

Abstract
Classical Hodgkin lymphoma is curable in the majority of cases with chemotherapy and/or radiation. However, 15–20% of patients ultimately relapse and succumb to their disease. Pathologically, classical Hodgkin lymphoma is characterized by rare tumor-initiating Reed-Sternberg cells surrounded by a dense immune microenvironment. However, the role of the immune microenvironment, particularly T and B cells, in either promoting or restricting Classical Hodgkin lymphoma growth remains undefined. Recent dramatic clinical responses seen using monoclonal antibodies against PD-1, a cell surface receptor whose primary function is to restrict T cell activation, have reignited questions regarding the function of the adaptive immune system in classical Hodgkin lymphoma. This review summarizes what is known regarding T cells, B cells, and immune checkpoints in classical Hodgkin lymphoma.
Introduction
Nearly two hundred years after Thomas Hodgkin’s initial description of “morbid experiences of the absorbent glands and spleen”,1 the underlying pathophysiology of this eponymous disease remains highly enigmatic. While it has been established that the malignant Reed-Sternberg (RS) cells of classical Hodgkin lymphoma (CHL) are of B cell origin,2,3 these cells comprise only a small percentage of CHL tumor bulk while the remaining tumor microenvironment is rich in T cells, non-malignant B cells, granulocytes, eosinophils, and stromal cells. The contribution of the immune microenvironment to CHL pathogenesis remains incompletely defined; however, the recent success of novel treatments aimed at amplifying anti-tumor T cell responses suggests a potential therapeutic role for the immune system in this disease.4,5 This review will highlight both the relative contribution of non-malignant T and B cells to the pathogenesis and prognosis of CHL as well as the role of negative regulatory immune checkpoints in CHL pathophysiology and therapeutic potential.
T cells in CHL: friends or foes?
The role of non-malignant T cells in CHL pathogenesis and treatment remains poorly understood. T cells are thought to suppress the development and growth of lymphomas; the increased incidence of lymphomas in patients receiving long-term immunosuppressants as well as immunodeficient mice supports this hypothesis.6–8 The presence of multiple tumor-infiltrating T cells “rosetting,” but failing to eliminate, malignant RS cells has been well-described in CHL and is highly suggestive of an ineffectual T cell response in this disease.9,10 This has been complemented by the demonstration of impaired proliferative responses to mitogenic stimuli in peripheral blood lymphocytes isolated from CHL patients.11
What explains the impaired T cell responses seen in CHL¿ First, the T cells that accumulate within the CHL microenvironment are largely skewed towards differentiation into either Th2 cells or regulatory T cells (Tregs).12–15 This accumulation is driven by a combination of selective recruitment as well as intratumoral functional reprogramming.16 RS cells produce a variety of Th2 and Treg-selective chemoattractants, including CCL17/TARC,17 CCL22,18 CCL5,19,20 IL-4, IL-5, IL-10, and IL-13.15,21,22 Production of these chemoattractants is associated with inferior responses to therapy.23,24 Additionally, RS cells secrete factors known to induce functional reprogramming of tumor-infiltrating T cells into Th2 cells and Tregs, such as galectin-1,25–28 macrophage migration inhibitory factor29 and IL-7.30 Stromal cells within the CHL microenvironment also recruit immunosuppressive myeloid-derived suppressor cells and Tregs by secreting factors such as indoleamine 2,3 dioxygenase (IDO)31 (Figure 1A).
Figure 1.

Suppression of anti-tumor T cell responses by the CHL microenvironment. (A) RS cells and stromal cells secrete cytokines, chemokines, and other soluble immunomodulatory factors, such as IL-10, CCL17/TARC, galectin-1, and indoleamine 2,3-dioxygenase (IDO) which both recruit Th2 and regulatory CD4+ T cells and favor the differentiation of tumor-infiltrating T cells into regulatory and Th2 cells via the induction of lineage specific transcription factors Gata3 (Th2) and FoxP3 (Treg). (B) RS cells evade recognition by CD8+ and CD4+ T cells by downregulating expression of MHC-I and MHC-II in the majority of cases. They also express ligands that activate negative regulatory receptors present on T cells, such as PD-1. Conversely, RS cells are able to derive growth signals from CD40L, which is present on the majority of T cells within the microenvironment and activates CD40 on RS cells, driving NF-κB signaling and RS cell proliferation.
Second, effector T cells in CHL display features of chronic ineffectual antigen encounter, a phenomenon known as T cell “exhaustion” characterized by the upregulation of negative regulatory receptors such as the immunoglobulin superfamily member Programmed Death 1 (PD-1; CD279). PD-1 upregulation was initially characterized in models of chronic viral infection32,33 but is also seen in multiple lymphomas, including diffuse large B-cell lymphoma and follicular lymphoma.34,35 In CHL, the expression of PD-1 on T cells is likely driven by constitutive upregulation of its ligands, PD-L1 and PD-L2, on RS cells36 (Figure 1B). Accordingly, the presence of PD-1+ T cells, both in the microenvironment and in the peripheral blood, is a negative prognostic factor in CHL.37,38
Finally, impaired anti-tumor immunity in CHL may be due to an inability of T cells to recognize RS cells. RS cells frequently lack expression of MHC-I and MHC-II, which are required for antigen recognition by CD8+ and CD4+ T cells, respectively. This can occur secondary to mutations, such as in the β2M39 and CIITA40,41 genes, or via epigenetic mechanisms at the CIITA promoter leading to decreased transcription.42 While T cells in CHL are rendered incapable of mediating anti-tumor responses, there is some evidence to suggest that they may actually support RS cell growth and survival. CHL has been noted to develop during the immune response to active viral infections, such as acute Epstein-Barr virus mediated mononucleosis,43 and during immune reconstitution following the initiation of antiretroviral therapy in HIV+ patients.44 Mechanistically, T cells in CHL can promote RS cell survival and proliferation via CD40/CD40 ligand-mediated alternative activation of NF-κB;45 this growth signal may be particularly important for the survival of RS cells, which have lost the ability to activate NF-κB through conventional B cell receptor-driven signals.46–48 The multiple mechanisms by which RS cells and the CHL microenvironment suppress immune responses are summarized in Figure 1; therapies aimed at breaking this pathological cycle of T cell fueled growth and immune evasion, primarily via checkpoint blockade, are discussed below.
B cells: innocent bystanders or active participants?
Less is known regarding the role of non-malignant B cells in CHL pathogenesis and response to therapy as compared to T cells. Non-malignant B cells are prevalent in lymphocyte-predominant Hodgkin lymphoma (LP-HL), a biologically distinct disease in which the tumor-initiating cells also express CD20; this form of Hodgkin lymphoma is frequently monitored and, when requiring therapy, can be successfully treated with radiation alone or single agent rituximab.49,50 In CHL, non-malignant B cells are also generally present in the microenvironment, likely due to the normal predominance of B cells within a non-malignant lymph node. However, their role in facilitating CHL growth is less established. Non-malignant B cells can easily be distinguished from RS cells, which lose expression of classical B cell antigens including CD20, CD79a, and PAX-5 due to mutations and/or epigenetic silencing.51 The effect of B cells within the CHL microenvironment is also not well established; B cell production of IL-10 may suppress anti-tumor T cell responses;52,53 on the other hand, non-malignant B cells may compete with RS cells for T cell-derived survival signals such as CD40L, and in this way suppress RS cell growth. In support of the latter hypothesis, gene expression signatures consistent with non-malignant B cells are associated with improved outcomes in CHL, although this may simply reflect low CHL tumor burden within an otherwise healthy LN.54–56
Targeting B cells within the tumor microenvironment with rituximab has shown some clinical activity, with an overall response rate of 22% as a single agent regardless of RS cell CD20 expression.57 In a phase 2 study of rituximab plus ABVD (adriamycin, bleomycin, vinblastine, and dacarbazine) in newly diagnosed CHL, five-year event-free and overall survival rates of 83% and 96% compared favorably with historical controls treated with ABVD therapy alone.58 The reasons for rituximab efficacy in CHL are likely to be multifactorial. It has demonstrated benefit in a subset of patients whose RS cells express CD20.59 In the majority of CHL cases, which lack CD20 expression on RS cells, rituximab may deplete CHL precursor cells, which have a memory-like B cell phenotype and express CD20.60 In a phase 2 study of rituximab plus ABVD (R-ABVD) in untreated, advanced stage CHL, circulating CD20+ clonal B cells were found in 21 out of 25 assayed patients, and clearance of these precursor cells following treatment with R-ABVD was associated with a reduced risk of relapse as compared to patients in whom clonal CD20+ cells persisted.61 Ultimately, randomized controlled trials currently underway evaluating R-ABVD versus ABVD in unselected CHL patients with early stage (clinicaltrials.gov identifier: 00992030) and advanced stage (clinicaltrials.gov identifier: 00654732) disease will provide insight into the value of depleting CD20+ malignant and non-malignant B cells in CHL.
Immune checkpoints: breaks in the action
Broadly speaking, immune checkpoints are a diverse group of proteins whose function is to restrict physiologic immune cell responses in order to limit damage to host tissues. These include members of the immunoglobulin superfamily such as CTLA-4, PD-1, and LAG-3.62 The essential role for negative regulators of the immune response was first established by the diffuse systemic immune hyperactivation and multisystem organ failure seen in mice lacking CTLA-4.63,64 Increasingly, malignant co-opting of immune checkpoints has emerged as a mechanism by which tumor cells can subvert immune surveillance and anti-tumor immunity.
Targeting of immune checkpoints, particularly with the anti-PD-1 antibodies nivolumab and pembrolizumab, has resulted in dramatic clinical responses in CHL,4,5 although the mechanisms by which these drugs induce an anti-tumor effect remain somewhat enigmatic. Furthermore, PD-1 represents only one of multiple immune checkpoints, all of which can promote immune evasion in CHL and might be amenable to therapeutic blockade. The specifics of individual immune checkpoints and their potential for therapeutic intervention are discussed below.
PD-1
PD-1, a costimulatory molecule within the immunoglobulin superfamily of receptors, was first established as a negative regulator of T cell activation based on the presence of a cytoplasmic inhibitory tyrosine-based ITIM motif, as well as the development of a lupus-like autoimmune disease in PD-1 knockout mice.65 Subsequently, PD-1 was found to be present on many tumor-infiltrating lymphocytes (TILs),66 and its ligand is upregulated in a variety of human cancers.67 Checkpoint-mediated immune evasion was established as a hallmark of CHL pathogenesis with the identification of amplifications of the 9p24 locus resulting in constitutive expression of PD-L1 and PD-L2 in more than 85% of CHL patients.36 Even in patients without genetic amplifications of PD-L1 or PD-L2, physiologic upregulation of these ligands likely occurs downstream of JAK/STAT signaling, IFNγ production or, in EBV-associated cases of CHL, expression of the viral-associated protein LMP1.67,68
In solid tumors, PD-1 blockade acts by promoting T cell activation via a variety of mechanisms. PD-1 blockade reverses SHP-2-mediated dephosphorylation of the proximal T cell receptor-associated kinase ZAP-70, leading to increased T cell activation.69 Furthermore, PD-1 blockade increases the dwell time of T cells on antigen-presenting and target cells, increasing the opportunity for a T cell to encounter its cognate antigen and successfully initiate an anti-tumor response.70 Indeed, the blockade of PD-1 increases the sensitivity of T cells to foreign antigens and increases effector function and cytokine production of both CD4+ and CD8+ T cells in models of both tumor and virally mediated chronic T cell exhaustion.71,72 PD-1 is thought to tune T cells during the effector, rather than priming, phase of T cell antigen encounter. This likely underlies the lower incidence of off-target, autoimmune-like adverse events associated with anti-PD1 as compared to anti-CTLA-4 therapy. Indeed, PD-1 knockout mice have a relatively mild, organ-specific autoimmune phenotype,65 and clinical PD-1 blockade does not induce the activation of peripheral blood T cells.73
Clinically in CHL, the reversal of PD-1 mediated T cell suppression using blocking monoclonal antibodies has resulted in impressive and durable remissions in patients with highly refractory disease. Nivolumab, a human IgG4 monoclonal antibody, elicited an overall response rate (ORR) of 87% and complete response (CR) rate of 17% in 23 patients with relapsed and refractory CHL whose disease had progressed after or were ineligible for autologous stem cell transplant.4 Pembrolizumab, also an IgG4 monoclonal antibody to PD-1, had an ORR of 65% with 16% complete remissions in 31 patients, all of whom had progressed or were ineligible for autologous stem cell transplant and had progressed on brentuximab vedotin.5 The median duration of response was not reached during the short follow-up time of less than one year in either study; however, recent data suggests that the majority of remissions have been durable for longer than one year.74
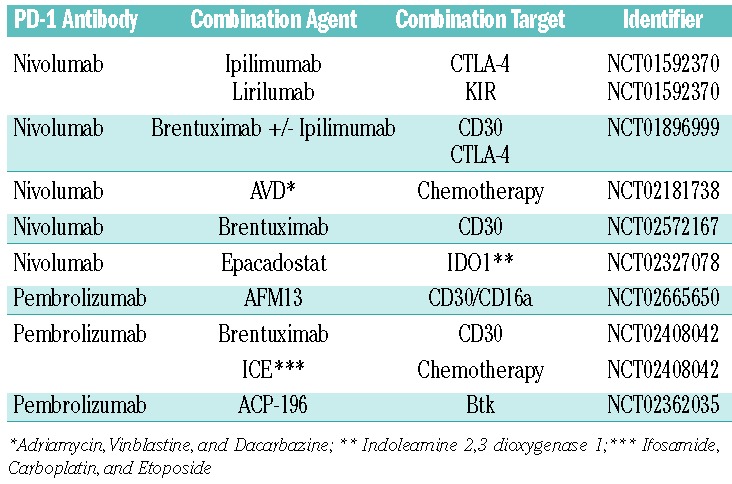
Objective biomarkers correlating with PD-1 response in CHL, however, have remained elusive. In some solid tumors, PD-L1 expression correlates with response to therapy,75–77 but this has not yet been demonstrated in CHL. Similarly, somatic mutation and neoantigen burden have been shown to correlate with anti-PD-1 response to therapy,78 but the mutational burden of CHL remains uncharacterized. The mechanism by which anti-PD-1 therapy promotes responses in CHL is likely to have implications in other types of lymphoma such as diffuse large B-cell lymphoma (DLBCL), in which PD-L1 expression on tumor cells was recently demonstrated to portend an adverse clinical outcome.79 Single agent studies of nivolumab and pembrolizumab in patients with relapsed/refractory disease (clinicaltrials.gov identifier: 02453594), in comparison with brentuximab vedotin (clinicaltrials.gov identifier: 02684292), as maintenance following autologous transplant (clinicaltrials.gov identifier: 02362997), and in relapsed patients following allogeneic transplant (clinicaltrials.gov identifier: 01822509) are currently underway. Single agent studies of antibodies targeting PD-L1 are also accuring patients (clinicaltrials.gov identifier: 01452334, clinicaltrials.gov identifier: 02603419). Finally, multiple trials combining PD-1 blockade with other checkpoint inhibitors, targeted agents, and chemotherapy are underway (Table 1). Currently, anti-PD-1 therapy has only been studied in highly refractory patients and has not yet been FDA approved for this indication. Furthermore, the role of anti-PD-1 therapy in untreated patients or those curable with autologous stem cell transplant (in which it is likely to be combined with chemotherapy) remains to be defined.
Table 1.
Clinical trials investigating combination strategies with checkpoint blockade in CHL.
CTLA-4
CTLA-4 was initially discovered as an additional member of the immunoglobulin superfamily involved in cell-cell interactions in 1987.80 Subsequently, CTLA-4 was shown to be a critical negative regulator of T cell activation based both on in vitro studies81,82 and in fatal lymphoproliferative disorders seen in mice lacking CTLA-4.64 The repression of immune responses by CTLA-4 occurs via a number of mechanisms. In effector T cells, CTLA-4 competes strongly with CD28 for effective costimulation by CD80/86, leading to impaired T cell costimulation and functional inactivation. CTLA-4 also impairs the “stop signal” initiated by T cells upon antigen encounter leading to impaired T cell activation.83 Finally, CTLA-4 induces trans-endocytosis of the costimulatory ligands CD80 and CD86, restricting opportunities for further T cell activation.84
Pre-clinical rationale for targeting CTLA-4 in CHL was seen shortly after CTLA-4 was characterized with histopathologic demonstrations of CTLA-4+ T cells infiltrating CHL tumors.85 The best evidence to support clinical activity of CTLA-4 blockade comes from a phase I trial of patients with malignancies progressing after allogeneic stem cell transplantation.86 Two complete remissions were seen out of 14 CHL patients treated in the study. A clinical trial of ipilimumab, nivolumab, or both in combination with brentuximab vedotin in patients with relapsed or refractory CHL is currently accruing patients (clinicaltrials.gov identifier: 01896999).
LAG-3
LAG-3 was discovered in 1990 and was initially reported to be a ligand for MHC-II.87,88 Subsequently it was determined that LAG-3, like PD-1, is upregulated on T cells during chronic antigen stimulation.89 LAG-3 suppresses CD4+ T cell expansion in response to antigen,90 and LAG-3 was found to be synergistic with CTLA-4 and PD-1 in mediating T cell suppression during chronic antigenic stimulation.91,92 Additionally, LAG-3 is important in promoting the function of regulatory T cells.93 As a result, antibodies to LAG-3 augment CD4+ T cell expansion94 and CD8+ T cell function95 while blocking peripheral Treg differentiation and function.96,97
In CHL, CD4+ T cells from patients with active disease were found to express significantly higher levels of LAG-3 as compared to patients in long-term remission, and expression of LAG-3 was associated with impaired T cell responses to EBV-associated viral antigens LMP1 and LMP2.12 Intriguingly, LAG-3 is also expressed on natural killer (NK) cells.98 Thus, LAG-3 upregulation may suppress antitumor immunity through effects on T cells, Tregs, and NK cells, and is an intriguing candidate for therapeutic targeting. Monoclonal antibodies to LAG-3 are currently in clinical development, with early phase studies demonstrating that a LAG-3 monoclonal antibody is well tolerated with objective responses both as a single agent and in combination with chemotherapy in solid tumors.99,100 Given the established synergy between LAG-3 and PD-1, both in double knockout mice101 and with dual blockade in mouse models,62 this may be an attractive target for combination therapy. A phase I study of the anti-LAG-3 antibody BMS-986016 is currently accruing patients (clinicaltrials.gov identifier: 02061761).
Checkpoint blockade in CHL: a mechanistic conundrum
While it is clear that checkpoint blockade produces clinical responses in the majority of CHL patients, the mechanism by which this occurs has not been fully characterized. As described above, checkpoint blockade enhances T cell activation by eliminating negative regulation of either T cell receptor signaling or positive costimulatory signals. In solid tumors, checkpoint blockade primarily augments CD8+ T cell responses to tumor antigens presented by MHC class I molecules on tumor cells. Correspondingly, anti-PD-1 activity correlates with the presence of CD8+ TILs at the invasive margin of the tumor.77 In the setting of checkpoint blockade, CD8+ T cells can recognize tumor antigens, including self-antigens for which T cell tolerance is incomplete, including those with restricted tissue expression, or tumor “neoantigens” produced by somatic mutations within tumor cells.102,103 Recent reports suggest that the somatic mutational and consequent neoantigen burden correlates with response to anti-CTLA-4 and anti-PD-1 therapy in mouse models78 as well as in patients with melanoma and non-small cell lung cancer,104,105 in which neoantigen-specific CD8+ T cell clonal expansion could be detected in the peripheral blood.
In CHL, however, there are multiple barriers to CD8+ T cell recognition of tumor antigens in the setting of checkpoint blockade. First, it is unclear whether the CHL somatic mutational burden generates sufficient neoantigens to drive anti-tumor responses. Median somatic mutational burdens vary widely across cancers,106 and correlate strongly with neoantigen burden. The mutational burden in CHL is not well established as sequencing efforts have thus far been hampered by the paucity of RS cells within CHL tumors, although this can be overcome by either flow cytometry or microdissection-based cell enrichment.39,107 Another intriguing option for assessment of mutation burden is via assessment of cell-free DNA, which can be detected in the serum of the majority of CHL patients,108 although it is not yet clear whether cell-free or circulating tumor DNA can be used for comprehensive whole exome sequencing. More importantly, the majority of CHL samples demonstrate a loss of beta-2 microglobulin, leading to an absence of MHC-I expression on RS cells.39 As CD8+ T cells require antigen presentation on MHC-I molecules for their effector function, they are highly unlikely to be the primary mediators of the anti-PD-1 response (Figure 2A).
Figure 2.

A model for anti-tumor immunity in the setting of checkpoint blockade. (A) In solid tumors, anti-tumor immunity is mediated primarily by CD8+ T cell responses that are amplified in the setting of PD-1 blockade. However, in CHL this is mitigated by downregulation of MHC-I in the majority of cases. (B) This may predispose RS cells to killing by NK cells, which also express PD-1. Similarly, RS cell downregulation of MHC-II may limit CD4+ T cell responses following checkpoint blockade, but CD4+ T cells can also be primed by other APCs within the CHL microenvironment that do express MHC-II. Additionally, checkpoint blockade may impair the immunosuppressive function of infiltrating regulatory T cells, increasing productive T cell activation.
It remains possible that CD4+ T cells could be major contributors to the anti-PD-1-mediated anti-tumor response in CHL. CD4+ T cells are able to mediate tumor rejection, both through the production of pro-inflammatory cytokines and via the recruitment and activation of innate effector cells, such as macrophages and NK cells. Both reversal of Th1 anergy and an increased IFNg-response signature are seen in in vitro models38 as well as in patients in response to anti-PD-1 therapy, suggesting that the amplification of effector CD4+ T cell responses may be important to the anti-PD-1 response. Whether CD4+ T cells exert anti-tumor immunity directly or through recruitment of innate effector cells has yet to be established. Arguing against a role of CD4+ T cells in mediating the anti-PD-1 response is the loss of MHC-II on RS cells in at least 40% of patients, and likely higher in patients with relapsed disease.40 In a minority of cases this likely results from gene fusions involving CIITA, a transactivator required for MHC-II synthesis.41 However, unlike CD8+ T cell function, which requires class I antigen presentation on tumor cells, CD4+ T cells could be primed in CHL by APCs in the microenvironment or draining lymph node, and so loss of MHC-II does not preclude a CD4+ T cell mediated effect in anti-PD-1 treated patients (Figure 2B). Furthermore, both class I and class II restricted neoantigens have been described with associated expansion of neoantigen-specific CD4+ as well as CD8+ T cells,109–111 suggesting that a neoantigen-specific CD4+ T cell response may be possible in CHL.
Checkpoint blockade may also induce anti-tumor responses in CHL in an effector T cell-independent fashion. PD-1 is expressed on NK cells as well as T cells,67,112 and PD-1 is upregulated on NK cells in models of chronic infection.113 PD-1 blockade may thus promote anti-tumor immunity by facilitating NK cell recognition of MHC-I deficient RS cells, and this effect has been seen in primary hematopoietic cancer cells114 (Figure 2A). Meanwhile, Tregs are actually activated by PD-1 ligand binding,115,116 suggesting that the suppression of Treg function may be another potential immunomodulatory effect of anti-PD-1 therapy (Figure 2B).
Finally, blockade of the PD-1/PD-L1 interaction may have cell autonomous effects on tumor growth, as suggested by a recent study demonstrating that blockade of PD-L1 reduces glucose consumption by tumors. This blockade simultaneously inhibits tumor cell growth and increases extracellular glucose availability permitting T cell activation, proliferation, and cytokine production.117
The lack of a defined mechanism of action for checkpoint blockade in Hodgkin lymphoma has resulted in the lack of biomarkers predicting response to therapy. Expression of PD-L1 is unlikely to predict response, as it is amplified in the overwhelming majority of patients treated with checkpoint inhibitors. A recent analysis of peripheral blood from patients treated with the anti-PD-1 antibody pembrolizumab demonstrated an increase in the absolute number of CD4+, CD8+, and NK cells with parallel gene expression profiles demonstrating an increased IFNg response signature,118 but whether these changes correlate with treatment response has not been established. Future investigations into the mechanism of response to checkpoint blockade should focus both on evaluating the extent to which known immunosuppressive features of RS cells and the CHL microenvironment affect response to checkpoint blockade, as well as identifying the effector cells responsible for mediating this response. These studies would include assessment of tumor mutational and neoantigen burden, MHC-I and MHC-II expression, intratumoral effector and regulatory T cells, and development of clonal CD4+ and CD8+ T cell responses in response to therapy (Table 2).
Table 2.
Potential biomarkers under investigation to predict response to checkpoint blockade in CHL.

Towards rational combination strategies in Hodgkin lymphoma
Despite the encouraging clinical responses seen with checkpoint blockade, and particularly with anti-PD-1 therapy, complete remissions to immunotherapy remain rare, with only 15–20% of patients achieving a complete remission to PD-1 blockade.5,74 This may be due to a variety of factors, both on RS cells and within the tumor microenvironment. Effective anti-tumor immune responses may not be feasible in the setting of restricted antigen expression, either due to epigenetic silencing or downregulation of antigen presentation machinery. Additionally, tumor-infiltrating Tregs and immunosuppressive tumor-associated macrophages may effectively negate anti-tumor responses even in the presence of checkpoint blockade.
Rational combination strategies may help to overcome these limitations and provide sustained remissions. Combinations of checkpoint inhibitors, including PD-1 and CTLA-4 blockade, are part of ongoing active clinical trials (clinicaltrials.gov identifier: 01896999, clinicaltrials.gov identifier: 01592370, clinicaltrials.gov identifier: 01592370). Combining checkpoint blockade with agonist antibodies against costimulatory molecules present on T cells, such as OX40 and 4–1BB, represents an intriguing strategy to overcome multiple mechanisms of immunosuppression known to be present within the CHL microenvironment, and agonist antibodies against OX40 and 4–1BB are currently being investigated in active clinical trials (clinicaltrials.gov identifier: 02205333, clinicaltrials.gov identifier: 01644968, clinicaltrials.gov identifier: 02253992, clinicaltrials.gov identifier: 01775631).119
An additional candidate for combination therapy with checkpoint blockade is the family of chromatin-modifying agents, including hypomethylating agents and histone deacetylase (HDAC) inhibitors. These agents mediate direct apoptosis of CHL cell lines in in vitro studies but have additional effects that may cooperate with checkpoint blockade to increase antitumor immunity. Hypomethylating agents may increase tumor antigen expression, leading to more diverse antigen-specific responses that can prevent immune escape.120 HDAC inhibition also suppresses RS production of multiple cytokines and chemokines favoring Th2 cell recruitment and differentiation. For example, the treatment of CHL cell lines with vorinostat was shown to reduce STAT-mediated production of Th2 polarizing cytokines IL-5, IL-10 and IL-13 as well as the Th2 recruiting chemokine TARC.121 These findings were paralleled in phase 2 studies of mocetinostat and panobinostat, in which treatment-induced decreases in TARC correlated with reductions in tumor burden and progression-free survival.122,123 HDAC inhibition can also reinvigorate exhausted T cells in CHL by upregulating OX40 on RS cells124 and by downregulating PD-1 expression on CD4+ and CD8+ T cells.125 Finally, HDAC inhibition may selectively deplete Tregs by suppressing FoxP3 expression and depleting intratumoral accumulation of myeloid-derived suppressor cells.126,127 The multiple pleiotropic effects of HDAC inhibition may collectively tip the balance towards deeper responses to checkpoint blockade.
Future directions
CHL remains an enigmatic disease in which components of the microenvironment, including T and B cells, may help feed or extinguish RS cell growth. The advent of checkpoint blockade has provided dramatic, durable clinical responses even in highly refractory cases, but many questions remain. What are the ultimate roles for T and B cell subsets in promoting and restricting CHL growth¿ What are the dominant immune checkpoints in suppressing antitumor immunity in CHL¿ Which immune cells serve as the primary effectors for checkpoint blockade therapy¿ The answers to these questions will undoubtedly lay the groundwork for rational combination strategies and hopefully result in an increased cure rate in this disease.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/7/794
References
- 1.Hodgkin. On some Morbid Appearances of the Absorbent Glands and Spleen. Med Chir Trans. 1832;17:68–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanzler H, Kuppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med. 1996;184(4):1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci USA. 1994;91(23):10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moskowitz CH, Ribrag V, Michot JM, et al. PD-1 Blockade with the Monoclonal Antibody Pembrolizumab (MK-3475) in Patients with Classical Hodgkin Lymphoma after Brentuximab Vedotin Failure: Preliminary Results from a Phase 1b Study (KEYNOTE-013). Blood. 2014;124(21):290 abstr. [Google Scholar]
- 6.Huang P, Westmoreland SV, Jain RK, Fukumura D. Spontaneous nonthymic tumors in SCID mice. Comp Med. 2011;61(3):227–234. [PMC free article] [PubMed] [Google Scholar]
- 7.Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. 2000;356(9244): 1795–1799. [DOI] [PubMed] [Google Scholar]
- 8.Penn I, Starzl TE. Immunosuppression and cancer. Transplant Proc. 1973;5(1):943–947. [PMC free article] [PubMed] [Google Scholar]
- 9.Payne SV, Jones DB, Wright DH. Reed-Sternberg-cell/lymphocyte interaction. Lancet. 1977;2(8041):768–769. [DOI] [PubMed] [Google Scholar]
- 10.Stuart AE, Williams AR, Habeshaw JA. Rosetting and other reactions of the Reed-Sternberg cell. J Pathol. 1977;122(2):81–90. [DOI] [PubMed] [Google Scholar]
- 11.Levy R, Kaplan HS. Impaired lymphocyte function in untreated Hodgkin’s disease. The N Engl J Med. 1974;290(4):181–186. [DOI] [PubMed] [Google Scholar]
- 12.Gandhi MK, Lambley E, Duraiswamy J, et al. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific CD8+ T-cell function in Hodgkin lymphoma patients. Blood. 2006;108(7):2280–2289. [DOI] [PubMed] [Google Scholar]
- 13.Ishida T, Ishii T, Inagaki A, et al. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res. 2006;66(11):5716–5722. [DOI] [PubMed] [Google Scholar]
- 14.Marshall NA, Christie LE, Munro LR, et al. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood. 2004;103(5): 1755–1762. [DOI] [PubMed] [Google Scholar]
- 15.Re D, Kuppers R, Diehl V. Molecular pathogenesis of Hodgkin’s lymphoma. J Clin Oncol. 2005;23(26):6379–6386. [DOI] [PubMed] [Google Scholar]
- 16.Tanijiri T, Shimizu T, Uehira K, et al. Hodgkin’s reed-sternberg cell line (KM-H2) promotes a bidirectional differentiation of CD4+CD25+Foxp3+ T cells and CD4+ cytotoxic T lymphocytes from CD4+ naive T cells. J Leukoc Biol. 2007;82(3):576–584. [DOI] [PubMed] [Google Scholar]
- 17.van den Berg A, Visser L, Poppema S. High expression of the CC chemokine TARC in Reed-Sternberg cells. A possible explanation for the characteristic T-cell infiltratein Hodgkin’s lymphoma. Am J Pathol. 1999;154(6):1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuppers R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009;9(1):15–27. [DOI] [PubMed] [Google Scholar]
- 19.Aldinucci D, Lorenzon D, Cattaruzza L, et al. Expression of CCR5 receptors on Reed-Sternberg cells and Hodgkin lymphoma cell lines: involvement of CCL5/Rantes in tumor cell growth and microenvironmental interactions. Int J Cancer. 2008;122(4):769–776. [DOI] [PubMed] [Google Scholar]
- 20.Fischer M, Juremalm M, Olsson N, et al. Expression of CCL5/RANTES by Hodgkin and Reed-Sternberg cells and its possible role in the recruitment of mast cells into lymphomatous tissue. Int J Cancer. 2003;107(2):197–201. [DOI] [PubMed] [Google Scholar]
- 21.Kapp U, Yeh WC, Patterson B, et al. Interleukin 13 is secreted by and stimulates the growth of Hodgkin and Reed-Sternberg cells. J Exp Med. 1999;189(12):1939–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skinnider BF, Mak TW. The role of cytokines in classical Hodgkin lymphoma. Blood. 2002;99(12):4283–4297. [DOI] [PubMed] [Google Scholar]
- 23.Plattel WJ, van den Berg A, Visser L, et al. Plasma thymus and activation-regulated chemokine as an early response marker in classical Hodgkin’s lymphoma. Haematologica. 2012;97(3):410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sauer M, Plutschow A, Jachimowicz RD, et al. Baseline serum TARC levels predict therapy outcome in patients with Hodgkin lymphoma. Am J Hematol. 2013;88(2):113–115. [DOI] [PubMed] [Google Scholar]
- 25.Kamper P, Ludvigsen M, Bendix K, et al. Proteomic analysis identifies galectin-1 as a predictive biomarker for relapsed/refractory disease in classical Hodgkin lymphoma. Blood. 2011;117(24):6638–6649. [DOI] [PubMed] [Google Scholar]
- 26.Ouyang J, Plutschow A, Pogge von Strandmann E, et al. Galectin-1 serum levels reflect tumor burden and adverse clinical features in classical Hodgkin lymphoma. Blood. 2013;121(17):3431–3433. [DOI] [PubMed] [Google Scholar]
- 27.Rodig SJ, Ouyang J, Juszczynski P, et al. AP1-dependent galectin-1 expression delineates classical hodgkin and anaplastic large cell lymphomas from other lymphoid malignancies with shared molecular features. Clin Cancer Res. 2008;14(11):3338–3344. [DOI] [PubMed] [Google Scholar]
- 28.Toscano MA, Bianco GA, Ilarregui JM, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8(8):825–834. [DOI] [PubMed] [Google Scholar]
- 29.Yaddanapudi K, Putty K, Rendon BE, et al. Control of tumor-associated macrophage alternative activation by macrophage migration inhibitory factor. J Immunol. 2013;190 (6):2984–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cattaruzza L, Gloghini A, Olivo K, et al. Functional coexpression of Interleukin (IL)-7 and its receptor (IL-7R) on Hodgkin and Reed-Sternberg cells: Involvement of IL-7 in tumor cell growth and microenvironmental interactions of Hodgkin’s lymphoma. Int J Cancer. 2009;125(5):1092–1101. [DOI] [PubMed] [Google Scholar]
- 31.Holmgaard RB, Zamarin D, Li Y, et al. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015;13(2):412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallimore A, Glithero A, Godkin A, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187(9):1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zajac AJ, Blattman JN, Murali-Krishna K, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188(12):2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen BJ, Chapuy B, Ouyang J, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. 2013;19(13):3462–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myklebust JH, Irish JM, Brody J, et al. High PD-1 expression and suppressed cytokine signaling distinguish T cells infiltrating follicular lymphoma tumors from peripheral T cells. Blood. 2013;121(8):1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116(17):3268–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chemnitz JM, Eggle D, Driesen J, et al. RNA fingerprints provide direct evidence for the inhibitory role of TGFbeta and PD-1 on CD4+ T cells in Hodgkin lymphoma. Blood. 2007;110(9):3226–3233. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto R, Nishikori M, Kitawaki T, et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood. 2008;111(6): 3220–3224. [DOI] [PubMed] [Google Scholar]
- 39.Reichel J, Chadburn A, Rubinstein PG, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood. 2015;125(7): 1061–1072. [DOI] [PubMed] [Google Scholar]
- 40.Diepstra A, van Imhoff GW, Karim-Kos HE, et al. HLA class II expression by Hodgkin Reed-Sternberg cells is an independent prognostic factor in classical Hodgkin’s lymphoma. J Clin Oncol. 2007;25(21):3101–3108. [DOI] [PubMed] [Google Scholar]
- 41.Steidl C, Shah SP, Woolcock BW, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471(7338):377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cycon KA, Mulvaney K, Rimsza LM, Persky D, Murphy SP. Histone deacetylase inhibitors activate CIITA and MHC class II antigen expression in diffuse large B-cell lymphoma. Immunology. 2013;140(2):259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Massey FC, Lane LL, Inbriglia JE. Acute infectious mononucleosis and Hodgkin’s disease occurring simultaneously in the same patient. J Am Med Assoc. 1953;151 (12):994–995. [DOI] [PubMed] [Google Scholar]
- 44.Biggar RJ, Jaffe ES, Goedert JJ, Chaturvedi A, Pfeiffer R, Engels EA. Hodgkin lymphoma and immunodeficiency in persons with HIV/AIDS. Blood. 2006;108(12):3786–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Sattarzadeh A, Diepstra A, Visser L, van den Berg A. The microenvironment in classical Hodgkin lymphoma: an actively shaped and essential tumor component. Semin Cancer Biol. 2014;24:15–22. [DOI] [PubMed] [Google Scholar]
- 46.Carbone A, Gloghini A, Gruss HJ, Pinto A. CD40 antigen expression on Reed-Sternberg cells. A reliable diagnostic tool for Hodgkin’s disease. Am J Pathol. 1995;146(3):780–781. [PMC free article] [PubMed] [Google Scholar]
- 47.Greaves P, Clear A, Owen A, et al. Defining characteristics of classical Hodgkin lymphoma microenvironment T-helper cells. Blood. 2013;122(16):2856–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gruss HJ, Hirschstein D, Wright B, et al. Expression and function of CD40 on Hodgkin and Reed-Sternberg cells and the possible relevance for Hodgkin’s disease. Blood. 1994;84(7):2305–2314. [PubMed] [Google Scholar]
- 49.Advani RH, Horning SJ, Hoppe RT, et al. Mature results of a phase II study of rituximab therapy for nodular lymphocyte-predominant Hodgkin lymphoma. J Clin Oncol. 2014;32(9):912–918. [DOI] [PubMed] [Google Scholar]
- 50.Parikh RR, Grossbard ML, Harrison LB, Yahalom J. Early-stage nodular lymphocyte-predominant Hodgkin lymphoma: the impact of radiotherapy on overall survival. Leuk Lymphoma. 2015. October 2:1–8. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ushmorov A, Leithauser F, Sakk O, et al. Epigenetic processes play a major role in B-cell-specific gene silencing in classical Hodgkin lymphoma. Blood. 2006;107(6): 2493–2500. [DOI] [PubMed] [Google Scholar]
- 52.Inoue S, Leitner WW, Golding B, Scott D. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006;66(15):7741–7747. [DOI] [PubMed] [Google Scholar]
- 53.Vassilakopoulos TP, Nadali G, Angelopoulou MK, et al. Serum interleukin-10 levels are an independent prognostic factor for patients with Hodgkin’s lymphoma. Haematologica. 2001;86(3):274–281. [PubMed] [Google Scholar]
- 54.Chetaille B, Bertucci F, Finetti P, et al. Molecular profiling of classical Hodgkin lymphoma tissues uncovers variations in the tumor microenvironment and correlations with EBV infection and outcome. Blood. 2009;113(12):2765–3775. [DOI] [PubMed] [Google Scholar]
- 55.Greaves P, Clear A, Coutinho R, et al. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of classical Hodgkin lymphoma is predictive of outcome. J Clin Oncol. 2013;31(2):256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steidl C, Lee T, Shah SP, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362(10):875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Younes A, Romaguera J, Hagemeister F, et al. A pilot study of rituximab in patients with recurrent, classic Hodgkin disease. Cancer. 2003;98(2):310–314. [DOI] [PubMed] [Google Scholar]
- 58.Younes A, Oki Y, McLaughlin P, et al. Phase 2 study of rituximab plus ABVD in patients with newly diagnosed classical Hodgkin lymphoma. Blood. 2012;119(18):4123–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rassidakis GZ, Medeiros LJ, Viviani S, et al. CD20 expression in Hodgkin and Reed-Sternberg cells of classical Hodgkin’s disease: associations with presenting features and clinical outcome. J Clin Oncol. 2002;20(5): 1278–1287. [DOI] [PubMed] [Google Scholar]
- 60.Jones RJ, Gocke CD, Kasamon YL, et al. Circulating clonotypic B cells in classic Hodgkin lymphoma. Blood. 2009;113(23): 5920–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kasamon YL, Jacene HA, Gocke CD, et al. Phase 2 study of rituximab-ABVD in classical Hodgkin lymphoma. Blood. 2012;119(18):4129–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wherry EJ, Ha SJ, Kaech SM, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–684. [DOI] [PubMed] [Google Scholar]
- 63.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–547. [DOI] [PubMed] [Google Scholar]
- 64.Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–988. [DOI] [PubMed] [Google Scholar]
- 65.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–151. [DOI] [PubMed] [Google Scholar]
- 66.Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114(8):1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. [DOI] [PubMed] [Google Scholar]
- 68.Green MR, Rodig S, Juszczynski P, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. 2012;18(6):1611–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Honda T, Egen JG, Lammermann T, Kastenmuller W, Torabi-Parizi P, Germain RN. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity. 2014;40(2):235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. [DOI] [PubMed] [Google Scholar]
- 72.Wei F, Zhong S, Ma Z, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci USA. 2013;110(27):E2480–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ansell S, Armand P, Timmerman JM, et al. Nivolumab in Patients (Pts) with Relapsed or Refractory Classical Hodgkin Lymphoma (R/R cHL): Clinical Outcomes from Extended Follow-up of a Phase 1 Study (CA209–039). Blood. 2015;126(23):583 abstr. [Google Scholar]
- 75.Herbst H, Foss HD, Samol J, et al. Frequent expression of interleukin-10 by Epstein-Barr virus-harboring tumor cells of Hodgkin’s disease. Blood. 1996;87(7):2918–2929. [PubMed] [Google Scholar]
- 76.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kiyasu J, Miyoshi H, Hirata A, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood. 2015;126(19):2193–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature. 1987;328(6127):267–270. [DOI] [PubMed] [Google Scholar]
- 81.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5): 405–413. [DOI] [PubMed] [Google Scholar]
- 83.Schneider H, Downey J, Smith A, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313(5795):1972–1975. [DOI] [PubMed] [Google Scholar]
- 84.Qureshi OS, Zheng Y, Nakamura K, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vandenborre K, Delabie J, Boogaerts MA, et al. Human CTLA-4 is expressed in situ on T lymphocytes in germinal centers, in cutaneous graft-versus-host disease, and in Hodgkin’s disease. Am J Pathol. 1998;152(4): 963–973. [PMC free article] [PubMed] [Google Scholar]
- 86.Bashey A, Medina B, Corringham S, et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood. 2009;113(7):1581–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baixeras E, Huard B, Miossec C, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. 1992;176(2):327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huard B, Mastrangeli R, Prigent P, et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc Natl Acad Sci USA. 1997;94(11):5744–5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blackburn SD, Shin H, Haining WN, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1): 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hannier S, Tournier M, Bismuth G, Triebel F. CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J Immunol. 1998;161(8):4058–4065. [PubMed] [Google Scholar]
- 91.Okazaki T, Okazaki IM, Wang J, et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med. 2011;208(2):395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. 2004;172(9):5450–5455. [DOI] [PubMed] [Google Scholar]
- 93.Huang CT, Workman CJ, Flies D, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21(4):503–513. [DOI] [PubMed] [Google Scholar]
- 94.Macon-Lemaitre L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology. 2005;115(2): 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Grosso JF, Kelleher CC, Harris TJ, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. 2007;117(11):3383–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Durham NM, Nirschl CJ, Jackson CM, et al. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PloS one. 2014;9(11): e109080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sega EI, Leveson-Gower DB, Florek M, Schneidawind D, Luong RH, Negrin RS. Role of lymphocyte activation gene-3 (Lag-3) in conventional and regulatory T cell function in allogeneic transplantation. PloS one. 2014;9(1):e86551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Triebel F, Jitsukawa S, Baixeras E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171(5): 1393–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brignone C, Escudier B, Grygar C, Marcu M, Triebel F. A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clin Cancer Res. 2009;15(19):6225–6231. [DOI] [PubMed] [Google Scholar]
- 100.Brignone C, Gutierrez M, Mefti F, et al. First-line chemoimmunotherapy in metastatic breast carcinoma: combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J Transl Med. 2010;8:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Castle JC, Kreiter S, Diekmann J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–1091. [DOI] [PubMed] [Google Scholar]
- 103.Matsushita H, Vesely MD, Koboldt DC, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348 (6230):124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gunawardana J, Chan FC, Telenius A, et al. Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet. 2014; 46(4):329–335. [DOI] [PubMed] [Google Scholar]
- 108.Oki Y, Neelapu SS, Fanale M, et al. Sequence Level Analysis of Hodgkin Lymphoma Clonotypes Detected in Peripheral Blood Using a Next-Generation Sequencing Approach. Blood. 2014;124(21):1610 abstr.25030064 [Google Scholar]
- 109.Linnemann C, van Buuren MM, Bies L, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21(1):81–85. [DOI] [PubMed] [Google Scholar]
- 110.Robbins PF, Lu YC, El-Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.van Rooij N, van Buuren MM, Philips D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Benson DM, Jr, Bakan CE, Mishra A, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116(13):2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Norris S, Coleman A, Kuri-Cervantes L, Bower M, Nelson M, Goodier MR. PD-1 expression on natural killer cells and CD8(+) T cells during chronic HIV-1 infection. Viral Immunol. 2012;25(4):329–332. [DOI] [PubMed] [Google Scholar]
- 114.Bellucci R, Martin A, Bommarito D, et al. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015;4(6):e1008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Park HJ, Park JS, Jeong YH, et al. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J Immunol. 2015;194(12):5801–5811. [DOI] [PubMed] [Google Scholar]
- 116.Yogev N, Frommer F, Lukas D, et al. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity. 2012;37(2):264–275. [DOI] [PubMed] [Google Scholar]
- 117.Chang CH, Qiu J, O’Sullivan D, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162(6):1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Armand P, Shipp MA, Ribrag V, et al. PD-1 Blockade with Pembrolizumab in Patients with Classical Hodgkin Lymphoma after Brentuximab Vedotin Failure: Safety, Efficacy, and Biomarker Assessment. Blood. 2015;126(23): [Google Scholar]
- 119.Batlevi CL, Matsuki E, Brentjens RJ, Younes A. Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol. 2016;13(1):25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cruz CR, Gerdemann U, Leen AM, et al. Improving T-cell therapy for relapsed EBV-negative Hodgkin lymphoma by targeting upregulated MAGE-A4. Clin Cancer Res. 2011;17(22):7058–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Buglio D, Georgakis GV, Hanabuchi S, et al. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood. 2008;112(4):1424–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Younes A, Oki Y, Bociek RG, et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: an open-label, single-arm, phase 2 trial. Lancet Oncol. 2011;12(13):1222–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Younes A, Sureda A, Ben-Yehuda D, et al. Panobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol. 2012;30(18):2197–2203. [DOI] [PubMed] [Google Scholar]
- 124.Buglio D, Khaskhely NM, Voo KS, Martinez-Valdez H, Liu YJ, Younes A. HDAC11 plays an essential role in regulating OX40 ligand expression in Hodgkin lymphoma. Blood. 2011;117(10):2910–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Oki Y, Buglio D, Zhang J, et al. Immune regulatory effects of panobinostat in patients with Hodgkin lymphoma through modulation of serum cytokine levels and T-cell PD1 expression. Blood Cancer J. 2014;4:e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim K, Skora AD, Li Z, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111(32):11774–11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shen L, Ciesielski M, Ramakrishnan S, et al. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PloS one. 2012;7(1): e30815. [DOI] [PMC free article] [PubMed] [Google Scholar]