

Abstract
Ips spp. bark beetles use ipsdienol, ipsenol, ipsdienone and ipsenone as aggregation pheromone components and pheromone precursors. For Ips pini, the short-chain oxidoreductase ipsdienol dehydrogenase (IDOLDH) converts (−)-ipsdienol to ipsdienone, and thus likely plays a role in determining pheromone composition. In order to further understand the role of IDOLDH in pheromone biosynthesis, we compared IDOLDH to its nearest functionally characterized ortholog with a solved structure: human L-3-hydroxyacyl-CoA dehydrogenase type II/ amyloid-β binding alcohol dehydrogenase (hHADH II/ABAD), and conducted functional assays of recombinant IDOLDH to determine substrate and product ranges and structural characteristics. Although IDOLDH and hHADH II/ABAD had only 35% sequence identity, their predicted tertiary structures had high identity. We found IDOLDH is a functional homo-tetramer. In addition to oxidizing (−)-ipsdienol, IDOLDH readily converted racemic ipsenol to ipsenone, and stereo-specifically reduced both ketones to their corresponding (−)-alcohols. The (+)-enantiomers were never observed as products. Assays with various substrate analogs showed IDOLDH had high substrate specificity for (−)-ipsdienol, ipsenol, ipsenone and ipsdienone, supporting that IDOLDH functions as a pheromone-biosynthetic enzyme. These results suggest that different IDOLDH orthologs and or activity levels contribute to differences in Ips spp. pheromone composition.
Keywords: ipsdienol, ipsenol, monoterpene, pheromone, short-chain dehydrogenase
Ipsdienol (2-methyl-6-methylene-2,7-octadien-4-ol) and ipsenol (2-methyl-6-methylene-7-octen-4-ol), both hydroxylation products of the monoterpene, myrcene (7-methyl-3-methylene-1,6-octadiene), are important semiochemicals in Ips spp. bark beetles (rev. 1). The enantiomeric ratios of these chemicals can be important for their semiochemical function. For example, racemic ipsdienol is produced by western North American Ips pini populations exposed to a myrcene atmosphere (2), but this mixture does not act as an aggregation pheromone. In contrast, males synthesizing pheromone produce ∼95% (enantiomeric excess) (−)-ipsdienol that serves as a potent aggregation pheromone. Eastern North American populations produce and respond to an ∼60% (−)/40% (+)-ipsdienol blend (3, 4).
For western North American I. pini, the pheromone-biosynthetic cytochrome P450 that hydroxylates myrcene to ipsdienol, CYP9T2, does not produce the natural enantiomeric blend of ‘pheromonal ipsdienol’ (5–7). This led to the suggestion that other enzymes ‘tune’ the enantiomeric ratios of ipsdienol produced by myrcene hydroxylases. One candidate enzyme, ipsdienol dehydrogenase (IDOLDH, GenBank JN653323.1), is produced in a pattern consistent with other pheromone-biosynthetic enzymes and stereospecifically oxidizes (−)-ipsdienol to ipsdienone (2-methyl-6-methylene-2,7-octadien-4-one) (8). The expression pattern and activity of IDOLDH strongly suggested that it serves to produce pheromonal ipsdienol.
IDOLDH is a member of the short-chain family of alcohol dehydrogenases (SDRs) with activity that places it in the E.C.1.1.1 group (8). These enzymes have very low sequence conservation, but retain the common Rossman fold tertiary structure (rev. 9). They catalyse redox reactions for a wide variety of endogenous or exogenous substrates, with individual enzymes displaying narrow to broad substrate ranges (10–13) which are strongly influenced by a substrate-binding loop near the C-terminus (14). Drosophila alcohol dehydrogenase is an SDR that is hypothesized to have evolved a preference for ethanol, consistent with that insect’s alcohol-rich environment (15), whereas SDRs from other insects tend to have broader substrate ranges, without a preference for ethanol (13, 16). Thus, insect SDRs are good candidates to study evolutionary pressures affecting functional divergence.
The need to provide the correct pheromone signal would place high pressure on IDOLDH and possibly other associated enzymes to serve the pheromone biosynthetic pathway, leading to narrow substrate range and high product specificity. In order to test the hypothesis that IDOLDH functions primarily in pheromone biosynthesis and therefore has a strong preference for monoterpenoid secondary alcohol substrates, we further characterized its substrate range and biochemical properties. We found that IDOLDH is a functional homo-tetramer with very high specificity for (−)-ipsdienol and (−)-ipsenol and their corresponding ketones. The ability to discriminate for these substrates appears to rely on their entire structure rather than small sub-regions. The observed narrow distribution of substrates and products is consistent with a role in chemical communication rather than general metabolism.
Materials and Methods
Materials
Fetal bovine serum was from Atlas, Grace’s 1x Insect Basal Medium and Hink’s 1x TNM-FH Media (Supplemented Grace’s Medium) were from Mediatech, Inc (Herndon, VA). Agarose Gel (4%) and 2x Grace’s Insect Media and Sf-900 II SFM (1X) media were from Gibco (Grand Island, NY). Sf9 cells were from Invitrogen. Ipsdienone and ipsenone were kind gifts from D. Vanderwel (U. Winnipeg). Neat ‘racemic’ ipsdienol (50% (+)-, 45% (−)- ipsdienol, 5% ipsdienone) was from Bedoukian Research, Inc. (Danbury, CT), and 80 mg/ml 2% (−): 96% (+)-ipsdienol and 98% (−): 1% (+)-ipsdienol in 1,3-butane diol were from PheroTech Inc (Contech, Vancouver, Canada). Cytochrome C, Dextran Blue, yeast alcohol dehydrogenase, geraniol, nerol, citral, (−)-menthone, 1,3-butanediol, β-estradiol, α-ketobutyrate, 4-methyl-2-pentanol, 4-methyl-3-penten-2-one, 4-methyl-2-pentanone, NADP+, NAD+, NADH, NADPH, PMSF and protease inhibitor cocktail were from Sigma (St. Louis, MO). 1-Propanol and 2-propanol were from Fisher. Sephadex G100-120 was from Pharmacia. 8β,9α-Ryanoid and 10-epi-ryanoid were prepared by Luc Ruest (Université de Sherbrooke).
Recombinant protein preparation
Recombinant IDOLDH was produced from a baculoviral vector in Sf9 (insect) cell culture, which has proven highly reliable for studies of pheromone biosynthetic enzymes (5–8, 17), essentially as described previously (8). Assays were done using recombinant IDOLDH recovered in three different ways from infected Sf9 cells: from crude culture media (8), cell homogenates (crude, infected cell lysate), or cell homogenates partially purified with (NH4)2SO4 (see below). Enzyme preparations were stored at 4°C no longer than a month until enzyme assays were done. Protein concentrations for all preparations were determined using the BCA Protein Assay Kit as per the manufacturer’s protocol (Pierce, Rockford, IL), except for those used in kinetic assays, which were determined using a modified Lowry assay (18, 19).
Recombinant protein purification
IDOLDH in lysates of infected cells was partially purified (PPIDOLDH) by (NH4)2SO4 precipitation. Briefly, 35 ml of IDOLDH was prepared as described above and solid (NH4)2SO4 added with stirring to 20% saturation. The samples were mixed by vortexing for 30 s and further mixed on a rotator for 16 h or overnight at 4°C. The samples were then centrifuged at 10,000 × g for 20 min at 4°C, and solid (NH4)2SO4 was added to the supernatant to a final concentration of 40% (40PPIDOLDH) and 60% as described above. The distribution of IDOLDH in pellets and supernatants at different (NH4)2SO4 concentrations was determined by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and western blotting using an IDOLDH-specific polyclonal antibody as described previously (8). To test for storage temperature stability, all PPIDOLDH was flash frozen in liquid nitrogen and stored at 4, −20 or −80°C for a week, then thawed at room temperature and assayed using the standard assay as described below. IDOLDH activity was obtained from averaged estimated initial velocities (N = 2) and an NADPH extinction coefficient of 6.22 mM−1. The ± spread was calculated and reported.
Gel permeation chromatography
The mass of the active form of IDOLDH recovered from infected cell lysates was estimated by gel permeation chromatography using a Sephadex G100-200 column calibrated with ∼7 mg/ml Dextran Blue, cytochrome c, haemoglobin and ∼1 mg/ml yeast alcohol dehydrogenase. Samples were prepared in aqueous 0.05% NaN3, 100 mM NaCl, applied to the top of the column, and collected in 0.75–1.0 ml fractions. The absorbances of Dextran Blue, cytochrome c and haemoglobin were read at 620, 551 and 410 nm, respectively, in a spectrophotometer. Fractions containing IDOLDH or yeast alcohol dehydrogenase were identified by enzymatic activity as described below (Standard assays). A standard curve was created using elution volume (Ve) relative to that of Dextran Blue (Vo) by plotting relative elution volume (Ve/Vo) versus log molecular weight of the standards.
pH dependence
The effect of pH on IDOLDH activity was measured using enzyme recovered from crude culture media to catalyse oxidation of ipsdienol to ipsdienone. Initial rates of reaction were measured at 25°C in 800 μl of assay buffer (100 mM sodium phosphate, 1.1 mM EDTA) (8) adjusted to the appropriate pH and 200 μM (15 μl) NADP+, 13 mM (2.5 μl) racemic ipsdienol and 1,300 μg (300 μl) of protein. Change of absorbance was measured spectrophotometrically at 340 nm (8). This experiment was performed once. The buffer preference of IDOLDH prepared from crude lysates was assayed using 0.1% gelatin in 0.1 M Tris HCl pH 7.5 or 8.0 (N = 2), or 0.1% gelatin in 0.1 M sodium phosphate pH 6.5, 7.0 and 7.5 (N = 2) as described below (Standard assays).
Metal ion requirement
The contribution of metal ions to IDOLDH activity was tested by incubation with the metalloenzyme inhibitor, 1, 10-phenanthroline. Approximately 40 μg (20 μl) of cell lysate containing IDOLDH was incubated with 3.0 mM 1, 10-phenanthroline in 180 μl of 0.1 M Tris HCl pH 7.5 or 0.1 M sodium phosphate buffer for 0–300 min, and then assayed for activity using standard conditions as outlined below (Standard assays).
Secondary structure prediction
Based on a blastp search using IDOLDH as query, the most similar characterized protein of known structure is human L-3-hydroxyacyl-CoA dehydrogenase type II/ amyloid-β binding alcohol dehydrogenase (hHADH II/ABAD; NP_004484.1) (8). A primary sequence alignment between IDOLDH and hHADH II/ADAB was done using Vector NTI (Informax, N. Bethesda, MD, USA) and CLUSTALW (20). Secondary structure predictions were done by the PSIPRED server (21).
Standard assays
Pyridine nucleotides, most substrates, and protein were kept on ice until assay. All other reagents were maintained and assays were conducted at 25°C.
Enzyme activity was determined spectrophotometrically with a 3 ml stirred cuvette system in a Hewlett Packard 8452A Diode-Array spectrophotometer to monitor NADPH formation or disappearance at 340 nm (22). Each ∼3 ml assay contained 2.0 ml of reaction buffer (0.1% gelatin in 66.7 mM sodium phosphate buffer pH 7.5), 1 ml of 0.167–8.33 mM NADP+ for oxidation or NADPH for reduction, 10–160 μl of substrate (see below) and 6–40 μg (5–20 μl, 40PPIDOLDH) or 20–100 μg (10–50 μl) of total recombinant protein (IDOLDH from cell lysate or yeast alcohol dehydrogenase) for oxidation or reduction reactions. Briefly, samples were prepared with reaction buffer, pyridine nucleotide and either enzyme or substrate and change in absorbance was recorded for ∼1–3 min to establish the baseline. Reactions were then initiated by the addition of either missing component (substrate or enzyme) and absorbance was recorded for a further 1–10 min until no change was visible. All assays for specific activity were run at least in duplicate unless otherwise noted or unless no reaction was seen with a particular protein preparation or substrate, in which case that assay was not repeated. Progress curves of reactions were plotted as product versus time and curves fit to a single exponential equation (8). Estimated initial velocities (in units OD/s) and an NADPH extinction coefficient of 6.22 mM−1 were used to calculate estimated specific activities (ESAs) in terms of nmol·min−1·mg−1 total protein. All data were plotted and analysed using Origin 5.0 software (Northampton, MA).
Normalization assays
Data were collected from multiple enzyme preparations with variable enzyme expression levels and recoveries. In order to make quantitative comparisons and test enzyme stability between preparations, ‘normalizing’ ESAs were determined at the beginning and end of each experimental session with the following parameters: 2 ml of 0.1% gelatin in 0.1 M sodium phosphate buffer pH 7.5, 1 ml of 2.5 mM NADP+, 200 μl of 0.33 mM (−)-ipsdienol and 20–100 μg (10–50 μl) of total IDOLDH lysate or 6–40 g (10 μl) of 40PPIDOLDH (where noted). Experimental ESA values were then divided by those of the normalization assay to calculate reported specific activities. Experimental determinates (N) are the number of independent enzyme preparations tested. In cases where two experimental determinations were obtained and the data were normalized to protein concentration, pooled into a single data set and a single fit of all the data was obtained and standard error of the estimate reported. When N > 2, averages of obtained specific activities and standard errors were reported.
Specific activities for various substrates were explored using the following, with concentrations listed being final concentrations in the assay: 0.33 M 1- or 2-propanol or ethanol; 17.6 μM (−)-ipsdienol in 1,3-butanediol, (+)-ipsdienol in 1,3-butanediol, racemic ipsenol, ipsdienone, ipsenone, (−)-menthone, nerol, geraniol, citral or myrcene, 24 μM or 3.4 mM 4-methyl-2-pentanol, 25 μM or 3.5 mM 4-methyl-2-pentanone or 3-penten-2-ol, 28 μM or 17.65 mM 4-methyl-3-penten-2-one, 31 μM or 5.11 mM 3-penten-2-ol or 90 mM acetone, (Fig. 1) each dissolved in assay buffer; and 21-22 μM 10-epi-921-dehydroryanodine, 8β,9α-dihydroxy-10-O-methyl-10-epiryanodine, or 20 μM β-estradiol, each dissolved in ethanol. Similarly, yeast alcohol dehydrogenase was assayed with 1 - or 2-propanol, or (−)-ipsdienol at the concentrations listed above.
Fig. 1.

Substrates and analogs tested with IDOLDH. Strong substrates showing specific activity (SA) > 27 nmol·min−1·mg−1 at <20 μM substrate, and weak substrates (SA of 10 to 27 nmol·min−1·mg−1 at between 17 and 31 μM substrate are in the upper and lower left panels, respectively, while poor or non-substrates are in the right panel.
Kinetic constants for oxidation or reduction reactions were determined by varying the substrate concentration from 2.2 μM to 17.6 μM. Separate additions of the substrate were added to the assay buffer with enzyme and cofactor (NADP+ at 0.866 or 8.66 mM for oxidation and NADPH at 0.167 mM for reduction) and tested using the standard assay. All assays to determine kinetic constants (Km and Vmax) were performed in duplicate.
Inhibition constants (KI) were determined for (−)-ipsdienol oxidation by IDOLDH by comparing velocities of reactions in the presence and absence of each putative inhibitor. Compounds were tested for inhibition at the following concentrations: 17.6 μM for ipsdienone,1, 3-butanediol, 8β,9α ryanoid, 10-epi ryanoid, nerol, geraniol, citral, (+)-ipsdienol, 33.3 mM 1-propanol and 2-propanol, 17.6 μM and 35.2 μM for (−)-menthone, and 39 μM for β-estradiol (Table II). Apparent inhibition constants and inhibitor dissociation constants were calculated by standard methods assuming competitive inhibition.
Table II.
Modulators of IDOLDH activity
| [(−)-Ipsdienol] (μM) | Inhibitor | [Inhibitor] (μM) | Specific activity (nmol·min−1·mg−1) |
KI | |
|---|---|---|---|---|---|
| without inhibitor | with inhibitor | ||||
| 16 a | Ipsdienone | 16 | 0.308 | 0.246 | 29 μM |
| 16 | Ipsdienone | 16 | 0.650 | 0.573 | 15 μM |
| 4 | (−)-Menthone | 16–35 | 0.330 | 0.228 | 30 μMb |
| 16 | 1-Propanol | 33,300 | 0.308 | 0.126 | 33.6 mM |
| 16 | 2-Propanol | 33,300 | 0.308 | 0.441 | 9.9 mM |
| 10 | β-Estradiol | 39 | 0.981 | 0.783 | 25 μMc |
| 16 | 1,3-Butanediol | 16 | 0.213 | 0.245 | — |
| 16 | 8β,9α Ryanoid | 16 | 0.194 | 0.195 | — |
| 16 | 10-Epi ryanoid | 16 | 0.194 | 0.197 | — |
| 16 | Nerol | 16 | 0.182 | 0.197 | — |
| 16 | Geraniol | 16 | 0.182 | 0.207 | — |
| 16 | Citral | 16 | 0.182 | 0.203 | — |
| 16 | (+)-Ipsdienol | 16 | 0.396 | 0.389 | — |
aSample contained 25 mM NADP instead of 2.5 mM NADP
bAverage value obtained from experiments with variable (−)-menthone concentrations
cExperiment done once.
Product assay and determination
IDOLDH prepared from infected cell lysates and uninfected Sf9 cell lysates (negative control) were assayed with ipsdienone or ipsenone in the presence of NADPH in one ml reactions. Assay buffer (962.5 μl), 100 μg of enzyme (20 μl of IDOLDH cell lysate, 1 to 1 dilution of Sf9 cell lysate and assay buffer), and 225 μM NADPH (15 μl of 15 mM prepared in water) were added to a small glass vial. The reactions were initiated by the addition of 67–73 μM substrate (2.5 μl of 27–29 mM substrate diluted in assay buffer). The reaction vials were capped and mixed by vortexing for 10 s before incubation at 25°C for 15 min.
Products were analysed by coupled GC-MS using a DB-5 capillary column, and the enantiomeric compositions of ipsdienol and ipsenol were determined by chiral separation using the same GC-MS system with a CycloSil-B (30 m × 0.25 mm internal diameter, 0.25 μm film thickness) column (J&W Scientific) essentially as described previously (8). The retention times and mass spectra of true standards were used for comparison.
Results
Active form
Gel permeation chromatography gave a single peak of IDOLDH activity with an estimated mass of 117 kDa as compared to known standards (Fig. 2). The estimated mass is ∼4 times higher than the 27.5 kDa observed in SDS–PAGE gels and predicted by conceptual translation of its open reading frame (8).
Fig. 2.
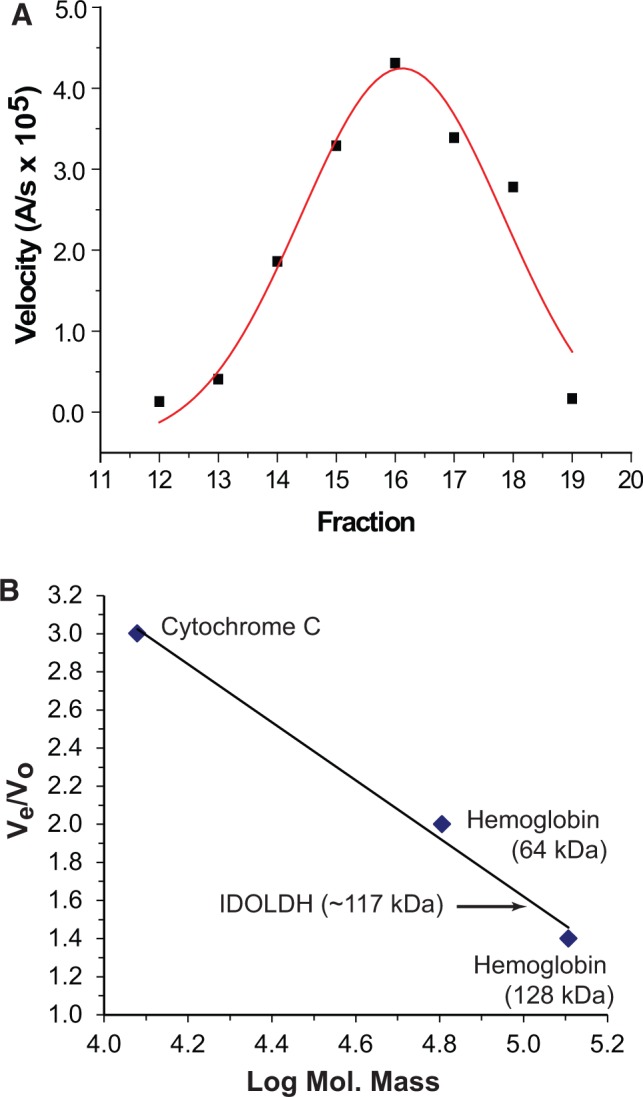
Gel permeation chromatography. (A) Activity profile of IDOLDH-catalysed oxidation of (−)-ipsdienol in fractions eluting from a Sephadex G100-200 column. Most activity eluted in Fraction 16. (B) Elution profile of standards and IDOLDH. The activity profile in panel A placed functional IDOLDH at 117 kDa.
pH dependence
IDOLDH activity was highest in mildly basic conditions (Fig. 3A). The enzyme was less active in acidic pH conditions. A narrow-range survey showed higher IDOLDH specific activity in solutions buffered with 0.1 M Tris HCl compared to those buffered with 0.1 M sodium phosphate (Fig. 3B).
Fig. 3.
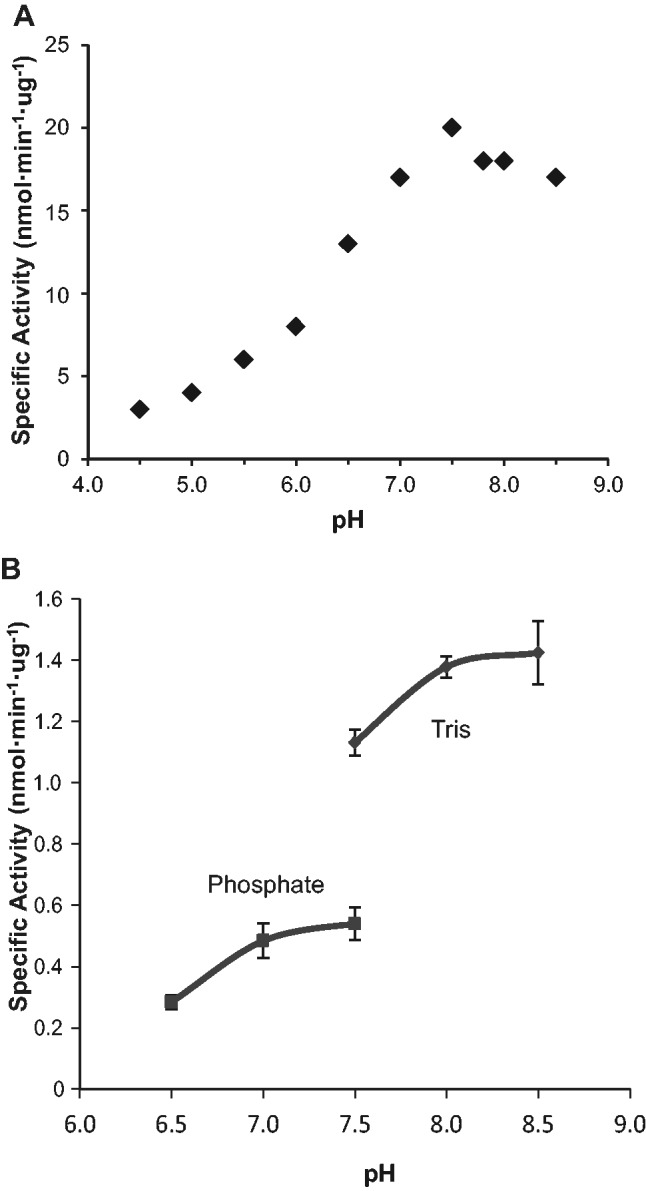
pH effects on activity. (A) pH titration curve of recombinant IDOLDH prepared from crude culture media. All reactions were done with 13 mM racemic ipsdienol and 2 mM NADP+. Values are from a single replicate. (B) Effect of buffer composition on activity of IDOLDH prepared from infected cell lysates. Oxidation of (−)-ipsdienol by IDOLDH was measured in phosphate or Tris based buffers. Values are means of two replicates, ± maxima and minima.
Metal ion requirement
IDOLDH use of metal ions was tested by incubation with the metalloenzyme inhibitor, 1, 10-phenanthroline. A slight, statistically insignificant (P > 0.07) increase of (−)-ipsdienol oxidation activity was observed after 40 μg IDOLDH was incubated for up to 5 h in 3.0 mM 1, 10-phenanthroline in 0.1 M Tris HCl pH 7.5 (specific activity of 614.3 ± 36.6 or 519.7 ± 30.6 nmol min−1·mg−1, respectively) or 0.1 M sodium phosphate buffer (specific activity of 524.7 ± 21.1 or 470.3 ± 6.5 nmol·min−1·mg−1, respectively).
Enzyme stability
Recombinant IDOLDH produced by baculovirus-infected Sf9 cells distributed to both cell culture media and the cell homogenates (8). Crude cell lysates were chosen for further characterization because they had a higher IDOLDH activity relative to total protein.
IDOLDH preparations from both crude culture media and infected cell lysates showed highest activity in the resuspended pellets of 40% (NH4)2SO4 cuts (40PPIDOLDH) compared to activity of 20% and 60% (NH4)2SO4 cuts (Supplementary Fig. S1). Forty and sixty percent ammonium sulphate purification yielded approximately a 7-fold purification by mass and 2.7 - and 2-fold higher specific activity (217 ± 0.5 and 159 ± 18 5 nmol·min−1·mg−1), respectively, compared to crude cell lysate preparations (81 5 ± 1 nmol·min−1·mg−1). Storage at −80°C and 4°C further stabilized 40PPIDOLDH with half activity still retained after 6 months at 4°C after initial freezing and thawing (data not shown). Over half of the activity was lost when IDOLDH from cell lysates or the 40% (NH4)2SO4 supernatant (40PPIDOLDH) were stored at −20°C for a week (data not shown). Furthermore, IDOLDH lysate-mediated reduction of ipsdienone was observed for only 0.5–1.0 min before loss of observable activity, whereas 40PPIDOLDH-mediated reduction of ipsdienone could be detected for almost 10 min. Over half of the activity of IDOLDH prepared from infected cell lysates was lost after a month of storage at 4°C but 40PPIDOLDH retained most to all of its activity under the same conditions.
Standard assays conducted at the beginning and end of an experimental session showed enzyme activity was stable through the experimental period and from day to day (data not shown). To be consistent, the specific activity obtained at the beginning of assay was always used to normalize ESA or Vmax values of the experiment.
Structure analysis
IDOLDH and hHADH II/ ABAD primary structures were 253 a.a., and 261 a.a., respectively, with 35% a.a. identity distributed throughout the sequences (Fig. 4). The hHADH II/ABAD secondary structure predicted by PSIPRED (21) was very similar to the X-ray crystal structure in the Protein Data Bank (ID 1U7T; not shown), suggesting that PSIPRED could reliably predict α-helices and model IDOLDH. The overall secondary structure predicted for IDOLDH and observed for hHADH II/ ABAD were very similar, with residues contributing to the typical Rossmann-fold motif for nucleotide binding (22–24) and the catalytic tetrad (Asn 121,Ser 155, Tyr 168, Lys 172 in hHADH II/ABAD), highly conserved (Fig. 4).
Fig. 4.

Structural comparison of short-chain dehydrogenases. ClustalW alignment of hHADH/ABAD (GenBank NP_004484.1 and western I. pini IDOLDH (wIpIDOLDH; CB408666.1). Conserved residues are in bold font. Gaps inserted to optimize the alignment are indicated by dashes. The substrate binding loops and the nucleotide-binding motifs are boxed. The catalytic tetrad residues are underlined.
IDOLDH activity
Conventional methods were used to compare specific activities based on the steady-state (initial velocity) portion of the progress curves. The specific activity of recombinant IDOLDH prepared from infected Sf9 cell lysates for (−)-ipsdienol oxidation in the presence of NADP+ was 427 ± 25 to nmol·min−1·mg−1 (Table I), ∼19-fold higher than preparations from culture media (8). IDOLDH also efficiently catalysed the oxidation of racemic ipsenol to ipsenone with a specific activity of 196 ± 8 nmol·min−1·mg−1. No activity was observed using (+)-ipsdienol as a substrate. The reduction reactions of ipsdienone and ipsenone using NADPH as coenzyme yielded the corresponding alcohols with specific activities of 50 ± 4 and 92 ± 7 to nmol·min−1·mg−1, respectively. GC analysis using an enantioselective column indicated ipsdienone was reduced to exclusively (−)-ipsdienol, while ipsenone was reduced to (−)-ipsenol; the (+)-enantiomers of these alcohols were never observed (Fig. 5). In general, the oxidation reaction appeared more favoured than the reduction reaction (Table I). (−)-Menthone was reduced by IDOLDH with a specific activity of ∼16 nmol·min−1·mg−1, however no activity was observed for other monoterpenoid chemicals, including nerol, geraniol and citral. Myrcene was neither oxidized nor reduced by IDOLDH under our assay conditions (Table I). Parallel tests using uninfected Sf9 cell lysates showed no activity on monoterpenoid alcohol substrates, and barely detectable activity on carbonyls (not shown).
Table I.
IDOLDH substrate profile
| Substratea | Concentration (μM) | Coenzyme | Specific activity (nmol·min−1·mg−1 ± SEE or std. err.b) | N |
|---|---|---|---|---|
| (−)-Ipsdienol | 17 | NADP+ | 427 ± 25 | 62 |
| (+)-Ipsdienol | 17 | NADP+ | 0.2 ± 0.2 | 1 |
| Racemic ipsenol | 17 | NADP+ | 196 ± 8 | 3 |
| Ipsdienone | 17 | NADPH | 50 ± 4 | 16 |
| Ipsenone | 17 | NADPH | 92 ± 7 | 24 |
| (−)-Menthone | 17 | NADPH | 16 ± 0.1 | 2 |
| Nerol | 17 | NADP+ | 2.6 ± 0.4 | 2 |
| Geraniol | 17 | NADP+ | 1.5 ± 0.5 | 2 |
| Citral | 17 | NADP+ | 2.5 ± 0.5 | 2 |
| Myrcene | 17 | NADP+ | 0.22 ± 0.1 | 1 |
| Myrcene | 17 | NADPH | 4.4 ± 0.2 | 2 |
| 1,3 Butenediol | 17 | NADP+ | 3.0 ± 0.6 | 3 |
| 4-Methyl-2-pentanol | 24 | NADP+ | 1.0 ± 2.0 | 2 |
| 4-Methyl-2-pentanol | 3,400 | NADP+ | 19 ± 0.1 | 2 |
| 4-Methyl-2-pentanone | 25 | NADPH | 5.9 ± 0.1 | 2 |
| 4-Methyl-2-pentanone | 3,500 | NADPH | 70.9 ± 0.2 | 2 |
| 4-Methyl-3-penten-2-one | 28 | NADPH | 3.9 ± 0.1 | 2 |
| 4-Methyl-3-penten-2-one | 17,650 | NADPH | 24 ± 1 | 2 |
| Acetone | 90,000 | NADPH | 41 ± 1 | 4 |
| 10-Epi-ryanoid | 21 | NADP+ | 3.0 ± 1 | 1 |
| 8β-9α-ryanoid | 22 | NADP+ | 5.4 ± 0.6 | 1 |
| 3-Penten-2-ol | 31 | NADP+ | 13.6 ± 1.0 | 2 |
| 3-Penten-2-ol | 5,110 | NADP+ | 48.8 ± 1.0 | 2 |
| β-Estradiol | 20 | NADP+ | 27.0 ± 0.8 | 1 |
Enzyme activity was determined by measuring changes in absorbance at 340 nm. The coenzyme concentration in all cases was 200 μM NADP+ or 167 μM NADPH. See Materials and Methods for standard assay conditions.
aOther substrates tested but showing no activity included α-ketobutyrate, 1 - and 2-propanol and ethanol.
bStandard error of the estimate (SEE) is a measure of the quality of the data with smaller values indicating a higher probability the model is correct. SEE is reported when N = 1, N = 2; Std. err. is reported when N ≥ 3.
Fig. 5.

Stereo-selective ketone reduction. GC traces of products extracted from reactions of IDOLDH prepared from infected cell lysates incubated with NADPH and either ipsdienone (A) or ipsenone (B). Traces of authentic racemic ipsdienol and ipsenol are inset in grey. IDOLDH produced only the minus enantiomer of each alcohol. Similar results were seen with IDOLDH prepared from culture media (data not shown). Control Sf9 cell lysate reactions showed a 1–2% reduction of ketone to corresponding alcohol (not shown).
In order to further map characteristics that influence substrate specificity, we tested monoterpenoid analogs with structures analogous to various portions of (−)-ipsdienol, (−)-ipsenol, ipsdienone and ipsenone, including 2-propanol, 4-methyl-2-pentanol, 3-penten-2-ol, acetone, 4-methyl-3-penten-2-one and 4-methyl-2-pentanone (Fig. 1). Of these, only 4-methyl-2-pentanol and 3-penten-2-ol served as oxidation substrates, whereas 4-methyl-3-penten-2-one and 4-methyl-2-pentanone served as very poor reduction substrates, but only at very high (3–17 mM) concentrations (Table I). No reaction could be detected when these substrates were assayed at micromolar concentrations.
The hHADH II/ABAD substrate, β-estradiol (24) was a poor oxidation substrate at 20 μM for IDOLDH, with a specific activity of 27 nmol·min−1·mg−1 (Table I). Additionally, 39 μM β-estradiol was an inhibitor of IDOLDH oxidation of (−)-ipsdienol and had a Ki of 25 μM (Table II). Kinetic and inhibitor reactions with β-estradiol finished earlier than reactions with other substrates (0.5–1 min versus 10 min, respectively). 3-Hydroxybutyrate was not a substrate for IDOLDH (data not shown).
Plots of velocity versus enzyme concentration were linear for (−)-ipsdienol and other monoterpenoid substrates (racemic ipsenol, ipsdienone and ipsenone; data not shown). Product versus time and velocity versus substrate concentration plots were hyperbolic. The product versus time plot was consistent with exponential relaxation kinetics due to product accumulation or substrate depletion and was the normal shape for the progress curve of an enzyme-catalysed reaction running to completion (not shown).
Kinetic values for various substrates are shown in Table III. In general, Km and specific activity values for the preferred substrate, (−)-ipsdienol, were in the µM and mid- nmol·min−1·μg−1 range, respectively, and did not change markedly over different NADP+ concentrations. By comparison, we found yeast ADH had apparent Km and specific activity values in the mM and low nmol·min−1·μg−1 range, respectively, for ethanol, comparable to the published values (24).
Table III.
Km and Vmax values of putative substrates
| Row | Substrate | [Constant Substrate] | Km ± SEE | Vmaxa | Normalized | Relative | Normalized |
|---|---|---|---|---|---|---|---|
| (μM) | (nmol ·min−1·mg−1) | Vmaxb | Vmaxc | Vmax/Km (uM−1) | |||
| 1 | (−)-Ipsdienol | 8.66 mM NADP | 13 ± 5 | 639 ± 94 | 1.68 | 1.26 | 0.129 |
| 2 | (−)-Ipsdienol | 0.866 mM NADP | 4 ± 2 | 523 ± 50 | 1.33 | 1.00 | 0.333 |
| 3 | (−)-Ipsdienold | 0.866 mM NADP | 527 ± 44 | 2,590 ± 70 | 1.18 | 0.89 | 0.00224 |
| 4 | (−)-Ipsdienole | 0.866 mM NADP | 22 ± 4 | 1,820 ± 90 | 1.07 | 0.80 | 0.0486 |
| 5 | Racemic Ipsenol | 0.866 mM NADP | 9 ± 4 | 230 ± 45 | 0.58 | 0.44 | 0.0644 |
| 6 | Ipsdienone | 0.167 mM NADPH | 26 ± 15 | 49 ± 19 | 0.21 | 0.16 | 0.00808 |
| 7 | Ipsdienonee | 0.167 mM NADPH | 2 ± 0.6 | 380 ± 10 | 0.18 | 0.14 | 0.0900 |
| 8 | Ipsenone | 0.167 mM NADPH | 3 ± 3 | 113 ± 28 | 0.4 | 0.30 | 0.133 |
| 9 | (−)-Menthone | 0.167 mM NADPH | 20 ± 10 | 28 ± 11 | 0.05 | 0.04 | 0.00250 |
| 10 | NADP | 17.6 μM (−)-Ipsdienol | 0.08 ± 0.04 | 517 ± 89 | 1.26 | 0.95 | 15.8 |
aVmax is expressed as specific activity
bCalculated by dividing the Vmax of the experimental by the specific activity of the normalization assay (see Normalization assay).
cCalculated by dividing the normalized Vmax of the experimental by the normalized Vmax of the preferred substrate, (−)-ipsdienol.
dAssays done with 40PPIDOLDH (IDOLDH partially purified with 40% ammonium sulphate) within the first week of protein preparation.
eAssays done with 40PPIDOLDH after the first week of protein preparation.
Ratios of apparent experimental specific activities per specific activity of the normalization assay were determined to directly compare kinetic rates of IDOLDH substrates. The normalized specific activity for the oxidation of (−)-ipsdienol was 6.3 times higher than that for the reduction of ipsdienone, while the oxidation of racemic ipsenol was only 1.5 times faster than the reduction of ipsenone (Table I). Both linear monoterpenoid ketones showed higher normalized specific activity values than that of menthone. In general, normalized specific activity/Km values for preferred substrates were in the sub-µM−1 range, while those for NADP+ were ∼100-fold higher than those of (−)-ipsdienol.
The kinetic constants for (−)-ipsdienol and ipsdienone were obtained for 40PPIDOLDH. Specific activity values for (−)-ipsdienol and ipsdienone were 4-5 and 8 X higher, respectively, when assayed with partially purified enzyme than with crude lysate (Table III, rows 3, 4). The Kms for (−)-ipsdienol and ipsdienone obtained when assayed with 40PPIDOLDH preparations over a week old were similar to those obtained from crude IDOLDH lysates (Table III). However, the Km for (−)-ipsdienol was an order of magnitude higher when tested with 40PPIDOLDH within the first week of preparation. Specific activity values for 40PPIDOLDH were consistently higher than those for unpurified IDOLDH (Table III, rows 2, 3, and rows 6, 7)
The action of various chemicals as inhibitors of (−)-ipsdienol oxidation by IDOLDH was also tested. Ipsdienone and (−)-menthone both inhibited the reaction with similar KIs ranging from 15 to 30 μM, respectively, whereas 1 - and 2-propanol had much higher KIs of 33.6 and 9.9 mM, respectively (Table II). Other chemicals, including 1,3 butandiol, nerol, geraniol, citral, (+)-ipsdienol and the ryanoids did not act as inhibitors (Table II).
Discussion
Monoterpene metabolism by animals is common, but monoterpene biosynthesis in animals appears limited to some insects that use monoterpenoid derivatives for communication (1) or defence (26). Furthermore, while plants often produce a range of structurally diverse monoterpenes, insect monoterpenoid producers typically synthesize and further modify only myrcene or geraniol (26). For I. pini, pheromone-biosynthetic males produce ipsdienol at daily rates exceeding 1% of their body mass (27). This significant metabolic load is accompanied by strong coordinate up-regulation of the mevalonate pathway (28) as well genes encoding geranyl diphosphate synthase/myrcene synthase (2) and myrcene hydroxylase (6), two committed steps in ipsdienol production. This tightly coordinated regulation implies significant pressure for these enzymes to serve the pathway. IDOLDH, with an expression pattern tightly coordinated with these other enzymes, and encoding an enzyme that specifically interacts with ipsdienol, can reasonably be expected to serve the same pathway—i.e pheromone biosynthesis (8).Here, we present biochemical evidence supporting the interpretation that IDOLDH has evolved into a pheromone-biosynthetic role.
Short-chain oxidoreductases (SDRs) catalysing the oxidation of alcohols and reduction of carbonyls form a large enzyme class. While their primary structures vary widely, often displaying as little as 15% sequence identity, they share highly conserved secondary and tertiary structures and often function as oligomers (rev. (9)). Structurally, IDOLDH conforms to precedents, showing common SDR sequence motifs with only 35% a.a. identity to the nearest characterized blast hit (8) while mapping onto its secondary structure with high confidence (Fig. 4). IDOLDH activity eluted from a gel permeation column at ∼117 kDa (Fig. 2). Given that the predicted monomer molecular mass is 27.5 kDa (8), this observation suggests that IDOLDH is active as a homo-tetramer. Activity was highest at mildly basic pH, consistent with an expected mildly basic midgut cell environment (29) where IDOLDH functions in vivo (8). There was an apparent preference for Tris-based buffers compared with phosphate buffers (Fig. 3), likely due to phosphate inhibition of nucleotide binding or unknown buffer component interactions with the enzyme and/or substrate. While our assay does not establish a requirement for metal ions, IDOLDH activity was not significantly affected by the metalloenzyme inhibitor, 1, 10-phenanthroline (Table I), suggesting metal ions are not required for activity.
Ammonium sulphate precipitation is commonly used to purify proteins and can, depending on the protein and assay conditions, denature and/or inhibit an enzyme (30). Ammonium sulphate did not inhibit IDOLDH activity and at higher concentrations effectively partially purified and increased both stability and shelf life of IDOLDH in infected cell lysates (Supplementary Fig. S1). Ipsdienone reduction reactions ordinarily lasting up to 1 min before loss of observable activity were prolonged to almost 10 min when assayed with IDOLDH lysates partially purified with 40 % ammonium sulphate (data not shown), allowing for a more detailed analysis of the reaction and illustrating the usefulness of this preparative step. Additionally, IDOLDH shelf life was improved with minimal or half activity lost from the starting rates after storage at 4°C for 1 or 6 months, respectively, in contrast to unpurified IDOLDH lysates, which lost half to all activity after storage at 4°C for 1 or 6 months, respectively (data not shown). These results suggest ammonium sulphate partial purification is an effective method to remove proteases that reduce IDOLDH stability. Ammonium sulphate precipitation also affected the kinetic properties of IDOLDH, with significant increases in both Km and specific activity compared to the unpurified enzyme (Table III, rows 2, 3). The specific activity value for 40PPIDOLDH also changed following a 1-week storage period (Table III, rows 3, 4). The reasons for these changes are not clear, but may be due to a combination of removing associated proteins and/or conformational changes with time.
Kinetic assay values for various substrates were compared to those of normalization assays with (−)-ipsdienol and NADP+ in order to reduce variability arising from different rounds of enzyme production and preparation. When the specific activity of an experiment is normalized to the specific activity of the normalization assay, we can directly compare the rates of reaction between alcohol and ketone substrates. Reduction reactions should be compared with the oxidation reactions with caution, as ketone reduction reactions were not fully saturated with NADPH. IDOLDH appeared readily saturated by NADP+, as indicated by the small change in Km and specific activity for the (−)-ipsdienol substrate over a 10-fold range of NADP+ concentrations (Table III, rows 1, 2). In general IDOLDH accelerates and prefers (−)-ipsdienol oxidation to ipsdienone more than racemic ipsenol oxidation to ipsenone as substrate specificity of (−)-ipsdienol versus racemic ipsenol is five times higher (0.333 μM−1 versus 0.0644 μM−1, respectively, Table III). The two alcohols differ only in the presence of a double bond α to the hydroxyl group; with ipsdienol being more conformationally restricted than ipsenol. Relocation of the hydroxyl to a terminal position removes activity as other myrcene-derivatives, including nerol, geraniol and citral, were not substrates.
Compared to the sharp specific activity of secondary alcohol oxidation, reduction of carbonyls showed a flatten specificity (Table I). The rate of acetone reduction was comparable to that of ipsdienone (at a 1,000-fold lower concentration), in contrast to the rates of 2-propanol and (−)-ipsdienol oxidation (Table I). However, IDOLDH appeared to favour ipsenone over ipsdienone as a substrate (Table I).
IDOLDH interacts with a variety of compounds in vivo, many of which are cofactors, substrates and inhibitors. ‘Classical’ SDRs, like IDOLDH (8), typically catalyse reactions through an ordered ‘bi-bi’ mechanism where the cofactor binds first and exits the enzyme last (rev. 9). The large specificity constant of NADP+ indicates the cofactor has an association rate constant with IDOLDH orders of magnitude greater than that of any terpene substrate tested with enzyme. This is in support of a mechanism where IDOLDH requires NADP+ to bind before it can bind the substrate and turnover a product. Amongst terpene substrates, kinetic and specificity constants clearly suggest (−)-ipsdienol and ipsenone are the preferred substrates of IDOLDH followed by racemic ipsenone, ipsdienone and (−)-menthone (Table III). Our exploration of structure-function relationships of potential substrates and inhibitors (Fig. 1) is a preliminary examination of IDOLDH’s biological activities with respect to terpene metabolism. None of the analogs was substrates at the concentrations used to assay known substrates, with extremely low activity at concentrations three orders of magnitude higher (Table I). The analogs also did not inhibit (−)-ipsdienol oxidation by IDOLDH (Table II). In general IDOLDH prefers a polar oxygen flanked by hydrophobic groups and functions as a specialist for secondary alcohol oxidation and a generalist for ketone reduction, as outlined below:
Both (−)-menthone and ipsdienone bound IDOLDH with dissociation constants similar to their Kms (Tables II and III) though (−)-menthone, a cyclic analog of ipsenone, had a normalized specific activity approximately an order of magnitude lower and specificity constant three times less (Table III). The Vmax for both (−)-menthone and ipsdienone are very similar (28 and 49 nmol·min−1·mg−1, respectively), however the normalized and relative specific activity values of the acyclic ketone are at least four times that of the cyclic ketone, suggesting ipsdienone is a far better substrate for IDOLDH. The Ki of 30 μM and normalized specific activity of 0.05 for (−)-menthone suggests that loss of conformational flexibility due to its ring structure reduces menthone’s binding affinity. The best terpene substrate for IDOLDH was (−)-ipsdienol, which is a linear monoterpenoid alcohol with a double C-C bond α to the hydroxyl group. Any structural change greatly reduced activity. The 6-methylene–7-ene portion of all identified strong IDOLDH substrates (Fig. 1) was very important as its removal (e.g. in the substrate analog 4-methyl-3-penten-2-one) abolished activity (Table I). Primary monoterpenoid alcohols (nerol and geraniol), as well as myrcene, did not inhibit ipsdienol oxidation (Table II), indicating that they were not substrates because they did not bind IDOLDH. Thus, the C4 position for the hydroxyl group was also very important for catalysis. Removing the C-C double bond α to the hydroxyl group (i.e. ipsenol) appeared to also reduce activity. The ipsenol analogs 2-propanol and 4-methyl-2-pentanol were not substrates, whereas the ipsdienol analog 3-penten-2-ol, with ∼5% relative velocity, was a poor substrate. In addition to illustrating the importance of the C-C double bond α to the hydroxyl group, these analogs further show that a larger molecule is required to realize catalysis. Oddly, 2-propanol stimulated IDOLDH oxidation of (−)-ipsdienol, while 1-propanol was a weak inhibitor (Ki = 33.6 mM; Table II). By comparison, the Kms of 2-propanol for horse and human alcohol dehydrogenase (E.C.1.1.1.1) are 268 mM and 560 mM, respectively (31, 32). These data suggest a small alcohol can bind IDOLDH but fails to form a productive transition state.
Interestingly, β-estradiol was a poor substrate though it is much larger than ipsdienol, whereas ryanoids were neither inhibitors nor substrates (Table II). β-Estradiol bound with an apparent Ki of 25 μM, comparable to the Km of 13 μM for ipsdienol (Table III) and the Ki of 15 μM for ipsdienone (as an inhibitor of ipsdienol oxidation; Table II), and thus likely binds IDOLDH as effectively as the reduction substrate, ipsdienone. These data suggest a binding/active site large enough to form intimate contacts with 10-carbon compounds but open on one side to allow partial binding of large compounds. It is interesting that the most extensive sequence divergence between IDOLDH and hHADH II/ABAD is in their putative substrate-binding loops (Fig. 4), consistent with most of the substrate selectivity being conferred by this structure (10). However, there is enough similarity between both enzymes for IDOLDH to use the hHADH II/ABAD substrate β-estradiol as a poor substrate (Tables I and III).
Insect SDRs are typically encoded in rapidly evolving gene families. While most have not been functionally characterized, it appears that closely related enzymes have similar, though not identical substrate profiles and are encoded by genes with sometimes very different transcription profiles, suggesting physiological advantages are gained through divergence (13, 33). IDOLDH showed a very strong bias towards ipsdienol-related monoterpenoid substrates, apparently relying on cues from their full structures to interact with them, and thus appears to have evolved to work with ipsdienone, ipsenone and the (−)-enantiomers of ipsdienol and ipsenol to the exclusion of other chemicals. This further supports the hypothesis that IDOLDH is a highly regulated, pheromone-biosynthetic enzyme. IDOLDH activity thus likely contributes to pheromone blend composition in different Ips populations and species.
Supplementary Data
Supplementary Data are available at JB Online.
Acknowledgements
We thank Luc Ruest (U. Sherbrooke) for preparing ryanoids, the Nevada Proteomics Center for GC-MS support, Sharon Young and Ryan Olsen for SDS–PAGE assistance, and Dr William H. Welch for invaluable help and insight throughout all stages of this work.
Funding
This research was supported by the United States National Science Fountation (1122248), the United States Department of Agriculture (2009-05200), the Nevada State Hatch/McIntyre-Stennis research awards, and the National Institute of General Medical Sciences division of the National Institutes of Health (8 P20 GM103440-11), and is a contribution of the Nevada Agricultural Experiment Station.
Conflict of Interest
IDOLDH is protected by US patents 8,404,825 and 8,945,890 B2, granted to the Board of Regents of the Nevada System of Higher Education on Behalf of the University of Nevada, Reno, and for which R.F.-T., G.J.B., and C.T. are co-authors. C.T. is a co-founder and officer in EscaZyme Biochemicals, LLC, which holds a license for the patent.
Glossary
Abbreviations
- a.a.
amino acid
- ADH
alcohol dehydrogenase
- hHADH II/ABAD
L-3-hydroxyacyl-CoA dehydrogenase type II/ amyloid-β binding alcohol dehydrogenase
- IDOLDH
ipsdienol dehydrogenase
- MDR
medium-chain dehydrogenase
- MOI
multiplicity of infection
- PPIDOLDH
partially purified IDOLDH
- SDR
short-chain oxidoreductase
- 40PPIDOLDH
IDOLDH recovered from a 40% (NH4)2SO4 cut
References
- 1.Blomquist GJ., Figueroa-Teran R., Aw M., Song M., Gorzalski A., Abbott N.L., Chang E., Tittiger C. (2010) Pheromone production in bark beetles. Insect Biochem. Mol. Biol. 40, 699–712 [DOI] [PubMed] [Google Scholar]
- 2.Lu F. (1999) Origin and Endocrine Regulation of Pheromone Biosynthesis in the Pine Bark Beetles, Ips pini (Say) and Ips paraconfusus Lanier (Coleoptera: Scolytidae. Ph.D., University of Nevada, Reno [Google Scholar]
- 3.Domingue M., Starmer W., Teale S. (2006) Genetic control of the enantiomeric composition of ipsdienol in the Pine Engraver, Ips pini. J. Chem. Ecol. 32, 1005–1026 [DOI] [PubMed] [Google Scholar]
- 4.Domingue M., Teale S. (2008) The genetic architecture of pheromone production between populations distant from the hybrid zone of the pine engraver, Ips pini. Chemoecology 17, 255–262 [Google Scholar]
- 5.Sandstrom P., Ginzel M.D., Bearfield J.C., Welch W.H., Blomquist G.J., Tittiger C. (2008) Myrcene hydroxylases do not determine enantiomeric composition of pheromonal ipsdienol in Ips spp. J. Chem. Ecol. 34, 1584–1592 [DOI] [PubMed] [Google Scholar]
- 6.Sandstrom P., Welch W.H., Blomquist G.J., Tittiger C. (2006) Functional expression of a bark beetle cytochrome P450 that hydroxylates myrcene to ipsdienol. Insect Biochem. Mol. Biol. 36, 835–845 [DOI] [PubMed] [Google Scholar]
- 7.Song M., Kim A.C., Gorzalski A.J., MacLean M., Young S., Ginzel M.D., Blomquist G.J., Tittiger C. (2013) Functional characterization of myrcene hydroxylases from two geographically distinct Ips pini populations. Insect Biochem. Mol. Biol. 43, 336–343 [DOI] [PubMed] [Google Scholar]
- 8.Figueroa-Teran R., Welch W.H., Blomquist G.J., Tittiger C. (2012) Ipsdienol dehydrogenase (IDOLDH): a novel oxidoreductase important for Ips pini pheromone production. Insect Biochem. Mol. Biol. 42, 81–90 [DOI] [PubMed] [Google Scholar]
- 9.Kavanagh K., Jörnvall H., Persson B., Oppermann U. (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 65, 3895–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarboe L. (2011) YqhD: a broad-substrate range aldehyde reductase with various applications in production of biorenewable fuels and chemicals. Appl. Microbiol. Biotechnol. 89, 249–257 [DOI] [PubMed] [Google Scholar]
- 11.Lin S.X., Shi R., Qiu W., Azzi A., Zhu D.W., Dabbagh H.A., Zhou M. (2006) Structural basis of the multispecificity demonstrated by 17β-hydroxysteroid dehydrogenase types 1 and 5. Mol. Cell. Endocrinol. 248, 38–46 [DOI] [PubMed] [Google Scholar]
- 12.Ye Q., Yan M., Yao Z., Xu L., Cao H., Li Z., Chen Y., Li S., Bai J., Xiong J., Ying H., Ouyang P. (2009) A new member of the short-chain dehydrogenases/reductases superfamily: purification, characterization and substrate specificity of a recombinant carbonyl reductase from Pichia stipitis. Bioresour. Technol. 100, 6022–6027 [DOI] [PubMed] [Google Scholar]
- 13.Mayoral J.G., Leonard K.T., Nouzova M., Noriega F.G., Defelipe L.A., Turjanski A.G. (2013) Functional analysis of a mosquito short-chain dehydrogenase cluster. Arch. Insect Biochem. Physiol. 82, 96–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman F., Sotriffer C., Evers A., Xiong G., Maser E. (2006) Structural aspects of oligomerization in 3a-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni: new approaches for efficient protein design in Enzymology and Molecular Biology of Carbonyl Metabolism (Weiner H., Plapp H., Lindahl and Maser E., eds) pp. 308–314, Purdue University Press, Lafayette, IN [Google Scholar]
- 15.Ashburner M. (1998) Speculations on the subject of alcohol dehydrogenase and its properties in Drosophila and other flies. BioEssays 20, 949–954 [DOI] [PubMed] [Google Scholar]
- 16.Zhang J., Dean A.M., Brunet F., Long M. (2004) Evolving protein functional diversity in new genes of Drosophila. Proc. Natl. Acad. Sci. U. S. A. 101, 16246–16250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song M., Delaplain P., Nguyen T.T., Liu X., Wickenberg L., Jeffrey C., Blomquist G.J., Tittiger C. exo-Brevicomin biosynthetic pathway enzymes from the Mountain Pine Beetle, Dendroctonus ponderosae. Insect Biochem. Mol. Biol. 2014. Oct;53:73–80 [DOI] [PubMed] [Google Scholar]
- 18.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. (1951) Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 19.Sutherland E.W., Cori C.F., Haynes R., Olsen N.S. (1949) Purification of the hyperglycemic-glycogenolytic factor from insulin and from gastric mucosa. J. Biol. Chem. 180, 825–837 [PubMed] [Google Scholar]
- 20.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., Thompson J.D., Gibson T.J., Higgins D.G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 21.Bryson K., McGuffin L.J., Marsden R.L., Ward J.J., Sodhi J.S., Jones D.T. (2007) Protein structure prediction servers at University College London. Nucleic Acids Res. 33, W36–W38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Decker L.A. (1977) Worthington Enzyme Manual. Worthington Biochemical Corporation, Lakewood, NJ [Google Scholar]
- 23.Joernvall H., Danielsson O., Hjelmqvist L., Persson B., Shafqat J. (1995) The alcohol dehydrogenase system. Adv. Exp. Med. Biol. 372, 281–294 [DOI] [PubMed] [Google Scholar]
- 24.Powell A.J., Read J.A., Banfield M.J., Gunn-Moore F., Yan S.D., Lustbader J., Stern A.R., Stern D.M., Brady R.L. (2000) Recognition of structurally diverse substrates by type II 3-hydroxyacyl-CoA dehydrogenase (HADH II)/Amyloid-β binding alcohol dehydrogenase (ABAD). J. Mol. Biol. 303, 311–327 [DOI] [PubMed] [Google Scholar]
- 25.Rossman M., Liljas A., Branden C., Banaszak J. (1975) Evolutionary and structural relationships among dehydrogenases in The Enzymes (Boyer P., ed.) pp. 61–102, Academic Press, New York [Google Scholar]
- 26.Burse A., Schmidt A., Frick S., Kuhn J., Gershenzon J., Boland W. (2007) Iridoid biosynthesis in Chrysomelina larvae: fat body produces early terpenoid precursors. Insect Biochem. Mol. Biol. 37, 255–265 [DOI] [PubMed] [Google Scholar]
- 27.Seybold S.J., Quilici D.R., Tillman J.A., Vanderwel D., Wood D.L., Blomquist G.J. (1995) De novo biosynthesis of the aggregation pheromone components ipsenol and ipsdienol by the pine bark beetles Ips paraconfusus Lanier and Ips pini (Say) (Coleoptera: Scolytidae). Proc. Natl. Acad. Sci. U. S. A. 92, 8393–8397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keeling C.I., Blomquist G.J., Tittiger C. (2004) Coordinated gene expression for pheromone biosynthesis in the pine engraver beetle, Ips pini (Coleoptera: Scolytidae). Naturwissenschaften 91, 324–328 [DOI] [PubMed] [Google Scholar]
- 29.Coppedge B.R., Jones J.M., Felton G.W., Stephen F.M. (1994) Examination of midgut proteinases of the adult Southern Pine Beetle (Coleoptera: Scolytidae). J. Entomol. Sci. 29, 457–465 [Google Scholar]
- 30.Englard S., Seifter S. (1990) Precipitation techniques. Meth. Enzymol. 182, 287–300 [DOI] [PubMed] [Google Scholar]
- 31.Ray M., Ray S. (1985) L-Threonine dehydrogenase from goat liver. Feedback inhibition by methylglyoxal. J. Biol. Chem. 260, 5913–5918 [PubMed] [Google Scholar]
- 32.Ditlow C.C., Holmquist B., Morelock M.M., Vallee B.L. (1984) Physical and enzymic properties of a class II alcohol dehydrogenase isozyme of human liver:.pi.-ADH. Biochemistry 23, 6363–6368 [DOI] [PubMed] [Google Scholar]
- 33.Mayoral J.G., Nouzova M., Navare A., Noriega F.G. (2009) NADP+-dependent farnesol dehydrogenase, a corpora allata enzyme involved in juvenile hormone synthesis. Proc. Natl. Acad. Sci. U. S. A. 106, 21091–21096 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.