

Abstract
The feature of imperfect complementary effect of miRNAs to mRNAs implies that miRNAs may simultaneously target different mRNAs to affect multiple aspects of tumorigenesis. In our previous results, we demonstrated that miR-182 was over-expressed in breast cancer cell lines and clinical tumor tissues and its up-regulation increased tumorigenicity and invasiveness by repressing a tumor suppressor RECK. In this study, we showed that overexpression miR-182 regulated actin distribution and filopodia formation to increase invasiveness of breast cancer cells. In addition, miR-182 enhanced cell cycle progression and proliferation. We further identified the E3 ubiquitin-protein ligase FBXW7 as a target gene of miR-182. We also demonstrated that miR-182-overexpressing cells were highly sensitive to hypoxia. Under hypoxic condition, HIF-1α and VEGF-A proteins were significantly upregulated in these cells. In addition, the conditioned medium of miR-182-overexpressing cells contained more VEGF-A than the control cells and induced angiogenesis more efficiently in vitro. All these effects could be counteracted by ectopic expression of FBXW7 in cells or neutralization of VEGF-A in the conditioned media by specific antibody. Finally, our data showed that miR-182 expression was inversely correlated with FBXW7 in breast tumor tissues. In conclusion, our study explores a novel mechanism by which miR-182 elevates HIF-1α expression to promote breast cancer progression.
Keywords: MiR-182, FBXW7, HIF-1α, VEGF-A, breast cancer
Introduction
MiRNAs are endogenous 21-24 nucleotide single-stranded noncoding RNAs, that bind to the 3’-untranslated region (3’-UTR) of multiple target mRNAs via imperfect complementary match to decrease protein expression by either translation repression or mRNA cleavage [1-3]. Recent studies demonstrated that miRNAs participate in diverse biological processes, including cellular proliferation, apoptosis, tissues homeostasis, organ development and human diseases. In cancers, miRNAs may function as tumor suppressors by inhibiting the expression of cellular oncogenes or tumor promoters by targeting tumor suppressor genes [4-7].
MiR-182 is a member of the miR-96-182-183 cluster. This miRNA cluster is located on the chromosome 7q32.2 and is highly expressed in the retina during embryo development [8]. The functional importance of miR-182 in the regulation of retina formation was further confirmed in animal study [8,9]. Recent investigation demonstrated that the miR-96-182-183 cluster also plays an important role in ear development [10]. The potential oncogenic activity of miR-182 was firstly shown in human melanoma [11]. MiR-182 was found to be up-regulated in melanoma and repressed tumor suppressors Forkhead box O3 (FOXO3) and microphthalmia-associated transcription factor (MITF) to protect cancer cell from apoptosis and to promote cell invasion. Subsequent studies confirmed that miR-182 was overexpressed in many cancers and revealed a number of downstream targets affected by miR-182 to promote tumorigenesis [12-19]. Overexpression of miR-182 has been reported in breast cancer and this miRNA affects cell survival, migration and the DNA damage response by inhibiting FOXO1, MIM (Missing in metastasis), MTSS1 (Metastasis suppressor 1), and BRCA1 (Breast cancer 1) [20-23]. Our previous results also demonstrated that miR-182 was overexpressed in breast cell lines and tumor tissues and its expression was associated with various clinicopathological features of breast cancer patients. More importantly, we identified the endogenous matrix metalloproteinase (MMP) inhibitor RECK as a novel target of miR-182 to enhance the aggressive phenotypes of cancer cells [24].
F-box and WD repeat domain-containing 7 (FBXW7) is a component subunit of the SKP1-cullin-F-box (SCF) ubiquitin protein ligases which mediate the ubiquitination and degradation of a number of oncoproteins including cyclin E, Notch, c-Myc etc [25-28]. The conserved C-terminus region of FBXW7 protein contains (1) eight WD40 domain for substrate binding, (2) F-box domain for the binding with Skp1 of the SCF complex and (3) D domain for FBXW7 dimerization [29,30]. The variable N-terminus contains different signal sequences that may localize three splicing isoforms (α, β and γ) to different cellular compartments to mediate their biological effects. Because most of the substrates of FBXW7 are oncoproteins, FBXW7 has been suggested to act as a tumor suppressor. Indeed, down-regulation, mutation or deletion of this gene was frequently found in different cancers. Inactivation of FBXW7 by mutation or deletion occurs only in a small portion (about 6-30%) of cancer patients [31-34] and many cancers like ovarian, lung and colon cancer rarely exhibit FBXW7 mutations [35-37]. Recent studies suggested that down-regulation of FBXW7 due to dysregulation of upstream signaling molecules and transcription factors (like p53, C/EBP-δ) or up-regulation of miRNAs seems to be a major cause of the reduction of FBXW7 protein in tumor tissues [38-42]. In this study, we investigated the effect of miR-182 on the proliferation and invasion of breast cancer and identified FBXW7 as a direct target of miR-182. In addition, we demonstrated for the first time that hypoxia-induced vascular endothelial growth factor-A (VEGF-A) production and angiogenesis was significantly enhanced in miR-182-overexpressing cells. Collectively, our results suggested that miR-182 is an oncogenic miRNA in breast cancer cells and promotes tumorigenesis by increasing proliferation, invasion and angiogenesis.
Materials and methods
Cell culture and treatment
H184B5F5/M10 human mammary epithelial cells were purchased from Bioresource Collection and Research Center (Hsinchu, Taiwan) and were cultured in Dulbecco’s modified Eagle’s medium/F-12 medium containing 10% fetal calf serum (FCS) and antibiotics. MCF-7 breast cancer cell was obtained from the same resource and were cultured in Dulbecco’s modified Eagle’s medium containing 10% FCS and antibiotics. EA. hy926 human endothelial cell line was kindly provided by Dr. Ming-Hong Tai (National Sun Yat-Sen University, Kaohsiung, Taiwan). For hypoxia study, cells were incubated in a hypoxic chamber with 1% O2 for different times before harvest for analysis.
Establishment of stable cell lines
H184B5F5/M10 normal human mammary epithelial cells were transfected with pCMV-miR-182 expression vector (Origene, Rockville, MD) for 48 h and selected in medium containing G418 (1 mg/ml) for 3 weeks. A stable cell line named as M10-miR-182 with highest miR-182 expression determined by real-time PCR (data not shown) was generated and was used for the comparison with the control cell line transfected with pCMV-MIR control vector. We also transfected miR-182 sponge construct into MCF-7 cell line, which expressed high level of miR-182, to inhibit miR-182 expression [43]. A stable cell line named as MCF-7-sponge with lowest miR-182 expression was established by continuous selection in medium containing puromycin (1 ug/ml) and was used for the comparison with the control cell line transfected with the control pGIPZ vector.
Transfection and electroporation
Transfection of cells was conducted by using the GeneIn reagent (Amsbio, Cambridge, MA) according to manufacturer’s instructions. FBXW7-FLAG expression vector was kindly provided by Dr. Welcker (Fred Hutchinson Cancer Research Center, USA). FBXW7 siRNA (Santa Cruz, Dallas, TX) was delivered by Neon Electroporation system (Thermo, Walthem, MA) according to manufacturer’s instructions.
Immunofluorescent staining
Cells were seeded on cover slides. After 24 h, cells were fixed in 4% paraformaldehyde in phosphoate-buffered saline (PBS) for 10 min, permeabilized with 1% Triton X-100 for 10 min and blocked with 5% bovine serum albumin in PBS for 30 min. Cells were washed twice in PBS, incubated with the Alexa-Fluor 594 Phalloidin (Invirtogen, Carlsbad, CA) for 30 min according to the manufacturer’s protocol and imaged with a Leica DMI 4000 fluorescent microscope (Wetzlar, Germany).
Transwell invasion assay
In vitro invasion assays were performed by using 24-well transwell units. The upper and lower units were separated by polycarbonate filters (pore size of 8 μm) and the upper unit was pre-coated with Matrigel (BD Bioscience, San Jose, CA). Cells (1×104) in 100 μl of serum-free medium were seeded onto upper wells. The lower units were filled with 10% FCS medium. After 24 h, cells on the upper part of the membrane were removed with a cotton swab. Invaded cells on the bottom surface of the membrane were fixed in formaldehyde, stained with Giemsa solution, and counted under a microscope.
Clonogenic and cell proliferation assay
5×103 cells were seeded on 6-well plates and the colonies grown on the plates after one week were stained with Giemsa solution, and quantified by using the Image ProPlus software. For proliferation assay, cells were seeded on 24-well plates and cell growth was assessed at 24, 48, 72 h after seeding. Cells were incubated with 200 ul of MTT reagent at 37°C for 4 h and dissolved by DMSO. The signal intensity was detected by using an ELISA reader with a wave length of 570 nm.
Flow cytometry
The stable cell lines were collected by cell dissociation buffer and fixed by 70% ethanol in -20°C overnight. In the next day, cells were stained by 25 ug propidium iodide in the presence of RNase 100 mg/ml for 30 min at 37°C. The percentage of cells in different phases of cell cycle was determined by flow cytometer.
Bioinformatics prediction
We used different public databases including miRWalk, TargetScan and Pictar to identify the crucial tumor suppressor genes that are targeted by miR-182 and confirmed the expression of these target genes in the stable cell lines H184B5F5/M10-miR-182 and MCF7-Sponge.
Reverse-transcription polymerase chain reaction (RT-PCR) and real-time PCR
Total RNA was isolated from cells by Total RNA Mini kit (Geneaid, New Taipei City, Taiwan) and reverse-transcribed into cDNA by M-MLV reverse transcriptase (Promega, Madison, WI). Real-time PCR was quantified by GoTaq qPCR master mix (Promega, Madison, WI). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control to check the efficiency of cDNA synthesis and PCR amplification. The sequence of primers used are FBXW7 forward: 5’-CGTTGCAGGGGCATACTAAT-3’ and FBXW7 reverse: 5’-ATGCAATTCCCTGTCTCCAC-3’; VEGF-A forward: 5’-AAGGAGGAGGGCAGAATCAT-3’ and VEGF-A reverse: 5’-ATCTGCATGGTGATGTTGGA-3’; VEGF-C forward: 5’-GCCAACCTCAACTCAAGGAC-3’ and VEGF-C reverse: 5’-CCCACATCTG TAGACGGACA-3’; VEGF-D forward: 5’-CATCCCATCGGTCCACTAGG-3’ and VEGF-D reverse: 5’-GGGCTGCACTGAGTTCTTTG-3’; GAPDH forward: 5’-AAGGCTGGGGCTCATTTGC-3’ and GAPDH reverse: 5’-GCTGATGATCTTGAGGCTGTTG-3’.
Western blotting analysis
Cellular lysates were harvested in lysis buffer containing protease inhibitors (50 mM Tris-HCl buffer, pH 7.4, 150 mM NaCl, 5 mM EDTA, 50 mM NaF, 1% Triton X-100, 1 mM phenylmethylsulphonyl fluoride, 1 mg/ml aprotinin, 2 µg/ml pepstatin A, 2 µg/ml leupeptin and 1 mM sodium pervanadate) at 4°C for 10 min. Cellular lysates were subjected to centrifugation at 13000 rpm at 4°C for 5 min and supernatant protein concentrations were normalized by BCA protein assay. Cellular proteins were boiled in SDS-PAGE sample buffer, separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose paper. Membranes were blocked with nonfat dry milk in Tris-buffered saline containing 0.05% Tween 20 (TBST) and then incubated with primary antibodies against FBXW7, Cyclin E (Santa Cruz, Dallas, TX), Notch (Abcam, Cambridge, UK), FLAG (Sigma, St. Louis, MO), HIF1-α and VEGF-A (Genetex, Irvine, CA) and actin (Millipore, Temecula, CA). After washing with TBST, the blots were incubated with secondary antibodies for 1 h, developed by ECL reagent and visualized under UVP iospectrum 600. For the detection of secreted VEGF-A, conditioned medium were collected from cells cultured in normoxia or hypoxia conditions for 24 h and were centrifuged to remove cell debris. The conditioned media were concentrated by Amicon Ultra-0.5 centrifugal filter with 3KDa (Millipore, Temecula, CA) and were boiled in SDS-PAGE sample buffer. After SDS-PAGE separation, secreted proteins were transferred to nitrocellulose paper and probed with VEGF-A antibody.
3’-UTR reporter and dual-luciferase assay
Cells were transfected with 3’-UTR reporter of FBXW7 (Genecopoeia, Rockville, MD). Mutations were introduced into the two predicted binding sites (ttgccaa) of miR-182 using QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) to mutate the two targeting sequence 5’-ttgccaaccattgccaa-3’ to 5’-ttgccGaccaGGgccaa-3’ (in which three nucleotides indicated by boldface were mutated). After 48 h, the medium was removed and the cells were washed by cold PBS. Cells were detached by 0.05% trypsin-EDTA and were collected by centrifuged at 1500 rpm for 5 min. Cells were then dissolved in Passive Lysis Buffer (Promega, Madison, WI) and 20 μl of cell lysate was transferred into luminometer plate. Luciferase activity and Renilla luciferase activity (as an internal control) were measured by a Luminometer (BERTHOLD, CentroPRO LB 962 Microplate Luminometer). FBXW7 3’-UTR reporter activity was normalized by Renilla luciferase activity and the results from three independent assays in different cell lines were compared.
Tube formation assay
For tube formation assay, 70 ul of ice cold matrigel (BD) was added to a pre-chilled 96-well and allowed to gelation at 37°C for 30 min. Conditioned medium were collected from cells cultured in normoxia or hypoxia conditions for 24 h and antibody neutralization was performed by the control IgG or anti-VEGF-A antibody. Finally, EA-hy926 cells (5×103/well) was seeded into plates and the images of tube formation was taken by Leica DMI 4000 phase-contrast microscope (Leica Microsystems, Germany) with contrast objective after 6 h. Nodule number, tube area and tube length were quantified and analyzed by using the NIH ImageJ software.
Breast tumor tissues
Forty-five paired normal adjacent and breast tumor tissues were collected at the Department of Surgery, Chung-Ho Memorial Hospital, Kaohsiung Medical University with approval from the Internal Review Board and informed consent from all patients. Detailed data of patient and tumor-related variables were obtained by reviewing the patients’ medical charts. All of the patients received primary surgical treatment and the pathologic features were shown in Table 1. Tissues were quickly placed into the RNAlater solution (Ambion) and were subjected for RNA isolation by using TRIzol reagent (Invitrogen). The expression of miR-182 and FBXW7 was determined by real-time RT-PCR as mentioned above.
Table 1.
Association of miR-182 with FBXW7 and clinicopathological parameters of breast cancer
| miR-182 | ||||
|---|---|---|---|---|
|
|
||||
| ≤1.5 | >1.5 | p value | ||
| FBXW7 | Normal | 5 | 6 | 0.01* |
| Reduced | 4 | 30 | ||
| Histological grade | I | 4 | 16 | 0.64 |
| II | 3 | 8 | ||
| III | 2 | 12 | 0.42 | |
| ER | + | 2 | 28 | 0.001** |
| - | 7 | 8 | ||
| PR | + | 1 | 25 | 0.001** |
| - | 8 | 11 | ||
| HER-2 | ≥3 | 7 | 24 | 0.66 |
| <3 | 2 | 10 | ||
Statistical analysis
Appropriate statistical analyses were performed by using Graphpad Prism 5 (Version 5.04, Graphpad Software, Inc., La Jolla, CA, USA). For statistical analyses, Student’s t-test and Chi-squared test were used. A p value less than 0.05 was considered statistically significant.
Results
Overexpression of miR-182 affects the morphology and actin distribution of breast cancer cells
We had previously demonstrated that miR-182 was overexpressed in breast tumor tissues [24]. Among the breast cancer cell lines tested, MCF-7 cells expressed the highest level of miR-182 [24]. To address the functional role of miR-182 in normal breast epithelial cells and cancer cells, we ectopically expressed miR-182 in normal H184B5F5/M10 breast epithelial cells to generate H184B5F5/M10-miR-182 clone. In addition, we established MCF-7-Sponge stable clone by overexpressing miR-182 sponge to inhibit endogenous miR-182 in MCF-7 cells. The miR-182 expression level was verified by real-time PCR (data not shown). H184B5F5/M10 cells showed a typical epithelial morphology while H184B5F5/M10-miR-182 cells exhibited spindle-like phenotype with loose cell-cell contact (Figure 1A). Although the morphology of MCF-7-Sponge cells did not show a significant difference to the parental MCF-7 cells that exhibited typical mammary epithelial morphology, we found that MCF-7-Sponge cells were tightly cohesive and the cell-cell contact was very close (Figure 1B). We investigated the actin distribution by phalloidin staining. As Figure 1C and 1D, H184B5F5/M10-miR-182 cells showed enriched filopodia and less stress fiber when compared to the control H184B5F5/M10 cells. Conversely, knockdown of miR-182 in MCF-7 cells increased stress fiber formation. Cell invasion assays confirmed that overexpression of miR-182 enhanced cell invasive ability in normal H184B5F5/M10 breast epithelial cells while inhibition of miR-182 reduced invasiveness of MCF-7 cells (Figure 1E and 1F). These data indicated that miR-182 overexpression changed cytoskeleton formation and enhanced motility of breast cancer cells.
Figure 1.
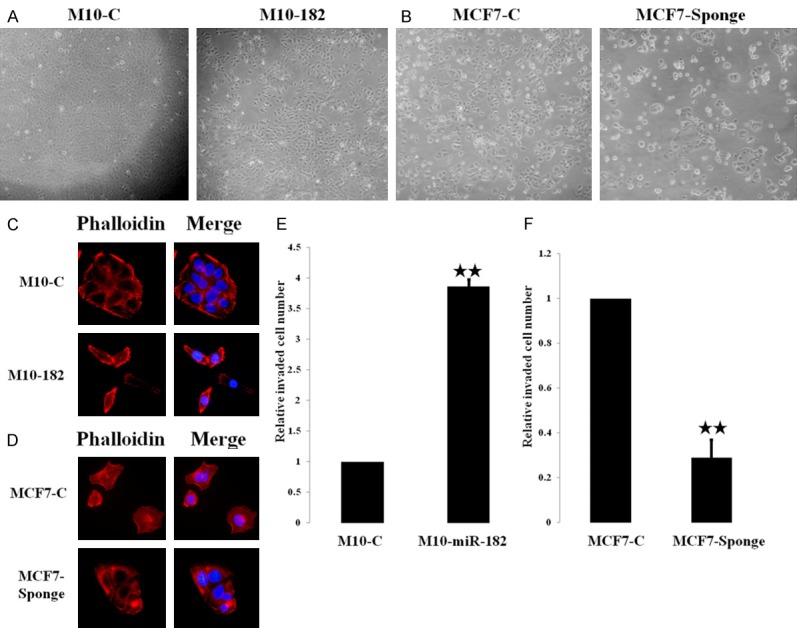
Overexpression of miR-182 affected the morphology and actin distribution of breast cancer cells. A. The morphology of H184B5F5/M10 and H184B5F5/M10-miR-182 cells. B. The morphology of MCF-7 and MCF7-Sponge cells. C. Immunofluorescent staining showed the alteration of cytoskeleton structure in H184B5F5/M10-miR-182 cells with enriched filopodia. D. The change of cytoskeleton structure in MCF-7 sponge cells compared to MCF-7 cells. E. H184B5F5/M10-miR-182 cells exhibited higher invasive ability than the parental H184B5F5/M10 cells. F. MCF7-Sponge cells showed reduced invasiveness than MCF-7 cells. **P<0.01.
MiR-182 enhanced cell cycle progression, proliferation and clonogenicity of breast cancer cells
We next investigated the effect of miR-182 on cell growth. Flow cytometry analysis demonstrated that significant increase of cells at the S phase and G2/M phases were found in H184B5F5/M10-miR-182 cells suggesting that overexpression of miR-182 enhanced cell cycle progression (Figure 2A). In consistent with this data, proliferation of H184B5F5/M10-miR-182 was higher than that of H184B5F5/M10 cells (Figure 2B). In addition, the clonogenicity of H184B5F5/M10-miR-182 was also increased (Figure 2C). On the contrary, inhibition of miR-182 in MCF-7 cells decreased the proportion of cells at S and G2/M phase that was accompanied with reduced cell proliferation and clonogenicity (Figure 2D-F). Our results suggested that miR-182 acted as an oncogenic miRNA and simultaneously enhanced growth and invasiveness of breast cancer cells.
Figure 2.

Overexpression of miR-182 increased cell cycle progression, proliferation and clonogenicity of breast cancer cells. A. Overexpression of miR-182 significantly increased the population of cells at the S and G2/M phase in H184B5F5/M10-miR-182 cells. B. Proliferation was significantly increased in H184B5F5/M10-miR-182 cells. C. H184B5/F5/M10-miR-182 cells exhibited higher clonogenicity than the parental H184B5F5/M10 cells. D. Knockdown of miR-182 reduced the population of cells at the S and G2/M phase in MCF-7 cells. E. Kncodown of miR-182 decreased the growth of MCF-7 cells. F. MCF7-Sponge cells showed reduced clonogenicity than that of MCF-7 cells. **P<0.01 and *P<0.05.
FBXW7 was targeted by miR-182
Because miR-182 is an oncogenic miRNA in breast cancer, we used bioinformatics analysis to search the tumor suppressor genes which could be targeted by this miRNA and identified FBXW7, a component of the E3 ubiquitin-protein ligase that mediates the degradation of various oncoproteins, as a potential target (Supplementary Figure 1). We analyzed the expression of FBXW7 in different breast cancer cell lines. The expression of FBXW7 mRNA and protein was down-regulated in MDA-MB-231 and SKBR3 cells possibly caused by transcriptional inhibition (Figure 3A). Interestingly, we found that the FBXW7 mRNA level was similar in normal H184B5F5/M10 breast epithelial cells and MCF-7 cells. However, the FBXW7 protein level was significantly reduced in MCF-7 cells suggesting that FBXW7 might be regulated via post-transcriptional regulation by miRNA (Figure 3A). Overexpression of miR-182 in H184B5F5/M10 cells reduced FBXW7 protein level but not mRNA (Figure 3B). Conversely, inhibition of miR-182 increased FBXW7 protein level in MCF-7 cells. We further verified miR-182 could target FBXW7 directly by using FBXW7 3’UTR reporter assay. As shown in Figure 3C, the FBXW7 3’UTR reporter activity of H184B5F5/M10-miR-182 cells was decreased by 30-40% when compared to the control cells and mutation of the two miR-182-binding sites abolished the miR-182-repressed reporter activity. We next transfected FBXW7 3’UTR reporter into MCF-7 cells which expressed high level of endogenous miR-182 and found that the reporter activity was repressed (Figure 3D). However, the activity of FBXW7 3’UTR reporter was reversed in MCF-7-Sponge cells indicating that miR-182 could direct target FBXW7 and suppression of miR-182 could relieve the inhibitory effect.
Figure 3.

FBXW7 was targeted by miR-182. A. FBXW7 mRNA and protein levels were detected in normal mammary epithelial cells and different breast cancer cell lines. B. Overexpression or knockdown of miR-182 could inhibit or increase the protein level of FBXW7 but not the mRNA level. C. The miR-182 targeting sequence in the FBXW7 3’UTR was shown and mutagenesis was performed to change the three nucleotides underlined (upper panel). Wild type or mutated FBXW7 3’UTR reporters were transfected into H184B5F5/M10 and H184B5F5/M10-miR-182 cells and the reporter activities were compared (left panel). In addition, the control reporter vector (pEZX) or FBXW7 3’UTR reporter vector was transfected into H184B5F5/M10-miR-182 cells and the reporter activities were compared (right panel). D. The control reporter vector (pEZX) or FBXW7 3’UTR reporter vector was transfected into MCF-7 or MCF-7 Sponge cells and the reporter activities were compared. ***P<0.001, **P<0.01 and *P<0.05.
Our data also demonstrated that reduction of FBXW7 by miR-182 in H184B5F5/M10 cells was accompanied with increase of FBXW7 degradation substrates cyclin E and Notch (Figure 4A). In contrast, inhibition of miR-182 in MCF-7 by miRNA sponge significantly increased FBXW7 protein level and reduced cyclin E and Notch (Figure 4A). When we depleted FBXW7 by siRNA in H184B5F5/M10 cells, we found the increase of cyclin E and Notch (Figure 4B). In addition, the increase of colony formation induced by miR-182 overexpression in H184B5F5/M10 cells was also reversed by FBXW7 (Supplementary Figure 2). Conversely, ectopic expression of FBXW7 induced the down-regulation of cyclin E and Notch in MCF-7 cells. These data suggested that miR-182 directly suppressed FBXW7 and consequently increased the oncoproteins (cyclin E and Notch) to promote cell growth and clonogenicity.
Figure 4.
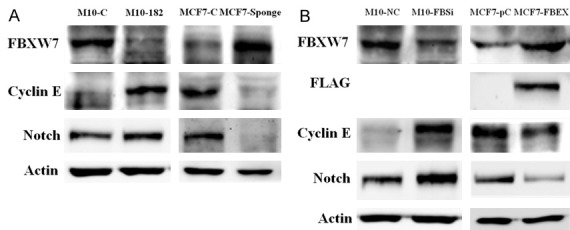
MiR-182 affected the protein level of two FBXW7 degradation substrates cyclin E and Notch. A. The protein level of FBXW7, cyclin E and Notch were investigated in different cell lines. B. Knockdown of FBXW7 by siRNA (FBSi) in H184B5F5/M10 cells or ectopic expression of FBXW7 (FBXW7 expression vector, FBEX) in MCF-7 cells modulated the protein level of cyclin E and Notch.
MiR-182 enhanced hypoxia-induced HIF-1α expression and angiogenesis
In addition to growth advantage, we tested the response of miR-182-overexpressing cells to extracellular stimuli. Under normoxia, H184B5F5/M10 cells did not express HIF-1α protein and overexpression of miR-182 also did not affect the HIF-1α protein level (Figure 5A). However, hypoxia induced a significant increase of HIF-1α in H184B5F5/M10-miR-182 cells while it only up-regulated HIF-1α marginally in H184B5F5/M10 cells (Figure 5A). This result suggested that H184B5F5/M10-miR-182 cells are very sensitive to hypoxia.
Figure 5.

MiR-182 enhanced hypoxia-induced HIF-1α expression and angiogenesis. A. H184B5F5/M10 and H184B5F5/M10-miR-182 cells were cultured in normoxia and 1% O2 for 24 h and the protein level of FBXW7 and HIF-1α was investigated by western blot analysis. B. H184B5F5/M10, H184B5F5/M10-miR-182 and H184B5F5/M10-miR-182 cells transfected with FBXW7 (182-FBXW7) were cultured in normoxia and 1% O2 for 24 h. Total RNAs were isolated and the expression of VEGF-A, -C, and -D was studied by real-time RT-PCR. C. Cells were treated as described in B and the protein level of HIF-1α, FBXW7 and secreted VEGF-A was detected by Western blot analysis. D. H184B5F5/M10 and H184B5F5/M10-miR-182 cells were cultured in 1% O2 for 24 h. The conditioned media were collected and were used to treat endothelial cells (lane 1 and 2). The conditioned media of hypoxia-treated H184B5F5/M10-miR-182 cells were also incubated with control IgG or anti-VEGF-A antibody before treating endothelial cells (lane 3 and 4). The conditioned media of H184B5F5/M10-miR-182 cells transfected with FBXW7 expression vector were collected to treat endothelial cells (lane 5). Results from three independent assays were expressed as Mean ± SE. **P<0.01 and *P<0.05.
It is well-know that HIF-1α induces VEGF expression to promote tumor angiogenesis. Because HIF-1α was significantly up-regulated in miR-182-verexpressing cells, we detected the mRNA level of several crucial angiogenesis factors including VEGF-A, -C and -D by real-time PCR in these cells. Among them, only VEGF-A was significantly up-regulated in hypoxia condition (Figure 5B). Overexpression of miR-182 promoted VEGF-A expression which could be blocked by re-expressing FBXW7 (Figure 5B). Similar to the change of mRNA level, miR-182 overexpression significantly enhanced hypoxia-induced VEGF-A production which could be suppressed by FBXW7 re-expression (Figure 5C). We further conducted the tube formation assay and measured the number of junctions in the tube network (number of nodes), the area of tube network enclosed by tube (tube area) and the linear length of the tubes (tube length) to test the angiogenic activity of the conditioned medium collected from parental and miR-182-overexpressing cells. As shown in Figure 5D, the conditioned medium of H184B5F5/M10-miR-182 cells was much more effective in the induction of tube formation than that of H184B5F5/M10 cells and this angiogenic activity was significantly inhibited by depletion of VEGF-A in the conditioned medium by neutralizing antibody (Figure 5D). Ectopic expression of FBXW7 in H184B5F5/M10-miR-182 cells also reduced VEGF-A production and the angiogenic activity of the conditioned medium.
Expression of miR-182 was inversely correlated with FBXW7
To further verify the association between miR-182 and FBXW7, we analyzed the expression of miR-182 and FBXW7 in forty-five paired breast tumor tissues by real-time PCR. Our data demonstrated that miR-182 up-regulation was correlated with FBXW7 down-regulation (P=0.01). Interestingly, a positive association between miR-182 and estrogen receptor (ER) and progesterone receptor (PR) was found, especially in ER and PR positive cases (Table 1).
Discussion
In our previous study, we demonstrated that miR-182 was over-expressed in breast cancer cell lines and tumor tissues [24]. To further elucidate the function of miR-182, we established stable cell lines with overexpression or knockdown of miR-182. We found that overexpression or knockdown of miR-182 significantly altered the morphology of breast cancer cell lines by altering cytoskeleton distribution. Furthermore, miR-182 significantly increased the proliferation, invasion and clonogenicity of breast cancer cell lines. These data suggested that miR-182 acts as an oncogenic miRNA in breast cancer cells and may affect multiple functions in cells.
We also identified an important target FBXW7 of miR-182 in breast cancer cells. FBXW7 is a crucial component of the SCF E3 ubiquitin-protein ligase that mediates the degradation of a number of oncoproteins. Although a previous study had demonstrated that miR-96 and miR-182, two members of the miR-96-182-183 cluster, could inhibit FBXW7 and INSIG-2 to up-regulate the sterol regulatory element-binding proteins to modulate intracellular lipid homeostasis [44], the contribution of the miR-182-FBXW7 axis in breast carcinogenesis is still unclear. We found that miR-182 inhibits FBXW7 which leads to increased cyclin E and Notch protein levels. This increase may be associated with cell proliferation and clonogenicity. To further confirm the correlation between miR-182 and FBXW7, we analyzed the expression of miR-182 and FBXW7 in forty-five paired breast tumor tissues by real-time PCR. Indeed, we found that miR-182 is negatively associated with FBXW7 (Table 1). Interestingly, we also found a positive correlation between miR-182 and ER and PR. It is possible that miR-182 expression may be regulated by these hormone receptor as suggested in previous studies [45,46].
Another important finding of our study is the hypersensitivity of miR-182-overexpressing cells to hypoxia. When tumor grows to a critical size, its central region becomes a hypoxic microenvironment that results from higher oxygen consumption and aberrant development of blood vessels in tumors. Hypoxia in cancer cells up-regulates the expression of HIF-1α to adapt this hypoxia stress and subsequently HIF-1α induces the production of angiogenic factors like VEGF-A to trigger angiogenesis to provide oxygen and nutrient and to sustain tumor growth. HIF-1α activity is regulated by prolyl hydroxylase enzymes (PHD). In the presence of oxygen, PHD enzymes hydroxylate HIF-1α and facilitates the binding of HIF-1α to the von Hippel-Lindau (VHL) E3 ubiquitin ligase complex that ubiquitinates and degrades HIF-1α. Under oxygen deprivation, PHD enzymes decrease HIF-α hydroxylation and results in HIF-α accumulation and nuclear translocation to activate gene transcription and angiogenesis [47,48]. We demonstrated that HIF-1α was not expressed in the H184B5F5/M10 and H184B5F5/M10-miR-182 cells under normoxia condition. However, miR-182 overexpression could significantly increase HIF-α protein under hypoxia and this induction was repressed by re-expression of FBXW7. These data suggested that HIF-1α is the degradation substrate of FBXW7 and miR-182 could potentiate the hypoxia-induced HIF-α by suppressing FBXW7. Our hypothesis is also supported by two previous studies showing that FBXW7 is involved in the degradation of HIF-α [49,50].
Among the angiogenic factors investigated, only VEGF-A was up-regulated by miR-182 in hypoxia condition. We also demonstrated that the dramatic increase of HIF-α in miR-182-overexpressing cancer cells produced a large amount of VEGF-A which triggered tube formation of endothelial cells more efficiently in vitro. Moreover, we showed that the angiogenic activity was inhibited by depletion of VEGF-A in the conditioned medium or ectopic expression of FBXW7 to antagonize the miR-182 inhibition. Recently, a study demonstrated that knockdown of miR-182 in colon cancer cells could inhibit several genes including HIF-α and VEGF-A [51]. Results of our study support the notion that miR-182 controls the HIF-α/VEGF-A axis to enhance tumor angiogenesis.
However, the crosstalk between HIF-α and miR-182 is more complex than expected. In hepatocellular carcinoma (HCC), hypoxia induces HIF-α-dependent miR-182 expression and promotes angiogenesis by targeting RASA1 suggesting HIF-α is an upstream regulator of miR-182 [52]. Another study also indicated that miR-182 was regulated by HIF-α in prostate cancer and targeted PHD2 and FIH1, two negative regulator of HIF-α. Therefore, overexpression of miR-182 increased HIF-α protein level [53]. Results of our and others studies imply a positive feedback loop may exist in breast cancer cells to amplify miR-182 and HIF-α signaling in the cells. Collectively, we conclude that miR-182 acts as an oncogenic miRNA in breast cancer and may target multiple genes to promote breast tumorigenesis.
Acknowledgements
This study was supported by the grants from National Science Council (NSC 104-2320-B-400-027) and from Department of Health and Welfare (CA-105-PP-15, CA-103-PP-09, and MOHW105-TDU-B-212-134007-Health and welfare surcharge of tobacco products) of Republic of China.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Esquela-Kerscher A, Slack FJ. Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 2.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 3.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–4662. doi: 10.1242/dev.02073. [DOI] [PubMed] [Google Scholar]
- 6.Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 7.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu S, Witmer PD, Lumayag S, Kovacs B, Valle D. MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J Biol Chem. 2007;282:25053–25066. doi: 10.1074/jbc.M700501200. [DOI] [PubMed] [Google Scholar]
- 9.Jin ZB, Hirokawa G, Gui L, Takahashi R, Osakada F, Hiura Y, Takahashi M, Yasuhara O, Iwai N. Targeted deletion of miR-182, an abundant retinal microRNA. Mol Vis. 2009;15:523–533. [PMC free article] [PubMed] [Google Scholar]
- 10.Sacheli R, Nguyen L, Borgs L, Vandenbosch R, Bodson M, Lefebvre P, Malgrange B. Expression patterns of miR-96, miR-182 and miR-183 in the development inner ear. Gene Expr Patterns. 2009;9:364–370. doi: 10.1016/j.gep.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Segura MF, Hanniford D, Menendez S, Reavie L, Zou X, Alvarez-Diaz S, Zakrzewski J, Blochin E, Rose A, Bogunovic D, Polsky D, Wei J, Lee P, Belitskaya-Levy I, Bhardwaj N, Osman I, Hernando E. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc Natl Acad Sci U S A. 2009;106:1814–1819. doi: 10.1073/pnas.0808263106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang L, Mao P, Song L, Wu J, Huang J, Lin C, Yuan J, Qu L, Cheng SY, Li J. miR-182 as a prognostic marker for glioma progression and patient survival. Am J Pathol. 2010;177:29–38. doi: 10.2353/ajpath.2010.090812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mihelich BL, Khramtsova EA, Arva N, Vaishnav A, Johnson DN, Giangreco AA, Martens-Uzunova E, Bagasra O, Kajdacsy-Balla A, Nonn L. miR-183-96-182 cluster is overexpressed in prostate tissue and regulates zinc homeostasis in prostate cells. J Biol Chem. 2011;286:44503–44511. doi: 10.1074/jbc.M111.262915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Z, Liu J, Segura MF, Shao C, Lee P, Gong Y, Hernando E, Wei JJ. MiR-182 overexpression in tumourigenesis of high-grade serous ovarian carcinoma. J Pathol. 2012;228:204–215. doi: 10.1002/path.4000. [DOI] [PubMed] [Google Scholar]
- 15.Weeraratne SD, Amani V, Teider N, Pierre-Francois J, Winter D, Kye MJ, Sengupta S, Archer T, Remke M, Bai AH, Warren P, Pfister SM, Steen JA, Pomeroy SL, Cho YJ. Pleiotropic effects of miR-183~96~182 converge to regulate cell survival, proliferation and migration in medulloblastoma. Acta Neuropathol. 2012;123:539–552. doi: 10.1007/s00401-012-0969-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Li J, Shen J, Wang C, Yang L, Zhang X. MicroRNA-182 downregulates metastasis suppressor 1 and contributes to metastasis of hepatocellular carcinoma. BMC Cancer. 2012;12:227. doi: 10.1186/1471-2407-12-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H, Du L, Wen Z, Yang Y, Li J, Wang L, Zhang X, Liu Y, Dong Z, Li W, Zheng G, Wang C. Up-regulation of miR-182 expression in colorectal cancer tissues and its prognostic value. Int J Colorectal Dis. 2013;28:697–703. doi: 10.1007/s00384-013-1674-0. [DOI] [PubMed] [Google Scholar]
- 18.Yang MH, Yu J, Jiang DM, Li WL, Wang S, Ding YQ. microRNA-182 targets special AT-rich sequence-binding protein 2 to promote colorectal cancer proliferation and metastasis. J Transl Med. 2014;12:109. doi: 10.1186/1479-5876-12-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Wang X, Wang Z, Tang H, Fan H, Guo Q. miR-182 promotes cell growth and invasion by targeting forkhead box F2 transcription factor in colorectal cancer. Oncol Rep. 2015;33:2592–8. doi: 10.3892/or.2015.3833. [DOI] [PubMed] [Google Scholar]
- 20.Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem. 2009;284:23204–23216. doi: 10.1074/jbc.M109.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K, Weinstock DM, Gorospe M, Harris AL, Helleday T, Chowdhury D. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell. 2011;41:210–220. doi: 10.1016/j.molcel.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishnan K, Steptoe AL, Martin HC, Wani S, Nones K, Waddell N, Mariasegaram M, Simpson PT, Lakhani SR, Gabrielli B, Vlassov A, Cloonan N, Grimmond SM. MicroRNA-182-5p targets a network of genes involved in DNA repair. RNA. 2013;19:230–242. doi: 10.1261/rna.034926.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lei R, Tang J, Zhuang X, Deng R, Li G, Yu J, Liang Y, Xiao J, Wang HY, Yang Q, Hu G. Suppression of MIM by microRNA-182 activates RhoA and promotes breast cancer metastasis. Oncogene. 2014;33:1287–1296. doi: 10.1038/onc.2013.65. [DOI] [PubMed] [Google Scholar]
- 24.Chiang CH, Hou MF, Hung WC. Up-regulation of miR-182 by beta-catenin in breast cancer increases tumorigenicity and invasiveness by targeting the matrix metalloproteinase inhibitor RECK. Biochim Biophys Acta. 2013;1830:3067–3076. doi: 10.1016/j.bbagen.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 25.Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 26.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoeck JD, Jandke A, Blake SM, Nye E, Spencer-Dene B, Brandner S, Behrens A. Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c-Jun. Nat Neurosci. 2010;13:1365–1372. doi: 10.1038/nn.2644. [DOI] [PubMed] [Google Scholar]
- 28.Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499–1502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 30.Tan Y, Sangfelt O, Spruck C. The Fbxw7/hCdc4 tumor suppressor in human cancer. Cancer Lett. 2008;271:1–12. doi: 10.1016/j.canlet.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 31.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, Mueller-Holzner E, Corcoran M, Dagnell M, Nejad SZ, Nayer BN, Zali MR, Hansson J, Egyhazi S, Petersson F, Sangfelt P, Nordgren H, Grander D, Reed SI, Widschwendter M, Sangfelt O, Spruck C. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 32.Kemp Z, Rowan A, Chambers W, Wortham N, Halford S, Sieber O, Mortensen N, von Herbay A, Gunther T, Ilyas M, Tomlinson I. CDC4 mutations occur in a subset of colorectal cancers but are not predicted to cause loss of function and are not associated with chromosomal instability. Cancer Res. 2005;65:11361–11366. doi: 10.1158/0008-5472.CAN-05-2565. [DOI] [PubMed] [Google Scholar]
- 33.Lee JW, Soung YH, Kim HJ, Park WS, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. Mutational analysis of the hCDC4 gene in gastric carcinomas. Eur J Cancer. 2006;42:2369–2373. doi: 10.1016/j.ejca.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 34.Byrd KN, Huey B, Roydasgupta R, Fridlyand J, Snijders AM, Albertson DG. FBXW7 and DNA copy number instability. Breast Cancer Res Treat. 2008;109:47–54. doi: 10.1007/s10549-007-9623-7. [DOI] [PubMed] [Google Scholar]
- 35.Sgambato A, Cittadini A, Masciullo V, Di Salvatore M, Graziani C, Rettino A, Valdivieso P, Scambia G, Bianchino G, Zupa A, Improta G, Cifarelli RA. Low frequency of hCDC4 mutations in human primary ovarian cancer. Gynecol Oncol. 2007;105:553–555. doi: 10.1016/j.ygyno.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 36.Woo Lee J, Hwa Soung Y, Young Kim S, Woo Nam S, Sang Park W, Young Lee J, Jin Yoo N, Hyung Lee S. Somatic mutation of hCDC4 gene is rare in lung adenocarcinomas. Acta Oncol. 2006;45:487–488. doi: 10.1080/02841860500400979. [DOI] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–779. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- 39.Balamurugan K, Wang JM, Tsai HH, Sharan S, Anver M, Leighty R, Sterneck E. The tumor suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. EMBO J. 2010;29:4106–4117. doi: 10.1038/emboj.2010.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Y, Sengupta T, Kukreja L, Minella AC. MicroRNA-223 regulates cyclin E activity by modulating expression of F-box and WD-40 domain protein 7. J Biol Chem. 2010;285:34439–34446. doi: 10.1074/jbc.M110.152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lerner M, Lundgren J, Akhoondi S, Jahn A, Ng HF, Akbari Moqadam F, Oude Vrielink JA, Agami R, Den Boer ML, Grander D, Sangfelt O. MiRNA-27a controls FBW7/hCDC4-dependent cyclin E degradation and cell cycle progression. Cell Cycle. 2011;10:2172–2183. doi: 10.4161/cc.10.13.16248. [DOI] [PubMed] [Google Scholar]
- 42.Zhou C, Shen L, Mao L, Wang B, Li Y, Yu H. miR-92a is upregulated in cervical cancer and promotes cell proliferation and invasion by targeting FBXW7. Biochem Biophys Res Commun. 2015;458:63–69. doi: 10.1016/j.bbrc.2015.01.066. [DOI] [PubMed] [Google Scholar]
- 43.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeon TI, Esquejo RM, Roqueta-Rivera M, Phelan PE, Moon YA, Govindarajan SS, Esau CC, Osborne TF. An SREBP-responsive microRNA operon contributes to a regulatory loop for intracellular lipid homeostasis. Cell Metab. 2013;18:51–61. doi: 10.1016/j.cmet.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munagala R, Aqil F, Vadhanam MV, Gupta RC. MicroRNA ‘signature’ during estrogen-mediated mammary carcinogenesis and its reversal by ellagic acid intervention. Cancer Lett. 2013;339:175–184. doi: 10.1016/j.canlet.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McDermott AM, Miller N, Wall D, Martyn LM, Ball G, Sweeney KJ, Kerin MJ. Identification and validation of oncologic miRNA biomarkers for luminal A-like breast cancer. PLoS One. 2014;9:e87032. doi: 10.1371/journal.pone.0087032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 48.Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011;2:1117–1133. doi: 10.1177/1947601911423654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cassavaugh JM, Hale SA, Wellman TL, Howe AK, Wong C, Lounsbury KM. Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J Cell Biochem. 2011;112:3882–3890. doi: 10.1002/jcb.23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flugel D, Gorlach A, Kietzmann T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood. 2012;119:1292–1301. doi: 10.1182/blood-2011-08-375014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li L, Sarver AL, Khatri R, Hajeri PB, Kamenev I, French AJ, Thibodeau SN, Steer CJ, Subramanian S. Sequential expression of miR-182 and miR-503 cooperatively targets FBXW7, contributing to the malignant transformation of colon adenoma to adenocarcinoma. J Pathol. 2014;234:488–501. doi: 10.1002/path.4407. [DOI] [PubMed] [Google Scholar]
- 52.Du C, Weng X, Hu W, Lv Z, Xiao H, Ding C, Gyabaah OA, Xie H, Zhou L, Wu J, Zheng S. Hypoxia-inducible MiR-182 promotes angiogenesis by targeting RASA1 in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34:67. doi: 10.1186/s13046-015-0182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, Zhang D, Wang X, Yao X, Ye C, Zhang S, Wang H, Chang C, Xia H, Wang YC, Fang J, Yan J, Ying H. Hypoxia-inducible miR-182 enhances HIF1alpha signaling via targeting PHD2 and FIH1 in prostate cancer. Sci Rep. 2015;5:12495. doi: 10.1038/srep12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.