

Abstract
The spiro[pyrrolidine-3, 3´-oxindole] moiety is present as a core in number of alkaloids with substantial biological activities. Here in we report design and synthesis of a library of compounds bearing spiro[pyrrolidine-3, 3´-oxindole] motifs that demonstrated exceptional inhibitory activity against the proliferation of MCF-7 breast cancer cells. The synthesis involved a one pot Pictet Spengler-Oxidative ring contraction of tryptamine to the desired scaffolds and occurred in 1:1 THF and water with catalytic trifluoroacetic acid and stoichiometric N-bromosuccinimide as an oxidant. Phenotypic profiling indicated that these molecules induce apoptotic cell death in MCF-7 cells. Target deconvolution with most potent compound 5l from the library, using chemical proteomics indicated histone deacetylase 2 (HDAC2) and prohibitin 2 as the potential cellular binding partners. Molecular docking of 5l with HDAC2 provided insights pertinent to putative binding interactions.
Globally, breast cancer is one of the most common cause of fatalities in women. Nearly 12% of the world wide women population is affected by this debilitating disease. In 2012, 25% of cancer diagnosed women suffered from breast cancer. In 2008, it inflicted fatalities to nearly 0.5 million people all over the world1. Treatments for breast cancer involved mono and combination drug therapies, surgical and radiation techniques, novel targeted therapies and cancer vaccines2,3,4. Variety of breast cancer drugs such as Tamoxifen™, Letrozole™, Docetaxyl™ and etc. are being prescribed to the patients as preventive and curative treatments5,6,7. However undue toxicity and side effects in these medicines spoil their efficacy8,9,10. Consequently discovering novel,puissant small molecules as potential anti breast cancer drugs and with better safety profile is the need of the hour. Interestingly natural products provide a healthy source for such compounds. Among variety of natural product scaffolds the spiro[pyrrolidine-3, 3´-oxindole] scaffold forms the basic architectural motif in a plethora of natural as well as non-natural compounds that demonstrates pronounced anticancer activities. It belonged to a family of natural products that were first isolated from plants Apocynaceae and Rubiacae family11. The essential elements of these class of compounds is a pyrrolidine ring spiro fused at the 3rd position of the oxindole moiety with diverse substitution on both the pyrrolidine and oxindole rings. For example Spirotryptostatin A and B,1 and 2 inhibits tubulin polymerization and induces cell cycle inhibition of cancer cells at G2/M phase and spirooxindole MI-5, 3, demonstrated novel type of inhibition of p53-MDM2 protein-protein interaction that is critical for modulating tumor suppressing ability of the p53-proteins (Fig. 1)12,13,14. These interesting therapeutic attributes of spiro[pyrrolidine-3, 3´-oxindole] scaffolds, make them attractive synthetic targets. There has been quite a few elegant chiral and achiral synthesis for this class of compound15. For example intramolecular Mannich reaction of tryptamine or a tryptophan-derived oxindole and an aldehyde, oxidative ring contraction of tetrahydro-β-carbolines, dipolar cycloaddition reactions of azomethineylides with oxindolylidene 3-ylidene acetate and intramolecular Heck reaction followed by trapping of an η3-allylpalladium species by a tethered nitrogen nucleophile16,17,18,19. There are also few one-pot synthesis to access this class of compounds, involving cycloaddition strategies20,21.
Figure 1.

Representative spiro[pyrrolidine-3, 3´-oxindole] natural and non-natural bioactive compounds, Spirotryptostatin A (1) (inhibits tubulin polymerization), Spirotryptostatin B (2) (inhibits cancer cells at G2/M phase of the cell cycle) and MI-5 (3) (inhibits p53-MDM2 protein-protein interaction).
Here we report design of a library of spiro[pyrrolidine-3, 3´-oxindole] as potential anti breast cancer molecule that are synthesized via one pot Pictet Spengler-Oxidative ring contraction strategy of tryptamines mediated by N-bromosuccinimide (NBS) (Fig. 2). The compounds exhibited substantial inhibition in MCF-7 breast cancer cells where we could identify quite a few analogs with low micromolar EC50 values. Phenotypic profiling indicated that these compounds induce cell death through apoptosis (Fig. 2). A unique polymer technology based proteomics strategy divulged histone deacetylase 2 (HDAC2) and prohibitin 2 as the potential cellular binding partners (Fig. 2). Molecular docking of the most active compound in the library with HDAC2 revealed putative binding interaction that could be harnessed to achieve more potent molecules through target based approach.
Figure 2. It depicts a comparison of our one pot strategy against step wise linear sequence of Pictet-Spengler and oxidative ring contraction reaction of tryptamine with appropriate aldehydes.

One of the resulting compounds 5, inhibits proliferation of MCF-7 cells and the putative binding targets are HDAC2 and prohibitin 2.
Results and Discussion
Design
In the recent past, we had reported diversity oriented synthesis and phenotypic screening of spiropyrrolindole based potent antibreast cancer compounds 4, which exhibited inhibition of proliferation of MCF-7 cells with an IC50 of ~24–500 nM (Fig. 3)22. Preliminary structure activity relationship (SAR) studies indicated that the aromatic or heteroaromatic functionality on the pyrrole moiety played a crucial role in imparting the inhibitory activity. Next we scanned the literature and could identify several indole derivatives such as Chaetochosins G, Vinorelbine (NVB), Vincristine and Vinblastine to name a few that have exhibited potential activity against breast cancer (Fig. 3)23,24,25,26. These findings highlighted the importance of spiropyrroloindole framework in breast cancer. Subsequently we conceived a library of spiro[pyrrolidine-3, 3´-oxindole] 5. Taking a cue from the preliminary SAR of 4, the design focused on diversifying the aromatic functionality of the pyrrole domain of 5 (Fig. 3).
Figure 3.

The new spiro[pyrrolidine-3, 3´-oxindole] scaffold based compounds, 5a–o and other analogs were designed by drawing inspiration from natural products such as Chaetocochins G, compound 4 and Vinorelbine.
Chemistry
After close scrutiny of the existing strategies we realized that the oxidative ring contraction would be most amenable for our purpose as with modification it can provide the compounds in a combinatorial fashion. The original sequence involved Pictet Spengler reaction of tryptamine with appropriate aldehydes to afford tetrahydro-β-carboline, which subsequently undergo oxidative ring contraction in presence of water, appropriate acid and an oxidant to generate the spiro[pyrrolidine-3, 3´-oxindole]27,28,29,30. We envisioned a telescoping synthesis and subsequently remolded the sequence to make it a one pot procedure, where the intermediate tetrahydro-β-carboline need not be isolated. Tryptamine and p-tolualdehyde were used as model substrates. Variety of solvents such as tetrahydrofuran (THF), acetonitrile (ACN), 1, 4-dioxane, toluene, dichloromethane (DCM), ethylene glycol were used (Table 1, entry 1–7). All the reactions were started at 0 °C and were warmed to various temperatures ranging from rt → 100 °C. In most of the reactions the tetrahydro-β-carboline intermediate 4a was isolated along with the desired compound 5a. Finally the optimized protocolthat included one equivalent of tryptamine, one equivalent of p-tolualdehyde and 1.1 equivalent of N-bromosuccinimide in 1:1 THF and water with catalytic trifluoroacetic acid at 0 °C → rt afforded the desired compound 5a exclusively in 81% yield (Table 1).
Table 1. Reaction optimization of one pot Pictet Spengler-Oxidative ring contraction of tryptamine to furnish spiro[pyrrolidine-3, 3´-oxindole].
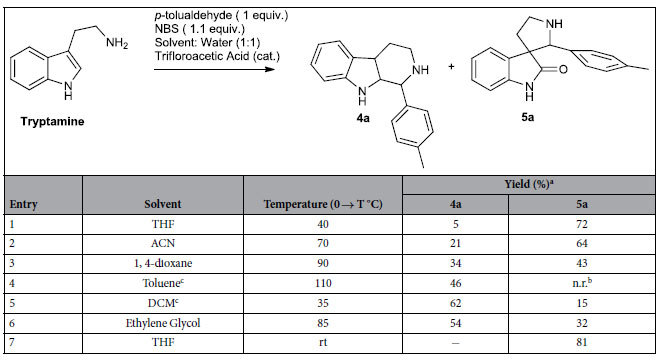
aIsolated yield.
bn. r.: no reaction.
cTetrabutyl ammonium bromide (TBAB).
With the optimized procedure in hand, next we explored the scope of the reaction with variety of aromatic aldehydes and tryptamine. The reactions were conducted in parallel in a custom made 20 mL, glass carousal and the compounds were purified using Flash Master Parallel (10 channel) Automated Flash Chromatography. The reaction condition was amenable to both electron rich and electron poor aromatic aldehydes, affording the desired compounds 5b-5m in 45–94% yield (Fig. 4). The electron poor aldehydes afforded the desired compounds 5b, d-e and 5m in relatively lower yields (45–60%) compared to the electron rich variant 5a/c and 5f-l (73–94%). Interestingly, during the reaction of 2, 5-trifluoromethyl benzaldehyde, the final product 5m was obtained along with the corresponding tetrahydro-β-carboline intermediate in ~1:1 ratio. This, indicated that during oxidative ring contraction of the corresponding tetrahydro-β-carboline intermediate, the C2-C7 bond migration depended on the nature of the substituent on the aromatic ring adjacent to the migrated bond and that electron withdrawing functionalities deter the migration (Fig. 4). Finally the versatility of the protocol was further emphasized by the synthesis of the heteroaromatic analogs 5n and o in 59 and 64% yield respectively.
Figure 4.

Synthesis of a library of spiro[pyrrolidine-3, 3´-oxindole] compounds 5b–o via one pot Pictet Spengler-oxidative ring contraction of tryptamine at room temperature with variety of aromatic aldehydes were executed.
In vitro screening and phenotypic profiling
To assess anti breast cancer potential of our library of compounds we screened them at 50 μM concentration against MCF-7 breast cancer cells using MTT colorimetric cell viability assay (Table 2). Etoposide, the standard breast cancer drug was used as a positive control31. From the compounds screened several displayed significant inhibition of the proliferation of MCF-7 cells, with EC50 values in low micromolar range (Table 2). Preliminary structure-activity relationships demonstrate that electron withdrawing groups are tolerated at meta and electron donating groups at ortho and para positions. The monosubstituted analogs are more potent than multiple substituted aromatic ring. The 2, 5-bistrifluoromethyl- and 3, 4-bismethoxy-aryl analogs retained very weak activity (25–32% inhibition) whilst the unsubstituted analog 5l exhibited best potency with an EC50 of 3.5 μM. It was interesting to observe that three of the most potent compounds 5e, i and l from our library did not inhibit the proliferation of healthy mammalian COS-7 and MCF10A cell lines.
Table 2. In vitro phenotypic activity of the library of spiro[pyrrolidine-3, 3´-oxindole] against MCF-7 and COS-7 cells.
| Entry | Compound | Percentage inhibitiona |
EC50(μM)b | ||
|---|---|---|---|---|---|
| MCF-7 | COS-7 | MCF10A | MCF-7 | ||
| 1 | 5a | 35.06 ± 0.09 | 11.73 ± 0.09 | − | − |
| 2 | 5b | 33.10 ± 0.12 | 20.73 ± 0.11 | − | − |
| 3 | 5c | 34.46 ± 0.11 | 28.80 ± 0.13 | − | − |
| 4 | 5d | 44.25 ± 0.17 | 9.22 ± 0.12 | − | − |
| 5 | 5e | 54.67 ± 0.08* | 3.56 ± 0.19* | 4.48 | 6.00* |
| 6 | 5f | 38.48 ± 0.18 | 34.87 ± 0.17 | − | − |
| 7 | 5g | 25.29 ± 0.24 | 2.39 ± 0.23 | − | − |
| 8 | 5h | 50.62 ± 0.24 | 25.20 ± 0.06 | − | − |
| 9 | 5i | 53.66 ± 0.06* | 15.60 ± 0.09* | 0.8 | 4.01* |
| 10 | 5j | 38.72 ± 0.15 | 7.52 ± 0.15 | − | − |
| 11 | 5k | 44.40 ± 0.09 | 7.14 ± 0.19 | − | − |
| 12 | 5l | 60.96 ± 0.09* | 15.42 ± 0.18* | 0.08 | 3.53* |
| 13 | 5m | 31.81 ± 0.11 | −0.51 ± 0.29 | − | − |
| 14 | Etoposide | 71.95 ± 0.11 | 77.65 ± 0.10 | − | − |
aAll data is the mean of one experiment conducted in triplicates.
bEC50 of 5e, 5i and 5l is the mean ± SD of three experiments.
*Unless otherwise indicated, the differences were considered to be statistically significant at P < 0.05. The analyses were performed using GraphPad Prism Software version 5.02 (GraphPad Inc., La Jolla, CA, USA).
Epithelial cell migration is an important characteristic of cancer cells. In a bid to evaluate if compound 5i and l, inhibit cells migration, MCF-7 cells were treated with it. The cells were photographed after 24 hours and the migration distance was measured. The data indicated that both the compounds, indeed inhibit cell migration of MCF-7 cells compared to the untreated cells. Again etoposide was used as positive control (refer supplementary information-page 75). Further, to determine whether apoptosis (or programmed cell death) was the mechanism of cell death, DNA-ladder assay was used. During apoptosis, fragmentation of nuclear proteins and DNA molecule result in chromatin condensation and formation of apoptotic bodies which is manifested as typical ladder fragmentation pattern when analyzed by gel electrophoresis32. However, at times during this experiment instead of ladder fragmentation pattern, smearing is also observed, which indicates necrosis or killing of cells by autolysis33. Experiments with 5i and 5l resulted in smearing of DNA fragments thereby indicating that both the compounds induce necrosis to MCF-7 cells. No DNA internucleosomal fragmentation was observed in cells treated with 0.2% DMSO (vehicle control) (Figure 1, supplementary information). Additionally necrosis was also confirmed by Annexin-PI FACS based assay (Figure 2–11, supplementary information).
Target deconvolution
Identifying cellular binding partners from a phenotypic screening is an onerous yet essential task. Recently proteomic tools are harnessed to achieve this. Herein we have identified cellular binding partners of 5l (the most active compound in our library against MCF-7) by employing a chemical proteomics method that involved our unique polymer technology34. During the process 5l was immobilized on a polymer (based on nitrocellulose) in aqueous buffer condition and was washed several times with 1X TBS, phosphate buffer. HPLC-UV based quantification of each buffer wash(refer supplementary information) indicated that 43% of 5l got retained in the matrix (Fig. 5).
Figure 5.

The retention characteristics of 5l on the nitrocellulose matrix is shown above. HPLC-UV based method indicated 43% of 5l got immobilized on the matrix.
Next, polymer immobilized 5l was incubated with MCF-7 cell lysate to facilitate drug-target interaction. After washing with 1X TBS, the 5l-polymer complex bound proteins were recovered by acetone precipitation, dissolved in 1X sample buffer, separated on a 4–12% gradient gel and the bands were digested by standard “in-gel” trypsin digestion method35. The target capture experiments involved mass spectrometry analysis of the gel slices in an Agilent 1260 infinity HPLC-Chip/MS system and ran in triplicates where a total of 1838 protein in control and 2184 proteins in 5l group were identified. The differential proteins (that are present in 5l but not in control group) which were common in all the three experiments were identified as the putative targets and are enlisted in the table below (Table 3).
Table 3. List of putative targets after target deconvolution.
| Entry | UniProt ID | Protein Description | Protein Class |
|---|---|---|---|
| 1 | Q92769 | Histone deacetylase 2 (HDAC2) | Nuclear Protein/Enzyme |
| 2 | Q99623 | Prohibitin 2 | Structural |
| 3 | Q9P2E9 | Ribosome-binding protein 1 | Receptor |
| 4 | Q08J23 | tRNA (cytosine (34)-C(5))-methyltransferase | Enzyme |
| 5 | Q5JTZ9 | Alanine—tRNA ligase, mitochondrial | Enzyme |
| 6 | Q13423 | NAD(P) transhydrogenase, mitochondrial | Enzyme |
| 7 | Q9NQC3 | Reticulon-4 | Growth Factor |
Of these seven proteins, Histone deacetylase 2 (HDAC2) and prohibitin2 (Table 3, entry 1 and 2) have been well demonstrated in scientific literature on their role in cancer. HDAC2 is a histone deacetylase enzyme that is over-expressed in various types of cancer including breast cancer36. Vorinostat, an HDAC inhibitor is an US-FDA approved drug for the treatment of lymphomas37. It is possible that 5l exerts its anti-cancer activity by inhibiting HDAC2. Recent studies highlight that prohibitin is involved in estrogen dependent breast cancer38. It is a mitochondrial protein and is considered essential for structural integrity of mitochondrial membrane and is also involved in pro-survival mechanisms. Compounds that bind to Prohibitin-2 has been shown to induce apoptosis39. Several other mitochondrial proteins such as NAD(P) transhydrogenase and Alanine-tRNA ligase (Table 3, entry 5 and 6) could have possibly co-eluted along with Prohibitin. However, this needs careful interpretation of not losing sight that these proteins have been observed in every experiment as a differential protein and thus could be 5l target on its own. Targets such as reticulon-4, Ribosome-binding protein 1, t-RNA (cytosine(34)-C(5))-methyltransferase (Table 3, entry 3, 4 and 7) needs further analysis as there is only a limited literature available on their role in any form of cancer or cellular toxicity. Hence HDAC2 and Prohibitin 2 are considered top priority targets for 5l (Table 3).
Molecular docking
Next, we conducted molecular docking of 5l with HDAC2 (Protein Data Bank [PDB] ID of 4LY1, this crystal structure is chosen as it has the best resolution (1.57 Å) among all HDAC2 crystal structures in PDB) to understand their putative binding interaction. Interestingly, it revealed several bonding and non-bonding interactions that could rationalize HDAC2 as the deconvoluted target for 5l in MCF-7. There is a hydrogen bond between HIS183 residue and 5l at a distance of 2.23Å. Several hydrophobic interactions were observed between 5l and amino acids PHE155, PHE210 and LEU276. Finally two pi-stacking between 5l and HDAC2 further strengthens their binding. The overall binding energy of 5l-HDAC2 complex is -6.35 kcal/mol (Fig. 6).
Figure 6.
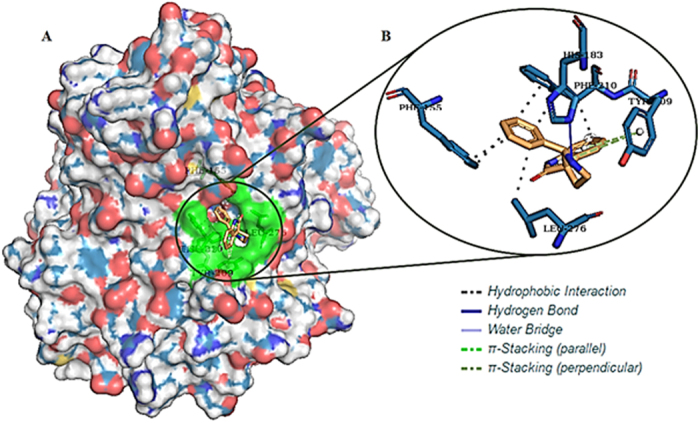
(A) 3D surface representation of HDAC2 is depicted, green color represents the active site residues with which our compound is binding. (B) Stick model represents the binding of 5l with active site residues.
Discussion
Herein we discussed design of a library of spiro[pyrrolidine-3, 3´-oxindole] inspired from natural and non-natural products which are efficacious against breast cancer. The library synthesis involved a one pot Pictet Spengler-oxidative ring contraction strategy. In general it is a two-step procedure involving Pictet Spengler reaction between tryptamine and appropriate aldehydes to afford β-carbolines followed by NBS-mediated oxidative ring contraction. After a thorough exploration of reaction conditions we were enabled to telescope these two steps into one, which provided access to these compounds in moderate to excellent yields. Interestingly no aromatic ring brominated product was isolated as the byproduct during the reaction. This further highlights the efficiency of this process. Phenotypic screening against MCF-7 revealed several active compounds with EC50 in low μmolar range (5e, i and l). Etoposide was used as the +ve control and the activity of the most potent compounds, were comparable to it. Further phenotypic profiling indicated that these compounds induce necrotic cell death to MCF-7. Identifying cellular targets from phenotypes is an essential yet extremely tedious effort. Various strategies such as affinity chromatography, activity based protein profiling, label-free techniques, expression cloning techniques and in silico approaches have been harnessed to achieve this40,41,42,43,44. Our target deconvolution strategy with the most active compound 5l included a unique polymer technology via nitrocellulose based polymer that deciphered HDAC2 and Prohibitin 2 as the potential cellular binding targets in MCF-7. Molecular modelling with HDAC2 and 5l divulged putative binding interaction. This effort exhibited an efficient early drug discovery process involving phenotypic screening.
Conclusion
In summary a novel compound 5l has been discovered via a facile one pot Pictet Spengler-oxidative ring contraction strategy that exhibited substantial inhibition against MCF-7 cells with low μmolar EC50. It belonged to a library of compounds based on spiro[pyrrolidine-3, 3´-oxindole] motif. The library design was inspired from natural and synthetic products with similar architecture that displayed efficacy against breast cancer cells. Phenotypic profiling indicated that it induces cell death by necrosis pathway. A novel proteomics based target deconvolution strategy divulged HDAC2 and prohibit 2 as putative targets. Interestingly compounds 5e, i and l selectively presented deleterious effects on MCF-7 and presented little or no effect on the corresponding primary cell line MCF10A. This is not surprising as these two cells are genetically different. Additionally it can also be hypothesized that HDAC2 which is the potential target for 5l, is over expressed in MCF-7 and not in MCF10A. Presently SAR studies involving 5l and related analogs, with HDAC2 as drug target (for breast cancer) is ongoing in our lab. The results will be reported in due course.
Methods
Chemical synthesis
All reactions were carried out in flame-dried tubes with magnetic stirring. Unless otherwise noted, all experiments were performed under argon atmosphere. All reagents were purchased from Sigma Aldrich, Acros or Alfa Aesar. Solvents were treated with 4 Å molecular sieves or sodium and distilled prior to use. 1H NMR and 13C NMR spectra were recorded with tetramethylsilane (TMS) as internal standard at ambient temperature unless otherwise indicated Bruker 500 MHz for 1 H NMR and 125 MHz for 13C NMR. Chemical shifts are reported in parts per million (ppm) and coupling constants are reported as Hertz (Hz). Splitting patterns are designated as singlet (s), broad singlet (bs), doublet (d), triplet (t). Splitting patterns that could not be interpreted or easily visualized are designated as multiple (m). The Mass Spectrometry analysis was done on the 6540 UHD Accurate-Mass Q-TOF LC/MS system (Agilent Technologies) equipped with Agilent 1290 LC system obtained by the Dept. of Chemistry, School of Natural Sciences, Shiv Nadar University, Uttar Pradesh 201314, India.
MCF10A was obtained from American Tissue Type Culture Collection (ATCC, Bethesda, MD). All cells were grown in a humidified atmosphere at 37 °C, with 5% CO2 in air.
Cell culture conditions
MCF-7 cells were maintained as monolayer in 25 cm2 culture flasks (T-25) at 37 °C in DMEM medium (GIBCO, St. Louis, USA) supplemented with high glucose, L-glutamine, pyridoxine hydrochloride, 110 mg/L sodium pyruvate, 3.7 g/L NaHCO3 and antibiotics (Penicillin-10,000 units/mL, Streptomycin −10,000 μg/mL, GIBCO, USA). Growth medium (pH 7.4) was prepared by adding 5% heat inactivated FBS (Fetal Bovine Serum, GIBCO, USA) and stored at 4 °C. The medium was changed 10–18 hrs prior to experiment, and cells were confluent at the time of experimentation. Cells were dislodged from flasks by gentle trypsinization containing 0.25% Trypsin. MCF-7 cells were regularly sub-cultured thrice a week in a seeding density of 40,000–50,000 cells/cm2area. Cell confluency = 70%
Cell Viability Assay (MTT Assay)
The MTT colorimetric viability assay was performed to evaluate the anticancer effect of compounds. 30,000 cells/100 μl of media were seeded per well and allowed to stretch and adhere overnight at 37 °C. Following day media was removed and 100 μl fresh media was added to the grown cells. Adhered cells were then treated with the compounds 5a-m dissolved in DMSO in triplicates at a standard concentration of 50 μM for 48 hours. There was one positive and one negative control along with 13 compounds. The results were measured all in triplicate in 45 wells of 96 well plate at each time point. Cytotoxic effect was evaluated using ability of live cells to cleave MTT ((3-[4, 5- dimethylthiazol-2-yl]-2, 5-diphenyl tetrazolium bromide)) (Sigma-Aldrich, St Louis, MO, USA), into formazan crystals. Post 48 hours of treatment, 10 μl of MTT (5 mg/ml in PBS) was added to the cells and incubated for 4 hours in dark at 37 °C. The violet colored crystals formed were then dissolved in 100 μl DMSO solvent (Dimethyl Sulfoxide, Sigma-Aldrich, St. Louis, MO, USA). The colorimetric assay was read by a MultiMode Plate Reader (Bio-Rad) at 595 nm. Percentage of Inhibition of drug was calculated by following formula:
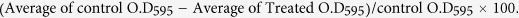 |
Cell Migration assays
MCF-7 cells were grown to confluence in six-well tissue culture plastic dishes to a density of ∼0.2 million cells/well. After 24 hrs of seeding, cells were treated with chemical compounds. The medium was removed and the cells were scratched by 200 μL pipette micro tip through the center of the plate. The cultures were washed to remove debris and re-cultured with DMEM containing 10% FBS for cell migration. The cells were imaged at 24 hrs and the migrated distance was measured. Migration rate was calculated by counting five to seven fields per image and the results presented as the average of three independent experiments performed over multiple days.
DNA fragmentation assay
The DMSO (dimethyl sulphoxide)–SDS (sodium dodecyl sulphate)–TE (Tris-EDTA) method: 2 million MCF-7cells were taken as starting material for DNA isolation. Cells were dislodged with Trypsin-EDTA and washed with 1 X PBS. DMSO was added directly to the cell pellet (100 μl) and mixed well by pipetting. TE buffer (pH 7.4) with 2% SDS was added into the mixture with same volume, followed by pipetting. The solution was centrifuged at 3000 rpm at 4 °C and 30 μL of the supernatant was loaded on agarose gel to check the effect of drugs on DNA molecule.
Annexin V/propidium iodide staining
The assay is based on the ability of the protein annexin V to bind to phosphatidylserine (PS) exposed on the outer membrane leaflet in apoptotic cells (PS also appears on the necrotic cell surface). The annexin V assay was performed following the manufacturer’s instructions (Annexin V-FITC kit,Alexa Fluor). Briefly, the cells were washed with PBS and suspended in pre-diluted binding buffer. The cell density was adjusted to 2 million and 195 μl of cell suspension were mixed with 5 μl Annexin V-FITC and incubated for 10 min in the dark at room temperature. The cells were washed with binding buffer, re-suspended in 195 μl binding buffer, counterstained with 10 μl of the 20 μg/ml propidium iodide stock (final concentration 1 μg/ml) and the data were acquired by BD FACS diva 7.0 at 6 h, 12 h and 24 h.
Protein Deconvolution
Immobilization of 5l
5l was provided as a lyophilized powder. A 100 mM stock of 5l was made in DMSO. The stock solution was diluted to 215 μM in 1 mM of phosphate buffer in pH 6.0 and incubated with the matrix for 16 hrs at room temperature. After incubation, the matrix was washed five times with Phosphate buffer. The amount of molecule in the phosphate buffer wash was quantified by HPLC-UV based method and amount of molecule retained on the matrix was calculated.
Affinity enrichment of target proteins on 5l immobilized matrix
Cell lysates were prepared in RIPA buffer and protein concentration was determined by Bradford method. Cell lysate was diluted with 1X TBS to a concentration of 1 mg/ml. The 5l immobilized matrix was incubated with 2 ml of cell lysate for 2 min with mild shaking. The matrix was washed with 1X TBS to remove unbound proteins and to reduce non-specific interactions. The molecule bound proteins were then recovered by acetone precipitation and was dissolved in 1X sample buffer. Proteins were separated on a 4–12% gradient gel and bands were digested by standard “in-gel” trypsin digestion methods.
Mass spectrometry analysis of proteins
Protein Reduction, Alkylation and Digestion
Clearly identified gel-bands were cut in typical size of 1 mm band from the gel and placed in 1.5 ml tubes. Gel slices were washed once with distil water and then twice, with 100 μL of 100 mM ABC/50% acetonitrile (ACN) solution by placing the bands in each respective solution at room-temperature for 30 minutes. After discarding the last wash gel-slices were incubated with 50 μL of 10 mM DTT at 56 °C for 10 minutes in a dry bath. Tubes were then cooled and then gel-slices were incubated with 50 μL of 100 mM IAA at 37 °C for 30 minutes. IAA solution was removed and gel slices were rinsed by incubating it with 100 mM ABC/50% ACN at 37 °C for 15 minutes. Rinse solution was removed and gel-slices were dehydrated by incubating them with 100 μl of 100% ACN for 10 minutes. ACN was removed and gel-slices were dried using a Speed Vac for 45–60 minutes at room temperature. Gel-slices were then incubated with 20–30 μl of 20 μg/ml trypsin solution for an hour at room temperature. 50 μl of 50 mM ABC was added to the gel-slices and completely covered tubes were placed at 37 °C for 12 hours for maximum trypsin digestion of the protein. Gel-slices were then incubated with 150 μl of HPLC water for 10 minutes. Solution was frequently mixed using vortex mixer. Supernatant was removed and saved after appropriate labelling. Gel-slices were then incubated with 100 μl of 50%ACN/5% TFA at 37 °C for 60 minutes to extract the peptides from gel-slices. Supernatant was collected and pooled with earlier collected supernatant. Supernatant was then concentrated at room temperature until moderate dryness. Dried peptides were dissolved in 5 μl of 0.1%TFA/HPLC Water solution and set-aside for mass-spectrometry analysis.
HPLC-CHIP-MS
Refer supplementary information.
Spectra Analysis and Protein Database Searches
Agilent Mass Hunter software was used for data acquisition and analysis of Total ion chromatograms. Protein searches were carried out using Morpheus software tools using the Human proteome database. Auto-validation protocol having <1% False Detection Rate was used in confirming the identity of protein. The peptide sequence was also analysed for variable modifications such as methionine oxidation, acetylation, cysteine sulfoxide, deamidation and pyro glutamic acid modification.
Molecular docking
Molecular docking was performed using Molecular Operating Environment (MOE) version 2014.0901, Chemical Computing Group Inc., Montreal, Canada45. The X-ray crystal structure of the Histone deacetylase 2 (HDAC2) was obtained from Protein Data bank (PDB). The PDB ID of the protein is 4LY1, this crystal structure is chosen as this is having the best resolution (1.57 Å) among all HDAC2 crystal structures. The structure was prepared using LigX module of the MOE. This preparation involved addition of hydrogen atoms and addition of any missing residues or atoms. Energy minimization is done using AMBER 10: EHT force fields. The 3D structure of the compound was constructed using the builder interface of the MOE program. Docking of the compound was done using MOE-DOCK module in MOE. The active site of the target protein is defined by the inhibitor present in the crystal structure. Triangle matcher is used as the placement methodology and London dG is used as a scoring methodology. The best pose was selected on the basis of the best score.
Additional Information
How to cite this article: Hati, S. et al. Spiro[pyrrolidine-3, 3´-oxindole] as potent anti-breast cancer compounds: Their design, synthesis, biological evaluation and cellular target identification. Sci. Rep. 6, 32213; doi: 10.1038/srep32213 (2016).
Supplementary Material
Acknowledgments
This work was supported by the Department of Biotechnology, India through pilot grant BT/Med/Pilot Project Cancer/2014.
Footnotes
Author Contributions S.H. and P.K.D. conducted the chemical synthesis and S.T. conducted the biological screening. R.A. conducted the molecular docking. S.S. and A.S. planned the biological and docking experiments respectively. R.S. conducted the protein deconvolution experiment. S. Sen. conceived and planned the project and wrote the manuscript.
References
- Irvin V. L. & Kaplan R. M. Screening Mammography & Breast Cancer Mortality: Meta-Analysis of Quasi-Experimental Studies. Plos One 9, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes M. D. et al. Intake and Survival after Breast Cancer. Journal of Clinical Oncology 28, 1467–1472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting B. & Michelle A. R. The Clinical Pharmacology of Anastrozole. European Oncology & Hematology 7, 106–108 (2011). [Google Scholar]
- Burstein H. J. et al. Adjuvant Endocrine Therapy for Women with Hormone Receptor-Positive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline Focused Update. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 32, 2255–2269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan V. C. Fourteenth Gaddum Memorial Lecture. A current view of tamoxifen for the treatment and prevention of breast cancer. British Journal of Pharmacology 110, 507–517 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingle J. N. et al. A randomized phase II trial of two dosage levels of letrozole as third-line hormonal therapy for women with metastatic breast carcinoma. Cancer 80, 218–224 (1997). [DOI] [PubMed] [Google Scholar]
- Araque Arroyo P., Ubago P. & Cancela D. Controversies in the management of adjuvant breast cancer with taxanes: Review of the current literature. Cancer Treatment Reviews 37, 105–110 (2011). [DOI] [PubMed] [Google Scholar]
- Yang G., Nowsheen S., Khaled A. & Georgakilas A. Toxicity and adverse effects of Tamoxifen and other anti-estrogen drugs. Pharmacology & Therapeutics. 139, 392–404 (2013). [DOI] [PubMed] [Google Scholar]
- Monnier A. Long term efficacy and safety of letrozole for the adjuvant treatment of early breast cancer in postmenopausal women: a review. Ther. Clin. Risk Manag. 5, 725–738 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyseng-Williamson K. A. & Fenton C. Docetaxel: a review of its use in metastatic breast cancer. Drugs 65 2513–2531 (2011). [DOI] [PubMed] [Google Scholar]
- Bindra J. S. The Alkaloids Vol. 14, (ed. Manske R. H. F.) 84–121(Academic Press, New York, 1973). [Google Scholar]
- Cui C.-B., Kakeya H. & Osada H. Novel mammalian cell cycle inhibitors, spirotryprostatins A and B, produced by Aspergillus fumigatus, which inhibit mammalian cell cycle at G2/M phase. Tetrahedron. 52, 12651–12666 (1996). [Google Scholar]
- Cui C. B. & Spirotryprostatin B. A novel mammalian cell cycle inhibitor produced by Aspergillus fumigatus”. J. Antibiot. 49, 832–835 (1996). [DOI] [PubMed] [Google Scholar]
- Wang S., Zhao Y., Bernard D., Aguilar A. & Kumar S. Targeting the MDM2 Protein-Protein Interaction for New Cancer Therapeutics. Top. Med. Chem. 8, 57–80 (2012). [Google Scholar]
- Marti C. & Carreira E. M. Construction of Spiro[pyrrolidine-3, 3´-oxindoles]-Recent Applications to the Synthesis of Oxindole Alkaloids. Eur. J. Org. Chem. 12, 2209–2219 (2003). [Google Scholar]
- Ban Y. & Oishi T. The Synthesis of 3-Spirooxindole Derivatives. III. Stereospecific Syntheses of rac-N-Methylrhynchophyllane for Stereochemistry of Rhynchophylline and Isorhynchophylline. Chem. Pharm. Bull. 11, 451–460 (1963). [DOI] [PubMed] [Google Scholar]
- Edmondson S. D. & Danishefsky S. J. Total Synthesis of Spirotryprostatin A. Angew. Chem. Int. Ed. 37, 1138–1140 (1998). [DOI] [PubMed] [Google Scholar]
- Grigg R., Sridharan V. & Thianpatanagul S. X=Y–ZH systems as potential 1,3-dipoles. Part 6. Metallo-1,3-dipoles. Cycloadditions of divalent metal complexes of glycine and alanine imines to electronegative olefins. J. Chem. Soc., Perkin Trans. 1, 1669–1675 (1986). [Google Scholar]
- Abelman M. M., Oh T. & Overman L. E. Intramolecular alkene arylations for rapid assembly of polycyclic systems containing quarternary centers. A new synthesis of spirooxindoles and other fused and bridged ring systems. J. Org. Chem. 52, 4130–4133 (1987). [Google Scholar]
- Antonchick A. P. et al. Highly enantioselective synthesis and cellular evaluation of spirooxindoles inspired by natural products. Nat. Chem. 2, 735–740 (2010). [DOI] [PubMed] [Google Scholar]
- Sebahar P. R. & Williams R. M. The Asymmetric Total Synthesis of (+)- and (−)-Spirotryprostatin B. J. Am. Chem. Soc. 122, 5666–5667 (2000). [Google Scholar]
- Surakanti. R. et al. Synthesis of Privileged Scaffolds by Using Diversity-Oriented Synthesis. Chem. Asian J. 8, 1168–1176 (2013). [DOI] [PubMed] [Google Scholar]
- Wang F. et al. Indole diketopiperazines from endophytic Chaetomiumsp 88194 induce breast cancer cell apoptotic death. Sci. Rep. 5, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty M. et al. Oral vinorelbine pharmacokinetics and absolute bioavailability study in patients with solid tumors.Ann. Oncol. 12, 143–1649 (2001). [DOI] [PubMed] [Google Scholar]
- Jordan M. A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Current medicinal chemistry. Anti-cancer agents 2, 1–17 (2002). [DOI] [PubMed] [Google Scholar]
- Goppi P. G. et al. Vinblastine, Bleomycin, and Methotrexate Chemotherapy plus Irradiation for Patients with Early-Stage, Favorable Hodgkin Lymphoma. Cancer 98, 2393–2401 (2011). [DOI] [PubMed] [Google Scholar]
- Pellegrini C., Strässler C., Weber M. & Borschberg H. J. Synthesis of the oxindole alkaloid (−)-horsfiline. Tetrahedron: Asymmetry 5, 1979–1992 (1994). [Google Scholar]
- Peterson A. C. & Cook J. M. Studies on enantiospecific synthesis of oxindole alkaloids. Tetrahedron Lett. 35, 2651–2654 (1994). [Google Scholar]
- Edmondson S. D., Danishefsky S. J., Sepp-Lorenzino L. & Rosen N. Total Synthesis of Spirotryprostatin A, Leading to the Discovery of Some Biologically Promising Analogues. J. Am. Chem. Soc. 121, 2147–2155 (1999). [Google Scholar]
- Yu. P. & Cook J. M. Diastereospecific synthesis of ketooxindoles. Potential intermediates for the synthesis of alstonisine as well as for Voachalotinerelated oxindole alkaloids. Tetlett. 38, 8799–8802 (1997). [Google Scholar]
- Pommier Y., Leo E., Zhang H. & Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17, 421–433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung M. C. Accelerated apoptotic DNA laddering protocol. BioTechniques 33(4), 734–736 (2002). [DOI] [PubMed] [Google Scholar]
- Kennedy C. L. et al. Programmed Cellular Necrosis Mediated by the Pore-Forming a-Toxin from Clostridium septicum. PlosPathog 5, 1–11 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena C. Inventor; Shantani Proteome Analytics Pvt. Ltd., assignee. Methods for Determining Protein Targets of A Ligand Without Ligand Derivatization. Provisional Patent Application Number 201621000681, Reference Number E-2/66/2-16-MUM. 2016 January 8.
- Shevchenko A., Tomas H., Havli J., Olsen J. V. & Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Prot. 1, 2856–2860 (2007). [DOI] [PubMed] [Google Scholar]
- Krusche C. A. et al. Histone deacetylase-1 and -3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Research and Treatment 90, 15–23 (2005). [DOI] [PubMed] [Google Scholar]
- Duvic. M. et al. Phase 2 trial of oral vorinostat (suberoylanilidehydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 1, 31–39 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushyar S., Jiang W. G. & Dart D. A. Unveiling the potential of prohibitin in cancer. Cancer Lett. 28, 316–322 (2015). [DOI] [PubMed] [Google Scholar]
- Moncunill-Massaguer C. et al. A novel prohibitin-binding compound induces the mitochondrial apoptotic pathway through NOXA and BIM upregulation. Oncotarget 6, 41750–41765 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S., Murata A., Shirakawa T. & Uesugi M. Biochemical target isolation for novices: affinity-based strategies. Chem. Biol. 17, 616–623 (2010). [DOI] [PubMed] [Google Scholar]
- Nomura D. K., Dix M. M. & Cravatt B. F. Activity-based protein profiling for biochemical pathway discovery in cancer. Nat. Rev. Cancer 10, 630–638 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomenick B. et al. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. USA 106, 21984–21989 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dorst B. et al. cDNA phage display as a novel tool to screen for cellular targets of chemical compounds. Toxicol. In Vitro. 24, 1435–1440 (2010). [DOI] [PubMed] [Google Scholar]
- Lounkine E. et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 486, 361–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE). A package for high throughput discovery, bioinformatics, and computer aided molecular design. Chemical Computing Group Inc., Montreal, QC, Canada. URL https://www. chemcomp.com/ (2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.