

Abstract
Motivation: Mutations in homologous proteins affect changes in the backbone conformation that involve a complex interplay of forces which are difficult to predict. Protein design algorithms need to anticipate these backbone changes in order to accurately calculate the energy of the structure given an amino acid sequence, without knowledge of the final, designed sequence. This is related to the problem of predicting small changes in the backbone between highly similar sequences.
Results: We explored the ability of the Rosetta suite of protein design tools to move the backbone from its position in one structure (template) to its position in a close homologous structure (target) as a function of the diversity of a backbone ensemble constructed using the template structure, the percent sequence identity between the template and target, and the size of local zone being considered in the ensemble. We describe a pareto front in the likelihood of moving the backbone toward the target as a function of ensemble diversity and zone size. The equations and protocols presented here will be useful for protein design.
Availability and implementation: PyRosetta scripts available at www.bioinfo.rpi.edu/bystrc/downloads.html#ensemble
Contact: bystrc@rpi.edu
1 Introduction
Computational protein design has been increasingly employed in novel protein engineering applications and has been met with considerable success for a number of applications. Protein design is typically defined as the problem of identifying amino acid sequences that are compatible with a given, input target protein structure. Some of the most notable successes of computational protein design involve enhancing the thermal stability of natural proteins (Dantas, 2003; Joachimiak, 2006; Malakauskas and Mayo, 1998), designing novel binding sites on natural proteins (Cochran, 2005; Guntas, 2010; Karanicolas, 2011; Kortemme, 2004), and de novo design of novel proteins (Dahiyat and Mayo, 1997; Kuhlman, 2003) and enzymes (Jiang, 2008; Rothlisberger, 2008). From a computational point of view, the most important considerations for developing a robust protein design workflow to best enable experimental success are the selection of a suitable energetic function to distinguish favorable conformations from unfavorable ones, and an efficient algorithm to search the conformational space.
One major limitation with traditional computational protein design is its reliance on the assumption that the protein backbone remains fixed. Computationally speaking, holding the backbone fixed simplifies the design process considerably by limiting the number of degrees of freedom in the system, but unfortunately the fixed backbone assumption has been shown to fail for a significant number of design applications (Friedland, 2008; Fu, 2007; Kapp, 2012; Mooers, 2003). Many natural proteins are structurally optimized for the wildtype sequence and hence introducing sequence mutations leads to the necessity of modeling backbone flexibility to achieve reliable design success (Kuhlman and Baker, 2000). One early approach to implicitly modeling backbone flexibility involves simply dampening the van der Waals repulsive energetic term to allow for small steric clashes (Desjarlais and Handel, 1999), but this approach can lead to overpacking of the protein core.
Many flexible backbone design approaches have moved towards paradigms that explicitly model discrete backbone conformations as ensembles of related backbone structures (Friedland and Kortemme, 2010). The major advantage of an ensemble composed of discrete states is that it can be easily incorporated into a traditional fixed backbone protein design algorithm with ease. The ensemble can be generated in a variety of different ways, many of which have been successfully used to design novel protein functions. For instance, one study generated ensembles simply by randomly perturbing the φ/ψ backbone dihedral angles and found that the profiles of sequences explored on these ensembles tended to reproduce the natural sequence profile for the original input structure and its homologs (Larson, 2002). Another group utilized normal mode analysis to generate an ensemble of helical peptides and successfully designed novel peptides to inhibit the anti-apoptotic receptor Bcl-xL (Fu, 2007). Molecular dynamics (MD) is also another accessible method for diversifying an initial structure and in one case was able to produce an ensemble that was able to explore sequence space in a couple of common folds similar to the sequence space explored by the corresponding homologous families (Ding and Dokholyan, 2006).
Recently, methods that explicitly model specific, naturally occurring protein motions have been used to generate biophysically relevant ensembles. One such method models so-called ‘backrub motions’ (Smith and Kortemme, 2008) which was inspired by the local motions observed within tripeptide segments of sub-Angstrom resolution crystal structures (Davis, 2006).
Backrub has generated ensembles that have successfully been employed to better recapitulate the side chain order parameters of NMR-determined structures (Friedland, 2008), model the structural and sequence diversity observed experimentally in ubiquitin (Friedland, 2009), and recapitulate the phage display-determined sequence library of the Herceptin-HER2 interface (Babor, 2012). Another potential application for modeling backbone motions is kinematic closure (KIC), a protocol for sampling protein loop motions inspired by inverse kinematics developments in robotics (Mandell, 2009). KIC has also been demonstrated to be able to produce ensembles that recapitulate the experimental library of the Herceptin-HER2 interface (Babor, 2012). Backrub and KIC are both implemented within the Rosetta software package giving them access to well-characterized energy functions (Leaver-Fay, 2011).
In this manuscript, we investigate the degree to which random ensemble generation using backrub, kinematic closure, and molecular dynamics, performed without using knowledge of any target homolog structure, samples backbone conformations that are similar to ones observed in close target homologs or in identical sequences solved under different experimental conditions. In this paper, ensembles that produce backbone conformations that are locally more similar to a given homolog are considered ‘improved.’ We consider how improvement scales with increased sampling of backbone conformational space, both in terms of increased ensemble size and backbone diversity sampled among the members of the ensemble. Furthermore, we will characterize improvement as a function of local radius, which defines the size of local zones or substructures of a protein. We comment on how these insights into ensemble generation can be utilized to improve current ensemble-dependent protein design algorithms.
2 Methods
2.1 Construction of the benchmark dataset
Entries in the non-redundant set of protein structures ‘PDBselect’ (Hobohm and Sander, 1994) were selected that had at least 60% sequence identity and aligned with no insertions or deletions, to best simulate a protein before and after computational design. Table 1 lists the 20 template/target pairs.
Table 1.
Protein target/template pairs that have gapless alignments used for assessment of target-blind template backbone diversification, sorted by target/template RMSD orig. = template/target
| Template | Target | Length | %ID | RMSD |
% |
%IM |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| orig. | min. | Helix | Sheet | MD | BR | KIC | Function | ||||
| 1gxuA | 1gxtA | 88 | 99 | 0.084 | 0.278 | 24 | 41 | 54 | 6 | 58 | Hydrogenase maturation protein hypf |
| 1gd0C | 1cgqC | 118 | 99 | 0.223 | 0.602 | 27 | 29 | 54 | 25 | 74 | Macrophage migration inhibitory factor |
| 1h4gB | 1h4hD | 207 | 99 | 0.261 | 0.594 | 7 | 60 | 55 | 15 | 50 | Xylanase |
| 1i0dB | 1dpmB | 331 | 100 | 0.280 | 0.533 | 44 | 14 | 3 | 43 | 50 | Phosphotriesterase |
| 1bwsA | 1e6uA | 314 | 99 | 0.295 | 0.748 | 43 | 16 | 49 | 38 | 54 | gdp-4-keto-6-deoxy-d-mannose epimerase/reductase |
| 1fuoA | 1furB | 456 | 100 | 0.329 | 1.218 | 58 | 6 | 51 | 22 | 61 | Fumarase c |
| 1h6xA | 1dyoB | 159 | 99 | 0.352 | 0.455 | 0 | 52 | 50 | 25 | 49 | Endo-1,4-beta-xylanase y |
| 1bplB | 1e40A | 290 | 92 | 0.396 | 1.012 | 30 | 21 | 13 | 21 | 53 | Alpha-1,4-glucan-4-glucanohydrolase |
| 1fr2A | 1bxiA | 83 | 96 | 0.475 | 0.776 | 54 | 0 | 36 | 41 | 68 | Colicin e9 immunity protein |
| 1k61D | 1le8B | 58 | 94 | 0.605 | 0.376 | 66 | 0 | 11 | 78 | 73 | Mating-type protein alpha-2 |
| 1idpC | 7stdC | 147 | 99 | 0.639 | 0.857 | 26 | 51 | 52 | 53 | 65 | Scytalone dehydratase |
| 1acd_ | 1a2dB | 131 | 98 | 0.648 | 0.915 | 12 | 58 | 13 | 41 | 66 | Adipocyte lipid binding protein |
| 1danT | 1a21B | 75 | 80 | 0.731 | 0.825 | 5 | 58 | 41 | 58 | 78 | Blood coagulation factor via light chain |
| 1frwA | 1h4eA | 185 | 99 | 0.750 | 0.875 | 32 | 22 | 13 | 46 | 78 | Molybdopterin-guanine dinucleotide biosynthesis |
| 1a6i_ | 1du7A | 193 | 95 | 0.811 | 2.213 | 73 | 0 | 11 | 46 | 76 | Tetracycline repressor protein class d |
| 1eaqB | 1h9dC | 125 | 98 | 0.823 | 0.873 | 6 | 35 | 11 | 41 | 69 | Runt-related transcription factor 1 |
| 1jcdC | 1eq7A | 50 | 78 | 0.850 | 1.095 | 96 | 0 | 14 | 89 | 89 | Major outer membrane lipoprotein |
| 1khoB | 1gygB | 370 | 85 | 1.063 | 1.009 | 40 | 15 | 15 | 53 | 60 | Clostridium perfringens alpha-toxin |
| 1fqkC | 1cipA | 317 | 78 | 1.126 | 1.226 | 45 | 13 | 47 | 52 | 66 | Chimera of guanine nucleotide-binding protein |
| 1im2A | 1do0F | 346 | 84 | 1.744 | 1.804 | 46 | 10 | 38 | 56 | 46 | ATP-dependent hsl protease ATP-binding subunit |
RMSD min., RMSD after energy minimizing template; %IM, percent of positions where the 8 Å local RMSD to target decreased by at least 10% in at least one of 40 ensemble members, considering only the 50% most divergent positions.
Additionally, three extra template/target pairs were selected for case study: 1foeB-1a4rA (1.6 Å RMSD, 72% ID), 1fr2A-1bxiA (0.77 Å RMSD, 96% ID), and 1adwA-1bawA (2.4 Å RMSD, 34% ID). These three pairs were selected because they span the RMSD and sequence identity ranges and served as useful guides for analyzing the effect of ensemble size and diversity on local substructural improvement. For the remainder of this manuscript, the first protein of the pair will be the starting template structure and the second will be the target structure.
2.2 Generating an ensemble from an initial scaffold
An ensemble in the context of this manuscript is a set of similar backbone conformations. An ensemble may be generated using molecular dynamics (MD) (Ding and Dokholyan, 2006), kinematic closure (KIC) (Mandell, 2009), or backrub moves (BR) (Smith and Kortemme, 2008). A scoring function or energy function is used to select plausible conformers from among the astronomical number of ways of perturbing a protein chain.
In this work we used BR, KIC and MD motions coupled with energy minimization to sample backbone conformational space, subject to the Rosetta Talaris2013 scoring function for BR and KIC within the PyRosetta software framework (Chaudhury, 2010), or the Amber94 forcefield for MD. PyRosetta provides Python bindings to several of the protocols implemented within Rosetta 3.5 (Leaver-Fay, 2011). MD ensembles were generated using the 2013 version of the Molecular Operating Environment (MOE) licensed by the Chemical Computing Group (Montreal, Canada).
For BR ensembles, each template was energy minimized in torsion space in order to center the ensemble on the nearest energy minimum (note that Rosetta can energy minimize as a function of Cartesian coordinates, or as a function of dihedral angles. The latter is called ‘torsion space’ minimization). To diversify the starting structure, we made random backrub movements at temperatures determined by a maximum RMSD value, where higher values meant higher initial temperatures. For each member of the template ensemble, a desired RMSD value was set, following a linear gradient up to the set maximum value. The maximum RMSD was set based on the template/target sequence identity. The homolog target structure was never used in template ensemble generation.
The starting structure was subjected to ten random backrub movements, followed by torsion space minimization. If the RMSD for an ensemble member structure to the starting structure reached the targeted, gradient-determined value, the simulation was ended. If the RMSD surpassed the desired value beyond a tolerance, the minimization and backrub moves were undone, the backrub temperature was decreased, and the process was repeated. If the RMSD was too low, ten more random backrub movements were performed. If 100 rounds of backrub/minimization were performed without reaching the desired RMSD, the temperature was increased to increase the speed at which the targeted RMSD value was reached.
The final structure for each ensemble member was minimized in Cartesian space to remove the small backbone angle distortions that result from backrub movements. Alternate-residue Cartesian minimizations were performed to keep the backbone from moving too much from its final conformation. First the even residues, and then the odd residues were minimized. The backrub method was used to construct the majority of the ensembles used in subsequent analyses.
For comparison purposes, ensembles were also generated using KIC and MD. The protocol for generating ensembles using KIC is the same as for backrub, except that a KIC PyRosetta mover is used in place of a backrub mover. The KIC mover is applied on randomly selected loops of length 3 to 12 residues, to mirror backrub movements which are performed on peptides of length 3-12 residues.
MD ensembles were constructed using MOE. The initial templates were first energy minimized using the Amber94 forcefield. MD was then performed using the Amber94 forcefield and an implicit distance-based dielectric solvation model. The system was heated to a temperature of 300K over 10 ps with structures being outputted every 0.01 ps. The RMSDs of these outputted structures to the original, energy minimized structure were then computed, and structures whose RMSDs were closest to the desired RMSD gradient explained above were selected as members of the template ensemble.
2.3 Evaluating the quality of the ensemble
The quality of an ensemble was defined as the fraction of positions that moved towards the target structure. Specifically, ‘improvement’ (abbreviated ‘%IM’) is the percentage of local substructures for a template whose RMSD to the corresponding target substructure was decreased by at least 10% in at least one of the de novo members of the ensemble. Local substructures were defined to be all alpha carbons within RÅ of a given alpha carbon, where R ranged from 6 to 25. In our view, an ensemble is of high quality if its local substructures contain any members that have significantly moved towards the unseen homolog. This manuscript does not evaluate the ability of various scoring functions to actually discriminate between these improved structures, as this is a separate problem. However, good homolog structure sampling is a necessary prerequisite to good structure prediction.
3 Results
Before we could analyze the effectiveness of utilizing backbone ensembles for computational protein design purposes, we needed to investigate the ensemble generation algorithm to ensure that outputted ensembles could sample the desired amount of backbone diversity while retaining energetically realistic structures. The structures in the ensemble must be nearly isoenergetic to prevent scoring biases in a protein design algorithm. Following this confirmation we attempted to characterize the two parameters that inform ensemble generation, namely the ensemble size N and the maximum RMSD of the ensemble B. Finally, we assessed the ability of this ensemble generation protocol to produce ensembles that would be suitable for protein design applications, using gapless, sequentially similar homologs as template-target prediction pairs.
3.1 Constructing the template ensemble
One of the primary aims of our work involves constructing an ensemble that can reliably sample backbone space around the initial backbone conformation of the starting template structure. To achieve this end, we developed a computational heuristic for constructing an ensemble of template structures with linearly increasing RMSD to the starting structure up to a desired maximum value. Backbone bond angle distortions introduced by the extensive BR and KIC moves were effectively removed by Cartesian space energy minimization (see Section 2). For example, 15% of the residues in the 1adwA ensemble exhibited distorted bond geometry after backrub moves. After energy minimization this number was reduced to 1%. After minimization, the measured energies of each member of the ensemble were found to be nearly equal, ensuring that the ensemble members occupy the same well in the energy landscape.
Figure 1 shows the RMSDs to the original template for ensembles of sizes 11 and 41 constructed with backrub (e.g. 40 diversified template structures plus the minimized starting structure) for the three case studies: 1foeB-1a4rA (maximum RMSD: 1.5 Å), 1fr2A-1bxiA (maximum RMSD: 0.5 Å), and 1adwA-1bawA (maximum RMSD: 2 Å). Minimizing the energy perturbs the linear gradient (light colors), especially for low target RMSD values.
Fig. 1.
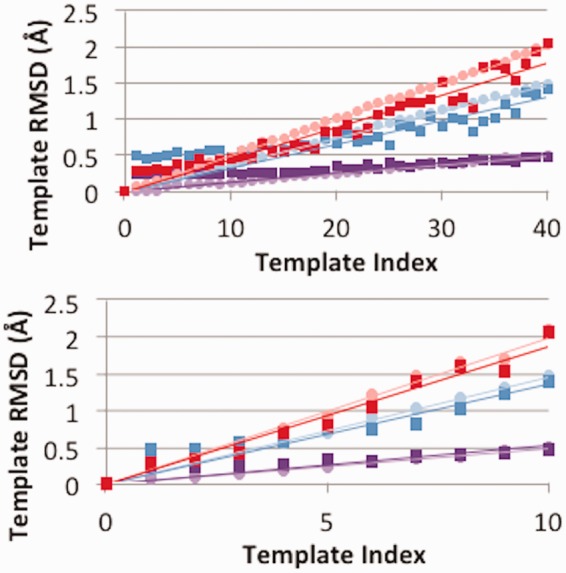
The RMSD values of perturbed template structures in an ensemble of size 41 (a) and size 11 (b) for three template-target pair case studies: 1foeB-1a4rA (blue, maximum RMSD: 1.5 Å), 1fr2A-1bxiA (violet, maximum RMSD: 0.5 Å), and 1adwA-1bawA (red, maximum RMSD: 2.0 Å). RMSDs before Cartesian minimization are depicted with light circles and RMSDs after Cartesian minimization are shown with dark boxes
Constructing ensembles on a linear RMSD gradient is inspired by the reasoning that such an ensemble explores increasingly dissimilar structural space more gradually. In a protein design application, oftentimes the design scheme has several designable positions and others that retain the wild-type amino acid. The extent to which backbone structural changes are expected is highly dependent on how much of the wildtype sequence is retained. Designed sequences that are highly similar to the original inputted sequence will likely fold to a backbone structure similar to the inputted structure, whereas increasingly dissimilar designed sequences will fold to increasingly different backbone structures, which inadvertently increases likelihood that they will not fold at all. A gradient-generated ensemble retains some structures that are only mildly dissimilar from the original starting structure, which will tend to favor wild type sequences, while still sampling some structures that are more structurally divergent and permissive of sequence changes. Traditionally, ensembles are generated by using a fixed simulation temperature, which generates de novo structures that all tend to exhibit the same amount of structural diversity. Depending on the temperature setting, this either enforces too much rigidity on the design or permits too much flexibility, which has the potential to introduce uncertainty in folding.
3.2 Characterizing ensemble generation parameters for case studies
In order to assess the utility of these ensembles for protein design purposes, we first studied the behavior of ensembles generated for three pairs of homologous proteins, henceforth referred to as ‘template/target’ pairs. An ensemble is generated using the template as a starting scaffold and is characterized by its ability to model varying local substructures of the target structure. For any given residue r between the template and the target, the local structural conformations of the ensemble members can be compared to the target local structure simply by superimposing the two regions and measuring the RMSD. An 8 Å local region, for example, is defined as all the residues that have at least one atom within 8 Å of any of the atoms of r in the template structure. A qualitative example of the local structural improvement scheme for 1e40A and 1bglB is displayed in Figure 2. This evaluation can be performed on all positions to ascertain whether the ensemble is exploring local substructural space more similar to the target. If ensembles are generating de novo structures that have local substructures that have moved closer to the target, then the idea that the ensemble is exploring biophysically relevant conformations is supported.
Fig. 2.

Template 1e40A (magenta tubes) and target 1bglB (orange tubes) locally least-squares superimposed with one of the backbone-diversified de novo structures displayed in thin gray string representation. If the gray string more closely resembles 1bglB, the unseen target, then the ensemble is deemed to have produced a locally improved structure
Two parameters contribute to the effectiveness of the ensemble at achieving this end: the number of structures in the ensemble (N) and the maximum RMSD from the original input template (B). First, we evaluated the effect of including more structures in the ensemble by finding the average %IM over all ensemble members for the three case studies (Fig. 3a). Not surprisingly, increasing N permits the ensemble to sample structural space closer to the target structure. This is simply due to the fact that more structures are being generated in the ensemble, which raises the probability of eventually generating a local substructure with improvement. The case study with the lowest template/target RMSD, 1fr2A-1bxiA with pairwise RMSD = 0.77 Å, was not able to generate improved local substructure for any of the tested values of N. However, template/target pairs with moderate (1foe-1a4r with RMSD = 1.6 Å) and high (1adw-1baw with RMSD = 2.4 Å) backbone divergence showed %IM that increased with N. Intuitively this is reasonable, since improvement is nearly impossible when the starting template structure is already very similar to the target structure. An ensemble size of 41 (40 de novo structures and the original minimized template) was used for the remainder of this analysis, although an ideal value of N would be the maximum value possible given computational constraints.
Fig. 3.

(a) Effect of the size of the template ensemble as measured by the ability to produce de novo local substructures with at least a 10% reduction in local RMSD to the corresponding target substructures, shown for the three case studies. (b) Effect of ensemble maximum RMSD on the ability of the ensemble to generate de novo local substructures with at least a 10% improvement over the original template substructures
Next we performed a similar analysis to explore the effect of changing the value of B, the maximum RMSD to the original, undiversified template (Fig. 3b). Increasing the value of B increases the level of diversity captured by the ensemble but also increases the size of the conformational space being sampled. Once again the 1fr2A-1bxi case study was unable to generate improved local substructures for the same reason given above. For the 1foeB-1a4rA case study, it appears that the optimal setting of B occurs at or near 1.5 Å, which was the closest setting to the actual pairwise RMSD between 1foeB and 1a4rA of 1.6 Å. A similar phenomenon was observed for 1adwA and 1bawA, which had an optimal value of B at 2 Å and a pairwise RMSD of 2.4 Å. The results from Figure 3 suggest that an optimal setting of B should closely follow the global pairwise RMSD between the template and target proteins. Unfortunately, this information will not be available when designing or predicting a protein in practice, since its structure is not known. However, sequence identity can be estimated from the number of designable sequence positions, and sequence identity in turn can be used as a means to estimate the expected pairwise RMSD. Sequence identity ID between the template and target may be used to estimate B according to the following function:
| (1) |
3.3 Analyzing the generality of using structural ensembles to sample improved local substructures
Having established that randomly generated ensembles for these aforementioned case studies can indeed sample local substructures closer to the corresponding target substructures, we wanted to investigate whether this process can be generalized to a wide range of protein design and prediction applications. To accomplish this, we assembled a database of 20 gapless, homologous protein pairs (Table 1) for further analysis. For comparison purposes, three sampling methods were used to generate diversity in the structural ensembles. We generated ensembles of size 40 for each of the 20 template-target pairs in Table 1 using BR, KIC, and MD, as described in Methods, in 10 replicates, comprising a total of 24 600 models including the 20 unperturbed templates. Table 1 reports the %IM across all local (8 Å) substructures on each of the 20 homolog pairs.
For individual target/template pairs, BR and MD fall within the standard error range, suggesting that any differences between these two methods are due to random energetic and sampling differences (Fig. 4). However, KIC significantly outperforms BR (P = 2e4) and MD (P = 3e7), as assessed using unweighted ANOVA. Although %IM was calculated by looking for improvement in a single ensemble member and the number of ensemble members that were improved was not counted in this metric, it should be pointed out that BR and MD tended to produce around 10 improved members for an ensemble of size 40, whereas KIC tended to produce 7 improved members.
Fig. 4.
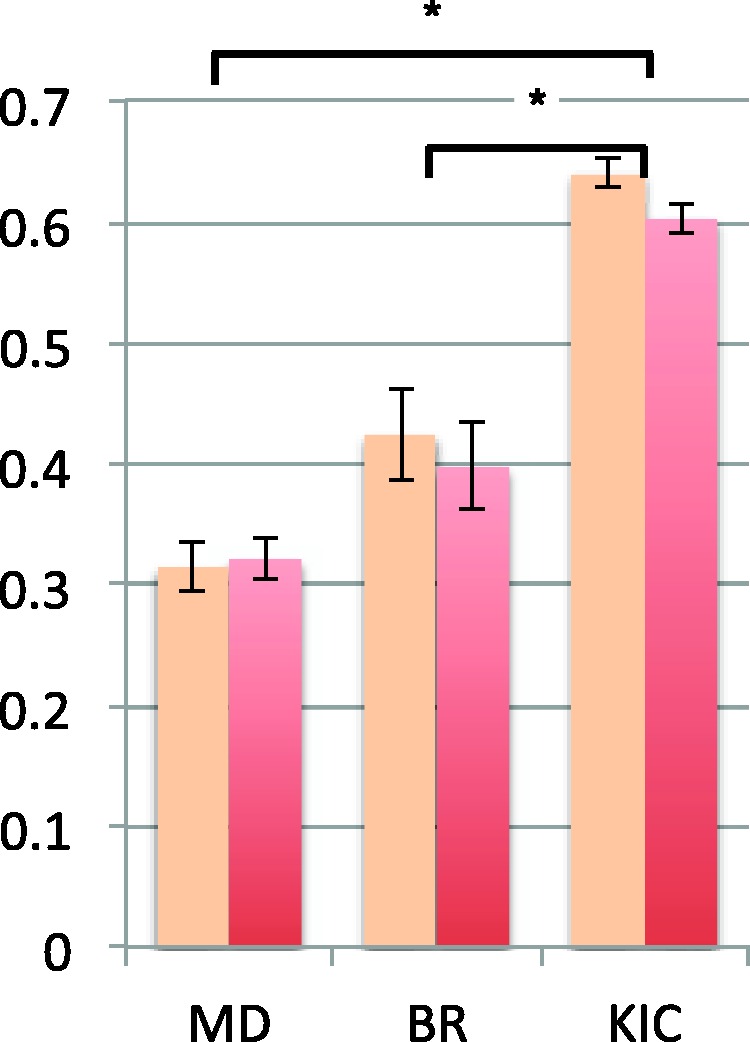
Comparison of ensemble generation methods using average improvement, per target (light color) and weighted average improvement, per target (dark color), weighted by sequence length. Error bars are average standard error over ten replicate ensembles. Stars (*) indicate statistical significance at the 0.01 level based on unweighted ANOVA
The standard deviations from native for each of the three backbone torsion angles is reported in Table 2. All three methods produced the same ‘gradient’ of global RMSDs, however, BR accomplished this diversity using smaller backbone angle shifts than MD and KIC. MD ensembles were energy minimized with the Amber94 forcefield whereas BR and KIC used the Rosetta Talaris2013 forcefield, and MD produced larger omega angle deviations, casting some suspicion on the ensembles generated by that method. KIC generated ensemble diversity using relatively large phi and psi angle shifts, but without the large omega deviations.
Table 2.
Standard deviations from native for backbone dihedral angles, over template ensembles produced by three sampling methods
| RMS deviation from native angles |
|||
|---|---|---|---|
| Method | Φ | Ψ | Ω |
| MD | 9.2 | 8.4 | 6.1 |
| BR | 4.7 | 4.6 | 1.7 |
| KIC | 9.1 | 10.2 | 1.6 |
These ensembles were characterized to ascertain whether local substructures close to the target structure could be sampled without knowledge of the target structure informing the construction of the ensemble. The results are shown in Figure 5. Similar trends can be seen on the benchmark results as were observed in the analysis of the three case studies. Not surprisingly, again there appears to be a clear trend towards ensembles with more members, denoted by the variable N.
Fig. 5.
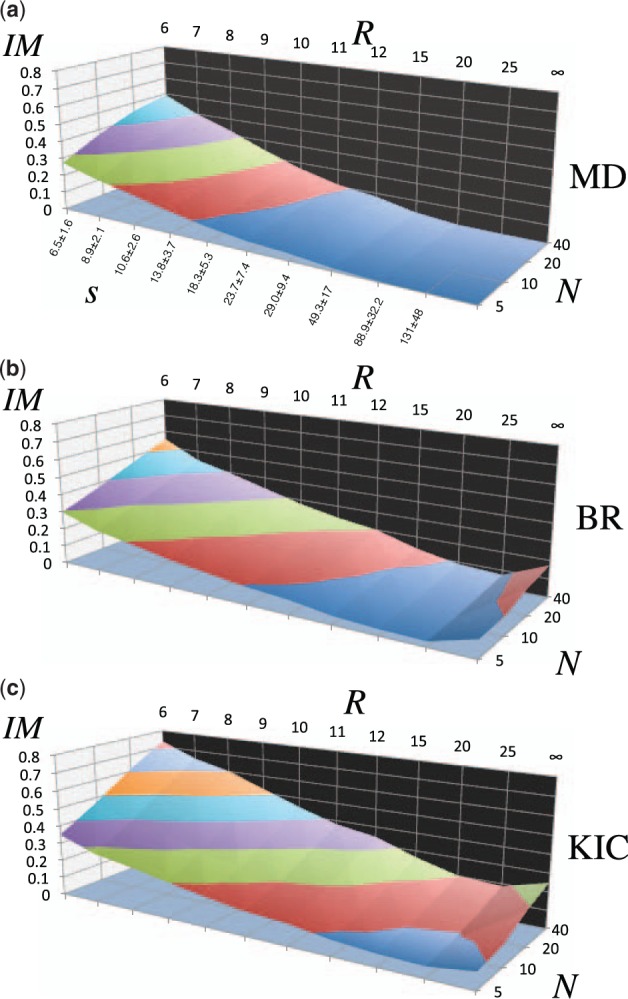
Improvement (%IM) of local substructures between the target structure and the template ensemble for a 20-member protein dataset (Table 1), averaged over 10 replicates. R is the radius of expansion that defines the local substructure and N is the number of de novo structures generated in the ensemble. s is the average number of Cα carbon atoms in the local substructure ± standard deviation is shown along the lower left in (a), for each radius R. Surfaces show the improvement for local substructures. Iso-lines illustrate the pareto front in the dimensions of N and R. (a) MD:Amber94 molecular dynamics ensembles. (b) BR: Rosetta backrub ensembles. (c) KIC: Rosetta kinematic closure ensembles
Much of the success of the previous analyses has depended on defining the local substructures adequately. The final aim of this work consists in evaluating the sensitivity of the improvement of local substructures on the radius R of expansion that defines the local substructures, also displayed in Figure 5. For this analysis, local structural improvement was evaluated for increasing radii around each position, up to a maximum of ∞ which represents the global structure. A constant ensemble RMSD value of 1 Å was used to generate the ensembles in Figure 5.
A clear trend can be seen that demonstrates that the percentage of de novo generated structures in ensembles of various sizes tends to increase as the radius that defines local substructures is decreased. Intuitively, the probability of generating globally improved de novo structures randomly without knowledge of the actual target structure should be essentially 0%, due to the large number of degrees of freedom in the conformational space of a protein. Decreasing the size of the region to be considered for an RMSD calculation increases the probability that improved de novo substructures would be sampled at random, due simply to the diminished number of degrees of freedom considered in smaller substructures. As we decreased the radius of the local region, the chances of randomly seeing a lower RMSD to the target grew steadily.
Counterintuitively, we observed that a significant percentage of templates were improved at the global level (R = ∞) by KIC and BR, but not by MD. Upon inspection of the three structures (out of 20) that improved at the global level, we found that largescale domain-domain motions were observed and that these motions were sometimes accurately predicted by the two Rosetta-based methods but not by AMBER. We believe that the trajectories of largescale motions are determined by the overall shape of the molecule and are not a unique result of the Rosetta energy function. In fact, global differences between crystal structures of a protein can be predicted by normal mode analysis (Brooks and Karplus, 1985), which models all interactions as simple harmonic functions. That the MD ensemble did not show such global improvements is probably an artifact of our choice of sampling method. We sampled at very short time intervals because AMBER trajectories quickly diverged.
The %IM value exhibits a pareto front in the two variables of R and N. The dependence of the desired minimum number of ensemble members N on the local radius R appears to be log-linear for the iso-lines in %IM. Similarly, the dependence of N on %IM for the iso-lines of R also appears to be log-linear. The results shown in Figure 5(c) for KIC were therefore fit to a log-bilinear equation, restricting to local radii between 7 and 10 Å, which are reasonable limits for energy calculations. The best fit gave the relation,
| (2) |
where (7 ≤ R ≤ 10) is the local radius, (0.3 ≤ %IM ≤ 0.8) is the desired percentage of local substructural improvement, and (N ≥ 5) is the desired minimum number of members to generate in the structural ensemble. To reproduce these results, the ensemble should be generated as described in Figure 1, along a linear gradient of RMSD whose maximum value B is predicted using Equation 1. B must be at least 1 Å, which is the approximate degree of variability seen between crystal structures of the same protein under different crystallization conditions.
An illustrative example of how to use this equation will help to demonstrate its potential utility. Assume that the protein designer establishes a goal of 80% local substructural improvement of the designable positions. Intuitively the protein designer reasons that 7 Å is an acceptable cutoff radius for accurate energy calculations. Our results suggest that such a researcher would need to generate at least N = 107 ensemble members using KIC. Suppose instead that the protein designer concludes that 8 Å is necessary for accurate energy calculations and is willing to accept 50% local structural improvement. According to the fit in Equation 2, an ensemble size of N = 25 would be acceptable for this goal.
4 Discussion
In this manuscript, we explored the effectiveness of using knowledge-free backbone ensembles for protein structure prediction applications using template-target close homologs as benchmarks. Generation of the ensembles was performed using a gradient-based approach whose behavior and characteristics were studied as a function of number of members of the ensemble and maximum ensemble diversity. Consequently, our results suggest that increasing the number of structures in the ensemble increases the likelihood of finding locally improved substructures. This probability is increased if the ensemble diversity extends to a certain optimal level, which may be adequately estimated based on the number of designable positions. These results are not surprising, however the analysis of the ensemble’s ability to generate improved local substructures randomly as a function of the radius of the local substructure is more interesting. A pareto relationship is observed between the intuitively known local radius, the number of ensemble members, and the desired percentage of local improvement. This relationship establishes how many template conformations should be sampled in order to generate a reasonable number of improved backbone structures, given a local radius.
The method of ensemble generation can have impacts on the biophysical relevance of the resulting ensemble. KIC clearly outperforms BR and MD in the task of finding the unseen target for 8 Å local regions. MD ensembles suffer from high ω values, presumably because the structure is being perturbed from ideal backbone geometry. The nature of BR and KIC moves prevents large perturbations of ω. Interestingly, KIC samples more diversity in φ and ψ than BR, even as the two methods produce ensembles with the same RMSD. BR move rotates all atoms between two randomly determined pivot Cα atoms as a group, whereas a KIC move modifies all of the backbone dihedrals. Since BR moves do not modify the internal structure of the loop spanning the two pivot atoms, it is probably comparatively more difficult for BR to sample diversity in well-packed regions of the protein, due to steric collisions. KIC can collapse and modify whole loops, which would increase the structural diversity explored in well-packed regions. KIC probably outperforms MD due to the fact that the heating step of the MD simulation imparts quick atomic accelerations at the beginning of the simulation, distorting ideal geometry. Diversity is generated very quickly in MD, requiring a sampling time of 0.01 ps to capture the small RMSD ensemble members. It should be noted that in some cases, diversity was generated so quickly that the lower end of the RMSD gradients had lower resolution, which probably contributed to the lower average performance of MD. It appears that KIC ensemble generation is superior to BR and MD, at least as carried out here, because of its greater backbone diversity and minimal distortion.
One important caveat is that our results do not imply that the scoring function is able to discriminate between optimal substructures. All that our results establish is that energy-guided but otherwise random backbone perturbations can and do sample the conformational shifts of unseen homologs. The challenge remains to develop and implement an energy function that can correctly identify the optimal de novo template substructure.
4.1 Target structure construction from the member substructures of a template ensemble
We have described and evaluated a method for ensemble generation that is inherently local in nature. Even if the improved members of the ensemble can be correctly identified by energy calculations, the task remains to build a single model from the separate pieces, since the optimal substructures are most likely dispersed across ensemble members. Local coordinates can be used to inform a construction of the global target structure by piecemeal incorporation of the coordinates of the optimal local substructures.
The results suggest an approach to protein design that should, in principle, predict global backbone changes that maximally reproduce the optimal local substructures. The algorithm might mirror the following protocol:
Generate an ensemble of N members using desired characteristics and Equation 2.
Perform synchronous protein design in the context of this ensemble.
Following design, calculate the energy of all local substructures as defined by the desired radius.
Construct the global target structure by energy minimization while restraining the local distances to their values in the optimal substructures.
All of these steps can be easily performed using the Rosetta and PyRosetta tools. Synchronous multiple template design is provided by Rosetta multi-state design. Ensemble generation and local substructural scoring can be accomplished using PyRosetta scripting, and has been incorporated in a new Rosetta graphical interface called InteractiveROSETTA (Schenkelberg and Bystroff, 2015). Step 4 can be performed using distance restraints in either Rosetta or PyRosetta in the context of an energy minimization using the harmonic restraint function provided by Rosetta.
Acknowledgements
The authors would like to acknowledge the Rosetta Commons community for their tireless efforts to produce and maintain the Rosetta protein modeling software, which was integral to this work. Furthermore, the authors would like to thank the PyRosetta developers that rendered Rosetta more usable for custom use in the form of Python bindings for the underlying C ++ Rosetta code.
Funding
This work was supported by NIH grant R01 GM099827 to C.B.
Conflict of Interest: none declared.
References
- Babor M. et al. (2012) Assessment of flexible backbone protein design methods for sequence library prediction in the therapeutic antibody Herceptin-HER2 interface. Prot. Sci., 20, 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B., Karplus M. (1985) Normal modes for specific motions of macromolecules: application to the hinge-bending mode of lysozyme. Proc. Natl. Acad. Sci., 82, 4995–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S. et al. (2010) PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics, 26, 689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran F.V. et al. (2005) Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. J. Am. Chem. Soc., 127, 1346–1347. [DOI] [PubMed] [Google Scholar]
- Dahiyat B.I., Mayo S.L. (1997) De Novo protein design: fully automated sequence selection. Science, 278, 82–87. [DOI] [PubMed] [Google Scholar]
- Dantas G. et al. (2003) A large scale test of computational protein design: folding and stability of nine completely redesigned globular proteins. J. Mol. Biol., 332, 449–460. [DOI] [PubMed] [Google Scholar]
- Davis I.W. et al. (2006) The backrub motion: how protein backbone shrugs when a sidechain dances. Struct., 14, 265–274. [DOI] [PubMed] [Google Scholar]
- Desjarlais J.R., Handel T.M. (1999) Side-chain and backbone flexibility in protein core design. J. Mol. Biol., 289, 305–318. [DOI] [PubMed] [Google Scholar]
- Ding F., Dokholyan N.V. (2006) Emergence of protein fold families through rational design. PLoS Comput. Biol., 2, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland G.D., Kortemme T. (2010) Designing ensembles in conformational and sequence space to characterize and engineer proteins. Curr. Opin. In Struct. Biol., 20, 377–384. [DOI] [PubMed] [Google Scholar]
- Friedland G.D. et al. (2009) A correspondence between solution-state dynamics of an individual protein and the sequence and conformational diversity of its family. PLoS Comput. Biol., 5, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland G.D. et al. (2008) A simple model of backbone flexibility improves modeling of side-chain conformational variability. J. Mol. Biol., 380, 757–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X. et al. (2007) Modeling backbone flexibility to achieve sequence diversity: the design of novel α-helical ligands for Bcl-xL. J. Mol. Biol., 371, 1099–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntas G. et al. (2010) Engineering a protein-protein interface using a computationally designed library. PNAS, 107, 19296–19301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobohm U., Sander C. (1994) Enlarged representative set of protein structures. Prot. Sci., 3, 522–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L. et al. (2008) De novo computational design of retro-aldol enzymes. Science, 319, 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachimiak L.A. et al. (2006) Computational design of a new hydrogen bond network and at least a 300-fold specificity switch at a protein−protein interface. J. Mol. Biol., 361, 195–208. [DOI] [PubMed] [Google Scholar]
- Kapp G.T. et al. (2012) Control of protein signaling using a computationally designed GTPase/GEF orthogonal pair. PNAS, 109, 5277–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanicolas J. et al. (2011) A de novo protein binding pair by computational design and directed evolution. Mol. Cell., 42, 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme T. et al. (2004) Computational redesign of protein-protein interaction specificity. Nat. Struct. Mol. Biol., 11, 371–379. [DOI] [PubMed] [Google Scholar]
- Kuhlman B., Baker D. (2000) Native protein sequences are close to optimal for their structures. PNAS, 97, 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B. et al. (2003) Design of a novel globular protein fold with atomic-level accuracy. Science, 302, 1364–1368. [DOI] [PubMed] [Google Scholar]
- Larson S.M. et al. (2002) Thoroughly sampling sequence space: large-scale protein design of structural ensembles. Prot. Sci., 11, 2804–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A. et al. (2011) Rosetta3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol., 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakauskas S.M., Mayo S.L. (1998) Design, structure and stability of a hyperthermophilic protein variant. Nat. Struct. Biol., 5, 470–475. [DOI] [PubMed] [Google Scholar]
- Mandell D.J. et al. (2009) Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods, 6, 551–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooers B.H.M. et al. (2003) Repacking the core of T4 lysozyme by automated design. J. Mol. Biol., 332, 741–756. [DOI] [PubMed] [Google Scholar]
- Rothlisberger D. et al. (2008) Kemp elimination catalysts by computational enzyme design. Nature, 453, 190–195. [DOI] [PubMed] [Google Scholar]
- Schenkelberg C.D., Bystroff C. (2015) InteractiveROSETTA: a graphical user interface for the PyRosetta protein modeling suite. Bioinformatics, btv492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.A., Kortemme T. (2008) Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J. Mol. Biol., 380, 742–756. 2008; [DOI] [PMC free article] [PubMed] [Google Scholar]