

Summary
Myocardial infarction results in compromised myocardial function with heart failure due to insufficient cardiomyocyte self-renewal1. Unlike lower vertebrates, mammalian hearts only have a transient neonatal renewal capacity2. Reactivating primitive reparative ability in the mature heart requires knowledge of the mechanisms promoting early heart repair. By testing an established Hippo-deficient heart regeneration model for renewal promoting factors, we found that Pitx2 expression was induced in injured, Hippo-deficient ventricles. Pitx2-deficient neonatal hearts failed to repair after apex resection while Pitx2-gain-of-function in adult cardiomyocytes conferred reparative ability after myocardial infarction. Genomic analyses indicated that Pitx2 activated genes encoding electron transport chain components and reactive oxygen species scavengers. A subset of Pitx2 target genes was cooperatively regulated with the Hippo effector, Yap. Furthermore, Nrf2, a regulator of antioxidant response3, directly regulated Pitx2 expression and subcellular localization. Pitx2 mutant myocardium had elevated reactive oxygen species levels while antioxidant supplementation suppressed the Pitx2-loss-of-function phenotype. These findings reveal a genetic pathway, activated by tissue damage that is essential for cardiac repair.
We used immunofluorescence staining to look for developmental transcription factors that were up-regulated in regenerating Hippo-deficient heart4. Paired-like homeodomain 2 (Pitx2) was enriched in border zone ventricular cardiomyocyte nuclei of adult Hippo-deficient hearts after myocardial infarction (MI) (Fig. 1a–c). Pitx2, encoding three isoforms (Pitx2a, Pitx2b, and Pitx2c), is mutated in Rieger Syndrome that is characterized by craniofacial, umbilical, and cardiac abnormalities5 and functions in left-right asymmetric organ development6. Notably, Pitx2 deficiency results in predisposition to atrial fibrillation (AF), a common human arrhythmia7,8. Pitx2c is the major isoform expressed in heart.
Figure 1.
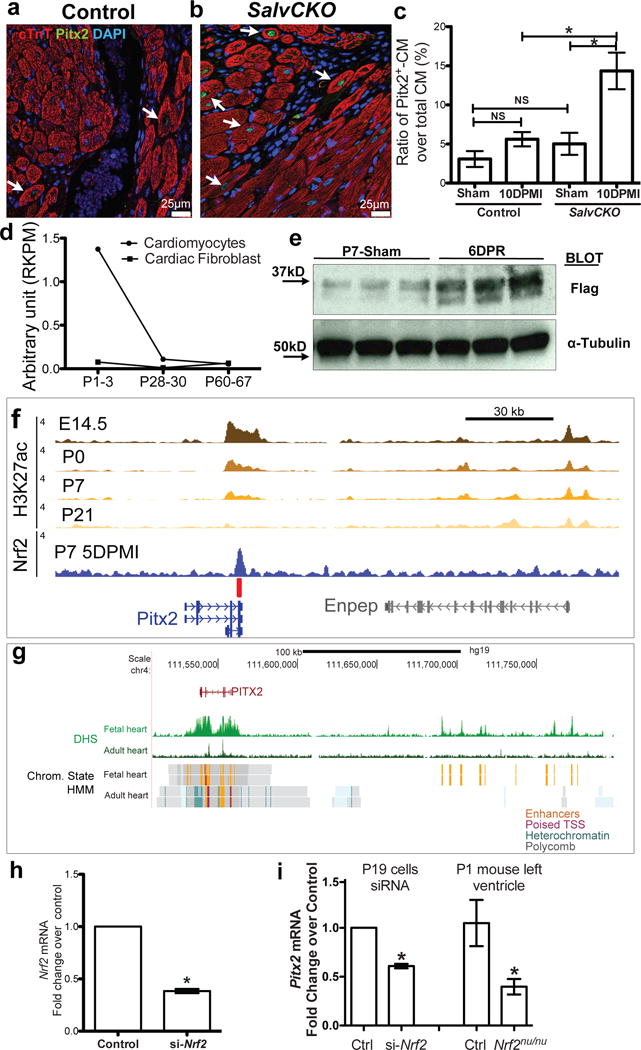
Pitx2 is induced in injured myocardial. (a–c) Border zone of SalvCKO (b) and control (a) hearts stained for Pitx2 (green), cTnT (red), and DAPI (blue) at 10 day-post-MI, with Pitx2+ cardiomyocyte ratio quantified in c, n=4. (d) Pitx2 expression showed by RNA-Seq, P, postnatal day. (e) Western blot of Flag and a-Tubulin in 5 DPR Pitx2flag ventricles, resected at P1. (f) Nrf2 directly binds to Pitx2 enhancer after LAD-O. The heart specific enhancers are marked by H3K27ac ChIP-Seq. red bar, Nrf2 binding element. (g) DHS-Seq and chromatin state tracks of fetal and adult human heart tissue. Orange color indicates active enhancer regions. (h) qPCR showed knocking-down of Nrf2 by siRNA in P19 cells, n=4. (i) qPCR of Pitx2 in P19 cells with siRNA targeting Nrf2, and Nrf2nu/nu heart, compared to controls, n=4. Mean ± S.E.M.; Statistical test, (c) one-way ANOVA plus Bonferroni post-test; (i, right part) Mann-Whitney; (h, i left part) see Methods; *, p<0.05; NS, not significant.
Available RNA-sequencing (RNA-Seq) data indicated that Pitx2 transcripts in cardiomyocytes dropped postnatally9 (Fig. 1d) while Western blot revealed Pitx2 protein induction after injury during regenerative stages (Fig. 1e). Consistent with reduced Pitx2 expression in adult hearts, active histone marks at the Pitx2 locus were reduced in adult hearts (Fig. 1f, g)10. Available Dnase I Hypersensitive sequencing (DHS) data revealed that Nrf2 binding-elements were enriched in the Pitx2 locus (data not shown). To evaluate whether Nrf2 activated Pitx2 after injury, we performed an Nrf2 Chromatin Immunoprecipitation Sequenceing (ChIP-Seq) experiment on hearts 4 days after postnatal day (P) 2 left anterior descending artery occlusion (LAD-O) and discovered Nrf2 binding at the Pitx2 locus (Fig. 1f). Nrf2 knockdown in P19 cells and Nrf2 loss-of-function in mice resulted in decreased Pitx2 mRNA expression supporting the conclusion that Nrf2 directly regulates Pitx2 after tissue injury (Fig. 1h, i).
We determined whether Pitx2, similarly to Yap, was required for neonatal heart regeneration2. Using muscle creatine kinase cre (MCKcre)11, we inactivated Pitx2 in cardiomyocytes and performed P1 apex resection. While control hearts regenerated as expected, MCKcre; Pitx2f/f (Pitx2CKO) hearts had increased scarring and reduced function (Fig. 2a–e). We injured Pitx2 mutant hearts by LAD-O at P1 and used both MCKcre and Myh6creERT to inactivate Pitx2 in myocardium. Pitx2 mutants failed to repair after LAD-O (Extended Data Fig. 1).
Figure 2.
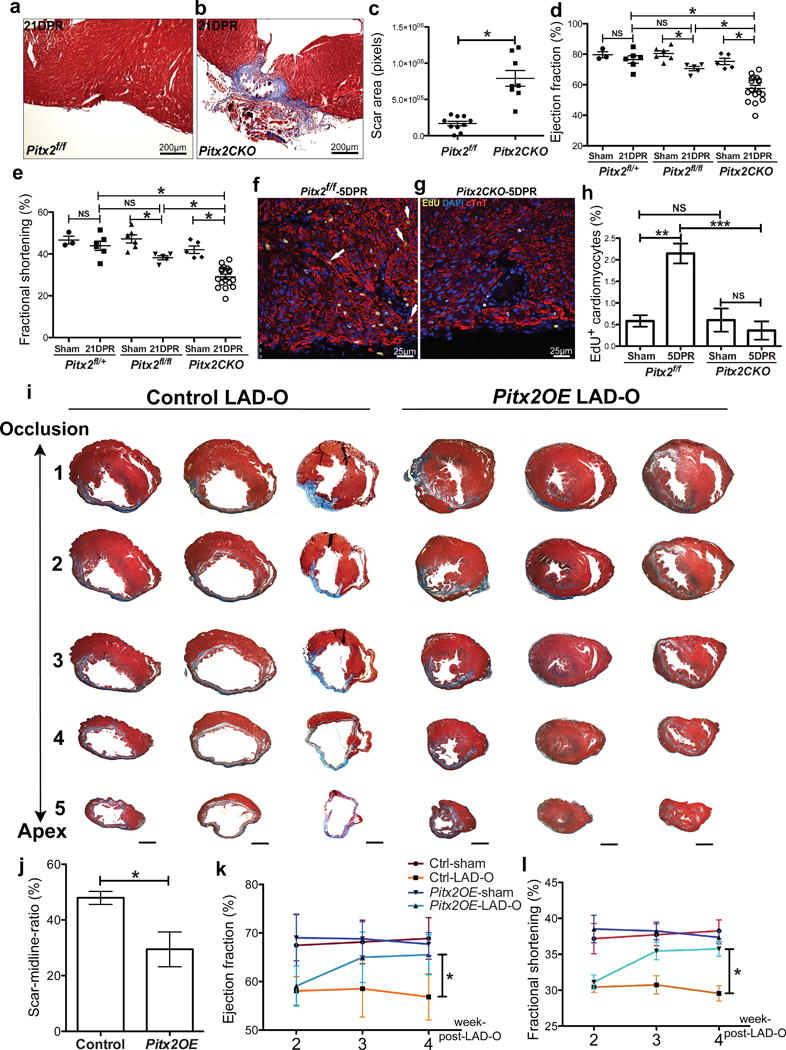
Pitx2 is required and sufficient to promote myocardial regeneration. (a–c) Trichrome-stained Pitx2f/f (a) and Pitx2CKO (b) apex at 21 DPR, with scar size quantified in c. (d, e) Echocardiography showed the ejection fraction (d) and fractional shorting (e) at 21 DPR. (f–h) 5 DPR Pitx2f/f (f) and Pitx2CKO (g) apical sections stained for EdU (yellow), cTnT (red), and DAPI (blue). Arrow, EdU+ cardiomyocyte. Cardiomyocyte proliferative ratio was quantified in h, n=4. (i) Serial transverse heart sections at 5 weeks post-LAD-O, performed at 8weeks. (j) Percentage of fibrotic left ventricular myocardium quantified at 5 weeks post-LAD-O, n=5. Scale bar, 1mm. (k, l) Ejection fraction (k) and fractional shortening (l) of LAD-O and sham hearts. Mean ± S.E.M.; Statistical test, (d, e) one-way ANOVA plus Bonferroni post-test; (c, h, j–l) Mann-Whitney; *, p<0.05; NS, not significant.
We examined cardiomyocyte proliferation in P1 apex resection model at 5 day-post-resection (DPR) by pulse-labeling and immunofluorescence of 5-ethynyl-2′-deoxyuridine (EdU). In Pitx2f/f controls, injury induced a threefold increase of EdU positive cardiomyocytes compared to sham that was absent in Pitx2CKO after injury, supporting the hypothesis that Pitx2, like Yap, is essential for neonatal heart regeneration by promoting proliferation and injury resistance (Fig. 2f–h).
To investigate whether Pitx2 is sufficient for adult cardiomyocyte repair, we generated Pitx2Gof, a cre-activated Pitx2c gain-of-function transgenic line (Extended Data Fig. 2a). Immunoblotting and qPCR showed elevated Pitx2c levels in Myh6cre-Ert;Pitx2Gof (Pitx2OE) ventricles (Extended Data Fig. 2b–d). LAD-O performed in 8-week-old mice after tamoxifen administration revealed that Pitx2OE hearts had reduced scar size (Fig. 2i, j)4. Heart morphology was comparable between controls (Myh6cre-Ert/+) and Pitx2OE after sham surgery (Extended Data Fig. 2e–g). Two weeks after LAD-O both Pitx2OE and controls showed decreased ejection fraction (EF) and fractional shortening (FS), however, Pitx2OE mice had functional recovery at 3 and 4 weeks-post-LAD-O (Fig. 2k, l). Non-regenerative stage P8 apex resections in control and Pitx2OE hearts revealed that Pitx2OE hearts had reduced scarring (Extended Data Fig. 2h–j) and improved function at 28 DPR (Extended Data Fig. 2k, l). EdU incorporation at 8 DPR showed increased cardiomyocyte S-phase entry in Pitx2OE hearts (Extended Data Fig. 2m–o).
Since Pitx2 was up-regulated in Hippo-deficient hearts, we tested whether Pitx2 was required for Hippo-deficient cardiomyocyte renewal. SalvCKO hearts regenerate efficiently after MI4. However, SalvCKO hearts that were also Pitx2 mutant, called double knock out (DKO), failed to regenerate (Fig. 3a–c). Twenty-eight days after P8 LAD-O, DKO hearts had a larger scar and compromised EF (Fig. 3d, e)4. Apex resection in non-regenerative P8 hearts also revealed the requirement for Pitx2 function in SalvCKO cardiomyocyte renewal (Extended Data Fig. 3).
Figure 3.
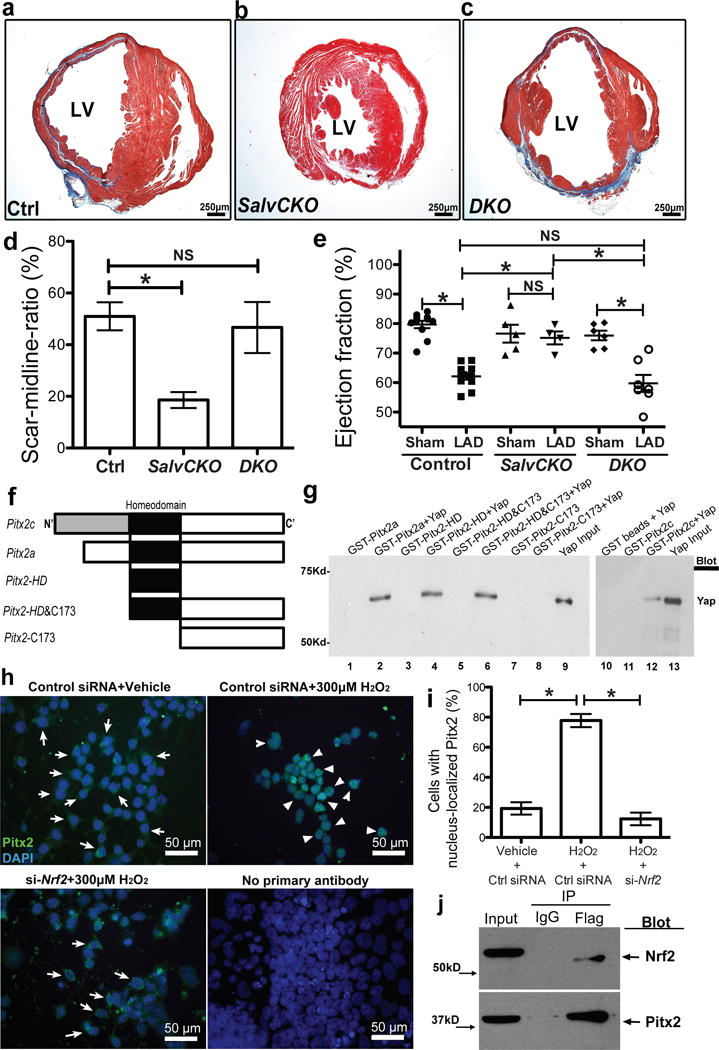
Pitx2 interacts with Yap in regenerating hearts, and its nuclear shuttling requires Nrf2. (a–d) Trichrome-stained control (Salvf/f;Pitx2f/f) (a), SalvCKO (b) and DKO (c) sections at 28 days after P8 LAD-O with scar size quantification (d), n=4. (e) Echocardiography showed ejection fraction. (f) Diagram of GST-Pitx2 constructs. (g) GST-Pitx2 pull-down assay. Yap was detected by Western blotting. (h–i) Immunofluorescent staining of Pitx2 (green) and DAPI (blue) in P19 cells after vehicle or H2O2 treatment, with control siRNA or siRNA targeting Nrf2. Arrow, cytoplasmic staining; arrowhead, nuclear staining. The ratio of cells with nucleus-localized Pitx2 over total cell number is quantified in i. (j) Co-IP of Flag in 5 DPR Pitx2flag ventricles, resected at P1, blotting of Nrf2 and Pitx2. Mean ± S.E.M.; Statistical test, (e) one-way ANOVA plus Bonferroni post-test; (d, i) Mann-Whitney; *, p<0.05; NS, not significant.
Available genomic footprinting data from cardiac DHS datasets can uncover sequence-specific transcription factor (TF)-DNA interactions in an unbiased fashion. Motifs for Pitx2 and Tead, the Yap DNA binding partner, were highly enriched in fetal heart footprints and often found in close proximity (Extended Data Fig. 4a, b). Genomic regions containing Pitx2 or Tead motifs were enriched for H3K4me1 chromatin marks indicating that Pitx2 or Tead binding regions were transcriptionally active. Regions containing both Pitx2 and Tead motifs showed globally increased transcriptional activity compared to Pitx2 motif only regions (Extended Data Fig. 4c, d).
The co-occurrence of TF binding motifs often indicates TF interactions. We tested whether Pitx2 was a Yap binding partner using purified Glutathione S-Transferase (GST) fusion proteins. In vitro binding assays with Pitx2 fusion peptides and full length Yap revealed that Yap bound the Pitx2 homeodomain (Fig. 3f, g; Extended Data Fig. 5a, b). We uncovered an in vivo interaction between endogenous Pitx2 and Yap using co-immunoprecipitation (co-IP) of endogenous cardiac proteins (Extended Data Fig. 5c). The Pitx2flag allele, previously generated by gene targeting in embryonic stem cells (ESC), expresses endogenous levels of FLAG-epitope-tagged Pitx2 from the Pitx2 locus7.
Immunofluorescence showed widespread distribution of Pitx2 in P19 cells and a cytoplasm-to-nucleus translocation upon H2O2 treatment (Fig. 3h, Extended Data Fig. 6a), similar to the Nrf2 response to oxidative stress (Extended Data Fig. 6b)3. Pitx2 nuclear translocation after H2O2 treatment depended on Nrf2 activity (Fig. 3h, i). In contrast, Nrf2 nuclear translocation after H2O2 treatment was intact in Pitx2null P19 cells indicating that Pitx2 was dispensable for Nrf2 response to reactive oxygen species (ROS) (Extended Data Fig. 6b, c). We found that Pitx2 interacts with Nrf2 in heart extracts, expressing endogenous protein levels (Fig. 3j). Co-IP using Nuclear-cytoplasmic fractionation of P19 cells, looking at endogenous proteins, indicated that Pitx2 binding to Nrf2 in the nucleus was increased after H2O2 treatment (Extended Data Fig. 6d, e). We also found less nuclear Pitx2 in Nrf2 mutant hearts after P1 apex resection (Extended Data Fig. 6f–h).
To solidify the connection between Nrf2, Pitx2, and Yap, we tested if Nrf2 was required for neonatal regeneration, as is the case for Pitx2 and Yap. MI at P2 revealed that Nrf2 null hearts were unable to regenerate indicating that induction of the antioxidant response is required for regeneration (Extended Data Fig. 7)12. Notably, Pitx2OE mice that were heterozygous for Nrf2nu allele failed to regenerate, suggesting that Pitx2 promotes the antioxidant response. It is also possible that Nrf2 is downstream of Pitx2 in certain contexts. We also made Pitx2OE mice that were heterozygous for a Yapfl allele13. Reducing Yap dosage compromised Pitx2OE heart regeneration in P8 resection model (Extended Data Fig. 8a–e).
To investigate Pitx2 target genes induced by injury, we harvested P1 resected ventricles from Pitx2f/f and Pitx2CKO hearts at 5 DPR and performed RNA-Seq (Extended Data Fig. 9a–d). We identified 1002 down-regulated genes in Pitx2 mutants (fdr ≤ 0.1, fold change ≥ 1.5). There was extensive overlap between up-regulated genes following apex resection in controls and down-regulated genes in 5DPR Pitx2CKO hearts, indicating that in the absence of Pitx2, a set of stress response genes, including oxidative stress response genes, fail to be activated (Extended Data Fig. 9a–d).
We examined the response of Pitx2 and antioxidant scavenger genes to H2O2 in Pitx2-null (Pitx2nu) ESCs since Pitx2 has been implicated in ROS response in skeletal muscle6,14. After H2O2 treatment, qPCR showed increased Pitx2, Gpx1, Mt1 and Mt2 expression in wild type ESCs, but not Pitx2nu ESCs (Extended Data Fig. 9f) supporting a critical role for Pitx2 in response to ROS. While ESCs had low endogenous Pitx2 levels, the mouse P19 embryonic carcinoma cell line expressed readily detectable Pitx2, primarily the Pitx2c isoform. H2O2 treated P19 cells increased Pitx2 and its target gene expression levels in a dose-dependent manner (Extended Data Fig. 9g, h).
To identify Pitx2 target genes, we performed P1 apex resection and harvested 5 DPR Pitx2flag ventricles and performed ChIP-Seq (Extended Data Fig. 9e). Overlay of down-regulated genes from Pitx2CKO RNA-Seq with Pitx2-binding peaks from ChIP-Seq revealed 505 direct Pitx2 targets. Gene Ontology (GO) analysis revealed enrichment in mitochondria, oxidation-reduction, ribosome, and respiratory chain (Fig. 4a, b).
Figure 4.
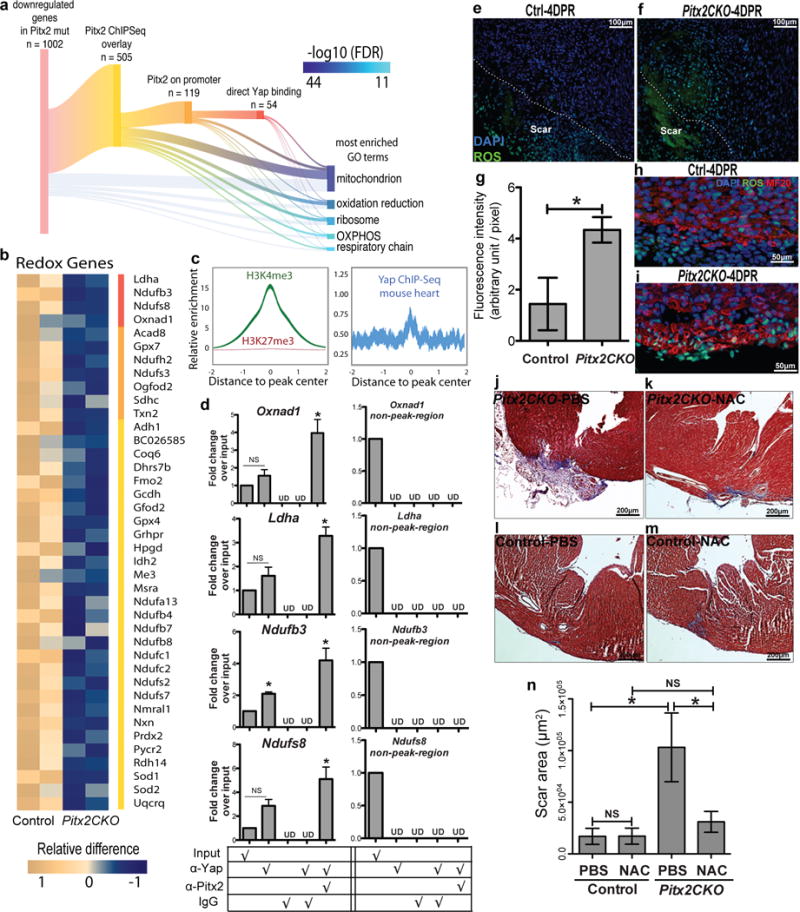
Pitx2 regulates redox balance in neonatal myocardium. (a) Sankey diagram shows direct target genes of Pitx2 from overlaying ChIP-Seq and RNA-Seq profiles. A subset of genes is further branched according to promoter binding activity of Pitx2 and Yap. Gene ontology analysis is performed on 1002 down-regulated genes in Pitx2CKO ventricles at 5 days after P1 resection. The GO terms are listed by significance in descending order. The branches indicate the source of genes for each term. OXPHOS, oxidative phosphorylation. (b) Heat map highlights mitochondrial genes directly targeted by Pitx2. Red bar, genes co-regulated by Pitx2 and Yap; orange, direct binding of Pitx2 on promoters. (c) Heart specific H3K4me3 ChIP-Seq and Yap ChIP-Seq read distribution with in 2 kb interval of promoter region of Pitx2 direct target genes. The width of the curve indicate 95% confidence interval. (d) ChIP-re-ChIP showing co-occupancy of Pitx2 and Yap at the regulatory regions of common target genes (in b, red bar), n=3. (e–i) ROS staining (green) of apical border zone in Pitx2CKO (f, i) and control (e, h), with fluorescent intensity quantified in g. MF20, red; DAPI, blue, n=4. (j–n) Trichrome at 21DPR showed apical scarring of Pitx2CKO (j, k) and control (l, m) hearts treated with PBS (j, l) or NAC (k, m), with scar area quantified in n, n=5. Mean ± S.E.M.; Statistical test, (n) one-way ANOVA plus Bonferroni post-test; (g) Mann-Whitney; (d) Mann-Whitney and Wilcoxon signed-rank test; *, p<0.05; NS, not significant.
Among Pitx2 targets were genes that protect the cell from elevated ROS, such as superoxide dismutase genes, Sod1 and Sod2, which reduce superoxide to H2O2 and the glutathione peroxidase, Gpx4, that removes H2O2, and peroxiredoxin-2 (Fig. 4b)15. Pitx2 regulates electron transport chain (ETC) complex-I components including Ndufb3, Ndufb4, and Ndufb7 and ETC complex-IV component, Cox7c (Fig.4b). Defective complex-I in human patients increases ROS sensitivity16. Pitx2 regulated 21.5% of its direct target genes through promoter binding revealed by enrichment of H3K4me3 chromatin marks for active promoters in Pitx2 peaks (Fig. 4c; 119/505 direct targets). 8-week-old heart H3K4me3 chromatin marks are enriched in the center of Pitx2 binding sites, supporting the hypothesis that Pitx2 promotes transcriptional activation17.
To determine whether Pitx2 and Yap regulate common target genes, we performed Yap ChIP-Seq on ventricles 5 days after P2 LAD-O and found Yap binding sites enriched in nearly half of Pitx2-targeted gene promoters (54/119; Fig. 4a, c). Comparison of Pitx2 ChIP-Seq, our Yap ChIP-Seq, and available Yap ChIP-Seq18–21, revealed 4 redox genes bound by both Pitx2 and Yap. ChIP-re-ChIP assay from heart extracts revealed Pitx2 and Yap were concurrently resident on these genes, indicating Yap and Pitx2 cooperatively activate the transcriptional response to oxidative stress. (Fig. 4d).
To investigate ROS activity in Pitx2f/f and Pitx2CKO apical border zones at 4 DPR, tissue sections were incubated with ROS-detecting reagent. Pitx2CKO hearts had elevated ROS in both cardiomyocytes and non-myocytes (Fig. 4e–i). To determine if elevated ROS contributed to scarring in 21 DPR Pitx2CKO hearts, we administrated N-Acetyl-L-cysteine (NAC) in Pitx2CKO neonates after apex resection. Daily NAC injections until 10 DPR decreased scar size in Pitx2CKO hearts (Fig. 4j–n).
Elevated ROS is a natural response to cardiac injury including ischemic damage (Extended Data Fig. 10)22,23. During the postnatal transition from glycolytic to oxidative metabolism, ROS is elevated in the heart and inhibits cardiomyocyte regeneration22. In regenerative-stage hearts, Pitx2 promotes regeneration by inhibiting ROS. Injury induces Pitx2 expression and activity through Nrf2-activated Pitx2 transcription and nuclear shuttling. In turn, Pitx2 activates ROS scavengers, protecting cells from oxidative damage, and ETC components. It is also possible that Nrf2 also acts downstream of Pitx2 in some contexts. Thus, Pitx2 is essential for cardiomyocyte response to injury and may prevent cell death.
We uncovered a Pitx2-Yap interaction important for Hippo-deficient cardiac regeneration. Pitx2 binds Yap and cooperatively activates genes maintaining redox balance. Pitx2 gain-of-function, sufficient to confer reparative capacity in adult cardiomyocytes, is repressed by Yap heterozygosity. This suggests that Pitx2 recruits Yap to target genes even in contexts when Hippo is active and the pool of nuclear Yap is relatively low. This mechanism may work in parallel to other mechanisms by which Yap protects the cell from ROS24.
Extended Data
Extended Data Figure 1.
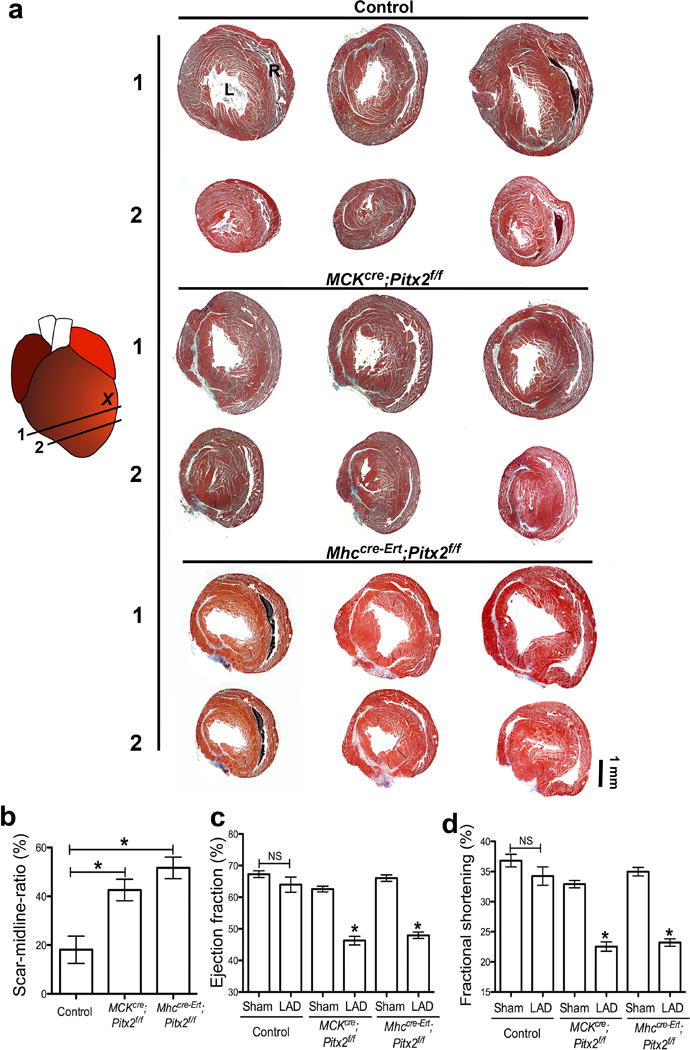
Pitx2 is required in neonatal myocardial regeneration after LAD-O. (a) Serial trichrome images of control (Pitx2f/f), MCKcre;Pitx2f/f, and Mhccre-Ert;Pitx2f/f 21 days after LAD-O at P2. (b) Percentage of fibrotic left ventricular myocardium quantified at 3 weeks post-LAD-O, n=4. (c, d) Ejection fraction (c) and fractional shortening (d) of LAD-O and sham hearts. L, left ventricle; R, right ventricle. Mean ± S.E.M.; Statistical test, (c–d) one-way ANOVA plus Bonferroni post-test; (b) Mann-Whitney. *, p<0.05; NS, not significant.
Extended Data Figure 2.
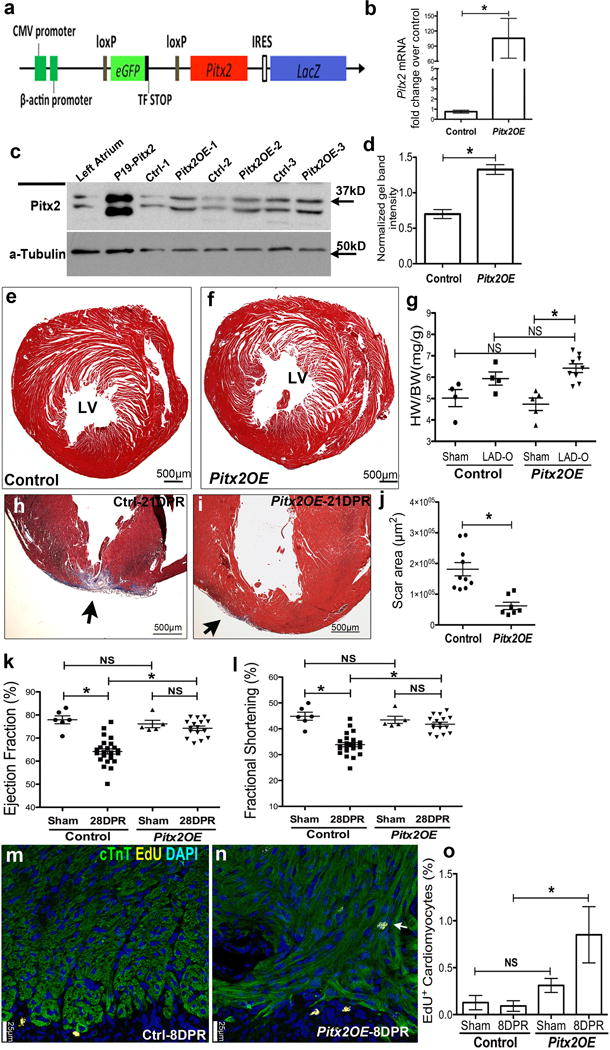
Pitx2 promotes myocardial regeneration after apex resection at P8. (a) Schematic of Pitx2-expressing construct (Pitx2Gof). (b–d) Pitx2Gof was crossed with Mhccre-Ert strain to generate Mhccre-Ert/+;pitx2Gof (Pitx2OE), after Tamoxifen treatment, qPCR (b, n=4), western (c, d, n=3) showed the over-expression of Pitx2 in Pitx2OE ventricles. (e–f) Trichrome-stained cross sections from 13 weeks old sham hearts of control (e) and Pitx2OE (f), with tamoxifen administrated at 7–8 weeks old. (g) Heart weight over body weight ratio of adult sham and LAD-O hearts. (h–j) Apex resection of Pitx2OE (i) and control (Mhccre-Ert/+) (h) hearts at P8 followed by trichrome staining at 28 DPR, scar area was quantified in j. (k, l) Echocardiography showed ejection fraction (k) and fractional shortening (l) at 28 DPR. (m–o) EdU labeling of Pitx2OE (n) and control (m) apical area, 8 days after P8 resection, sections were stained for cTnT (green), EdU (yellow), and DAPI (blue). Arrow indicates EdU-labeled cardiomyocytes, with quantification in o, n=4. Mean ± S.E.M.; Statistical test, (g, k, l) one-way ANOVA plus Bonferroni post-test; (b, d, j, o) Mann-Whitney. *, p<0.05; NS, not significant.
Extended Data Figure 3.
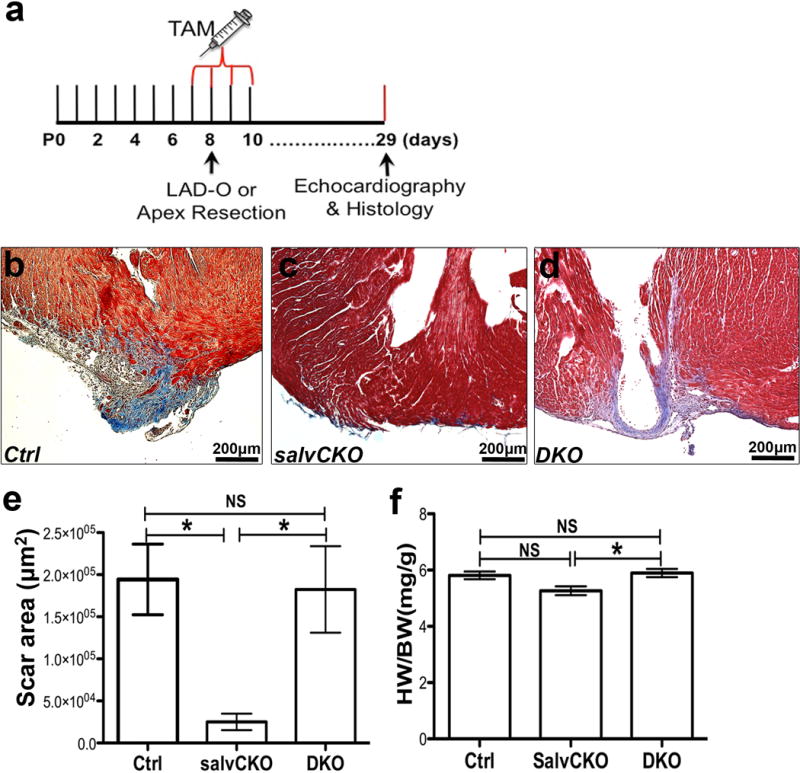
Pitx2 is required for Hippo deficient heart regeneration. (a) Schematic study plan for figure 3a–e. (b–e) Trichrome-stained apical areas of control (b), SalvCKO (c) and DKO (d) hearts 21 days after P8 apex resection. Scar area was quantified in e. (f) Heart weight to body weight ratio of sham hearts at 28 days after tamoxifen administration. N number, see Methods. Mean ± S.E.M.; Statistical test, (e, f) Mann-Whitney. *, p<0.05; NS, not significant.
Extended Data Figure 4.
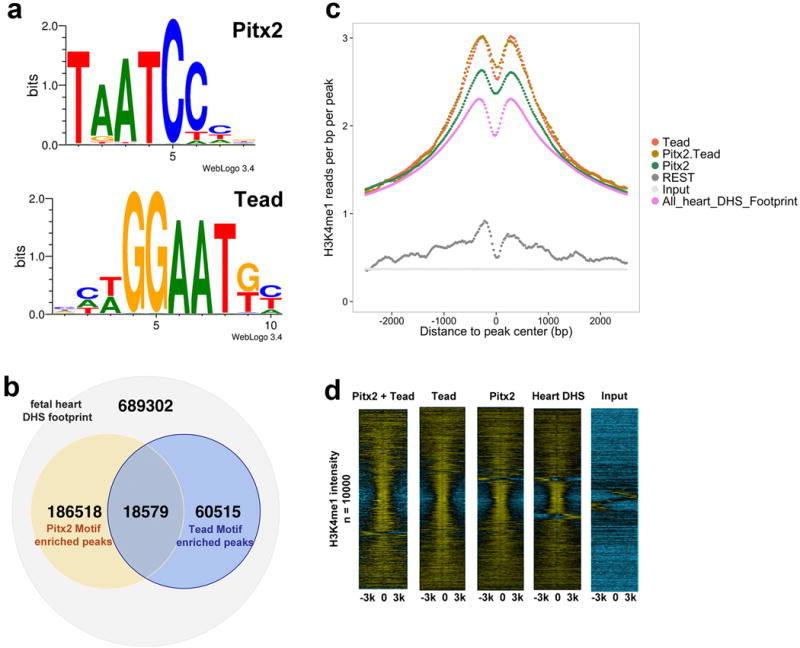
Co-occurrence of Pitx2 and Tead DNA-binding motifs in fetal heart enhancers. (a) Consensus Pitx2 and Tead motifs. (b) Pitx2 and Tead motif co-occurrence in fetal heart Dnase I Hypersensitive (DHS) peaks. (c) Aggregate plot of H3K4me1 in fetal heart ChIP-Seq reads within 6 kb range of DHS peaks. (d) Heat-map of fetal heart H3K4me1 ChIP-Seq or input read density in 6 kb regions of DHS peaks. DHS peaks were centered on Pitx2 motif, Tead motif, Pitx2-Tead motifs, or randomly selected. The read density was in log2 scale; negative values, blue color; positive values, yellow color.
Extended Data Figure 5.
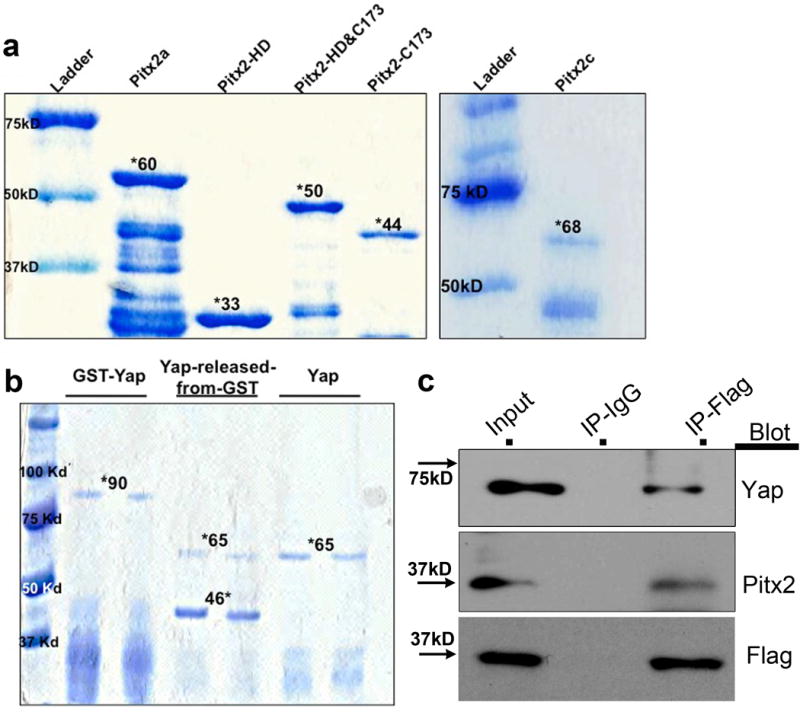
Generation of GST-tagged proteins and interaction between Pitx2 and Yap in vivo. (a) The mouse Pitx2a, Pitx2c, and truncated proteins were purified and run on 10% SDS-gel, coomassie blue staining showed the GST fusion protein band with correct size (marked by *). (b) Coomassie blue staining of the purified GST-Yap, Yap cut by prescission protease and pure Yap protein. (c) Co-IP of Flag in Pitx2flag ventricles at 5 DPR, and blotting of Yap, Pitx2, and Flag.
Extended Data Figure 6.
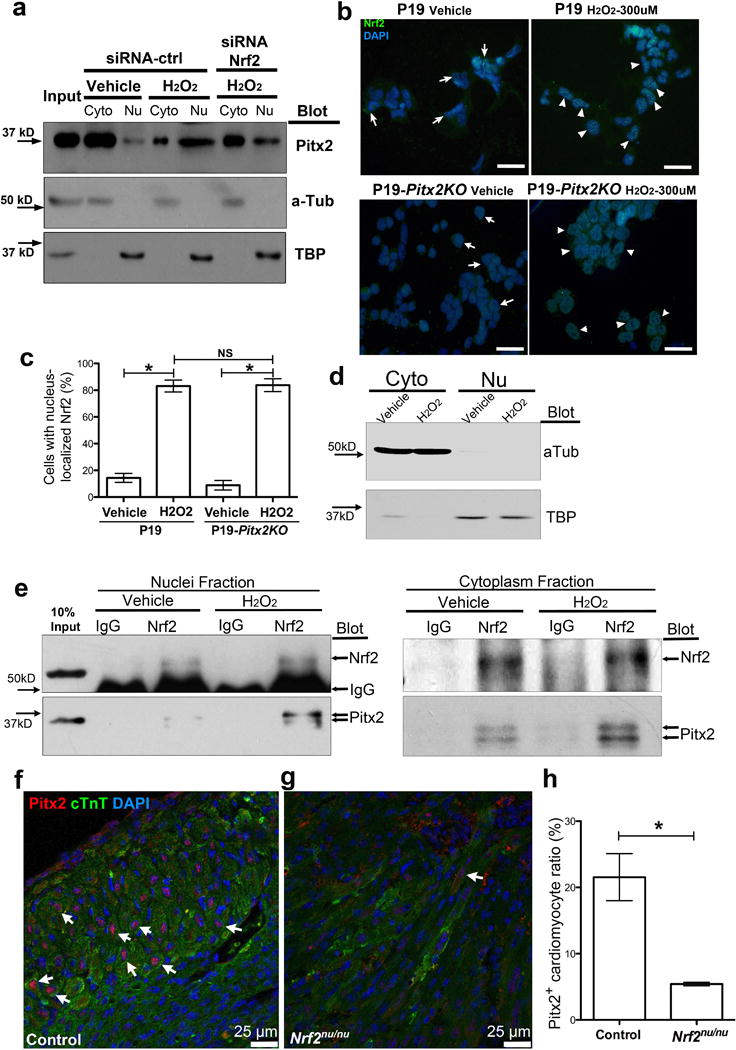
Nuclei-shuttling of Nrf2 is independent of Pitx2. (a) Blotting of Pitx2, α-Tubulin, and TATA-binding protein (TBP) of P19 cell fraction after H2O2, with or without Nrf2 siRNA treatment. (b) Immunofluorescent staining of Nrf2 (green) in P19 and P19-Pitx2KO cells after vehicle or H2O2 treatment. DAPI, blue. Scale bar, 50 μm. (c) The ratio of cells with nuclear Nrf2 over total cell number, n=6. (d) Blotting of α-Tubulin and TBP to show cell fraction of P19 cells used in (e). (e) Co-IP Nrf2 from nuclear and cytoplasmic fraction of P19 cells after vehicle or H2O2 treatment, blotting shows Nrf2 and Pitx2. (f–h) 4 DPMI control (C57BL6) (f) and Nrf2nu/nu (g) cross-sections stained for Pitx2 (red), cTnT (green), and DAPI (blue), with the ratio of cardiomyocytes with nuclei-localized Pitx2 quantified in h, n=4. Arrows, Pitx2+ cardiomyocyte. Mean ± S.E.M.; Statistical test, (c) one-way ANOVA plus Bonferroni post-test; (h) Mann-Whitney. *, p<0.05; NS, not significant.
Extended Data Figure 7.
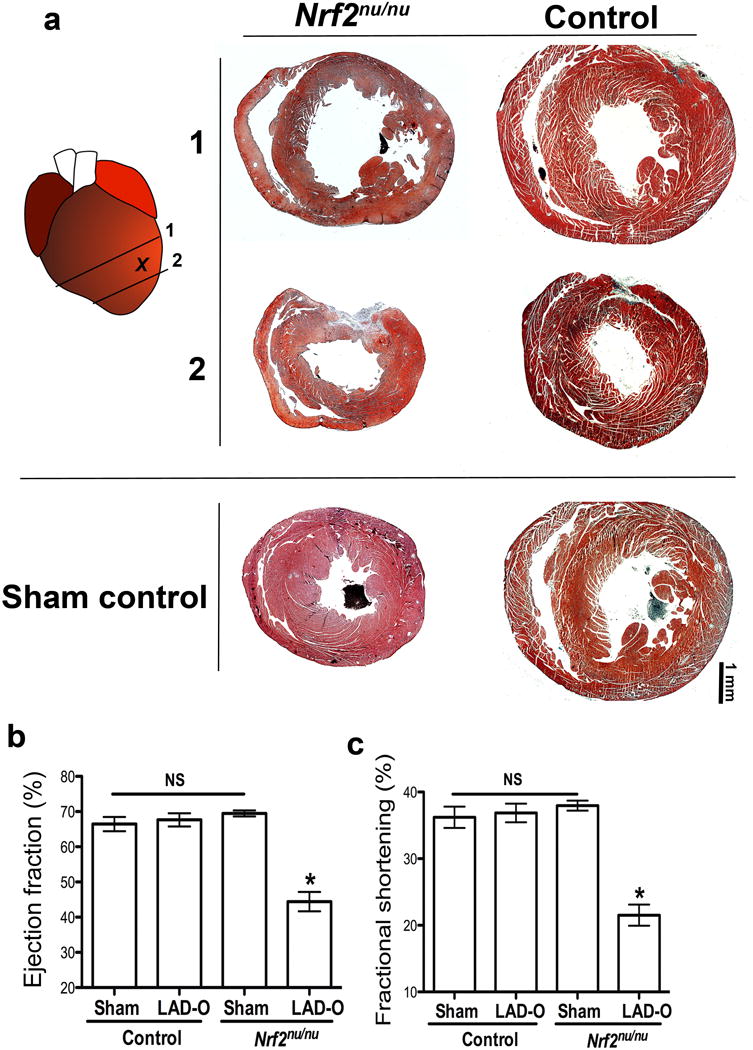
Nrf2 is required for neonatal myocardial regeneration. (a) Trichrome images of Nrf2nu/nu and control heart (C57BL6) at 21 days after P2 LAD-O, along with sham controls. (b, c) Ejection fraction (b) and fractional shortening (c) of LAD-O and sham hearts. Mean ± S.E.M.; Statistical test, (b, c) Mann-Whitney. *, p<0.05; NS, not significant.
Extended Data Figure 8.
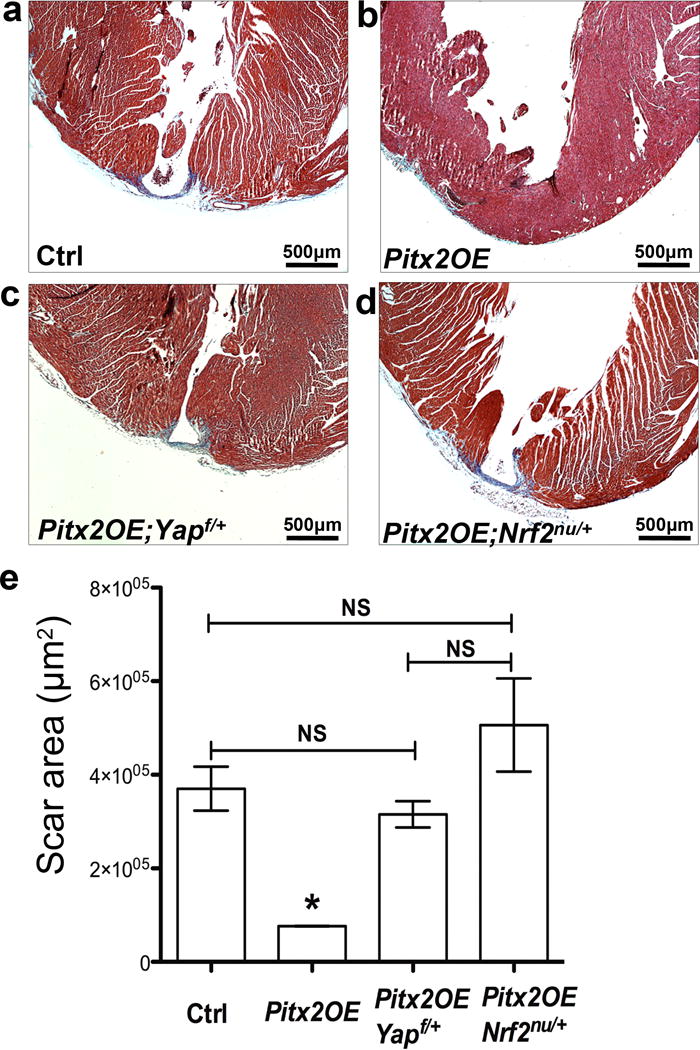
Yap and Nrf2 are essential for Pitx2-induced myocardial regeneration. (a–d) Trichrome staining showing apical scarring of different groups at 28 DPR, apex resection was performed at P8. (e) Quantification of scar area, n=4. Mean ± S.E.M.; Statistical test, (e) Mann-Whitney. *, p<0.05 compared to other three groups; NS, not significant.
Extended Data Figure 9.
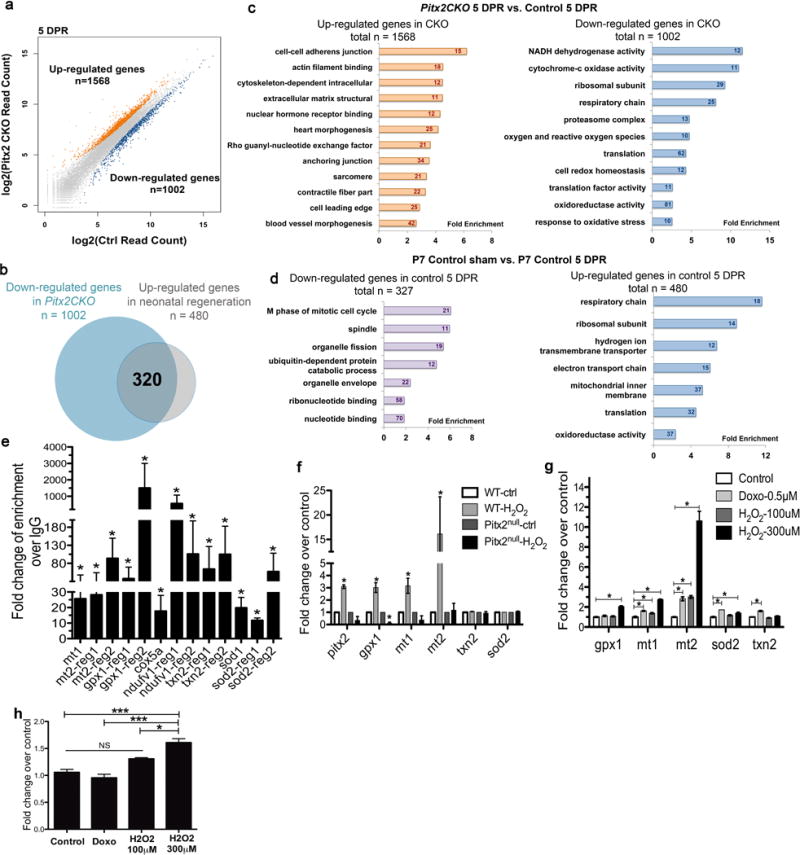
Pitx2 regulates antioxidant scavenger genes. (a) Overall change of genes in Pitx2CKO compared to control. (b) Up-regulated genes in 5 DPR control over wild type sham heart (n=480) overlaid with down-regulated genes in 5 DPR Pitx2CKO over 5 DPR control heart (n=1002). (c) GO-analysis of genes up-regulated (left) and down-regulated (right) in Pitx2CKO ventricles over controls at 5 DPR. (d) GO-analysis of genes up-regulated (right) and down-regulated (left) in 5 DPR control ventricles over age matching sham hearts. (e) ChIP-qPCR confirming the binding of Pitx2 to the regulatory regions of target genes, n=4. (f) qPCR detecting Pitx2 and antioxidant genes in wild type and pitx2nu/nu embryonic stem cells after vehicle or H2O2 treatment, n=4. (g) qPCR of antioxidant genes in P19 cells after doxorubicin or H2O2 treatment, n=5. (h) qPCR of Pitx2 in P19 cells after doxorubicin or H2O2 treatment, n=5. Mean ± S.E.M.; Statistical test, (e–h) Mann-Whitney. *, p<0.05; ***, p<0.001; NS, not significant.
Extended Data Figure 10.
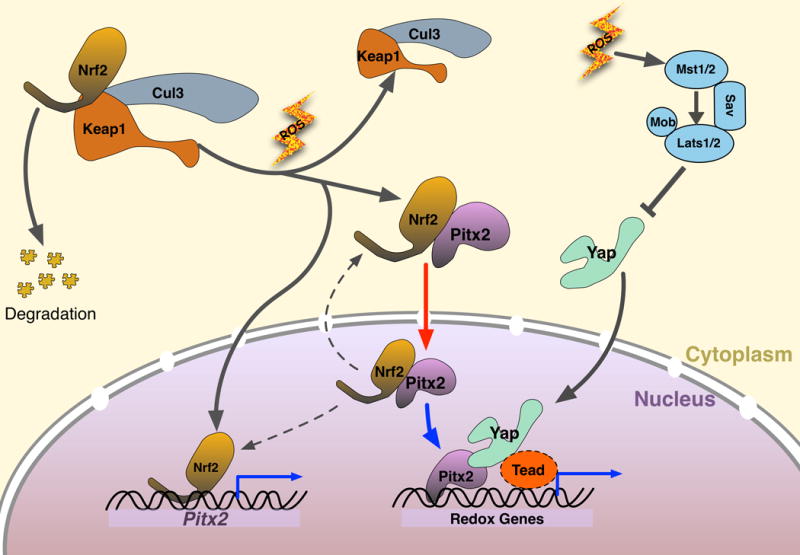
Mechanism model of Pitx2, Nrf2, and Yap responding to oxidative stress. When oxidative stress is low, Nrf2 is sequestered in cytoplasm by its degradation complex (Cul3, Keap1), and Pitx2 stays either in cytoplasm or at low expression level. When redox balance is disturbed by ROS, Nrf2 breaks away from degradation complex, and enter nuclei to up-regulate Pitx2, Nrf2 also binds cytoplasmic Pitx2 and shuttle it to nuclei, where Pitx2 and Yap co-regulate their common targets including critical antioxidant genes. In wild type adult mouse heart, active Yap is maintained at a low level, even after ischemic injury, thus not able to repair myocardium efficiently. When Pitx2 is over-expressed in cardiomyocytes, sufficient amount of Pitx2 will cooperate with low level of resident active Yap to induce the expression of beneficial antioxidant scavengers in a synergetic pattern, rendering protection to injured myocardium. Red arrow, supported by in vitro evidence; Blue arrows, supported by in vivo evidence.
Supplementary Material
Acknowledgments
The project was supported in part by IDDRC grant number 1U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. This project was supported by the Mouse Phenotyping Core at Baylor College of Medicine with funding from the NIH (U54 HG006348). The project was also supported by grants from the National Institutes of Health (NIH)[DE 023177 and HL 118761 to J.F.M., DE 13941 to B.A.A], the Vivian L. Smith Foundation [J.F.M.]. J.F.M. was supported by Transatlantic Network of Excellence Award LeDucq Foundation Transatlantic Networks of Excellence in Cardiovascular Research 14CVD01: “Defining the genomic topology of atrial fibrillation”. G.T. was supported by AHA 13POST17040027 [G.T.]. P.C.K was supported by German Research Foundation (DFG) (KA4018/1-1).
Footnotes
Author contributions
J.F.M. and G.T. conceived the project and designed the experiments. G.T., P.C.K., Y.M., M.R, T.R.H., L.L. performed experiments and analyzed data. Z.S. and B.A.A. provided reagents and performed experiments. E.N.O. provided transgenic animal model. M.Z. and G.T. performed bioinformatics and statistical analyses. J.F.M supervised the project and analyzed data. G.T. and J.F.M. wrote the manuscript.
Sequencing dataset generated in this manuscript can be assessed at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wfolwmwevdkltcd&acc=GSE70413 Series# GSE70413
References
- 1.Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol. 2013;14:529–541. doi: 10.1038/nrm3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Porrello ER, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. doi:331/6020/1078 [pii] 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itoh K, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heallen T, et al. Hippo signaling impedes adult heart regeneration. Development. 2013;140:4683–4690. doi: 10.1242/dev.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semina EV, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 6.Lu MF, Pressman C, Dyer R, Johnson RL, Martin JF. Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature. 1999;401:276–278. doi: 10.1038/45797. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, et al. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc Natl Acad Sci U S A. 2010;107:9753–9758. doi: 10.1073/pnas.0912585107. doi:0912585107 [pii] 10.1073/pnas.0912585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirchhof P, et al. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circulation Cardiovascular genetics. 2011;4:123–133. doi: 10.1161/CIRCGENETICS.110.958058. [DOI] [PubMed] [Google Scholar]
- 9.Giudice J, et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nature communications. 2014;5:3603. doi: 10.1038/ncomms4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nord AS, et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell. 2013;155:1521–1531. doi: 10.1016/j.cell.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruning JC, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2:559–569. doi: 10.1016/s1097-2765(00)80155-0. doi:S1097-2765(00)80155-0 [pii] [DOI] [PubMed] [Google Scholar]
- 12.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xin M, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. 2013;110:13839–13844. doi: 10.1073/pnas.1313192110. doi:1313192110 [pii] 10.1073/pnas.1313192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.L’Honore A, et al. Redox regulation by Pitx2 and Pitx3 is critical for fetal myogenesis. Dev Cell. 2014;29:392–405. doi: 10.1016/j.devcel.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 15.Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655–673. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 16.Larsson NG, Clayton DA. Molecular genetic aspects of human mitochondrial disorders. Annual review of genetics. 1995;29:151–178. doi: 10.1146/annurev.ge.29.120195.001055. [DOI] [PubMed] [Google Scholar]
- 17.Yue F, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zanconato F, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218–1227. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galli GG, et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol Cell. 2015;60:328–337. doi: 10.1016/j.molcel.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein C, et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015;11:e1005465. doi: 10.1371/journal.pgen.1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morikawa Y, et al. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in Hippo-deficient mice. Sci Signal. 2015;8:ra41. doi: 10.1126/scisignal.2005781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puente BN, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chouchani ET, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao D, et al. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nature communications. 2014;5:3315. doi: 10.1038/ncomms4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.