


Abstract
Rad26, a DNA dependent ATPase that is homologous to human CSB, has been well known to play an important role in transcription coupled DNA repair (TCR) in the yeast Saccharomyces cerevisiae. Sen1, a DNA/RNA helicase that is essential for yeast cell viability and homologous to human senataxin, has been known to be required for transcriptional termination of short noncoding RNA genes and for a fail-safe transcriptional termination mechanism of protein-coding genes. Sen1 has also been shown to protect the yeast genome from transcription-associated recombination by resolving RNA:DNA hybrids naturally formed during transcription. Here, we show that the N-terminal non-essential region of Sen1 plays an important role in TCR, whereas the C-terminal nonessential region and the helicase activity of Sen1 are largely dispensable for the repair. Unlike Rad26, which becomes completely dispensable for TCR in cells lacking the TCR repressor Spt4, Sen1 is still required for efficient TCR in the absence of Spt4. Also unlike Rad26, which is important for repair at many but not all damaged sites in the transcribed strand of a gene, Sen1 is required for efficient repair at essentially all the damaged sites. Our results indicate that Sen1 plays a more direct role than Rad26 in TCR.
INTRODUCTION
Nucleotide excision repair (NER) is a highly conserved ‘cut-and-patch’ DNA repair pathway that removes bulky and/or helix-distorting DNA lesions, such as UV induced cyclobutane pyrimidine dimers (CPDs) (1,2). Transcription coupled repair (TCR) is a subpathway of NER that is dedicated to rapid removal of DNA lesions in the transcribed strand of actively transcribed genes. Global genomic repair (GGR) is the other subpathway of NER that removes lesions throughout the genome including the non-transcribed strand of actively transcribed genes. The two NER subpathways share most factors during the repair process, but differ in the initial damage recognition step.
TCR is triggered by an RNA polymerase, which is not a damage sensor per se but provides damage recognition specificity by a mechanism referred to as ‘recognition by proxy’ (3). In Escherichia coli, the transcription-repair coupling factor Mfd, a DNA-stimulated ATPase, binds to and displaces a lesion-stalled RNA polymerase by pushing it forward and concurrently recruits the NER machinery to the lesion site (4–7). Upon binding to ATP, Mfd also directly binds to DNA in a manner that the DNA wraps around the protein (8). The DNA binding activity may anchor Mfd for carrying out its transcription-repair coupling function (9). UvrD, a DNA helicase, has also been shown to facilitate TCR in E. coli (10). Unlike Mfd, UvrD forces the RNA polymerase to backtrack, thereby exposing DNA lesions for access of the repair machinery.
In eukaryotic cells, TCR occurs in RNA polymerase II (RNAP II) transcribed genes (11–13). A certain level of TCR has also been shown to occur in RNA polymerase I transcribed genes in the yeast Saccharomyces cerevisiae (14). It has been known for over two decades that Rad26, a DNA-dependent ATPase that is homologous to the human Cockayne syndrome B (CSB) protein, plays an important role in TCR in S. cerevisiae (15). It was initially assumed that Rad26 may facilitate TCR by serving as a transcription-repair coupling factor, like Mfd in E. coli. However, we and others have found that Rad26 is completely or partially dispensable for TCR in yeast cells lacking a number of TCR repressors, including Rpb4 (16), Spt4 (17), Spt5 (18,19) and any subunit of the 5-subunit RNAP II associated factor complex (PAFc) (20). Rpb4 is a non-essential subunit of RNAP II and forms a subcomplex with Rpb7, another subunit of RNAP II (21–23). Spt4 and Spt5 are transcription elongation factors that form a complex (24). PAFc accompanies RNAP II throughout a gene and is involved in numerous transcription-related processes (25). It appears that the coordinated interactions of these TCR repressors with RNAP II hold the RNAP II complex in a closed conformation that is highly competent for transcription elongation but intrinsically repressive to TCR (19). The dispensability of Rad26 for TCR in the absence of a TCR repressor suggests that, instead of serving as a transcription-repair coupling factor, Rad26 may facilitate TCR indirectly by antagonizing the TCR repressors (2,12).
Sen1 is a yeast ATPase-helicase that is the homolog of human senataxin, a protein implicated in ataxia ocular apraxia 2 (AOA2) (26) and an autosomal dominant form of juvenile amyotrophic lateral sclerosis (ALS4) (27). Sen1 is essential for yeast cell viability and has been shown to be required for expression, maturation and termination of diverse classes of non-protein-coding RNAs (28–31). Sen1 has also been shown to directly bind to nascent protein-coding RNAs (32), affect RNAP II distribution (33), and provide a fail-safe transcriptional termination mechanism of protein-coding genes (34). Furthermore, Sen1 has been shown to protect the genome from transcription-associated recombination by resolving RNA:DNA hybrids (R-loops) naturally formed during transcription (34,35).
A role for Sen1 in TCR was suggested by yeast-two-hybrid and immuno-pulldown assays, which showed that Sen1 interacts with Rad2 (36), a single-stranded DNA endonuclease that is essential for incision on the 3′ side of a lesion during NER (both GGR and TCR) (2). Rad2 interacts with and stabilizes TFIIH (37), a 10-subunit complex that is essential for both transcription initiation and NER (38). The Rad2-TFIIH interaction seems to be well conserved, as XPG, the human homolog of Rad2, also interacts with and stabilizes TFIIH in human cells (39). To date, whether Sen1 is implicated in TCR has not been directly tested. Here, we present evidence that Sen1 plays a more direct role than Rad26 in TCR.
MATERIALS AND METHODS
Yeast plasmids and strains
Plasmid vectors used in this study are pRS415 and pRS416, which bear LEU2 and URA3 as selection markers, respectively (40). SEN1 gene fragments containing the native promoter and 3′ terminator and encoding the wild type, truncated or point mutant Sen1 proteins (Figure 1B) were amplified by PCR and inserted between the SacI and XhoI sites of the vectors. Plasmid pKS212, which bears a 1 kb EcoRI-XhoI fragment of the RPB2 gene on the Bluescript pKS+ vector (41), was used for generating strand-specific RNA probes for the RPB2 gene.
Figure 1.

Sen1 domains. (A) Schematic of Sen1 protein. Numbers indicate amino acid residue positions. The regions that interact with Rad2, Rnt1 and Rpb1, and with Nab3 are indicated by the bars below the schematic. (B) Sen1 proteins with truncations and point mutations used for analyses of UV induced DNA lesions. Yeast-two-hybrid interactions of some of the point mutations with Rad2, Rnt1 and Rpb1 are shown on the right. Based on (36,47,48).
All yeast strains used in this study were derivatives of BJ5465 (MATa ura3-52 trp1 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1.6R can1) (42). Deletions of RAD2, RAD7, RAD26 and SPT4 genes were done by using procedures as described previously (16,19). To delete the genomic SEN1 gene which is essential for cell viability, yeast cells were first transformed with a pRS416-based plasmid encoding SEN1. The genomic SEN1 gene was then deleted using standard procedures. To create yeast cells specifically expressing the truncated and point mutant Sen1, the genomic SEN1-deleted cells were transformed with pRS415-based plasmids encoding the different Sen1 proteins. The transformants were cultured on plates containing uracil but not leucine to select for the LEU2 plasmids and allow the loss of the URA3 plasmid. The colonies were replica-plated onto plates containing 5-fluoroorotic acid, which is toxic to cells with a functional URA3 gene (43), to select for cells that had lost the pRS416-based plasmid. The loss of the pRS416-based plasmid was confirmed by PCR.
Tests of UV sensitivity
Yeast cells were grown at 30°C in synthetic dextrose (SD) medium to saturation, and 10-fold serial dilutions were made. The diluted yeast cells were spotted onto YPD (1% yeast extract, 2% peptone and 2% dextrose) plates, irradiated with appropriate doses of UV (254 nm) and incubated at 30°C in the dark. The plates were photographed after all the viable cells were able to form distinct colonies in a spot of well-diluted cells (3–5 days). The UV sensitivities were assessed by evaluating the numbers of distinct colonies (rather than by the overall cell growth intensities) in the spots.
Repair analysis of UV induced CPDs
Yeast cells were grown at 30°C in SD medium to late log phase (A600 ≈ 1.0), washed with ice-cold H2O, resuspended in 2% dextrose and irradiated with 120 J/m2 of UV. One‐tenth volume of a stock solution containing 10% yeast extract and 20% peptone was added to the irradiated cell suspension, and the cells were incubated in the dark at 30°C. At different times of the repair incubation, aliquots were removed and the genomic DNA was isolated using a hot SDS procedure as described previously (44).
To analyze repair of CPDs at specific sites, a nucleotide-resolution method was used (45,46). One of the most obvious advantages of using the nucleotide-resolution method is that repair rates at different sites of a DNA fragment can be resolved on the same DNA sequencing gel, and thus a difference of repair among different sites or regions, where different NER mechanisms may be active, can be unambiguously identified. Briefly, the RPB2 gene fragment was released from the genomic DNA by restriction digestion (DraI) and the DNA was incised at all CPD sites by using an excess amount of T4 endonuclease V (Epicentre). The RPB2 fragments were fished out from other genomic fragments by using biotinylated oligonucleotides and streptavidin magnetic beads. The RPB2 fragments were 3′ end labeled with [α-32P]dATP and Sequenase Version 2.0 (Affymetrix), resolved on sequencing gels and exposed to PhosphorImager screens. The intensities of gel bands corresponding to CPD sites were quantified by using the Quantity One software (Bio-Rad). Only GGR but not TCR is operative in the region that is over 40 nucleotides upstream of the transcription start site of the RPB2 gene. For analyzing TCR in the transcribed strand of the RPB2 gene in GGR-deficient rad7Δ cells, the band intensities in the region that is over 40 nucleotides upstream stream of the transcription start site were used to normalize signals of different gel lanes. The time required for repairing 50% of CPDs (t1/2) were calculated by linear or second-order polynomial regression of CPDs remaining at different time points of the repair incubation.
As GGR is operative at all sites along a gene, it is impossible to accurately normalize the signals among different gel lanes by using the signal intensities at certain CPD sites on a sequencing gel. We therefore also used a Southern blot assay to analyze GGR in the non-transcribed strand of the RPB2 gene. Briefly, 0.2 μg of genomic DNA was digested with Acc65I and PvuI to release the 4.5 kb RPB2 fragment. Half of each sample was mock-treated and the other half was treated with an excess amount of T4 endonuclease V to incise at CPDs. The DNA was electrophoresed in 0.8% alkaline agarose gels, blotted to positively charged Nylon membranes (Hybond-N+, GE Healthcare Life Sciences). The non-transcribed strand of the RPB2 gene was hybridized with [α-32P] UTP labeled RNA probes. The probes were synthesized by cleaving plasmid pKS212 (41) with EcoRI and incubating the linearized plasmid with rNTPs, [α-32P] UTP and T3 RNA polymerase (New England Biolabs) under conditions recommended by the manufacturer. The probe-hybridized membranes were exposed to PhosphorImager screens and the bands were quantified by using the Quantity One software. The CPD content in the fragment was calculated by using the equation – ln(RFa/RFb), where RFa and RFb represent the signal intensities of the intact restriction fragments of the T4 endonuclease V and mock-treated DNA, respectively.
RESULTS
The N-terminal non-essential region of Sen1 is important for TCR, whereas the C-terminal nonessential region plays a minor role in the repair process
Sen1 is essential for yeast cell viability. The minimal essential region of Sen1 corresponds to the ATPase-helicase domain and one of the two flanking nuclear localization sequences (47) (Figure 1A). To determine whether Sen1 plays a role in TCR, we analyzed repair of UV induced CPDs in the transcribed strand of the RPB2 gene in rad7Δ and rad7Δ rad26Δ cells expressing Sen1 proteins with truncations or point mutations (Figure 2). Rad7 is essential for GGR, but completely dispensable for TCR (2). Therefore, the overall TCR and Rad26-independent TCR can be unambiguously analyzed in rad7Δ and rad7Δ rad26Δ cells, respectively. A nucleotide resolution method, which utilizes biotinylated oligonucleotides and streptavidin magnetic beads to facilitate fishing-out and strand-specific end-labeling of specific DNA fragments, was used for the analysis (45,46). As expected, TCR was fast in the transcribed strand of the RPB2 gene in rad7Δ (SEN1+) cells (Figures 2A, 3A and 4A). As only GGR but not TCR is operative in the region that is over 40 nucleotides upstream of the RPB2 gene, no apparent repair can be seen in this upstream region in rad7 cells (Figure 2A, marked with the open bar at the bottom-left; Figure 3A). Therefore, the band intensities in this upstream region can be used to unambiguously normalize the signals of different gel lanes. rad7Δ rad26Δ cells showed decreased TCR in the coding region of the RPB2 gene, except for certain sites especially those in the short region (∼50 nucleotides) immediately downstream of the transcription start site (Figures 2E and 3).
Figure 2.

DNA sequencing gels showing repair of CPDs in the 1.1 kb DraI fragment of the transcribed strand of the RPB2 gene in rad7Δ and rad7Δ rad26Δ cells expressing the wild type and different Sen1 mutants. Samples were from unirradiated (U) and UV irradiated cells at different times (h) of repair incubation. The top strong band corresponds to the full-length restriction fragment of the RPB2 gene containing no DNA lesion. Bands below the top band correspond to CPD sites. Decrease of band intensities with time reflects repair. Approximate nucleotide positions relative to the transcription start site of the RPB2 gene are indicated on the left of (A). Open bar at the bottom-left of (A) marks the upstream region of the RPB2 gene where only GGR but not TCR is operative and thus is not repaired in rad7Δ cells. The band intensities in this upstream region can be used to normalize signals in different lanes of the gels. Bracket on the left of (E) indicates the region where robust Rad26-independent TCR occurs.
Figure 3.

Time (h) required for repairing 50% (t1/2) of CPDs at individual sites of the transcribed strand of the RPB2 gene in rad7Δ and rad7Δ rad26Δ cells expressing the wild type and different Sen1 mutants. The nucleotide numbers are relative to the transcription start site of the RPB2 gene. The t1/2 values were calculated by linear or second-order polynomial regression of the CPDs remaining at different repair times.
Figure 4.

Plots showing percent CPDs remaining in the coding region of the transcribed strand of the RPB2 gene in (A) rad7Δ, (B) rad7Δ rad26Δ, (C) rad7Δ Spt4Δ and (D) rad7Δ rad26Δ spt4Δ cells expressing the wild type (red symbols) and different Sen1 mutants (black symbols). The means (± SD) of CPDs remaining at each indicated repair time in cells expressing the N-, and N- and C-terminal truncated Sen1 (sen1-NΔ/NΔ+CΔ, which include sen1-975-2231, sen1-1089-2231, sen1-1004-1907 and sen1-1089-1929) were plotted in blue. The TCR rates in the sen1-NΔ/NΔ+CΔ cells were significantly slower than those in wild type cells (P < 0.01, Student's t-test).
Truncation of the N- or N- and C-terminal non-essential regions of Sen1 significantly reduced TCR at essentially all sites in the RPB2 gene, including those in the short region immediately downstream of the transcription start site (Figure 2, compare B and C with A, and F and G with E; Figure 3; Figure 4A and B; Supplementary Figure S1). Indeed, TCR at most sites of the RPB2 gene in rad7Δ sen1-1004-1907, rad7Δ sen1-1089-1929 and rad7Δ sen1-1089-2231 cells were even significantly slower than that in rad7Δ rad26Δ cells (Figure 2, compare B and C with E; Figure 3; Figure 4A and B). Truncation of the entire C-terminal nonessential region of Sen1 caused a mild TCR deficiency (Figure 2, compare D with A, and H with E; Figure 3; Figure 4A and B). These results indicate that the N-terminal non-essential region of Sen1 plays a more important role in TCR than the C-terminal non-essential region.
The N-terminal 565 residues of Sen1 have been shown by yeast-two-hybrid assays to be involved in interactions with Rad2, Rpb1 (the largest subunit of RNAP II) and Rnt1 (an RNA processing factor) (48). A number of point mutations in the N-terminal region of Sen1 were shown to abolish all or some of the interactions (48). We found that truncation of the N-terminal 565 residues caused a TCR deficiency (not shown) that is similar to that caused by truncation of the N-terminal 975 residues (Supplementary Figure S1, panels C and G). However, E58K, K128E, R197E and R302W mutations of Sen1, which were shown to abolish interaction with Rad2, Rnt1 and/or Rpb1 (48) (Figure 1B), caused no significant TCR deficiency (not shown).
Rpb9, a non-essential subunit of RNAP II, has been shown to be required for Rad26-independent TCR (16). To determine the functional interactions of Sen1 and Rpb9 during TCR, we attempted to create rad7Δ rpb9Δ and rad7Δ rad26Δ rpb9Δ cells expressing the N- or N- and C-terminal non-essential region truncated Sen1. However, these cells appear to be inviable. Therefore, Rpb9 and the N-terminal non-essential region of Sen1 may complement with each other for cell viability.
The ATPase-helicase activity of Sen1 plays a minor role in TCR
The E1597 residue is located in the ATPase-helicase domain of Sen1 and forms an intra-domain salt bridge with R1641 (47). The E1597K mutation has been shown to abolish its ATPase-helicase activity and compromise transcription termination (28,33,49,50). Interestingly, this mutation only caused a mild TCR deficiency (Figure 4A and B; Supplementary Figure S1, compare D with A, and H with E), indicating that the helicase activity of Sen1 plays a minor role in TCR. In agreement with this notion, the G1747D mutation, which also abolishes its ATPase-helicase activity (51), also caused a mild TCR deficiency (similar to that caused by the E1597K mutation) (not shown).
To examine if the ATPase-helicase activity of Sen1 acts synergistically or additively with its N-terminal non-essential region in TCR, we attempted to create yeast cells expressing Sen1 with the N-terminal 975 residue truncation and the E1597K or G1747D mutation. However, Sen1 with both the truncation and point mutation appears to be lethal for the cell.
Deletion of SPT4 cannot fully restore TCR in cells expressing the N- or N- and C-terminal truncated Sen1
In agreement with previous reports showing that deletion of SPT4 can fully restore TCR in cells lacking Rad26 (17), the TCR rate in rad7Δ rad26Δ spt4Δ cells was similar to that in rad7Δ cells (compare Figures 2A, 2E and 5E; Figure 4A, B and D). However, TCR rates in rad7Δ rad26Δ spt4Δ cells expressing the N- or N- and C-terminal truncated Sen1 were much slower than those in rad7Δ rad26Δ spt4Δ (SEN1+) (Figure 5, compare F and G with E; Supplementary Figure S2, compare E and F with D) and rad7Δ (SEN1+) (compare Figure 5F and G with Figure 2A; Figure 4, compare A with D) cells, indicating that TCR cannot be fully restored by deleting Spt4 in cells expressing the truncated Sen1. Also, TCR rates in rad7Δ spt4Δ cells expressing the N- or N- and C-terminal truncated Sen1 were significantly slower than those in rad7Δ spt4Δ (SEN1+) (Figure 5, compare B and C with A; Supplementary Figure S2, compare B and C with A) and rad7Δ (SEN1+) (compare Figure 5B and C with Figure 2A; Figure 4, compare A and C) cells, further indicating that deletion of SPT4 cannot fully restore TCR in the Sen1 truncation mutant cells. These results suggest that Sen1 plays a more direct role than Rad26 in TCR, and is not completely dispensable for the repair even in the absence of the TCR repressor Spt4.
Figure 5.

DNA sequencing gels showing repair of CPDs in the transcribed strand of the RPB2 gene in rad7Δ spt4Δ and rad7Δ rad26Δ spt4Δ cells expressing wild type and different Sen1 mutants. See Figure 2 for more explanations of the labels.
The N- and C-terminal truncations of Sen1 do not significantly affect GGR
To determine if Sen1 also plays a role in GGR, we analyzed repair of CPDs at individual sites in the non-transcribed strand of the RPB2 gene in cells expressing the N- and C-terminal non-essential region truncated Sen1. Cells with the truncated Sen1 showed essentially the same repair rate as those expressing the wild type Sen1 (Figure 6), indicating that the truncations do not significantly affect GGR.
Figure 6.
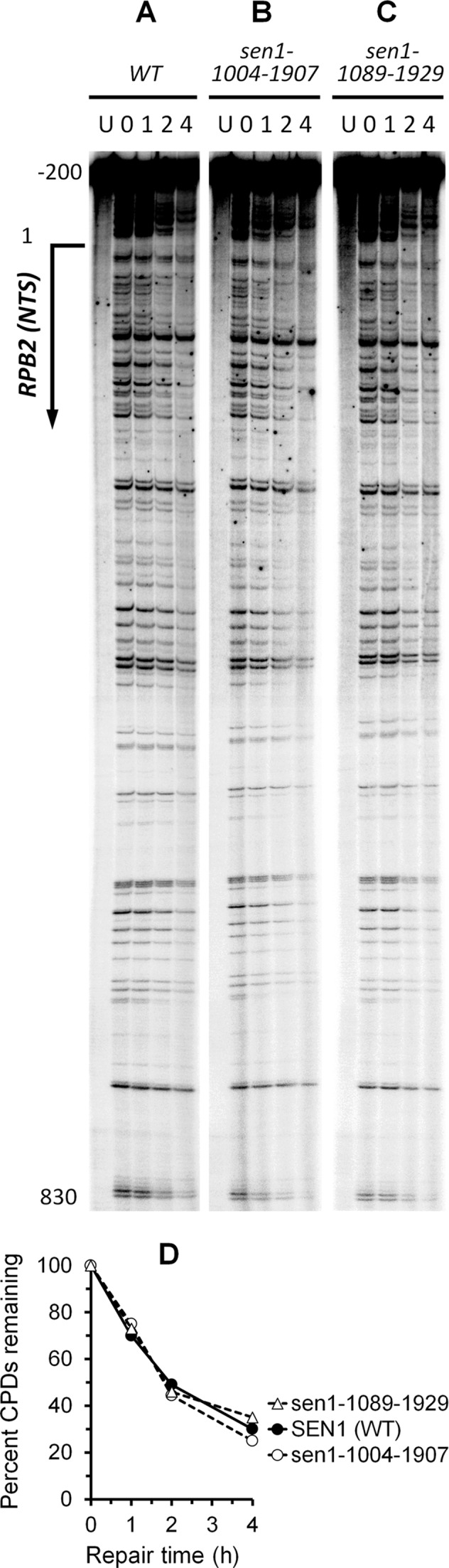
(A–C) DNA sequencing gels showing repair of CPDs in the 1.1 kb DraI fragment of the non-transcribed strand of the RPB2 gene in cells expressing wild type Sen1 (WT), and sen1-1004-1907 and sen1-1089-1929 mutants. (D) Plot showing percent CPDs remaining in the non-transcribed strand of the RPB2 gene in the wild type and mutant cells. Repair rates in cells expressing sen1-1089-1929 and sen1-1004-1907 mutants were not significantly different from those expressing the WT (Student's t-test). Error bars (SD) for the wild type and Sen1 mutants overlap and are not shown for clarity.
As GGR is operative at all sites along a gene, it is impossible to accurately normalize the signals among different gel lanes by using the signal intensities at certain CPD sites on a sequencing gel. We therefore further analyzed GGR by measuring overall repair of CPDs in the 4.5 kb Acc65I-PvuI fragment of the non-transcribed strand of the RPB2 gene by using Southern blot. Cells with the N- and C-terminal truncated Sen1 showed essentially the same repair rate as those expressing the wild type Sen1 (WT) (Figure 7), further indicating that the truncations do not significantly affect GGR.
Figure 7.

(A) Southern blot showing repair of CPDs in the 4.5 kb (Acc65I-PvuI) fragment of the non-transcribed strand of the RPB2 gene in cells expressing WT, and sen1-1004-1907 and sen1-1089-1929 mutants. (B) Plot showing percent CPDs remaining in the non-transcribed strand of the RPB2 gene in the wild type and mutant cells. The values shown are averages of two experiments (line bars indicate the range of the two experimental values).
Epistatic interactions of SEN1 with RAD26, RAD7 and RAD2
To determine if the role of Sen1 in DNA repair is limited to TCR, we analyzed epistatic interactions of SEN1 with RAD7, RAD26 and RAD2. In agreement with previous reports (15,16,52), deletion of RAD26 did not significantly increase the UV sensitivity of otherwise wild type cells (Figure 8, compare SEN1 samples in panels A and B), but moderately increased the UV sensitivities of rad7Δ cells (Figure 8, compare SEN1 samples in panels D and E). The sen1-1089-1929 mutant increased UV sensitivities of all the yeast strains tested (Figure 8), indicating that this mutant has deficiencies not only in TCR but also in a non-NER pathway that is implicated in repair and/or tolerance of UV induced DNA lesions. How the sen1-1089-1929 mutant is implicated in the non-NER pathway was not pursued in this study. All the other N- or N- and C-terminal truncated Sen1 mutants did not increase the UV sensitivities of rad26Δ, rad2Δ or otherwise wild type cells (Figure 8A, B and C), but increased the UV sensitivities of rad7Δ and rad7Δ rad26Δ cells (Figure 8D and E). This indicates that the deficiencies of the latter Sen1 truncation mutants in repairing UV lesions are limited to TCR. The sen1-E1597K (Figure 8E and data not shown) and sen1-G1747D (not shown) mutants did not significantly increase the UV sensitivities of all the yeast strains tested, indicating that the ATPase-helicase activity of Sen1 does not play a major role in NER (TCR or GGR) or other DNA repair pathways responsible for removal and/or tolerance of UV lesions.
Figure 8.

Effects of Sen1 truncation and point mutations on UV sensitivities of yeast cells with different NER subpathways operative. Panels show growth of 10-fold serially diluted yeast cells spotted on plates after different doses of UV irradiation. The plates were photographed after all the viable cells were able to form distinct colonies in a spot of well-diluted cells (3–5 days). The UV sensitivities were assessed by evaluating the numbers of distinct colonies (rather than by the overall cell growth intensities) in the spots.
DISCUSSION
We showed here that Sen1 plays an important role in TCR. While deletion of SPT4 in rad26Δ cells can fully restore TCR, the deletion can only partially restore TCR in cells expressing the N- or N- and C-terminal non-essential region truncated Sen1. Therefore, unlike Rad26, which appears to facilitate TCR solely by antagonizing TCR repressors, Sen1 may facilitate TCR not only by antagonizing TCR repressors but also through a more direct mechanism.
In view of the yeast-hybrid studies showing that Sen1 interacts with Rpb1, the largest subunit of RNAP II and Rad2, an essential NER factor (36,48), it is tempting to propose that Sen1 may be a true transcription-repair coupling factor in yeast, like Mfd in E. coli. However, although deletion of the N-terminal 565 residues of Sen1 caused a TCR deficiency, point mutations in this region that were shown to abolish Sen1 interactions with Rpb1 and/or Rad2 did not seem to cause any TCR defect. One possibility is that the interactions are not important for TCR. Alternatively, unlike the yeast-two-hybrid situations, the point mutations may not abolish Sen1 interactions with Rpb1 and/or Rad2 under native conditions. Also, while the ATPase activity of Mfd is essential for its TCR activity in E. coli (7), the ATPase activity of Sen1 seems to play a minor role in the yeast TCR. Therefore, the functional mechanism of Sen1 in yeast TCR may not be identical to that of Mfd in E. coli TCR. Furthermore, the helicase activity of UvrD appears to be required for its TCR function in E. coli (10). Sen1 may also behave differently from UvrD in TCR.
The ATPase-helicase domain of Sen1 is essential for cell viability. Although the helicase activity of Sen1 plays a minor role in TCR, the importance of this domain for this repair pathway remains to be elucidated. In addition to its helicase activity, this domain may play a structural role in TCR. The residual TCR in rad7Δ and rad7Δ rad26Δ cells expressing the N- or N- and C-terminal truncated Sen1 might be accomplished by the ATPase-helicase domain. The partially restored TCR in rad7Δ spt4Δ and rad7Δ rad26Δ spt4Δ cells expressing the truncated Sen1 might also be due to the ATPase-helicase domain. If this is the case, all TCR subpathways, Rad26-dependent and independent, may be eventually dependent on Sen1. Identification and characterization of potential TCR-deficient mutations in the ATPase-helicase domain of Sen1 may be greatly informative for addressing the question.
It has been well-known that Sen1 is a subunit of the Nrd1-Nab3-Sen1 complex (53). Sen1 directly interacts with Nab3 but not with Nrd1 (31,36,54,55). Yeast-two-hybrid assays have shown that the C-terminal nonessential region of Sen1 that interacts with Nab3 includes residues 1890–2092 (55) (Figure 1A). We found here that truncation of the whole C-terminal non-essential region (residues 1859–2231) of Sen1 caused a minor TCR deficiency. It is therefore likely that the interaction of Sen1 with Nab3 (and indirectly with Nrd1) may not play a major role in TCR.
In short, we identified Sen1 as a new TCR facilitator in yeast. How Sen1 functions in TCR remains to be elucidated. Also, if its homolog senataxin plays a similar role in TCR in human cells remains to be tested.
Supplementary Material
Acknowledgments
The authors thank their laboratory members for inspiring discussion.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB-1244019]. Funding for open access charge: National Science Foundation [MCB-1244019].
Conflict of interest statement. None declared.
REFERENCES
- 1.Scharer O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013;5:a012609. doi: 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tatum D., Li S. In: DNA Repair - On the Pathways to Fixing DNA Damage and Errors. Storici F, editor. InTech; 2011. pp. 97–122. [Google Scholar]
- 3.Sancar A., Lindsey-Boltz L.A., Unsal-Kacmaz K., Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 4.Deaconescu A.M., Chambers A.L., Smith A.J., Nickels B.E., Hochschild A., Savery N.J., Darst S.A. Structural basis for bacterial transcription-coupled DNA repair. Cell. 2006;124:507–520. doi: 10.1016/j.cell.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 5.Howan K., Smith A.J., Westblade L.F., Joly N., Grange W., Zorman S., Darst S.A., Savery N.J., Strick T.R. Initiation of transcription-coupled repair characterized at single-molecule resolution. Nature. 2012;490:431–434. doi: 10.1038/nature11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park J.S., Marr M.T., Roberts J.W. E. coli Transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell. 2002;109:757–767. doi: 10.1016/s0092-8674(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 7.Selby C.P., Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 8.Selby C.P., Sancar A. Structure and function of transcription-repair coupling factor. I. Structural domains and binding properties. J. Biol. Chem. 1995;270:4882–4889. doi: 10.1074/jbc.270.9.4882. [DOI] [PubMed] [Google Scholar]
- 9.Selby C.P., Sancar A. Structure and function of transcription-repair coupling factor. II. Catalytic properties. J. Biol. Chem. 1995;270:4890–4895. doi: 10.1074/jbc.270.9.4890. [DOI] [PubMed] [Google Scholar]
- 10.Epshtein V., Kamarthapu V., McGary K., Svetlov V., Ueberheide B., Proshkin S., Mironov A., Nudler E. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature. 2014;505:372–377. doi: 10.1038/nature12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanawalt P.C., Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 12.Li S. Transcription coupled nucleotide excision repair in the yeast Saccharomyces cerevisiae: The ambiguous role of Rad26. DNA Repair (Amst) 2015;36:43–48. doi: 10.1016/j.dnarep.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Vermeulen W., Fousteri M. Mammalian transcription-coupled excision repair. Cold Spring Harb. Perspect. Biol. 2013;5:a012625. doi: 10.1101/cshperspect.a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conconi A., Bespalov V.A., Smerdon M.J. Transcription-coupled repair in RNA polymerase I-transcribed genes of yeast. Proc. Natl. Acad. Sci. U.S.A. 2002;99:649–654. doi: 10.1073/pnas.022373099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Gool A.J., Verhage R., Swagemakers S.M., van de Putte P., Brouwer J., Troelstra C., Bootsma D., Hoeijmakers J.H. RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J. 1994;13:5361–5369. doi: 10.1002/j.1460-2075.1994.tb06871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S., Smerdon M.J. Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J. 2002;21:5921–5929. doi: 10.1093/emboj/cdf589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jansen L.E., den Dulk H., Brouns R.M., de Ruijter M., Brandsma J.A., Brouwer J. Spt4 modulates Rad26 requirement in transcription-coupled nucleotide excision repair. EMBO J. 2000;19:6498–6507. doi: 10.1093/emboj/19.23.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding B., LeJeune D., Li S. The C-terminal repeat domain of Spt5 plays an important role in suppression of Rad26-independent transcription coupled repair. J. Biol. Chem. 2010;285:5317–5326. doi: 10.1074/jbc.M109.082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W.T., Giles C., Li S.S. Insights into how Spt5 functions in transcription elongation and repressing transcription coupled DNA repair. Nucleic Acids Res. 2014;42:7069–7083. doi: 10.1093/nar/gku333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tatum D., Li W., Placer M., Li S. Diverse roles of RNA polymerase II-associated factor 1 complex in different subpathways of nucleotide excision repair. J. Biol. Chem. 2011;286:30304–30313. doi: 10.1074/jbc.M111.252981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Armache K.J., Kettenberger H., Cramer P. Architecture of initiation-competent 12-subunit RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6964–6968. doi: 10.1073/pnas.1030608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armache K.J., Mitterweger S., Meinhart A., Cramer P. Structures of complete RNA polymerase II and its subcomplex, Rpb4/7. J. Biol. Chem. 2005;280:7131–7134. doi: 10.1074/jbc.M413038200. [DOI] [PubMed] [Google Scholar]
- 23.Bushnell D.A., Kornberg R.D. Complete, 12-subunit RNA polymerase II at 4.1-A resolution: implications for the initiation of transcription. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6969–6973. doi: 10.1073/pnas.1130601100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartzog G.A., Fu J. The Spt4-Spt5 complex: a multi-faceted regulator of transcription elongation. Biochim. Biophys. Acta. 2013;1829:105–115. doi: 10.1016/j.bbagrm.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaehning J.A. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim. Biophys. Acta. 2010;1799:379–388. doi: 10.1016/j.bbagrm.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreira M.C., Klur S., Watanabe M., Nemeth A.H., Le Ber I., Moniz J.C., Tranchant C., Aubourg P., Tazir M., Schols L., et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004;36:225–227. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y.Z., Bennett C.L., Huynh H.M., Blair I.P., Puls I., Irobi J., Dierick I., Abel A., Kennerson M.L., Rabin B.A., et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am. J. Hum. Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinmetz E.J., Brow D.A. Repression of gene expression by an exogenous sequence element acting in concert with a heterogeneous nuclear ribonucleoprotein-like protein, Nrd1, and the putative helicase Sen1. Mol. Cell. Biol. 1996;16:6993–7003. doi: 10.1128/mcb.16.12.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ursic D., Himmel K.L., Gurley K.A., Webb F., Culbertson M.R. The yeast SEN1 gene is required for the processing of diverse RNA classes. Nucleic Acids Res. 1997;25:4778–4785. doi: 10.1093/nar/25.23.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasmussen T.P., Culbertson M.R. The putative nucleic acid helicase Sen1p is required for formation and stability of termini and for maximal rates of synthesis and levels of accumulation of small nucleolar RNAs in Saccharomyces cerevisiae. Mol. Cell. Biol. 1998;18:6885–6896. doi: 10.1128/mcb.18.12.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinmetz E.J., Conrad N.K., Brow D.A., Corden J.L. RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature. 2001;413:327–331. doi: 10.1038/35095090. [DOI] [PubMed] [Google Scholar]
- 32.Creamer T.J., Darby M.M., Jamonnak N., Schaughency P., Hao H., Wheelan S.J., Corden J.L. Transcriptome-wide binding sites for components of the Saccharomyces cerevisiae non-poly(A) termination pathway: Nrd1, Nab3, and Sen1. PLoS Genet. 2011;7:e1002329. doi: 10.1371/journal.pgen.1002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinmetz E.J., Warren C.L., Kuehner J.N., Panbehi B., Ansari A.Z., Brow D.A. Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol. Cell. 2006;24:735–746. doi: 10.1016/j.molcel.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 34.Rondon A.G., Mischo H.E., Kawauchi J., Proudfoot N.J. Fail-safe transcriptional termination for protein-coding genes in S. cerevisiae. Mol. Cell. 2009;36:88–98. doi: 10.1016/j.molcel.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mischo H.E., Gomez-Gonzalez B., Grzechnik P., Rondon A.G., Wei W., Steinmetz L., Aguilera A., Proudfoot N.J. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell. 2011;41:21–32. doi: 10.1016/j.molcel.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ursic D., Chinchilla K., Finkel J.S., Culbertson M.R. Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res. 2004;32:2441–2452. doi: 10.1093/nar/gkh561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Habraken Y., Sung P., Prakash S., Prakash L. Transcription factor TFIIH and DNA endonuclease Rad2 constitute yeast nucleotide excision repair factor 3: implications for nucleotide excision repair and Cockayne syndrome. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10718–10722. doi: 10.1073/pnas.93.20.10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Compe E., Egly J.M. TFIIH: when transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 2012;13:343–354. doi: 10.1038/nrm3350. [DOI] [PubMed] [Google Scholar]
- 39.Ito S., Kuraoka I., Chymkowitch P., Compe E., Takedachi A., Ishigami C., Coin F., Egly J.M., Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol. Cell. 2007;26:231–243. doi: 10.1016/j.molcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 40.Sikorski R.S., Boeke J.D. In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- 41.Sweder K.S., Hanawalt P.C. Preferential repair of cyclobutane pyrimidine dimers in the transcribed strand of a gene in yeast chromosomes and plasmids is dependent on transcription. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10696–10700. doi: 10.1073/pnas.89.22.10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones E.W. Tackling the protease problem in Saccharomyces cerevisiae. Methods Enzymol. 1991;194:428–453. doi: 10.1016/0076-6879(91)94034-a. [DOI] [PubMed] [Google Scholar]
- 43.Boeke J.D., LaCroute F., Fink G.R. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- 44.Li S., Smerdon M.J. Nucleosome structure and repair of N-methylpurines in the GAL1-10 genes of Saccharomyces cerevisiae. J. Biol. Chem. 2002;277:44651–44659. doi: 10.1074/jbc.M206623200. [DOI] [PubMed] [Google Scholar]
- 45.Li S., Waters R. Nucleotide level detection of cyclobutane pyrimidine dimers using oligonucleotides and magnetic beads to facilitate labelling of DNA fragments incised at the dimers and chemical sequencing reference ladders. Carcinogenesis. 1996;17:1549–1552. doi: 10.1093/carcin/17.8.1549. [DOI] [PubMed] [Google Scholar]
- 46.Li S., Waters R., Smerdon M.J. Low- and high-resolution mapping of DNA damage at specific sites. Methods. 2000;22:170–179. doi: 10.1006/meth.2000.1058. [DOI] [PubMed] [Google Scholar]
- 47.Chen X., Muller U., Sundling K.E., Brow D.A. Saccharomyces cerevisiae Sen1 as a model for the study of mutations in human senataxin that elicit cerebellar ataxia. Genetics. 2014;198:577–590. doi: 10.1534/genetics.114.167585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chinchilla K., Rodriguez-Molina J.B., Ursic D., Finkel J.S., Ansari A.Z., Culbertson M.R. Interactions of Sen1, Nrd1, and Nab3 with multiple phosphorylated forms of the Rpb1 C-terminal domain in Saccharomyces cerevisiae. Eukaryot. Cell. 2012;11:417–429. doi: 10.1128/EC.05320-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeMarini D.J., Winey M., Ursic D., Webb F., Culbertson M.R. SEN1, a positive effector of tRNA-splicing endonuclease in Saccharomyces cerevisiae. Mol. Cell. Biol. 1992;12:2154–2164. doi: 10.1128/mcb.12.5.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim M., Vasiljeva L., Rando O.J., Zhelkovsky A., Moore C., Buratowski S. Distinct pathways for snoRNA and mRNA termination. Mol. Cell. 2006;24:723–734. doi: 10.1016/j.molcel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 51.Porrua O., Libri D. A bacterial-like mechanism for transcription termination by the Sen1p helicase in budding yeast. Nat. Struct. Mol. Biol. 2013;20:884–891. doi: 10.1038/nsmb.2592. [DOI] [PubMed] [Google Scholar]
- 52.Verhage R.A., van Gool A.J., de Groot N., Hoeijmakers J.H., van de Putte P., Brouwer J. Double mutants of Saccharomyces cerevisiae with alterations in global genome and transcription-coupled repair. Mol. Cell. Biol. 1996;16:496–502. doi: 10.1128/mcb.16.2.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porrua O., Libri D. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat. Rev. Mol. Cell Biol. 2015;16:190–202. doi: 10.1038/nrm3943. [DOI] [PubMed] [Google Scholar]
- 54.Conrad N.K., Wilson S.M., Steinmetz E.J., Patturajan M., Brow D.A., Swanson M.S., Corden J.L. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics. 2000;154:557–571. doi: 10.1093/genetics/154.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nedea E., Nalbant D., Xia D., Theoharis N.T., Suter B., Richardson C.J., Tatchell K., Kislinger T., Greenblatt J.F., Nagy P.L. The Glc7 phosphatase subunit of the cleavage and polyadenylation factor is essential for transcription termination on snoRNA genes. Mol. Cell. 2008;29:577–587. doi: 10.1016/j.molcel.2007.12.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.