

Abstract
Action potential initiation and propagation in myelinated axons require ion channel clustering at axon initial segments (AIS) and nodes of Ranvier. Disruption of these domains after injury impairs nervous system function. Traditionally, injured CNS axons are considered refractory to regeneration, but some recent approaches challenge this view by showing robust long-distance regeneration. However, whether these approaches allow remyelination and promote the reestablishment of AIS and nodes of Ranvier is unknown. Using mouse optic nerve crush as a model for CNS traumatic injury, we performed a detailed analysis of AIS and node disruption after nerve crush. We found significant disruption of AIS and loss of nodes within days of the crush, and complete loss of nodes 1 week after injury. Genetic deletion of the tumor suppressor phosphatase and tensin homolog (Pten) in retinal ganglion cells (RGCs), coupled with stimulation of RGCs by inflammation and cAMP, dramatically enhanced regeneration. With this treatment, we found significant reestablishment of RGC AIS, remyelination, and even reassembly of nodes in regions proximal, within, and distal to the crush site. Remyelination began near the retina, progressed distally, and was confirmed by electron microscopy. Although axons grew rapidly, remyelination and nodal ion channel clustering was much slower. Finally, genetic deletion of ankyrinG from RGCs to block AIS reassembly did not affect axon regeneration, indicating that preservation of neuronal polarity is not required for axon regeneration. Together, our results demonstrate, for the first time, that regenerating CNS axons can be remyelinated and reassemble new AIS and nodes of Ranvier.
SIGNIFICANCE STATEMENT We show, for the first time, that regenerated CNS axons have the capacity to both remyelinate and reassemble the axon initial segments and nodes of Ranvier necessary for rapid and efficient action potential propagation.
Keywords: axon, ion channel, myelin, regeneration
Introduction
Axons of the mammalian CNS fail to regenerate after traumatic injury due to disruption of intrinsic growth pathways and the presence of growth inhibitory molecules in the CNS environment (Park et al., 2010; Cregg et al., 2014; Schwab and Strittmatter, 2014). Nevertheless, recent studies have uncovered several pathways whose manipulation can promote significant CNS axon regeneration. For example, inflammation in the vitreous of the eye or knock-out of growth inhibitory genes in retinal ganglion cells (RGCs) increases RGC survival and induces axon regeneration through the optic nerve after a crush injury. More specifically, activation of neutrophils by intravitreal injection of zymosan or recruitment of resident and nonresident myeloid cells after intravitreal injection of β(1,3) glucan, a dectin-1 ligand, induces axon regeneration past the injury site (Yin et al., 2003; Kurimoto et al., 2013; Baldwin et al., 2015). Alternatively, deletion of phosphatase and tensin homolog (PTEN) or SOCS3 increases RGC survival and promotes axon regeneration in the optic nerve (Park et al., 2008; Smith et al., 2009; Sun et al., 2011). Remarkably, combinatorial approaches, such as pairing the knock-out of PTEN with intravitreal administration of zymosan and cAMP analogs, or codeletion of both PTEN and SOCS3 can synergistically increase axon regeneration (Sun et al., 2011; de Lima et al., 2012; Kurimoto et al., 2013).
Despite several indications of simple physiological responses after CNS regeneration (de Lima et al., 2012; Jin et al., 2015; Li et al., 2015; Bei et al., 2016), full restoration of function requires remyelination and the presence of the excitable domains (e.g., axon initial segments [AISs] and nodes of Ranvier) necessary for the initiation and rapid propagation of action potentials. We previously showed that optic nerve crush disrupts the AIS in RGCs due to proteolysis of the ankyrinG- (ankG) and βIV spectrin-based AIS cytoskeleton (Schafer et al., 2009); loss of these cytoskeletal proteins disrupts AIS Na+ channel clustering and neuronal polarity (Hedstrom et al., 2008; Sobotzik et al., 2009).
Because it is unclear whether regenerating CNS axons reestablish AIS or whether they can be remyelinated and reassemble nodes of Ranvier, we used optic nerve crush as a model of traumatic CNS injury together with a combinatorial approach for axon regeneration to answer this question. Although optic nerve crush resulted in loss of both AISs and nodes of Ranvier throughout the optic nerve, we found that regenerating axons can rebuild their AIS, be remyelinated, and reassemble nodes of Ranvier. However, the process is very protracted compared with development, begins near the retina, and is less efficient with increasing distance from RGCs. Furthermore, using a conditional knock-out mouse, we found that RGCs do not require ankG to regenerate their axons, suggesting that reassembly of the AIS and maintenance of the initial neuronal polarity are not a prerequisite for axon regeneration. Together, our results provide the first definitive evidence that regenerating CNS axons can become remyelinated and reassemble the functional domains necessary for rapid and efficient action potential propagation.
Materials and Methods
Animals.
C57BL/6J mice were purchased from The Jackson Laboratory. Ank3f/f mice were described previously (Ho et al., 2014). These animals were housed at Baylor College of Medicine and given access to food and drink ad libitum. Ptenf/f mice were described previously (Park et al., 2008). These animals were housed at the Boston Children's Hospital and given access to food and water ad libitum. All surgical and experimental procedures were conducted in accordance with protocols approved by the Baylor College of Medicine Institutional Animal Care and Use Committees, the Boston Children's Hospital Animal Care and Use Committees, and the National Institutes of Health Guide for the care and use of laboratory animals. For crush experiments examining degeneration, we used only male mice. For regeneration experiments using Ptenf/f and Ank3f/f mice, we used a combination of male and female mice.
Axon regeneration.
Unless otherwise stated, all experiments involving regeneration (both treatment and saline-injected controls) were performed using Ptenf/f mice. To delete the Pten gene, Ptenf/f mice received an intravitreal injection of an adeno-associated virus (Vector Biolabs; AAV2, 1012 GC/ml) expressing Cre recombinase. Two weeks after virus injection, mice were subjected to an optic nerve crush for 5 s and, to augment axon regeneration, received an intravitreal injection of zymosan (Sigma; 12.5 μg/μl, sterilized before use) along with the cAMP analog CPT-cAMP (Sigma; 50 μm, 3 μl) immediately afterward. For longer survival time points (6 and 12 weeks), animals were given an additional dose of zymosan at half the original concentration plus CPT-cAMP at the original concentration into the vitreous every 3 weeks. To delete the ANK3 gene (corresponding to the protein ankG), Ank3f/f mice were administered an intravitreal injection of an adeno-associated virus (Vector Biolabs; AAV2, 1013 GC/ml, 1:2 dilution in sterilized 1× PBS, 2 μl) expressing either GFP alone (control) or Cre recombinase and GFP. Four weeks after injection of AAV, mice were subjected to optic nerve crush. To facilitate axon regeneration in Ank3f/f mice, immediately after injury, mice were given an intravitreal injection of Curdlan, a particulate β(1,3)-glucan (Wako Chemicals; 25 μg/μl). Preparation of Curdlan was previously described (Baldwin et al., 2015).
Immunofluorescence labeling.
Immunostaining was performed on 12- or 16-μm-thick optic nerve sections and free-floating flat-mount retinas. Animals were killed with an overdose of isoflurane. Optic nerves and flat-mounted whole retinas were immediately removed and placed in 4% PFA for 1 h at 4°C. Optic nerves were transferred to 20% sucrose in 0.1 m PB and left overnight at 4°C followed by 30% sucrose overnight at 4°C. Optic nerves were then embedded in OCT and stored at −80°C. Optic nerves were sectioned on a Cryostar NX70. Sections were placed on 1% bovine gelatin-coated coverslips and, after drying, were washed in 0.1 m PB for 5 min at room temperature, and blocked at room temperature with 0.1 m PB, 0.3% Triton X-100, and 10% goat serum (PBTGS) for 1 h. Primary antibodies (excluding GAP-43) were diluted in PBTGS and placed on sections overnight at room temperature. Sections were washed three times in PBTGS. Secondary antibodies were diluted in PBTGS and placed on sections for 2 h at room temperature. Sections were then washed once with PBTGS, twice in 0.1 m PB, air dried, and mounted in mounting medium (KPL) for visualization. GAP-43 immunostaining was performed on 16-μm-thick longitudinal sections of optic nerve mounted on 1% bovine gelatin-coated coverslips. Sections were washed in 1× TBS for 5 min at room temperature followed by immersion in methanol for 10 min. Blocking was done at room temperature with 1× TBS and 10% donkey serum. Primary antibodies were diluted in TBS2T, 5% donkey serum, and 2% BSA (solution A) overnight at 4°C. Sections were then washed in TBS2T for 1 h at 4°, followed by solution A for 1.5 h at room temperature and TBS2T for 1 h at room temperature. Secondary antibody was diluted in solution A and placed on sections for 2 h at room temperature. Sections were washed twice in TBS2T for 30 min, once in 1×TBS for 30 min at room temperature, air dried, and mounted in mounting medium (KPL) for visualization. Free-floating flat-mount retinas were moved from 4% PFA to 0.1 m PB for 5 min and blocked in PBTGS for 1 h. Primary antibodies were diluted in PBTGS and 3 mm sodium azide. Retinas were incubated in primary antibody for 5 d at room temperature and washed 3 times in PBTGS for 10 min at. Secondary antibodies were diluted in PBTGS and 3 mm sodium azide and placed on tissue for 3 h. Tissue was washed once for 10 min in PBTGS and 2× for 10 min in 0.1 m PB. Flat-mounts were mounted on microscope slides with the ganglion cell layer facing up. Sections were air dried and mounted in mounting medium (KPL) for visualization.
Electron microscopy.
To identify regenerating axons, we injected HRP-conjugated CTB (3 μl, 0.2% List Biological) 6 weeks after optic nerve crush in control mice (no crush), or in mice in which we injected with either saline or AAV-Cre+zymosan+cAMP into the eye as indicated above. After allowing 4 d for transport of the tracer, mice were perfused with 2.5% glutaraldehyde and 0.5% PFA in 0.1 m PB. Following dissection, a 500-μm-thick transverse segment of the optic nerve 2.5 mm distal to the crush site was left overnight in 0.1 m PB. To detect the tracer, we used tetramethylbenzidine (TMB) as a substrate for HRP histochemistry. The optic nerve was immersed in TMB working solution (TMB, Vector Laboratories) for 1 h and in DAB-costaining solution 30 min. Tissues were washed in 0.1 m cacodylate buffer, pH 7.4, and postfixed for 1 h in 1% osmium tetroxide containing 1.5% potassium ferrocyanide in distilled water followed by washing in distilled water and staining in 1% uranylacetate for 1 h. Tissues were then washed in water, dehydrated in graded ethanol infiltrated with propyleneoxide for 3 h, and polymerized at 60° for 48 h. Ultrathin cross-sections (70 nm) were collected on copper grids and contrasted with lead citrate. Microscopy was performed using a JEOL 1200EX electron microscope. Images were recorded digitally at 3000×. Images were collected from optic nerves from 3 mice in each group: control, saline, and Pten deletion/zymosan/cAMP treatment groups.
Antibodies.
Affinity purified rabbit anti-βIV spectrin antibodies were previously described (Yang et al., 2004); AB_2315635. Anti-Nav1.6 (K87A/10; AB_2184197), Anti-Caspr (K65/35; AB_2083496), Anti-AnkG (106/36; AB_10673030) mouse monoclonal antibodies were purchased from the University of California Davis/National Institutes of Health monoclonal antibody resource Neuromab. Anti-GFP was purchased from Abcam (AB_300798). Anti-neurofilament-M (NF-M) was purchased from Millipore (AB_91201) and Santa Cruz Biotechnology (AB_628005). Anti-GAP-43 was custom-made in sheep as described previously (Benowitz et al., 1988).
Imaging and analysis.
Imaging was performed on a Zeiss AxioObserver Z1 fluorescent microscope with an apotome attachment using Axiovision and Zen software. Image analysis was performed using Fiji. For quantification of nodes of Ranvier, z-stacks were taken at 100× (1.4 NA oil immersion). For a single optic nerve, 4 z-stacks were taken along the length of the optic nerve: 325 μm proximal to the crush site (−325 μm); at the crush site (0 μm), 325 μm distal to the crush site (+325 μm), and 975 μm distal to the crush site (+975 μm). A minimum of three optic nerves, two longitudinal sections per nerve, was used per condition. Nodes were counted only if they included both βIV spectrin immunoreactivity and a flanking pair of Caspr-labeled paranodes. Heminodes were not counted in our analysis. For quantification of AIS, z-stacks were taken at 100×. For a single flat-mount retina, two z-stacks were taken in opposite quadrants 2 mm away from the optic disk. A minimum of three retinas was used per condition. AISs were counted if they were labeled for βIV spectrin and failed to display the characteristic puncta typical of the lobular dendritic process emanating from amacrine cells (Van Wart et al., 2005). For quantification of regenerating axons in Ank3f/f mice, 5 z-stacks were taken at 20× (0.8 NA) along the length of the optic nerve: 0.3, 0.6, 0.9, 1.2, and 1.5 mm distal to the crush site. A minimum of three optic nerves, two longitudinal sections per nerve, was used per condition. Axons were counted if they were immunostained for either GAP-43 or GFP. Axon counts were normalized using a previously described method (Baldwin et al., 2015).
Statistics.
Statistics were performed using GraphPad Prism 6. For two group comparisons, Student's t tests with a Mann–Whitney post hoc test was used. For multiple comparisons, one-way ANOVA with a Tukey's post hoc test was used. Data are presented as the mean ± SD unless otherwise indicated. p < 0.05 was considered significant.
Results
AIS and nodes of Ranvier are disrupted after optic nerve crush
To determine the time course and extent of AIS and node of Ranvier disruption following CNS traumatic injury, we examined these domains at 12 h, 24 h, 7 d, and 30 d after optic nerve crush. Optic nerve crush caused immediate and progressive RGC axon degeneration that began at the crush site and progressed both proximally and distally away from the crush (Fig. 1A–D). Immunostaining with antibodies against NF-M revealed rapid fragmentation (within 24 h; Fig. 1A,B) of the axonal cytoskeleton that progressed until only remnants of these cytoskeletons were detected in both proximal and distal regions by 7 d after crush injury (Fig. 1C). Thus, optic nerve crush results in axon degeneration in regions both proximal and distal to the injury.
Figure 1.
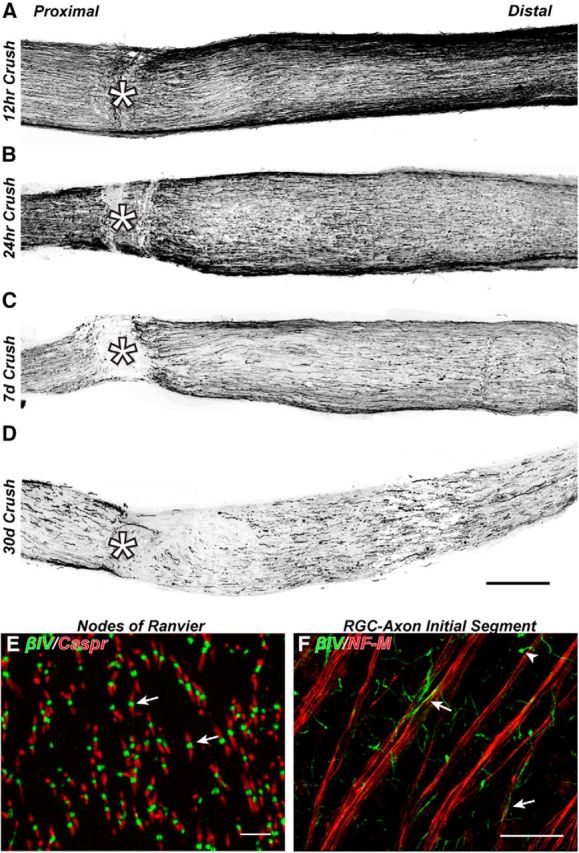
Optic nerve crush results in both proximal and distal axon degeneration. A–D, Optic nerves immunostained for NF-M to label axons 12 h, 24 h, 7 d, and 30 d after optic nerve crush. *Crush site. E, Naive mouse optic nerve immunostained for βIV spectrin to label nodes of Ranvier (green, arrow) and Caspr to label flanking paranodes (red). F, Flat-mount of mouse retina immunostained for βIV spectrin to label AIS (green, arrow) and NF-M to label axons (red). Scale bars: A–D, 200 μm; E, 5 μm; F, 30 μm.
To identify nodes of Ranvier and AISs, we immunolabeled optic nerves and retinas using antibodies against βIV spectrin, a cytoskeletal protein that is specifically enriched at these sites (Fig. 1E,F, arrows) and is a potent substrate for calpain-mediated proteolysis after injury (Schafer et al., 2009). We used antibodies against Caspr, a cell adhesion molecule and primary component of the paranodal junctions flanking nodes of Ranvier, to further define nodes (Fig. 1E) and as a surrogate for myelination because paranodal Caspr clustering requires myelination (Eisenbach et al., 2009). We measured the density of nodes per FOV in regions proximal (−325 μm), within (0 μm), and distal to the crush site (+325 and +975 μm). We included in this analysis only βIV spectrin-labeled nodes of Ranvier flanked by Caspr-labeled pairs of paranodal junctions; heminodes (i.e., clusters of βIV spectrin flanked only by a single paranode) were not counted. We found rapid degeneration of nodes of Ranvier within the crush site such that at all times after crush injury nodes were not detected (Fig. 2B,F,J,N,R). At 12–24 h after crush and in regions immediately proximal and distal to the crush (−325 μm and +325 μm, respectively), we found approximately half of nodes degenerated (Fig. 2A,E,C,G,Q,S). In contrast, regions that were farther from the crush site (+975 μm) showed nodes that were unaffected by the crush (Fig. 2D,H,T).
Figure 2.
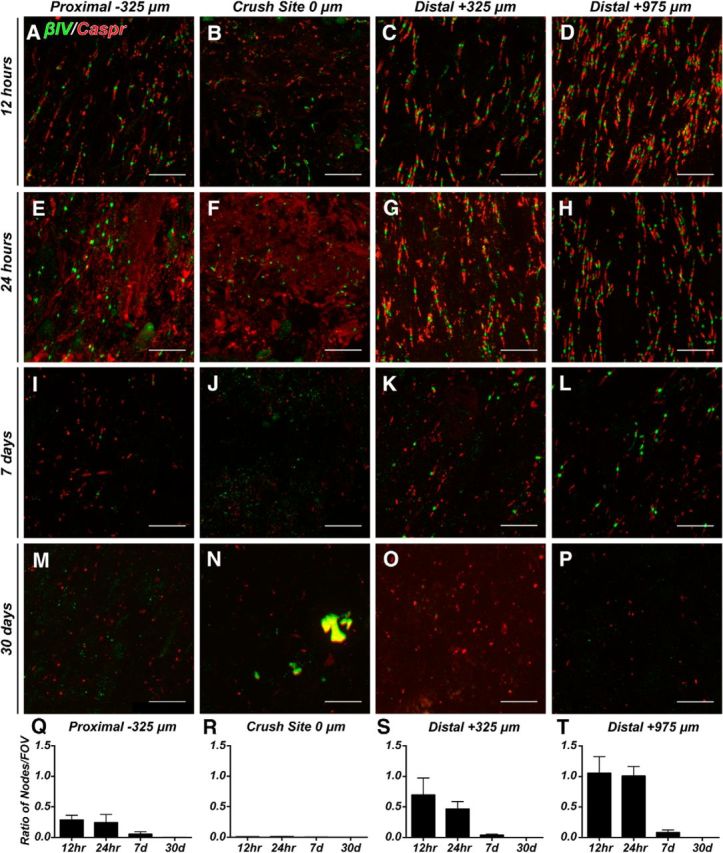
Nodes of Ranvier degenerate after optic nerve crush. A–P, Representative images of optic nerves immunostained for βIV spectrin (green) and caspr (red) at the indicated time points after nerve crush and distances from the crush site. Q–T, The ratio of nodes of Ranvier per FOV in crushed optic nerve to uninjured control optic nerve in areas proximal (−325 μm), within (0 μm), and distal (+325 and +975 μm) to the crush site. Scale bars, 10 μm.
At 7 d after optic nerve crush, we rarely detected any nodes of Ranvier in regions close to the crush site (−325 and +325 μm; Fig. 2I,K), but some nodes could still be detected in areas farther from the crush (+975 μm; Fig. 2L), indicating that the disruption of nodes begins at the crush and moves in both proximal and distal directions, with the degree of disruption depending on the distance from the crush. Furthermore, by mechanisms that remain unknown, proximal axons degenerate more quickly than axons distal to the crush. By 30 d after injury, we were unable to detect either nodes or paired clusters of Caspr in any region of the crushed optic nerve (Fig. 2M–P). Together, these observations show that optic nerve crush results in rapid disruption of nodes of Ranvier and that by 30 d after crush there is no evidence of regeneration or node reassembly in the absence of any treatments.
To determine the time course and extent of RGC AIS disruption, we labeled AIS at 1, 2, 3, 7, and 30 d after optic nerve crush. Retinal flat-mounts revealed two βIV spectrin-labeled structures: AISs in the proximal axon of RGCs (Fig. 3A–F, arrows) and a morphologically distinct lobular dendritic process found in AII amacrine cells (Fig. 3A–F, arrowheads) (Wu et al., 2011); in our analysis, we only counted RGC AIS. We found that RGC AIS degeneration began within days of the crush, with a significant loss of AISs per FOV detected by 7 d after injury (Fig. 3G) and significant shortening of AISs within 3 d of the crush (Fig. 3H). Previous studies have shown that ∼50% and ∼20% of RGCs survive 7 and 14 d after optic nerve crush, respectively (Park et al., 2008; Puyang et al., 2016). We measured a 70% reduction in the number of AISs 1 week after optic nerve crush (Fig. 3G), consistent with the idea that AISs are actively disrupted by calpain-dependent proteolysis before cell death. Furthermore, those AISs that were detected at 7 and 30 d were not significantly different in length from control RGCs, suggesting that surviving RGCs may have intact AISs. Together, these observations demonstrate that the excitable domains necessary for rapid and efficient action potential propagation are rapidly disrupted following optic nerve crush.
Figure 3.
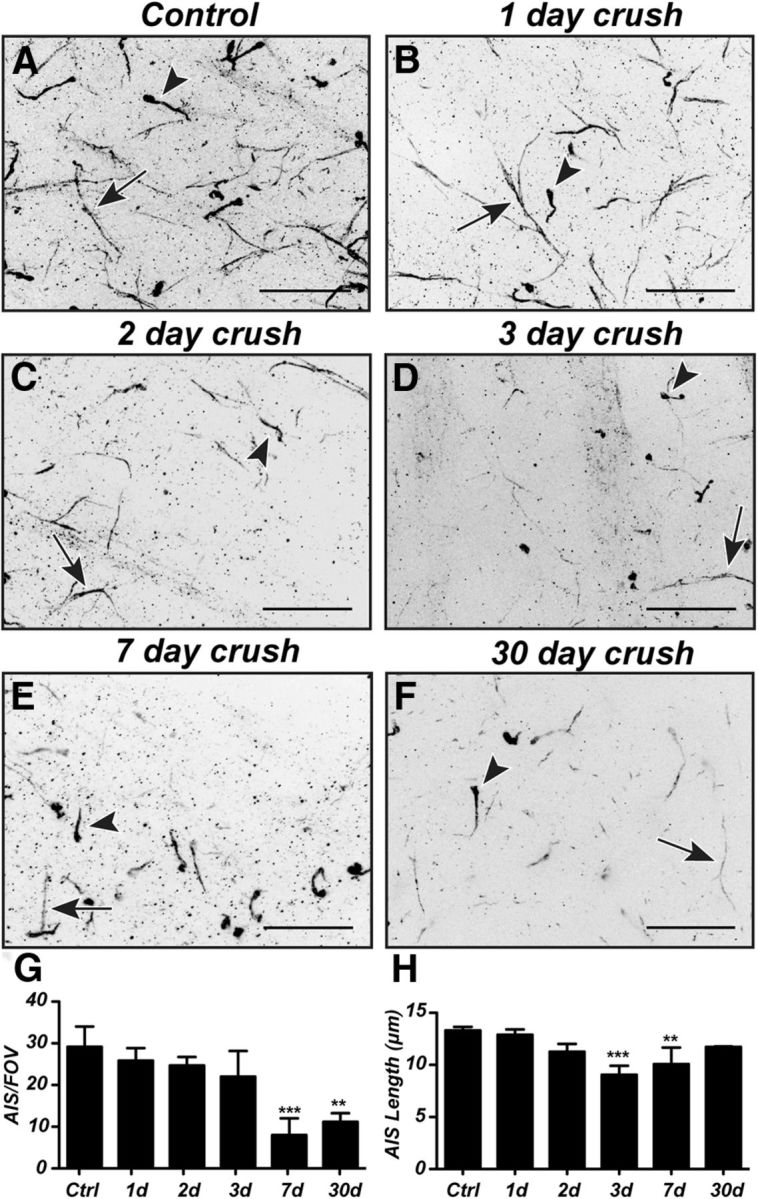
AISs of RGCs degenerate after optic nerve crush. A–F, Representative images of flat-mounts of retina immunostained for βIV spectrin reveal RGC AIS (arrow) and AII amacrine cell lobular dendritic processes (arrowhead) in control (A) and after optic nerve crush (B–F). G, H, Quantification of AIS degeneration (G) and changes in AIS length (H) after optic nerve crush. Scale bars: A–F, 20 μm. **p < 0.01 (unpaired t test with Mann–Whitney post test). ***p < 0.001 (unpaired t test with Mann–Whitney post test).
Regenerating CNS axons are remyelinated and reassemble nodes of Ranvier
Nodes of Ranvier are essential for rapid and efficient action potential propagation, depend on myelination, and their reassembly will be an important component of any therapeutic strategy aimed at neural repair. To determine whether regenerating CNS axons can be remyelinated and can reassemble nodes of Ranvier, we coupled optic nerve crush with knock-out of Pten and administration of both zymosan and CPT-cAMP. This combinatorial approach results in significant axon regeneration distal to the lesion site (Fig. 4A–C; crush site indicated by an asterisk) (Kurimoto et al., 2010) and has been reported to reinnervate downstream thalamic targets, resulting in recovery of basic visual responses, such as depth perception, the optokinetic response, and circadian photoentrainment (de Lima et al., 2012). Apparent axon regeneration can be detected in regions of the optic nerve proximal to the nerve crush within 2 weeks of injury (Fig. 4A). However, because many of the axons are degenerating, it is not possible to unequivocally distinguish between regenerating and degenerating axons at this time point. Nevertheless, by 6 and 12 weeks after injury, profound axon regeneration beyond the crush site can be seen in the treated nerves, whereas saline-injected controls have few, if any, regenerating axons that extend past the crush site (Fig. 4B,C). By 12 weeks after injury, some regenerating axons had abnormal trajectories, including axonal “U-turns” in both proximal and distal regions of the nerve (see Fig. 7A,B, arrows). These incorrect trajectories likely reflect the absence of guidance cues that keep axons on track during normal development. U-turns of regenerating optic nerve axons have been described previously (Pernet et al., 2013).
Figure 4.

Axons regenerate after genetic deletion of PTEN coupled with intravitreal injection of zymosan and CTP-cAMP (Cre+Zymo+cAMP). A–C, Optic nerves immunostained for NF-M in control saline-injected and Cre+zymosan+cAMP-treated mice at 2 weeks (A), 6 weeks (B), and 12 weeks (C) after crush. *Crush site. Scale bar, 200 μm.
Figure 7.

Regenerating CNS axons can be remyelinated and reassemble paranodal junctions. A, B, Proximal (A) and distal (D) regions of optic nerve immunostained for NF-M showing examples of “U-turn” axons (arrows) 12 weeks after optic nerve crush in Cre+zymosan+cAMP mice. C, D, Proximal optic nerve immunostained for Nav1.6 (C) or AnkG (D) to label nodes of Ranvier (red, arrow) and Caspr to label flanking paranodes (green) 12 weeks after optic nerve crush in Cre+zymosan+cAMP mice. E–H, Optic nerve immunostained for Caspr (green) and NF-M (red) to label paranodes and axons, respectively, in Cre+zymosan+cAMP mice 12 weeks after optic nerve crush. I, Quantification of Caspr-labeled paranodes along regenerating optic nerve in saline-treated and Cre+zymosan+cAMP-treated mice 12 weeks after optic nerve crush. J, K, Electron microscopy of control (J) and regenerating axons (K) showing the electron dense reaction product from HRP-CTB histochemistry (asterisks) in the axoplasm of myelinated axons. L, The number of regenerating CTB-HRP-labeled myelinated axons per nerve. N = 3 mice per group. ***p < 0.001 (unpaired t test). Scale bars: A, B, 5 μm; C, D, 10 μm; E, 300 μm; F–H, 5 μm; J, K, 0.5 μm.
We used Caspr, an essential component of the paranodal axoglial junction, as a robust indicator of remyelination and proper axon-glia interactions in the regenerating optic nerve. In addition, we used clustering of βIV spectrin, a component of all mature nodes of Ranvier, flanked by Caspr-labeled paranodes as an indicator of node of Ranvier reassembly. Analysis of the optic nerve proximal to the crush site 2 weeks after injury revealed little or no remyelination or reassembly of nodes in either the treated RGCs or saline-treated controls (Fig. 5A,G); saline-treated controls are not shown but were qualitatively and quantitatively (Figs. 5G,H, 6G,H) similar to the results obtained with crush alone (Fig. 2). However, at 6 and 12 weeks after crush, we observed robust remyelination and reassembly of nodes of Ranvier in regions proximal to the crush site in the regenerating optic nerve (Fig. 5C,E, arrows, G). Paranodes were well formed, and nodes contained βIV spectrin, ankG, and Nav1.6 Na+ channels (Figs. 5C,E, 7C,D, arrows). Similar to proximal regions, the crush site showed no remyelination or clustering of βIV spectrin at 2 weeks after crush (Fig. 5B,H); but by 6 and 12 weeks after crush, we observed robust remyelination and reassembly of nodes of Ranvier (Fig. 5D,F, arrows, H).
Figure 5.
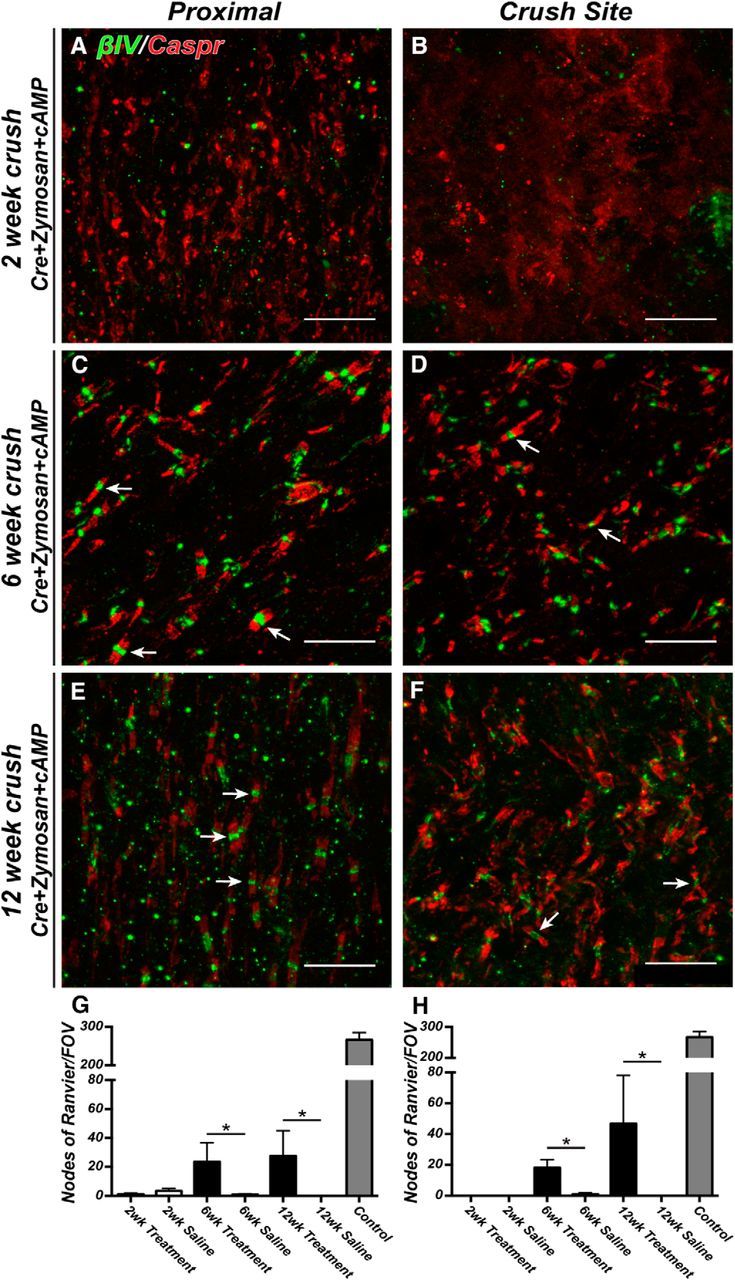
Nodes of Ranvier are reassembled in regenerating CNS axons. A–F, Representative images of optic nerves immunostained for βIV spectrin (green, arrows) and Caspr (red) proximal to the crush (A, C, E) and at the crush site (B, D, F). Optic nerves were examined at 2, 6, and 12 weeks after crush. G, H, The ratio of nodes of Ranvier per FOV in crushed optic nerve to uninjured control optic nerve at 2, 6, and 12 weeks after crush. Measurements were made proximal to the crush site (−325 μm) and within the crush site (0 μm). Treatment = Cre+zymosan+cAMP. Scale bars: A–F, 10 μm. *p < 0.05 (unpaired t test with Mann–Whitney post test).
Figure 6.
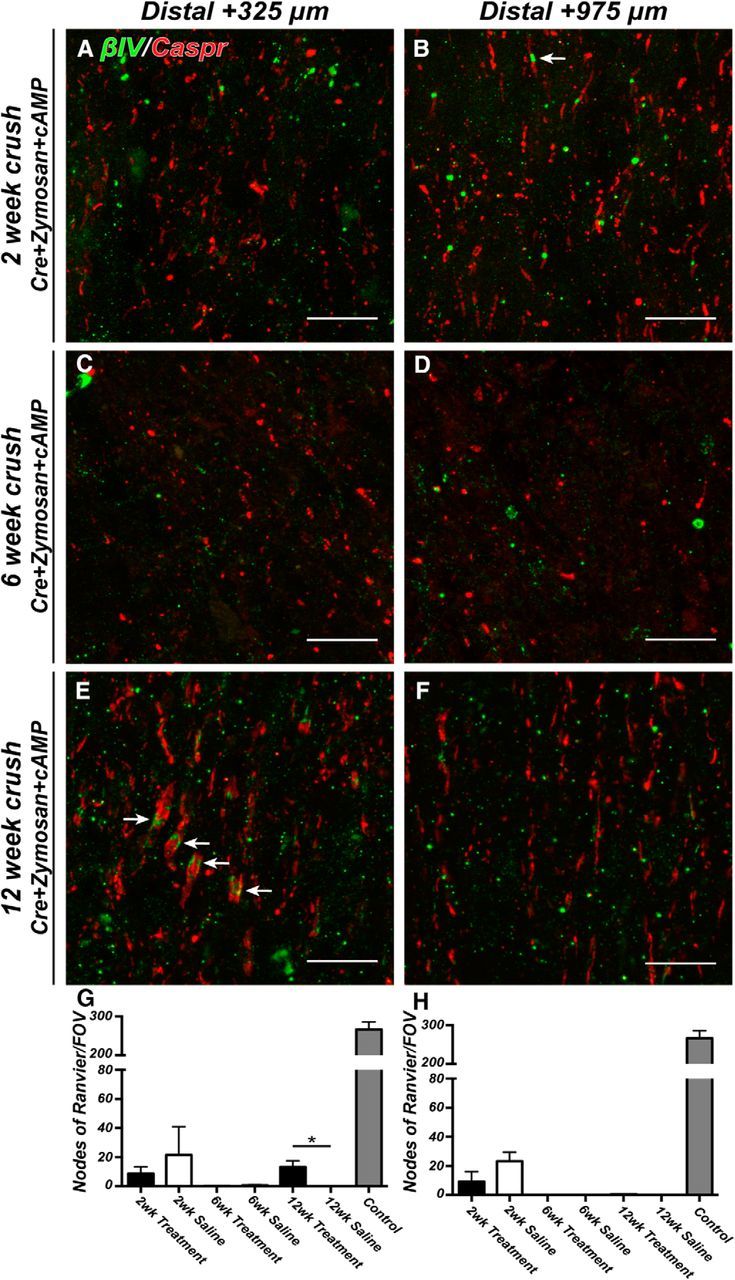
Nodes of Ranvier are reassembled in regenerating CNS axons distal to the crush site. A–F, Representative images of optic nerves immunostained for βIV spectrin (green, arrows) and Caspr (red) distal to the crush (A, C, E: 325 μm; B, D, F: 925 μm). Optic nerves were examined at 2, 6, and 12 weeks after crush. G, H, The ratio of nodes of Ranvier per FOV in crushed optic nerve to uninjured control optic nerve at 2, 6, and 12 weeks after crush. Measurements were made distal to the crush site (+325 and +975 μm). Treatment = Cre+zymosan+cAMP. Scale bars: A–F, 10 μm. *p < 0.05 (unpaired t test with Mann–Whitney post test).
Distal to the crush site, the regenerating optic nerve revealed very few nodes of Ranvier and paranodal junctions 2 weeks after crush injury (Fig. 6A,B arrow, G,H). We speculate that the few nodes observed distal to the crush site reflect those that had not yet degenerated because, by 6 weeks after crush, we did not detect any Caspr-labeled paranodes or βIV spectrin-labeled nodes (Fig. 6C,D,G,H), despite the robust axon regeneration seen at this time point (Fig. 4B). By 12 weeks after nerve crush, however, we found Caspr-labeled paranodal junctions and βIV spectrin clustering at sites +325 μm distal to the crush site (Fig. 6E, arrows, G), though still not in regions that were farther from the crush site (+975 μm) (Figs. 6F,H, 7I). Together, these data indicate that, despite rapid and extensive regeneration of optic nerve axons (Figs. 4, 7E–H), both remyelination and node of Ranvier reassembly begin at the proximal optic nerve and progress toward distal regions only with increasing time. This view is consistent with the observation that, 12 weeks after optic nerve crush, paranodal junctions were detected more frequently near the proximal end of the optic nerve (near the crush site) than in regions more distal to the crush (Fig. 7F,G, arrows, I), although we observed clear instances of appropriate axoglial contacts ∼3 mm from the eye indicating remyelination of NF-M-labeled axons; however, those contacts far from the eye were rarely seen as pairs of adjacent paranodes characteristic of mature nodes of Ranvier (compare Fig. 7F,H, arrows). Together, these results indicate that remyelination and reassembly of nodes occur in an anterograde manner from proximal to distal locations along the optic nerve. These observations predict that waiting even longer periods of time should result in the detection of nodes in regions > +975 μm from the crush site.
To further demonstrate regeneration and remyelination in treated nerves, we performed intravitreal injections of HRP-conjugated cholera toxin B (CTB-HRP) 6 weeks after optic nerve crush in mice that also received intravitreal injections of saline or AAV-Cre+zymosan+cAMP. We then performed electron microscopy 2.5 mm distal to the crush site and identified CTB-HRP labeled axons by the presence of an electron dense precipitate. We observed a labeling efficiency of ∼4% in both uninjured controls and regenerating optic nerves (Figs. 7J, 7K, respectively; labeled axons are indicated by an asterisk); we rarely detected CTB-HRP-labeled axons in the saline-injected mice. Quantification of the number of regenerating axons showed a dramatic and significant increase in the number of labeled myelinated axons in optic nerves of treated mice. Together, these observations demonstrate that regenerating CNS axons can be remyelinated.
AISs are reassembled in regenerating CNS axons
Flat-mounts of retinas from uninjured mice normally show axon fascicles that course across the surface of the retina where they enter the optic nerve head (Fig. 8A). At 12 weeks after optic nerve crush, saline-injected controls have dramatically fewer axons, likely due to the death of RGCs (Fig. 8B) (Park et al., 2008). In contrast, PTEN-deficient RGCs stimulated with zymosan and cAMP (treated RGCs) have a much higher density of axons. Unlike the uninjured and saline-injected controls, axons of treated RGCs also had abnormal and tortuous trajectories across the surface of the retina (Fig. 8C). As shown above, optic nerve crush causes a dramatic loss of AISs (Fig. 3) (Schafer et al., 2009). Because AISs initiate action potentials and regulate neuronal polarity (Rasband, 2010), an essential component of neural repair must be the reestablishment of AISs in regenerating axons. To determine whether regenerated CNS axons can reassemble AISs, we immunostained flat-mounts of retinas using antibodies against βIV spectrin and NF-M (to label axons) at 2, 6, and 12 weeks after optic nerve crush. At 2 weeks after crush we did not measure any difference in the number of AISs between saline-injected or treated RGCs (Fig. 8L), although the number of AISs in both saline-injected and treated retinas was dramatically reduced compared with uninjured controls (Fig. 8F,L). However, there was a significant increase (or preservation) in the length of AIS of treated RGCs compared with saline-injected controls (Fig. 8M). At 6 and 12 weeks after optic nerve crush, we found very few AISs in saline-injected controls (Fig. 8D,G,L), and AIS length was not significantly different at any time point (Fig. 8M). In contrast, at both 6 and 12 weeks after optic nerve crush, treated RGCs had significantly more AISs (Fig. 8E,H, arrows, L), and the average lengths of these AIS were significantly increased compared with saline-injected controls (Fig. 8M); nevertheless, AISs of varying lengths were present along the axons of treated RGCs (Fig. 8I–K). Thus, regenerating CNS axons can reassemble their AISs.
Figure 8.
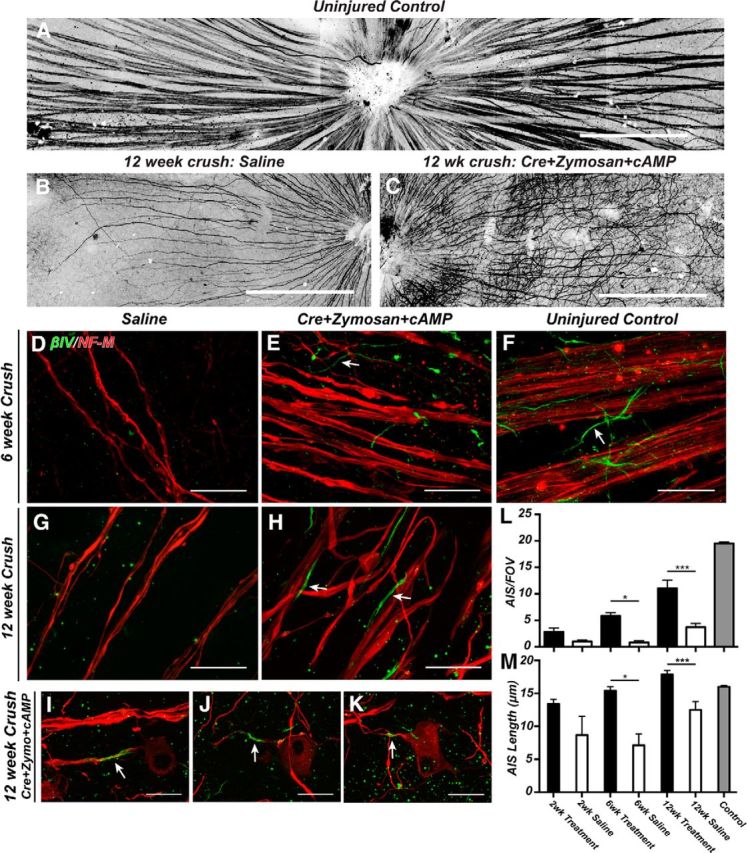
AISs are reassembled in regenerating CNS axons. A–C, Flat-mounts of uninjured control (A), 12 week saline-treated (B), and 12 week Cre+zymosan+cAMP-treated retinas (C) immunostained for NF-M to label axons. D–K, Immunostaining of retinas using antibodies against βIV spectrin (green) and NF-M (red) to label axons in saline-treated (D, G) and Cre+zymosan+cAMP-treated (E, H–K), retinas at 6 weeks (D, E) and 12 weeks (G–K) after optic nerve crush. F, Uninjured controls. L, The number of AISs per FOV after optic nerve crush. M, AIS length after optic nerve crush. Treatment = Cre+zymosan+cAMP. Labels on x-axis of M apply to the x-axis of L. Scale bars: A–C, 300 μm; D–K, 20 μm. *p < 0.05 (unpaired t test with Mann–Whitney post test). ***p < 0.001 (unpaired t test with Mann–Whitney post test).
AnkyrinG is not required for CNS axon regeneration
Neurons and RGCs are highly polarized cells with distinct input (somatodendritic) and output (axonal) domains. Although ankG is required to maintain neuronal polarity, it is not required for initial axon specification during development (Galiano et al., 2012). However, it is not known whether regenerating neurons require ankG (and by extension an AIS) to reestablish neuronal polarity, or if they revert to a state where axon specification and growth occurs independent of ankG. To determine whether ankG is required for axon regeneration, we used a different regeneration paradigm to avoid the need for simultaneous removal of both PTEN and ankG. Instead, we used intravitreal injection of β(1,3)-glucan (Curdlan) at the time of optic nerve crush, which results in robust axon regeneration past the crush site by 2 weeks after injury (Fig. 9A,B) (Baldwin et al., 2015). To eliminate ankG, we injected AAV-Cre-GFP (or AAV-GFP as a control) in Ank3f/f mice (Ho et al., 2014). At 4 weeks after AAV injection, many axons were labeled with GFP and nodes of Ranvier could be easily identified (Fig. 9C). In contrast to AAV-GFP injected Ank3f/f mice, nodes of Ranvier in AAV-Cre-GFP-injected Ank3f/f mice lacked axonal ankG immunostaining (Fig. 9C, arrows), whereas paranodal oligodendroglial ankG remained intact (Fig. 9C, arrowheads) (Chang et al., 2014). We assessed axon regeneration by immunostaining for GAP-43 (Fig. 9D) or GFP (Fig. 9F). Both cases showed similar levels of axon regeneration past the crush site (Fig. 9E,G). Thus, ankG (and an AIS) is not required for axon regeneration.
Figure 9.

AnkG is not required for axon regeneration. A, GAP-43 immunostatining of optic nerve after intravitreal injection of β-1,3-glucan (Curdlan) or saline, 2 weeks after optic nerve crush. B, Quantification of GAP-43-labeled axons in mice administered either Curdlan or saline 2 weeks after optic nerve crush. C, Immunostaining of Ank3f/f mouse optic nerves after intravitreal injection of either AAV-GFP or AAV-Cre-GFP using antibodies against ankG (red), Caspr (blue), and GFP (green). Arrows indicate nodes of Ranvier. Red arrowheads indicate paranodal ankG in the AAV-Cre-GFP injected mice. D, F, Optic nerves from Ank3f/f mice treated with Curdlan and either AAV-GFP or AAV-Cre-GFP and immunostained with GAP-43 (D) or GFP (F) 2 weeks after optic nerve crush. E, G, Quantification of GAP-43 (E) or GFP (G) positive axons in regenerating axons in Ank3f/f mice administered curdlan and either AAV-GFP or AAV-Cre-GFP 2 weeks after optic nerve crush. Scale bars: A, D, 300 μm; F, 200 μm. ****p < 0.0001 (unpaired t test with Mann–Whitney post test).
Discussion
Many CNS injuries or diseases cause axon injury or degeneration that disrupts the circuits necessary for proper nervous system function. Traditionally, these injuries have been viewed as irreversible. Furthermore, mammalian axons are frequently myelinated and subdivided into functional domains that include the AISs and nodes of Ranvier; these domains are also essential for proper nervous system function. Here, using optic nerve crush as a model of CNS traumatic injury, we showed that these domains are rapidly disassembled after injury. Disruption of AISs and nodes depends on calpain-dependent proteolysis of the unique ankyrin/spectrin cytoskeleton found at these locations (Schafer et al., 2009). In some neurons, calpains are activated by injury-induced Ca2+ influx through P2X7 receptors (Del Puerto et al., 2015). The dismantling of these excitable domains may be a neuroprotective response to decrease excitotoxicity, rather than a simple maladaptive response. In any case, a major therapeutic goal in translational neuroscience is to develop strategies that promote not only axon regeneration but also restore function through remyelination and the reestablishment of excitable domains.
The optic nerve has emerged as a favorite model system to study CNS axon regeneration due to its simplicity, accessibility, and relevance for diseases, such as glaucoma and traumatic injury (Benowitz et al., 2015). The discovery of pathways that control the intrinsic growth capacity of RGCs has led to manipulations that promote robust axon regeneration following optic nerve crush (Yin et al., 2006; Park et al., 2008; Kurimoto et al., 2010; Sun et al., 2011; de Lima et al., 2012; Belin et al., 2015). Despite these advances, neither remyelination nor reestablishment of nodes or AISs has been demonstrated in any model of CNS axon regeneration. De Lima et al. (2012) reported the presence of myelinated axons associated with robust optic nerve regeneration but did not systematically distinguish between injured axons that had not yet degenerated and regenerating axons, whereas Bei et al. (2016) recently reported that, although codeletion of Pten and Socs3 permitted robust axon regeneration following an optic tract injury close to the superior collicus in immature mice, they failed to observe any remyelination. Furthermore, functional recovery required treatment with the K+ channel blocker 4-aminopyridine. In a similar vein, regenerated axons in the dorsal columns can established connectivity with brainstem targets but fail to myelinate (Alto et al., 2009).
Although we did not evaluate functional recovery in our studies, previous experiments using the same treatment and injury model showed some recovery of depth perception, the optokinetic response, and circadian photoentrainment (de Lima et al., 2012). Moreover, through the use of CTB-HRP as an anatomical tracer, our EM results confirm that regenerating axons can be remyelinated under these conditions. Thus, our results are strikingly different from those of Bei et al. (2016) and may reflect different capacities for remyelination depending on the location of the CNS injury or method of inducing regeneration. Furthermore, inducing inflammation can promote distinct levels of remyelination (Setzu et al., 2006), and the different injury models may result in different immune responses, thereby impacting remyelination of regenerating axons. Alternatively, the absence of remyelination and node reassembly near the superior collicus may reflect regional heterogeneity of oligodendrocyte progenitor cells and their intrinsic capacity to differentiate and myelinate axons instead of differences in the RGC axons themselves. Thus, the efficacy of repair strategies will likely depend on the method of stimulating axon regeneration and/or the anatomical location of the injury, making it very important to evaluate anatomically diverse CNS injury/regeneration models for remyelination.
During mouse optic nerve development, myelination and node of Ranvier formation is very rapid and happens over a period of 2–3 weeks (Rasband et al., 1999; Susuki et al., 2013). Despite the long-distance axonal growth reported in this and other studies (de Lima et al., 2012; Baldwin et al., 2015), the remyelination and node reassembly we observed in our treated mice took months and failed to even reach the optic chiasm by 3 months after the crush injury. Thus, therapeutic strategies aimed at CNS neural repair must be combinatorial and promote not only axon regeneration, but also the differentiation of oligodendrocyte progenitor cells to perform the subsequent remyelination and clustering of ion channels at nodes of Ranvier. Experimental therapies aimed at promoting remyelination in multiple sclerosis (Mi et al., 2007; Juarez et al., 2015; Dietz et al., 2016) should also be evaluated in CNS regeneration models like the one reported here as they may accelerate functional recovery after axons regenerate.
Our experiments are the first to examine reestablishment of a molecularly defined AIS in regenerating CNS axons. In response to injury, we observed significant reductions in the number and length of AISs. In contrast, the proregeneration treatment induced significant increases in the number and length of RGC AISs. These results are similar to those observed in other injury models: AISs shorten after focal cortical stroke and traumatic brain injury (Baalman et al., 2013; Hinman et al., 2013). In contrast to RGCs that show little or no spontaneous recovery of AIS, in surviving cortical regions adjacent to a stroke new AIS and even supernumerary axons can be seen (Hinman et al., 2013). Similarly, after chronic transection of peripheral axons, spinal motoneurons develop supernumerary axons (Havton and Kellerth, 1987). In our model, we observed a dramatic increase in the number of axons and AISs in response to PTEN deletion and treatment with zymosan and a cAMP analog. However, we never observed the development of supernumerary axons (data not shown).
In mature neurons, ankG is required to maintain neuronal polarity; when ankG is lost, somatodendritic proteins enter into the axon, and the former axon develops spine-like protrusions that contain both postsynaptic proteins, including scaffolds and neurotransmitter receptors (Hedstrom et al., 2008; Sobotzik et al., 2009). Importantly, ankG is not required for axon specification (Galiano et al., 2012). Using ankG conditional knock-out mice and a proregenerative treatment, we found that elimination of ankG neither promotes nor inhibits regeneration: neurons with and without ankG were equally responsive to treatments. We suggest that regenerating RGCs may revert to a more immature state that supports growth and axon specification. On the other hand, these studies do not address whether axons with a preserved AIS are more, or less, responsive to proregenerative treatments. A combinatorial approach that involves preservation of the AIS by inhibiting its proteolysis (perhaps through blocking the calcium-dependent proteases responsible for ankG disruption) combined with treatments that promote axon regeneration will be required to fully determine whether the AIS can play a role.
In conclusion, to the best of our knowledge, the results presented here are the first demonstration of new nodes of Ranvier and remyelination in a CNS regeneration model. These results demonstrate the capacity for regenerated CNS axons to be remyelinated and reestablish their excitable domains. Furthermore, they provide additional justification for continued optimism and investment in the search for therapies that promote the functional recovery of axons.
Footnotes
This work was supported by National Institutes of Health Grants NS069688 and NS044916 to M.N.R. and Grant EY024481 to L.B., TIRR Foundation to M.N.R., and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to L.B., R.J.G., and M.N.R. S.d.L. was supported in part by Science Without Borders program/Council for Research and Technology of Brazil postdoctoral fellowship.
The authors declare no competing financial interests.
References
- Alto LT, Havton LA, Conner JM, Hollis ER, 2nd, Blesch A, Tuszynski MH. Chemotropic guidance facilitates axonal regeneration and synapse formation after spinal cord injury. Nat Neurosci. 2009;12:1106–1113. doi: 10.1038/nn.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baalman KL, Cotton RJ, Rasband SN, Rasband MN. Blast wave exposure impairs memory and decreases axon initial segment length. J Neurotrauma. 2013;30:741–751. doi: 10.1089/neu.2012.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin KT, Carbajal KS, Segal BM, Giger RJ. Neuroinflammation triggered by beta-glucan/dectin-1 signaling enables CNS axon regeneration. Proc Natl Acad Sci U S A. 2015;112:2581–2586. doi: 10.1073/pnas.1423221112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei F, Lee HH, Liu X, Gunner G, Jin H, Ma L, Wang C, Hou L, Hensch TK, Frank E, Sanes JR, Chen C, Fagiolini M, He Z. Restoration of visual function by enhancing conduction in regenerated axons. Cell. 2016;164:219–232. doi: 10.1016/j.cell.2015.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin S, Nawabi H, Wang C, Tang S, Latremoliere A, Warren P, Schorle H, Uncu C, Woolf CJ, He Z, Steen JA. Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron. 2015;86:1000–1014. doi: 10.1016/j.neuron.2015.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Apostolides PJ, Perrone-Bizzozero N, Finklestein SP, Zwiers H. Anatomical distribution of the growth-associated protein GAP-43/B-50 in the adult rat brain. J Neurosci. 1988;8:339–352. doi: 10.1523/JNEUROSCI.08-01-00339.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, He Z, Goldberg JL. Reaching the brain: advances in optic nerve regeneration. Exp Neurol. 2015 doi: 10.1016/j.expneurol.2015.12.015. pii:S0014--4886(15)30141-2. [DOI] [PubMed] [Google Scholar]
- Chang KJ, Zollinger DR, Susuki K, Sherman DL, Makara MA, Brophy PJ, Cooper EC, Bennett V, Mohler PJ, Rasband MN. Glial ankyrins facilitate paranodal axoglial junction assembly. Nat Neurosci. 2014;17:1673–1681. doi: 10.1038/nn.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregg JM, DePaul MA, Filous AR, Lang BT, Tran A, Silver J. Functional regeneration beyond the glial scar. Exp Neurol. 2014;253:197–207. doi: 10.1016/j.expneurol.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Puerto A, Fronzaroli-Molinieres L, Perez-Alvarez MJ, Giraud P, Carlier E, Wandosell F, Debanne D, Garrido JJ. ATP-P2X7 receptor modulates axon initial segment composition and function in physiological conditions and brain injury. Cereb Cortex. 2014;25:2282–2294. doi: 10.1093/cercor/bhu035. [DOI] [PubMed] [Google Scholar]
- de Lima S, Koriyama Y, Kurimoto T, Oliveira JT, Yin Y, Li Y, Gilbert HY, Fagiolini M, Martinez AM, Benowitz L. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc Natl Acad Sci U S A. 2012;109:9149–9154. doi: 10.1073/pnas.1119449109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz KC, Polanco JJ, Pol SU, Sim FJ. Targeting human oligodendrocyte progenitors for myelin repair. Exp Neurol. 2016 doi: 10.1016/j.expneurol.2016.03.017. pii:S0014-4886(16)30060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbach M, Kartvelishvily E, Eshed-Eisenbach Y, Watkins T, Sorensen A, Thomson C, Ranscht B, Barnett SC, Brophy P, Peles E. Differential clustering of Caspr by oligodendrocytes and Schwann cells. J Neurosci Res. 2009;87:3492–3501. doi: 10.1002/jnr.22157. [DOI] [PubMed] [Google Scholar]
- Galiano MR, Jha S, Ho TS, Zhang C, Ogawa Y, Chang KJ, Stankewich MC, Mohler PJ, Rasband MN. A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell. 2012;149:1125–1139. doi: 10.1016/j.cell.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havton L, Kellerth JO. Regeneration by supernumerary axons with synaptic terminals in spinal motoneurons of cats. Nature. 1987;325:711–714. doi: 10.1038/325711a0. [DOI] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman JD, Rasband MN, Carmichael ST. Remodeling of the axon initial segment after focal cortical and white matter stroke. Stroke. 2013;44:182–189. doi: 10.1161/STROKEAHA.112.668749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TS, Zollinger DR, Chang KJ, Xu M, Cooper EC, Stankewich MC, Bennett V, Rasband MN. A hierarchy of ankyrin-spectrin complexes clusters sodium channels at nodes of Ranvier. Nat Neurosci. 2014;17:1664–1672. doi: 10.1038/nn.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D, Liu Y, Sun F, Wang X, Liu X, He Z. Restoration of skilled locomotion by sprouting corticospinal axons induced by co-deletion of PTEN and SOCS3. Nat Commun. 2015;6:8074. doi: 10.1038/ncomms9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez LA, Lu RQ, He D. Oligodendrocyte progenitor programming and reprogramming: toward myelin regeneration. Brain Res. 2016;1638(Pt B):209–20. doi: 10.1016/j.brainres.2015.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto T, Yin Y, Omura K, Gilbert HY, Kim D, Cen LP, Moko L, Kügler S, Benowitz LI. Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. J Neurosci. 2010;30:15654–15663. doi: 10.1523/JNEUROSCI.4340-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto T, Yin Y, Habboub G, Gilbert HY, Li Y, Nakao S, Hafezi-Moghadam A, Benowitz LI. Neutrophils express oncomodulin and promote optic nerve regeneration. J Neurosci. 2013;33:14816–14824. doi: 10.1523/JNEUROSCI.5511-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, He Q, Wang H, Tang X, Ho KW, Gao X, Zhang Q, Shen Y, Cheung A, Wong F, Wong YH, Ip NY, Jiang L, Yung WH, Liu K. Injured adult retinal axons with Pten and Socs3 co-deletion reform active synapses with suprachiasmatic neurons. Neurobiol Dis. 2015;73:366–376. doi: 10.1016/j.nbd.2014.09.019. [DOI] [PubMed] [Google Scholar]
- Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, Wong WM, Wang L, Su H, Chu TH, Guo J, Zhang W, So KF, Pepinsky B, Shao Z, Graff C, Garber E, Jung V, Wu EX, Wu W. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13:1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Kanter JL, He Z. PTEN/mTOR and axon regeneration. Exp Neurol. 2010;223:45–50. doi: 10.1016/j.expneurol.2009.12.032. [DOI] [PubMed] [Google Scholar]
- Pernet V, Joly S, Jordi N, Dalkara D, Guzik-Kornacka A, Flannery JG, Schwab ME. Misguidance and modulation of axonal regeneration by Stat3 and Rho/ROCK signaling in the transparent optic nerve. Cell Death Dis. 2013;4:e734. doi: 10.1038/cddis.2013.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puyang Z, Feng L, Chen H, Liang P, Troy JB, Liu X. Retinal ganglion cell loss is delayed following optic nerve crush in NLRP3 knockout mice. Sci Rep. 2016;6:20998. doi: 10.1038/srep20998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010;11:552–562. doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci. 1999;19:7516–7528. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Jha S, Liu F, Akella T, McCullough LD, Rasband MN. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci. 2009;29:13242–13254. doi: 10.1523/JNEUROSCI.3376-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab ME, Strittmatter SM. Nogo limits neural plasticity and recovery from injury. Curr Opin Neurobiol. 2014;27:53–60. doi: 10.1016/j.conb.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setzu A, Lathia JD, Zhao C, Wells K, Rao MS, Ffrench-Constant C, Franklin RJ. Inflammation stimulates myelination by transplanted oligodendrocyte precursor cells. Glia. 2006;54:297–303. doi: 10.1002/glia.20371. [DOI] [PubMed] [Google Scholar]
- Smith PD, Sun F, Park KK, Cai B, Wang C, Kuwako K, Martinez-Carrasco I, Connolly L, He Z. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009;64:617–623. doi: 10.1016/j.neuron.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobotzik JM, Sie JM, Politi C, Del Turco D, Bennett V, Deller T, Schultz C. AnkyrinG is required to maintain axo-dendritic polarity in vivo. Proc Natl Acad Sci U S A. 2009;106:17564–17569. doi: 10.1073/pnas.0909267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, Zhang K, Yeung C, Feng G, Yankner BA, He Z. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susuki K, Chang KJ, Zollinger DR, Liu Y, Ogawa Y, Eshed-Eisenbach Y, Dours-Zimmermann MT, Oses-Prieto JA, Burlingame AL, Seidenbecher CI, Zimmermann DR, Oohashi T, Peles E, Rasband MN. Three mechanisms assemble central nervous system nodes of Ranvier. Neuron. 2013;78:469–482. doi: 10.1016/j.neuron.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A, Boiko T, Trimmer JS, Matthews G. Novel clustering of sodium channel Na(v)1.1 with ankyrin-G and neurofascin at discrete sites in the inner plexiform layer of the retina. Mol Cell Neurosci. 2005;28:661–673. doi: 10.1016/j.mcn.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Wu C, Ivanova E, Cui J, Lu Q, Pan ZH. Action potential generation at an axon initial segment-like process in the axonless retinal AII amacrine cell. J Neurosci. 2011;31:14654–14659. doi: 10.1523/JNEUROSCI.1861-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lacas-Gervais S, Morest DK, Solimena M, Rasband MN. BetaIV spectrins are essential for membrane stability and the molecular organization of nodes of Ranvier. J Neurosci. 2004;24:7230–7240. doi: 10.1523/JNEUROSCI.2125-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23:2284–2293. doi: 10.1523/JNEUROSCI.23-06-02284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Henzl MT, Lorber B, Nakazawa T, Thomas TT, Jiang F, Langer R, Benowitz LI. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat Neurosci. 2006;9:843–852. doi: 10.1038/nn1701. [DOI] [PubMed] [Google Scholar]