

Abstract
Prostatic diseases such as prostate cancer and benign prostatic hyperplasia are highly prevalent among men. The number of studies focused on the abundance and roles of intrinsically disordered proteins in prostate cancer is rather limited. The goal of this study is to analyze the prevalence and degree of disorder in proteins that were previously associated with the prostate cancer pathogenesis and to compare these proteins to the entire human proteome. The analysis of these datasets provides means for drawing conclusions on the roles of disordered proteins in this common male disease. We also hope that the results of our analysis can potentially lead to future experimental studies of these proteins to find novel pathways associated with this disease.
Keywords: intrinsically disordered proteins, posttranslational modifications, prostate cancer, protein-protein interaction, proteomics, proteome, unfoldome
INTRODUCTION
Prostate cancer is the most prevalent form of cancer in males. It is estimated that in 2016, it will account for 180 890 (21%) of new cancer cases. It is also estimated that 26 120 (8%) of all cancer deaths which will occur in the United States male population in the year 2016 will be caused by prostate cancer.1 Although there has been progress in the detection and treatment of prostate cancer, it is clear that more research is needed to make the diagnosis more reliable. The need for the constant improvement of the diagnostics tools is given, for example, by a recently established effect of obesity (measured in terms of the body mass index) on the predictive performance of the well-established and widely used biomarker, prostate-specific antigen (PSA), and PSA-related markers for prostate cancer.2 These and similar facts alone should be sufficient to spark the scientific community's interest in studying more on possible causes of this very common male disease. Sometimes, a new approach needs to be taken to further the advancements in biomedical research.
One of such approaches is considering roles of intrinsically disordered proteins, or IDPs, which are proteins that lack a stable secondary and tertiary structure and have been shown to constitute a noticeable part of any proteome of interest.3,4,5,6,7,8,9,10,11,12,13,14,15 At the primary sequence level, IDPs are characterized by noticeable compositional biases, being noticeably depleted in order-promoting amino acids (Cys, Trp, Tyr, Phe, Ile, Leu, Val, and Asn) and being found to be enriched with disorder-promoting residues (Pro, Arg, Gly, Gln, Ser, Glu, Lys, and Ala).3,16,17,18 This peculiar physicochemical nature of these proteins, therefore, does not favor spontaneous folding into well-defined globular structures, and they, in turn, remain “floppy.”19,20 Among other important features, this “floppiness” of IDPs provides them with an ability to be promiscuous binders and be involved in regulation and control of various signaling processes, being controlled themselves at multiple levels.4,5,21,22 Furthermore, intrinsically disordered protein regions (IDPRs) are often targeted for posttranslational modifications (PTMs)23,24,25,26 and are known to be involved in a myriad of biological processes.4,5,16,18,20,27,28,29,30,31,32 Proteins that are involved in multiple functions and processes are sometimes referred to as “moonlighting” proteins, and many of these moonlighting proteins are shown to be either completely disordered or possess long IDPRs.33 Often, IDPs are specifically compartmentalized, e.g., IDPs and proteins with IDPRs in various nuclear membrane-less organelles.34 Since cells in various cancers, not just of the prostate, grow and divide in an uncontrolled manner, it would be reasonable to assume that these moonlighting proteins with the multiple disordered regions have a significant role in many oncological processes. The validity of this hypothesis was demonstrated in earlier bioinformatics study where the majority of human cancer-associated proteins (HCAP) were shown to contain long IDPRs (i.e., regions possessing ≥30 consecutive disordered residues).35 More generally, a D2 (disorder in disorders) concept was introduced to emphasize that many proteins related to various human diseases such as cancer, neurodegeneration, diabetes, cardiovascular disease, and amyloidosis are intrinsically disordered.36
This two-part study was dedicated to the analysis of the prevalence and functionality of disordered proteins in prostate cancer cells and to compare them to the abundance and functionality of IDPs in the human proteome as a whole. The present paper, which represents the first part of this two-part study, reports the results of the global bioinformatics analysis of the disorderedness of proteins that were previously shown to be associated with the prostate cancer pathogenesis and compares these proteins with the entire human proteome. The second part continues this analysis, being focused on more detailed characterization of the prostate cancer-associated proteins from KEGG database.
MATERIALS AND METHODS
Acquisition of protein datasets
UniProt IDs of human proteins were obtained from the reviewed UniProt release 2015–09.37 From those, reviewed IDs without uncommon amino acid codes (J, X, O, U, B, and Z) and proteins shorter than 30 amino acids were removed. The remaining proteins totaled to 20 120 and were considered as the human proteome set.
To obtain an experimentally validated dataset of proteins that were already shown to experimentally be involved in prostate cancer, we used results of a proteomic study that reported on the characterization of a total of 359 proteins, including 17 potential biomarkers of prostate cancer from prostatic cell lines, entered into the prostate cancer proteomics (PCP) database (http://ef.inbi.ras.ru).38 This database is described as a multilevel informational database that was created using the results of a proteomic study of human prostate carcinoma and benign hyperplasia tissues, and of some human-cultured cell lines. Prostate cancer-related proteins entered to this database were first separated by 2D electrophoresis and subsequently identified by mass-spectrometry.38 Proteins are extensively annotated using data from published articles and existing databases and contain direct Internet links to the information in the NCBI and UniProt databases.38 Although this PCP website was originally designed in Russian, it can be toggled into English. The PCP database consists of 7 interrelated modules that contain proteomic data from different cell lines.38 The seven modules that were used in our analysis are prostate proteins (hyperplasia, cancer), LNCAP (IPG-2DE), LNCAP (IEF-2DE), proteins of Rhabdomyosarcoma A-204, normal human myoblasts proteins, PC3, and BPH-1. The gene names were retrieved from the PCP database, and then were used as identifiers to find corresponding human proteins in UniProtKB. Some gene product names could not be mapped to UniProt IDs and were not included in our analysis. Once these UniProt IDs were found, obvious duplicates were removed, which decreased this dataset to 291 proteins. Since the PCP dataset had 7 modules that were interrelated, there were many repeated UniProt IDs including those corresponding to various isoforms. Therefore, a series of programs were written to obtain the unique representatives for subsequent analysis. After parsing the files, a dataset consisted of 251 proteins was assembled. This dataset included 196 reviewed UniProt proteins and 55 unreviewed TrEMBL proteins. The isoforms (designated with a dash and a number in their UniProt IDs) fell into the unreviewed category. Collectively, dataset containing 196 UniProt-reviewed proteins from the PCP database is referred to as the “Russian dataset” in our analysis and corresponding files.
An independent search of KEGG database resource (http://www.genome.jp/kegg/)39 conducted in September 2015 provided us with a set of 48 proteins experimentally shown to be involved in prostate cancer. This dataset referred to as the “KEGG dataset” was used in a global analysis of abundance of intrinsic disorder in this set of prostate cancer-related proteins described in section of Results. A new search of KEGG database conducted on May 28, 2016, gave 40 additional proteins with experimentally validated connection to prostate cancer. The resulting set of 88 proteins constituted the “extended KEGG dataset”, and proteins from these datasets were subjected to a more focused and detailed analysis of their predisposition for intrinsic disorder in the companion paper (Supplementary Table 1 (197KB, tif) ).
Illustrative representation of the 2×2 contingency table to find significantly associated GO terms
Proteins from Russian and KEGG datasets that were not found in the whole human proteome dataset were filtered out. For comparison purpose, a modified human proteome dataset was generated from which proteins included into Russian and KEGG datasets were excluded. Since transmembrane domains of membrane proteins are typically enriched in hydrophobic, order-promoting residues, Russian, KEGG, and human proteome datasets were further divided into two categories: membrane and nonmembrane. Membrane and nonmembrane proteins were classified using the QuickGO gene association file (gene_association.goa_human.gz, updated on September 14, 2015), and 41 Cellular Component Gene Ontology (GO) terms with ‘integral’ and ‘membrane’ on their names.40,41
Functional annotation using gene ontology (GO) terms
The GO terms for the human proteome were downloaded on September 14, 2015, from QuickGO (https://www.ebi.ac.uk/QuickGO/). The resulting file shows all of the biological processes (P), molecular functions (F), and cellular component (C) for all of the human proteins in UniProt format. All of the GO terms that are specific for annotating the cellular component were downloaded from the Gene Ontology Consortium (http://www.geneontology.org).
Evaluation of intrinsic disorder propensity and disorder-based functionality
Intrinsic disorder propensities of human proteins were evaluated using PONDR-FIT, PONDR® VLXT, and PONDR® VSL2 algorithms.42,43,44,45,46 For each protein, after obtaining an average disorder score by each predictor, all three predictor-specific average scores were averaged again to generate a per-protein intrinsic disorder score. The use of consensuses for evaluation of intrinsic disorder is motivated by empirical observations that this leads to an increase in the predictive performance compared to the use of a single predictor.47,48,49 The dataset average disorder scores (AVG scores) were then calculated for human membrane and nonmembrane proteins based on the corresponding per-protein scores, and those AVG scores were used to prepare contingency tables.
The UniProt IDs in the set of human proteins were then used to match the disorder scores to the UniProt IDs of proteins in Russian and KEGG datasets to generate disorder scores for all of the proteins in our datasets. Since the human proteome disorder scores were only retrieved for the SwissProt reviewed human proteins, only the disorder propensities of the reviewed proteins in the Russian dataset were analyzed.
Contingency table constructions, statistical test, and common GO term search
Fisher's exact test was used to find significance of the frequency (number of proteins) that were observed in the prostate cancer protein datasets (Russian and KEGG) to have an average disorder score calculated based on scores all three disorder predictors (PONDR-FIT, PONDR® VSL2, and PONDR® VLXT) greater than or equal to the AVG score of the entire human proteome. These data were used to construct the 2 × 2 contingency tables (Supplementary Table 1 (197KB, tif) represents an example of such a contingency table), so the subsequent statistical analyses can be performed.50 The null (H0) and research hypotheses (H1) shown below for both membrane and nonmembrane subsets of all datasets were tested using α = 0.01 as the statistical significance level.
H0: a/(a + b) ≤ c/(c + d)
H1: a/(a + b) > c/(c + d)
IBM SPSS 22 software (IBM Corporation, USA) was used to perform the Fisher's exact test whereas the R statistical package was used to create figures to present the data.
Similar approach was also used to find GO terms significantly associated with intrinsically disordered proteins in all datasets. Here, after categorization of the protein sets in contingency tables, Fisher's exact test was performed to find GO terms significantly associated with proteins possessing per-protein intrinsic disorder score greater than AVG score in Russian or KEGG datasets separately when those were compared with the whole human proteome. In these studies, membrane and nonmembrane proteins were treated separately. Fisher's exact test P values were retained through Python 3 programming, and its module, scipy.
Next, significant GO terms were analyzed using AmiGo2.51 First, GO terms with P values smaller than 0.05 or 0.1 were obtained for KEGG or Russian dataset versus whole human proteome, separately. Those GO terms were uploaded to visualization server, and the resulting GO term maps were downloaded in the SVG format. These maps included significant GO terms specific for the protein in KEGG or Russian dataset and were used to find the intersection or common GO terms present in both datasets. Those intersection GO terms were analyzed using REVIGO.52
Evaluation of intrinsic disorder propensity and disorder-based functionality
For prostate cancer-related proteins from the KEGG dataset, we analyzed the per-residue disorder propensities by PONDR-FIT, PONDR® VLXT, PONDR® VSL2 algorithms,42,43,44,45,46 and by the PONDR® VL3 predictor that possesses high accuracy in finding long IDPRs.53 We also used a consensus approach MobiDB,54,55 applied a binary disorder classifier charge-hydropathy plot (CH-plot) that evaluates the predisposition of a given protein to be ordered or disordered as a whole,18,56 predicted potential disorder-based binding sites using the ANCHOR algorithm,57,58 looked at the functional disorder using D2P2 database,59 and analyzed interactivity of these proteins by STRING.60
The MobiDB database (http://mobidb.bio.unipd.it/),54,55 that generates consensus disorder scores by aggregating the output from ten predictors, such as two versions of IUPred (IUPred-long and IUPred-short),61 three versions of ESpritz (ESpritz-DisProt, ESpritz-NMR, and ESpritz-XRay),62 two versions of DisEMBL (DisEMBL-465 and DisEMBL-HL),63 JRONN,64 PONDR® VSL2B,46,65 and GlobPlot.66
A CH-plot represents an input protein as a point within the 2D graph where the mean Kate-Doolittle hydrophobicity and the mean absolute net charge are used as the X- and Y-coordinates, respectively. In the corresponding CH-plot, fully structured proteins and fully disordered proteins can be separated by a boundary line. All proteins located above this boundary line are highly likely to be extended whereas proteins located below this line are likely to be compact.18,56
In addition to CH-plot, another binary disorder predictor, cumulative distribution function (CDF) analysis was used.56 It summarizes the per-residue disorder predictions by plotting PONDR scores against their cumulative frequency, which allows ordered and disordered proteins to be distinguished on the basis of the distribution of prediction scores.56 At any given point on the CDF curve, the ordinate gives the proportion of residues with a PONDR score less than or equal to the abscissa. The optimal boundary that provided the most accurate order-disorder classification was shown to represent seven points located in the 12th through 18th bins.56 Thus, in the CDF analysis, order-disorder classification is based on whether a CDF curve of a given protein is above (ordered) or below (disordered) a majority of boundary points.56
Disorder evaluations together with important disorder-related functional information were retrieved from the D2P2 database (http://d2p2.pro/),59 which is a database of predicted disorder for a large library of proteins from completely sequenced genomes.59 D2P2 database uses outputs of IUPred,61 PONDR® VLXT,42 PrDOS,67 PONDR® VSL2B,46,65 PV2,59 and ESpritz.62 The database is further supplemented by data concerning location of various curated posttranslational modifications and predicted disorder-based protein-binding sites.
Additional functional information for these proteins was retrieved using Search Tool for the Retrieval of Interacting Genes; STRING, http://string-db.org/, which based on predicted and experimentally validated information on the interaction partners of a protein of interest generates a network of predicted associations.60 In the corresponding network, the nodes correspond to proteins whereas the edges show predicted or known functional associations. Seven types of evidence are used to build the corresponding network where they are indicated by the differently colored lines: a green line represents neighborhood evidence; a red line - the presence of fusion evidence; a purple line - experimental evidence; a blue line – co-occurrence evidence; a light blue line - database evidence; a yellow line – text mining evidence; and a black line – co-expression evidence.60 In our analysis, the most stringent criteria were used for selection of interacting proteins by choosing the highest cutoff of 0.9 as the minimal required confidence level.
Finally, potential disorder-based protein-binding sites of prostate cancer-related proteins from the KEGG dataset were identified by the ANCHOR algorithm.57,58 This algorithm utilizes the pair-wise energy estimation approach originally used by IUPred.61,68 This approach acts on the hypothesis that long regions of disorder include localized potential-binding sites which are not capable of folding on their own due to not being able to form enough favorable intrachain interactions, but can obtain the energy to stabilize via interaction with a globular protein partner.57,58
Evaluation of interactability of prostate cancer-related proteins in the KEGG dataset
Interactability of human proteins related to prostate cancer from the KEGG dataset was further evaluated by the APID (Agile Protein Interactomes DataServer) web server (http://apid.dep.usal.es).69 APID has information on 90 379 distinct proteins from more than 400 organisms (including Homo sapiens) and on the 678 441 singular protein-protein interactions. For each protein–protein interaction (PPI), the server provides currently reported information about its experimental validation. For each protein, APID unifies PPIs found in five major primary databases of molecular interactions, such as BioGRID,70 Database of Interacting Proteins (DIP),71 Human Protein Reference Database (HPRD),72 IntAct,73 and the Molecular Interaction (MINT) database,74 as well as from the BioPlex (biophysical interactions of ORFeome-based complexes)75 and from the protein databank (PDB) entries of protein complexes.76 This server provides a simple way to evaluate the interactability of individual proteins in a given dataset and also allows researchers to create a specific protein-protein interaction network in which proteins from the query dataset are engaged. Of 88 prostate cancer-related proteins in the KEGG dataset, APID was able to find protein-protein-related information on 87 proteins. No such information was available for the 3-oxo-5-alpha-steroid 4-dehydrogenase 2 (SRD5A2, UniProt ID: P31213).
RESULTS
Abundance of intrinsic disorder in Russian and KEGG datasets of prostate cancer-related proteins
There were 20 120 proteins in the whole human proteome dataset used. This dataset was separated into membrane proteins and nonmembrane proteins:
Membrane proteins: 5193
Nonmembrane proteins: 14 927.
The Russian dataset included 196 unique proteins that were mined from the PCP database. This dataset was then separated into membrane proteins and nonmembrane proteins:
Membrane proteins: 6
Nonmembrane proteins: 190.
The KEGG dataset contained 48 proteins, 4 of which were classified as membrane and remaining 44 as nonmembrane proteins.
Peculiarities of distribution of disorder within the members of these six datasets are shown in Figure 1 in a form of DSFIT vs DSVSL2 plots where DSFIT and DSVSL2 correspond to the mean disorder scores calculated for query proteins using PONDR-FIT and PONDR® VSL2 algorithms, respectively.
Figure 1.
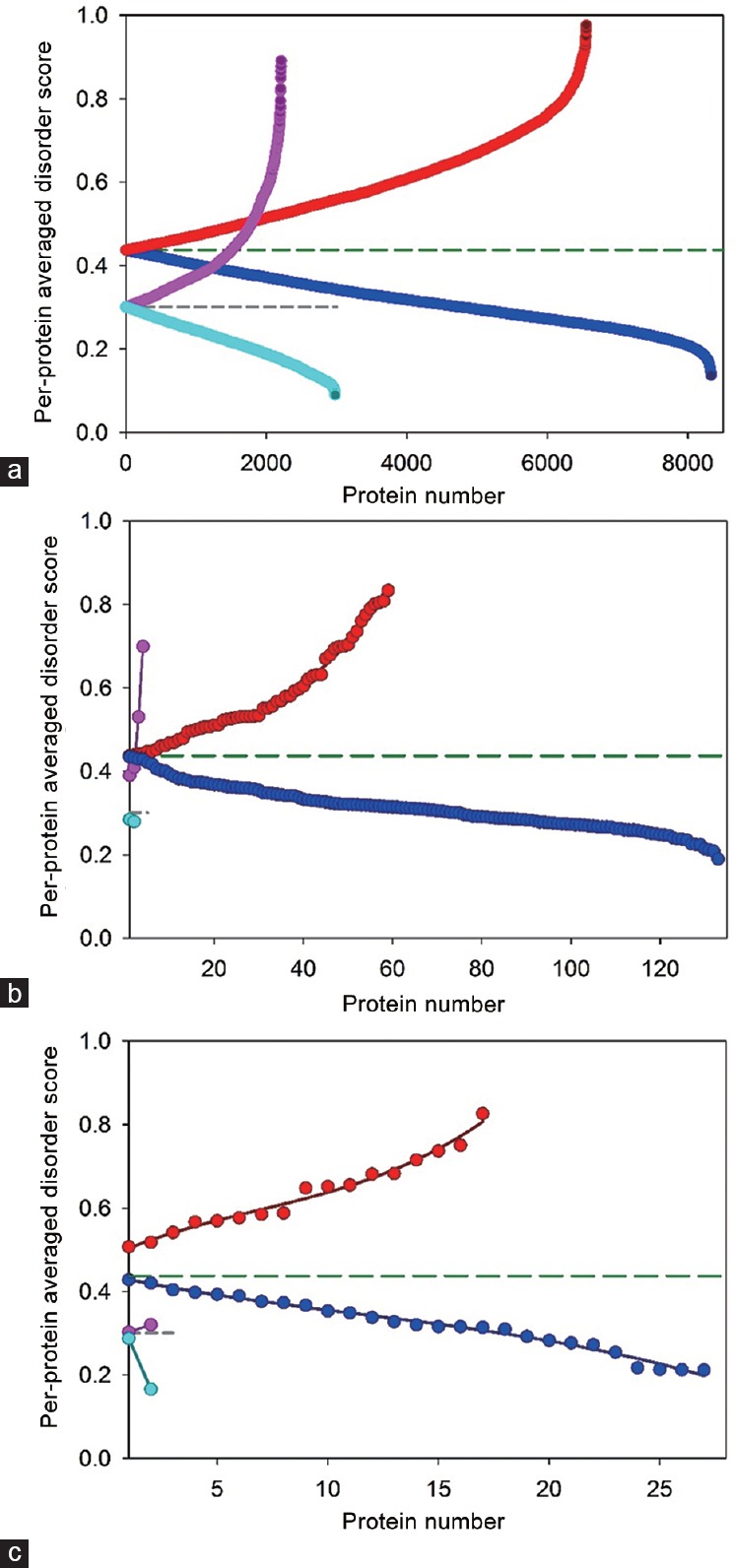
Abundance of intrinsic disorder in six datasets analyzed in this study - (a) human membrane (red and blue circles) and nonmembrane proteins (pink and cyan circles); (b) human membrane (red and blue circles) and nonmembrane prostate cancer-related proteins (pink and cyan circles) from Russian dataset; (c) human membrane (red and blue circles) and nonmembrane prostate cancer-related proteins (pink and cyan circles) from KEGG-dataset. Data for all these sets are shown in a form of proteins with per-protein disorder score above (red and pink circles) or below AVG (blue and cyan circles). AVGs for nonmembrane and membrane proteins are shown as medium dashed dark green and short dashed dark gray lines.
To avoid redundancy in the subsequent statistical analysis of GO terms, the membrane and nonmembrane subsets of the whole human proteome datasets were adjusted to exclude prostate cancer-associated proteins. Supplementary Tables 2 (153.9KB, tif) –5 (153.9KB, tif) show the resulting 2 × 2 contingency tables and the corresponding results are further shown in Figure 2 where they are depicted as a bar-graph.
Figure 2.

Bar graphs presenting data from Supplementary Tables 2 (153.9KB, tif) –5 (153.9KB, tif) . These graphs represent analyzed datasets that were split up into the membrane and nonmembrane groups, in which each bar represents either data for the human proteome, or the Russian (PCP) dataset (a), or KEGG dataset (b) within their respective group. The red section of each bar represents the fraction of proteins that have per-protein average disorder score ≥ AVG and the blue sections represent the fraction of proteins that have per-protein average disorder score < AVG.
2×2 contingency table of the membrane KEGG dataset and membrane proteins in whole human proteome (membrane AVG: 0.301)
2×2 contingency table of the non-membrane KEGG dataset and nonmembrane proteins in whole human proteome (non-membrane AVG: 0.437).
2×2 contingency table of the membrane Russian dataset and membrane proteins in whole human proteome
2×2 contingency table of the non-membrane Russian dataset and nonmembrane proteins in whole human proteome
We recognize that the values of some cells in the 2 × 2 contingency tables (Supplementary Materials (127.5KB, pdf) ) are rather low, indicating that it is too risky to draw any solid statistical conclusions when there are not enough samples, especially when comparison is done between very small and very large samples. Furthermore, disorder propensities of proteins analyzed in this study were evaluated using a set of standard disorder predictors that are characterized by the accuracy of 80%–85%. This indicates that the confidence of conclusions outlined below (especially for the cases having very limited samples) is further influenced by the limited accuracy of predictors. Therefore, data presented below should be taken only as indication of potential tendencies in the disorder predisposition and not as the final statistically significant conclusions.
Characterization of human proteins involved in the prostate cancer pathway
The average disorder scores between all three predictors (PONDR® VSL2, PONDR® VLXT, and PONDR-FIT) were calculated for nonadjusted subsets of membrane and nonmembrane proteins in whole human proteome. These average disorder scores of 0.301 and 0.437 for the membrane and nonmembrane dataset, respectively, were then used to perform the statistical analysis of the proportion of the disordered proteins in the KEGG and Russian datasets compared to the proportion of the disordered proteins in the entire human proteome.
The one-sided Fisher's exact test statistic value for the membrane proteins in Russian dataset is 0.363, and therefore, H0 cannot be rejected at the significance level of α = 0.01. This means that in the membrane subset of Russian dataset, the proportion of proteins that have per-protein disorder scores ≥ to the AVG score is not significantly different from human membrane proteins that have per-protein disorder scores ≥ to the AVG score.
On the other hand, the one-sided Fisher's exact test statistic value for the nonmembrane proteins from the Russian dataset is <0.001. Therefore, we reject H0 at the significance level of α = 0.01. From these data, we conclude that the fraction of the nonmembrane proteins that have per-protein disorder scores exceeding the AVG score is significantly higher in the Russian dataset of prostate cancer-associated proteins than in the human proteome. This indicates that proteins with high intrinsic disorder levels are found significantly more often in the nonmembrane Russian dataset than in the whole human proteome, suggesting that higher abundance of IDPs/IDPRs can be related to cancer development. This observation is in agreement with the results of earlier studies on the high abundance of intrinsic disorder in cancer-related proteins.35
For the KEGG membrane proteins versus dataset of human membrane proteins, we cannot reject the H0 hypothesis at α = 0.05. Therefore, there is no statistically significant difference between the proportions of KEGG membrane proteins with the per-protein disorder scores ≥ AVG and human membrane proteins with the per-protein disorder scores ≥ AVG. Similarly, for the KEGG nonmembrane proteins versus human nonmembrane proteins, we also cannot reject the Ho hypothesis at α = 0.05. Although this analysis revealed that the intrinsic disorder propensities of the prostate cancer-related proteins in the KEGG dataset are not significantly different from the disorder predispositions of proteins in human proteome, we conducted more detailed disorder-oriented analysis of proteins from the extended KEGG dataset to illustrate peculiarities of disorder distribution in these proteins and to see how IDPRs can be related to function and pathology (see Supplementary Materials (127.5KB, pdf) ).
Of the 17 potential biomarkers of prostate cancer outlined in Shishkin et al. paper,38 only 12 were included in the reviewed protein dataset analyzed in our study. All of these 12 potential biomarkers are in the nonmembrane subset members of which are characterized by the per-protein disorder scores below the AVG disorder score. Furthermore, the well-known prostate cancer biomarker PSA was also present in our set of nonmembrane prostate cancer-related proteins characterized by the disorder scores below the AVG disorder score of human nonmembrane proteins. These observations suggest that potential biomarkers of prostate cancer are characterized by lower disorder levels than an average human protein.
Finding GO terms significantly associated with intrinsically disordered prostate cancer-related proteins in the KEGG and Russian datasets
GO terms are based on three structured ontologies that are designed for consistent functional descriptions of proteins in a species-independent manner. These terms show the relations of the query proteins to the biological processes they are involved in, the molecular functions they conduct, and the cellular components where they can be found at. Obviously, because GO terms are specifically designed as general terms of functional classification of proteins, these terms are applicable to any annotated protein (not only to proteins found in various pathologies but also to the normally functioning proteins). However, GO terms can also be used to find a correlation between protein intrinsic disorder and functionality. In fact, it has been shown in several previous studies that some GO terms are preferentially found to be associated with IDPs whereas other GO terms would be more suitable for characterization of ordered proteins. Our analysis was conducted to show that many of the GO terms ascribed to the IDPs associated with prostate cancer describe disorder-related functions. This observation reemphasizes the importance of intrinsic disorder for these proteins.
GO term analysis using AmiGo251 did not produce noticeable number of disorder-associated GO terms when the P value threshold of less than 0.05 was used. Since we wanted to see which functions can be assigned to the intrinsically disordered proteins related to prostate cancer, we decided to use a loosen P value threshold of less than 0.1 to find disorder associated GO terms (Supplementary Table 6 (1.4MB, tif) ). One should keep in mind that although the use of the loosen P value (P < 0.1) does not provide statistically significant data, the corresponding analysis generates a notable statistical trend that can be used to look for weakly significant correlations between intrinsic disorder and function. Among those GO terms with tentatively significant correlation to intrinsic disorder, four process-oriented GO terms – GO: 0006915 (apoptotic process), GO: 0008219 (cell death), GO: 0012501 (programmed cell death), and GO: 0016265 (death) – were related with programmed cell death through REVIGO52 (Supplementary Figure 1a (515.2KB, pdf) ). Moreover, those four chosen GO terms showed a simple map through AmiGo2 visualize tool (Supplementary Figure 1b (515.2KB, pdf) ).
Prostate cancer-related proteins from the KEGG and Russian datasets with disorder-associated common Gene Ontology terms
Functions of intrinsically disordered proteins related to prostate cancer. (a) REVIGO analysis of the cellular process GO terms with P < 0.1. (b) AmiGo2 result for all GO terms with P value less than 0.1. Highlighted are the programmed cell death-related cellular process GO terms.
Bioinformatics analysis of the prostate cancer-related proteins in the KEGG dataset
To gain information on the disorder status of prostate cancer-related proteins in the KEGG dataset and on the potential functional roles of their predicted IDPRs, we looked at them using a set of disorder predictors of PONDR family (PONDR-FIT, PONDR® VSL2, PONDR® VL3, and PONDR® VLXT), a binary disorder predictor CH-plot, a consensus disorder evaluating internet tools MobiDB and D2P2, a platform STRING for finding potential interaction partners of a protein of interest, and a tool for predicting the disorder-based binding sites (ANCHOR). Some of these results are summarized in Supplementary Table 7 (259.9KB, pdf) and Figure 3.
Figure 3.
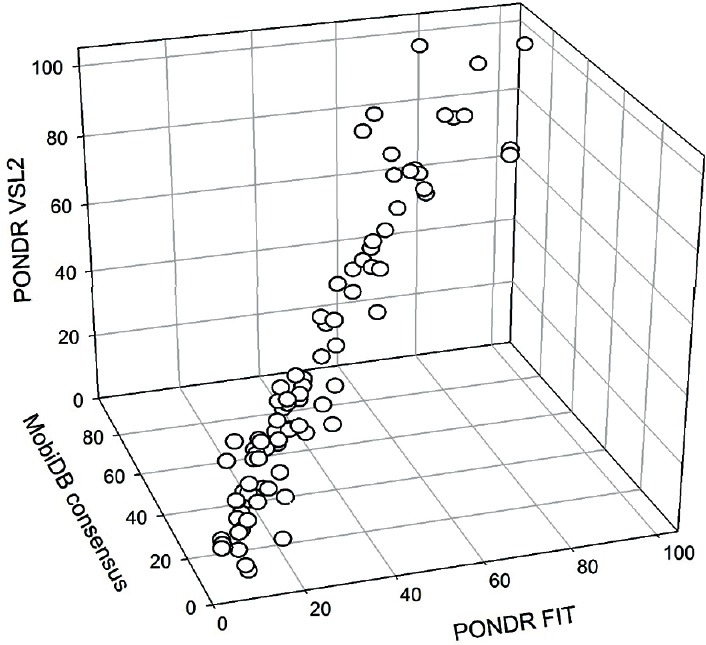
3D representation of the results of evaluation of disorder levels in human prostate cancer-related proteins from the extended KEGG dataset. Here, the percentages of residues in these proteins predicted to be disordered by PONDR-FIT and PONDR® VSL2 are compared with the predicted percentages of disordered residues predicted by the MobiDB platform that aggregates the output from ten disorder predictors. The overall goal of this plot is to show the overall agreement between the outputs of different disorder predictors used in this study.
Characterization of human proteins involved in the prostate cancer pathway
Figure 3 illustrates that disorder predictions generated for these KEGG proteins by PONDR-FIT, PONDR® VSL2, and MobiDB in a form of per-protein disorder scores (DSs) or the percentage of disordered residues (per-protein content of disordered residues, CDRs, for PONDR-FIT and MobiDB) are generally agreed. Furthermore, from data shown in Supplementary Table 7 (259.9KB, pdf) , it is clearly seen that many proteins in this dataset are predicted to be moderately or highly disordered using the classification of proteins as highly ordered, moderately disordered, or highly disordered, if their CDR <10%, 10% ≤ CDR <30%, and CDR ≥30%, respectively.77 In fact, according to these criteria, the KEGG dataset includes 34 (38.6%), 38 (43.2%), and 16 (18.2%) highly disordered, moderately disordered, and highly ordered proteins, respectively. This suggests that almost 82% of prostate cancer-related proteins in this dataset are very noticeably disordered.
Supplementary Table 7 (259.9KB, pdf) shows that although the disorder propensities of these proteins are spread over a wide range (e.g., by PONDR-FIT, from 3% of disordered residues in PI3K-α to 100% of such residues in BAD), the vast majority of them possess sizable IDPRs containing at least 10 consecutive residues predicted to be disordered, with many of these proteins having several such regions. In fact, only eleven proteins from the KEGG dataset were shown not to have such regions, and the remaining 77 proteins possessed 293 IDPRs (i.e., on average, each of these 77 proteins is expected to contain 3.81 such regions).
Often, IDPRs contain local regions with a strong tendency to become ordered at interaction with specific binding partners. Therefore, these regions might undergo coupled folding and binding, as shown for many of them by the NMR studies.78,79,80,81,82,83 Furthermore, such local short segments of order located within long disordered regions were shown to often coincide with the potential-binding sites.84 Therefore, a number of computational tools for finding such molecular recognition features (MoRFs) were developed (e.g., a tool for predicting short binding regions with high α-helix-forming propensity, α-MoRFs85,86 or the more general ANCHOR algorithm for finding potential disorder-based binding sites,57,58 which are termed below AIBS for ANCHOR-identified binding sites). Curiously, in earlier studies, a systematic application of such computational tools indicated that α-MoRFs are likely to play important roles in protein-protein interactions involved in signaling events.85 Our analysis revealed that the majority of proteins in the KEGG dataset (71 of 88) are predicted to have at least one AIBS, and that many of these proteins are expected to have multiple such binding regions each (Supplementary Table 7 (259.9KB, pdf) ). Furthermore, AIBSs are found in almost each KEGG proteins that have at least one disordered region, and many of these proteins are shown to contain multiple disorder-based binding sites, with 34 and 30 AIBSs being found in CREB-binding protein (Q92793) and histone acetyltransferase p300 (Q09472), respectively. In fact, we found 472 AIBSs in 71 human proteins associated with prostate cancer, suggesting that on average, each of these protein contains >6.6 disorder-based binding sites. Supplementary Table 7 (259.9KB, pdf) also shows that the length of AIBSs is ranging from 6 to 150 residues, and the overall content of residues involved in the disorder-based interactions ranges from 0% to 70.8%.
The presence of more than one AIBS in a protein suggests that many prostate cancer-related proteins in the KEGG dataset commonly utilize disorder for their interactions with binding partners, and that these proteins are involved either in the polyvalent interactions using multiple binding sites to interact with one binding partner or in scaffolding-like interactions using multiple binding sites to interact with multiple binding partners. The wide spread of lengths of identified AIBSs also suggests the presence of multiple disorder-based binding mechanisms (ranging from local folding-on-binding of short regions to wrapping around binding mode to global binding-induced folding of large regions).
We established that many of the proteins in the KEGG dataset are predicted to have noticeable amounts of intrinsic disorder. Furthermore, this analysis revealed that 14 of these proteins (P05019, P14625, P36402, P46527, Q00987, Q92934, Q99801, Q9UJU2, P38936, P07900, Q92569, P08238, Q9Y6K9, and Q02930) are expected to be disordered as a whole according to the CH-plot, i.e., are located above the boundary line separating compact and extended disordered proteins, and 16 more of these proteins, being located below this boundary, are found in its proximity (P04085, P04637, P25963, P60484, Q09472, Q92793, Q9NQB0, P62993, Q9Y243, P01127, Q9GZP0, P27986, O00459, Q9HCS4, Q96BA8, and P18848). Analogous analysis using another binary predictor, cumulative distribution function (CDF) plot (where proteins expected be disordered or ordered as a whole are found based on the position of their CDF curve relative to the boundary separating mostly ordered and disordered proteins), revealed that 39 are expected to be mostly disordered (O00716, P04085, P04637, P05019, P06400, P10275, P10415, P16220, P25963, P36402, P46527, P49841, Q00987, Q01094, Q07889, Q09472, Q12778, Q14209, Q92793, Q92934, Q99801, Q9NQB0, Q8WYR1, P38936, O43889, Q9UJU2, P01127, O00459, P15056, Q07890, Q04206, Q9Y6K9, Q9HCS4, Q02930, Q8TEY5, Q68CJ9, Q70SY1, Q96BA8, and P18848) since the majority of their CDF curves are located below the boundary, whereas the CDF curves of 11 additional proteins (O14920, P01308, P10398, P14625, P24864, P36507, Q02750, P07900, O15530, P27986, and Q92569) follows boundary almost exactly, suggesting that these proteins are definitely not ordered as whole, as their overall disorder status is “undecided”. It was pointed out that combined analysis of protein disorder status using CH-plot and CDF analysis simultaneously can provide additional important information on the classification of protein disorder. This combined approach is known as the CH-CDF analysis,87,88,89 and it is based on the presence of a principle difference between the sensitivity of the CH-plot and the CDF analysis to different types of disorder. Here, the CH-plot can discriminate proteins with substantial amount of extended disorder (random coils and pre-molten globules) from proteins with compact conformations (molten globule-like and rigid well-structured proteins) whereas the CDF analysis may discriminate all disordered conformations, including molten globules and mixed proteins containing both disordered and ordered regions, from rigid well-folded proteins. Therefore, the CH-CDF analysis represents a computational tool to discriminate proteins with extended disorder from potential molten globules and mixed proteins containing comparable amounts of ordered and disordered regions.87,88,89 Therefore, based on the combination of outputs of their CH-plot and CDF analyses, proteins can be classified as follows: proteins predicted to be disordered by CH-plots, but ordered by CDF; ordered proteins (i.e., proteins predicted as ordered by both tools); putative molten globules or mixed proteins (i.e., proteins predicted to be disordered by CDF, but compact by CH-plot); and proteins with extended disorder (i.e., proteins predicted to be disordered by both methods).87,88,89 Based on this classification, there are 13 prostate cancer-associated proteins with extended disorder (P05019, P14625, P36402, P46527, Q00987, Q92934, Q99801, Q9UJU2, P38936, P07900, Q92569, Q9Y6K9, and Q02930) and there are at least 29 putative native molten globules or mixed proteins containing comparable amounts of ordered and disordered regions (O00716, P04085, P04637, P06400, P10275, P10415, P16220, P25963, P49841, Q01094, Q07889, Q09472, Q12778, Q14209, Q92793, Q9NQB0, Q8WYR1, O43889, P01127, O00459, P15056, Q07890, Q04206, Q9HCS4, Q8TEY5, Q68CJ9, Q70SY1, Q96BA8, and P18848).
Furthermore, our analysis not only indicated the disorder status of these 88 proteins but also showed that disorder is crucial for functionality of many of the prostate cancer-related proteins. This conclusion follows from the analysis of the data generated for each protein in the KEGG dataset by the D2P2 that, in the visually attractive form, provides an access to the precomputed disorder predictions59 generated by PONDR® VLXT,42 IUPred,61 PONDR® VSL2B,46,65 PrDOS,67 ESpritz,62 and PV259 and also provides information on the curated cites of various posttranslational modifications and on the location of predicted potential disorder-based binding sites. Our analysis clearly shows that many of human prostate cancer-related proteins from the KEGG dataset are predicted to have disordered regions of various lengths, often possess numerous potential disorder-based binding motifs (Supplementary Table 7 (259.9KB, pdf) ) and contain multiple sites of various posttranslational modifications (PTMs). The finding that the IDPRs of these proteins have a multitude of PTMs is in agreement with the well-known fact that phosphorylation23 and many other enzymatically catalyzed PTMs are preferentially located within the IDPRs.23,24,25,26
The interactivity of prostate cancer-associated proteins from the KEGG dataset was further evaluated by the STRING computational platform that provides information on both experimentally validated and predicted interactions of query proteins.60 The corresponding STRING-generated protein-protein interaction (PPI) networks of these proteins (data not shown) indicate that all KEGG proteins are predicted to have very well-developed interactomes. This observation suggests that these proteins can serve as hubs in their functional PPI networks. These findings are in accord with earlier observations that intrinsic disorder plays an important role in functionality of hubs, defining their ability to be promiscuous binders engaged in interactions with a multitude of often unrelated partners.90,91,92,93,94,95,96 Furthermore, previous studies showed that many hubs are intrinsically disordered or contain functional IDPRs, and that the partners of ordered hubs are preferentially intrinsically disordered.90,91,92,93,94,95,96 It is likely that this binding promiscuity of the prostate cancer-related proteins can be at least partially attributed to the fact that many of them have numerous AIBSs. Also, this astonishing capability of mostly ordered, hybrid, and mostly disordered proteins from the KEGG dataset to be heavily connected hubs represents a major hurdle for the development of drugs targeting these proteins.
DISCUSSION
Interaction network of prostate cancer-related proteins
Results of the application of the APID server for evaluation of the interactivity of the 87 prostate cancer-related proteins from the KEGG dataset (as it was mentioned, no protein-protein interaction-related information was available for the 3-oxo-5-alpha-steroid 4-dehydrogenase 2 [SRD5A2, UniProt ID: P31213]) clearly showed that each of these proteins is known to be engaged in multiple protein-protein interactions (PPIs) (Supplementary Table 7 (259.9KB, pdf) ). In fact, the number of PPIs ranges from 4 (TCF7L1 and INSRR) to >1,000 (e.g., p53, a 393-residue-long protein for which the content of disordered residues predicted PONDR FIT (CDRFIT) is 54.96%, and EGFR with the length of 1021 residues and CDRFIT of 9.1%, interact with 1072 and 1031 partners, respectively). In fact, 81 proteins in this dataset are able to interact with more than 10 partners each (Supplementary Table 7 (259.9KB, pdf) ). This observation suggests that the vast majority of the prostate cancer-related proteins from the KEGG dataset can be considered as hub proteins. Figure 4 shows that the interactivity of these proteins is not correlated with their length (Figure 4a) or their intrinsic disorder content (Figure 4b), suggesting that these two features do not directly determine the ability of a prostate cancer-related protein to be involved in many PPIs and to serve as a hub. The aforementioned lack of a correlation between the interactability of a given protein and its disorder content seems to be in contradiction with the claim that the ability of a protein to be engaged in many PPIs relies on intrinsic disorder. However, it is known that some hub proteins can be entirely disordered, other hubs may contain both ordered and disordered regions, and still other hubs can be highly structured.97 However, the binding regions of the partner proteins of ordered hubs were found to be intrinsically disordered.98,99 These observations suggested two primary mechanisms by which disorder is utilized in protein-protein interaction networks, namely, one disordered region binding to many partners and many disordered region binding to one partner.91,92,93,94,97,100,101
Figure 4.
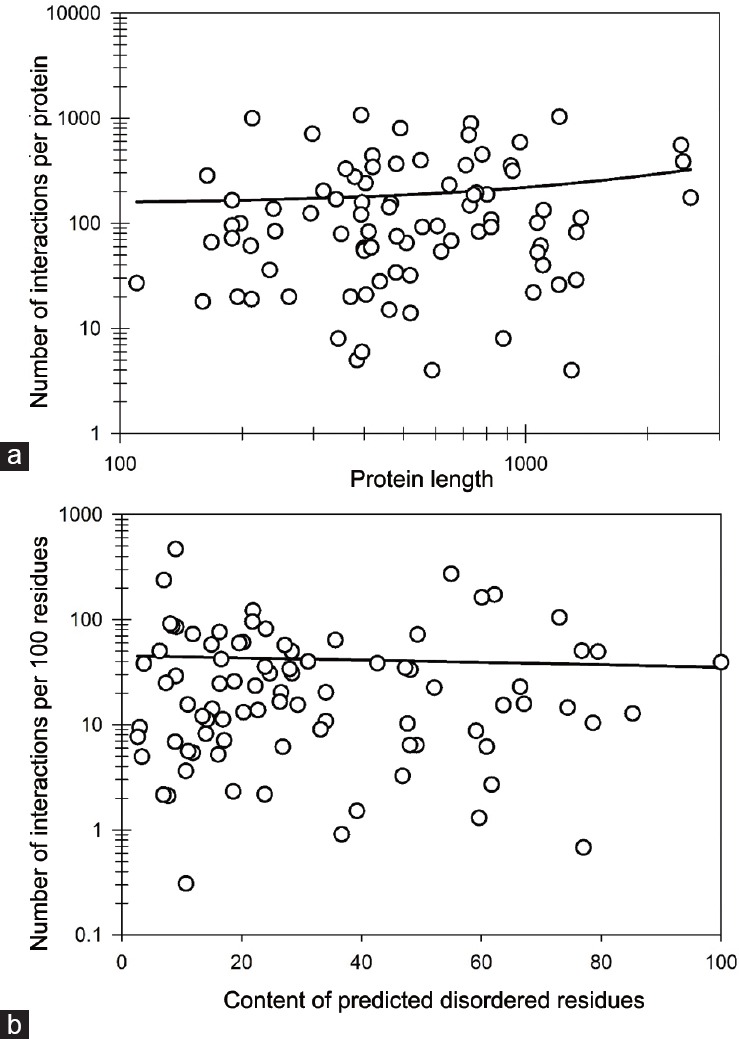
Characterization of the interactability of prostate cancer-related proteins from the extended KEGG dataset based on the results of their analysis by APID server. (a) Correlation between the number of PPIs found by APID for individual proteins and protein length. Note logarithmic scale of this plot. (b) Correlation between interactability (evaluated as the number of PPIs per 100 residues of a given protein) and intrinsic disorder (measured as PONDR FIT-based content of predicted disordered residues in a query protein). Note semi-logarithmic scale of this plot.
As it follows from brief description of several highly disordered proteins related to prostate cancer, it is not uncommon to find them to be involved in interaction with each other. To understand how common this phenomenon is, we used the ability of the APID web server (http://apid.dep.usal.es) to build a specific PPI network between proteins included in a query list.69 Figure 5 represents the results of application of this tool to prostate cancer-related proteins from the KEGG dataset and shows that all proteins with known interactions are involved in the formation of a common interactive cluster, where each prostate cancer-related protein interacts with at least one other prostate cancer-related protein. Figure 5 shows results of this analysis in a form of a grid, where each node corresponds to a protein from the KEGG dataset, and where PPIs are shown as corresponding edges, thickness of which reflects the reliability of a given interaction. The resulting prostate cancer-related interactome clearly shows that almost all proteins currently known to be related to the pathogenesis of this disease are talking to each other. Therefore, both internal (interactions with other prostate cancer-related proteins) and external connectivities (interaction with other proteins) are high for many prostate cancer-related proteins.
Figure 5.

Evaluation of the inter-set interactivity of prostate cancer-related proteins from the KEGG dataset using the APID web server (http://apid.dep.usal.es). This tool builds a PPI network between proteins included in a query list. Results are shown in a form of a grid, where each node corresponds to a protein from the KEGG dataset, and where PPIs are shown as corresponding edges, thickness of which reflects the reliability of a given interaction.
Functionality of intrinsic disorder in a major player, androgen receptor
Androgen receptor (AR, CDRFIT is 42.7%) is one of the steroid hormone receptors. AR is a 920 residue-long ligand-activated transcription factor controlling expression of various eukaryotic genes and affecting proliferation and differentiation of cells in target tissues. AR plays a key role in the development and progression of prostate cancer and contributes to the development of resistance to androgen deprivation leading to the formation of the castration-resistant form of prostate cancer.102 There are several functional regions/domains in the AR. For example, the most AR transcriptional activity is due to the N-terminal domain (NTD) transcriptional activation domain whereas interaction with DNA is attributed to the central DNA-binding domain that contains 2 zinc-finger motifs. Nuclear localization upon activation is driven by a short, flexible, hinge located after the DNA-binding domain. Finally, interaction of AR with ligands is conducted through its C-terminal ligand-binding domain (LBD).
Structural information is available for the LBD (residues 659–920) and the N-terminal peptide, AR20–30.103 Analysis of the solution structure of AR NTD (residues 1–537) by circular dichroism revealed that this domain has a relatively limited amount of stable secondary structure.104 The lack of stable structure in NTD is further supported by the results of evaluation of disorder predisposition in this protein as shown in Supplementary Table 7 (259.9KB, pdf) . As typical for IDPs and hybrid proteins containing ordered domains and IDPRs, AR has several regions with compositional biases, such as Gln-rich region (residues 58–120), poly-Gln regions (residues 58–80, 86–91, and 195–199), a poly-Pro region (residues 374–383), and poly-Ala and poly-Gly regions (residues 398–404 and 451–473, respectively). There are four alternatively spliced isoforms of this protein, where in comparison with the canonical form, isoform 2 misses residues 1–532 and has a GPYGDMR → MILWLHS substitution at position 533–539; isoform 3 has a substitution ARKLKKLGNLKLQEEG → EKFRVGNCKHLKMTRP (residues 629–644) and misses region 645–920; finally, in the isoform 4, residues 649–920 are missing and region 630–648 has a RKLKKLGNLKLQEEGEASS → AVVVSERILRVFGVSEWLP substitution. As follows from our analysis, the majority of regions affected by alternative splicing is predicted to be disordered.
CONCLUSIONS
This study shows that intrinsically disordered proteins and proteins with long disordered regions are commonly found in prostate cancer. Many of these proteins are predicted to be moderately or highly disordered using either the per-protein disorder score (DS) for classification of proteins as highly ordered (DS <0.1), moderately disordered (0.1≤ DS <0.3), and highly disordered (DS ≥0.3) or looking at the per-protein content of disordered residues (CDRs), with proteins being classified as highly ordered, moderately disordered, or highly disordered, if their CDR <10%, 10% ≤ CDR <30%, and CDR ≥30%, respectively. Functions of these proteins are regulated by various posttranslational modifications. Furthermore, many of these proteins are promiscuous binders and contain numerous disorder-based binding sites. We also show that irrespectively of their disorder status (i.e., irrespectively of being mostly ordered, mixed, or mostly disordered), these proteins are characterized by an astonishing capability to be heavily connected hubs involved in a broad range of interactions. This binding promiscuity might represent a major hurdle for the development of drugs targeting these proteins.
Although our analysis revealed that potential biomarkers of prostate cancer are characterized by lower disorder levels than an average human protein, in our view, there is no contradiction between these observations and our discussion of the fact that IDPs and proteins with long disordered regions are commonly found in prostate cancer, where they might play a number of important roles. In fact, what we are showing is that intrinsically disordered regions are commonly present among prostate cancer-related proteins. Furthermore, the fact that disorder might be more commonly found in the whole human proteome than in a set of proteins related to prostate cancer does not mean that IDPs are not important for progression of this disease. In addition to the information on how many IDPs are associated with prostate cancer, a very important consideration is who these cancer-related IDPs are and what they do. For example, AR, PTEN, and p53 are known as major players in prostate cancer development (and, as a matter of fact, PTEN and p53 are involved in the development of many other pathological conditions). Loss of function of these three proteins (all of which have long disordered regions) contributes greatly to disease progression. Another example is given by NKX3.1, which is also a very disordered protein, whose loss is associated with the prostate cancer development. Therefore, we think that our data make an important contribution to the field by bringing attention to IDPs potentially related to prostate cancer.
AUTHOR CONTRIBUTIONS
KSL collected datasets, conducted computational analysis, participated in data analysis, and drafted the manuscript. IN participated in collecting datasets, conducted computational analysis, participated in data analysis, conducted statistical analysis, and drafted the manuscript. ROS participated in data analysis and contributed to drafting the manuscript. VNU conceived the idea of the study, designed and coordinated experiments, analyzed data, conducted computational analysis, and drafted the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declared lack of any competing financial interest.
Supplementary information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Zhu Y, Han CT, Zhang GM, Liu F, Ding Q, et al. Effect of body mass index on the performance characteristics of PSA-related markers to detect prostate cancer. Sci Rep. 2016;6:19034. doi: 10.1038/srep19034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uversky VN. Dancing protein clouds: the strange biology and chaotic physics of intrinsically disordered proteins. J Biol Chem. 2016;29:6681–8. doi: 10.1074/jbc.R115.685859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Habchi J, Tompa P, Longhi S, Uversky VN. Introducing protein intrinsic disorder. Chem Rev. 2014;114:6561–88. doi: 10.1021/cr400514h. [DOI] [PubMed] [Google Scholar]
- 6.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–71. [PubMed] [Google Scholar]
- 7.Peng Z, Yan J, Fan X, Mizianty MJ, Xue B, et al. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol Life Sci. 2015;72:137–51. doi: 10.1007/s00018-014-1661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–45. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30:137–49. doi: 10.1080/07391102.2012.675145. [DOI] [PubMed] [Google Scholar]
- 10.Tompa P, Dosztanyi Z, Simon I. Prevalent structural disorder in E. coli and S. cerevisiae proteomes. J Proteome Res. 2006;5:1996–2000. doi: 10.1021/pr0600881. [DOI] [PubMed] [Google Scholar]
- 11.Tompa P, Davey NE, Gibson TJ, Babu MM. A million peptide motifs for the molecular biologist. Mol Cell. 2014;55:161–9. doi: 10.1016/j.molcel.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 12.Vincent M, Whidden M, Schnell S. Quantitative proteome-based guidelines for intrinsic disorder characterization. Biophys Chem. 2016;213:6–16. doi: 10.1016/j.bpc.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Charon J, Theil S, Nicaise V, Michon T. Protein intrinsic disorder within the Potyvirus genus: from proteome-wide analysis to functional annotation. Mol Biosyst. 2016;12:634–52. doi: 10.1039/c5mb00677e. [DOI] [PubMed] [Google Scholar]
- 14.Skupien-Rabian B, Jankowska U, Swiderska B, Lukasiewicz S, Ryszawy D, et al. Proteomic and bioinformatic analysis of a nuclear intrinsically disordered proteome. J Proteomics. 2016;130:76–84. doi: 10.1016/j.jprot.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Pancsa R, Tompa P. Structural disorder in eukaryotes. PLoS One. 2012;7:e34687. doi: 10.1371/journal.pone.0034687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 17.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, et al. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–56. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–27. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 19.Uversky VN. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta. 2013;1834:932–51. doi: 10.1016/j.bbapap.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Uversky VN. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 2013;22:693–724. doi: 10.1002/pro.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uversky VN. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev. 2011;40:1623–34. doi: 10.1039/c0cs00057d. [DOI] [PubMed] [Google Scholar]
- 22.Uversky VN. Intrinsic disorder-based protein interactions and their modulators. Curr Pharm Des. 2013;19:4191–213. doi: 10.2174/1381612811319230005. [DOI] [PubMed] [Google Scholar]
- 23.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–49. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pejaver V, Hsu WL, Xin F, Dunker AK, Uversky VN, et al. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 2014;23:1077–93. doi: 10.1002/pro.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zerze GH, Mittal J. Effect of O-linked glycosylation on the equilibrium structural ensemble of intrinsically disordered polypeptides. J Phys Chem B. 2015;119:15583–92. doi: 10.1021/acs.jpcb.5b10022. [DOI] [PubMed] [Google Scholar]
- 26.Kurotani A, Sakurai T. In silico analysis of correlations between protein disorder and post-translational modifications in algae. Int J Mol Sci. 2015;16:19812–35. doi: 10.3390/ijms160819812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–82. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 28.Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Protein Chem. 2002;62:25–49. doi: 10.1016/s0065-3233(02)62004-2. [DOI] [PubMed] [Google Scholar]
- 29.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–56. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 31.Uversky VN. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell Mol Life Sci. 2003;60:1852–71. doi: 10.1007/s00018-003-3096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tompa P, Schad E, Tantos A, Kalmar L. Intrinsically disordered proteins: emerging interaction specialists. Curr Opin Struct Biol. 2015;35:49–59. doi: 10.1016/j.sbi.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 33.Tompa P, Szasz C, Buday L. Structural disorder throws new light on moonlighting. Trends Biochem Sci. 2005;30:484–9. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 34.Meng F, Na I, Kurgan L, Uversky VN. Compartmentalization and functionality of nuclear disorder: intrinsic disorder and protein-protein interactions in intra-nuclear compartments. Int J Mol Sci. 2015;17:24. doi: 10.3390/ijms17010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–84. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 36.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 37.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32:D115–9. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shishkin SS, Kovalyov LI, Kovalyova MA, Lisitskaya KV, Eremina LS, et al. “Prostate cancer proteomics” database. Acta Naturae. 2010;2:95–104. [PMC free article] [PubMed] [Google Scholar]
- 39.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–60. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Binns D, Dimmer E, Huntley R, Barrell D, O’Donovan C, et al. QuickGO: a web-based tool for gene ontology searching. Bioinformatics. 2009;25:3045–6. doi: 10.1093/bioinformatics/btp536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huntley RP, Sawford T, Mutowo-Meullenet P, Shypitsyna A, Bonilla C, et al. The GOA database: gene Ontology annotation updates for 2015. Nucleic Acids Res. 2015;43:D1057–63. doi: 10.1093/nar/gku1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, et al. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform Ser Workshop Genome Inform. 1999;10:30–40. [PubMed] [Google Scholar]
- 44.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, et al. Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol. 2005;3:35–60. doi: 10.1142/s0219720005000886. [DOI] [PubMed] [Google Scholar]
- 46.Obradovic Z, Peng K, Vucetic S, Radivojac P, Dunker AK. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005;61(Suppl 7):176–82. doi: 10.1002/prot.20735. [DOI] [PubMed] [Google Scholar]
- 47.Walsh I, Giollo M, Di Domenico T, Ferrari C, Zimmermann O, et al. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics. 2015;31:201–8. doi: 10.1093/bioinformatics/btu625. [DOI] [PubMed] [Google Scholar]
- 48.Fan X, Kurgan L. Accurate prediction of disorder in protein chains with a comprehensive and empirically designed consensus. J Biomol Struct Dyn. 2014;32:448–64. doi: 10.1080/07391102.2013.775969. [DOI] [PubMed] [Google Scholar]
- 49.Peng Z, Kurgan L. On the complementarity of the consensus-based disorder prediction. Pac Symp Biocomput. 2012:176–87. [PubMed] [Google Scholar]
- 50.Pietrosemoli N, Garcia-Martin JA, Solano R, Pazos F. Genome-wide analysis of protein disorder in Arabidopsis thaliana: implications for plant environmental adaptation. PLoS one. 2013;8:e55524. doi: 10.1371/journal.pone.0055524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, et al. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–9. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS one. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Domenico T, Walsh I, Martin AJ, Tosatto SC. MobiDB: a comprehensive database of intrinsic protein disorder annotations. Bioinformatics. 2012;28:2080–1. doi: 10.1093/bioinformatics/bts327. [DOI] [PubMed] [Google Scholar]
- 55.Potenza E, Domenico TD, Walsh I, Tosatto SC. MobiDB 2.0: an improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res. 2015;43(Database issue):D315–20. doi: 10.1093/nar/gku982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, et al. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 57.Meszaros B, Simon I, Dosztanyi Z. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol. 2009;5:e1000376. doi: 10.1371/journal.pcbi.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dosztanyi Z, Meszaros B, Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–6. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, et al. D (2) P (2): database of disordered protein predictions. Nucleic Acids Res. 2013;41:D508–16. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–8. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–4. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 62.Walsh I, Martin AJ, Di Domenico T, Tosatto SC. ESpritz: accurate and fast prediction of protein disorder. Bioinformatics. 2012;28:503–9. doi: 10.1093/bioinformatics/btr682. [DOI] [PubMed] [Google Scholar]
- 63.Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, et al. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–9. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 64.Yang ZR, Thomson R, McNeil P, Esnouf RM. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics. 2005;21:3369–76. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
- 65.Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31:3701–8. doi: 10.1093/nar/gkg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ishida T, Kinoshita K. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007;35:W460–4. doi: 10.1093/nar/gkm363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dosztanyi Z, Csizmok V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 2005;347:827–39. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 69.Alonso-Lopez D, Gutierrez MA, Lopes KP, Prieto C, Santamaria R, et al. APID interactomes: providing proteome-based interactomes with controlled quality for multiple species and derived networks. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw363. pii: gkw363. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015;43:D470–8. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, et al. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–51. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, et al. Human protein reference database-2009 update. Nucleic Acids Res. 2009;37:D767–72. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012;40:D841–6. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40:D857–61. doi: 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, et al. The BioPlex network: a systematic exploration of the human interactome. Cell. 2015;162:425–40. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rose PW, Prlic A, Bi C, Bluhm WF, Christie CH, et al. The RCSB protein data bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015;43:D345–56. doi: 10.1093/nar/gku1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rajagopalan K, Mooney SM, Parekh N, Getzenberg RH, Kulkarni P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem. 2011;112:3256–67. doi: 10.1002/jcb.23252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bienkiewicz EA, Adkins JN, Lumb KJ. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1) Biochemistry. 2002;41:752–9. doi: 10.1021/bi015763t. [DOI] [PubMed] [Google Scholar]
- 79.Chi SW, Kim DH, Lee SH, Chang I, Han KH. Pre-structured motifs in the natively unstructured preS1 surface antigen of hepatitis B virus. Protein Sci. 2007;16:2108–17. doi: 10.1110/ps.072983507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee H, Mok KH, Muhandiram R, Park KH, Suk JE, et al. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J Biol Chem. 2000;275:29426–32. doi: 10.1074/jbc.M003107200. [DOI] [PubMed] [Google Scholar]
- 81.Ramelot TA, Gentile LN, Nicholson LK. Transient structure of the amyloid precursor protein cytoplasmic tail indicates preordering of structure for binding to cytosolic factors. Biochemistry. 2000;39:2714–25. doi: 10.1021/bi992580m. [DOI] [PubMed] [Google Scholar]
- 82.Sayers EW, Gerstner RB, Draper DE, Torchia DA. Structural preordering in the N-terminal region of ribosomal protein S4 revealed by heteronuclear NMR spectroscopy. Biochemistry. 2000;39:13602–13. doi: 10.1021/bi0013391. [DOI] [PubMed] [Google Scholar]
- 83.Zitzewitz JA, Ibarra-Molero B, Fishel DR, Terry KL, Matthews CR. Preformed secondary structure drives the association reaction of GCN4-p1, a model coiled-coil system. J Mol Biol. 2000;296:1105–16. doi: 10.1006/jmbi.2000.3507. [DOI] [PubMed] [Google Scholar]
- 84.Garner E, Romero P, Dunker AK, Brown C, Obradovic Z. Predicting Binding regions within disordered proteins. Genome Inform Ser Workshop Genome Inform. 1999;10:41–50. [PubMed] [Google Scholar]
- 85.Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, et al. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–70. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 86.Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, et al. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46:13468–77. doi: 10.1021/bi7012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mohan A, Sullivan WJ, Jr, Radivojac P, Dunker AK, Uversky VN. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst. 2008;4:328–40. doi: 10.1039/b719168e. [DOI] [PubMed] [Google Scholar]
- 88.Xue B, Oldfield CJ, Dunker AK, Uversky VN. CDF it all: consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009;583:1469–74. doi: 10.1016/j.febslet.2009.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang F, Oldfield C, Meng J, Hsu WL, Xue B, et al. Subclassifying disordered proteins by the CH-CDF plot method. Pac Symp Biocomput. 2012:128–39. [PubMed] [Google Scholar]
- 90.Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–84. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 91.Patil A, Nakamura H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006;580:2041–5. doi: 10.1016/j.febslet.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 92.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, et al. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ekman D, Light S, Bjorklund AK, Elofsson A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol. 2006;7:R45. doi: 10.1186/gb-2006-7-6-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dosztanyi Z, Chen J, Dunker AK, Simon I, Tompa P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res. 2006;5:2985–95. doi: 10.1021/pr060171o. [DOI] [PubMed] [Google Scholar]
- 95.Singh GP, Ganapathi M, Sandhu KS, Dash D. Intrinsic unstructuredness and abundance of PEST motifs in eukaryotic proteomes. Proteins. 2006;62:309–15. doi: 10.1002/prot.20746. [DOI] [PubMed] [Google Scholar]
- 96.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, et al. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998:473–84. [PubMed] [Google Scholar]
- 97.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets: the roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–48. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 98.Bustos DM, Iglesias AA. Intrinsic disorder is a key characteristic in partners that bind 14-3-3 proteins. Proteins. 2006;63:35–42. doi: 10.1002/prot.20888. [DOI] [PubMed] [Google Scholar]
- 99.Radivojac P, Vucetic S, O’Connor TR, Uversky VN, Obradovic Z, et al. Calmodulin signaling: analysis and prediction of a disorder-dependent molecular recognition. Proteins. 2006;63:398–410. doi: 10.1002/prot.20873. [DOI] [PubMed] [Google Scholar]
- 100.Singh GP, Dash D. Intrinsic disorder in yeast transcriptional regulatory network. Proteins. 2007;68:602–5. doi: 10.1002/prot.21497. [DOI] [PubMed] [Google Scholar]
- 101.Singh GP, Ganapathi M, Dash D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins. 2007;66:761–5. doi: 10.1002/prot.21281. [DOI] [PubMed] [Google Scholar]
- 102.Anantharaman A, Friedlander TW. Targeting the androgen receptor in metastatic castrate-resistant prostate cancer: a review. Urol Oncol. 2015 doi: 10.1016/j.urolonc.2015.11.003. Doi: 10.1016/j.urolonc.2015.11.003. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 103.He B, Gampe RT, Jr, Kole AJ, Hnat AT, Stanley TB, et al. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol Cell. 2004;16:425–38. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 104.Davies P, Watt K, Kelly SM, Clark C, Price NC, et al. Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J Mol Endocrinol. 2008;41:301–14. doi: 10.1677/JME-08-0042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Illustrative representation of the 2×2 contingency table to find significantly associated GO terms
2×2 contingency table of the membrane KEGG dataset and membrane proteins in whole human proteome (membrane AVG: 0.301)
2×2 contingency table of the non-membrane KEGG dataset and nonmembrane proteins in whole human proteome (non-membrane AVG: 0.437).
2×2 contingency table of the membrane Russian dataset and membrane proteins in whole human proteome
2×2 contingency table of the non-membrane Russian dataset and nonmembrane proteins in whole human proteome
Characterization of human proteins involved in the prostate cancer pathway
Prostate cancer-related proteins from the KEGG and Russian datasets with disorder-associated common Gene Ontology terms
Functions of intrinsically disordered proteins related to prostate cancer. (a) REVIGO analysis of the cellular process GO terms with P < 0.1. (b) AmiGo2 result for all GO terms with P value less than 0.1. Highlighted are the programmed cell death-related cellular process GO terms.
Characterization of human proteins involved in the prostate cancer pathway