

Abstract
Differentiated embryonic chondrocyte expressed gene 2 (DEC2; BHLHE41/Sharp1) is a helix-loop-helix (bHLH) transcription factor, and its deregulation has been observed in several tumors. However, this gene’s effects on tumor progression are controversial, and its roles in gastric cancer (GC) remain unclear. In the present study, we found that DEC2 expression level is lower in GC tissues compared with adjacent non-tumor tissues, and negatively correlated with tumor invasion, lymph node metastasis, TNM stage, and poor survival of GC patients. Positive clinical correlations of DEC2 with EMT regulator, E-cadherin, were also observed in the tissue sections. Overexpression of DEC2 inhibits cell proliferation and EMT in vitro, as well as tumor growth and metastasis in vivo. DEC2 expression also induces cell apoptosis. Furthermore, the anti-metastatic effect of DEC2 was mediated by inhibiting ERK/NF-κB/EMT axis. After treatment with ERK1/2 chemical inhibitor (U0126), DEC2’s inhibitory effect on ERK/NF-κB/EMT was further decreased. Collectively, these data helped to characterize DEC2, which might be a potential molecular target for diagnostic and therapeutic approaches for GC.
Keywords: Differentiated embryonic chondrocyte expressed gene 2 (DEC2), gastric cancer (GC), proliferation, tumor metastasis, tumor suppressive gene, epithelial-mesenchymal transition (EMT)
Introduction
Gastric cancer (GC) is one of the most common malignancies, which is difficult to detect in the early stages; it is regarded as the second leading cause of cancer-related deaths worldwide [1,2]. Although mortality due to GC has declined in recent years primarily according to the improvements in endoscopic detection [3], early diagnosis and effective treatment of GC remain challenging. Therefore, it is essential to elucidate the underlying molecular mechanisms of initiation and metastasis and to develop novel therapeutic approaches.
Differentiated embryonic chondrocyte expressed gene 2 (DEC2), also known as BHLHE41, Sharp1, belongs to the basic helix-loop-helix (bHLH) transcription factor family. This gene is ubiquitously expressed in human tissues and involved in various biological phenomena [4-8]. Importantly, recent studies analyzing the relationships between DEC2 expression and clinicopathological features in cancer patients have indicated that DEC2 is a novel molecular marker of progression in various cancers, including pancreatic cancer and endometrial carcinoma [9,10]. However, DEC2 may act as either a tumor suppressor or carcinogenic agent. In breast cancer, it was reported that DEC2 significantly inhibited cell proliferation and metastasis but contributed to anti-apoptotic effects [11-13]. In addition, DEC2 impaired epithelial-mesenchymal transition (EMT)-associated metastasis of human endometrial and pancreatic cancers [9,14], it has been confirmed that DEC2 competes with SP1 for the SP1 binding site of the TWIST1 promoter [14]. In contrast, it was reported DEC2 increased proliferation of breast cancer [15] and invasiveness of osteosarcomas [16], but had anti-apoptotic effects in human oral cancers [17]. All of these results suggest that the aberrant expression of DEC2 may play an important role in tumor progression, even though the exact function of DEC2 in cancer malignancy is controversial. It is essential to investigate whether DEC2 is involved in the progression and metastasis of GC.
In this study, we first investigated the biological function of DEC2 in gastric cancer. Immunohistochemistry results showed that DEC2 expression levels are lower in GC tissues compared with adjacent non-tumor tissues. By manipulating DEC2 expression in GC cells, we determined that DEC2 inhibited cell proliferation and epithelial-mesenchymal transition (EMT) in vitro, as well as tumor growth and metastasis in vivo. Meanwhile, we confirmed that DEC2 induced GC cell apoptosis. Furthermore, we observed that DEC2 inhibits EMT-associated GC progression via regulating ERK/NF-κB/EMT axis. These results reveal that DEC2 functions as a tumor suppressor, which inhibits tumor growth and metastasis in GC.
Materials and methods
Patients and ethics statement
A total of 49 specimens were collected from GC patients admitted to Jinan Central Hospital of Shandong University from 2007 to 2014. All patients had not received treatment before sample collection. The tumor stage was clinically and histologically categorized basing on the guidelines described by the sixth edition of The American Joint Committee on Cancer. The clinicopathological features of patients are shown in Table 1. Patients from 2008 to 2011 were contacted by phone to check upon their health status and the last censor date was on January 31th, 2014. Written informed consent was acquired from each patient for this study. The study methodologies conformed to the standards set by the Declaration of Helsinki. The research protocol and consent program were approved by Central Hospital Affiliated to Shandong University Medical Institutional Ethical Committee.
Table 1.
Association of DEC2 or E-cadherin expression with the clinicopathological characteristics of GC
| Characteristics | Case (49) | DEC2 expression | P value | E-cadherin expression | P value | ||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| High (11) | Low (38) | High (13) | Low (36) | ||||
| Age (years) | |||||||
| ≤50 | 7 | 2 | 5 | 0.646 | 2 | 5 | 0.608 |
| >50 | 42 | 9 | 33 | 11 | 31 | ||
| Gender | |||||||
| Female | 15 | 4 | 11 | 0.716 | 8 | 7 | 0.011* |
| Male | 34 | 7 | 27 | 5 | 29 | ||
| Tumor size (diameter) | |||||||
| ≤4 | 22 | 7 | 15 | 0.185 | 8 | 14 | 0.202 |
| >4 | 27 | 4 | 23 | 5 | 22 | ||
| Differentiation | |||||||
| I | 32 | 8 | 24 | 0.725 | 9 | 23 | 0.504 |
| II-III | 17 | 3 | 14 | 4 | 13 | ||
| Tumor invasion (AJCC) | |||||||
| T1-T2 | 14 | 8 | 6 | 0.001* | 9 | 5 | 0.000* |
| T3-T4 | 35 | 3 | 32 | 4 | 31 | ||
| Lymphatic metastasis | |||||||
| Absent | 17 | 7 | 10 | 0.033* | 8 | 9 | 0.038* |
| Present | 32 | 4 | 28 | 5 | 27 | ||
| TNM stage | |||||||
| I-II | 23 | 10 | 13 | 0.001* | 10 | 13 | 0.021* |
| III-IV | 26 | 1 | 25 | 3 | 23 | ||
Statistically significant difference; AJCC, American Joint Committee on Cancer.
Cell culture
Human gastric cancer cell lines (BGC823, MGC803, HGC27, and MKN-45) were obtained from the Cell Bank of The Chinese Academy of Sciences (Shanghai, China). Non-malignant gastric epithelial cells GES-1 were purchased from Cwbiotech Company (Beijing, China). MGC803 and GES-1 cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Hyclone, USA); BGC823, HGC27, and MKN-45 cells were cultured in RPMI 1640 medium (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone, USA). All cells were incubated at 37°C in a humidified atmosphere with 5% CO2. The ERK1/2 inhibitor, U0126, was purchased from a commercial source (Selleck, Houston, TX, USA).
Cell transfection
The DEC2 vector (GeneCopoeia, Rockville, MD, USA) was prepared with full length human DEC2 cDNA linked to the Pez-lv105 vector in order to induce DEC2 over-expression. An empty vector was used as the control. Transfection was performed using the Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer’s instructions. Human DEC2 cDNA was subcloned into a pGV-puro lentiviral vector containing the puromycin resistance gene for establishment of stable cell lines (Genechem, China). Stable cell lines were selected by incubating puromycin containing medium for 48 h after transfection. The resulting stably transfected cell lines were collected after four weeks.
Human DEC2 shRNA linked with Lentivirus pGLV/H1/GFP vector (Genechem Co., Ltd., Shanghai, China) was used to induce DEC2-silence in the cells. Cells transfected with Lentivirus pGLV/H1/GFP (NC) vector served as negative control. Stable cell lines were selected by incubating puromycin containing medium for 48 h after transfection. The resulting stably transfected cell lines were collected after four weeks. shRNA sequences are: shDEC2, 5’-CTGGACTATTCCTCTTTGTAT-3’; shNC, 5’-GTTCTCCGAACGTGTCACGT-3’.
Cell proliferation assay in vitro
Cell proliferation was evaluated using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) assay. According to the manufacturer’s instructions, Cells were plated in 96-well plates at a started number of 5×103 cells/well. Then, 10 μl CCK8 solution was added to each well followed by incubation at 37°C for 2 h. Sample absorbances were measured at 450 nm for six days using a microplate reader (Bio-Rad, Hercules, CA, USA). The ratio of optical density (OD) value of each group using the cells on Day 1 as standard at the indicated time points are presented. Each experiment was performed in triplicate.
EdU incorporation assay
Transfected GC cells seeded in 96-well plates were incubated with EdU (5 μM, Ribobio, Guang zhou, China) for 2 h before fixation in 4% paraformaldehyde. After permeabilization with 0.5% Triton X-100, 100 μl 1x Apollo® staining reaction liquid was added to GC cells at 37°C for 30 min, the cells were counterstained with DAPI, and imaged using a fluorescence microscope (Olympus).
Annexin V/PI staining assay for apoptosis
Apoptosis assay were performed with an Annexin V-FITC apoptosis detection kit (BD Biosciences, USA), according to the manufacturer’s instructions. Briefly, the transfected cells were washed twice with cold PBS and resuspended to a concentration of 1×106 cells/mL. Cells were then incubated with 100 μl binding buffer containing 2.5 μl Annexin V-FITC and 1 μl PI for 15 min at room temperature in the dark. Following this incubation period, the samples were analyzed by flow cytometry (BD, San Diego, CA, USA).
Migration and invasion assay in vitro
Cell migration was performed in 24-well transwell plates with 8 μm-pore polycarbonate membranes (Costar, Corning, MA, USA). Matrigel invasion assay was performed using membranes coated with Matrigel matrix (BD Science, Sparks, MD, USA). The transfected cells (1×105 cell/ml) suspended in serum-free medium were added to the upper chamber and incubated for 24 h at 37°C for migration assay and 36 h for invasion assay. Migrated or Invasive cells were fixed with 100% methanol for 15 min, then stained with 0.1% Crystal violet-Solution for 20 min, and five representative fields from sample were counted under a light optic microscope (Olympus). Each experiment was performed in triplicate. For wound healing assay, transfected cells were seeded into 6-well plates and grown to confluence in complete medium. The confluence monolayers were scratched by a 10 μl pipette tip after which the wounded monolayers were washed by PBS to remove cell debris and cultured in FBS-free media. Photos were taken under a microscope at 0 h and 48 h.
Quantitative real-time RT-PCR
Total RNA was isolated using the Trizol reagent (Invitrogen) following the manufacturer’s instructions. First strand complementary DNAs were prepared using the Reverse Transcription Reaction Kit (Takara). Quantitative real-time PCR was analyzed on the Applied Biosystems 7300 Real-Time PCR System. The relative amount mRNA was calculated using the 2(-ΔΔCt) method after normalization to GAPDH mRNA levels. All the samples were performed in triplicates in each experiment. PCR reaction was performed using DEC2 primers: 5’-GCATGAAACGAGACGACACC-3’ (forward), 5’-ATTTCAGATGTTCAGGCAGT-3’ (reverse); GAPDH primers: 5’-AGAAGGCTGGGGCTCATTTG-3’ (forward) and 5’-AGGGGCCATCCACAGTCTTC-3’ (reverse).
Protein extraction and western blot assay
Cells were lysed in RIPA buffer (Beyotime, Jiangsu, China) supplemented with protease inhibitors. Total protein was quantified by BCA protein assay (Thermo Scientific, Rockford, USA). Proteins were separated by 10% SDS-PAGE gel and transferred to PVDF membranes (Millipore, Boston, MA, USA). Subsequently, the membrane was blocked and probed overnight at 4°C with primary antibodies including anti-DEC2 (1:500; Sigma), anti-bcl-2 (1:500; Epitomics), anti-Bax (1:500; Epitomics), anti-survivin (1:1000; Epitomics), anti-ERK (1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-p-ERK (1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-NF-κB/p65 (1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-N-cadherin (1:10000; Epitomics), anti-E-cadherin (1:1000; Epitomics), anti-vimentin (1:2500; Epitomics) and anti-GAPDH (1:2500; Proteintech Group, Inc., Wuhan, China). After incubation with HRP-conjugated secondary antibodies (1:10000, Proteintech Group, Inc., Wuhan, China), the blots were exposed to FluorChemE system (Cell Biosciences, Santa Clara, USA).
Immunofluorescence analysis
The transfected cells were seeded in 24-well plates and cultured overnight. Then the cells were washed with PBS twice and fixed with 4% paraformaldehyde for 20 min. After permeabilization with 0.5% Triton X-100, cells were blocked with normal goat serum for 1 h, subsequently incubated with primary antibodies overnight, anti-E-cadherin (1:500; Epitomics), anti-vimentin (1:1000; Epitomics). After 1 h of incubation at 37°C with Alexa Fluor® 488 conjugated goat anti-rabbit or Alexa Fluor® 594 conjugated anti-mouse IgG antibodies (1:200 dilution, life technologies, Invitrogen, USA), the cells were counterstained with DAPI and imaged using a fluorescence microscope (Olympus).
Proliferation and metastasis assays in vivo
All animal care and experimental procedures in present study were approved by the Animal Ethics Committee of the Medical School of Shandong University, in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, Jinan, Shandong Province, China. In addition, the ARRIVE guidelines were followed [18].
Four-to six-week-old female BALB/c nu/nu mice were purchased from Beijing HFK Bioscience (Beijing, China). Animals were housed in specific-pathogen free environment with controlled temperature and humidity conditions. To induce ectopic tumor, 2×106 cells suspending in 100 μl medium were subcutaneously injected in right flank of the mice (n = 6). Tumor dimensions were measured with vernier caliper. The volume of tumor nodules was calculated by the formula twice weekly: length × width2 ×0.5. After four weeks, tumor nodules were surgically excised and weighed and subjected to further analysis. To ascertain the role of DEC2 in tumor metastasis, 1×106 cells suspending in 100 μl medium were intravenously injected into the lateral tail vein of nude mice. The mice were sacrificed at 36 days after injection and lungs were harvested for counting the number of metastasis nodules and being fixed in 10% neutral-buffered formalin for further examination. The mice were killed in a CO2 cage, and the tumors were extracted by standard surgery.
Immunohistochemistry
IHC staining was performed using a standard immunoperoxidase staining procedure. Briefly, paraffin sections were cut to a thickness of 4 μm and mounted on silanized slides. Next, slides were dewaxed in xylene and hydrated in graded alcohol solutions. After antigen retrieval with heat treatment in 10 mM sodium citrate buffer (PH 8.0), the slides were incubated with the primary antibodies against anti-DEC2 (1:200, Sigma), anti-E-cadherin (1:500, Epitomics). Following a final wash, the slides were incubated with secondary antibody (KIT-5010, Max Vision, Maixin, Bio, China), and visualized by incubation with 3, 3-diaminobenzidine solution. Slides incubated with normal mouse or rabbit IgG instead of primary antibodies were used as negative control. The nucleus was counterstained with hematoxylin.
Immunohistochemical analysis was performed by two independent investigators concurrently. The assessment was described by percentage of stained tumor cells and staining intensity by counting 5 random fields at a magnification of 400×. The percentage of staining tumor cells was scored as follows: (0 = none; 1 = less than 25%; 2 = 25-75%; 3 = greater than 75%). The staining intensity of positive tumor cell was scored as followed: (0 = none; 1 = weak; 2 = intermediate; 3 = strong). The overall amount of protein present in each tumor was then expressed as the product or total score of the proportion and intensity scores for negative and positive tumor cells (ranges = 0-9, respectively). The cut point for positive was a score ≥4, and that for a negative score was <4.
Statistical analysis
SPSS version 19.0 (SPSS, Chicago, IL, USA) was used for statistical analysis. Data were expressed as mean ± SD, or mean ± SEM. DEC2 levels in tumors and paired non-tumor tissues were compared using the paired Student’s t-test. The associations between the expression of DEC2 and clinicopathologic features were analyzed using Pearson’s chi-squared test. Kaplan-Meier plots and log-rank tests were used for survival analysis. P<0.05 was considered statistically significant.
Results
Expression of DEC2 is significantly downregulated in GC and clinical relative to the expression of E-cadherin
A total of 49 primary human GC specimens were collected to detect DEC2 expression by immunohistochemical analysis and analyzed the associations with clinicopathological characteristics. As shown in Figure 1A and Table 1, in adjacent normal tissues, positive DEC2 expression was displayed in the cell nucleus and cytoplasm, and DEC2-high expression tissues accounted for 67.35% (33/49) of all tissues. Among the tested tumor tissues, DEC2-high expression tissues accounted for 22.45% (11/49), and the positive expression was only displayed in the cell cytoplasm. Significantly, overexpression of DEC2 was observed in GC tissues with early stage (Figure 1B and 1C).
Figure 1.
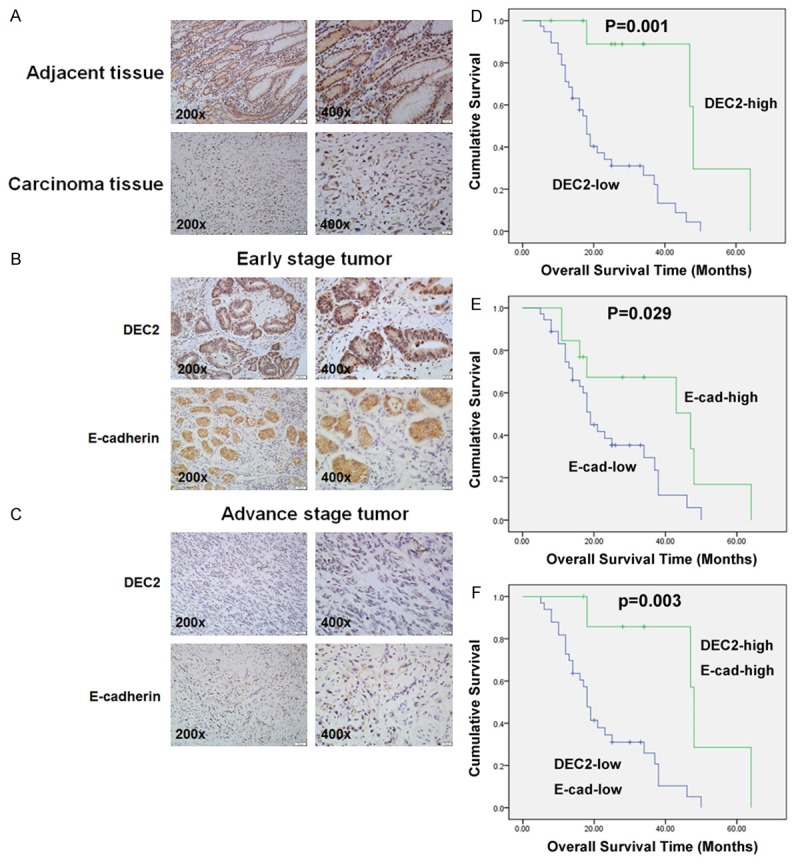
Immunohistochemical expression of DEC2 and the EMT-related marker, E-cadherin. A. Immunohistochemical staining of DEC2 and E-cadherin in primary lesion and adjacent normal gastric mucosa. B, C. Expression of DEC2 and E-cadherin in tumor specimens. D. Kaplan-Meier analysis of the correlation between DEC2 expression and overall survival in GC patients (Long-rank test). E. Kaplan-Meier analysis of the correlation between E-cadherin expression and overall survival in GC patients (Long-rank test). F. Kaplan-Meier analysis of the correlation between DEC2-high/E-cadherin-high group and DEC2-low/E-cadherin-low group and overall survival in GC patients (Long-rank test).
The correlation between the DEC2 expression levels and clinicopathological characteristics was summarized in Table 1. The data demonstrated that DEC2 expression was significantly correlated with tumor invasion (P = 0.001), lymph node metastasis (P = 0.033), and TNM stage (P = 0.001). As shown in Table 1, we found that the epithelial marker, E-cadherin, expression levels are lower (13/49) in the 49 tumor tissues, and E-cadherin expression significantly correlated with gender (P = 0.011), tumor invasion (P = 0.000), lymph node metastasis (P =0.038), and TNM stage (P = 0.021). Furthermore, the decreased E-cadherin expression correlated with lower DEC2 expression (r = 0.563, P<0.001) (Figure 1B, 1C and Table 2).
Table 2.
Correlation between the expression of DEC2 and E-cadherin
| DEC2 expression | P value | r value | ||
|---|---|---|---|---|
|
| ||||
| High (11) | Low (38) | |||
| E-cadherin expression | ||||
| High | 8 | 5 | <0.001* | 0.563 |
| Low | 3 | 33 | ||
Statistically significant difference; AJCC, American Joint Committee on Cancer.
Importantly, we examined the prognosis significance of DEC2 and E-cadherin in GC patients. Kaplan-Meier analysis showed that the overall survival of DEC2 or E-cadherin high-expression group was higher than the corresponding low-expressing group, respectively (Figure 1D and 1E, DEC2, P = 0.001; E-cadherin, P = 0.029). In addition, the overall survival of patients with DEC2+/E-cadherin + expression was much higher than that of group with DEC2-/E-cadherin - expression (Figure 1F, P = 0.003).
Overexpression of DEC2 inhibits GC cell proliferation and promotes cell apoptosis in vitro
To investigate the biological functions of DEC2 in GC development, we examined DEC2 expression in normal gastric cell line (GES-1) and GC cell lines (HGC27, MGC803, BGC823, MKN-45) using western blot and real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR). Our results show that DEC2 expression levels were much lower in GC cells than in normal gastric cells (Figure 2A-C). MGC803 and MKN-45 cells with endogenous low DEC2 expression were selected to be transfected with DEC2 vector, leading to significantly upregulation of DEC2 (Figure 2D-F). CCK8 and EdU incorporation assays were performed to evaluate whether DEC2 may affect the cell proliferation in GC cells. As shown by CCK8 assay, DEC2 overexpression significantly inhibited GC cells viability (Figure 2G and 2H). As shown by 5-ethynyl-2’-deoxyuridine (EdU) incorporation assay, MGC803 and MKN-45 cells transfected with DEC2 vector proliferated at a lower rate than those with corresponding vector transfection (Figure 2I).
Figure 2.

DEC2 significantly suppresses cell proliferation and promotes apoptosis in GC cell lines. A-C: DEC2 expression as shown by western blot and qRT-PCR in normal gastric cell line and GC cell lines. D-F: DEC2 expression in MGC803 and MKN-45 cells transfected with DEC2 vector and empty vector (control) assessed by western blot and qRT-PCR. G, H: Cell viability of MGC803 and MKN-45 cells were measured by CCK8 assay. I: Comparison of proliferation ratio of MGC803 and MKN-45 cells transfected with DEC2 vector and empty vector by EdU incorporation assay. J. The expressions of apoptotic-related proteins, bcl-2, Bax, and survivin, were detected in GC cells using western blot. K. GC cells transfected with DEC2 were stained with Annexin V-FITC/PI and analyzed by flow cytometry. The percentage of apoptotic cells is shown in Supplementary Figure 1C. The error bars indicate ± SEM. *P<0.05; **P<0.01 by Student’s t-test. All the results were repeated thrice.
The effect of DEC2 on apoptosis was investigated by flow cytometry. Compared with control cells, the apoptotic rate was significantly increased in DEC2-transfected cells (Figure 2K and Supplementary Figure 1C). To confirm this, we further analyzed the expression of apoptotic-related proteins using western blot. As shown in Figure 2J, compared with control cells, the expression of anti-apoptotic protein bcl-2 decreased while the apoptotic protein Bax increased in DEC2-trasfected cells. In addition, expression of survivin, a member of the inhibitor of apoptosis gene family, decreased after transfection with DEC2 vector. Quantitative results of western blot are shown in Supplementary Figure 1A and 1B.
Next, DEC2 shRNA vector decreased DEC2 expression in HGC27 cells with endogenous high DEC2 expression (Supplementary Figure 1D and 1E). Silence of DEC2 increased cell proliferation rate, significantly (Supplementary Figure 1F). In addition, the expression of anti-apoptotic protein bcl-2 and survivin increased while the apoptotic protein Bax decreased in HGC27 cells transfected with DEC2 shRNA vector (Supplementary Figure 1G and 1H). These data indicate that exogenous DEC2 could inhibit proliferation in GC cells and induce GC cell apoptosis.
Overexpression of DEC2 inhibits GC cell migration and invasion
Wound healing and Transwell assays were performed to investigate DEC2’s effect on cancer cell migration and invasion in vitro. The wound healing assay showed that increased DEC2 expression in MGC803 and MKN-45 cells was associated with significantly slower wound closure (Figure 3A). Transwell assays ± Matrigel demonstrated that MGC803 and MKN-45 cells transfected with DEC2 vector had lower invasive activity than control cells (Figure 3B), while silence of DEC2 increased HGC27 cells migration and invasion (Figure 6E). These results indicate that DEC2 inhibits GC cell proliferation and motility in vitro.
Figure 3.

DEC2 inhibits GC cell migration and invasion. A. Wound healing assays were used to examine MGC803 and MKN-45 cells’ motility following transfection with DEC2 and corresponding control vector, respectively. Wound closure percentages are shown in the right panel. B. Migration and invasion assays were performed using MGC803 and MKN-45 cells transfected with DEC2 and corresponding control vector, respectively. Quantitative results are shown in the bottom panel. The error bars indicate ± SEM. *P<0.05; **P<0.01 by Student’s t-test. All the results were repeated thrice.
Figure 6.

DEC2 inhibits EMT through inactivation of ERK/NF-κB/E-cadherin pathway. A, D. Western blot results of NF-κB p65, p-ERK, and E-cadherin expression levels in DEC2/MGC803, DEC2/MKN-45, DEC2-sh/HGC27 and their corresponding control cells. Quantitative results are shown in Supplementary Figure 1. B, E. Western blot results of NF-κB p65, p-ERK, and E-cadherin expression levels in DEC2/MGC803, DEC2/MKN-45, DEC2-sh/HGC27 and their corresponding control cells after treated with ERK inhibitor U0126 (10 μM). C. Comparisons of the migration and invasion ability in stable transfected DEC2/MGC803 and DEC2/MKN-45 cells after treated with ERK inhibitor U0126 (10 μM) or not. Quantitative results are shown in the right panel. F. Comparisons of the migration and invasion ability in DEC2-sh/HGC27 and their corresponding control cells after treated with ERK inhibitor U0126 (10 μM) or not. Quantitative results are shown in the bottom panel. The error bars indicate ± SEM. *P<0.05; **P<0.01 by Student’s t-test. All the results were repeated thrice.
DEC2 suppresses GC tumor growth and metastasis in vivo
To address whether DEC2 suppressed tumor growth in vivo, stable transfected DEC2/MKN-45 and DEC2-sh/HGC27 and the corresponding control cells were injected into the right flank subcutaneous tissues of nude mice in order to induce formation of ectopic tumors. In contrast to their corresponding control cells, the tumor volumes were smaller in mice with injection of DEC2/MKN-45 cells and massive in mice with injection of DEC2-sh/HGC27 cells at the designated time points (Figure 4A, 4B, 4I and 4J). At day 36, when compared with their corresponding control groups, the tumor weights in DEC2/MKN-45 group were significantly decreased and that in DEC2-sh/HGC27 group were increased (Figure 4C and 4K). Additionally, expressions of DEC2, E-cadherin, and Bax were higher, while bcl-2 expression was lower in DEC2/MKN-45 groups compared with their corresponding control groups (Figure 4G and 4H). Unfortunately, expressions of DEC2, E-cadherin, Bax and bcl-2 were not markedly altered in DEC2-sh/HGC27 group (data not shown).
Figure 4.

DEC2 suppresses GC cell proliferation and metastasis in vivo. A, B, I, J. Tumor volume. C, K. Weight are shown after subcutaneous injection with DEC2/MKN-45, DEC2-sh/HGC27 and their corresponding control cells, respectively. D, L. Lung metastasis in mice after intravenous injection with DEC2/MKN-45, DEC2-sh/HGC27 and their corresponding control cells by H&E staining. E, M. The number of tumor macrometastatic nodules in the lungs of mice after intravenous injection with DEC2/MKN-45, DEC2-sh/HGC27 and their corresponding control cells, respectively. F. Expression of DEC2 and E-cadherin in metastasis nodules of DEC2/MKN-45-treated mice lungs were determined by immunohistochemistry. G, H. DEC2, E-cadherin, bcl-2, and Bax expression were determined in three pairs of xenograft tumors of group NC and group DEC2 overexpression by western blot. “C” means group NC, “D” means group DEC2 overexpression. n = 6. Quantitative results are shown in the right panel. The error bars indicate ± SEM. *P<0.05; **P<0.01 by Student’s t-test.
Next, to evaluate whether DEC2 inhibits distant metastasis in vivo, mice were implanted with DEC2/MKN-45 and DEC2-sh/HGC27 and corresponding control cells via the tail vein, respectively. At day 36, hematoxylin-eosin (HE) staining showed that less metastatic foci were observed in the lungs of DEC2/MKN-45-treated mice (Figure 4D) and more metastatic foci were observed in the lungs of DEC2-sh/HGC27-treated mice (Figure 4L). Obviously, less numbers of lung metastatic nodules could be counted in DEC2/MKN-45 group and more numbers of lung metastatic nodules could be counted in DEC2-sh/HGC27 group, compared with corresponding control groups (Figure 4E and 4M). In addition, DEC2 and E-cadherin expression were increased in metastasis nodules of mice lungs in DEC2/MKN-45 groups, which was consistent with the results in vitro (Figure 4F). Unfortunately, expressions of DEC2, E-cadherin in metastasis nodules of mice lungs were not markedly altered in DEC2-sh/HGC27 group (data not shown).
Effects of DEC2 on epithelial to mesenchymal transition process
Metastasis is a complex multistep process and EMT is regarded as an important step during the tumor development. To ascertain whether DEC2 regulates metastasis through modulation of EMT in GC, we first observed morphological changes, which are indicative of EMT. As shown, DEC2 impaired the mesenchymal morphology in stable transfected DEC2/MGC803 and DEC2/MKN-45 cells, while the mesenchymal morphology was not markedly altered in DEC2-sh/HGC27 cells (Figure 5A).
Figure 5.
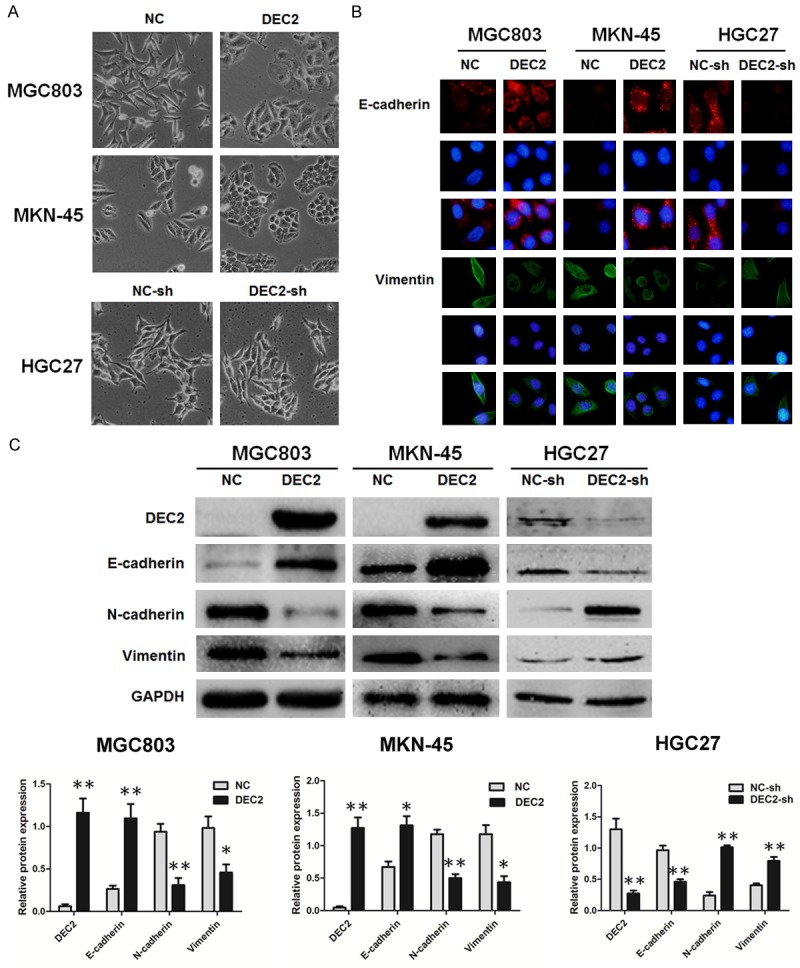
DEC2 inhibits EMT in gastric cancer cells. A. The morphological changes of MGC803, MKN-45 and HGC27 cells after up or down regulation of DEC2 under light microscope (magnification ×100). B. Immunofluorescence staining results of epithelial marker E-cadherin and mesenchymal marker vimentin in stable transfected DEC2/MGC803, DEC2/MKN-45, DEC2-sh/HGC27 cells and their corresponding control cells (magnification ×400). C. Comparison of EMT-related proteins in stable transfected DEC2/MGC803, DEC2/MKN-45, DEC2-sh/HGC27 cells and their corresponding control cells. Quantitative results are shown in the bottom panel. The error bars indicate ± SEM. *P<0.05; **P<0.01 by Student’s t-test. All the results were repeated thrice.
Next, we detected EMT-related proteins by immunofluorescence staining and western blot assay in those cells (Figure 5B, 5C). As anticipated, DEC2 overexpression significantly reduced N-cadherin and vimentin but increased E-cadherin expression in MGC803 and MKN-45 cells, whereas silence of DEC2 got the opposite results. These data suggest a functional role of DEC2 in repressing EMT, and this repression might result in the decrease of gastric cancer cell invasion and metastasis ability.
DEC2 inhibits EMT-associated metastasis via inactivation of ERK/NF-κB pathway
Recent studies reported that NF-κB plays an important role in the induction and maintenance of EMT [19]. To clarify the signaling pathway involved in EMT-associated metastasis in GC cells, the influence of DEC2 on NF-κB expression was examined. As anticipated, the expression of NF-κB p65 was decreased in stable transfected DEC2/MGC803 and DEC2/MKN-45 cells compared with their respective control cells (Figure 6A). It was reported the activation of ERK1/2 contributes to the regulation of NF-κB [20], and NF-κB was reported to trigger the progression of EMT by activating different molecules [21,22]. Next, we analyzed the expression of the upstream molecule, ERK1/2, and downstream molecule, E-cadherin. Our data indicated that ERK1/2 signaling was inhibited, and E-cadherin expression was increased in stable transfected DEC2/MGC803 and DEC2/MKN-45 cells, when compared with their control cells (Figure 6A). DEC2 knockdown markedly stimulated the phosphorylation of ERK and NF-κB p65, whereas decreased E-cadherin expression in HGC27 cells (Figure 6D). As shown in Figure 6B, U0126 treatment further inhibited the expression of NF-κB p65 and the EMT marker. In addition, after treated with U0126, the migration and invasion ability of stable transfected DEC2/MGC803 and DEC2/MKN-45 cells was further decreased (Figure 6C). In contrast, pretreatment of the cells with U0126 blocked activating effects of DEC2 knockdown on ERK/NF-κB, migration and EMT in DEC2-sh/HGC27 cells (Figure 6E and 6F). But DEC2 express-ion was not markedly altered after U0126 treatment regardless of up or down regulation of DEC2 (data not shown). These results provide direct evidence confirming the role of ERK/NF-κB/E-cadherin pathway in the inhibitory effects of DEC2 on EMT-associated metastasis.
Discussion
The bHLH proteins are closely associated with developmental events, including lineage commitment and cellular differentiation [23]. Based on sequence alignment and functional domain analyses, DEC2 and its structurally related protein, DEC1, constitute a new class of bHLH transcription factors [4,24]. In addition to cell differentiation, DEC proteins are shown to play important roles in immune response, regulation of molecular clock, and carcinogenesis [5,6,23,25-27]. Especially, it has been reported that DEC1 and DEC2 were dysregulated in various types of cancers, including breast, pancreatic, and human endometrial cancer, as well as oesophageal squamous cell carcinoma, [11,14,27-30], but had opposite roles in these cancers [9,29]. Our previous studies showed that DEC1 expression level was higher in gastric cancer tumors than in adjacent normal tissues [31,32]. Because there are differences in constructs of C-terminus between DEC1 and DEC2 [33,34], they may have differential functions in tumor progression. Furthermore, DEC2 is a negative regulator of DEC1 expression [35]. We assume that DEC2 has a role opposite to DEC1 and acts as a tumor suppressor in gastric cancer.
In this study, we firstly evaluated DEC2 expression in human GC tumor tissues and cell lines, confirming that DEC2 expression was downregulated in GC tumor tissues when compared to non-tumor tissues. As anticipated, DEC2 expression patterns were opposite to DEC1 among paired tumor-normal samples from GC samples. It was reported DEC2 expression level in human endometrial cancer was higher in cases at stage IA than in cases at more than stage IB [14]. Similarly, in our study, high DEC2 nuclear expression was more frequently detected in early stage cases than in advanced stage cases. Thereby, DEC2 may serve as a marker of tumor staging. In addition, DEC2 expression was correlated with clinicopathological factors including tumor invasion, lymph node metastasis, and TNM stage. These results indicate that DEC2 may serve as a potential diagnostic biomarker for GC.
The dysregulation of DEC2 appears to be negatively associated with cancer initiation and progression [9,12,13,17,36]. DEC2 has been shown to regulate the pro-apoptotic factor [17], inhibit cell proliferation, and repress the expression of cyclin D1 [12]. Consistent with previous results, we found that exogenous DEC2 suppressed GC cell proliferation in vitro and in vivo; DEC2 overexpression also induced apoptosis in MGC803 and MKN-45 cells. On the other side, functioning as a tumor suppressor, DEC2 inhibited tumor metastasis and invasiveness by regulating EMT-related markers and VEGF expression [9,36]. Recently, it was reported that DEC2 was regulated by p63 metastasis suppressor and inhibited triple-negative breast cancer aggressiveness through inhibition of HIF-1α and HIF-2α [13]. In the present study, we found exogenous DEC2 suppressed GC cell migration and invasion after analyzing results from transwell assay in vitro; we also found that DEC2 inhibited lung metastasis in nude mice. These results confirm the tumor-suppressive role of DEC2 in GC cells and provide evidence for the potential usefulness of DEC2 for GC therapy.
EMT is a dynamical process by which immotile epithelial cells lose the epithelial characteristic of cell-cell adhesion and acquire mesenchymal features, thereby gaining increased motility and invasiveness. EMT is thus regarded as a common molecular mechanism promoting tumor metastasis; many proteins contribute to this process [37]. SLUG, TWIST1, and E-cadherin were typical regulators of EMT. SLUG could enhance invasion and migration by deregulation of E-candherin [38,39]. TWIST1 also affected EMT progression by regulating E-cadherin expression [40,41]. A previous study reported that DEC2 inhibited TGF-β1-induced EMT progress by regulating SLUG, rather than SNAIL [9]. A recent report indicated that DEC2 competes with SP1 for binding to TWIST1 promoter [14]. Accordingly, we hypothesized that DEC2 is involved in EMT-mediated metastasis in GC via regulation of E-cadherin. We found that DEC2 expression was closely associated with EMT marker levels, including upregulation of epithelial marker E-cadherin. Both in vitro and in vivo studies showed exogenous introduction of DEC2 inhibited GC cell invasiveness. Our results suggest that DEC2 likely participates in EMT-associated GC progression.
In addition to the DEC2-related functions described above, the potential DEC2 mechanisms involved in EMT were investigated. Although recent studies have shown that DEC2 impaired the EMT process by negatively regulating promoter activities of TWIST1 and Slug [9,14], we demonstrated that DEC2 regulates the EMT process by inactivating the ERK/NF-κB signaling pathway. Increasing evidence has demonstrated that NF-κB plays a vital role in EMT induction and maintenance [19,42,43], activation of the ERK1/2 positively contributes to the regulation of NF-κB activity [20], and blockade of ERK/NF-κB has been reported to inhibit EMT process [20,44]. Our results demonstrated the presence of DEC2 inhibitory effect on the ERK/NF-κB/EMT pathway. After treatment with U0126, the inhibitory effect of DEC2 on ERK/NF-κB/EMT was further decreased. These results support the hypothesis that DEC2 inhibits GC metastasis, at least partly, via inactivation of the ERK/NF-κB/EMT pathway.
In conclusion, we are the first to demonstrated several novel findings: 1) DEC2 expression is downregulated in GC tissues, especially in poorly differentiated tissues, and this downregulation appears to correlate with disease progression, and 2) DEC2 inhibits GC cell proliferation and metastasis in vitro and in vivo, the inhibiting effects are attributed to a negative influence on EMT-associated metastasis through inactivation of ERK/NF-κB/EMT pathway in GC. Collectively, our results indicate that targeting the DEC2/ERK/NF-κB/EMT axis may serve as a novel diagnostic and therapeutic approach for managing GC patients.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 81000869, 81272588, 31671468 and 81602593), 973 Project grant (2012CB966503 and 2012CB966504), Shandong Provincial Natural Science Foundation of China (No. ZR2015HM018 and ZR2012HM061), and the key projects of science and technology of Jinan City (No. 2013020).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Danaei G, Vander Hoorn S, Lopez AD, Murray CJ, Ezzati M Comparative Risk Assessment collaborating group (Cancers) Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors. Lancet. 2005;366:1784–1793. doi: 10.1016/S0140-6736(05)67725-2. [DOI] [PubMed] [Google Scholar]
- 3.Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn. 2005;233:706–720. doi: 10.1002/dvdy.20345. [DOI] [PubMed] [Google Scholar]
- 4.Rossner MJ, Dorr J, Gass P, Schwab MH, Nave KA. SHARPs: mammalian enhancer-of-split- and hairy-related proteins coupled to neuronal stimulation. Mol Cell Neurosci. 1997;10:460–475. [PubMed] [Google Scholar]
- 5.Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, Kato Y, Honma K. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. 2002;419:841–844. doi: 10.1038/nature01123. [DOI] [PubMed] [Google Scholar]
- 6.Liu Z, Li Z, Mao K, Zou J, Wang Y, Tao Z, Lin G, Tian L, Ji Y, Wu X, Zhu X, Sun S, Chen W, Xiang C, Sun B. Dec2 promotes Th2 cell differentiation by enhancing IL-2R signaling. J Immunol. 2009;183:6320–6329. doi: 10.4049/jimmunol.0900975. [DOI] [PubMed] [Google Scholar]
- 7.Ozaki N, Noshiro M, Kawamoto T, Nakashima A, Honda K, Fukuzaki-Dohi U, Honma S, Fujimoto K, Tanimoto K, Tanne K, Kato Y. Regulation of basic helix-loop-helix transcription factors Dec1 and Dec2 by RORalpha and their roles in adipogenesis. Genes Cells. 2012;17:109–121. doi: 10.1111/j.1365-2443.2011.01574.x. [DOI] [PubMed] [Google Scholar]
- 8.Sato F, Bhawal UK, Yoshimura T, Muragaki Y. DEC1 and DEC2 Crosstalk between Circadian Rhythm and Tumor Progression. J Cancer. 2016;7:153–159. doi: 10.7150/jca.13748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sato F, Kawamura H, Wu Y, Sato H, Jin D, Bhawal UK, Kawamoto T, Fujimoto K, Noshiro M, Seino H, Morohashi S, Kato Y, Kijima H. The basic helix-loop-helix transcription factor DEC2 inhibits TGF-beta-induced tumor progression in human pancreatic cancer BxPC-3 cells. Int J Mol Med. 2012;30:495–501. doi: 10.3892/ijmm.2012.1037. [DOI] [PubMed] [Google Scholar]
- 10.Yunokawa M, Tanimoto K, Nakamura H, Nagai N, Kudo Y, Kawamoto T, Kato Y, Hiyama E, Hiyama K, Nishiyama M. Differential regulation of DEC2 among hypoxia-inducible genes in endometrial carcinomas. Oncol Rep. 2007;17:871–878. [PubMed] [Google Scholar]
- 11.Liu Y, Sato F, Kawamoto T, Fujimoto K, Morohashi S, Akasaka H, Kondo J, Wu Y, Noshiro M, Kato Y, Kijima H. Anti-apoptotic effect of the basic helix-loop-helix (bHLH) transcription factor DEC2 in human breast cancer cells. Genes Cells. 2010;15:315–325. doi: 10.1111/j.1365-2443.2010.01381.x. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Shen Q, Kim HT, Bissonnette RP, Lamph WW, Yan B, Brown PH. The rexinoid bexarotene represses cyclin D1 transcription by inducing the DEC2 transcriptional repressor. Breast Cancer Res Treat. 2011;128:667–677. doi: 10.1007/s10549-010-1083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montagner M, Enzo E, Forcato M, Zanconato F, Parenti A, Rampazzo E, Basso G, Leo G, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature. 2012;487:380–384. doi: 10.1038/nature11207. [DOI] [PubMed] [Google Scholar]
- 14.Asanoma K, Liu G, Yamane T, Miyanari Y, Takao T, Yagi H, Ohgami T, Ichinoe A, Sonoda K, Wake N, Kato K. Regulation Mechanism of TWIST1 Transcription by BHLHE40 and BHLHE41 in Cancer Cells. Mol Cell Biol. 2015;35:4096–109. doi: 10.1128/MCB.00678-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Sato H, Suzuki T, Yoshizawa T, Morohashi S, Seino H, Kawamoto T, Fujimoto K, Kato Y, Kijima H. Involvement of c-Myc in the proliferation of MCF-7 human breast cancer cells induced by bHLH transcription factor DEC2. Int J Mol Med. 2015;35:815–820. doi: 10.3892/ijmm.2014.2042. [DOI] [PubMed] [Google Scholar]
- 16.Hu T, He N, Yang Y, Yin C, Sang N, Yang Q. DEC2 expression is positively correlated with HIF-1 activation and the invasiveness of human osteosarcomas. J Exp Clin Cancer Res. 2015;34:22. doi: 10.1186/s13046-015-0135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Y, Sato F, Bhawal UK, Kawamoto T, Fujimoto K, Noshiro M, Seino H, Morohashi S, Kato Y, Kijima H. BHLH transcription factor DEC2 regulates pro-apoptotic factor Bim in human oral cancer HSC-3 cells. Biomed Res. 2012;33:75–82. doi: 10.2220/biomedres.33.75. [DOI] [PubMed] [Google Scholar]
- 18.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. J Pharmacol Pharmacother. 2010;1:94–99. doi: 10.4103/0976-500X.72351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strippoli R, Benedicto I, Perez Lozano ML, Cerezo A, Lopez-Cabrera M, del Pozo MA. Epithelial-to-mesenchymal transition of peritoneal mesothelial cells is regulated by an ERK/NF-kappaB/Snail1 pathway. Dis Model Mech. 2008;1:264–274. doi: 10.1242/dmm.001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, Wang Y, Lai YJ, LaBaff AM, Wu TJ, Lin BR, Yang MH, Hortobagyi GN, Hung MC. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72:1290–1300. doi: 10.1158/0008-5472.CAN-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma H, Gao L, Li S, Qin J, Chen L, Liu X, Xu P, Wang F, Xiao H, Zhou S, Gao Q, Liu B, Sun Y, Liang C. CCR7 enhances TGF-beta1-induced epithelial-mesenchymal transition and is associated with lymph node metastasis and poor overall survival in gastric cancer. Oncotarget. 2015;6:24348–24360. doi: 10.18632/oncotarget.4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kreider BL, Benezra R, Rovera G, Kadesch T. Inhibition of myeloid differentiation by the helix-loop-helix protein Id. Science. 1992;255:1700–1702. doi: 10.1126/science.1372755. [DOI] [PubMed] [Google Scholar]
- 24.Shen M, Yoshida E, Yan W, Kawamoto T, Suardita K, Koyano Y, Fujimoto K, Noshiro M, Kato Y. Basic helix-loop-helix protein DEC1 promotes chondrocyte differentiation at the early and terminal stages. J Biol Chem. 2002;277:50112–50120. doi: 10.1074/jbc.M206771200. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Llordella M, Esensten JH, Bailey-Bucktrout SL, Lipsky RH, Marini A, Chen J, Mughal M, Mattson MP, Taub DD, Bluestone JA. CD28-inducible transcription factor DEC1 is required for efficient autoreactive CD4+ T cell response. J Exp Med. 2013;210:1603–1619. doi: 10.1084/jem.20122387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato F, Muragaki Y, Zhang Y. DEC1 negatively regulates AMPK activity via LKB1. Biochem Biophys Res Commun. 2015;467:711–716. doi: 10.1016/j.bbrc.2015.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bi H, Li S, Qu X, Wang M, Bai X, Xu Z, Ao X, Jia Z, Jiang X, Yang Y, Wu H. DEC1 regulates breast cancer cell proliferation by stabilizing cyclin E protein and delays the progression of cell cycle S phase. Cell Death Dis. 2015;6:e1891. doi: 10.1038/cddis.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong VC, Ko JM, Qi RZ, Li PJ, Wang LD, Li JL, Chan YP, Chan KW, Stanbridge EJ, Lung ML. Abrogated expression of DEC1 during oesophageal squamous cell carcinoma progression is age- and family history-related and significantly associated with lymph node metastasis. Br J Cancer. 2011;104:841–849. doi: 10.1038/bjc.2011.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Y, Sato F, Yamada T, Bhawal UK, Kawamoto T, Fujimoto K, Noshiro M, Seino H, Morohashi S, Hakamada K, Abiko Y, Kato Y, Kijima H. The BHLH transcription factor DEC1 plays an important role in the epithelial-mesenchymal transition of pancreatic cancer. Int J Oncol. 2012;41:1337–1346. doi: 10.3892/ijo.2012.1559. [DOI] [PubMed] [Google Scholar]
- 30.Inaguma S, Riku M, Hashimoto M, Murakami H, Saga S, Ikeda H, Kasai K. GLI1 interferes with the DNA mismatch repair system in pancreatic cancer through BHLHE41-mediated suppression of MLH1. Cancer Res. 2013;73:7313–7323. doi: 10.1158/0008-5472.CAN-13-2008. [DOI] [PubMed] [Google Scholar]
- 31.Zheng Y, Shi X, Wang M, Jia Y, Li B, Zhang Y, Liu Q, Wang Y. The increased expression of DEC1 gene is related to HIF-1alpha protein in gastric cancer cell lines. Mol Biol Rep. 2012;39:4229–4236. doi: 10.1007/s11033-011-1209-0. [DOI] [PubMed] [Google Scholar]
- 32.Jia YF, Xiao DJ, Ma XL, Song YY, Hu R, Kong Y, Zheng Y, Han SY, Hong RL, Wang YS. Differentiated embryonic chondrocyte-expressed gene 1 is associated with hypoxia-inducible factor 1alpha and Ki67 in human gastric cancer. Diagn Pathol. 2013;8:37. doi: 10.1186/1746-1596-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen M, Kawamoto T, Yan W, Nakamasu K, Tamagami M, Koyano Y, Noshiro M, Kato Y. Molecular characterization of the novel basic helix-loop-helix protein DEC1 expressed in differentiated human embryo chondrocytes. Biochem Biophys Res Commun. 1997;236:294–298. doi: 10.1006/bbrc.1997.6960. [DOI] [PubMed] [Google Scholar]
- 34.Fujimoto K, Shen M, Noshiro M, Matsubara K, Shingu S, Honda K, Yoshida E, Suardita K, Matsuda Y, Kato Y. Molecular cloning and characterization of DEC2, a new member of basic helix-loop-helix proteins. Biochem Biophys Res Commun. 2001;280:164–171. doi: 10.1006/bbrc.2000.4133. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Xie M, Song X, Gragen S, Sachdeva K, Wan Y, Yan B. DEC1 negatively regulates the expression of DEC2 through binding to the E-box in the proximal promoter. J Biol Chem. 2003;278:16899–16907. doi: 10.1074/jbc.M300596200. [DOI] [PubMed] [Google Scholar]
- 36.Sato F, Bhawal UK, Kawamoto T, Fujimoto K, Imaizumi T, Imanaka T, Kondo J, Koyanagi S, Noshiro M, Yoshida H, Kusumi T, Kato Y, Kijima H. Basic-helix-loop-helix (bHLH) transcription factor DEC2 negatively regulates vascular endothelial growth factor expression. Genes Cells. 2008;13:131–144. doi: 10.1111/j.1365-2443.2007.01153.x. [DOI] [PubMed] [Google Scholar]
- 37.Samant RS, Shevde LA. NMI and EMT. Oncoscience. 2014;1:476–477. doi: 10.18632/oncoscience.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao J, Klausen C, Qiu X, Cheng JC, Chang HM, Leung PC. Betacellulin induces Slug-mediated down-regulation of E-cadherin and cell migration in ovarian cancer cells. Oncotarget. 2016 doi: 10.18632/oncotarget.7591. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang P, Yu Z, Zhang K, Wang Y, Ma Z, Zhang S, Chen D, Zhou Y. Slug down-regulation by RNA interference inhibits invasion growth in human esophageal squamous cell carcinoma. BMC Gastroenterol. 2011;11:60. doi: 10.1186/1471-230X-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Hou J, Wang T, Xie Q, Deng W, Yang JY, Zhang SQ, Cai JC. N-Myc-interacting protein (NMI) negatively regulates epithelial-mesenchymal transition by inhibiting the acetylation of NF-kappaB/p65. Cancer Lett. 2016;376:22–33. doi: 10.1016/j.canlet.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 43.Huber MA, Beug H, Wirth T. Epithelial-mesenchymal transition: NF-kappaB takes center stage. Cell Cycle. 2004;3:1477–1480. doi: 10.4161/cc.3.12.1280. [DOI] [PubMed] [Google Scholar]
- 44.Han D, Wu G, Chang C, Zhu F, Xiao Y, Li Q, Zhang T, Zhang L. Disulfiram inhibits TGF-beta-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-kappaB/Snail pathway. Oncotarget. 2015;6:40907–40919. doi: 10.18632/oncotarget.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.