

Abstract
A series of ferrous-heme 2,6-dimethylphenyl isocyanide (DIMPI) and ferrous-heme mononitrosyl complexes have been synthesized and characterized. The heme portion of the complexes studied is varied with respect to the nature of the axial ligand, including complexes, where it is covalently tethered to the porphyrinate periphery. Reduced heme complexes, [(F8)FeII], [(PPy) FeII], [(PIm)FeII], and [(PImH)FeII], where F8 = tetrakis(2,6-difluorophenyl)-porphyrinate and PPy, PIm, and PImH are partially fluorinated tetraaryl porphyrinates with covalently appended axial base pyridyl/imidazolyl or histamine moieties, were employed; PImH is a new construct. Room temperature addition of DIMPI to these iron(II) complexes affords the bis-isocyanide species [(F8)FeII-(DIMPI)2] in the case of [(F8)FeII], while for the other hemes, mono-DIMPI compounds are obtained, [(PPy)FeII-(DIMPI)] [(2)-DIMPI], [(PIm)FeII-(DIMPI)] [(3)-DIMPI], and [(PImH) FeII-(DIMPI)] [(4)-DIMPI]. The structures of complexes (3)-DIMPI and (4)-DIMPI have been determined by single crystal X-ray crystallography, where interesting H…F(porphryinate aryl group) interactions are observed. 19F-NMR spectra determined for these complexes suggest that H…F(porphyrinate aryl groups) attractions also occur in solution, the H atom coming either from the DIMPI methyl groups or from a porphyinate axial base imidazole or porphyrinate pyrrole. Similarly, we have used nitrogen monoxide to generate ferrous-nitrosyl complexes, a five-coordinate species for F8, [(F8)FeII-(NO)], or low-spin six-coordinate compounds [(PPy)FeII-(NO)], [(PIm)FeII-(NO)], and [(PImH)FeII-(NO)]. The DIMPI and mononitrosyl complexes have also been characterized using UV–Vis, IR, 1H-NMR, and EPR spectroscopies.
Keywords: Ferrous heme, Heme isocyanide, X-ray structures, Heme nitrosyl, H…F(porphyrinate) interactions
Graphical abstract
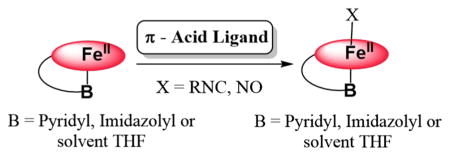
Introduction
Heme containing proteins participate in critical and diverse biological functions which include electron transfer, catalysis, and signaling. For the latter two subjects, small molecule diatomic gases are often involved, such as O2, NO, and CO [1, 2]. There exist classes of proteins which serve to discriminate between these molecules for purposes, including detection, signaling, and/or function [3, 4]. For molecular oxygen, roles include storage transfer, or activation of O2 for substrate oxidation or oxygenation chemistries [5]. Nitric oxide (nitrogen monoxide) is a signaling molecule [6] such as in its interaction with the heme center in guanylate cyclase, wherein binding leads to a signaling cascade resulting in smooth muscle relaxation [7–14]. Carbon monoxide is also a diatomic gas which is biosynthesized through heme O2-activation chemistry (i.e., in heme oxygenases [15–17]); CO can also act in biological signaling via heme protein binding [18].
In the history of the study of O2 interactions with hemoproteins, the investigation of the binding of diatomic surrogate ligands, mainly CO and NO, has received considerable attention. These have been utilized as structural models, but also are useful in the study of ligand binding dynamics and electronic structure of the ligated reduced hemes [19–23]. For example, CO bound hemes are amenable to vibrational spectrosopic analyses, along with CO photoejection and CO rebinding study [21, 24]. As well, reduced hemes with NO bound are active for EPR spectroscopic interrogation. The replacement of CO with isocyanide (RNC:) ligands has also been found to be a useful probe to investigate vibrational spectroscopy and binding kinetics or heme-ligand photodissociation and time-resolved rebinding. The strong isocyanide N–C triple-bond stretching vibration can be monitored, whereas variation in the size or nature of the isocyanide R-group, e.g., R = aryl vs −Me or −tBu, provides insights concerning steric effects or issues of small ligand binding to iron relative to the size or shape of a protein active-site pocket [25–30].
One of our research group’s major foci has been and continues to be the study of dioxygen binding and reduction at heme-copper heterobinuclear metal ion centers [31]. We seek to determine how neighboring copper-ligand moieties influence the binding of O2 to hemes, and in a complementary manner, see how hemes affect O2 binding to copper ion in varying ligand environments. Then, as such synthetic heme-O2-Cu assemblies can be compared and related to the active-site chemistry of heme-copper oxidases which bind and reduce O2 to two water molecules (while also translocating protons through a mitochondrial membrane which downstream facilitates ATP biosynthesis), we are interested in elucidating detailed insights into the O–O reductive cleavage process, as a function of the exact nature (structure and electronics/bonding behavior) of the heme, the copper ligand and the source of electrons (E° value) and protons (pKa). Additional factors include heme or copper-ligand electron-donating ability (and thus the FeIII/FeII and/or CuII/CuI E° value), nature of porphyrinate peripheral groups, and/or copper-ligand denticity and their possible steric influences or affects upon the entire heme-O2-Cu(ligand) structure, for example the Fe…Cu distance in the heme-O2-Cu assembly [31–35].
A specific example of such a synthetic construct is shown in Fig. 1, where also an added heme axial ‘base’, dicyclohexylimidazole (DCHIm), is present [35, 36]. As anticipated, the structural, spectroscopic properties and reactivity of this and other such assemblies significantly depend on the detailed nature of the heme, the copper ligand, the axial base, etc., as mentioned above [31, 36].
Fig. 1.

Synthetic steps involved in generation of a low-spin heme-peroxo-copper complex
As such, it is critical that complementary investigations be carried out on surrogate ligand binding to the various components of our assemblies. One such aspect is the investigation of O2, CO, NO, and/or RNC binding to varying designed porphyrinoids, wherein the axial base ligand is varied between a weak O-donor (as solvent), such as tetrahydrofuran (THF), or N-donor ligands, such as DCHIm (Fig. 2), or a covalently linked imidazolyl or pyridyl ligand. To better our understanding of the chemistry of full heme-O2-Cu assemblies, it is very useful to understand the structural aspects, physical properties, and reactivity patterns of just these heme-containing moieties with varying axial ligand base, with O2, CO, NO, and RNCs. In fact, some of this information has been obtained and previously published, in particular, for heme-O2 (FeIII-superoxide) complexes and for four of the five species, as shown in Fig. 2 [37, 38]. In addition, some of the related (heme)FeII-CO and (heme)FeII-NO compounds have also been described [37]. Here, we report on advances made from the study of new (heme)FeII derived adducts with DIMPI along with nitric oxide, using the F8, PIm, PPy, and a brand new heme PImH possessing a covalently linked histamine moiety, see Fig. 2 and Eqs. 3 and 4. New insights have been obtained based on the X-ray structures and physical properties which are described and also compared with the corresponding adducts using F8, which does not incorporate a tethered axial ligand:
| (3) |
| (4) |
Fig. 2.

Ferric heme-superoxo complexes previously characterized and a new five-coordinate ferrous heme
Materials and methods
All chemicals and solvents were purchased as commercially available analytical grade unless otherwise specified. Tetrahydrofuran (THF, inhibitor free) was dried over sodium/benzophenone ketyl, and purified by distillation under argon. Pentane was dried by distillation over calcium hydride. Toluene was used after passing through a 60 cm long column of activated alumina (Innovative Technologies), under argon. 2,6-Dimethylphenyl isocyanide (DIMPI) was purchased from Sigma Aldrich. ·NO gas was obtained from Matheson Gases and purified following methods previously described in the literature [39]. A three-way syringe was used for the addition of ·NO gas to all metal complex solutions. Preparation and handling of air-sensitive compounds was performed under an argon atmosphere using the standard Schlenk techniques or in an MBraun Labmaster 130 inert atmosphere (less than 1 ppm O2, less than 1 ppm H2O) drybox filled with nitrogen. Deoxygenation of all solvents was accomplished by either repeated freeze/pump/thaw cycles or bubbling with argon for 45–60 min.
Instrumentation: Benchtop UV–Vis measurements were carried out using a Hewlett Packard 8453 diode array spectrophotometer equipped with HP Chemstation software and a Unisoku thermostated cell holder for low-temperature experiments. A 10 mm path length quartz cell cuvette modified with an extended glass neck with a female 14/19 joint, and stopcock was used to perform all UV–Vis experiments. ESI–MS were acquired using a Finnigan LCQ Duo ion-trap mass spectrometer equipped with an electrospray ionization source (Thermo Finnigan, San Jose, CA). The heated capillary temperature was 250 °C, and the spray voltage was 5 keV. Spectra were recorded continuously after injection. Infrared (IR) spectra were obtained on solid samples using a Thermo Scientific Nicolet Nexus 670 Fourier transform IR (FT-IR) spectrophotometer with ATR attachment. 1H-NMR and 19F-NMR spectra were acquired using a Bruker 300-MHz NMR spectrometer. Chemical shifts were reported as δ (ppm) values relative to an internal standard (tetramethylsilane) and the residual solvent proton peak. Electron paramagnetic resonance (EPR) spectra were recorded with a Bruker EMX spectrometer equipped with a Bruker ER 041 × G microwave bridge and a continuous-flow liquid helium cryostat (ESR900) coupled to an Oxford Instruments TC503 temperature controller. Spectra were obtained at 8 K under nonsaturating microwave power conditions (ν = 9.428 GHz, microwave power = 0.201 mW, modulation amplitude = 10 G, microwave frequency = 100 kHz, and receiver gain = 5.02 × 103). EPR spectra were simulated using the Easy Spin (see ESI).
The compounds (F8)FeII (1) [35, 40, 41], (PPy)FeII (2) [37, 38], and (PIm)FeII (3) [37, 42] were synthesized as previously described.
Synthesis of (PImH)FeII (4)
The synthesis of complex (PImH)FeII (4) involves multiple steps,, as shown in Scheme 1, where F6(NHCOCH2CH2Br) TPPH2 [43] and LN3Tr [44] were synthesized as previously described.
Scheme 1.

Steps involved in the synthesis of the (PImH)FeII (4) complex
PImTr
To a 50 mL round bottom flask containing 1.8 g (2.0 mmol) of F6(NHC(O)CH2CH2Br)TPPH2 and 0.7 mL of diisopropylethyl amine was added to 1.7 g (4 mmol) of LN3Tr in 15 mL of THF. The resulting mixture was heated to reflux overnight. After cooling to room temperature, the solvent was removed under vacuum. The solid residue was re-dissolved in dichloromethane and washed several times with distilled water, from which the organic layer was dried over anhydrous sodium sulfate. Column chromatography of the crude material on alumina (ethyl acetate:hexane = 97:3, Rf =0.4) yielded 2.3 g (1.9 mmol, 65 %) of the desired product as a purple solid. ESI–MS (m/z): 1236.11 (M+H)+ (Fig. S1); 1H-NMR (CD3CN, 300 MHz; δ, ppm, RT): 8.88–8.78 (m, 8H, pyrrole-H), 8.73 (s, 1H, pyridine-H), 8.29 (d, 1H, pyridine-H), 8.02 (d, 1H, pyridine-H), 7.91–7.73 (m, 4H, Ar–H, pyridine-H), 7.47–7.19 (m, 20H, Ar–H, Tr-H), 6.94-6.92 (m, 6H, Ar–H), 6.87 (s, 1H, Imidazole-H), 6.04 (s, 1H, Imidazole-H), 1.68 (t, 2H, (–CH2–)), 1.49 (t, 2H, (–CH2–)), 1.32 (t, 2H, (–CH2–)), 1.20 (t, 2H, (–CH2–)), 1.04 (s, 2H, (–CH2–)py), and −2.87 (s, 2H, NHpyrrole) (Fig. S2).
PImTr-d8
The pyrrole deuterated porphyrin ligand PImTr-d8 was prepared using a procedure identical to that described above for PImTr, but employing the pyrrole deuterated porphyrin F6(NH2)-d8 [36] instead of F6(NH2).
[(PImH)FeIII-Cl]
The ligand PImTr (1.3 g, 1.05 mmol) was dissolved in 20 mL THF under an argon atmosphere. Iron(II) chloride tetrahydrate (7 g, 55.2 mmol) was added, and the solution was heated to reflux at 60 °C under argon for 3 h. After cooling to room temperature, the solution was exposed to air and stirred for 3 h. The solvent was removed by rotary evaporation, and the residue obtained was re-dissolved in 100 mL CH2Cl2 followed by filtering the insoluble solid present. The solution was stirred with HCl (1 M, 100 mL) for 3 h and then neutralized using solid NaHCO3. The organic layer was washed with 100 mL saturated NaHCO3, and then, brine solution dried over anhydrous MgSO4. The desired product was purified by column chromatography (silica, CH2Cl2:MeOH = 95:5, Rf = 0.4). Yield: 0.8 g, 72 %. UV–Vis spectrum [λmax, nm] in THF: 419, 527, 553 (Fig. S3). ESI–MS (m/z): 1047.01 (M−Cl−)+ (Fig. S4) 1H-NMR (300 MHz, THF, δ, ppm, RT): 82 (s, br, pyrrole-H). EPR spectra (X-band spectrometer, ν = 9.428 GHz): g = 6.0, 1.98 in THF at 7 K.
[(PImH)FeIII-Cl]-d8
The pyrrole deuterated heme–FeIII [(PImH)FeIII-Cl]-d8 was prepared using an identical procedure to that described above for [(PImH)FeIII-Cl], but employing pyrrole deuterated porphyrin PImTr-d8 instead of PImTr. 2H NMR (THF, 300 MHz, δ, ppm, RT): 82 (s, br, pyrrole-D) and (δ, ppm, −90 °C): 126.0 (s, br, pyrrole-D), shown in Fig. S5.
(PImH)FeII
The degased solution of [(PImH)FeIII-Cl] (500 mg, 0.5 mmol) in 120 mL CH2Cl2 was added to a degassed 50 mL saturated Na2S2O4 (aq) solution under an argon atmosphere. The two solutions were mixed using argon bubbling for 30 min in an additional funnel. The reaction mixture was allowed to sit for 20 min until the two layers separated. The organic layer was separated and passed through anhydrous Na2SO4 powder loaded in a filter tube (one end connecting to the additional funnel and the other end connecting to a Schlenk flask) under an argon atmosphere. Then, the solvent was removed and dried in vacuo for 3 h. The resulting solid was kept in glove box. Yield: 437.1 mg, 93 %. UV–Vis spectrum [λmax, nm] in THF: 419, 525, 552. 19F-NMR (THF-d8, 282 MHz; δ, ppm): −109.7, −110.3, −111.0, −111.6, −112.4.
(PImH)FeII-d8
The pyrrole deuterated heme–FeII (PImH) FeII-d8 was prepared using an identical procedure to that described above for (PImH)FeII, but employing pyrrole deuterated porphyrin [(PImH)FeIII-Cl]-d8 instead of [(PImH) FeIII-Cl]. 2H NMR (300 MHz, THF): δ 57.0 (s, 1H, pyrrole-D), 49.0 (s, 1H, pyrrole-D), 19.0 (s, 1H, pyrrole-D), 15.7 (s, 1H, pyrrole-D), 8.3 (s, 4H, (pyrrole-D). 2H-NMR (THF, 300 MHz; δ, ppm, −90 °C): 9.80 (Fig. S6).
[(F8)FeII-(DIMPI)2], (1)-DIMPI
In the drybox, to a solution of (F8)FeII (1) (10.0 mg, 0.024 mmol) in THF (5 mL) was added 2,6-dimethylphenyl isocyanide (6.3 mg, 0.048 mmol). After stirring the reaction mixture for 30 min, the solvent was removed under vaccum to yield a red solid. The crude solid obtained was further dissolved in THF and layered with pentane to obtain a very fine crystalline material. UV–Vis spectrum [λmax, nm] in THF: 430, 527. 1H-NMR (THF-d8, 300 MHz; δ, ppm): 10.45 (pyrrole-H), 2.43 (s, –CH3 (DIMPI)), 7.2 (m, ArH (DIMPI)); 19F-NMR (THF-d8, 282 MHz; δ, ppm): −109 (d). FT-IR spectrum (solid): νCN = 2124 cm−1.
[(PPy)FeII-(DIMPI)], (2)-DIMPI
In the dry box, to the THF solution of (PPy)FeII, (2) (10.0 mg, 0.011 mmol) in a 10 mL Schlenk flask, we added one equivalent of DIMPI (1.5 mg, 0.012 mmol) and the reaction mixture was stirred for half an hour. The solvent was removed under vacuum to yield a deep red colored solid, which was further recrystallized by dissolving in a minimal amount of THF and layering it with pentane to obtain the fine crystalline material. UV–Vis spectrum [λmax, nm] in THF: 430, 534. FT-IR spectrum (solid): νCN = 2104 cm−1.
[(PIm)FeII-(DIMPI)], (3)-DIMPI
This complex was synthesized in a similar manner to complex (2)-DIMPI. X-ray quality crystals were obtained from the solution of MeTHF/pentane. UV–Vis spectrum [λmax, nm] in THF: 430, 532. 1H-NMR (THF-d8, 300 MHz; δ, ppm): 9.1 (pyr-role-H), 7.2 (m, ArH (DIMPI)), 2.43 (s, –CH3 (DIMPI)); 19F-NMR (THF-d8, 282 MHz; δ, ppm): −110.6 (d), −110.8 (d), −111.0 (d), −111.7 (d). FT-IR spectrum (solid): νCN = 2098 cm−1.
[(PImH)FeII-(DIMPI)], (4)-DIMPI
This complex was also synthesized in a similar manner as the aforementioned complexes, (2)-DIMPI and (3)-DIMPI. X-ray quality crystals were obtained from a solution of Toluene/pentane. UV–Vis spectrum [λmax, nm] in THF: 430, 533. 1H-NMR (THF-d8, 300 MHz; δ, ppm): 9.78 (pyrrole-H), 7.2 (m, ArH (DIMPI)), 2.27 (m, –CH3 (DIMPI)); 19F-NMR (THF-d8, 282 MHz; δ, ppm): −110.55 (d), −110.8 (d), −110.9 (m), −111.6 (d). FT-IR spectrum (solid): νCN = 2112 cm−1
[(F8)FeII-NO], (1)-NO [45]
The ferrous mononitrosyl complex (1)-NO was generated by bubbling excess NO gas through the THF solution of (F8)FeII (1) (2 mM) under argon atmosphere at room temperature. After the reaction mixture stirred for 2 h, the solvent was removed under vacuum to obtain a dark red solid. A highly pure material was obtained by dissolving red solid in the minimum amount of THF and layering it with pentane inside the dry box. UV–Vis spectrum [λmax, nm] in THF: 408, 547. 1H-NMR (THF-d8, 300 MHz; δ, ppm): 6.9 (br, pyrrole-H); 19F-NMR (THF-d8, 282 MHz; δ, ppm): −106 (br). FT-IR spectrum (solid): νNO = 1688 cm−1. EPR spectra (X-band spectrometer, ν = 9.428 GHz): g = 2.09, 2.02, 1.99 (hyperfine) in THF at 7 K.
[(PPy)FeII-NO], (2)-NO [46]
A method similar to that used to synthesize complex (1)-NO was used to make complex (2)-NO. Excess of NO gas was bubbled through the 2 mM THF solution of (PPy)FeII (2). UV–Vis spectrum [λmax, nm] in THF: 417, 543. 1H-NMR (THF-d8, 300 MHz; δ, ppm): 8.0 (pyrrole-H). FT-IR spectrum (solid): νNO = 1648 cm−1. EPR spectra (X-band spectrometer, ν = 9.428 GHz): g = 2.07, 2.01 (br-hyperfine), 1.98 in THF at 7 K.
[(PIm)FeII-NO], (3)-NO [46]
This complex was prepared in the same manner as complexes (1)-NO and (2)-NO. UV–Vis spectrum [λmax, nm] in THF: 423, 542. 1H-NMR (THF-d8, 300 MHz; δ, ppm): 8.8 (pyrrole-H); 19F-NMR (THF-d8, 282 MHz; δ, ppm): −106.2 (br), −107.9 (br), −110.9 (br). FT-IR spectrum (solid): νNO = 1650 cm−1. EPR spectra (X-band spectrometer, ν = 9.428 GHz): g = 2.07, 2.00 (hyperfine), 1.97 in THF at 7 K.
[(PImH)FeII-NO], (4)-NO
Synthesis of this complex was accomplished as mentioned above for the preparation of (1)-NO. UV–Vis spectrum [λmax, nm] in THF: 425, 541. 1H-NMR (THF-d8, 300 MHz; δ, ppm): 9.65 (pyrrole-H); 19F-NMR (THF-d8, 282 MHz; δ, ppm): −104.9 (br), −108.1 (br). FT-IR spectrum (solid): νNO = 1650 cm−1. EPR spectra (X-band spectrometer, ν = 9.428 GHz): g = 2.06, 1.99 (hyperfine), 1.96 in THF at 7 K.
X-ray crystal structure determination
X-ray structure determination of (3)-DIMPI and (4)-DIMPI was performed at the X-ray diffraction facility at Johns Hopkins University. CIF files have been deposited with the Cambridge Crystallographic Data Centre (CCDC). CCDC 1455862 and 1455863 contain the supplementary crystallographic data for this article. These data can be obtained free of charge from the CCDC via http://www.ccdc.cam.ac.uk/data_request/cif.
All reflection intensities were measured at 110(2) K using a SuperNova diffractometer (equipped with Atlas detector) with Cu Kα radiation (λ = 1.54178 Å) under the program CrysAlisPro (version 1.171.36.32 Agilent Technologies, 2013). The program CrysAlis-Pro (version 1.171.36.32 Agilent Technologies, 2013) was used to refine the cell dimensions and for data reduction. The structures were solved with the program SHELXS-2013 (Sheldrick 2013) and were refined on F2 with SHELXL-2013 (Sheldrick 2013). Analytical numeric absorption correction based on a multifaceted crystal model was applied using CrysAlisPro (version 1.171.36.32 Agilent Technologies, 2013). The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). The H atoms were placed at calculated positions using the instructions AFIX 23, AFIX 43, or AFIX 137 with isotropic displacement parameters having values 1.2 or 1.5 times Ueq of the attached C or N atoms.
Crystals of (3)-DIMPI were obtained from a MeTHF solution of complex and layered with pentane, while crystals of (4)-DIMPI were obtained from saturated solution of toluene. The structure of (3)-DIMPI is partly disordered. Some unresolved electron density—i.e., a very disordered lattice methyl THF solvent molecule—has been taken out in the final refinement (SQUEEZE details are provided in the CIF file, Spek, 2009) [47]. In addition, the imidazole/amide arm may be slightly disordered, but the disorder is not significant enough to model it in the final refinement. The structure (4)-DIMPI is partly disordered. One difluorophenyl group and the lattice toluene solvent molecule are found to be disordered over two orientations, and the occupancy factors of the major components of the disorder refine to 0.561(16) and 0.722(4), respectively (see the Electronic Supplementary Material, i.e., the appropriate CIF file).
Results and discussion
Stable heme–isocyanide complex formation
DIMPI reacts immediately with the reduced synthetic ferrous-heme complexes, [(F8)FeII], [(PPy)FeII], [(PIm)FeII], and [(PImH)FeII], to yield six-coordinate low-spin ferrous-heme isonitrile species, as shown in Schemes 2 and 3.
Scheme 2.
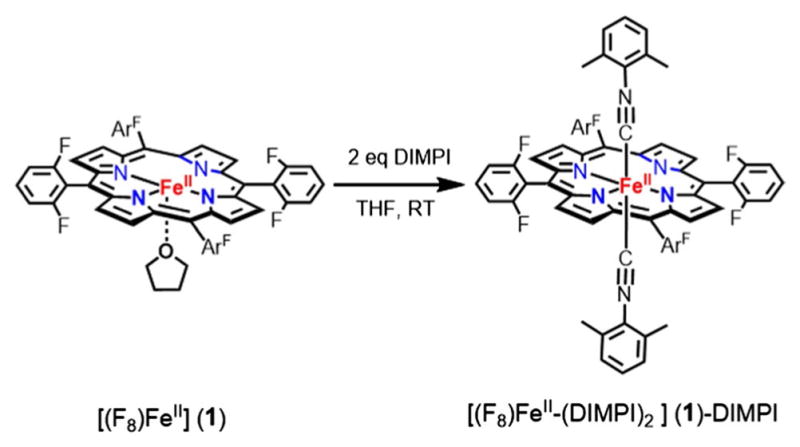
Generation of bis-isocyanide ferrous-heme complex; (1)-DIMPI at room temperature
Scheme 3.

Generation of six-coordinate ferrous-heme isocyanide complexes; (2)-, (3)-, and (4)-DIMPI
Generation of bis-isocyanide-porphyrin complex [(F8) FeII-(DIMPI)2]
When one equivalent DIMPI is added to a THF solution of [(F8)FeII] at room temperature, a new UV–Vis peak at 430 nm is observed, but the absorption at 422 nm characteristic of the starting complex still remains. However, the addition of another equivalent of DIMPI leads to the full formation of the 430 nm peak in the Soret region (Fig. 3; Scheme 2). Additional DIMPI added to the solution does not change the UV–Vis spectral features. Based on these observations, we postulate that two DIMPI molecules are bound to the iron(II) center, as also confirmed by integration of peaks in the 1H-NMR spectrum of [(F8)FeII-(DIMPI)2]. Similar UV–Vis spectral features were observed for a structurally characterized bis-isocyanide iron(II) complex with tetraphenylporphyrin (TPP), [(TPP)FeII-(tBuNC)2] [48]. The reactivity of DIMPI with reduced synthetic hemes is similar to that in general and previously observed for carbon monoxide (CO) as the heme ligand [37, 49]. Complex (1)-DIMPI was further characterized by FT-IR spectroscopy, where a single ν(C≡N) stretch is observed at 2124 cm−1 (vide infra, Fig. 6; Table 3), as would be expected for this highly symmetric compound, even with the presence of two DIMPI ligands per molecule. In addition, the 19F-NMR spectrum of (1)-DIMPI shows one sharp absorbance at −109.0 ppm for the o-difluoro substituted phenyl rings of the F8 porphyrin [50].
Fig. 3.

UV–Vis spectroscopic data for the ferrous DIMPI and NO complexes of (1), (2), (3), and (4) in THF at room temperature. black—reduced Fe(II) species; red—Fe(II)-DIMPI and blue—Fe(II)-NO complexes
Fig. 6.
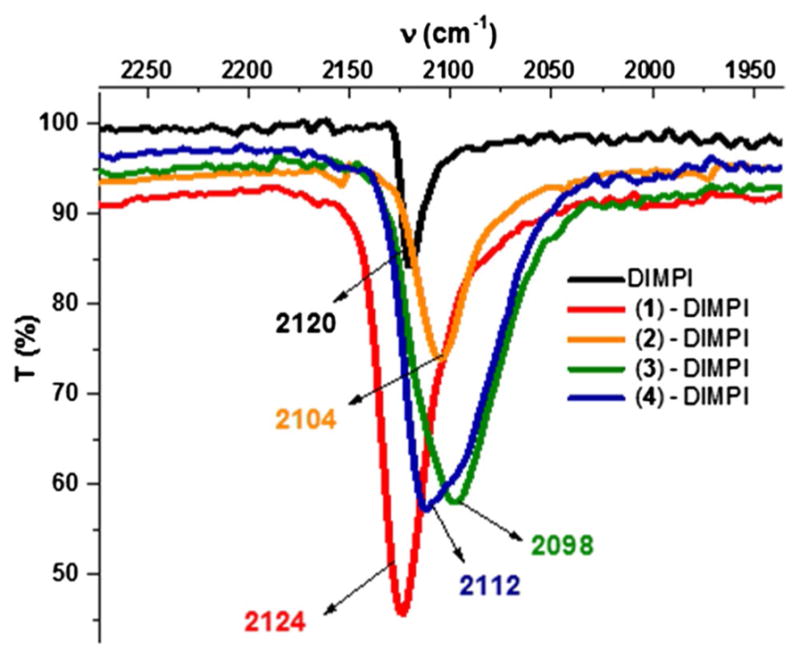
Solid-state FT-IR spectra for Fe(II)-DIMPI complexes of (1), (2), (3), and (4)
Table 3.
Properties of ferrous-heme-DIMPI and ferrous-heme-NO model complexes
| Compound | UV–Vis (nm) | IR (cm−1) ν(C≡N)/ν(N–O) | EPR (g values) |
|---|---|---|---|
| [(F8)FeII-(DIMPI)2] | 430 | 2124 | Silent |
| [(PPy)FeII-(DIMPI)] | 430 | 2104 | Silent |
| [(PIm)FeII-(DIMPI)] | 430 | 2098 | Silent |
| [(PImH)FeII-(DIMPI)] | 430 | 2112 | Silent |
| [(F8)FeII-NO] | 408 | 1688 | 2.09/2.01/1.99 |
| [(PPy)FeII-NO] | 417 | 1648 | 2.08/2.00/1.97 |
| [(PIm)FeII-NO] | 423 | 1650 | 2.07/2.00/1.97 |
| [(PImH)FeII-NO] | 425 | 1650 | 2.07/1.99/1.95 |
Generation of six-coordinate (PPy/PIm/PImH) iron(II)-DIMPI complexes
[(PPy)FeII] and [(PIm)FeII] in THF solution at RT exhibit different structures in this solvent, as previously deduced by the observation of the positions of NMR spectroscopic pyrrole resonances. [(PPy)FeII] and [(PIm)FeII] are both five-coordinate high spin, the former at all temperatures between RT and −90 °C. However, at low temperature, [(PIm)FeII] is six-coordinate low spin (S = 0), with the pyrrole resonances appearing in the diamagnetic region, indicating that both the imidazolyl group and a THF solvent molecule act as axial ligands [37]. The new complex [(PImH)FeII] behaves similarly; at room temperature, it is five-coordinate high spin, where the tethered imidazolyl is axially bound to the Fe(II) center. This results in an asymmetry, and four different pyrrole resonances appear in a ratio of 4:2:1:1 (see Fig. S6). By contrast, at lower temperatures, [(PImH)FeII] forms a six-coordinate low spin (S = 0) species, again postulated to have both a THF and imidazole bound to the Fe(II) center, as δpyrrole = 9.80 ppm, Fig. S6.
The addition of one equivalent of DIMPI solution to each of the reduced Fe(II) porphyrinates ([(PPy)FeII], [(PIm) FeII] and [(PImH)FeII]) in THF at room temperature leads to a substantial change in the UV–Vis Soret region, with the formation of a band at 430 nm in all three cases. Additional equivalents of DIMPI do not yield any change in the UV–Vis spectra (Fig. 3; Scheme 3). This may indicate that only one DIMPI molecule is bound to the iron(II) center axially, while the covalently linked axial base imidazole/pyridine is coordinated to the iron(II) center giving an overall six-coordinate low-spin ferrous-DIMPI complex. These conclusions are borne out by the X-ray structures determined for complexes (3)-DIMPI and (4)-DIMPI (see below, Fig. 4).
Fig. 4.

Displacement ellipsoid plots (50 % probability level) of [(PIm)FeII-(DIMPI)] (left; 3-DIMPI) and [(PImH) FeII-(DIMPI)] (right; 4-DIMPI), in both the cases showing the imidazolyl and 2,6-dimeth-ylphenyl isocyanide ligands bound to Fe(II) center. Lattice solvent molecules and H atoms have been omitted for the sake of clarity. Selected bond lengths and bond angles are reported in Table 2
We have also carried out experiments to determine binding constants of DIMPI with the ferrous hemes with covalently attached axial ligand bases. Titrations with DIMPI were performed using (PPy)FeII (2), (PIm)FeII (3), and (PImH)FeII (4), and isosbestic behavior was seen for all the titrations (Figs. S9–S11). A plot of the absorbance at 430 nm versus [DIMPI] (Fig. S9–S11, in the ESI) reaches a maximum at ~1 equiv of DIMPI, and no further spectral changes are observed with the addition of more DIMPI. Assuming that DIMPI reversibly binds to the ferrous-heme complexes (PPy)FeII (2), (PIm)FeII (3), and (PImH)FeII (4) under equilibrium conditions, a good fit of the data can be obtained with a model for a one-to-one binding isotherm. This fit gives association constants (Ka) of 2.29 × 107, 1.19 × 107, and 1.29 × 107 M−1, for (2), (3), and (4), respectively. These values are comparable with those measured for DIMPI binding to hemoglobin (Hb) and myoglobin (Mb) [Ka = 1.0 × 108 M−1] [51]. The binding constant for complex (2)–DIMPI is twofold greater than (3)–DIMPI and (4)–DIMPI, which is consistent with a less strong binding of the pyridine axial base in (2)–DIMPI when compared to the imidazole and histamine containing complexes.
Crystal structures of isocyanide complexes
To understand the similarities and differences in the coordination geometry of these iron(II)-DIMPI complexes, crystal structures of (3)-DIMPI and (4)-DIMPI were determined. The structures are shown in Fig. 4, and important structural parameters are listed in Tables 1 and 2. In both structures, the geometry around the iron is octahedral, where the fifth ligand is the covalently linked imidazole (or pyridine), while the sixth ligand is the isocyanide (DIMPI). The metal center is in the porphyrinate plane. The observed bond distances and angles for both indicate very little strain within the linker arm. A slight perturbation from the expected linear Fe–C–N bond angle is seen for complex (3)-DIMPI, with a ∠Fe–C–N value of 173.8(4)°. This is notably less linear when compared with (4)-DIMPI, where ∠Fe–C–N, 179.3(3)° is nearly linear. This difference could arise due to crystal packing effects [52]. The Fe–C(DIMPI) bond distances are ca. 1.82 Å for both (3)-DIMPI and (4)-DIMPI. The Fe–N(imidazole) bond distances are ca. 2.02 Å, while the average Fe–N(porphyrin) bond distances are 1.99 Å, similar to our previously reported [(F8)FeII]·2THF complex [53]. Both structures are well ordered except the difluorophenyl group and toluene solvent molecule in complex (4)-DIMPI (see “Materials and methods”). These appear to be disordered over two orientations and can rotate slightly from perpendicularity with respect to the porphyrin plane. Lehnert and co-workers have previously published on a crystal structure of the zinc(II) analogue, a five-coordinate complex with the PIm ligand [46].
Table 1.
Crystallographic data for complex [(PIm)FeII-(DIMPI)] and [(PImH)FeII-(DIMPI)]
| Compounds | (3)-DIMPI | (4)-DIMPI·Toluene |
|---|---|---|
| Formula weight (g/mol) | 1106.89 | 1271.13 |
| T (K) | 110(2) | 110(2) |
| Crystal shape | small dark red plate (0.11 × 0.05 × 0.02 mm3) | dark red plate (0.15 × 0.08 × 0.03 mm3) |
| Space group | Triclinic, P-1 (no. 2) | Monoclinic, P21/c (no. 14) |
| a (Å) | 12.3852(6) | 12.4643(2) |
| b (Å) | 12.5816(6) | 12.3010(2) |
| c (Å) | 19.3657(12) | 39.9319(8) |
| α (°) | 104.424(5) | 90 |
| β (°) | 95.293(4) | 93.9552(18) |
| γ (°) | 111.823(5) | 90 |
| V (Å3) | 2655.1(3) | 6107.91(19) |
| Z | 2 | 4 |
| Dx (g cm−3) | 1.385 | 1.382 |
| μ (mm−1) | 2.897 | 2.601 |
| Absorption correction range | 0.797–0.947 | 0.759–0.940 |
| (sin θ/λ)max (Å−1) | 0.60 | 0.60 |
| Total, unique, and observed reflections | 24,276, 9448, 6441 | 40,278, 11,998, 8909 |
| Rint | 0.0467 | 0.0597 |
| GOF | 1.041 | 1.021 |
| R1/wR2 [I > 2σ(I)] | 0.0703/0.1902 | 0.0501/0.1089 |
| R1/wR2 | 0.1038/0.2151 | 0.0760/0.1220 |
| Δρmax, Δρmin, rms | 1.126, −0.478, 0.083 | 0.644, −0.483, 0.054 |
Table 2.
Selected bond lengths (Å) and bond angles (°) for (3)-DIMPI and (4)-DIMPI. The proposed H-bonds are also listed
| Compound: | (3)-DIMPI Bond length (Å) | Compound: | (4)-DIMPI.Toluene Bond length (Å) |
|---|---|---|---|
| Fe–N1 | 1.998 (4) | Fe–N1 | 1.994 (2) |
| Fe–N2 | 2.000 (3) | Fe–N2 | 1.991 (2) |
| Fe–N3 | 1.990 (4) | Fe–N3 | 1.995 (2) |
| Fe–N4 | 2.001 (3) | Fe–N4 | 1.985 (2) |
| Fe–N5 | 2.034 (4) | Fe–N8 | 2.024 (2) |
| Fe–C56 | 1.824 (4) | Fe–C59 | 1.835 (3) |
| N8–C56 | 1.163 (6) | N11–C59 | 1.166 (3) |
| Compound: | (3)-DIMPI Bond angle (°) | Compound: | (4)-DIMPI·Toluene Bond angle (°) |
|---|---|---|---|
| N1–Fe–N5 | 91.38 (15) | N1–Fe–N8 | 85.99 (8) |
| N2–Fe–N5 | 89.77 (15) | N2–Fe–N8 | 89.90 (9) |
| N3–Fe–N5 | 88.85 (15) | N3–Fe–N8 | 91.10 (8) |
| N4–Fe–N5 | 88.58 (14) | N4–Fe–N8 | 89.03 (9) |
| N5–Fe–C56 | 176.03 (16) | N8–Fe–C59 | 177.61 (9) |
| Fe–C56–N8 | 173.8 (4) | Fe–C59–N11 | 179.3 (3) |
| Weak C–H…F interaction | Bond length (Å)/bond Angles (°) | Weak C–H…F interaction | Bond length (Å)/bond angles (°) |
|---|---|---|---|
| F1…H64methyl (C64) | 2.731/(133.93) | F1…H35py (C35) | 3.089/(129.88) |
| F2…H7pyrrole (C7) | 3.490/(95.04) | F1…H40imid (C40) | 2.914/(135.45) |
| F3…H8pyrrole (C8) | 2.966/(95.57) | F2…H67methyl (C67) | 3.148/(146.75) |
| F4…H39imid (C39) | 3.117/(153.44) | F3…H12pyrrole (C12) | 3.163/(91.25) |
| F5…H2pyrrole (C2) | 2.926/(100.23) | F4…H8pyrrole (C8) | 2.883/(101.78) |
| F6…H41imid (C41) | 2.991/(166.53) | F5…H17pyrrole (C17) | 2.903/(100.89) |
| F6…H66methyl(C66) | 2.539/(176.34) | ||
| N9H…N7pyridine | 1.986/(173.74) |
Other potentially important (from a structural perspective) observations obtained from the crystal structures of both complexes are that weak but noticeable intramolecular CH…F interactions occur, as shown in Fig. 5 and listed in Table 2. The observed range of C–H…F interactions for our new structures lie between 2.73 and 3.08 Å, which is greater than the sum of reported Van der Waals radii for hydrogen and fluorine (approximately 2.3–2.5 Å) [54]. Based on reported structural observations and DFT calculations [55, 56], the very strong H-bonding ability of fluorine gives rise to such C–H…F interactions which vary between ~2.7 and 3.1 Å, similar to what is observed for (3)-DIMPI, and (4)-DIMPI. Such literature examples of organic compounds with longer distance H…F interactions occur even, where the CH…F angle lies in the range between 130° and 145°, and even in some cases, it is close to 100° [55, 56]. Further comparisons may be made to examples of non-bonded CH…O contacts made between an O atom in an Fe(IV)-oxo complex with surrounding ligand methyl group H atoms; there, short CH…O distances are observed (2.3 to 2.7 Å), while very acute CH…O angles are present (~100°–109°) [57, 58].
Fig. 5.

Crystal structures showing weak intramolecular CH…F interaction identified from the green lines shown. (Left) (3)-DIMPI and (Right) (4)-DIMPI. See text for further discussion
With these precedents in mind, we postulate that in the solid-state structure of (3)-DIMPI, a CH…F interaction occurs between the methyl group on the DIMPI ligand (H64C) and F1 from the proximate o-F aryl porphyrinate substituent (see the green line in the middle top of the (3)-DIMPI structure in Fig. 5, left, and Fig. S7); here, the CH…F distance and angle are 2.73 Å and 134°, respectively (Table 2). A far closer to linear interaction occurs between F6 and H41 from the ligated imidazolyl group with a bond distance and angle of 2.99 Å and 167°, respectively (Table 2; Fig. 5, bottom right). Notice F6 and F5, on the same porphyrinate aryl group both H-bond, the latter to the pyrrole hydrogen atom, (H2), this F5…H2C interaction is likely synergistic with the F6…H41 interaction. Envisioning the iron atom as a kind-of pseudo center of symmetry, the F3 and F4 atoms on the left-hand o-F2-phenyl ring form two H-bonds, between F3…H8pyrrole (C8) and F4…H39imid (C39) (Table 2). The overall result is that the H-bonding interactions formed by the four F atoms just described fixes the orientation of the imidazolyl axial ligand, which is already close to perpendicular to the porphyrinate plane, to be nearly coplanar with both the aryl rings containing these F3, F4, F5, and F6 atoms. Notice how in viewing the structure of (3)-DIMPI (Fig. 5, left) that these two aryl rings and the imidazolyl ring seem to lie in the plane of the sheet. This imidazole orientation could also lead to a close interaction of F2…H7pyrrole (C7) in the solution state (see 19F-NMR spectroscopy, below), whereas in solid-state structure, this distance seems too long for this interaction to occur (Table 2).
Similarly, for (4)-DIMPI, we observe close to the linear CH…F interaction between a methyl group H atom on the DIMPI ligand (H66) and F6 from the o-F porphyrinate aryl substituent, shown by the green dotted line on the lower left side of Fig. 5, right; here, the CH…F distance and angle are 2.54 Å and 176°, respectively (Table 2; Fig. 5-right, also see Fig. S8). The other methyl group on the DIMPI ligand likewise forms an H-bond, with the F2 atom on the aryl group to the lower right side of (4)-DIMPI, as shown in Fig. 5, right. To the left side (Fig. 5, right), the F5 atom that on the same ring as F6 is involved in H-bonding to a pyr-role H atom (H17) (Table 2). For F1, on the right-hand aryl group, we postulate that multiple H…F interactions occur, to H40 from the ligated imidazolyl group as well as H35 from the dangling free pyridyl group (see the green lines to the upper right of the (4)-DIMPI structure in Fig. 5, Right); here, the CH…F distances and angles are 2.91 Å and 135° (F1…H40), and 3.08 Å and 130° (F1…H35), respectively. Tilting of the o-F2-aryl group containing F3 and F4 allows for two H-bonds to form to pyrrole H atoms (Table 2). Thus, all six fluorine atoms in (4)-DIMPI may participate in H-bonding. In part, we postulate that these solid-state interactions persist in solution, as indicated by 19F-NMR spectroscopy, where almost all of the fluorine resonances exist as doublets due to H-atom coupling, as described below.
Another H-bond observed in this X-ray structure of (4)-DIMPI is a very strong one, between the H atom on the uncoordinated N atom of the imidazolyl group, to the N atom of the dangling pyridine (Fig. 5-Right). Here, the N9H…N7pyridine angle is near linear (173.7°) with a N9H… N7 distance of 1.986 Å (and where N9…N7 = 2.863 Å) (Table 2).
Complexes (2)-DIMPI, (3)-DIMPI, and (4)-DIMPI were also characterized by FT-IR spectroscopy (Fig. 6; Table 3). The IR spectrum for (1)-DIMPI shows a single sharp ν(C≡N) band (2124 cm−1), corresponding to the absorption for both isonitrile ligands in [(F8)FeII-(DIMPI)2], which is shifted 4 cm−1 higher in energy relative to the stretching frequency of the uncomplexed ligand. The shift to higher energy is relatively small and could be attributed to a ligand ( ) to metal (dσ) interaction, consistent with the expected behavior for an isonitrile ligand acting as a σ-donor to a metal center. [(TPP)FeII-(tBuNC)2] [48], as mentioned above, was structurally characterized, but no IR data were given.
On the other hand, for all three porphyrin Fe(II)-DIMPI complexes with covalently attached axial bases (pyridine/imidazole/histamine moieties), we observe a shift in the C≡N bond stretch of the isonitrile ligand to lower energy with respect to the uncomplexed ligand. The ν(C≡N) band for (2)-DIMPI is 2104 and 16 cm−1 lower in energy compared to that of free DIMPI, and also 20 cm−1 lower in energy with respect to (1)-DIMPI. For (3)-DIMPI, the value of ν(C≡N) is 2098 cm−1, which is 23 cm−1 lower in energy with free ligand. In (4)-DIMPI [ν(C≡N) band (2112 cm−1)], we again observe a lower energy C≡N bond stretch compared to free DIMPI, but only by a small amount (Δν(C≡N) = −8 cm−1) [59]. The shift to lower energy in all these cases could be due to the presence of a more strongly donating axial base trans to the isonitrile ligand. This will most likely decrease the σ-donation to the Fe(II) from the isocyanide ligand while increasing backbonding to the ligand C≡N π* orbitals from the Fe(II) dπ orbitals.
Diamagnetic 1H-NMR spectra were observed for all Fe(II)-DIMPI (S = 0, d6) complexes. The pyrrole hydrogens of the Fe(II)-DIMPI porphyrinates resonate between 8.5 and 9.5 ppm compared with the starting reduced high-spin five-coordinated paramagnetic Fe(II) (d6) species ranging in δpyrole = 12–58 ppm [37]. We also observed a singlet δ 2.65 ppm for the DIMPI o-methyl protons and a multiplet for the aromatic protons of bound DIMPI at δ 7.2–7.5 ppm; both the sets of peaks are slightly shifted downfield from what is observed for free DIMPI.
In the 19F-NMR for bis-isocyanide complex, [(F8)FeII-(DIMPI)2]/(1)-DIMPI, we observe one sharp singlet peak at −109.0 ppm, as shown in Fig. 7. Therefore, unlike what we have suggested (and described above) for what is observed in the X-ray crystal structures for the superstructured hemes [(3)-DIMPI and (4)-DIMPI], there are no observed F-atom interactions or coupling to porphyrin or isocyanide H atoms.
Fig. 7.
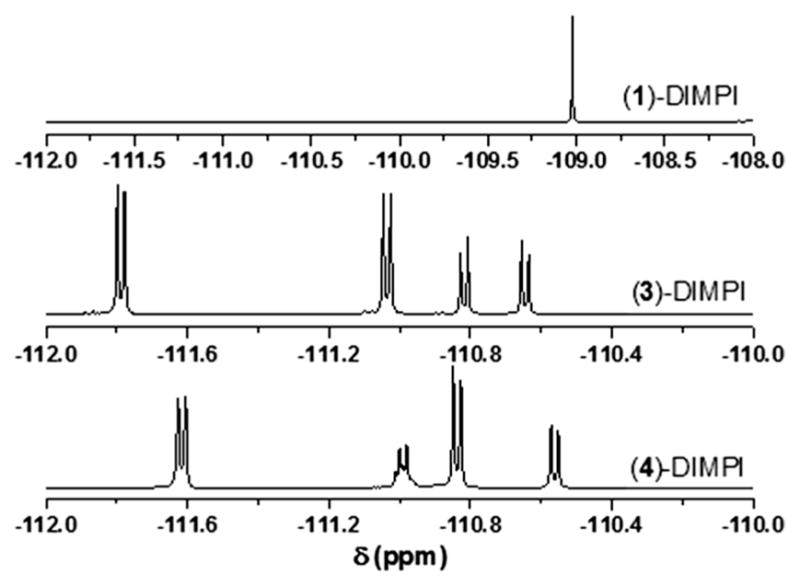
19F-NMR of (1)-DIMPI (top), (3)-DIMPI (middle), and (4)-DIMPI (bottom) complexes in THF at room temperature
On the other hand, we observed CH…F coupling for (3)-DIMPI and (4)-DIMPI (Fig. 7) with peaks for the ortho F-atom resonances on the porphryinate aryl groups occurring between −110 and −112 ppm [50]. In the case of (3)-DIMPI, doublet peaks, proposed to be due to 19F resonances coupled to an S = 1/2 H atom, are observed at −111.7 and −111.0 ppm, and both these appear to integrate to two F atoms. Our suggested assignments are as follows: (a) one of the two upfield (more negative delta value) doublets corresponds to F4 and F6 coupling to imidazole H atoms H39 and H41, which spatially line up rather well (see Table 2; Fig. 5). (b) The other upfield doublet represents 2 o-fluorine H-bonds with pyrrole H atoms, possibly F3 and F5 with pyrrole H atoms H8 and H2, and these also seem to be in very closely matching chemical environments. (c) One of the two absorptions, −110.8 or −110.6 corresponds to a single o-fluorine atom (F2) also coupling to a pyrrole CH7 atom, but by symmetry, this is in a different chemical environments then for the interactions discussed just above for F5 and F3. (d) Then, the other upfield absorption is a unique interaction of F1 with the DIMPI methyl group H atoms (CH64…F1).
Similarly, for (4)-DIMPI, we observe four ortho F-atom resonances, three doublets, and one multiplet in 19F-NMR spectroscopy (Fig. 7). The peaks at −111.6 and −110.8 ppm both integrate to two fluorine atoms, which we propose are due to (a) fluorine atoms F6 and F2 coupling with the DIMPI methyl H atoms H66A and H67C (see Table 2; Fig. 5), (b) while the other generally upfield doublet represents two fluorine atom being in the same chemical environments due to the coupling of F atoms F3 and F4 from the same porphyrinate phenyl ring, forming H-bonds with the pyrrole H atoms H8 and H12. If (or probably when) the aryl ring with F3 and F4 tilts the other way, then symmetry-related switching of H-bonding to pyrrole H atoms may (or probably does) occur, but the result is the same type of CH…F H-bonding. Furthermore, the doublet resonance at −110.5 ppm should correspond to F5 coupling to the pyrrole H atom H17. This leaves the multiplet peak at −110.9 ppm which likely arises due to the H-bonding interaction and magnetic coupling of F1 to two H atoms, the imidazole H atom H40 and the dangling pyridine H atom H35 (see Table 2; Fig. 5). During the dynamic behavior expected in solution, the dangling pyridyl ring may break its H35…F1 interaction, and twist over to the other side (to the left in Fig. 5, right), as the imidazole bound to Fe also twists to maintain the very strong imidazole N–H to pyridine nitrogen hydrogen bond); the pyridyl H35 would now H-bond to F5 (instead of F1); furthermore, F5 may then gain an H-bond to H40. All these motions would leave the 19F-NMR shifts and couplings unchanged.
As mentioned, for (1)-DIMPI, we do not observe any DIMPI ligand H atom coupling to F atoms of the F8 heme. In addition, we do not observe any such couplings for the compound (THF)(F8)FeII-CO (unpublished observation). We suggest that when there is a tethered axial ligand, such as in PIm or PImH, there are constraints in the movement or rotation of the axial ligand, which thereby allow for these weaker imidazole-H…F interactions to be observed. Further studies may be warranted.
Stable heme–Fe–nitrosyl formation
In this study, we have also investigated the reactivity of [(F8)FeII], [(PPy)FeII], and [(PIm)FeII], and the newly synthesized [(PImH)FeII] towards nitric oxide (NO). All iron (II) complexes form an NO-adduct at room temperature by bubbling NO(g) through the solution of each of the reduced iron complexes, as shown in Scheme 4. We have systematically characterized Fe(II)–NO complexes using UV–Vis, FT-IR, 1H-NMR, 19F-NMR spectroscopy, and low-temperature EPR spectroscopy, to access the binding properties of our covalently tethered N-donor ligands.
Scheme 4.
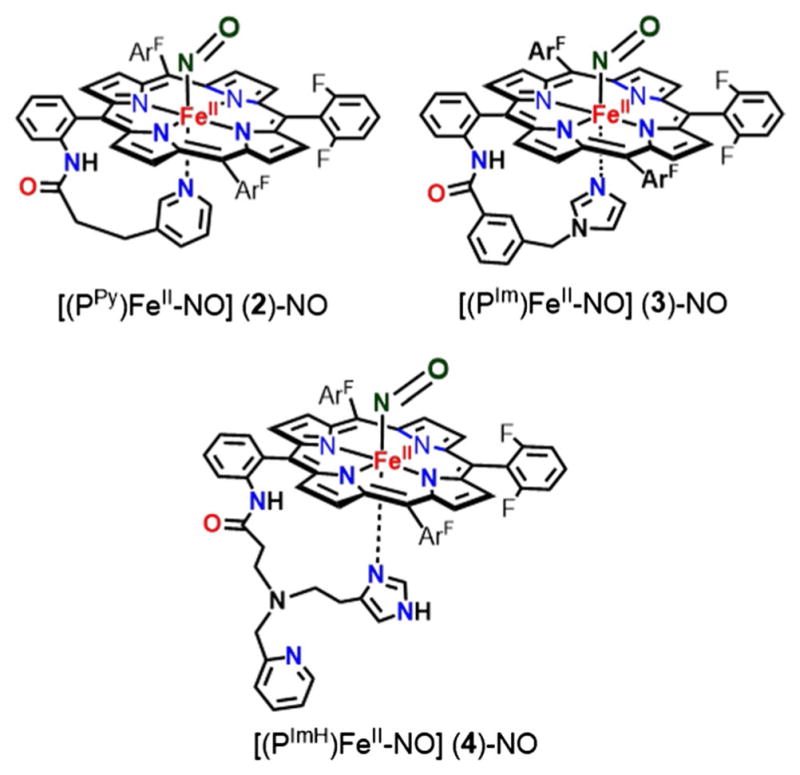
Six coordinate ferrous-heme mononitrosyl complexes of (PPy)FeII, (PIm)FeII, and (PImH)FeII
In-depth studies have been done by Lehnert and coworkers [14, 46] using synthetic heme porphyrins with tethered N-donor ligands which indicate a direct correlation between the coordination geometry of the iron center and the observed spectroscopic properties obtained from UV–Vis, IR, and EPR. For five-coordinate (5C) heme nitrosyls, the Soret band (UV–Vis) is typically about 405 nm, whereas in six-coordinate (6C) porphyrinoids, where the proximal ligand (N-donor) is bound to the Fe center, the Soret λmax shifts to ~426 nm. Similarly, in IR spectroscopy, 5C and 6C ferrous-heme mononitrosyl species have distinct N–O stretching modes. For 5C complexes, the N–O stretch typically lies between 1675 and 1700 cm−1, whereas for 6C complexes, the N–O stretch occurs at ~1630 cm−1. Low-temperature EPR spectroscopy studies conducted by several authors reveal interesting differences between the 5C and 6C iron(II) porphyrin NO adducts [60, 61]. Hyperfine lines resulting from the bound nitrogen of NO are observed at the lowest g value (g min) in 5C ferrous-heme nitrosyls. The coordination of the proximal nitrogen atom in 6C ferrous-heme nitrosyls causes a broadening in the EPR spectrum at g-mid resulting from the hyperfine lines of the bound NO and the trans-N donor ligand. Based on spectroscopic data available from the literature, our [(F8)FeII-NO] complex forms a typical 5C ferrous-heme mononitrosyl complex with a Soret band λmax at 408 nm in the UV–vis region at room temperature (Fig. 3), along with a characteristic N–O stretching band ν(N–O) at 1680 cm−1 in its IR spectrum [45, 62]. An additional evidence for the 5C nitrosyl complex can be seen in its EPR spectrum, which displays g values at 2.09, 2.02, and 1.99, with three hyper-fine splittings at g(min) (Fig. 8) [45, 62]. In the 19F-NMR, a broad o-phenyl fluorine signal is observed at −106.0 ppm.
Fig. 8.
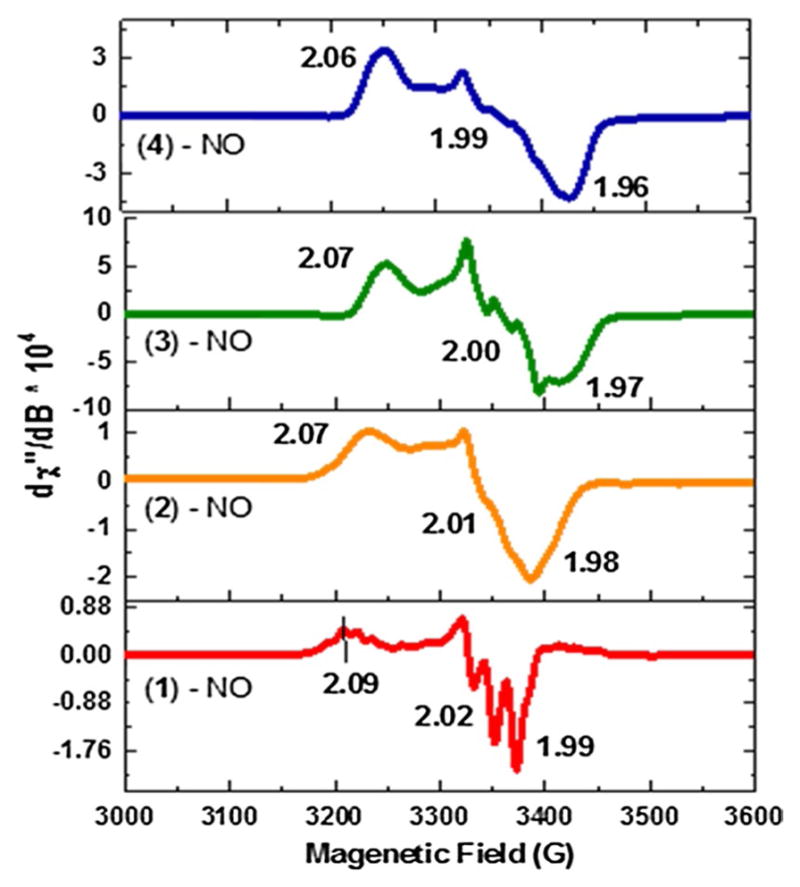
X-band EPR at 8 K in THF for ferrous-heme-NO complexes. [(F8)FeII-NO] (red, 5C species), while (2), (3), and (4) form 6C species. [(PPy)FeII-NO] (orange, 6C), [(PIm)FeII-NO] (green, 6C), and [(PImH)FeII-NO] (blue, 6C). These spectra were analyzed further using an EPR simulation computer program, and the results of those fits, giving g values and hyperfine coupling constants, are given in the ESI (Fig. S12)
In the case of [(PPy)FeII-NO], the Soret band absorption is observed at 417 nm (Fig. 3), which lies in between that known for 5C and 6C iron-nitrosyl complexes. This indicates that in solution at room temperature, the proximal pyridine is weakly bound to the iron center to give 6C species. Further evidence comes from the low-temperature EPR spectrum of [(PPy)FeII-NO], shown in Fig. 8, Fig. S12 and Table 3, which clearly resembles the spectra of other 6C low-spin heme-Fe(II)-nitrosyl complexes [46], lowering the temperature allows for stronger binding of the pyridyl group, as would be expected. The spectrum shows small, unresolved hyperfine splitting at g(mid) due to the presence of the proximal pyridine ligand (g = 2.07, 2.01, 1.98). The lack of UV–Vis spectral features at 400 and 470 nm, as well as EPR data for [(PPy)FeII-NO] confirms that in solution, it forms a 6C species, but where the pyridyl group is weakly bound to the iron center at room temperature.
On the other hand, complexes [(PIm)FeII-NO] and [(PImH)FeII-NO] form very stable 6C iron(II) nitrosyl species. In the UV–vis region, the observed Soret bands shift to 423 nm for (3)-NO and 425 nm for (4)-NO (Fig. 3) which match very well with reported 6C iron(II) nitrosyl complexes [46]. To further investigate the strength of the proximal (imidazole) ligand binding to the iron center in (3)-NO and (4)-NO, the EPR spectra of both complexes were recorded. The observed g values are 2.07, 2.00, and 1.97 for (3)-NO and 2.06, 1.99 and 1.96 for (4)-NO (Fig. 8; Fig. S12; Table 3). For both species, the hyperfine pattern is on g(mid) and the hyperfine lines are not well resolved. In addition, the stretching frequency, ν(N–O), for complexes (3)-NO and (4)-NO is the same at 1650 cm−1. This lower stretching frequency is due to the binding of the N-donor ligand (Imidazole) trans to the NO, which weakens the Fe–NO σ-bond [63]. Interestingly, these frequencies are higher in energy compared to a similar ferrous-heme nitrosyl with a free axial base, [Fe(To-F2PP)(MI)(NO)] (MI = methyl-imidazole; ν(N–O) = 1624 cm−1). The trend observed is in line with a previous study by Scheidt [64], further suggesting that the tethered axial ligand bases bind to the heme more weakly than would or does a freely added (or present) base. The IR data match closely with reported work by Lehnert and co-workers [46] and indicate that the benzyl-imidazole linker and the histamine linker impede the binding of the proximal N-donor ligand when compared to the free base, but still allow for the formation of very stable 6C complexes at room temperature.
In 1H-NMR, the pyrrole hydrogen atoms resonate at 8.8 and 9.65 ppm for (3)-NO and (4)-NO, respectively. While in 19F-NMR, the o-fluorine atoms resonate at −106, −107, and −110 ppm for (3)-NO and −105 and −108 ppm for (4)-NO, as shown in Fig. 9. Here, peaks are very broad compared with all the Fe(II)-DIMPI complexes, where peaks were very sharp, and displayed visible H–F coupling interactions.
Fig. 9.
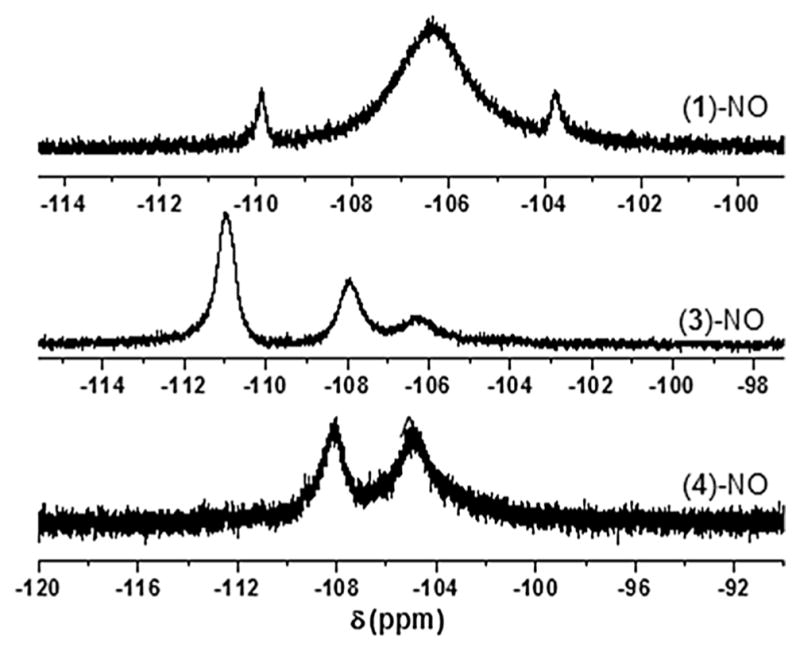
19F-NMR of (1)-NO (top), (3)-NO (middle), and (4)-NO (bottom) complexes in THF at room temperature
Summary
In summary, we have described the synthesis of a new porphyrin with a covalently tethered histamine type ligand, which binds to the iron center. These new ferric [(PImH)FeIII-Cl] and ferrous [(PImH)FeII] hemes have been thoroughly characterized by various spectroscopic methods. Using this newly designed heme and a family of other covalently tethered axial ligands (pyridine/imidazole) synthesized in our lab, we have explored their reactivity towards 2,6-dimethyl-phenyl isocyanide (DIMPI) and nitric oxide (NO). Towards this aim, we have generated and characterized the six coordinate ferrous-heme complexes; [(PPy)FeII(DIMPI)], [(PIm) FeII(DIMPI)], and [(PImH)FeII(DIMPI)], which have UV–Vis, IR, and EPR properties that are clearly distinguishable from those of [(F8)FeII(DIMPI)2]. The X-ray structures reveal a significant contribution from H-bonding between porphryinate meso-phenyl ortho-fluorine atoms, and these have been described. These are emphasized in large part, because 19F-NMR spectroscopy clearly indicates that most if not all of these interactions are maintained in solution. We have also characterized several ferrous-heme mononitrosyl complexes by multinuclear NMR, UV–Vis, EPR, and solid-state FT-IR spectroscopy. At room temperature, [(PIm) FeII] and [(PImH)FeII] form very stable six-coordinate ferrous iron-NO complexes. [(F8)FeII] forms a five-coordinate ferrous-heme nitrosyl complex, while [(PPy)FeII] appears to be somewhere between 5 coordinate and 6 coordinate due to its weakly binding axial pyridyl ligand.
As mentioned, the impetus for synthesizing ferrous-heme porphyrinates using the (PPy), (PIm), and (PImH) ligand systems is to utilize these porphyrins in our ongoing research into modeling the active-site chemistry of cytochrome c oxidase. An understanding of the nature of reactions and structures, i.e., coordination numbers, ligation preferences (e.g., pyridyl vs imidazolyl vs solvent THF), and other bonding or structural aspects can help to better understand the types of structures obtained in heme-O2-copper chemistry, and also inform the design of hemes utilized for such studies.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01 GM 060353 to K.D.K).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00775-016-1369-4) contains supplementary material, which is available to authorized users.
References
- 1.Ghosh A, editor. The smallest biomolecules: diatomics and their interactions with heme proteins. Elsevier; Amsterdam: 2008. [Google Scholar]
- 2.Zhu Y, Silverman RB. Biochemistry. 2008;47:2231–2243. doi: 10.1021/bi7023817. [DOI] [PubMed] [Google Scholar]
- 3.Aono S. Dalton Trans. 2008:3137–3146. doi: 10.1039/b802070c. [DOI] [PubMed] [Google Scholar]
- 4.Ohta T, Kitagawa T. Inorg Chem. 2005;44:758–769. doi: 10.1021/ic0486318. [DOI] [PubMed] [Google Scholar]
- 5.De Montellano PRO. Cytochrome P-450: structure, mechanism, and biochemistry. Springer; 2005. [Google Scholar]
- 6.Walker FA. J Inorg Biochem. 2005;99:216–236. doi: 10.1016/j.jinorgbio.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y, Brandish PE, Ballou DP, Marletta MA. Proc Natl Acad Sci USA. 1999;96:14753–14758. doi: 10.1073/pnas.96.26.14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poulos TL. Curr Opin Struct Biol. 2006;16:736–743. doi: 10.1016/j.sbi.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Ford PC, Bandyopadhyay S, Lim MD, Lorkovic IM. In: The smallest biomolecules: diatomics and their interactions with heme proteins. Ghosh A, editor. Elsevier; Amsterdam: 2008. pp. 66–91. [Google Scholar]
- 10.Tennyson AG, Lippard SJ. Chem Biol. 2011;18:1211–1220. doi: 10.1016/j.chembiol.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Traylor TG, Sharma VS. Biochemistry. 1992;31:2847–2849. doi: 10.1021/bi00126a001. [DOI] [PubMed] [Google Scholar]
- 12.Schopfer MP, Wang J, Karlin KD. Inorg Chem. 2010;49:6267–6282. doi: 10.1021/ic100033y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toledo JC, Jr, Augusto O. Chem Res Toxicol. 2012;25:975–989. doi: 10.1021/tx300042g. [DOI] [PubMed] [Google Scholar]
- 14.Hunt AP, Lehnert N. Acc Chem Res. 2015;48:2117–2125. doi: 10.1021/acs.accounts.5b00167. [DOI] [PubMed] [Google Scholar]
- 15.Wilks A, Ikeda-Saito M. Acc Chem Res. 2014;47:2291–2298. doi: 10.1021/ar500028n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsui T, Unno M, Ikeda-Saito M. Acc Chem Res. 2010;43:240–247. doi: 10.1021/ar9001685. [DOI] [PubMed] [Google Scholar]
- 17.Schuller DJ, Wilks A, De Montellano PRO, Poulos TL. Nat Struct Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 18.Watkins CC, Boehning D, Kaplin AI, Rao M, Ferris CD, Snyder SH. Proc Natl Acad Sci USA. 2004;101:2631–2635. doi: 10.1073/pnas.0308695100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsen RW, Mikšovská J. Coord Chem Rev. 2007;251:1101–1127. [Google Scholar]
- 20.Vos MH. Biochim Biophys Acta. 2008;1777:15–31. doi: 10.1016/j.bbabio.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Spiro TG, Soldatova AV, Balakrishnan G. Coord Chem Rev. 2013;257:511–527. doi: 10.1016/j.ccr.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vos MH, Liebl U. Biochim Biophys Acta. 2015;1847:79–85. doi: 10.1016/j.bbabio.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 23.Liebl U, Lambry JC, Vos MH. Biochim Biophys Acta. 2013;1834:1684–1692. doi: 10.1016/j.bbapap.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 24.Spiro TG, Wasbotten IH. J Inorg Biochem. 2005;99:34–44. doi: 10.1016/j.jinorgbio.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 25.Bandyopadhyay D, Walda KN, Grogan TM, Magde D, Traylor TG, Sharma VS. Biochemistry. 1996;35:1500–1505. doi: 10.1021/bi9518149. [DOI] [PubMed] [Google Scholar]
- 26.Derbyshire ER, Marletta MA. J Biol Chem. 2007;282:35741–35748. doi: 10.1074/jbc.M705557200. [DOI] [PubMed] [Google Scholar]
- 27.Evans JP, Kandel S, De Montellano PRO. Biochemistry. 2009;48:8920–8928. doi: 10.1021/bi9011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucas HR, Karlin KD. Metal Ions Life Sci. 2009;6:295–361. doi: 10.1039/BK9781847559159-00295. [DOI] [PubMed] [Google Scholar]
- 29.Blouin GC, Schweers RL, Olson JS. Biochemistry. 2010;49:4987–4997. doi: 10.1021/bi1001745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blouin GC, Olson JS. Biochemistry. 2010;49:4968–4976. doi: 10.1021/bi100172c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hematian S, Garcia-Bosch I, Karlin KD. Acc Chem Res. 2015;48:2462–2474. doi: 10.1021/acs.accounts.5b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim E, Helton ME, Wasser IM, Karlin KD, Lu S, Huang H-W, Moenne-Loccoz P, Incarvito CD, Rheingold AL, Honecker M, Kaderli S, Zuberbühler AD. Proc Natl Acad Sci USA. 2003;100:3623–3628. doi: 10.1073/pnas.0737180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halime Z, Kieber-Emmons MT, Qayyum MF, Mondal B, Gandhi T, Puiu SC, Chufan EE, Sarjeant AAN, Hodgson KO, Hedman B, Solomon EI, Karlin KD. Inorg Chem. 2010;49:3629–3645. doi: 10.1021/ic9020993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halime Z, Kotani H, Li Y, Fukuzumi S, Karlin KD. Proc Natl Acad Sci USA. 2011;108:13990–13994. doi: 10.1073/pnas.1104698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kieber-Emmons MT, Qayyum MF, Li Y, Halime Z, Hodgson KO, Hedman B, Karlin KD, Solomon EI. Angew Chem Int Ed. 2012;51:168–172. doi: 10.1002/anie.201104080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Bosch I, Adam SM, Schaefer AW, Sharma SK, Peterson RL, Solomon EI, Karlin KD. J Am Chem Soc. 2015;137:1032–1035. doi: 10.1021/ja5115198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Sharma SK, Karlin KD. Polyhedron. 2013;58:190–196. doi: 10.1016/j.poly.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma SK, Rogler PJ, Karlin KD. J Porphyrins Phthalo-cyanines. 2015;19:352–360. doi: 10.1142/S108842461550025X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schopfer MP, Mondal B, Lee D-H, Sarjeant AAN, Karlin KD. J Am Chem Soc. 2009;131:11304–11305. doi: 10.1021/ja904832j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chufan EE, Puiu SC, Karlin KD. Acc Chem Res. 2007;40:563–572. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- 41.Ghiladi RA, Kretzer RM, Guzei I, Rheingold AL, Neuhold Y-M, Hatwell KR, Zuberbühler AD, Karlin KD. Inorg Chem. 2001;40:5754–5767. doi: 10.1021/ic0105866. [DOI] [PubMed] [Google Scholar]
- 42.Garcia-Bosch I, Sharma SK, Karlin KD. J Am Chem Soc. 2013;135:16248–16251. doi: 10.1021/ja405739m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim E, Shearer J, Lu S, Moeenne-Loccoz P, Helton ME, Kaderli S, Zuberbühler AD, Karlin KD. J Am Chem Soc. 2004;126:12716–12717. doi: 10.1021/ja045941g. [DOI] [PubMed] [Google Scholar]
- 44.Kamaraj K, Kim E, Galliker B, Zakharov LN, Rheingold AL, Zuberbühler AD, Karlin KD. J Am Chem Soc. 2003;125:6028–6029. doi: 10.1021/ja034263f. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Schopfer MP, Puiu SC, Sarjeant AAN, Karlin KD. Inorg Chem. 2010;49:1404–1419. doi: 10.1021/ic901431r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berto TC, Praneeth VKK, Goodrich LE, Lehnert N. J Am Chem Soc. 2009;131:17116–17126. doi: 10.1021/ja904368n. [DOI] [PubMed] [Google Scholar]
- 47.Spek A. Acta Crystallogr Sect D. 2009;65:148–155. doi: 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jameson GB, Ibers JA. Inorg Chem. 1979;18:1200–1208. [Google Scholar]
- 49.Kretzer RM, Ghiladi RA, Lebeau EL, Liang H-C, Karlin KD. Inorg Chem. 2003;42:3016–3025. doi: 10.1021/ic020521i. [DOI] [PubMed] [Google Scholar]
- 50.Song B, Yu B-S. Bull Korean Chem Soc. 2003;24:981–985. [Google Scholar]
- 51.Wood MA, Dickinson K, Willey GR, Dodd GH. Biochem J. 1987;247:675–678. doi: 10.1042/bj2470675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.A reviewer suggested the bending may be due to a pseudo Jahn-Teller effect arising from a slight weakening of the Fe-N5(imidazole) bond in (3)-DIMPI compared to that in (4)-DIMPI (see Table 2); see Kitagawa T, et al. J Phys Chem B. 2005;109:21110–21117. doi: 10.1021/jp052158h.
- 53.Thompson DW, Kretzer RM, Lebeau EL, Scaltrito DV, Ghiladi RA, Lam K-C, Rheingold AL, Karlin KD, Meyer GJ. Inorg Chem. 2003;42:5211–5218. doi: 10.1021/ic026307b. [DOI] [PubMed] [Google Scholar]
- 54.Pauling L. J Am Chem Soc. 1932;54:3570–3582. [Google Scholar]
- 55.Shimoni L, Glusker JP. In: Science of crystal structures: highlights in crystallography. Hargittai I, Hargittai B, editors. Springer International Publishing; Cham: 2015. pp. 187–203. [Google Scholar]
- 56.Kryachko E, Scheiner S. J Phys Chem A. 2004;108:2527–2535. [Google Scholar]
- 57.Rohde J-U, In J-H, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 58.England J, Guo Y, Farquhar ER, Young VG, Münck E, Que L. J Am Chem Soc. 2010;132:8635–8644. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.As suggested by a reviewer, the room-temperature molecular structures for (3)-DIMPI and (4)-DIMPI may be dynamic with respect to Fe-C-N bending; the IR band observed for these complexes do seem to be asymmetric, and composed of two bands, possibly two conformers
- 60.Lehnert N, Scheidt WR, Wolf MW. In: Nitrosyl complexes in inorganic chemistry, biochemistry and medicine ii. Mingos DMP, editor. Springer; New York: 2014. pp. 155–223. [Google Scholar]
- 61.Hayes RG, Ellison MK, Scheidt WR. Inorg Chem. 2000;39:3665–3668. doi: 10.1021/ic000159c. [DOI] [PubMed] [Google Scholar]
- 62.Wasser IM, Huang H-W, Moeenne-Loccoz P, Karlin KD. J Am Chem Soc. 2005;127:3310–3320. doi: 10.1021/ja0458773. [DOI] [PubMed] [Google Scholar]
- 63.Praneeth VKK, Neese F, Lehnert N. Inorg Chem. 2005;44:2570–2572. doi: 10.1021/ic050144k. [DOI] [PubMed] [Google Scholar]
- 64.Scheidt WR, Brinegar AC, Ferro EB, Kirner JF. J Am Chem Soc. 1977;99:7315–7322. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.