

Significance
Multiple system atrophy (MSA) is a fatal neurodegenerative disorder associated with the accumulation of alpha-synuclein (α-syn) aggregates in oligodendrocytes. There is currently no treatment to slow down the aggressive neurodegenerative process. C-terminal truncation of α-syn promotes the formation of oligomers and aggregates that, in turn, mediate neurotoxicity in synucleinopathies including MSA. We present here an in vivo proof of concept of the ability of the caspase-1 inhibitor VX-765 to mitigate α-syn pathology and provide neuroprotection in a transgenic mouse model of MSA through reduction of α-syn C-terminal truncation. These findings suggest that VX-765, a well-tolerated drug in a 6 wk-long phase II trial in patients with epilepsy, is a promising candidate to achieve disease modification in MSA by limiting α-syn accumulation.
Keywords: alpha-synuclein, multiple system atrophy, caspase-1, truncation
Abstract
Multiple system atrophy (MSA) is a sporadic orphan neurodegenerative disorder. No treatment is currently available to slow down the aggressive neurodegenerative process, and patients die within a few years after disease onset. The cytopathological hallmark of MSA is the accumulation of alpha-synuclein (α-syn) aggregates in affected oligodendrocytes. Several studies point to α-syn oligomerization and aggregation as a mediator of neurotoxicity in synucleinopathies including MSA. C-terminal truncation by the inflammatory protease caspase-1 has recently been implicated in the mechanisms that promote aggregation of α-syn in vitro and in neuronal cell models of α-syn toxicity. We present here an in vivo proof of concept of the ability of the caspase-1 inhibitor prodrug VX-765 to mitigate α-syn pathology and to mediate neuroprotection in proteolipid protein α-syn (PLP-SYN) mice, a transgenic mouse model of MSA. PLP-SYN and age-matched wild-type mice were treated for a period of 11 wk with VX-765 or placebo. VX-765 prevented motor deficits in PLP-SYN mice compared with placebo controls. More importantly, VX-765 was able to limit the progressive toxicity of α-syn aggregation by reducing its load in the striatum of PLP-SYN mice. Not only did VX-765 reduce truncated α-syn, but it also decreased its monomeric and oligomeric forms. Finally, VX-765 showed neuroprotective effects by preserving tyrosine hydroxylase-positive neurons in the substantia nigra of PLP-SYN mice. In conclusion, our results suggest that VX-765, a drug that was well tolerated in a 6 wk-long phase II trial in patients with epilepsy, is a promising candidate to achieve disease modification in synucleinopathies by limiting α-syn accumulation.
Multiple system atrophy (MSA) is a sporadic adult-onset orphan neurodegenerative disorder clinically characterized by a combination of parkinsonism, cerebellar impairment, and autonomic dysfunction (1). The cytopathological hallmark of MSA is the accumulation of alpha-synuclein (α-syn) aggregates in oligodendrocytes, forming glial cytoplasmic inclusions (GCIs) (2, 3).
The 14-kDA protein α-syn can exist in vitro as an unfolded monomer; although other oligomeric species have been reported (4). Full-length α-syn undergoes several posttranslational modifications such as phosphorylation, tyrosine nitration, and truncation, any of which could promote the formation of toxic α-syn aggregates (5–7). Although the precise toxic species of α-syn have not been firmly established, several studies point to α-syn oligomerization and aggregation as a mediator of neurotoxicity in synucleinopathies (6, 8–10). Hence, decreasing aggregation might be an effective approach to disease modification. Among the mechanisms that promote aggregation of α-syn, C-terminal truncation has been identified as an enhancer/promoter of α-syn oligomerization and fibrillization (11–14). Accordingly, inhibiting α-syn truncation could alter the disease course in MSA (and other synucleinopathies) (15) by decreasing α-syn oligomerization and aggregation. Interestingly, the inflammatory protease caspase-1 cleaves α-syn at Asp121, promoting its aggregation into amyloid fibrils similar to those previously found both in vitro and in vivo (16). In turn, the caspase-1 inhibitor prodrug VX-765 decreases α-syn truncation and aggregation in vitro and rescues cells from α-syn–induced toxicity (16). Therefore, VX-765 could exert neuroprotective effects on MSA pathogenesis by reducing α-syn cleavage, hence limiting its toxicity and its ability to form aggregates. VX-765 is an orally active, well-tolerated, brain-penetrant prodrug that is hydrolyzed by esterases in vivo to produce a potent and selective caspase-1 inhibitor (17, 18), an activity supported by the in vivo demonstration of reduced cleavage of pro–Interleukin-1β (IL-1β) to its activated form IL-1β, a proinflammatory process under control of caspase-1 (18–20).
Here we show that the brain-penetrant VX-765 mitigates progressive synucleinopathy and neurodegeneration in a transgenic mouse model of MSA.
Results
VX-765 Prevents Motor Impairments in Transgenic MSA Proteolipid Protein α-Syn Mice.
Transgenic MSA proteolipid protein α-syn (PLP-SYN) mice display progressive motor impairment with aging, as shown with an increased number of errors on the traversing beam task (21). Motor performance of wild-type (WT) mice was not affected by VX-765 treatment (100 mg·kg·d over 11 wk; P = 0.99), whereas VX-765-treated PLP-SYN mice showed significant improvement in the traversing beam task compared with placebo-treated PLP-SYN mice (P = 0.01) (Fig. 1A).
Fig. 1.

VX-765 treatment reversed α-syn–induced pathology in PLP-SYN mice by decreasing α-syn load in the striatum and rescuing motor performance. (A) Placebo-treated PLP-SYN mice produced more errors per step compared with VX-765–treated PLP-SYN mice in the challenging beam test. (B–F) Representative immunoblot levels of oligomeric (D), monomeric (E), and truncated (F) α-syn in placebo- (B) and VX-765– (C) treated PLP-SYN mice. (G) Analysis of human α-syn mRNA levels in VX-765–treated PLP-SYN mice compared with placebo-treated mice. (H–M) Immunohistochemical analysis of α-syn load (H–J) and insolubility (K–M) in the striatum of placebo- (I and K) and VX-765– (J and L) treated PLP-SYN mice. In all panels, n = 8 per experimental group—except for mRNA α-syn (G), n = 5 for placebo-treated and n = 6 for VX-765–treated PLP-SYN mice. Error bars indicate SE. *P < 0.05, **P < 0.01, ***P < 0.001. FL, full length; Hmw, high molecular weight; PK, proteinase-K; Trc, truncated.
VX-765 Decreases α-Syn Burden in the Striatum of PLP-SYN Mice.
GCIs are the cytopathological hallmark of MSA (3). PLP-SYN mice overexpress α-syn under the PLP promoter, leading to the formation of GCIs (21, 22). To investigate whether reducing C-terminal truncation affects α-syn load in PLP-SYN mice, we first measured the quantity of α-syn in the striatum (Fig. 1) and the cortex (Fig. 2) of PLP-SYN mice by Western blot. VX-765 treatment decreased both oligomeric (–48%, P < 0.05) and monomeric α-syn (–33%, P = 0.001) (Fig. 1 B–E) in the striatum but not in the cortex (P = 0.72 and P = 0.23, respectively; Fig. 2 A–D) of PLP-SYN mice. Interestingly, VX-765–treated mice had a 53% decrease in α-syn truncation in the striatum (P = 0.05) (Fig. 1 B, C, and F), whereas no significant effect was found on the formation of the C-terminally truncated protein in the cortex of PLP-SYN mice (P = 0.4) (Fig. 2 A, B, and E). The amount of oligomeric α-syn in the striatum positively correlated with truncated α-syn (ρ = 0.73, P < 0.05) in the placebo group, whereas VX-765 treatment abolished this correlation in PLP-SYN mice (ρ = –0.08, P = 0.84) (Fig. S1).
Fig. 2.

(A–E) Representative immunoblot levels of oligomeric (C), monomeric (D), and truncated (E) α-syn in the cortex showing no significant difference between placebo- (A) and VX-765– (B) treated PLP-SYN mice. (F–K) α-syn immunohistochemistry assessing the load (F–H) and insolubility (I–K) of α-syn in placebo- (G and I) and VX-765– (H and J) treated PLP-SYN mice. In all panels, n = 8 per experimental group. Error bars indicate SE. FL, full length; Hmv, high molecular weight; PK, proteinase-K; Trc, truncated.
Fig. S1.

Individual correlation plots of truncated α-syn and full-length α-syn levels in the striatum of PLP-SYN mice receiving placebo (black circles) or VX-765 (red circles). Circle size reflects the load of high-molecular weight α-syn as measured by Western blot.
To further assess the effect of VX-765 treatment on α-syn load, we then used quantitative PCR (qPCR) to measure if VX-765 treatment has an effect on α-syn mRNA expression in PLP-SYN mice. Analysis of striatum samples showed no difference in mRNA levels of human α-syn (P = 0.08) (Fig. 1G) between groups, thus indicating no effect of VX-765 treatment on the transcription of human α-syn.
We then assessed the density of α-syn inclusions in PLP-SYN mice and whether VX-765 affects α-syn aggregate solubility using immunohistochemistry on adjacent sections, with or without proteinase-K (PK) pretreatment, in the striatum (Fig. 1 H–M) and in the cortex (Fig. 2 H–M). VX-765–treated PLP-SYN mice showed a significant decrease in the density of α-syn–immunopositive GCIs in the striatum (–40%, P < 0.001) (Fig. 1 H–J) but not in the cortex (P = 0.1) (Fig. 2 H–J) compared with placebo PLP-SYN mice. The amount of PK-resistant α-syn aggregates was also significantly lowered in the striatum of VX-765–treated PLP-SYN compared with placebo PLP-SYN mice (–21%, P < 0.05) (Fig. 1 K–M) but not in the cortex (P = 0.17) (Fig. 2 K–M).
VX-765 Decreases Activated IL-1β Levels in the Striatum of PLP-SYN Mice.
We then measured pro–IL-1β and IL-1β levels in the striatum and cortex of PLP-SYN mice (Fig. 3) to further assess the effect of VX-765 treatment within the targeted structures. We first measured the quantity of pro–IL-1β in the striatum (Fig. 3 A–C) and the cortex (Fig. 3 E–G) of PLP-SYN mice by Western blot. VX-765 treatment had no significant effect on the amount of pro–IL-1β in the striatum (P = 0.23) and cortex (P = 0.1) (Fig. 3 C and G). Interestingly, VX-765–treated mice had decreased IL-1β levels in the striatum (60%, P < 0.001) (Fig. 3 A, B, and D), whereas no significant effect was found on the formation of IL-1β in the cortex of PLP-SYN mice (P = 0.52) (Fig. 3 E, F, and H).
Fig. 3.

Representative immunoblot levels of pro–IL-1β (A–C and E–G) and IL-1β (A, B, D–F, and H) in the striatum (A–D) and cortex (E–H) showing pro–IL-1β in placebo- (A) and VX-765– (B) treated PLP-SYN mice in the striatum and placebo- (E) and VX-765– (F) treated PLP-SYN mice in the cortex. (C and E) Pro–IL-1β levels were not different between groups in striatum and cortex. (D) VX-765–treated PLP-SYN mice showed lower IL-1β levels in the striatum than placebo-treated PLP-SYN mice, whereas (H) no differences between groups was observed in the cortex. In all panels, n = 8 per experimental group. Error bars indicate SE. **P < 0.01.
VX-765 Protects Tyrosine Hydroxylase Neurons in the Substantia Nigra Pars Compacta of PLP-SYN Mice.
Oligodendroglial α-syn overexpression in PLP-SYN mice induces a loss of tyrosine hydroxylase (TH)-positive neurons in the substantia nigra pars compacta (SNc) (21, 23). Accordingly, post hoc analysis of stereological counts of dopaminergic neurons in the SNc revealed a significant loss of TH-positive neurons (–39%, P < 0.05) in placebo PLP-SYN mice compared with placebo WT mice [significant effect of treatment (P < 0.05) and interaction between genotype and treatment (P < 0.05)] (Fig. 4 A–C). More importantly, VX-765 treatment reduced dopaminergic neuron loss in the SNc of PLP-SYN mice, as demonstrated by a 41% difference in TH-positive neuron counts compared with placebo-treated PLP-SYN mice (P < 0.01; Fig. 4 A–C). The analysis of Nissl counts confirmed a significant effect of VX-765 treatment (P < 0.05; Fig. S2).
Fig. 4.

PLP-SYN mice treated with VX-765 showed no loss of TH-positive neurons in the SN. (A and B) Representative nigral sections from placebo- (A) and VX-765– (B) treated PLP-SYN mice. (C) Statistical analysis of TH+ stereological counting showing a loss of nigral TH+ neurons in placebo-treated PLP-SYN mice compared with VX-765–treated PLP-SYN mice, as well as placebo- and VX-765–treated WT mice. In all panels, n = 8 per experimental group. Error bars indicate SE. *P < 0.05, placebo PLP-SYN vs. VX-765 PLP-SYN; #P < 0.05, placebo PLP-SYN vs. VX-765 WT; $P < 0.05, placebo PLP-SYN vs. placebo WT.
Fig. S2.

Statistical analysis of Nissl counts showing a significant effect of VX-765 treatment compared with placebo. In all panels, n = 8 per experimental group. Error bars indicate SE. *P < 0.05, treatment vs. placebo.
VX-765 Crosses the Blood–Brain Barrier in Both WT and PLP-SYN Mice.
Because no other preclinical study showed brain penetrance of VX-765, we quantified the amount of VX-765 in the brain of WT and PLP-SYN mice. Mass spectrometry analysis showed that the brain content of VX-765 in treated animals is identical between WT and PLP-SYN mice (P = 0.86; Fig. S3).
Fig. S3.
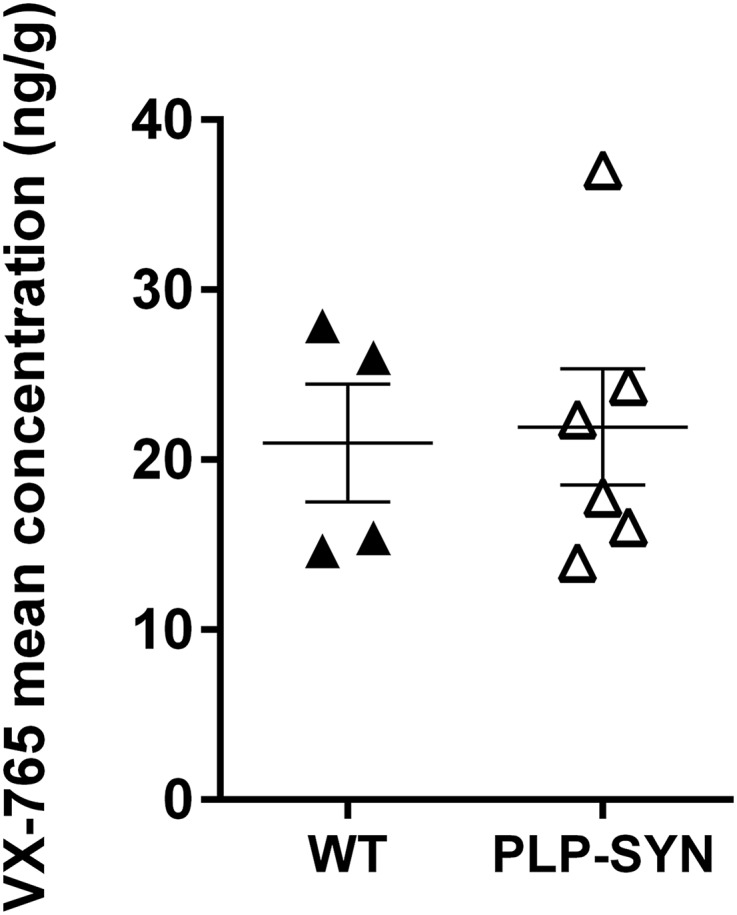
Analysis of VX-765 brain levels in VX-765–treated WT (n = 4) and PLP-SYN mice (n = 6).
Discussion
In the current study, PLP-SYN mice, a transgenic mouse model of MSA, and age-matched WT mice were treated for a period of 11 wk with VX-765 or placebo. VX-765 prevented motor deficits in PLP-SYN mice compared with placebo controls. More importantly, VX-765 was also able to limit the progressive toxicity of α-syn aggregation by reducing its load in the striatum of PLP-SYN mice. Not only did VX-765 reduce truncated α-syn, but it also decreased its monomeric and oligomeric forms without altering the transcription of α-syn. Finally, VX-765 showed neuroprotective effects by preserving TH-positive neurons in the SNc of PLP-SYN mice.
Transgenic models have been developed to support studies on the underlying mechanisms of MSA pathogenesis and preclinical drug screening. These models are based on overexpression of α-syn in oligodendrocytes and replicate several aspects of MSA pathology (24, 25). The PLP-SYN mouse model used in this study displays motor deficits, neuroinflammation, and loss of TH-positive neurons in the SNc in addition to the presence of α-syn inclusions in oligodendrocytes (23, 26). We show here that these mice also show C-terminal truncated α-syn.
Intracellular α-syn inclusions are the pathological hallmark of several neurodegenerative disorders known as synucleinopathies that include dementia with Lewy bodies (DLB), Parkinson’s disease (PD) and MSA (27). Most of the work done to assess α-syn toxicity and to describe the relationships between α-syn burden, spreading, and disease severity has, however, been done in PD models (28, 29). The precise mechanism by which α-syn aggregate formation leads to neurodegeneration remains unclear (28, 30). Recent research efforts have focused on limiting α-syn–induced neurodegeneration by inhibiting α-syn oligomerization and aggregation (31–35). Several studies have shown that C-terminal truncated α-syn is prone to form fibrils (36–39). In turn, α-syn fibrillization is toxic when overexpressed in animal models of PD (14, 40). More importantly, C-terminal truncation elicits the production of toxic α-syn aggregates and promotes neurodegeneration (10–14, 34, 38–47). Some studies have shown that C-terminally truncated α-syn is found in GCIs in MSA (48, 49) as well as in Lewy bodies of PD and DLB patient brains (11, 12, 39, 50, 51). Several proteases such as calpain, matrix metalloproteases (MMPs), cathepsin D, and plasmin have been implicated in α-syn truncation, subsequently resulting in increased levels of protein aggregates; however, none has been established as a major producer of C-terminally truncated α-syn in vivo, especially in response to inflammation (12, 39, 42, 52–55).
VX-765 is a prodrug that in vivo produces a potent and selective inhibitor of caspase-1, an inflammatory protease that has recently been shown to cleave α-syn in its disordered C-terminal region following residue Asp-121 (16). This same study showed that VX-765 decreases C-terminal truncation and aggregate formation in vitro. Here, we demonstrate the ability of VX-765 to mitigate MSA-like neuropathology together with a concomitant reduction in C-terminal truncation and aggregation of α-syn as well as dopaminergic neurodegeneration in PLP-SYN mice.
Recent efforts targeting α-syn truncation have shown that the overexpression of a calpain-specific inhibitor reduces α-syn aggregation and other neuropathological features in the [A30P]α-syn–Thy-1 PD mouse model (44), whereas immunotherapy directed against the C-terminal region of α-syn proved to be beneficial in the mThy1–α-syn PD mouse model (45) and the transgenic DLB mouse model using the PDGF β promoter (34). These studies have shown that targeting α-syn truncation in vivo decreases α-syn aggregation and neurotoxicity. Interestingly, Games et al. (45) reported that decreasing α-syn truncation and the resultant effects could well be explained by blocking α-syn propagation from neurons.
We cannot rule out the possibility that the preservation of dopaminergic neurons reported here might also involve caspase-1–dependent mechanisms other than the inhibition of α-syn truncation and the resultant decrease in oligomeric species (e.g., a reduced proinflammatory response due to decreased IL-1β activation) (56).
VX-765 treatment also reduced monomeric α-syn in oligodendrocytes. This might be due to the decrease in truncated and oligomeric α-syn load, which allowed the clearance systems in oligodendrocytes to better handle the overexpressed monomeric α-syn. Truncated and oligomeric α-syns are both products of monomeric α-syn modification (4, 6, 30, 38, 51). Thus, a marked decrease in both forms might well be secondary to the decrease in monomeric α-syn. This might not be the case with VX-765 treatment, as it cancelled the correlation between truncated and oligomeric α-syn observed in placebo-treated mice. Moreover, VX-765 treatment had no effect on human α-syn mRNA levels, ruling out the potential implication of VX-765 on α-syn expression.
VX-765 treatment had no effect on α-syn levels in the cortex compared with the striatum, where it decreased truncated α-syn levels. Such difference was also found for IL-1β levels, as VX-765 only decreased IL-1β levels in the striatum but not in the cortex. VX-765 is an inhibitor of caspase-1, the primary cleaving and activating mechanism of IL-1β from its precursor, pro–IL-1β (17–19). These results further support the hypothesis that the effects of VX-765 in the brain are structure and region dependent. Moreover, we also show the ability of VX-765 to pass the blood–brain barrier and its presence in the brain. Future studies should focus on measuring the bioavailability of VX-765 in several structures, thereby assessing if VX-765 efficacy is structure/region-dependent in the brain. More importantly, studies assessing α-syn pathology in the brain should assess the structure-dependent weight/contribution of different proteases known to cleave α-syn in the brain, thereby leading to a better understanding of the effect of protease inhibitors in the brain. In this regard, we have previously shown a region-dependent alteration/expression of MMPs in the putamen and cortex of MSA patients (55). MMPs have been previously shown to cleave α-syn, thereby producing truncated forms (52, 54).
We here present an in vivo proof of concept of the ability of the caspase-1 inhibitor prodrug VX-765 to mitigate α-syn pathology and to mediate neuroprotection in an MSA mouse model. Our results show that VX-765, a drug that was well tolerated in a phase II trial in patients with epilepsy (20), is a promising candidate to achieve disease modification in synucleinopathies by limiting α-syn accumulation.
Materials and Methods
Animals.
Mice expressing human WT α-syn in oligodendrocytes under the control of the proteolipid promoter (PLP-SYN) were previously generated on a C57BL/6 background (22). For histopathological and biochemical analysis, PLP-SYN (n = 16) and WT littermates (n = 16) aged 6 wk at the beginning of the treatment period were randomly allocated into two groups, placebo (8 WT, 8 PLP-SYN) and VX-765 (8 WT, 8 PLP-SYN). For the qPCR and VX-765 brain dosing experiments, PLP-SYN (n = 11) and WT littermates (n = 8) aged 6 wk were randomly allocated to groups and treated accordingly with placebo (4 WT, 5 PLP-SYN) and VX-765 (4 WT, 6 PLP-SYN). All experiments were performed in accordance with French guidelines (87-848, Ministère de l’Agriculture et de la Forêt) and the European Community Council Directive (2010/63/EU) for the care of laboratory animals. All experiments were approved by The Institutional Animal Care and Use Committee of Bordeaux (CE50, license #50120100-A). Mice were maintained in a temperature- and humidity-controlled room on a 12:12 light–dark cycle with food and water ad libitum.
Pharmacological Treatment.
Mice were treated via gavage (VetTech solutions Ltd., Dosing Catheter: 4.5fg, length 60 mm) once a day with VX-765 (MedKoo Biosciences), which was prepared daily and dissolved in deionized water containing 0.5% methylcellulose and 0.1%Tween-80 at a dose of 100 mg/kg. The same solution without VX-765 was administered to the placebo group. VX-765 is an orally absorbed prodrug of VRT-043198, a potent and selective inhibitor of caspases in the ICE/caspase-1 subfamily of cysteine proteases. VX-765 is converted to the cell permeable inhibitor VRT-043198 in vivo by the action of plasma and liver esterases. Although brain penetrance of the active drug is modest, it has been shown to inhibit caspase-1 in mouse brain at the doses used here (57). This dose is lower than the dose that was well-tolerated over a 6-wk period in a phase II clinical trial in patients with epilepsy (20).
Behavioral Test.
Motor coordination and balance were assessed with a modified version of the traversing beam task that was adapted from a previously described method (58). This test measures the ability to traverse a narrow beam to reach a goal box.
Tissue Processing.
At the end of the 11-wk treatment period, mice were anesthetized with pentobarbital (100 mg/kg i.p.) and intracardially perfused with 0.9% saline. Brains were quickly removed and cut in half between the two hemispheres. The right hemisphere was frozen directly for biochemical analysis, and the left hemisphere was postfixed for 5 d in 4% (wt/vol) PFA, then cryoprotected in 20% (wt/vol) sucrose in 0.1 M PBS, frozen on powdered dry ice, and stored at –80 °C.
qPCR.
Striatum tissue samples were homogenized in Trireagent (Euromedex). RNA was isolated using a chloroform/isopropanol protocol (59) and processed and analyzed following an adaptation of published methods (60). cDNA was synthesized using RevertAid Premium Reverse Transcriptase (Fermentas). qPCR reactions were done with LightCycler 480 SYBR Green I Master using a LightCycler 480 II system (Roche). For the determination of the reference genes, the Genorm method was used (61), and the two reference genes were Eef1a1 and NONO. The relative level of expression was calculated using the comparative (2–∆∆CT) method (61). Primer sequences for human α-syn were as follows: forward, aaaggaccagttgggcaagaa; reverse, atccacaggcatatcttccag.
VX-765 Brain Assay with Mass Spectrometry.
The brain was homogenized in water using an IKA T25 Ultra-Turrax. A volume of 400 µL of acetonitrile was added to a 100-µL aliquot of homogenate, then vortexed 10 min using a multi-Reax (Dutcher), and centrifuged. Supernatant was evaporated to dryness using a Turbovap LV evaporator (Biotage) and resuspended with 100 µL of initial mobile phase. VX-765 was quantified using linear regression with 1/X weighing, and a range from 0.33 to 150 ng/g brain was used for calibration. We injected 10 µL extract into the liquid chromatography with tandem mass spectrometry detection analytical system, which consists of a Waters ACQUITY UPLC System with an Acquity UPLC BEH Shield RP18 1.7 µm, 2.1 × 50 mm column, and a reversed phase gradient over a run time of 5 min. Initial conditions consisted of mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile) with a column temperature of 50 °C and a flow rate of 0.500 mL/min. The gradient conditions ramped from 10% B to 80% B between 0.5 and 2.5 min and maintained up to 3.0 min, then ramped to 100% and maintained up to 3.5 min, and ramped to 10 and maintained up to 5.0 min for re-equilibration. The MS analysis was performed on a Waters XEVO TQ-S Mass Spectrometer operating in positive ion electrospray multiple reaction monitoring mode. Multiple reaction monitoring transition was m/z 509.1 > 243 for VX-765, which displayed a mean retention time around 2.0 min.
Immunoblotting.
For Western blot analysis, patches were taken from the motor cortex and striatum of PLP-SYN. Protein extraction and sample preparation was done as previously described (62). A total of 30 μg of protein were loaded and run on 4–15% gradient gels (Bio-Rad Laboratories) to measure oligomeric forms of α-syn using anti-human–specific antibodies Syn-211 (1:1,000, Thermo Fisher Scientific). Truncated forms of α-syn were assessed on 18% SDS/PAGE gels and incubated with human-specific antibody Syn-204 (1:500, Abcam). For IL-1β and pro–IL-1β levels, proteins were run on 12% SDS/PAGE gels and incubated with anti–IL-1β (1:1,000; R&D) as previously described (63). Proteins were normalized to actin (1:2,000; Sigma) or tubulin (1:4,000; Sigma) as a loading control.
Histopathological Analysis.
We collected 40 μm free-floating coronal sections for histopathological analysis. To assess the solubility of α-syn inclusions in oligodendrocytes, sections from PLP-SYN mice were first incubated with and without PK at 10 μg/mL (Sigma-Aldrich) for 10 min at room temperature as previously described (21, 64). PK-treated and nontreated adjacent sections were then processed for α-syn immunohistochemistry using the primary antibody against α-syn (clone LB509, Invitrogen Laboratories, 1:500) as previously described (21). For TH immunostaining, every fourth section was processed for TH immunohistochemistry with the primary antibody against TH (MAB318, Millipore, 1:10000) as previously described (21).
Quantification.
The distribution of α-syn–immunopositive inclusions was assessed in the cortex and striatum of PLP-SYN mice as previously described (21). For SNc TH counts, stereological sampling was performed as previously described (64).
Statistical Analysis.
Data were compared between placebo and VX-765–treated PLP-SYN mice (behavior, biochemical analysis of α-syn load and pro–IL-1β/IL-1β a levels, mRNA expression of human α-syn, and histopathological assessment of α-syn inclusions) and between WT and PLP-SYN mice (brain assay of VX-765) by using t tests. If data were not normally distributed, a Mann–Whitney U test was used instead. Histopathological data for TH and Nissl staining were analyzed using a two-way ANOVA with genotype and treatment as independent variables. ANOVAs were followed by post hoc t tests corrected for multiple comparison by the method of Bonferroni whenever appropriate. A Pearson correlation was performed between striatal levels of truncated and oligomeric α-syn. Statistical analyses were performed with Sigma Plot 12 software (Systat Software Inc.). For all statistical tests, the level of significance was set at P < 0.05. All data are expressed as mean ± SEM.
Acknowledgments
The authors thank Dr. Elisabeth Normand, Melissa Deshors, and Florian Roguet for the maintenance of the PLP-SYN colony and animal care. The Université de Bordeaux and Centre National de la Recherche Scientifique provided infrastructural support. This work was supported by the French Research Agency (ANR-14-RARE-0001-01), under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases, and Grant LABEX BRAIN ANR-10-LABX-43. Q.Q.H. acknowledges funding from the NIH (Grants 1R21NS07988 and 5R01GM111639).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1609291113/-/DCSupplemental.
References
- 1.Gilman S, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670–676. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spillantini MG, et al. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251(3):205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- 3.Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome) J Neurol Sci. 1989;94(1-3):79–100. doi: 10.1016/0022-510x(89)90219-0. [DOI] [PubMed] [Google Scholar]
- 4.Fauvet B, et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287(19):15345–15364. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rochet JC, Conway KA, Lansbury PT., Jr Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry. 2000;39(35):10619–10626. doi: 10.1021/bi001315u. [DOI] [PubMed] [Google Scholar]
- 6.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14(1):38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muntané G, Ferrer I, Martinez-Vicente M. α-synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience. 2012;200:106–119. doi: 10.1016/j.neuroscience.2011.10.042. [DOI] [PubMed] [Google Scholar]
- 8.Cremades N, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149(5):1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auluck PK, Caraveo G, Lindquist S. α-Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Annu Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- 10.Winner B, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci USA. 2011;108(10):4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W, et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci USA. 2005;102(6):2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu CW, et al. A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: Implications for pathogenesis of Parkinson disease. J Biol Chem. 2005;280(24):22670–22678. doi: 10.1074/jbc.M501508200. [DOI] [PubMed] [Google Scholar]
- 13.Hoyer W, Cherny D, Subramaniam V, Jovin TM. Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry. 2004;43(51):16233–16242. doi: 10.1021/bi048453u. [DOI] [PubMed] [Google Scholar]
- 14.Ulusoy A, Febbraro F, Jensen PH, Kirik D, Romero-Ramos M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur J Neurosci. 2010;32(3):409–422. doi: 10.1111/j.1460-9568.2010.07284.x. [DOI] [PubMed] [Google Scholar]
- 15.Fernagut PO, et al. Multiple system atrophy: A prototypical synucleinopathy for disease-modifying therapeutic strategies. Neurobiol Dis. 2014;67:133–139. doi: 10.1016/j.nbd.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 16.Wang W, et al. Caspase-1 causes truncation and aggregation of the Parkinson disease-associated α-synuclein. Proc Natl Acad Sci USA. 2016;113:9587–9592. doi: 10.1073/pnas.1610099113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boxer MB, Shen M, Auld DS, Wells JA, Thomas CJ. 2010. A small molecule inhibitor of Caspase 1, probe reports from the NIH Molecular Libraries Program (NIH, Bethesda, MD) [PubMed]
- 18.Wannamaker W, et al. (S)-1-((S)-2-[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther. 2007;321(2):509–516. doi: 10.1124/jpet.106.111344. [DOI] [PubMed] [Google Scholar]
- 19.Noe FM, et al. Pharmacological blockade of IL-1β/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol Dis. 2013;59:183–193. doi: 10.1016/j.nbd.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 20.Vertex P. 2011. Vertex Announces Completion of Phase 2 Study of VX-765 in People with Epilepsy who did not Respond to Previous Treatment. Available at investors.vrtx.com/releasedetail.cfm?ReleaseID=555967.
- 21.Fernagut PO, et al. Age-related motor dysfunction and neuropathology in a transgenic mouse model of multiple system atrophy. Synapse. 2014;68(3):98–106. doi: 10.1002/syn.21719. [DOI] [PubMed] [Google Scholar]
- 22.Kahle PJ, et al. Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 2002;3(6):583–588. doi: 10.1093/embo-reports/kvf109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stefanova N, et al. Oxidative stress in transgenic mice with oligodendroglial alpha-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am J Pathol. 2005;166(3):869–876. doi: 10.1016/s0002-9440(10)62307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernagut PO, Tison F. Animal models of multiple system atrophy. Neuroscience. 2012;211:77–82. doi: 10.1016/j.neuroscience.2011.09.044. [DOI] [PubMed] [Google Scholar]
- 25.Stefanova N, Tison F, Reindl M, Poewe W, Wenning GK. Animal models of multiple system atrophy. Trends Neurosci. 2005;28(9):501–506. doi: 10.1016/j.tins.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Stefanova N, et al. Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: Implications for multiple system atrophy. Mov Disord. 2007;22(15):2196–2203. doi: 10.1002/mds.21671. [DOI] [PubMed] [Google Scholar]
- 27.Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- 28.Recasens A, Dehay B. Alpha-synuclein spreading in Parkinson’s disease. Front Neuroanat. 2014;8:159. doi: 10.3389/fnana.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Recasens A, et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75(3):351–362. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 30.Conway KA, et al. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc Natl Acad Sci USA. 2000;97(2):571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savolainen MH, et al. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol Dis. 2014;68:1–15. doi: 10.1016/j.nbd.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bieschke J, et al. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci USA. 2010;107(17):7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levin J, et al. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014;127(5):779–780. doi: 10.1007/s00401-014-1265-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masliah E, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011;6(4):e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Myöhänen TT, et al. A prolyl oligopeptidase inhibitor, KYP-2047, reduces α-synuclein protein levels and aggregates in cellular and animal models of Parkinson’s disease. Br J Pharmacol. 2012;166(3):1097–1113. doi: 10.1111/j.1476-5381.2012.01846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 1998;436(3):309–312. doi: 10.1016/s0014-5793(98)01146-6. [DOI] [PubMed] [Google Scholar]
- 37.Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci USA. 2000;97(9):4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murray IV, et al. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry. 2003;42(28):8530–8540. doi: 10.1021/bi027363r. [DOI] [PubMed] [Google Scholar]
- 39.Dufty BM, et al. Calpain-cleavage of alpha-synuclein: Connecting proteolytic processing to disease-linked aggregation. Am J Pathol. 2007;170(5):1725–1738. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci. 2007;27(12):3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michell AW, et al. The effect of truncated human alpha-synuclein (1-120) on dopaminergic cells in a transgenic mouse model of Parkinson’s disease. Cell Transplant. 2007;16(5):461–474. doi: 10.3727/000000007783464911. [DOI] [PubMed] [Google Scholar]
- 42.Mishizen-Eberz AJ, et al. Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. J Neurochem. 2003;86(4):836–847. doi: 10.1046/j.1471-4159.2003.01878.x. [DOI] [PubMed] [Google Scholar]
- 43.Tsigelny IF, et al. Dynamics of alpha-synuclein aggregation and inhibition of pore-like oligomer development by beta-synuclein. FEBS J. 2007;274(7):1862–1877. doi: 10.1111/j.1742-4658.2007.05733.x. [DOI] [PubMed] [Google Scholar]
- 44.Diepenbroek M, et al. Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-Synuclein processing, aggregation and synaptic impairment in [A30P]αSyn transgenic mice. Hum Mol Genet. 2014;23(15):3975–3989. doi: 10.1093/hmg/ddu112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Games D, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci. 2014;34(28):9441–9454. doi: 10.1523/JNEUROSCI.5314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daher JP, et al. Conditional transgenic mice expressing C-terminally truncated human alpha-synuclein (alphaSyn119) exhibit reduced striatal dopamine without loss of nigrostriatal pathway dopaminergic neurons. Mol Neurodegener. 2009;4:34. doi: 10.1186/1750-1326-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tofaris GK, et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): Implications for Lewy body disorders. J Neurosci. 2006;26(15):3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gai WP, Power JH, Blumbergs PC, Culvenor JG, Jensen PH. Alpha-synuclein immunoisolation of glial inclusions from multiple system atrophy brain tissue reveals multiprotein components. J Neurochem. 1999;73(5):2093–2100. [PubMed] [Google Scholar]
- 49.Tong J, et al. Brain alpha-synuclein accumulation in multiple system atrophy, Parkinson’s disease and progressive supranuclear palsy: A comparative investigation. Brain. 2010;133(Pt 1):172–188. doi: 10.1093/brain/awp282. [DOI] [PubMed] [Google Scholar]
- 50.Campbell BC, et al. The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J Neurochem. 2001;76(1):87–96. doi: 10.1046/j.1471-4159.2001.00021.x. [DOI] [PubMed] [Google Scholar]
- 51.Baba M, et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol. 1998;152(4):879–884. [PMC free article] [PubMed] [Google Scholar]
- 52.Choi DH, et al. Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. J Biol Chem. 2011;286(16):14168–14177. doi: 10.1074/jbc.M111.222430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim KS, et al. Proteolytic cleavage of extracellular α-synuclein by plasmin: implications for Parkinson disease. J Biol Chem. 2012;287(30):24862–24872. doi: 10.1074/jbc.M112.348128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sung JY, et al. Proteolytic cleavage of extracellular secreted alpha-synuclein via matrix metalloproteinases. J Biol Chem. 2005;280(26):25216–25224. doi: 10.1074/jbc.M503341200. [DOI] [PubMed] [Google Scholar]
- 55.Bassil F, et al. Region-specific alterations of matrix metalloproteinase activity in multiple system atrophy. Mov Disord. 2015;30(13):1802–1812. doi: 10.1002/mds.26329. [DOI] [PubMed] [Google Scholar]
- 56.Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maroso M, et al. Interleukin-1β biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics. 2011;8(2):304–315. doi: 10.1007/s13311-011-0039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fleming SM, et al. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24(42):9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 60.Bustin SA, et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 61.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 62.Dehay B, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci USA. 2012;109(24):9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mingam R, et al. In vitro and in vivo evidence for a role of the P2X7 receptor in the release of IL-1 beta in the murine brain. Brain Behav Immun. 2008;22(2):234–244. doi: 10.1016/j.bbi.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fernagut PO, et al. Behavioral and histopathological consequences of paraquat intoxication in mice: Effects of alpha-synuclein over-expression. Synapse. 2007;61(12):991–1001. doi: 10.1002/syn.20456. [DOI] [PMC free article] [PubMed] [Google Scholar]