

Abstract
Most small-molecule drug candidates fail before entering the market, frequently because of unexpected toxicity. Often, toxicity is detected only late in drug development, because many types of toxicities, especially idiosyncratic adverse drug reactions (IADRs), are particularly hard to predict and detect. Moreover, drug-induced liver injury (DILI) is the most frequent reason drugs are withdrawn from the market and causes 50% of acute liver failure cases in the United States. A common mechanism often underlies many types of drug toxicities, including both DILI and IADRs. Drugs are bioactivated by drug-metabolizing enzymes into reactive metabolites, which then conjugate to sites in proteins or DNA to form adducts. DNA adducts are often mutagenic and may alter the reading and copying of genes and their regulatory elements, causing gene dysregulation and even triggering cancer. Similarly, protein adducts can disrupt their normal biological functions and induce harmful immune responses. Unfortunately, reactive metabolites are not reliably detected by experiments, and it is also expensive to test drug candidates for potential to form DNA or protein adducts during the early stages of drug development. In contrast, computational methods have the potential to quickly screen for covalent binding potential, thereby flagging problematic molecules and reducing the total number of necessary experiments. Here, we train a deep convolution neural network—the XenoSite reactivity model—using literature data to accurately predict both sites and probability of reactivity for molecules with glutathione, cyanide, protein, and DNA. On the site level, cross-validated predictions had area under the curve (AUC) performances of 89.8% for DNA and 94.4% for protein. Furthermore, the model separated molecules electrophilically reactive with DNA and protein from nonreactive molecules with cross-validated AUC performances of 78.7% and 79.8%, respectively. On both the site- and molecule-level, the model’s performances significantly outperformed reactivity indices derived from quantum simulations that are reported in the literature. Moreover, we developed and applied a selectivity score to assess preferential reactions with the macromolecules as opposed to the common screening traps. For the entire data set of 2803 molecules, this approach yielded totals of 257 (9.2%) and 227 (8.1%) molecules predicted to be reactive only with DNA and protein, respectively, and hence those that would be missed by standard reactivity screening experiments. Site of reactivity data is an underutilized resource that can be used to not only predict if molecules are reactive, but also show where they might be modified to reduce toxicity while retaining efficacy. The XenoSite reactivity model is available at http://swami.wustl.edu/xenosite/p/reactivity.
Short abstract
Electrophilically reactive metabolites often drive drug toxicity by conjugating to biological macromolecules, including DNA and protein. A deep neural network accurately predicts both atom- and molecule-level reactivity.
Introduction
Most small-molecule drug candidates fail before entering the market,1 frequently because of unexpected toxicity.1,2 Often, toxicity is detected only late in drug development, because many types of toxicities, especially idiosyncratic adverse drug reactions (IADRs), are particularly hard to predict and detect.3,4 Moreover, drug-induced liver injury (DILI) is the most frequent reason drugs are withdrawn from the market and causes 50% of acute liver failure cases in the United States.5
A common mechanism often underlies many types of drug toxicity, including both DILI and IADRs. Drugs are bioactivated by drug-metabolizing enzymes into reactive metabolites, which then conjugate to sites in proteins or DNA to form adducts. DNA adducts are often mutagenic and may alter the reading and copying of genes and their regulatory elements, causing gene dysregulation and even triggering cancer.5−7 Similarly, protein adducts can disrupt their normal biological functions and induce harmful immune responses.5,8,9
Metabolites are reactive because of their chemical properties and are often generally classified as soft or hard electrophiles based on polarizability and their preferential reaction with targeted nucleophiles.10 Soft electrophiles such as epoxides or Michael acceptors have low electron density at multiple sites, while hard electrophiles such as carbocations or saturated aldehydes have a localized site with low electron density.10−12 Soft electrophiles tend to react with soft nucleophiles, such as cysteine residues within glutathione (GSH) or protein, whereas hard electrophiles generally react with hard nucleophiles, such as purine and pyrimidine bases in DNA or lysine and histidine residues within protein.5,10,11,13−15 Despite these general rules, it remains a challenge to predict the reactivity of small molecules and their likelihood for modifying DNA and proteins.
Conjugation between small molecules and nucleophilic GSH (soft) or cyanide (hard) is commonly used in screening studies to identify molecules capable of forming adducts. Detecting GSH or cyanide adducts is easier than detecting protein or DNA adducts, and serves as a proxy for reactivity to macromolecules in experimental studies.16,17 Moreover, GSH is a physiologically relevant trapping agent, which reaches millimolar levels in cells and protectively conjugates with many reactive molecules.18
However, GSH and cyanide are only imperfect proxies for protein and DNA. Proteins and DNA are structurally complex macromolecules and likely have correspondingly complex reactivities with a diverse set of soft and hard nucleophilic sites. Therefore, we expect some molecules will react with protein or DNA, but not efficiently react with GSH or cyanide. These molecules are of special concern, because they do not react with GSH or cyanide, and consequently are likely to be missed by many standard reactivity experiments. Computational modeling could provide a complementary strategy for detecting molecules likely to be reactive, and therefore prone to causing DILI or IADRs, including those molecules missed by standard screening assays. Others have proposed QSAR models to predict GSH reactivity, yet these models are of limited value, as they are focused on very limited structural groups.19 In contrast, we recently published a computational model that predicted GSH reactivity toward a diverse set of chemicals at both the site and molecule level.20
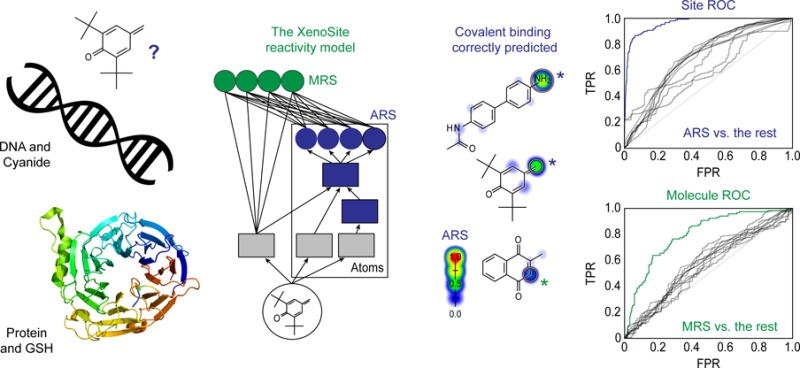
Here, we extended our previously published GSH reactivity model to also predict reactivity with cyanide, protein, and DNA. First, we extracted a structurally diverse, literature-derived database of molecules known to bind DNA and protein, as well as the simple nucleophilic traps cyanide and GSH (Figure 1). Second, we labeled the site of conjugation on each molecule, known as its site of reactivity (SOR). Third, we used a deep convolution neural network to accurately predict these SORs in cross-validated experiments. Fourth, we transformed SOR scores to accurate molecule-level electrophilic reactivity scores that accurately predict whether molecules will conjugate to DNA or protein. Fifth, we applied these molecule reactivity scores to calculate DNA and protein selectivity scores to estimate the fraction of molecules that are reactive to DNA and protein but not cyanide or GSH.
Figure 1.

Examples of the four types of sites of electrophilic reactivity modeled. Predictions by the XenoSite reactivity model are indicated on the left with a colored shading gradient and white circle for each known site of reactivity. These predictions range from zero to one, indicating the probability that an atom is reactive with each of the four nucleophilic targets. From top to bottom, a cyanide conjugation of a nefazodone metabolite,21 a DNA conjugation of N-acetylbenzidine,22 a glutathione (GSH) conjugation of menadione,23 and a protein conjugation of a butylated hydroxytoluene metabolite.24 DNA and protein are inherently structurally diverse, and thus cartoonized macromolecules are depicted, with the rest of each macromolecule represented by “Xx”.
Of course, ultimately, the XenoSite reactivity model will be connected to models of drug metabolism to be most useful. Although out of the scope of this study, combined metabolism and reactivity models would be able to predict both bioactivation and subsequent toxicity of metabolites. Nonetheless, this study takes a significant step toward effectively managing the IADR and DILI risk of new medications with computational modeling.
Results and Discussion
In the initial section, we summarize a systematic effort to optimize the structure and training of the model, with the goal of choosing the best method for predicting reactivity. The following sections then investigate the performance of the final optimized model. First, we evaluated the model’s cross-validated atom reactivity scores (ARS) by testing SOR classification performance within reactive molecules. Second, we compared ARS performance to that of atom-level quantum chemical reactivity indices. Third, we assessed ARS performance on an external test set. Fourth, we calculated the accuracy of the model’s cross-validated molecule reactivity scores (MRS) at predicting molecule reactivity. Fifth, we compared MRS performance to molecule-level quantum chemical reactivity indices. Sixth, we use the model to estimate the number of high throughput screening molecules that are reactive with macromolecules (DNA and protein) but are not flagged by small-molecule trapping agents (GSH and cyanide).
Model Optimization
Several experiments demonstrate how each of the innovations in our modeling approach improves performance. These experiments are briefly discussed here, and further details and data are available in Supporting Information. First, we hypothesized that jointly modeling several types of reactivity in a multitask learning model would improve predictions on the smaller data sets.25,26 Indeed, the multitask model outperformed the individual modeling approach at predicting cyanide and protein SOR (Figure S2). This is likely because the cyanide and protein reactivity tasks are the most difficult and, therefore, benefit most from integrated modeling. The cyanide data set is difficult because it is small, and the protein data set is both small and includes the most diverse mechanisms. The data reported herein reflects modeling all four types of reactivity together in a multitask network, instead of building separate models for each task.
Second, a modular input layer was used to group related descriptors (such as the identities of all atoms a certain depth away), rather than a traditional three-layer neural network structure. We hypothesized that building explicit chemical knowledge into the model structure could reduce the total number of parameters in the model, thereby creating a simpler model with better generalizability. This possibility was inspired by several examples of modular neural networks in the literature.27−32 In fact, the modular structure did enable reduction of the total number of weights in the model by 50%, while retaining the same performance (Figure S3). This weight-reduced, modularly structured model outperformed a traditionally structured model with the same number of weights. Third, we found that including quantum chemical descriptors did not improve performance compared to a topological-descriptor-only model (Figure S4), so we did not include the quantum chemical descriptors in the final model. Construction of this topological-descriptor-only model was inspired by our previous model of GSH reactivity, which primarily relied upon topological descriptors rather than quantum chemical descriptors.20 A common critique of neural networks is their opacity compared to more transparent methods with easily interpretable weights, like a logistic regressor. In response to this critique and to gain insight into the inner workings of our model, a permutation sensitivity analysis was performed (Figure S5). We have previously used this approach to expose the structure of similar models.20,33,34 Details of the permutation sensitivity analysis are available in the Supporting Information. Fourth and finally, we found that including the negative epoxides in the training data substantially improved the model’s ability to identify reactive epoxides (Figure S6).
Accuracy at Predicting Sites of Reactivity
A central objective of this study was to accurately predict SORs: the specific atom(s) within reactive molecules that covalently bind to nucleophilic sites within DNA and protein. The XenoSite reactivity model gives four reactivity scores to each atom (ARS) within a test molecule, each of which ranged from zero to one, and represented the probability that an atom is reactive with cyanide, DNA, GSH, or protein, respectively. Within a reactive molecule, a well-performing model should assign reactive atoms with higher scores than nonreactive atoms. The magnitude of the SOR values and identity of the target(s) of the reactive molecule sheds light on preferential chemistries leading to adduction and structures of the adducts. Such knowledge is relevant for reactive metabolite identification under experimental conditions and further study on the consequences of adducts on normal biological processes. Furthermore, SOR predictions indicate the probability of reactive hot spots leading to unfavorable adductions, and such knowledge could be leveraged to engineer rational modifications to reduce a molecule’s reactivity and potentially its toxicity.
The accuracy of the ARS scores was assessed using 10 replicates of 10-fold cross-validation, where groups of related molecules are held out in the same fold. Each replicate yielded very similar results, so for brevity we reported the results from the first one. Performance was quantified by two metrics. First, we calculated the average site area under the curve (AUC) by computing the area under the receiver operating characteristic (ROC) curve (AUC) for each molecule and averaging the AUCs for each molecule in the data set.20,34 Second, we calculated the top-two metric, which is a standard for site of metabolism predictions. This approach considers a molecule correctly predicted if any of its SOR are predicted in the first or second rank positions.34−38 Reactivity indices drawn from the quantum modeling literature are another method for predicting atom reactivity,39−43 and thus, they provide an important point of comparison to our ARS for predicting SOR.
XenoSite’s cross-validated ARS predicted reactive atoms with average site AUC accuracies of 96.6%, 89.8%, 92.8%, and 94.4%, and top-two accuracies of 83.9%, 80.6%, 80.9%, and 84.2%, for cyanide, DNA, GSH, and protein, respectively (Figure 2). These performances are very accurate, especially when compared to the current standard of atom-level reactivity indices (listed in Table 1) from quantum simulations.39−43 Consistent with our previous model of GSH reactivity, ARS outperformed all reactivity indices tested across all four nucleophilic targets.20 For example, for predicting DNA SOR, the best performing descriptor by both metrics was πS(r), with an average site AUC of 60.9% and a top-two accuracy of 27.6%, which are both significantly lower than ARS’s corresponding performances of 89.8% and 80.6%. The machine learning approach we adopt here is strikingly more accurate than quantum chemical measures of reactivity.
Figure 2.

Atom reactivity scores (ARS) accurately identified sites of reactivity (SORs). The average site AUC (top left) and top-two (top center) metrics were computed for each of the four target nucleophiles and used to assess the cross-validated ARS for 1364 reactive molecules extracted from the Accelrys Metabolite Database (AMD) with their SORs labeled. ARS outperformed all quantum chemical descriptors tested for each target nucleophile. Bottom, from the latest AMD release, an external test of 14 molecules was extracted and SOR(s) labeled for GSH. Performance is equivalent to that of the cross-validated predictions as measured by both the average site AUC (bottom left) and the top-two (bottom center) metrics. Right, two example molecules from the external test set are visualized with their predictions, indicated by the colored shading. Each site of reactivity is labeled with a white circle. Top right, an antimalarial drug candidate metabolite,44 and bottom right, gefitinib.45
Table 1. Quantum Chemical Reactivity Descriptors.
| Atom-Level Descriptors | |
|---|---|
| DN(r) | nucleophilic delocalizability |
| DE(r) | electrophilic delocalizability |
| πS(r) | self-polarizability |
| Molecule-Level Descriptors | |
|---|---|
| ELUMO | energy of the lowest unoccupied molecular orbital |
| EHOMO | energy of the highest occupied molecular orbital |
| max[DN(r)] | maximum atom nucleophilic delocalizability |
| max[DE(r)] | maximum atom electrophilic delocalizability |
| max[πS(r)] | maximum atom self-polarizability |
As further evidence of generalizability, the model was applied to an external test of 14 molecules reactive with GSH that were collected from a newer version of the AMD than that used in training (Figure 2). The model predicted SOR on this test set with an average site AUC accuracy of 93.6%, matching the cross-validated performance of 92.8%. The top-two external test set performance was 64.3%, somewhat lower than the cross-validated performance of 80.9%. The lower performance is well explained by the high variance of the estimate, because there are only a few molecules in the set. Moreover, the performance of the model is still substantially better than the quantum chemical descriptors, which are indistinguishable from zero performance on the test molecules. Depictions of all 14 external test set molecules with their GSH ARS and SOR are available in the Supporting Information, alongside their most closely similar molecule from the training set (Figure S7).
Accuracy at Identifying Reactive Molecules
Another central goal is to accurately discriminate between reactive and nonreactive molecules. XenoSite predicted four reactivity scores for each molecule (MRS), each of which ranged from zero to one, and represented the probability that the molecule is reactive with cyanide, DNA, GSH, or protein, respectively. The accuracies of MRS were measured by 10-fold cross-validation, with performance quantified by the area under the ROC curve (molecule AUC). The MRS scores were reasonably accurate in separating reactive and nonreactive molecules (90.3%, 78.7%, 77.7%, and 79.8% for cyanide, DNA, GSH, and protein, respectively, Figure 4). In contrast, the performances of the best reactivity indices using traditional quantum descriptors were much lower: 84.9% (−EHOMO for cyanide), 73.7% (max[DN(r)] for DNA), 62.9% (πS(r) for GSH), and 65.1% (max[DN(r)]) for protein). For each nucleophilic target, MRS outperformed all quantum chemical descriptors.
Figure 4.

Molecule reactivity scores (MRS) predict some molecules significantly more reactive with biological macromolecules than nucleophilic traps used in standard screening assays. Discordant predictions between the protein and GSH models (left), and between the DNA and cyanide models (right), are visualized. As indicated both by the colored shading (corresponding to atom reactivity scores) and by the MRS listed below, for each molecule the biological macromolecule (protein or DNA) prediction is significantly greater than the corresponding standard trapping agent (GSH or cyanide) prediction. Experimentally known sites of reactivity are labeled with white circles. Upper left pair: a metabolite of 6-nitrochrysene (MRSPRO: 0.42, MRSGSH: 0.29), an environmental pollutant.50 Bottom left pair: N-acetoxy-sulfamethoxazole (MRSPRO: 0.51, MRSGSH: 0.38), a metabolite that may mediate the toxicity of the antibiotic sulfamethoxazole.51 Upper right pair: a metabolite of lucidin-3-O-primeveroside (MRSDNA: 0.68, MRSCN: 0.26), a component of a food dye known to be carcinogenic in rats.52 Bottom right pair: 7H-dibenzo(c,g)carbazole-3,4-dione (MRSDNA: 0.64, MRSCN: 0.30), an environmental multispecies carcinogen.53
The model’s MRS scores were superior to other methods, yet still were lower accuracy than the ARS scores. This discrepancy was likely due to the presence of more noise in the molecule-level data than in the atom-level data. The ascertainment bias in the literature means that many of the negative molecules are actually reactive. When extracting nonreactive molecules, we assumed that molecules were not reactive to a nucleophile if they were not reported to be reactive to that nucleophile by any reactions in the AMD. This assumption was not solid evidence of molecules’ nonreactivity, because many studies do not test for reactivity. By contrast, the atom-level reactivity data were all drawn from experiments that tested for reactivity and thus were much less noisy with many fewer false negatives. A similar drop from atom- to molecule-level accuracy was observed in our previous studies with the literature derived data, including the GSH reactivity model that was a precursor to this work.20,34 In the future, we plan to improve the data by experimentally testing the reactivity of “nonreactive” molecules that nonetheless receive high MRS. We expect that some of these false positives will be shown to be in fact reactive and that this effort will validate the models and improve the training data.
Figure 3.

Molecule reactivity scores (MRS) accurately identified reactive molecules. A number of prediction methods were compared by their performance at identifying reactive molecules. Each nonreactive molecule is structurally similar to a reactive molecule. To measure performance, the area under the ROC curve was calculated (molecule AUC) . For each target nucleophile, MRS outperformed all quantum chemical descriptors tested. Right, four example molecules are visualized with their protein and GSH reactivity predictions indicated by colored shading. From top to bottom, a TRPV1 antagonist metabolite,46 a nitroso metabolite of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (formed during cooking of meat),47 a nicotine iminium ion metabolite (reactive with cyanide),48 and a benoxaprofen acyl glucuronide metabolite (a nonsteroidal anti-inflammatory drug withdrawn due to hepatotoxicity).49 Sites of reactivity are indicated by white circles.
Molecules Missed by Standard Screening Assays
Experimental studies with cyanide and GSH are traditional screening tools to trap hard and soft electrophilic reactive molecules, respectively, and thus provide potential insights on possible reactions between the reactive molecules and biological macromolecules.16,17 Importantly, cyanide and GSH possess only a single type of nucleophilic site, while biologically relevant macromolecules often contain both hard and soft nucleophiles with an array of different structures and hence different chemistries. Consequently, nucleophilic trapping assays using cyanide and GSH may not adequately reflect all possible reactions observed within biological macromolecules and thus fail to detect potentially toxic electrophiles.
We estimated how frequently molecules will react with macromolecules (protein and DNA) but not with trapping agents (GSH and cyanide) commonly used in experimental screens. We calculated the probability that each molecule will form adducts to either DNA or protein, but neither cyanide nor GSH. The DNA probability was computed by taking the product of its probabilities of reacting with DNA (MRSDNA) and not reacting with cyanide (1 – MRSCN) or GSH (1 – MRSGSH). The protein probability was computed by taking the product of its probabilities of reacting with protein (MRSPRO) and not reacting with cyanide (1 – MRSCN) or GSH (1 – MRSGSH). Next, we summed up the probabilities for all molecules with respect to each macromolecule. These scores are well scaled probabilities, so this sum is a valid estimate of the total number of molecules selectively reacting with each macromolecule and not the traditional nucleophilic traps, and hence those that would be missed by standard screening approaches. For the entire data set of 2803 molecules, this approach yielded totals of 257 and 227 molecules predicted to be reactive only with DNA and protein, respectively, and not cyanide or GSH. This finding suggests that in our data set 9.2% of DNA reactive molecules and 8.1% of protein reactive molecules would be missed by standard screening assays with cyanide and GSH. Though not the majority of molecules, this result suggests there are shortcomings of using small-molecule trapping agents to infer reactivity with macromolecules. Computationally modeling could fill a gap here, by identifying these problematic molecules when screening cannot (Figure 4). The experimental validation of specific missed reactive molecules and these estimates is outside the scope of this study but is planned in our future work.
Model Limitations
For predicting reactive metabolites, this study’s approach is limited by not modeling their metabolic activation, which may precede reactions with nucleophilic traps. Nevertheless, models for metabolic reactions are rapidly maturing as evidenced by our work34,35,54−56 and others,57−59 and it is conceivable that metabolism and reactivity models could be combined for a more biologically relevant prediction of risks poised by drugs and other compounds. An additional caveat is that some reactions in the database are likely missing reactive intermediates. For example, acyl glucuronides are predicted somewhat reactive, yet are known not to be reactive themselves. Instead, only after acyl migration and ring opening do acyl glucuronides form short-lived, reactive open-chain aldehyde intermediates that can lead to covalent binding to molecules.60 There are likely other uncertainties where intermediate reactive metabolites are not present in the database, as they may be so reactive that they are too short-lived to be observed experimentally. Missing intermediates is a limitation of the data, but is both a strength and weakness of our method. On the plus side, we implicitly model some types of rearrangements that yield reactive species, and in doing so, expand the utility of the model. Nevertheless, this process leads to motifs that are not reactive themselves being predicted reactive. Therefore, we are actually modeling a combination of intrinsic reactivity and potential for covalent binding. A final caveat is that the domain of applicability of XenoSite is likely limited to drug-like molecules. We do not expect the model to generalize well to molecules outside of this domain, such as to inorganic molecules. Precise assessment of the domain of applicability is a critical question for future work. We plan to directly test this with prospective experiments, which is much more convincing than computational strategies for assessing the applicability domain.
Conclusion
This study establishes that SOR modeling accurately predicts a key driver of drug toxicity: covalent binding to DNA and protein. The XenoSite reactivity model identifies SOR within reactive molecules with AUC performances of 96.6%, 89.8%, 92.8%, and 94.4%, for cyanide, DNA, GSH, and protein, respectively. Compared to building separate models, collectively modeling reactivity for all four nucleophilic targets enabled knowledge transfer between tasks, improving SOR predictions for cyanide and protein. For cyanide, DNA, GSH, and protein, the model separates reactive and nonreactive molecules with, respectively, 90.3%, 78.7%, 77.7%, and 79.8% AUC. These predictions of molecular reactivity can highlight potentially toxic molecules that might be missed in the early stages of drug development. Ultimately, to accurately predict reactive metabolites, both metabolism and reactivity must be modeled. While there has been significant progress on metabolism modeling,34,35,54−59,61 reactivity modeling has lagged far behind. By accurately modeling reactivity across a broad range of chemical space for biologically relevant macromolecules, such as DNA and protein, this study contributes a fundamental component of an integrative model of metabolism and reactivity.
Methods
Site of Reactivity Training Data
We assembled a training data set from the December 2014 release of the literature-derived Accelrys Metabolite Database (AMD). From 2489 total reactions, molecules were extracted based on reported reactions with cyanide, DNA, GSH, or protein. For each target nucleophile, the reactive atom(s) within each reactive molecule were marked based on the structures of the starting and product molecules, using an automated algorithm constructed using the RDKit python library.62 Topologically equivalent atoms were found using the Pybel python library, and atoms equivalent to reactive atoms were themselves labeled as reactive.63 The final data set included, in total, 1364 electrophilic molecules with their reactive atoms and SORs marked. This data set was composed of 51, 145, 1059, and 120 molecules known to be reactive with cyanide, DNA, GSH, or protein, respectively. For each of the four reactive nucleophilic targets, the atoms across the whole data set were labeled as reactive or nonreactive. Some atoms were marked reactive with more than one nucleophile.
Metabolically related but nonreactive molecules were extracted from the reaction network of each reactive molecule. Metabolic sibling and parent molecules were extracted from this network, while excluding molecules themselves known to be reactive. We also included 63 naturally occurring64 and known nonreactive65 epoxides as nonreactive molecules. While epoxides are generally quite reactive, they can be stable in certain cases; however, a shortcoming of our previous method for predicting GSH reactivity was that it predicted all epoxides reactive.20 These molecules were added to mitigate this shortcoming.
A total of 1439 molecules were labeled nonreactive, with 103, 248, 1269, and 255 labeled nonreactive to cyanide, DNA, GSH, and protein, respectively. At the molecule-level, for each of the four reactive nucleophilic targets, molecules across the entire data set were labeled as reactive or nonreactive. Some molecules were marked reactive or nonreactive with more than one nucleophile.
To enable replication of our work, we included all AMD molecule registry numbers in the Supporting Information, in addition to the SMILES strings of the nonreactive epoxides. Unfortunately, the AMD license precluded us from disseminating the rest of the molecule structures themselves.
External Site of Reactivity Test Data
After the training data were assembled, a new version of the AMD was released (June 2015) with several new reactions, which was used as an external data set. We filtered out all molecules already labeled as reactive in the training data. This process left only 14 new molecules that reacted with GSH and no new molecules reacting with cyanide, DNA, or protein. These 14 molecules were completely withheld from model training and optimization decisions, and were only tested with the final model.
Quantum Chemical Reactivity Indices
Several quantum chemical descriptors were calculated from self-consistent field computations with MOPAC, a quantum chemistry package, using the semiempirical method PM7 and the COSMO implicit solvent model.66,67 These included descriptors that have been previously proposed as reactivity indices, on both the atom- and molecule-level (Table 1).68−70 Atom-level descriptors include the nucleophilic (DN(r)) and electrophilic (DE(r)) delocalizabilities, which are also known as fukui reactivity indices, as well as self-polarizability (πS(r)). The maximum of each of these (max[DN(r)], max[DE(r)], and max[πS(r)]) were used as molecule descriptors, which also included the energies of the lowest unoccupied and highest occupied molecular orbitals (ELUMO and EHOMO). The performances of the final reactivity model—which only used topological descriptors—were compared to those of the quantum chemical indices.
Topological Descriptors
The reactivity model used 15 molecule-level and 194 atom-level topological descriptors, each of which describes a chemical property. An in-house python script calculated these descriptors from the structure of each molecule. This script derives these descriptors from several sources, such as each molecule’s connectivity distance matrix, periodic table properties, or motif patterns defined by Pybel.63 The majority of our topological descriptors have been shown to be useful for the XenoSite metabolism, reactivity, and epoxidation models.20,34,35 In this study, we used an expanded set of topological descriptors, which slightly improved performance in comparison to the previous set of topological descriptors (Figure S1). The full list of descriptors is available in the Supporting Information (Tables S1 and S2), as well as a list of new topological descriptors added for this project.
Combined Atom- and Molecule-Level Reactivity Model
We collectively modeled reactivity to cyanide, DNA, GSH, and protein at both the atom- and molecule-level by building a deep neural network using an in-house machine learning python library. The architecture included a molecule layer, an input layer, two hidden layers, and two output layers (Figure 5). The atom output layer computed atom-level SOR predictions against each nucleophile, while the molecule output layer computed molecule reactivity scores (MRS) for each nucleophile. The corresponding scores predicted the chances that a test molecule is reactive against that nucleophile.
Figure 5.
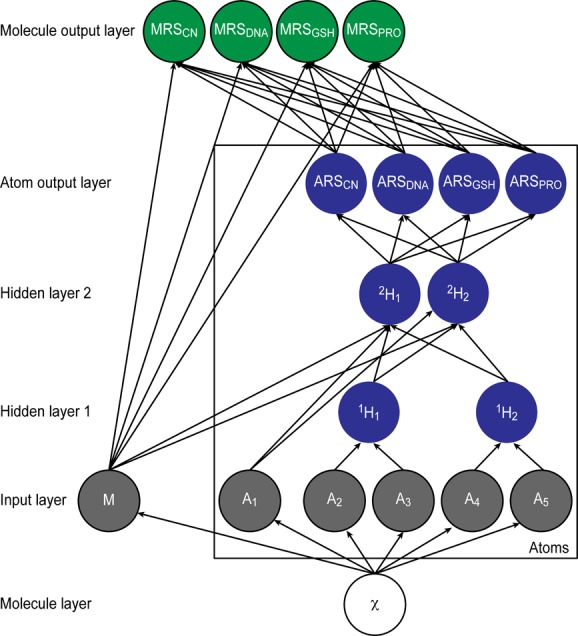
The structure of the XenoSite reactivity model. This diagram illustrates how information flowed through the model, which consisted of one input layer, two hidden layers, and two output layers. By simultaneously modeling all types of reactivity, the model was able to transfer knowledge between related tasks, thereby improving performance substantially over independent models. The model computed atom-level predictions for reactivity to each of four nucleophilic targets: cyanide (ARSCN), DNA (ARSDNA), GSH (ARSGSH), and protein (ARSPRO), collectively referred to as atom reactivity scores (ARS). Additionally, the model computed molecule reactivity scores (MRS): MRSCN, MRSDNA, MRSGSH, and MRSPRO, which predicted the chances of a molecule’s reactivity to each of the four nucleophilic targets, respectively. From the structure of an input model χ, 15 molecule-level and 194 atom-level descriptors were calculated. Some chemically related descriptors, such as neighbor atom identities, were grouped in the first hidden layer (with 30 nodes). Grouped and ungrouped nodes were inputted into the second hidden layer (with 17 nodes), which outputted four atom-level scores. Finally, for each of the four nucleophilic targets, the respective MRS was computed from the top five ARS for each of the four nucleophilic targets, corresponding to the scores of the five atoms predicted within a molecule to be the most reactive to each nucleophile, as well as all molecule-level descriptors. The diagram is condensed and displays one representative molecule input node, five atom input nodes, and two nodes for each hidden layer. The molecule input node is a chemical structure; all other nodes are vectors of real numbers computed from nodes or layers from which there are incoming connections.
We trained this network in two stages. First, the atom-level network was trained to produce atom reactivity scores (ARS). For this process, each atom was considered a possible SOR. A numerical vector was associated with each atom, which contained all of the descriptors for that atom. A binary target vector indicated if each atom was a SOR for each of the nucleophilic targets. For molecules of unknown reactivity against each nucleophilic target, the appropriate values were empty, neither one nor zero. Using gradient descent on the cross-entropy error, the weights of the network were trained such that SOR scored higher ARS than other atoms. Four ARS were produced for each atom, each ranging from zero to one, and representing the probability that the atom will be reactive with cyanide, DNA, GSH, or protein.
Second, the molecule-output nodes were trained to compute MRS. For this stage, each molecule was considered possibly reactive, and a numerical vector of descriptors was associated with each molecule. Descriptors included all molecule-level descriptors, as well as the top five ARS for each of the four nucleophilic targets, corresponding to the scores of the five atoms predicted within a molecule to be the most reactive to each nucleophile. For each molecule, a binary target vector (with unknown components empty) indicated if the molecule was reactive to each of the nucleophilic targets. For each nucleophile, a logistic regressor found a scoring function that gave reactive molecules high MRS and nonreactive molecules low MRS, ranging from zero to one.
Acknowledgments
We are grateful to the developers of the open-source chemoinformatics tools Open Babel and RDKit. Research reported in this publication was supported by the National Library of Medicine of the National Institutes of Health under Award Number R01LM012222. Computations were performed using the facilities of the Washington University Center for High Performance Computing, which were partially funded by National Institutes of Health Grant Numbers 1S10RR022984-01A1 and 1S10OD018091-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank both the Department of Immunology and Pathology at the Washington University School of Medicine and the Washington University Center for Biological Systems Engineering for their generous support of this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00162.
AMD Registry Numbers file lists the reaction and molecule registry numbers of the reactivity data sets, extracted from the December 2014 AMD release (ZIP)
AMD data, molecule-level and atom-level descriptors; performances of previous topological descriptors and new topological descriptors; individual vs multitarget trainings effect of modular neural network; effect of adding quantum chemical descriptors; descriptors driving prediction performance; effect of adding unreactive epoxides; visualization of external test set (PDF)
The authors declare no competing financial interest.
The new version of this article has updated Figures 1, 2, 3, 4, 5, and TOC/abstract files.
Supplementary Material
References
- McKim J. M. Jr. Building a tiered approach to in vitro predictive toxicity screening: a focus on assays with in vivo relevance. Comb. Chem. High Throughput Screening 2010, 13, 188. 10.2174/138620710790596736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMasi J. A. Success rates for new drugs entering clinical testing in the United States. Clin. Pharmacol. Ther. 1995, 58, 1–14. 10.1016/0009-9236(95)90066-7. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab. Pharmacokinet. 2011, 26, 3–14. 10.2133/dmpk.DMPK-10-RV-062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I.; Landis J. Can the pharmaceutical industry reduce attrition rates?. Nat. Rev. Drug Discovery 2004, 3, 711–716. 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Srivastava A.; Maggs J.; Antoine D.; Williams D.; Smith D.; Park B.. Adverse Drug Reactions; Springer: Berlin, 2010; pp 165–194. [Google Scholar]
- Lutz W. K. In vivo covalent binding of organic chemicals to DNA as a quantitative indicator in the process of chemical carcinogenesis. Mutat. Res., Rev. Genet. Toxicol. 1979, 65, 289–356. 10.1016/0165-1110(79)90006-X. [DOI] [PubMed] [Google Scholar]
- Gelboin H. V. Benzo [alpha] pyrene metabolism, activation and carcinogenesis: role and regulation of mixed-function oxidases and related enzymes. Physiol. Rev. 1980, 60, 1107–1166. [DOI] [PubMed] [Google Scholar]
- Knowles S. R.; Uetrecht J.; Shear N. H. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet 2000, 356, 1587–1591. 10.1016/S0140-6736(00)03137-8. [DOI] [PubMed] [Google Scholar]
- Numata K.; Kubo M.; Watanabe H.; Takagi K.; Mizuta H.; Okada S.; Kunkel S. L.; Ito T.; Matsukawa A. Overexpression of suppressor of cytokine signaling-3 in T cells exacerbates acetaminophen-induced hepatotoxicity. J. Immunol. 2007, 178, 3777–3785. 10.4049/jimmunol.178.6.3777. [DOI] [PubMed] [Google Scholar]
- LoPachin R. M.; Gavin T. Molecular mechanisms of aldehyde toxicity: a chemical perspective. Chem. Res. Toxicol. 2014, 27, 1081–1091. 10.1021/tx5001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B. K.; Kitteringham N. R.; Maggs J. L.; Pirmohamed M.; Williams D. P. The role of metabolic activation in drug-induced hepatotoxicity. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 177–202. 10.1146/annurev.pharmtox.45.120403.100058. [DOI] [PubMed] [Google Scholar]
- Stachulski A. V.; et al. The Generation, Detection, and Effects of Reactive Drug Metabolites. Med. Res. Rev. 2013, 33, 985–1080. 10.1002/med.21273. [DOI] [PubMed] [Google Scholar]
- Attia S. M. Deleterious effects of reactive metabolites. Oxid. Med. Cell. Longevity 2010, 3, 238–253. 10.4161/oxim.3.4.13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerberick G. F.; Vassallo J. D.; Bailey R. E.; Chaney J. G.; Morrall S. W.; Lepoittevin J.-P. Development of a peptide reactivity assay for screening contact allergens. Toxicol. Sci. 2004, 81, 332–343. 10.1093/toxsci/kfh213. [DOI] [PubMed] [Google Scholar]
- Dennehy M. K.; Richards K. A.; Wernke G. R.; Shyr Y.; Liebler D. C. Cytosolic and nuclear protein targets of thiol-reactive electrophiles. Chem. Res. Toxicol. 2006, 19, 20–29. 10.1021/tx050312l. [DOI] [PubMed] [Google Scholar]
- Ma S.; Subramanian R. Detecting and characterizing reactive metabolites by liquid chromatography/tandem mass spectrometry. J. Mass Spectrom. 2006, 41, 1121–1139. 10.1002/jms.1098. [DOI] [PubMed] [Google Scholar]
- Meneses-Lorente G.; Sakatis M. Z.; Schulz-Utermoehl T.; De Nardi C.; Watt A. P. A quantitative high-throughput trapping assay as a measurement of potential for bioactivation. Anal. Biochem. 2006, 351, 266–272. 10.1016/j.ab.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Montero D.; Tachibana C.; Rahr Winther J.; Appenzeller-Herzog C. Intracellular glutathione pools are heterogeneously concentrated. Redox Biol. 2013, 1, 508–513. 10.1016/j.redox.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.; Jensen N.; O’Brien P. J. Structure-activity relationships for thiol reactivity and rat or human hepatocyte toxicity induced by substituted p-benzoquinone compounds. J. Appl. Toxicol. 2008, 28, 608–620. 10.1002/jat.1312. [DOI] [PubMed] [Google Scholar]
- Hughes T. B.; Miller G. P.; Swamidass S. J. Site of Reactivity Models Predict Molecular Reactivity of Diverse Chemicals with Glutathione. Chem. Res. Toxicol. 2015, 28, 797–809. 10.1021/acs.chemrestox.5b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argoti D.; Liang L.; Conteh A.; Chen L.; Bershas D.; Yu C.-P.; Vouros P.; Yang E. Cyanide trapping of iminium ion reactive intermediates followed by detection and structure identification using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Chem. Res. Toxicol. 2005, 18, 1537–1544. 10.1021/tx0501637. [DOI] [PubMed] [Google Scholar]
- DeMarini D.; et al. Urinary mutagenicity as a biomarker in workers exposed to benzidine: correlation with urinary metabolites and urothelial DNA adducts. Carcinogenesis 1997, 18, 981–988. 10.1093/carcin/18.5.981. [DOI] [PubMed] [Google Scholar]
- Loughlin A. F.; Skiles G. L.; Alberts D. W.; Schaefer W. H. An ion exchange liquid chromatography/mass spectrometry method for the determination of reduced and oxidized glutathione and glutathione conjugates in hepatocytes. J. Pharm. Biomed. Anal. 2001, 26, 131–142. 10.1016/S0731-7085(01)00402-2. [DOI] [PubMed] [Google Scholar]
- Mitchell M. D.; Elrick M. M.; Walgren J. L.; Mueller R. A.; Morris D. L.; Thompson D. C. Peptide-based in vitro assay for the detection of reactive metabolites. Chem. Res. Toxicol. 2008, 21, 859–868. 10.1021/tx700344m. [DOI] [PubMed] [Google Scholar]
- Collobert R.; Weston J.. A unified architecture for natural language processing: Deep neural networks with multitask learning. Proceedings of the 25th International Conference on Machine Learning, July 5–9, 2008, Helsinki;International Machine Learning Society, 2008; pp 160–167.
- Pan S. J.; Yang Q. A survey on transfer learning. Knowledge and Data Engineering, IEEE Transactions on 2010, 22, 1345–1359. 10.1109/TKDE.2009.191. [DOI] [Google Scholar]
- Azam F.Biologically inspired modular neural networks. Ph.D. thesis, Virginia Polytechnic Institute and State University, 2000. [Google Scholar]
- Anand R.; Mehrotra K.; Mohan C. K.; Ranka S. Efficient classification for multiclass problems using modular neural networks. Neural Networks, IEEE Transactions on 1995, 6, 117–124. 10.1109/72.363444. [DOI] [PubMed] [Google Scholar]
- Osherson D. N.; Weinstein S.; Stob M.. Computational Neuroscience; MIT Press: Cambridge, MA, 1993; pp 369–377. [Google Scholar]
- Auda G.; Kamel M. Modular neural network classifiers: A comparative study. J. Intell. Rob. Syst. 1998, 21, 117–129. 10.1023/A:1007925203918. [DOI] [Google Scholar]
- Lu B.-L.; Ito M.. Biological and Artificial Computation: From Neuroscience to Technology; Springer: Berlin, 1997; pp 330–339. [Google Scholar]
- Balakin K. V.; Ekins S.; Bugrim A.; Ivanenkov Y. A.; Korolev D.; Nikolsky Y. V.; Ivashchenko A. A.; Savchuk N. P.; Nikolskaya T. Quantitative structure-metabolism relationship modeling of metabolic N-dealkylation reaction rates. Drug Metab. Dispos. 2004, 32, 1111–1120. 10.1124/dmd.104.000364. [DOI] [PubMed] [Google Scholar]
- Hunter A.; Kennedy L.; Henry J.; Ferguson I. Application of neural networks and sensitivity analysis to improved prediction of trauma survival. Comp. Meth. Progr. Biom. 2000, 62, 11–19. 10.1016/S0169-2607(99)00046-2. [DOI] [PubMed] [Google Scholar]
- Hughes T. B.; Miller G. P.; Swamidass S. J. Modeling epoxidation of drug-like molecules with a deep machine learning network. ACS Cent. Sci. 2015, 1, 168–180. 10.1021/acscentsci.5b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretzki J.; Matlock M.; Swamidass S. J. XenoSite: Accurately predicting CYP-mediated sites of metabolism with neural networks. J. Chem. Inf. Model. 2013, 53, 3373–3383. 10.1021/ci400518g. [DOI] [PubMed] [Google Scholar]
- Cruciani G.; Baroni M.; Benedetti P.; Goracci L.; Fortuna C. G. Exposition and reactivity optimization to predict sites of metabolism in chemicals. Drug Discovery Today: Technol. 2013, 10, e155–e165. 10.1016/j.ddtec.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Zamora I.; Afzelius L.; Cruciani G. Predicting drug metabolism: a site of metabolism prediction tool applied to the cytochrome P450 2C9. J. Med. Chem. 2003, 46, 2313–2324. 10.1021/jm021104i. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Gloriam D. E.; Zaretzki J.; Breneman C.; Olsen L. SMARTCyp: A 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Med. Chem. Lett. 2010, 1, 96–100. 10.1021/ml100016x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultinck P.; Van Neck D.; Acke G.; Ayers P. W. Influence of electron correlation and degeneracy on the Fukui matrix and extension of frontier molecular orbital theory to correlated quantum chemical methods. Phys. Chem. Chem. Phys. 2012, 14, 2408–2416. 10.1039/c2cp22543c. [DOI] [PubMed] [Google Scholar]
- Fukui K.; Yonezawa T.; Shingu H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. 10.1063/1.1700523. [DOI] [Google Scholar]
- Roy R.; Krishnamurti S.; Geerlings P.; Pal S. Local softness and hardness based reactivity descriptors for predicting intra-and intermolecular reactivity sequences: carbonyl compounds. J. Phys. Chem. A 1998, 102, 3746–3755. 10.1021/jp973450v. [DOI] [Google Scholar]
- Morell C.; Grand A.; Toro-Labbé A. New dual descriptor for chemical reactivity. J. Phys. Chem. A 2005, 109, 205–212. 10.1021/jp046577a. [DOI] [PubMed] [Google Scholar]
- Chattaraj P. K.; Maiti B.; Sarkar U. Philicity: a unified treatment of chemical reactivity and selectivity. J. Phys. Chem. A 2003, 107, 4973–4975. 10.1021/jp034707u. [DOI] [Google Scholar]
- Srivastava A.; Ramachandran S.; Hameed S. P.; Ahuja V.; Hosagrahara V. P. Identification and mitigation of a reactive metabolite liability associated with aminoimidazoles. Chem. Res. Toxicol. 2014, 27, 1586–1597. 10.1021/tx500212c. [DOI] [PubMed] [Google Scholar]
- Surve P.; Ravindran S.; Acharjee A.; Rastogi H.; Basu S.; Honrao P. Metabolite characterization of anti-cancer agent gefitinib in human hepatocytes. Drug Metab. Lett. 2014, 7, 126–136. 10.2174/1872312808666140317154110. [DOI] [PubMed] [Google Scholar]
- Bylund J.; Petersson C.; Lindgren A.; Olofsson S.; Czene S. Metabolic profiling of TRPV1 antagonists of the benzothiazole amide series: implications for in vitro genotoxicity assessment. Xenobiotica 2013, 43, 201–210. 10.3109/00498254.2012.708459. [DOI] [PubMed] [Google Scholar]
- Peng L.; Turesky R. J. Mass spectrometric characterization of 2-amino-1-methyl-6-phenylimidazo [4, 5-b] pyridine N-oxidized metabolites bound at Cys34 of human serum albumin. Chem. Res. Toxicol. 2011, 24, 2004–2017. 10.1021/tx2003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aislaitner G.; Bello A.; Tan S.; Hutt A.; Marriott C.; Gorrod J. Metabolism of (−)-(S)-nicotine in the isolated perfused rabbit lung. Eur. J. Drug Metab. Pharmacokinet. 1997, 22, 395–402. 10.1007/BF03190976. [DOI] [PubMed] [Google Scholar]
- Dong J. Q.; Liu J.; Smith P. C. Role of benoxaprofen and flunoxaprofen acyl glucuronides in covalent binding to rat plasma and liver proteins in vivo. Biochem. Pharmacol. 2005, 70, 937–948. 10.1016/j.bcp.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Li E. E.; Heflich R. H.; Bucci T. J.; Manjanath M. G.; Blaydes B. S.; Delclos K. B. Relationships of DNA adduct formation, K-ras activating mutations and tumorigenic activities of 6-nitrochrysene and its metabolites in the lungs of CD-1 mice. Carcinogenesis 1994, 15, 1377–1385. 10.1093/carcin/15.7.1377. [DOI] [PubMed] [Google Scholar]
- Nakamura H.; Uetrecht J.; Cribb A. E.; Miller M.; Zahid N.; Hill J.; Josephy P.; Grant D.; Spielberg S. In vitro formation, disposition and toxicity of N-acetoxy-sulfamethoxazole, a potential mediator of sulfamethoxazole toxicity. J. Pharmacol. Exp. Ther. 1995, 274, 1099–1104. [PubMed] [Google Scholar]
- Ishii Y.; Inoue K.; Takasu S.; Jin M.; Matsushita K.; Kuroda K.; Fukuhara K.; Nishikawa A.; Umemura T. Determination of lucidin-specific DNA adducts by liquid chromatography with tandem mass spectrometry in the livers and kidneys of rats given lucidin-3-O-primeveroside. Chem. Res. Toxicol. 2012, 25, 1112–1118. 10.1021/tx300084p. [DOI] [PubMed] [Google Scholar]
- Xue W.; Siner A.; Rance M.; Jayasimhulu K.; Talaska G.; Warshawsky D. A metabolic activation mechanism of 7 H-dibenzo [c, g] carbazole via O-quinone Part 2: Covalent adducts of 7 H-dibenzo [c, g] carbazole-3, 4-dione with nucleic acid bases and nucleosides. Chem. Res. Toxicol. 2002, 15, 915–921. 10.1021/tx0200156. [DOI] [PubMed] [Google Scholar]
- Zaretzki J.; Boehm K. M.; Swamidass S. J. Improved Prediction of CYP-Mediated Metabolism with Chemical Fingerprints. J. Chem. Inf. Model. 2015, 55, 972–982. 10.1021/ci5005652. [DOI] [PubMed] [Google Scholar]
- Zaretzki J.; Browning M. R.; Hughes T. B.; Swamidass S. J. Extending P450 Site-of-Metabolism Models with Region-Resolution Data. Bioinformatics 2015, 31, 1966–1973. 10.1093/bioinformatics/btv100. [DOI] [PubMed] [Google Scholar]
- Matlock M. K.; Hughes T. B.; Swamidass S. J. XenoSite server: a web-available site of metabolism prediction tool. Bioinformatics 2015, 31, 1136–1137. 10.1093/bioinformatics/btu761. [DOI] [PubMed] [Google Scholar]
- Kim D. N.; Cho K.-H.; Oh W. S.; Lee C. J.; Lee S. K.; Jung J.; No K. T. EaMEAD: Activation energy prediction of cytochrome P450 mediated metabolism with effective atomic descriptors. J. Chem. Inf. Model. 2009, 49, 1643–1654. 10.1021/ci900011g. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Olsen L. Predicting drug metabolism by cytochrome P450 2C9: comparison with the 2D6 and 3A4 isoforms. ChemMedChem 2012, 7, 1202–1209. 10.1002/cmdc.201200160. [DOI] [PubMed] [Google Scholar]
- Rudik A.; Dmitriev A.; Lagunin A.; Filimonov D.; Poroikov V. SOMP: web-server for in silico prediction of sites of metabolism for drug-like compounds. Bioinformatics 2015, 31, 2046–2048. 10.1093/bioinformatics/btv087. [DOI] [PubMed] [Google Scholar]
- Regan S. L.; Maggs J. L.; Hammond T. G.; Lambert C.; Williams D. P.; Park B. K. Acyl glucuronides: the good, the bad and the ugly. Biopharm. Drug Dispos. 2010, 31, 367–395. 10.1002/bdd.720. [DOI] [PubMed] [Google Scholar]
- Dang N. L.; Hughes T. B.; Krishnamurthy V.; Swamidass S. J. A simple model predicts UGT-mediated metabolism. Bioinformatics 2016, btw350. 10.1093/bioinformatics/btw350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum G. RDKit: Open-source cheminformatics. http://www.rdkit.org.
- O’Boyle N. M.; Morley C.; Hutchison G. R. Pybel: a Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 1–5. 10.1186/1752-153X-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Contelles J.; Molina M. T.; Anjum S. Naturally occurring cyclohexane epoxides: sources, biological activities, and synthesis. Chem. Rev. 2004, 104, 2857–2900. 10.1021/cr980013j. [DOI] [PubMed] [Google Scholar]
- Delaine T.; Ponting D. J.; Niklasson I. B.; Emter R.; Hagvall L.; Norrby P.-O.; Natsch A.; Luthman K.; Karlberg A.-T. Epoxyalcohols: Bioactivation and Conjugation Required for Skin Sensitization. Chem. Res. Toxicol. 2014, 27, 1860–1870. 10.1021/tx500297d. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. MOPAC: a semiempirical molecular orbital program. J. Comput.-Aided Mol. Des. 1990, 4, 1–103. 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- Hostaš J.; Řezáč J.; Hobza P. On the performance of the semiempirical quantum mechanical PM6 and PM7 methods for noncovalent interactions. Chem. Phys. Lett. 2013, 568-569, 161–166. 10.1016/j.cplett.2013.02.069. [DOI] [Google Scholar]
- Karelson M.; Lobanov V. S.; Katritzky A. R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 1996, 96, 1027–1044. 10.1021/cr950202r. [DOI] [PubMed] [Google Scholar]
- Cronin M. T.; Madden J. C.. In Silico Toxicology: Principles and Applications; Royal Society of Chemistry: Liverpool, 2010. [Google Scholar]
- Schwöbel J. A.; Koleva Y. K.; Enoch S. J.; Bajot F.; Hewitt M.; Madden J. C.; Roberts D. W.; Schultz T. W.; Cronin M. T. Measurement and estimation of electrophilic reactivity for predictive toxicology. Chem. Rev. 2011, 111, 2562–2596. 10.1021/cr100098n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.