

Acute lymphoblastic leukemia (ALL) comprises multiple entities with distinct clinico-biological features and treatment response. Among prognostic factors, cytogenetics remains one of the most discriminating parameters and is currently used to determine risk and treatment stratification. However, approximately 20–25% of childhood B-cell precursor (BCP)-ALL cases are genetically unclassified. In 2009, Den Boer ML et al. identified a subtype of high-risk BCP-ALL that lacks recurrent cytogenetic aberrations but exhibits a gene expression profile similar to BCR-ABL1-positive ALL.1 Notably, the prevalence of BCR-ABL1-like ALL seems to increase with age, from about 10% of childhood ALL to more than 25% in adolescents and young adults.2 Interestingly, it was shown that BCR-ABL1-like ALL were characterized by kinase-activating alterations, suggesting the potential interest of targeted therapy.2
We report the case of an adolescent with refractory BCP-ALL and NUP214-ABL1 fusion. Her disease was marked by multiple treatment failures and efficacy, although transient, of tyrosine kinase inhibitor (TKI), suggesting the need for further evaluation in this rare and hardly curable subtype of BCP-ALL.
A 15 year-old girl with no previous medical history presented in our pediatric department in February 2013 for fatigue, pale skin, weakness and tumour syndrome (lymphadenopathy and hepatosplenomegaly). The blood count showed hemoglobin at 9.2 g/dL, platelet count at 58 000/mm3 and hyperleukocytosis at 260 000/mm3 with 95% of blast cells. The bone marrow aspirate was of high cellularity with 93% of peroxidase negative staining blast cells. Immunophenotyping confirmed the diagnosis of BCP-ALL with the expression of CD34, HLA-DR and B-lymphoid markers CD19, CD10, CD20, CD22 and intracytoplasmic μ chain (Pre-B ALL). Cytogenetics analyses revealed a normal female karyotype 46,XX. Molecular studies did not show any of the following recurrent rearrangements: ETV6-RUNX1, TCF3-PBX1, BCR-ABL1 fusions or KMT2A (MLL) rearrangements.
First-line treatment was conducted according to EORTC 58081 guidelines. Evaluation at day 8 showed corticoid resistance. The patient was therefore treated in the very high risk (VHR) group. Induction therapy included daunorubicin, vincristine, cyclophosphamide, asparaginase and corticoid and triple intrathecal chemotherapy by methotrexate, cytarabine and corticoid.
End point evaluation at day 35 showed induction failure with a minimal residual disease (MRD) above 10−2 (Online Supplementary figure S1). She underwent a myeloablative conditioning using busulfan, fludarabine and thiotepa and received genoidentical bone marrow transplantation in October 2013. Evaluation at day 100 showed cytological, phenotypic and molecular complete remission (CR).
Unfortunately, the patient relapsed in April 2014 with 12% and 71% blast cells in peripheral blood and bone marrow, respectively. Second-line treatment with bortezomib, dexamethasone and vincristine according to BOREALL protocol was unsuccessful with 77% of blast cells in bone marrow at day 22. The patient was then placed on exclusive palliative care with oral chemotherapy using purinethol and methotrexate in August 2014.
In March 2015, the patient was hospitalised for sepsis. New laboratory investigations revealed combined bone marrow and central nervous system involvement. In May 2015, a single nucleotide polymorphism (SNP) array (Cytoscan High-Density, Affymetrix®) was finally performed on the bone marrow sample from diagnosis (Figure 1A). Analysis revealed small deletions targeting TBL1XR1 (3q26), LEF1 (4q25), IKZF1 (7p12, exon 1 and exons 4-8; confirmed by multiplex ligation-dependent probe amplification), CDKN2A/B (9p21), SERP2 (13q14), C20orf94 (20p12), RUNX1 (21q22) and VPREB1 (22q11), but also an amplification in 9q34 limited by NUP214 and ABL1 (Figure 1B and Online Supplementary Table S1), suggestive of a NUP214-ABL1 fusion. All abnormalities appeared to be present in the whole leukemic population. Ligation-dependent RT-PCR confirmed the fusion of NUP214 exon 32 with ABL1 exon 3 (Figure 1C,D).3
Figure 1.
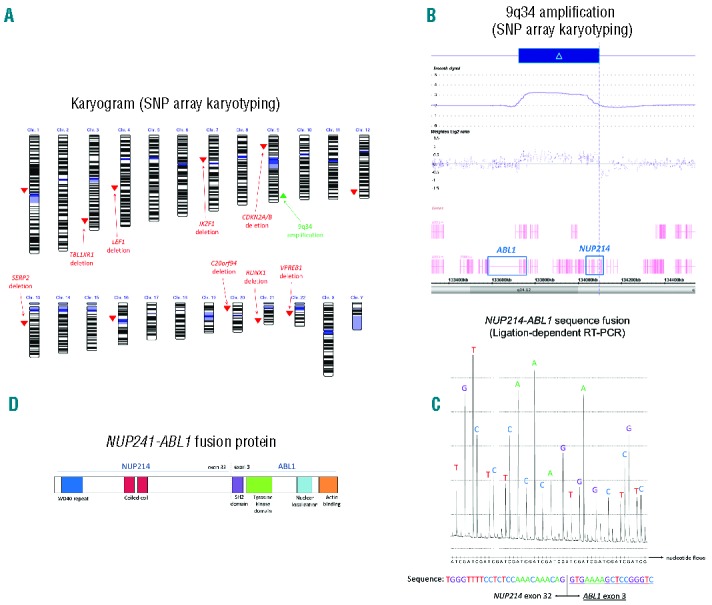
(A) Results of SNP array karyotyping. Genetic losses are shown in the color red. Amplification is show in green. (B) 9q34 amplification delimited by NUP214 and ABL1 (picture from Chromosome Analysis suite®). CN =copy number. (C) NUP214-ABL1 fusion sequence determined by ligation-dependent RT-PCR. (D) Schematic representation of the NUP214-ABL1 fusion protein.
The fusion retains the N-terminal region of NUP214, a component of the nuclear pore complex mediating nucleocytoplasmic transport, with the C-terminal region of the tyrosine kinase ABL1 (Figure 1D). Notably, the NUP214-ABL1 fusion is a recurrent abnormality found in approximately 6% of T-ALL.4 It is the second most prevalent fusion gene involving ABL1 in malignant hemopathies.5 However, only a few cases have been described in BCP-ALL. In vitro studies showed that NUP214-ABL1 fusion was sensitive to TKIs (imatinib, dasatinib),6 but clinical experience remains limited.7,8
Considering the potential responsiveness to TKIs, it was decided to start a new treatment with dasatinib 100 mg per day (D1 to D23) in combination with vincristine (D1, D8, D15 and D22) and dexamethasone (D1, D2, D8, D9, D15, D16, D22 and D23). Finally, the patient achieved a second CR in August 2015, and the improvement of her clinical condition allowed her to make her return to school. Dasatinib was then continued and the patient received one donor lymphocyte infusion in order to render her MRD negative. Unfortunately, the disease finally eluded treatment in October 2015.
Identifying BCR-ABL1-like ALL is of great interest in order to propose targeted therapy to chemotherapy and improve the CR rate and survival of patients with this type of leukemia. High-resolution SNP array karyotyping as well as ligation-dependent RT-PCR appears to be a very useful method to detect such cases when gene expression profiling is unavailable.9 Roberts et al. reported the case of a 12 year-old male with BCP-ALL and a NUP214-ABL1 fusion.2 Together these data define a rare subtype of BCP-ALL which could benefit from TKI therapy. Although the disease evaded treatment in the case presented herein, TKI in combination with chemotherapy rapidly decreased tumour burden. Notably, the same chemotherapy without TKI did not have any effect on the disease. We think the use of TKIs should be discussed as a first-line treatment in combination with standard chemotherapy in ABL1-rearranged BCP-ALL, with an objective of bone marrow transplantation. In routine practice, patients with BCP-ALL without recurrent cytogenetic or molecular abnormalities (also called “B-other”) should be studied rapidly, especially in adolescents and young adults or in poor responders to corticoids and/or standard chemotherapy. New trials should take into account BCR-ABL1-like ALL in order to evaluate the use of the different available TKIs and their optimal dose, and to assess whether adding TKIs to current therapy will improve the outcome of such patients. Interestingly, most of the NUP214-ABL1 fusions described in literature (in T-ALL, but also in the case of BCP-ALL from Roberts et al.2) involve ABL1 exon 2 with different exons of NUP214 (from exon 23 to 34).4,5 The case presented herein involves ABL1 exon 3. As a consequence, the fusion NUP214-ABL1 does not retain the SH3 domain involved in protein-protein interactions and the regulation of tyrosine kinase activity which could explain different cellular effects as well as special behavior.10
Footnotes
The online version of this letter has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts KG, Li Y, Payne-Turner D, et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N Engl J Med. 2014;371(11):1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruminy P, Marchand V, Buchbinder N, et al. Multiplexed targeted sequencing of recurrent fusion genes in acute leukaemia. Leukemia [Internet]. 2015. [cited 2015 Dec 9]; Available from: http://www.nature.com/doifinder/10.1038/leu.2015.177 [DOI] [PubMed]
- 4.Graux C, Cools J, Melotte C, et al. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet. 2004;36(10):1084–1089. [DOI] [PubMed] [Google Scholar]
- 5.De Braekeleer E, Douet-Guilbert N, Rowe D, et al. ABL1 fusion genes in hematological malignancies: a review: ABL1 fusion genes in leukemia. Eur J Haematol. 2011;86(5):361–371. [DOI] [PubMed] [Google Scholar]
- 6.De Keersmaecker K, Versele M, Cools J, et al. Intrinsic differences between the catalytic properties of the oncogenic NUP214-ABL1 and BCR-ABL1 fusion protein kinases. Leukemia. 2008;22(12):2208–2216. [DOI] [PubMed] [Google Scholar]
- 7.Deenik W, Beverloo HB, van der Poel-van de Luytgaarde SC, et al. Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia. 2009;23(3):627–629. [DOI] [PubMed] [Google Scholar]
- 8.Stergianou K, Fox C, Russell NH. Fusion of NUP214 to ABL1 on amplified episomes in T-ALL–implications for treatment. Leukemia. 2005;19(9):1680–1681. [DOI] [PubMed] [Google Scholar]
- 9.Lengline E, Beldjord K, Dombret H, et al. Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica. 2013;98(11): e146–e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith KM, Yacobi R, Van Etten RA. Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell. 2003;12(1):27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]