rigid caged Garcinia xanthone framework but lacks both the hydrophobic side chains at the periphery of the A ring of GBA (shown in green circles in Fig. 6A) and the C29 carboxylic acid group (shown in yellow circles in Fig. 6A). In addition, we synthesized DAP-19, in which the carboxylic acid of GBA has been converted to a morpholine amide. The incorporated morpholine amide unit was projected to occupy an available binding region around residue K435 and concomitantly to form an additional hydrogen bond between K435 and the morpholine oxygen, thereby increasing the binding to Hsp90β (Fig. 6B). To test these GBA derivatives for binding to Hsp90β, we assessed their ability to compete with Bio-GBA. We found that MAD28, which lacks the critical hydrophobic arms and has reduced ability to form H bonds, did not compete efficiently with Bio-GBA for binding to Hsp90β in cell lysate (Fig. 6C). In contrast, DAP-19 competed more effectively than did unlabeled GBA itself. These results were confirmed using the LUMIER assay to quantify Bio-GBA binding to Hsp90β (Fig. 6D).
Fig. 6.

Model-based structural modifications of GBA increase or decrease binding to Hsp90β. (A) Drug schematic indicates key structural differences between GBA, MAD28, and DAP-19. Hydrophobic side chains at the periphery of the A ring of GBA are marked with green dashed circles. Substituents at the C29 center are marked with a yellow dashed circle. (B) Predicted binding mode of DAP-19 (shown in blue stick representation) against Hsp90β (shown in pale cyan). The morpholine amide group at C29 is marked with a yellow dashed circle. (C) HEK293 cells were lysed and treated with unlabeled GBA, DAP-19, or MAD28 at various concentrations. Endogenous Hsp90β was then isolated from cell lysates with Bio-GBA and streptavidin beads. Compared with unlabeled GBA, DAP-19 more effectively blocked Hsp90β binding to Bio-GBA, whereas MAD28 was less effective. (D) Data obtained from LUMIER analysis are consistent with the results obtained by Western blot (C). Error bars represent SDs. *P < 0.05 relative to DMSO.
SI Materials and Methods
Drug Synthesis.
GBA and MAD28 were synthesized as previously described (43, 52).
Synthesis of DAP-19.
To a solution of GBA (20.0 mg, 31.8 µmol) and HATU (14.5 mg, 38.2 µmol) in dry MeCN (3 mL) was added DIPEA (22.2 µL, 127.2 µmol) via syringe at 0 °C. After the reaction mixture stirred for 15 min, morpholine (3.3 µL, 38.2 µmol) was added via syringe and the reaction mixture was stirred at room temperature for 2 h. The solvent was removed and the crude mixture was diluted with ethyl acetate (30 mL). The organic solution was washed with saturated NH4Cl (aqueous) (10 mL) and brine (10 mL) and dried over anhydrous Na2SO4 (solid). The organic phase was concentrated in vacuo and the residue was purified by column chromatography (silica gel, EtOAc/hexane 1:1) to afford DAP-19 (17.0 mg, 77%) as a yellow solid.
TLC.
Rf = 0.25 (silica gel, EtOAc/hexane 1:1).
[α]D25 = −183.16 (c 0.5, CHCl3).
1H NMR.
(500 MHz, CDCl3) δ 12.85 (s, 1H), 7.58–7.52 (m, 1H), 6.67 (d, J = 10.2 Hz, 1H), 5.49–5.39 (m, 2H), 5.13–4.99 (m, 2H), 3.65–3.51 (m, 5H), 3.47–3.40 (m, 3H), 3.36–3.14 (m, 5H), 2.51 (d, J = 9.3 Hz, 1H), 2.41–2.35 (m, 1H), 2.31–2.27 (m, 1H), 2.06–2.01 (m, 1H), 1.77–1.61 (m, 20H), 1.45–1.40 (m, 3H), 1.38–1.35 (m, 1H), 1.25 (s, 3H) ppm.
13C NMR.
(125 MHz, CDCl3) δ 203.53, 179.02, 169.84, 161.78, 157.85, 157.59, 135.56, 133.27, 133.17, 131.96, 131.72, 124.77, 123.85, 122.73, 122.29, 115.92, 107.72, 102.85, 100.57, 91.14, 83.68, 82.98, 81.52, 67.24, 66.85, 49.05, 47.11, 46.41, 42.15, 41.39, 30.18, 29.49, 28.84, 27.93, 25.78, 25.71, 25.46, 22.83, 21.81, 20.76, 18.22, 17.69 ppm.
High-resolution mass spectrometry.
(m/z): calculated for C42H52NO8+ [M + H]+: 698.3687; found: 698.3682.
Discussion
Current Hsp90 inhibitors being evaluated in the clinic lack the ability to distinguish between cytosolic chaperone isoforms (55). Recently, Blagg and colleagues identified an inhibitor binding to the Hsp90 CTD with moderate specificity for Hsp90α (34). Our data identify GBA as a bioprobe demonstrating Hsp90β-specific inhibition, with a 10-fold binding preference for Hsp90β compared with Hsp90α. Further, GBA does not bind to either the endoplasmic reticulum or mitochondrial Hsp90 paralogs GRP94 and TRAP1, respectively, making it an essentially pure Hsp90β inhibitor. This observation was confirmed through multiple binding assays using tagged, purified, and endogenous Hsp90 proteins. Brief exposure of SKBR3 breast cancer cells to GBA results in the loss of Hsp90 client protein expression, confirming an important role specifically for Hsp90β in these cells. Unlike most other NTD-directed Hsp90 inhibitors, brief exposure to GBA does not markedly induce the cytoprotective chaperones Hsp70 and Hsp27, and GBA is able to pull down Hsp90β that is complexed to diverse client proteins. Our data suggest that GBA may recognize multiple Hsp90 conformational states, unlike most NTD- or CTD-targeted inhibitors. This hypothesis is supported by the ability of GBA to pull down various cochaperone–Hsp90 complexes associated with both ATP-dependent (e.g., Aha1, Cyp40) and ATP-independent (Cdc37, CHIP, FKBP52) Hsp90 conformations. Further study is necessary to understand why other cochaperones identified with either ATP-dependent (e.g., p23) or ATP-independent (e.g., Hop) Hsp90 conformational states are not present in GBA pulldowns, but our findings are consistent with unique cochaperone impacts on Hsp90 conformation in and around the GBA-binding site.
These characteristics of GBA are inconsistent with binding to previously identified regions in the Hsp90 NTD or CTD (56, 57). Indeed, several NTD- and CTD-directed inhibitors did not compete with GBA for binding to Hsp90β, consistent with the possibility that GBA recognizes a unique region in Hsp90. Using a series of CTD and NTD truncation mutants, we localized the GBA-binding region to a portion of the Hsp90 MD not previously identified as a drug-binding site. Molecular modeling of this region identified a pocket in Hsp90β composed of amino acids 368 to 453 able to accommodate GBA and stabilize its interaction via a number of hydrophobic and ionic interactions. Importantly, the equivalent region in Hsp90α was unable to do so, providing an explanation of GBA’s unique isoform specificity and identifying this MD pocket as an additional druggable domain in Hsp90. The location of the GBA-binding pocket relative to the domain organization of Hsp90 is shown in Fig. S4.
Fig. S4.
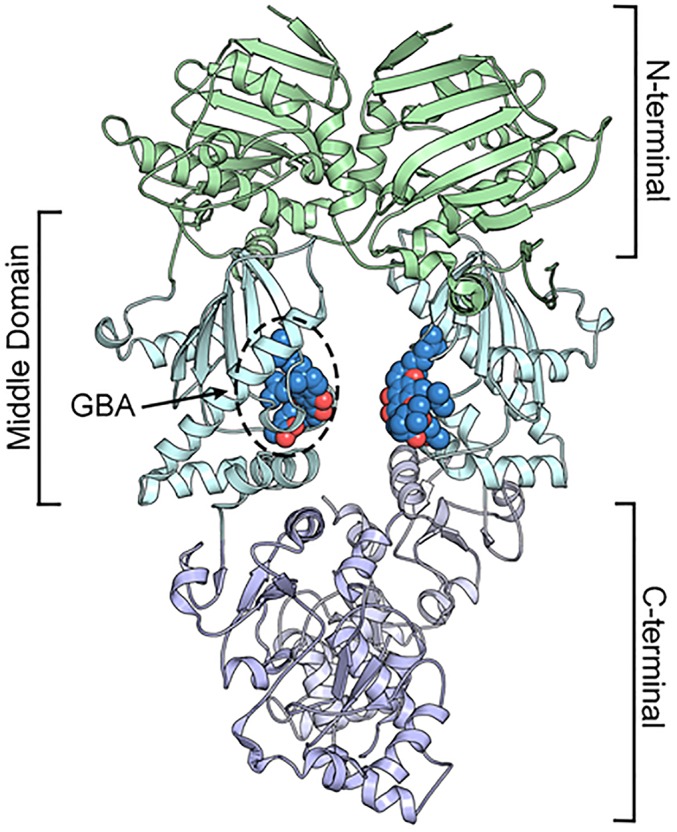
Structure of the Hsp90β dimer showing the N-terminal, middle, and C-terminal domains and the GBA-binding site in the MD.
Finally, based on our molecular docking model, we made chemical modifications to GBA that either improved (e.g., DAP-19) or impaired (e.g., MAD28) interaction with Hsp90β, thus providing further support for our proposed GBA-binding pose and proof of concept that this region in Hsp90β is pharmacologically tractable. Although additional studies will be necessary to more precisely identify the mechanism by which GBA disrupts Hsp90 conformational dynamics to inhibit its chaperone function, our data identify GBA as both a bioprobe with which to interrogate the cellular consequences of specifically inhibiting Hsp90β as well as a lead compound to identify novel and specific isoform-selective Hsp90β inhibitors with possible application to cancer and other diseases.
Materials and Methods
Reagents.
GBA (43), Bio-GBA (58), and MAD28 (52–54, 59) were synthesized as previously described. The synthesis and spectroscopic characterization of DAP-19 are shown in SI Materials and Methods and Figs. S5–S7. Geldanamycin was obtained from the Developmental Therapeutics Program, National Cancer Institute. STA-9090 and STA-7346 were obtained from Synta Pharmaceuticals. SNX-2112 and SNX-2112 affinity resin were a kind gift from Timothy Haystead, Duke University, Durham, NC. Antibodies used were as follows: anti-FLAG (Sigma-Aldrich), anti-TRAP1 (kind gift from Sara Felts, Mayo Clinic, Rochester, MN), anti-Hsp90α (Cell Signaling), anti-Hsp90β (Pierce), anti-HA (Sigma-Aldrich), anti-ErbB2 (Neomarkers), anti–phospho-Akt, (Cell Signaling), anti-AKT (Cell Signaling), anti-CDK4 (Santa Cruz), anti-GR (Santa Cruz), anti–α-tubulin (Calbiochem), anti-AR (Santa Cruz), anti-Raf1 (Neomarkers), anti-Hop (Enzo Life Sciences), anti-Aha1 (Rockland), anti-Cdc37 (Cell Signaling), anti-Cyp40 (Abcam), anti-FKBP52 (Abcam), anti-p23 (Enzo Life Sciences), and anti-CHIP (Cell Signaling).
Fig. S5.

Route of synthesis of DAP-19 from GBA.
Fig. S7.

13C NMR spectrum of DAP-19.
Fig. S6.

1H NMR spectrum of DAP-19.
Cell Lines and Transfection.
SKBR3 and 22Rv1 cells were purchased from the American Type Culture Collection. HEK293 cells were purchased from Invitrogen. Cells were cultured in DMEM containing 10% (vol/vol) FBS in incubators held at 37 °C and 5% CO2. Cells were washed with PBS and lysed in TNES buffer (50 mM Tris, 0.1% Nonidet P-40, 2 mM EDTA, 100 mM NaCl) with added protease and phosphatase inhibitor (Roche). Lysates were centrifuged at 16,000 × g for 10 min, and protein content in the clarified supernatant fraction was quantified using the BSA protein assay kit (Pierce). HEK293 cells were transfected with various plasmids (see below) using X-tremeGENE 9 (Roche) transfection reagent for 16 h following the manufacturer’s protocol. Cells were then lysed as above and subjected to further analysis.
Affinity Purification and Western Blot.
For affinity purification, 600 to 800 μg of total protein was precleared with streptavidin beads, treated with biotinylated drug (Bio-GBA or STA-7346) for 1 h at 4 °C, and incubated with streptavidin beads for an additional hour at 4 °C with rotation. SNX-2112 affinity resin was added to precleared lysate. Streptavidin bead or SNX-2112 affinity resin complexes were washed three times with TNES buffer, and proteins were eluted in 4× sample loading buffer [250 mM Tris⋅HCl, 40% (vol/vol) glycerol, 5% (vol/vol) SDS, 250 mM DTT, 0.1% bromophenol blue], subjected to SDS/PAGE, and transferred to nitrocellulose. Membranes were blocked for 1 h with 5% milk, and primary antibodies were added in 5% milk and incubated overnight at 4 °C. Secondary antibodies coupled to HRP were incubated for 1 h at room temperature, and signals were detected with the SuperSignal Detection System (Pierce).
Plasmids.
The following plasmids were generated as previously described (8): FLAG-Hsp90α, FLAG-Hsp90β, FLAG-TRAP-1, 3×F(LAG)-Hsp90α, 3×F-Hsp90β, HA-Hsp90α, HA-Hsp90β, HA-Hsp90β_S365N, HA-Hsp90β_369SSA (the triple mutant L369S, I370S, E371A), 3×F-Hsp90β_c200, 3×F-Hsp90β_c218, 3×F-Hsp90β_c236, 3×F-Hsp90β_c276, 3×F-Hsp90β_c315, 3×F-Hsp90β_c369, 3×F-Hsp90β_c453, 3×F-Hsp90β_c553, 3×F-Hsp90β_c598, 3×F-Hsp90β_c692, and 3×F-Hsp90β_268-642.
Drug Binding Using a Modified Luminescence-Based Mammalian Interactome Procedure.
To determine the relative binding strength of Bio-GBA to Hsp90 isoforms and their mutants, we followed a modified version of the LUMIER method, as previously described (8). Briefly, HEK293 cells were transfected with each 3×F-Hsp90 expression plasmid. Eighteen hours later, transfected cells were harvested with cold TGNET buffer (50 mM Tris⋅HCl, pH 7.5, 5% glycerol, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100). Streptavidin-coated 96-well plates (Pierce) were incubated with Bio-GBA, blocked with TGNET buffer + 3% BSA, and washed. An equal amount of fresh protein lysate was added simultaneously to a streptavidin plate and to an anti-FLAG–coated plate (Sigma-Aldrich). Both plates were incubated for 2 h at 4 °C and washed, and anti–FLAG-HRP (Sigma-Aldrich) was added to each well. The plates were again incubated at 4 °C for 2 h and washed. Finally, the plates were read on a plate reader to measure HRP luminescence activity (Pierce). The relative interaction strength was determined by dividing the light values of the Bio-GBA plate by those of the anti-FLAG plate. Each assay was repeated three times with four replicates each time. SDs are represented by error bars. A two-tailed t test was used to determine statistical significance. All calculations were performed in Excel (Microsoft).
Computer Modeling of GBA Docking to Hsp90β.
To determine the binding site of GBA in the MD of Hsp90β, an in-house molecular docking tool named iFitDock (60) was used to perform a global (blind) docking over the whole protein surface. The structure of Hsp90β (PDB ID code 3PRY) was prepared with the Protein Preparation Wizard (61) in the Maestro suite (https://www.schrodinger.com/maestro) with default settings. A large docking box with the size of 40 × 50 × 80 Å was designed to completely wrap the protein and be used to generate scoring grids with DOCK 6.5 (62). The initial coordinates of GBA and DAP-19 were generated by using Chem3D 14.0 (63) and minimized with the MM2 force field, and their atomic partial charges were assigned by means of the Gasteiger–Marsili method (64). The protein structure was taken as rigid and the other parameters were set as default in docking simulations. As a result, we identified a distinct binding site (and the only binding site) on Hsp90β that was able to accommodate GBA and DAP-19. Binding affinities for all of the binding modes of small molecules to protein were estimated by using the MM-GBSA method (65, 66) integrated in iFitDock, and the most reliable binding mode for each small molecule was selected according to the favorable binding affinity and our visual inspection from the conserved docking modes.
Acknowledgments
This work was supported in part by the Intramural Research Program, Center for Cancer Research, National Cancer Institute Grant Z01 SC010074-12 (to L.N.); National Institutes of Health Grants CA 133002 (to E.A.T.) and GM101467 (to P.A.J.); and a grant from the Cancer Research Coordinating Committee (CRC-15-380737) (to E.A.T.). Work at the Center for Theoretical Biological Physics was sponsored by the National Science Foundation (Grants PHY-1427654 and MCB-1214457) and the Cancer Prevention and Research Institute of Texas. F.B. was also supported by Welch Foundation Grant C-1792.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1606655113/-/DCSupplemental.
References
- 1.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11(7):515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 2.Krukenberg KA, Street TO, Lavery LA, Agard DA. Conformational dynamics of the molecular chaperone Hsp90. Q Rev Biophys. 2011;44(2):229–255. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des. 2013;19(3):347–365. doi: 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao YH, et al. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009;28(42):3689–3701. doi: 10.1038/onc.2009.229. [DOI] [PubMed] [Google Scholar]
- 5.Minet E, et al. Hypoxia-induced activation of HIF-1: Role of HIF-1α-Hsp90 interaction. FEBS Lett. 1999;460(2):251–256. doi: 10.1016/s0014-5793(99)01359-9. [DOI] [PubMed] [Google Scholar]
- 6.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin Cancer Res. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sreedhar AS, Kalmár E, Csermely P, Shen YF. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004;562(1–3):11–15. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 8.Prince TL, et al. Client proteins and small molecule inhibitors display distinct binding preferences for constitutive and stress-induced HSP90 isoforms and their conformationally restricted mutants. PLoS One. 2015;10(10):e0141786. doi: 10.1371/journal.pone.0141786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel PD, et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013;9(11):677–684. doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altieri DC. Mitochondrial HSP90s and tumor cell metabolism. Autophagy. 2013;9(2):244–245. doi: 10.4161/auto.22527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu G, Lee AS. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J Cell Physiol. 2015;230(7):1413–1420. doi: 10.1002/jcp.24923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panaretou B, et al. Activation of the ATPase activity of Hsp90 by the stress-regulated cochaperone Aha1. Mol Cell. 2002;10(6):1307–1318. doi: 10.1016/s1097-2765(02)00785-2. [DOI] [PubMed] [Google Scholar]
- 13.Prodromou C, et al. The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000;19(16):4383–4392. doi: 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 15.Sun L, Prince T, Manjarrez JR, Scroggins BT, Matts RL. Characterization of the interaction of Aha1 with components of the Hsp90 chaperone machine and client proteins. Biochim Biophys Acta. 2012;1823(6):1092–1101. doi: 10.1016/j.bbamcr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berl) 2004;82(8):488–499. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 17.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5(10):761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 18.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: An open and shut case for treatment. Biochem J. 2008;410(3):439–453. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 19.Ali MM, et al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440(7087):1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roe SM, et al. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37) Cell. 2004;116(1):87–98. doi: 10.1016/s0092-8674(03)01027-4. [DOI] [PubMed] [Google Scholar]
- 21.Prodromou C, et al. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90(1):65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 22.Chandarlapaty S, et al. SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase–dependent cancers. Clin Cancer Res. 2008;14(1):240–248. doi: 10.1158/1078-0432.CCR-07-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ying W, et al. Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol Cancer Ther. 2012;11(2):475–484. doi: 10.1158/1535-7163.MCT-11-0755. [DOI] [PubMed] [Google Scholar]
- 24.Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61(10):4003–4009. [PubMed] [Google Scholar]
- 25.Donnelly A, Blagg BS. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem. 2008;15(26):2702–2717. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anyika M, McMullen M, Forsberg LK, Dobrowsky RT, Blagg BS. Development of noviomimetics as C-terminal Hsp90 inhibitors. ACS Med Chem Lett. 2015;7(1):67–71. doi: 10.1021/acsmedchemlett.5b00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buckton LK, Wahyudi H, McAlpine SR. The first report of direct inhibitors that target the C-terminal MEEVD region on heat shock protein 90. Chem Commun (Camb) 2016;52(3):501–504. doi: 10.1039/c5cc03245h. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. Lancet Oncol. 2013;14(9):e358–e369. doi: 10.1016/S1470-2045(13)70169-4. [DOI] [PubMed] [Google Scholar]
- 29.Hong DS, et al. Targeting the molecular chaperone heat shock protein 90 (HSP90): Lessons learned and future directions. Cancer Treat Rev. 2013;39(4):375–387. doi: 10.1016/j.ctrv.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Bhat R, Tummalapalli SR, Rotella DP. Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors. J Med Chem. 2014;57(21):8718–8728. doi: 10.1021/jm500823a. [DOI] [PubMed] [Google Scholar]
- 31.Duerfeldt AS, et al. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134(23):9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muth A, et al. Development of radamide analogs as Grp94 inhibitors. Bioorg Med Chem. 2014;22(15):4083–4098. doi: 10.1016/j.bmc.2014.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel HJ, et al. Structure-activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum Hsp90 paralog Grp94. J Med Chem. 2015;58(9):3922–3943. doi: 10.1021/acs.jmedchem.5b00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu W, et al. KU675, a concomitant heat-shock protein inhibitor of Hsp90 and Hsc70 that manifests isoform selectivity for Hsp90α in prostate cancer cells. Mol Pharmacol. 2015;88(1):121–130. doi: 10.1124/mol.114.097303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chantarasriwong O, Batova A, Chavasiri W, Theodorakis EA. Chemistry and biology of the caged Garcinia xanthones. Chemistry. 2010;16(33):9944–9962. doi: 10.1002/chem.201000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chi Y, et al. An open-labeled, randomized, multicenter phase IIa study of gambogic acid injection for advanced malignant tumors. Chin Med J (Engl) 2013;126(9):1642–1646. [PubMed] [Google Scholar]
- 37.Gu H, et al. Gambogic acid mediates apoptosis as a p53 inducer through down-regulation of mdm2 in wild-type p53-expressing cancer cells. Mol Cancer Ther. 2008;7(10):3298–3305. doi: 10.1158/1535-7163.MCT-08-0212. [DOI] [PubMed] [Google Scholar]
- 38.Huang GM, Sun Y, Ge X, Wan X, Li CB. Gambogic acid induces apoptosis and inhibits colorectal tumor growth via mitochondrial pathways. World J Gastroenterol. 2015;21(20):6194–6205. doi: 10.3748/wjg.v21.i20.6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li C, et al. Gambogic acid promotes apoptosis and resistance to metastatic potential in MDA-MB-231 human breast carcinoma cells. Biochem Cell Biol. 2012;90(6):718–730. doi: 10.1139/o2012-030. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, et al. Methyl jasmonate sensitizes human bladder cancer cells to gambogic acid-induced apoptosis through down-regulation of EZH2 expression by miR-101. Br J Pharmacol. 2014;171(3):618–635. doi: 10.1111/bph.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yi T, et al. Gambogic acid inhibits angiogenesis and prostate tumor growth by suppressing vascular endothelial growth factor receptor 2 signaling. Cancer Res. 2008;68(6):1843–1850. doi: 10.1158/0008-5472.CAN-07-5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yue Q, et al. Proteomic analysis revealed the important role of vimentin in human cervical carcinoma HeLa cells treated with gambogic acid. Mol Cell Proteomics. 2016;15(1):26–44. doi: 10.1074/mcp.M115.053272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guizzunti G, Batova A, Chantarasriwong O, Dakanali M, Theodorakis EA. Subcellular localization and activity of gambogic acid. ChemBioChem. 2012;13(8):1191–1198. doi: 10.1002/cbic.201200065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davenport J, et al. Gambogic acid, a natural product inhibitor of Hsp90. J Nat Prod. 2011;74(5):1085–1092. doi: 10.1021/np200029q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang L, et al. Gambogic acid inhibits Hsp90 and deregulates TNF-α/NF-κB in HeLa cells. Biochem Biophys Res Commun. 2010;403(3–4):282–287. doi: 10.1016/j.bbrc.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 46.Matthews SB, et al. Characterization of a novel novobiocin analogue as a putative C-terminal inhibitor of heat shock protein 90 in prostate cancer cells. Prostate. 2010;70(1):27–36. doi: 10.1002/pros.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sidera K, Patsavoudi E. HSP90 inhibitors: Current development and potential in cancer therapy. Recent Patents Anticancer Drug Discov. 2014;9(1):1–20. [PubMed] [Google Scholar]
- 48.Chen B, Piel WH, Gui L, Bruford E, Monteiro A. The HSP90 family of genes in the human genome: Insights into their divergence and evolution. Genomics. 2005;86(6):627–637. doi: 10.1016/j.ygeno.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 49.Song HY, Dunbar JD, Zhang YX, Guo D, Donner DB. Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. J Biol Chem. 1995;270(8):3574–3581. [PubMed] [Google Scholar]
- 50.Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J Biol Chem. 2003;278(48):48330–48338. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- 51.Matts RL, et al. A systematic protocol for the characterization of Hsp90 modulators. Bioorg Med Chem. 2011;19(1):684–692. doi: 10.1016/j.bmc.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elbel KM, et al. A-ring oxygenation modulates the chemistry and bioactivity of caged Garcinia xanthones. Org Biomol Chem. 2013;11(20):3341–3348. doi: 10.1039/c3ob40395e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bai F, et al. The Fe-S cluster-containing NEET proteins mitoNEET and NAF-1 as chemotherapeutic targets in breast cancer. Proc Natl Acad Sci USA. 2015;112(12):3698–3703. doi: 10.1073/pnas.1502960112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Theodoraki MA, et al. Spontaneously-forming spheroids as an in vitro cancer cell model for anticancer drug screening. Oncotarget. 2015;6(25):21255–21267. doi: 10.18632/oncotarget.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peterson LB, Eskew JD, Vielhauer GA, Blagg BSJ. The hERG channel is dependent upon the Hsp90α isoform for maturation and trafficking. Mol Pharm. 2012;9(6):1841–1846. doi: 10.1021/mp300138n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hadden MK, Lubbers DJ, Blagg BS. Geldanamycin, radicicol, and chimeric inhibitors of the Hsp90 N-terminal ATP binding site. Curr Top Med Chem. 2006;6(11):1173–1182. doi: 10.2174/156802606777812031. [DOI] [PubMed] [Google Scholar]
- 57.Matts RL, et al. Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem Biol. 2011;6(8):800–807. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chantarasriwong O, et al. Evaluation of the pharmacophoric motif of the caged Garcinia xanthones. Org Biomol Chem. 2009;7(23):4886–4894. doi: 10.1039/b913496d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tisdale EJ, Slobodov I, Theodorakis EA. Unified synthesis of caged Garcinia natural products based on a site-selective Claisen/Diels-Alder/Claisen rearrangement. Proc Natl Acad Sci USA. 2004;101(33):12030–12035. doi: 10.1073/pnas.0401932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bai F, et al. Free energy landscape for the binding process of Huperzine A to acetylcholinesterase. Proc Natl Acad Sci USA. 2013;110(11):4273–4278. doi: 10.1073/pnas.1301814110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 62.Lang PT, et al. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA. 2009;15(6):1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mills N. ChemDraw Ultra 10.0 CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. www.cambridgesoft.com. Commercial Price: $1910 for download, $2150 for CD-ROM; Academic Price: $710 for download, $800 for CD-ROM. J Am Chem Soc. 2006;128(41):13649–13650. [Google Scholar]
- 64.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron. 1980;36(22):3219–3228. [Google Scholar]
- 65.Hawkins GD, Cramer CJ, Truhlar DG. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J Phys Chem. 1996;100(51):19824–19839. [Google Scholar]
- 66.Zou X, Sun Y, Kuntz ID. Inclusion of solvation in ligand binding free energy calculations using the generalized-born model. J Am Chem Soc. 1999;121(35):8033–8043. [Google Scholar]