

Abstract
Juvenile myelomonocytic leukemia is a clonal malignant disease affecting young children. Current cure rates, even with allogeneic hematopoietic stem cell transplantation, are no better than 50%–60%. Pre-clinical research on juvenile myelomonocytic leukemia is urgently needed for the identification of novel therapies but is hampered by the unavailability of culture systems. Here we report a xenotransplantation model that allows long-term in vivo propagation of primary juvenile myelomonocytic leukemia cells. Persistent engraftment of leukemic cells was achieved by intrahepatic injection of 1×106 cells into newborn Rag2−/−γc−/− mice or intravenous injection of 5×106 cells into 5-week old mice. Key characteristics of juvenile myelomonocytic leukemia were reproduced, including cachexia and clonal expansion of myelomonocytic progenitor cells that infiltrated bone marrow, spleen, liver and, notably, lung. Xenografted leukemia cells led to reduced survival of recipient mice. The stem cell character of juvenile myelomonocytic leukemia was confirmed by successful serial transplantation that resulted in leukemia cell propagation for more than one year. Independence of exogenous cytokines, low donor cell number and slowly progressing leukemia are advantages of the model, which will serve as an important tool to research the pathophysiology of juvenile myelomonocytic leukemia and test novel pharmaceutical strategies such as DNA methyltransferase inhibition.
Introduction
Juvenile myelomonocytic leukemia (JMML) is a malignant myeloproliferative disorder of infancy and early childhood with an aggressive clinical course. Clinical symptoms are caused by hematopoietic insufficiency and excessive proliferation of leukemic monocytes and granulocytes, leading to hepatosplenomegaly, lymphadenopathy, skin rash and respiratory failure.1–3 JMML is caused by hyperactivation of the RAS signaling pathway due to acquired activating mutations in the KRAS, NRAS or PTPN11 genes,4–7 or due to acquired loss of heterozygosity of the constitutionally deficient NF1 gene in patients with neurofibromatosis type 1 or of the CBL gene in the Noonan-like “CBL syndrome”.8–13 JMML is rapidly fatal unless allogeneic hematopoietic stem cell transplantation (HSCT) is performed, but even this approach is burdened with a significant risk of recurrence.14,15
A serious obstacle to research into JMML is the lack of suitable experimental models, impeding the development and pre-clinical evaluation of novel therapeutic approaches. Primary JMML leukemia cells cannot be maintained in culture as they differentiate and become apoptotic.16 An immortalized cell line derived from JMML cells has not yet been successfully established.17 The generation of induced pluripotent stem cell lines originating from JMML cells was reported, but conceptually such systems are limited by their artificial nature and the risk of further transformation during reprogramming.18 Several of the “canonical” JMML mutations that deregulate the RAS signaling pathway were studied in genetically engineered mouse models, successfully inducing myeloproliferative disorders in the experimental animals.19–28 Those were, however, still murine leukemias, and critical disease characteristics of JMML such as recurrent monosomy 7 or elevated fetal hemoglobin are not simulated in transgenic systems.
Xenotransplantation into murine hosts offers the unique possibility of basic and translational research into living primary JMML cells, while at the same time propagating and multiplying this precious clinical material. However, earlier attempts at JMML xenotransplantation were compromised by difficult leukemia cell engraftment (presumably owing to residual natural killer cell activity of the host strains) or rapid demise of engrafted animals within a few weeks, and not all reports documented the xenologous engraftment of long-term leukemia-initiating cells via successful serial transplantation.29–31 In addition, the experiments depended on high input cell numbers (up to 5×107 cells), a considerable practical obstacle concerning the limited availability of primary clinical JMML material, and on costly repeated application of human granulocyte-macrophage colony-stimulating factor (GM-CSF).
Here we report the suitability of the Rag2−/−γc−/− mouse strain for the reproduction of primary human JMML in recipient animals. The system is characterized by good phenotypic imitation of typical disease features, long duration of xenologous engraftment, quantitative expansion of leukemic cell material outside the human organism, and the possibility of retransplantation to further expand cell numbers and extend the duration of experiments without additional input of cryopreserved material. Not least, the data support the stem cell character of long-term leukemia-initiating cells in JMML.
Methods
Primary cells
Human cells were collected after obtaining informed consent from parents or legal guardians and approval from institutional review committees. Samples from JMML patients were collected in the context of the European Working Group of MDS in Childhood (EWOG-MDS). Clinical information is provided in Online Supplementary Table S1. Single cell suspensions obtained from mashed spleens were subjected to density gradient centrifugation (Ficoll) to separate and cryopreserve mononuclear cells (MNC). Where indicated, MNC were depleted from CD3+ T cells (MACS immunobeads, Miltenyi; <0.15% remaining T cells). Cord blood was obtained from healthy newborns and CD34+ cells were enriched by the MACS technique (Miltenyi; purity >90%).
Xenotransplantation
All experiments were approved by local authorities and followed the German “Tierversuchsgesetz”. Rag2−/−γc−/− BALB/c mice32 were maintained in a specific pathogen-free environment. Newborn mice were irradiated with 2.5 Gy within their first four days of life. Eight hours after irradiation, JMML MNC were thawed and 1×106 viable cells were injected intrahepatically (30 μl). Alternatively, 5-week old mice were irradiated with 3 Gy and transplanted intravenously with 5×106 viable MNC. Single cell suspensions were obtained from bone marrow (BM), spleen and blood. Liver, kidney and lung were digested with collagenase D and DNase (Roche) followed by density gradient centrifugation. For serial transplantation, 1–4×106 BM cells from engrafted mice (containing 60%–70% human cells) were injected into recipients.
Flow cytometry
Cell suspensions were subjected to red blood cell lysis and stained with antibodies listed in Online Supplementary Table S2. Cytometric Bead Array kits (human inflammatory and Th1/Th2 cytokines; BD) were used according to the manufacturer’s instructions. A FACSCalibur (BD) was used; analyses were performed using FlowJo (FlowJo) and Cyflogic (Cyflo). The gating strategy is shown in Online Supplementary Figure S1.
Immunohistochemistry
Organs were fixed in 4% buffered formalin, and sternums were decalcified. After paraffin-embedding, sections were deparaffinized in xylene and graded alcohols. H&E and chloracetate esterase staining followed standard protocols. Immunohistochemical staining was performed after specific antigen retrieval in “low pH target retrieval solution” (Dako) for 30 min. Primary and secondary antibodies are listed in Online Supplementary Table S2. The EnVision FLEX System or the APK5005 system were used for visualization (Dako). Sections were counterstained with hematoxylin (Dako) and mounted.
Genetic analysis
Human-specific PCR for PTPN11 was performed on hematopoietic cells isolated from murine organs (forward primer ATCCGACGTGGAAGATGAGA, reverse primer TCAGAGGTAGGATCTGCACAGT). Human HL60 cells and hematopoietic cells from non-transplanted mice were used as positive and negative controls. PCR products were sequenced bidirectionally (BigDye Terminator kit, Life Technologies; ABI 3730xl or 3130xl capillary sequencers).
Pyrosequencing
Human-specific PTPN11 PCR products were generated as above using a biotinylated reverse primer and pyrosequenced on a Pyromark Q24 (Qiagen) using sequencing primer ACATCAAGATTCAGAACACT. The wild-type/mutant allelic ratio of PTPN11 point mutations was calculated using PyroMark Q24 software v.2.0 (Qiagen).
Statistical analysis
Charts show mean values and standard errors of the mean (SEM). Mann-Whitney test, Kaplan-Meier analysis and Mantel-Cox log rank test were used (Statview 4.1 software). P<0.05 was considered statistically significant.
Results
Xenotransplantation of human JMML cells into Rag2−/−γc−/− mice results in leukemic engraftment
We chose Rag2 and interleukin-2 receptor gamma chain double-deficient mice (Rag2−/−γc−/−) as recipients for the JMML xenografts. The genetic defect of this strain leads to near-complete abolishment of residual T cell, B cell, and natural killer cell activity,32 a prerequisite for successful JMML xenotransplantation.31 The mice were transplanted with MNC isolated from splenectomy preparations of 5 children with JMML. Flow cytometry showed that the pre-transplantation MNC samples consisted of a median of 21% CD34+ stem/progenitor cells (range 1%–65%), 44% CD33+ myeloid cells (range 18%–89%), 13% CD3+ T cells (range 3%–23%) and 27% CD19+ B cells (range 9%–31%). The cellular viability upon thawing and the cell composition of xenotransplanted material from individual patients is shown in Online Supplementary Figure S2. Based on our own previous experience with a xenotransplantation system for healthy human hematopoiesis using the same host strain,33 we started by using intrahepatic injection of graft cells into newborn mice (1×106 viable JMML MNC per mouse) as route of transplantation. To see if the procedure could be simplified and make the model less dependent on the timely birth of pups, we also transplanted 5-week old mice via conventional intravenous injection; these mice received 5×106 JMML MNC to compensate for the higher body weight at that age. The xenografted cells were monitored in all mice by biweekly collection of blood and flow cytometry of human CD45+ cells. Whereas most animals were sacrificed at elected time points (ranging from 10 to 20 weeks after transplantation) for phenotype analysis and harvest of JMML cells, a subset of mice was euthanized only when in poor condition so as to learn about the natural disease course.
We defined the level of human engraftment in a given murine organ as the proportion of human CD45+ cells within the total population of murine and human CD45+ cells. Following an accepted convention in xenotransplantation models,30,34 the occurrence of 0.5% or more human CD45+ cells in the murine BM was scored as successful engraftment. Using these definitions, JMML MNC from 4 children (Patients #1, #2, #3 and #5) engrafted into recipient mice with an overall leukemic engraftment rate of 58/82 mice (Figure 1), not counting 16 animals with non-leukemic T-cell engraftment (see below). Transplantation from one patient was unsuccessful (Patient #4, n=9 recipient mice) (Figure 1). In total, 64% (58/91) mice engrafted. Levels of human engraftment were variable and there was no correlation between percentage of human CD45+ cells and time from transplantation or condition of the mice. However, we cannot exclude the possibility that leukemic engraftment in mice might have reached higher levels in some mice if sacrificed later.
Figure 1.
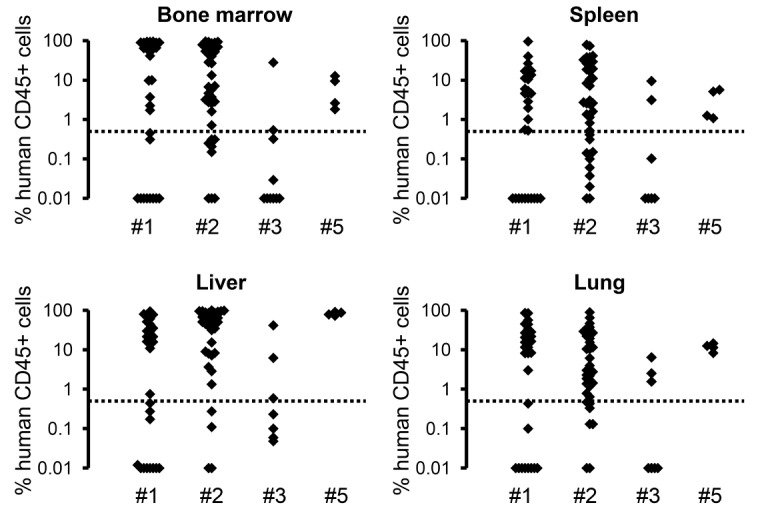
Sustained engraftment of xenotransplanted juvenile myelomonocytic leukemia (JMML) cells in Rag2−/−γc−/− mice. Spleen mononuclear cells (MNC) from JMML Patients #1 to #5 were transplanted into sublethally irradiated mice (1×106 cells per mouse via intrahepatic injection or 5×106 cells per mouse via intravenous injection). Hematopoietic cells were obtained from indicated organs at 7–37 weeks post transplant. Human cell engraftment as assessed by flow cytometry of CD45+ cells is shown for animals transplanted from Patient #1 (n=31 mice), Patient #2 (n=37 mice), Patient #3 (n=10 mice) and Patient #5 (n=4 mice). The level of human engraftment was defined as proportion of human CD45+ cells within the total population of murine and human CD45+ cells. Cells from Patient #4 consistently failed to engraft (n=9 mice). The dotted line represents the definition of successful engraftment (≥0.5% human CD45+ cells).
To confirm the presence of JMML cells and rule out the possibility that co-transplanted healthy hematopoietic stem cells were engrafted into recipient mice, human CD45+ cells were isolated from BM of 43 recipient mice after transplantation from Patients #1 and #2 and used for sequence analysis of the PTPN11 gene. In all mice, the JMML-related mutation (PTPN11 c. G181T and c. C215T, respectively) was detected in infiltrating cells.
Sustained leukemic engraftment in Rag2−/−γc−/− xenotransplanted mice recapitulates characteristic features of human JMML
To analyze the leukemic phenotype of xenografted animals, BM, spleen, liver, lung, and kidney of all mice were evaluated for human cell infiltration by flow cytometry, histopathology and immunohistochemistry. We observed consistent involvement of BM, spleen, liver and, importantly, lung. The kidney was unaffected in all animals. Flow cytometry revealed a strong predominance of human myeloid CD33+ cells in infiltrated murine organs (Figure 2A). The number of immature CD34+ cells was highest in BM and liver, reflecting the sites of perinatal hematopoiesis. Mature myeloid CD13+ cells were most abundant in lung. Only a minor proportion of human cells were B or T lymphocytes. On gross examination, significant splenomegaly was observed in mice with successful xenologous leukemic engraftment as opposed to non-engrafted animals (Figure 2B). Together, these features closely resemble JMML in children.
Figure 2.
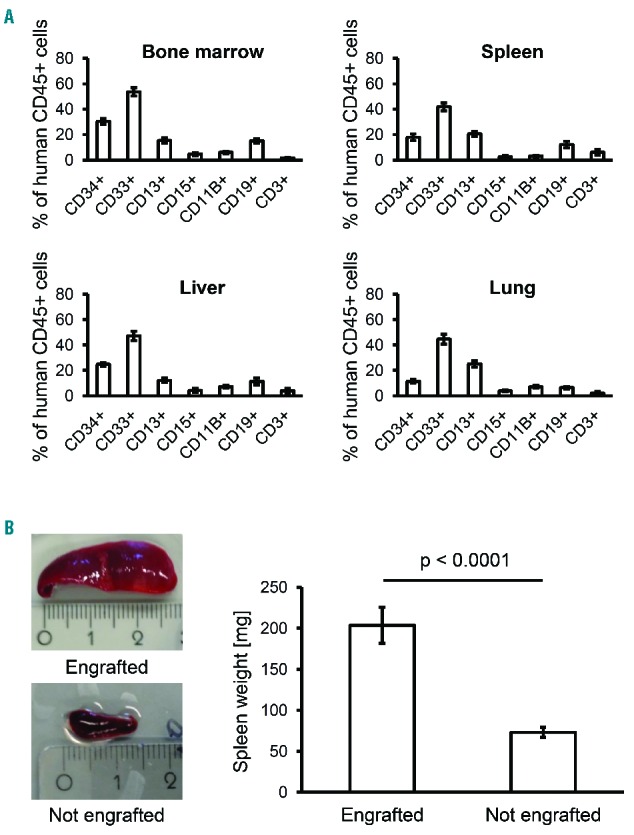
Organ infiltration and human cell subpopulations after xenotransplantation of juvenile myelomonocytic leukemia (JMML) cells in Rag2−/−γc−/− mice. (A) Hematopoietic cells were obtained from indicated organs at 7–37 weeks after transplantation from 4 patients with JMML (n=58 mice). Cell subpopulations were assessed by flow cytometry with antibodies to human CD45, CD34, CD33, CD13, CD15, CD11B, CD19 and CD3. Bars indicate mean value and standard error. (B) Representative example of splenomegaly in mice with JMML cell engraftment (top) and normal spleen size in non-engrafted mice (bottom). The spleen weight of 49 engrafted and 17 non-engrafted mice was measured (right panel).
A total of 19 mice was informative for survival analysis. These mice were euthanized only at terminal disease and fulfilled the criteria of leukemic engraftment outlined above (Figure 3). Leukemia established in the recipient mice by JMML-initiating cells led to death of host animals at 51–224 days post transplantation whereas the survival of non-engrafted or non-transplanted mice was not reduced.
Figure 3.
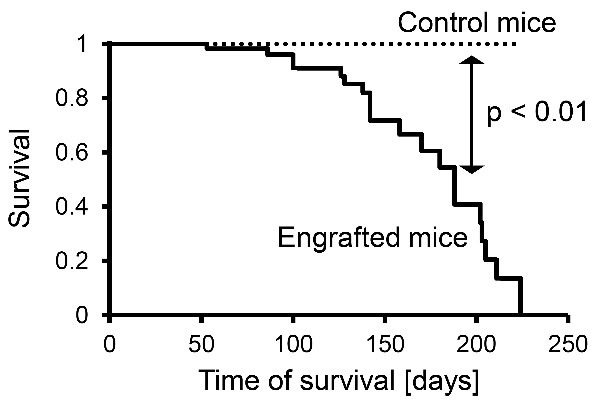
Xenologous engraftment of juvenile myelomonocytic leukemia (JMML) cells in Rag2−/−γc−/− mice results in decreased survival. Of 58 mice with xenologous JMML cell engraftment after transplantation, 39 were sacrificed at elected time points (not included in this Figure) and 19 were informative for survival analysis (solid line). Survival of non-transplanted control mice was unaffected (dotted line, n=5) (P=0.0004, Mantel-Cox log rank test).
The histopathology of JMML-xenotransplanted mice showed that the BM, spleen and liver were infiltrated by a predominant population of differentiating myelomonocytic cells (Figure 4A). Immature forms with blast-like appearance were detected to a lesser degree. Leukemic infiltrates were focal and displaced the normal murine hematopoiesis in BM and spleen or the murine hepatocytes in liver. Some parts of the organs were completely destroyed by the leukemic foci, while other parts remained unaffected (Figure 4B). Consistent with flow cytometry and the observation that many animals developed respiratory distress after xenologous engraftment, histopathology demonstrated human cell infiltration also in the lung (Figure 4A). BM immunohistochemistry showed that human CD34+ immature cells resided closer to the endosteal niches whereas more mature cells (lysozyme-positive or CD68+) were located towards the medulla. Not all cells with leukemic morphology stained positive for human CD45. However, mouse-specific antibodies excluded the murine origin of these cells (Figure 4A). We assume that such cells represent CD45-negative JMML progeny, for example early erythroid progenitors. We noted that several mice transplanted with JMML cells from Patient #2 carried a predominant blast cell population, while the majority of mice receiving cells from this donor showed the usual infiltration with differentiating myelomonocytic cells. This suggests the outgrowth of an acute myeloid leukemia (AML)-like subclone in single animals.
Figure 4.
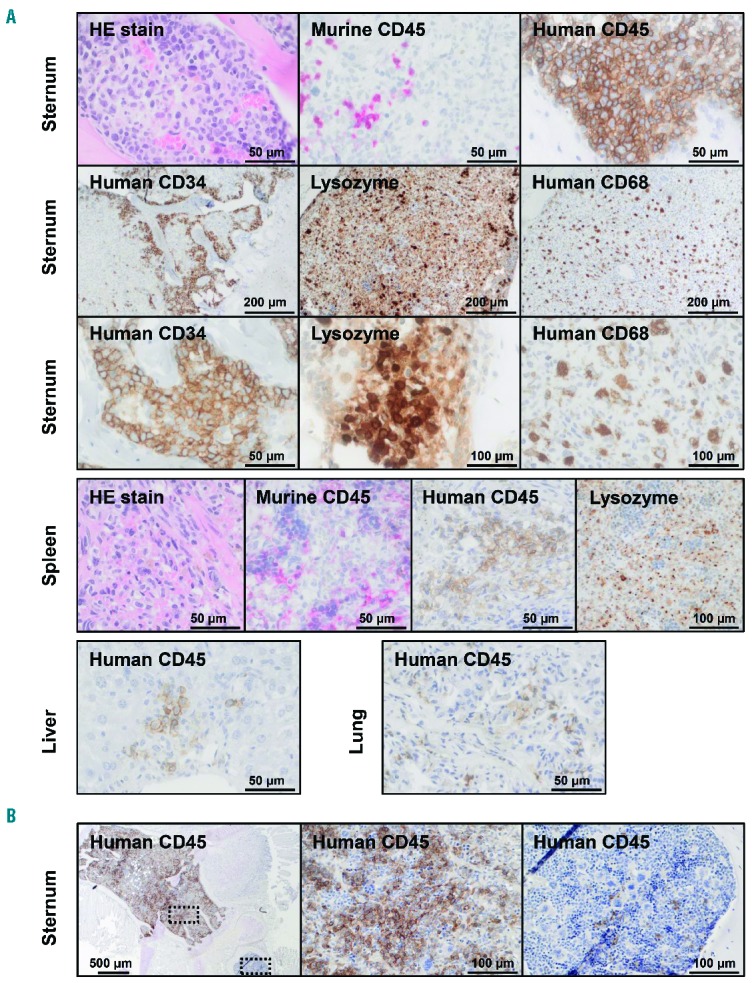
Histopathology demonstrates human myelomonocytic infiltration in murine tissues. (A) Histology and immunohistochemistry of bone marrow, spleen, liver and lung at time of terminal illness revealed leukemic infiltration with differentiating myelomonocytic cells that replaced regular murine tissue. Immunostaining with antibodies to murine CD45 and human CD45 confirmed the human origin of the leukemic cells (top panel, first row). Immature CD34-positive cells were located in the peritrabecular region and had a blast-like appearance. By contrast, lysozyme- and CD68-positive, more differentiated myelomonocytic cells were found in the center of the medullary cavity (top panel, second and third row). The murine spleen was infiltrated by myelomonocytic cells positive for human CD45 and lysozyme but negative for murine CD45 (middle panel). Human myelomonocytic cells were also found in murine liver and lung (bottom panel). (B) Focal displacement of murine hematopoiesis by human CD45-positive myelomonocytic cells. Dotted frames indicate areas with higher magnification shown on the right.
To determine if the two transplantation techniques resulted in different disease phenotypes, we prospectively compared 6 mice after neonatal intrahepatic transplantation (Online Supplementary Figure S3) with 5 mice transplanted intravenously at 5-weeks of age (Online Supplementary Figure S4). For the purpose of this experiment, the mice were killed only at terminal disease. We found no difference in engraftment levels in BM, spleen, liver, or lung (Online Supplementary Figures S3A and S4A). The length of survival was identical between the intrahepatic (Online Supplementary Figure S3B) and the intravenous (Online Supplementary Figure S4B) group. Likewise, the differentiation profile of infiltrating cells was the same in both groups (Online Supplementary Figures S3C and S4C).
To compare JMML engraftment with non-leukemic xenologous hematopoiesis, we transplanted human CD34+ cells derived from umbilical cord blood of a healthy newborn into 7 Rag2−/−γc−/− mice (Online Supplementary Figure S5). Contrary to mice transplanted with JMML cells, and in line with previous observations,33 the human cells found in these mice were predominantly B cells and myeloid differentiation was barely detectable. The mice did not develop organomegaly and their survival after transplantation was no shorter.
The Rag2−/−γc−/− xenotransplantation model is independent of exogenous stimulation with GM-CSF
A hallmark feature of JMML progenitor cells is their hypersensitivity to GM-CSF,17,35–37 and previous JMML xenograft models depended on continuous administration of human GM-CSF.30 To analyze the effect of exogenous GM-CSF in our model, we xenotransplanted 9 newborn mice, 4 of which received weekly injections of 5 μg human GM-CSF beginning eight weeks after transplantation (Figure 5A). We observed no difference to 5 unstimulated mice regarding human CD45+ or CD34+ cell content in bone marrow or spleen even when GM-CSF treatment was continued for as long as 20 weeks after transplant (Figure 5B). Histopathology did not reveal any noteworthy differences either. As expected, higher proportions of human CD33+ myeloid cells were noted in mice treated with GM-CSF (Figure 5B). It appears that paracrine secretion of human cytokines by differentiating monocytes is sufficient to sustain the JMML-initiating cells in the Rag2−/−γc−/− microenvironment. Accordingly, we detected the human cytokines interleukin-8, tumor necrosis factor-alpha and interferon-gamma in the serum of engrafted mice (Online Supplementary Figure S6).
Figure 5.
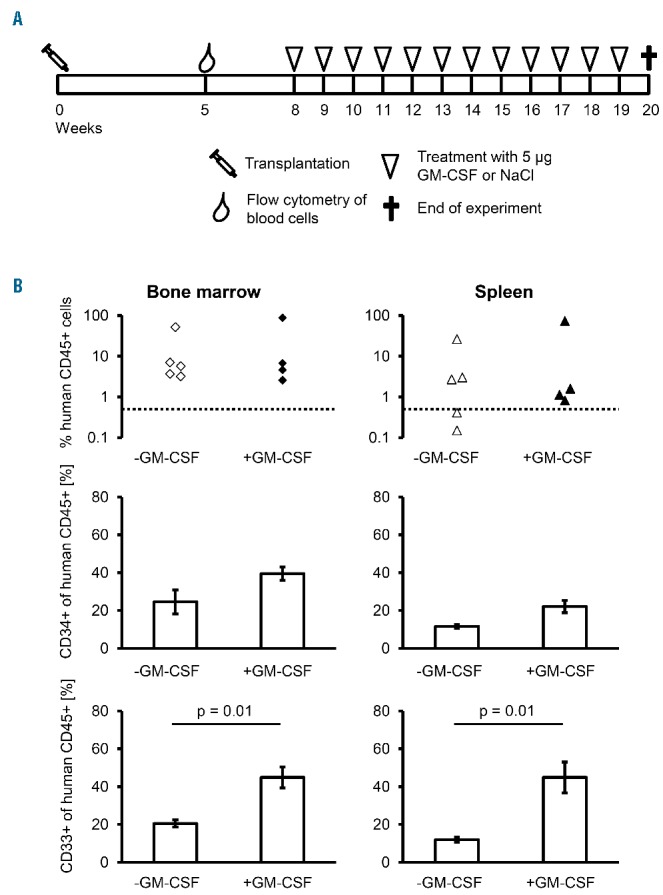
Application of exogenous granulocyte-macrophage colony-stimulating factor (GM-CSF) increases myeloid differentiation without affecting overall engraftment. (A) Schematic diagram of the experimental set up. Nine mice were transplanted intrahepatically with 1×106 juvenile myelomonocytic leukemia (JMML) mononuclear cells of Patient #2. Five weeks later, human cell engraftment was confirmed by flow cytometry of CD45+ peripheral blood cells. Mice were divided into two experimental groups matched for level of engraftment in peripheral blood. Four mice received weekly injections of 5 μg recombinant human GM-CSF, while saline was administered in 5 mice. Applications were started eight weeks after transplantation. The animals were analyzed 12 weeks later. (B) The human CD45+ cell engraftment, the proportion of CD34+ progenitor cells and the proportion of CD33+ myeloid cells were determined in bone marrow (left) and spleen (right). Levels of human CD45+ and CD34+ cells were comparable between untreated (open symbols) and treated (filled symbols) animals. The proportion of CD33+ cells was significantly higher in bone marrow and spleen of treated animals (P=0.01, Mann-Whitney test).
Graft-versus-host disease originating from T lymphocytes may overwhelm the leukemic engraftment of individual JMML samples
Whereas the xenotransplantation of unfractionated spleen MNC from Patients #1, #2 and #5 invariably led to myeloid leukemic engraftment in recipient mice, the spleen MNC from Patient #3 caused massive T lymphocyte infiltration of all organs and rapid death within 22–36 days after transplantation (Online Supplementary Figure S7). Flow cytometry with human-specific antibodies confirmed the human origin of these cells. Upon genetic analysis, the JMML-specific PTPN11 mutation was undetectable, indicating that co-transplanted non-leukemic T cells had expanded in the animals. When CD3+ lymphocytes were depleted from spleen MNC prior to transplantation using immunomagnetic beads, regular JMML cell engraftment but no T-cell expansion or graft-versus-host disease occurred in the recipient animals (see Figure 1, Patient #3). Hence, a T-cell depletion step may be required for successful xenotransplantation depending on the individual cell composition of the clinical material.
JMML-initiating cells are serially retransplantable and re-establish disease in mice
To assess the self-renewal capacity of long-term JMML-initiating cells, serial transplantations were performed. Seventeen weeks after xenotransplantation with JMML cells from Patient #1, BM was obtained from 2 mice and injected into 9 mice as second-generation xenograft. Successful leukemic engraftment was observed in all 9 mice (Figure 6A). The survival of recipient mice was similar to that of primary recipients (Figure 6B). The secondary recipients developed splenomegaly and showed predominant myeloid infiltration (Figure 6C). The overall level of organ infiltration with human cells and the lineage distribution of human cell progeny were comparable to first-generation xenograft mice.
Figure 6.

Analysis of secondary recipient mice after serial xenotransplantation. (A) Seventeen weeks after xenotransplantation with juvenile myelomonocytic leukemia (JMML) cells from Patient #1, bone marrow (BM) cells from 2 mice were re-transplanted into 9 second-generation mice (1×106 cells per animal). The secondary recipient animals were sacrificed for analysis when terminally sick (165–328 days after transplantation). The level of human engraftment was defined as proportion of human CD45+ cells within the total population of murine and human CD45+ cells. Open triangles indicate first-generation (donor) mice; closed diamonds, second-generation recipients. The dotted line represents the definition of successful engraftment (≥0.5% human CD45+ cells). (B) Secondary recipient mice (solid line) had significantly reduced survival compared to untransplanted control mice (dotted line, n=5) (P<0.01, Mantel-Cox log rank test). (C) Hematopoietic cells were obtained from indicated organs and cell subpopulations were assessed by flow cytometry with antibodies to human CD45, CD34, CD33, CD13, CD15, CD11B, CD19 and CD3. Bars indicate mean value and standard error.
At ten weeks after xenotransplantation with JMML MNCs from Patient #2, 8 secondary recipients were transplanted with BM harvested from 4 mice. Four of the secondary recipients showed successful engraftment. BM cells from these mice were then used for tertiary transplantation and led to engraftment in 8 of 8 mice (Online Supplementary Figure S8). Again, the level of leukemic organ infiltration and length of survival were comparable to first-generation xenograft mice. Mutation analysis demonstrated that the leukemia-specific PTPN11 mutation was invariably present when human cells were retrieved from serially engrafted mice, regardless of infiltrated organ or graft generation (data not shown). This ruled out the possibility that the leukemic clone might have been lost after repeated xenotransplantation and that long-lived non-leukemic progenitors might have prevailed. In addition, quantitative pyrosequencing was employed to compare the mutant allele fraction between source material (spleen MNC) and purified human CD45+ cells retrieved from BM cells of serially xenografted mice (Figure 7). We observed that close to 100% of human cells were of leukemic origin in primary, secondary, or tertiary recipients.
Figure 7.
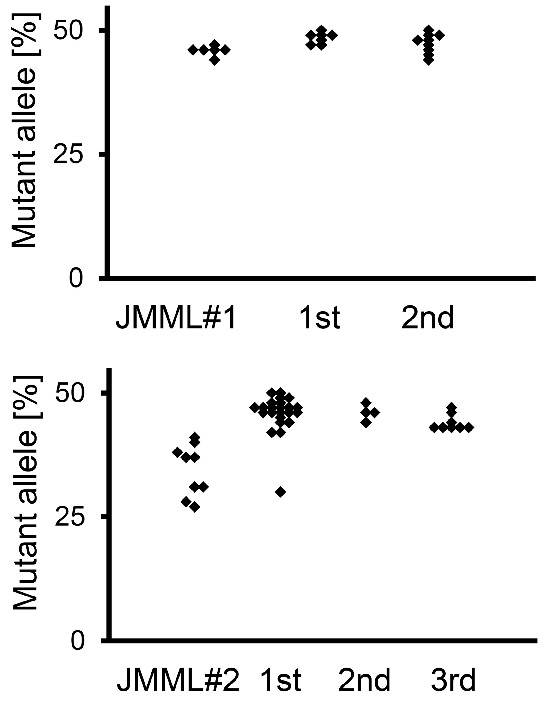
(right). Leukemic allele frequency was maintained during serial transplantations. Pyrosequencing was performed on human CD45+ cells obtained from murine bone marrow to determine the mutant allele frequency of PTPN11 c.C215T (Patient #1, upper panel) and PTPN11 c.G181T (Patient #2, lower panel) in serially transplanted animals. Since the leukemic mutations were heterozygous, a 50% allele frequency corresponds to 100% leukemic cells.
Discussion
The need for a pre-clinical model of JMML that can be used for basic research, biomarker identification and drug testing prompted us to establish a xenotransplantation system for this leukemia in immunodeficient mice. Other investigators have previously xenografted JMML cells but reported various difficulties.29–31 Lapidot et al., using SCID mice as host strain, observed a rapid decline in well-being and cachexia as soon as 2–4 weeks after xenotransplantation of JMML cells.30 Their experiments involved continuous treatment of host mice with human GM-CSF. In the absence of exogenous human GM-CSF the leukemic cells did not engraft at all. Nakamura et al. reported highly variable levels of myeloid engraftment in NOD/SCID/γc−/− mice (average 18.7% CD33+ of human CD45+ cells in the bone marrow, range 9.4%–37.7%), although the number of cells transplanted was fairly high (107 cells per mouse).31 In addition, those previous reports on JMML xenotransplantation lacked a detailed analysis of the natural disease course of recipient mice as all animals were sacrificed no later than 12 weeks after transplantation.29–31 However, we felt that a system with a more chronic disease course would be desirable if JMML were to be modeled in experimental animals for pre-clinical research.
In an attempt to overcome the obstacles discussed above we chose Rag2−/−γc−/− mice as hosts and intrahepatic injection into newborn recipients as mode of transplantation. We favored this technique because of an earlier description by Traggiai and the documented suitability for transplantation of healthy cord blood-derived CD34+ cells.33,38 Adopting an intrahepatic strategy seemed especially appropriate for JMML since it is a disease of early childhood, frequently affects the liver and most probably originates from fetal hematopoietic cells, which appear to be supported better by neonatal than adult tissues.39,40 In addition, intravenous injection would be technically challenging in newborn mice. When we later compared the phenotype of mice transplanted intrahepatically with that of animals xenografted at older age via the intravenous route we found no significant differences in level of organ infiltration, length of survival after transplantation, or other aspects of the ensuing leukemia. Although we did not perform systematic titrations of input cell numbers, we believe that the intrahepatic technique might be the better choice if clinical samples with limited cell number were to be transplanted.
Juvenile myelomonocytic leukemia cells from 4 patients readily engrafted in the mice whereas transplantation from one child was unsuccessful. We can only speculate whether this failure relates to low amounts of JMML-initiating cells in the spleen MNC preparations (Online Supplementary Table S1) or to poor material quality (i.e. latency between splenectomy and cryopreservation). After successful engraftment, the xenotransplanted mice displayed symptoms similar to those observed in children with JMML, including hepatosplenomegaly, cachexia and pulmonary infiltration with respiratory distress. Detailed analysis of murine hematopoietic organs revealed focal infiltration by human myelomonocytic CD33+ cells. CD13 expression indicated the presence of cells at more mature stages of differentiation and was stronger in spleen and lung than BM and liver, consistent with the physiological route of myeloid differentiation. Immature CD34+ leukemic cells were located in the endosteal regions of bone indicating that they shared hematopoietic niches with their normal counterparts, similar to a recent observation in AML.41 The lineage composition of engrafted JMML cell progeny clearly differed from that of xenotransplanted cord blood-derived healthy CD34+ cells, where B cells predominated and only minor myeloid populations were observed. Importantly, xenotransplanted mice showed a chronic disease course with a median survival of more than 20 weeks. This makes it possible to evaluate pharmaceuticals with delayed activity, in particular the DNA-hypomethylating agent azacitidine which has recently gained clinical interest for use in JMML.42,43 In contrast to previous JMML xenotransplantation models, Rag2−/−γc−/− mice efficiently sustained engrafting JMML cells in the absence of exogenous human GM-CSF. Weekly application of human GM-CSF enhanced myeloid differentiation but did not influence the total level of engraftment or time to leukemia.
Serial transplantation of JMML cells confirmed the presence of long-term JMML-initiating cells with self-renewal capacity. Phenotype and disease kinetics were similar in primary, secondary and tertiary recipients. Importantly, typical morphology with differentiating myelomonocytic cells was preserved over time and no disease acceleration was observed. Serial transplantability is not only important to the scientific concept of leukemia-initiating cells, but is also a valuable tool for the expansion of primary JMML cells from a practical perspective. Using the Rag2−/−γc−/− JMML system and serial retransplantation we have maintained JMML cells in vivo for 1.5 years in total. In the process, unmanipulated clinical JMML material was expanded rather than consumed. This has not so far been feasible by in vitro culture.
In summary, we present a novel xenotransplantation model of JMML that closely mimics human disease. We are confident that the model will be useful to further characterize the JMML-initiating cell, amplify scarce and valuable clinical material, and complement the recently evolving early-phase clinical trials for novel pharmaceutical strategies such as epigenetic therapy.
Acknowledgments
We are grateful to A. Meier, N. Fischer and B. Stopp for technical assistance and to N. Krause, B. Müller and K. Thumm for animal care. We thank H. Pahl and H. Eibel for insightful discussions.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/5/597
Funding
German Research Foundation (CRC 992-C05 to CF and SPP1463 FL345/4-2 to CF) and Freiburg Institute for Advanced Studies (fellowship to ME).
References
- 1.Niemeyer CM, Arico M, Basso G, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood. 1997;89(10):3534–3543. [PubMed] [Google Scholar]
- 2.Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. 2015;125(7):1083–1090. [DOI] [PubMed] [Google Scholar]
- 3.Chang TY, Dvorak CC, Loh ML. Bedside to bench in juvenile myelomonocytic leukemia: insights into leukemogenesis from a rare pediatric leukemia. Blood. 2014;124(16):2487–2497. [DOI] [PubMed] [Google Scholar]
- 4.Flotho C, Valcamonica S, Mach-Pascual S, et al. RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia. 1999;13(1):32–37. [DOI] [PubMed] [Google Scholar]
- 5.Tartaglia M, Niemeyer CM, Fragale A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34(2):148–150. [DOI] [PubMed] [Google Scholar]
- 6.Kratz CP, Niemeyer CM, Castleberry RP, et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood. 2005;106(6):2183–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyauchi J, Asada M, Sasaki M, et al. Mutations of the N-ras gene in juvenile chronic myelogenous leukemia. Blood. 1994;83(8):2248–2254. [PubMed] [Google Scholar]
- 8.Bader JL, Miller RW. Neurofibromatosis and childhood leukemia. J Pediatr. 1978;92(6): 925–929. [DOI] [PubMed] [Google Scholar]
- 9.Stiller CA, Chessells JM, Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer. 1994;70(5):969–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Side LE, Emanuel PD, Taylor B, et al. Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1. Blood. 1998;92(1):267–272. [PubMed] [Google Scholar]
- 11.Steinemann D, Arning L, Praulich I, et al. Mitotic recombination and compound-heterozygous mutations are predominant NF1-inactivating mechanisms in children with juvenile myelomonocytic leukemia and neurofibromatosis type 1. Haematologica. 2010;95(2):320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loh ML, Sakai DS, Flotho C, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009;114(9): 1859–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niemeyer CM, Kang MW, Shin DH, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42(9):794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locatelli F, Nollke P, Zecca M, et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood. 2005;105(1):410–419. [DOI] [PubMed] [Google Scholar]
- 15.Bergstraesser E, Hasle H, Rogge T, et al. Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: a retrospective analysis and definition of response criteria. Pediatr Blood Cancer. 2007;49(5):629–633. [DOI] [PubMed] [Google Scholar]
- 16.Sakashita K, Kato I, Daifu T, et al. In vitro expansion of CD34(+)CD38(−) cells under stimulation with hematopoietic growth factors on AGM-S3 cells in juvenile myelomonocytic leukemia. Leukemia. 2015;29(3):606–614. [DOI] [PubMed] [Google Scholar]
- 17.Gualtieri RJ, Castleberry RP, Gibbons J, et al. Cell culture studies and oncogene expression in juvenile chronic myelogenous leukemia. Exp Hematol. 1988;16(7):613–619. [PubMed] [Google Scholar]
- 18.Gandre-Babbe S, Paluru P, Aribeana C, et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood. 2013;121(24):4925–4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacks T, Shih TS, Schmitt EM, et al. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7(3):353–361. [DOI] [PubMed] [Google Scholar]
- 20.Hawley RG, Fong AZ, Ngan BY, Hawley TS. Hematopoietic transforming potential of activated ras in chimeric mice. Oncogene. 1995;11(6):1113–1123. [PubMed] [Google Scholar]
- 21.MacKenzie KL, Dolnikov A, Millington M, Shounan Y, Symonds G. Mutant N-ras induces myeloproliferative disorders and apoptosis in bone marrow repopulated mice. Blood. 1999;93(6):2043–2056. [PubMed] [Google Scholar]
- 22.Araki T, Mohi MG, Ismat FA, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10(8):849–857. [DOI] [PubMed] [Google Scholar]
- 23.Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5(4):375–387. [DOI] [PubMed] [Google Scholar]
- 24.Kim A, Morgan K, Hasz DE, et al. Beta common receptor inactivation attenuates myeloproliferative disease in Nf1 mutant mice. Blood. 2007;109(4):1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cutts BA, Sjogren AK, Andersson KM, et al. Nf1 deficiency cooperates with oncogenic K-RAS to induce acute myeloid leukemia in mice. Blood. 2009;114(17):3629–3632. [DOI] [PubMed] [Google Scholar]
- 26.Naramura M, Nandwani N, Gu H, Band V, Band H. Rapidly fatal myeloproliferative disorders in mice with deletion of Casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proc Natl Acad Sci USA. 2010;107(37):16274–16279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Liu Y, Li Z, et al. Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood. 2010;116(26):5991–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang YI, You X, Kong G, et al. Loss of Dnmt3a and endogenous Kras cooperate to regulate hematopoietic stem and progenitor cell functions in leukemogenesis. Leukemia. 2015;29(9):1847–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iversen PO, Lewis ID, Turczynowicz S, et al. Inhibition of granulocyte-macrophage colony-stimulating factor prevents dissemination and induces remission of juvenile myelomonocytic leukemia in engrafted immunodeficient mice. Blood. 1997;90(12): 4910–4917. [PubMed] [Google Scholar]
- 30.Lapidot T, Grunberger T, Vormoor J, et al. Identification of human juvenile chronic myelogenous leukemia stem cells capable of initiating the disease in primary and secondary SCID mice. Blood. 1996;88(7):2655–2664. [PubMed] [Google Scholar]
- 31.Nakamura Y, Ito M, Yamamoto T, et al. Engraftment of NOD/SCID/gammac(null) mice with multilineage neoplastic cells from patients with juvenile myelomonocytic leukaemia. Br J Haematol. 2005;130(1):51–57. [DOI] [PubMed] [Google Scholar]
- 32.Goldman JP, Blundell MP, Lopes L, et al. Enhanced human cell engraftment in mice deficient in RAG2 and the common cytokine receptor gamma chain. Br J Haematol. 1998;103(2):335–342. [DOI] [PubMed] [Google Scholar]
- 33.Labi V, Bertele D, Woess C, et al. Haematopoietic stem cell survival and transplantation efficacy is limited by the BH3-only proteins Bim and Bmf. EMBO Mol Med. 2013;5(1):122–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Notta F, Mullighan CG, Wang JC, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469(7330):362–367. [DOI] [PubMed] [Google Scholar]
- 35.Gualtieri RJ, Emanuel PD, Zuckerman KS, et al. Granulocyte-macrophage colony-stimulating factor is an endogenous regulator of cell proliferation in juvenile chronic myelogenous leukemia. Blood. 1989;74(7):2360–2367. [PubMed] [Google Scholar]
- 36.Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77(5):925–929. [PubMed] [Google Scholar]
- 37.Freedman MH, Cohen A, Grunberger T, et al. Central role of tumour necrosis factor, GM-CSF, and interleukin 1 in the pathogenesis of juvenile chronic myelogenous leukaemia. Br J Haematol. 1992;80(1):40–48. [DOI] [PubMed] [Google Scholar]
- 38.Traggiai E, Chicha L, Mazzucchelli L, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004;304(5667):104–107. [DOI] [PubMed] [Google Scholar]
- 39.Wulf-Goldenberg A, Keil M, Fichtner I, Eckert K. Intrahepatic transplantation of CD34+ cord blood stem cells into newborn and adult NOD/SCID mice induce differential organ engraftment. Tissue Cell. 2012;44(2):80–86. [DOI] [PubMed] [Google Scholar]
- 40.Arora N, Wenzel PL, McKinney-Freeman SL, et al. Effect of developmental stage of HSC and recipient on transplant outcomes. Dev Cell. 2014;29(5):621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyd AL, Campbell CJ, Hopkins CI, et al. Niche displacement of human leukemic stem cells uniquely allows their competitive replacement with healthy HSPCs. J Exp Med. 2014;211(10):1925–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furlan I, Batz C, Flotho C, et al. Intriguing response to azacitidine in a patient with juvenile myelomonocytic leukemia and monosomy 7. Blood. 2009;113(12):2867–2868. [DOI] [PubMed] [Google Scholar]
- 43.Cseh A, Niemeyer CM, Yoshimi A, et al. Bridging to transplant with azacitidine in juvenile myelomonocytic leukemia: a retrospective analysis of the EWOG-MDS study group. Blood. 2015;125(14):2311–2313. [DOI] [PubMed] [Google Scholar]