

Summary
IL-6-NF-κB signaling is linked to aggressive cancer phenotypes. We report a previously unknown function of integrin-linked kinase (ILK) as a critical mediator of IL-6-NF-κB signaling in breast cancer cells, which provides a rationale for exploiting ILK inhibition as a therapeutic strategy.
Abstract
Substantial evidence has clearly demonstrated the role of the IL-6-NF-κB signaling loop in promoting aggressive phenotypes in breast cancer. However, the exact mechanism by which this inflammatory loop is regulated remains to be defined. Here, we report that integrin-linked kinase (ILK) acts as a molecular switch for this feedback loop. Specifically, we show that IL-6 induces ILK expression via E2F1 upregulation, which, in turn, activates NF-κB signaling to facilitate IL-6 production. shRNA-mediated knockdown or pharmacological inhibition of ILK disrupted this IL-6-NF-κB signaling loop, and blocked IL-6-induced cancer stem cells in vitro and estrogen-independent tumor growth in vivo. Together, these findings establish ILK as an intermediary effector of the IL-6-NF-κB feedback loop and a promising therapeutic target for breast cancer.
Introduction
Triple-negative breast cancers (TNBC), which lack the expression of HER2, estrogen receptor (ER) and progesterone receptor (PR), account for ~15–25% of all breast cancer cases. The lack of effective targeted therapies for this breast cancer subtype and its high recurrence rate underscore the urgency to understand mechanisms underlying its aggressive phenotype and to identify relevant therapeutic targets that can be translated into improvements in the characteristically poor outcomes of TNBC patients (1).
In TNBC, interleukin-6 (IL-6) is typically highly expressed and correlates with low survival rates (2). The deregulated autocrine and/or paracrine signaling of IL-6 in TNBC cells impart both survival signals and aggressive phenotypes, including increased metastatic potential (3) and drug resistance (4,5). IL-6 is a pleiotropic inflammatory cytokine that can activate three oncogenic pathways, namely JAK/Stat3, PI3K/Akt and Ras/MEK/ERK, as well as stimulate its own production through an auto-feedback loop that is tightly connected to the tumor microenvironment. Mechanistically, IL-6 activates the NF-κB pathway, which, in turn, can stimulate IL-6 production via direct activation of IL-6 transcription or indirect inhibition of Let-7, thereby constituting a positive feedback loop (6). The clinical relevance of this IL-6 inflammatory loop is illustrated by recent reports that it represents a key mechanism by which HER2-positive breast cancer cells develop trastuzumab resistance through the expansion of the cancer stem cell population (7), and that IL-6-mediated Notch1 activation promotes breast cancer metastasis to bone (8). Together, these findings underscore the strong association of high IL-6 serum levels with poor prognosis in breast cancer patients (9,10).
In this study, we report a previously unknown function of integrin-linked kinase (ILK) as a molecular switch in this IL-6-NF-κB feedback loop. Data from this and other laboratories have demonstrated the role of ILK versus the mTOR–Rictor complex (mTORC2) in mediating Ser473-Akt phosphorylation in a cell line- or cellular context-specific manner (11,12). For example, while mTORC2 acts as phosphoinositide-dependent kinase (PDK)2 in nonaggressive MCF-7 cells, ILK plays a major role in facilitating the phosphorylation of Akt at Ser473 in more aggressive MDA-MB-468 and MDA-MB-231 cells. Here, we show that exposure of MCF-7 cells to IL-6 through stable expression or exogenous administration led to increased ILK expression, accompanied by concomitant increases in Ser-473 Akt phosphorylation, and that this IL-6-induced Akt phosphorylation could be blocked by shRNA-mediated knockdown or pharmacological inhibition of ILK. Conversely, stable knockdown of IL-6 by shRNA in IL-6-producing MDA-MB-231 and SUM-159 cells suppressed ILK expression. Pursuant to these findings, we obtained evidence that IL-6-induced ILK expression through the Stat3-cyclin D1/cyclin-dependent kinase (CDK)2-E2F1 signaling cascade, and that ILK, in turn, activated Akt/NF-κB signaling, leading to increased IL-6 production. Disruption of this IL-6-NF-κB signaling loop via shRNA-mediated knockdown or pharmacological inhibition of ILK by a novel small-molecule inhibitor, T315 (13) blocked IL-6-induced cancer stem cell (CSC)-like properties and estrogen-independent tumor formation in vivo of MCF-7IL-6 cells. Together, these findings suggest that ILK represents a therapeutically relevant target in breast cancer in response to autocrine and/or paracrine IL-6 signaling.
Materials and methods
Agents, reagents, plasmids, siRNA and statistical considerations
Detailed descriptions of chemical and biochemical agents, plasmids, siRNA and antibodies used in this study, as well as extended details of statistical considerations are provided in the Supplementary Materials and Methods, available at Carcinogenesis Online.
Cell culture
MCF-7, MDA-MB-468 and MDA-MB-231 cells were purchased from the American Type Culture Collection (Manassas, VA), and maintained in RPMI 1640 medium (Life Technologies; Grand Island, NY) supplemented with 10% fetal bovine serum, 100U/ml penicillin, 100 µg/ml streptomycin and 50 µg/ml gentamycin B. SUM-159 cells were obtained from Asterand Bioscience (Detroit, MI), and maintained in Ham’s F-12 (Life Technologies), supplemented with 5% fetal bovine serum, insulin (5 µg/ml), and hydrocortisone (1 µg/ml), 100U/ml penicillin, 100 µg/ml streptomycin and 50 µg/ml gentamicin B. MCF-7IL-6 stable line was a kind gift from Nicholas J. Sullivan at The Ohio State University (14). All cell lines were used in less than 6 months of continuous passage after acquisition, tested for mycoplasma contamination using the LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich), and authenticated by the cell bank source using short tandem repeat profiling. MDA-MB-231TRE- shIL-6, SUM-159TRE-shIL-6 and, MDA-MB-231TRE-shILK, MDA-MB-468 TRE-shILK stable lines were built up via lentiviral infection and puromycin selection. MDA-MB-231TRE-shILK/CA-Akt and MDA-MB-231TRE-shILK/RelA stable lines were further built up via GFP-positive cell sorting after transfection. Cells were incubated at 37°C in a humidified incubator containing 5% CO2.
Transfection
Cells were transfected with plasmids or siRNAs using an Amaxa Nucleofection system (Amaxa Biosystems, Gaithersburg, MD) or Lipofectamine 2000 (Life Technologies, Carlsbad, CA) according to the manufacturers’ instructions.
Lentivirus preparation and infection of breast cancer cell lines
Lentiviral plasmids were cotransfected with Addgene 3rd Generation Packaging Systems (pMDLg/pRRE [#12251], pRSV-Rev [#12253] and pMD2.G [#12259]) in 293T cells according to a standard calcium phosphate transfection procedure from the manufacturer. The collection of viral particles for infection of target cells, and selection of stable clones by exposure to puromycin (0.5–2 µg/ml) and G418 (250 µg/ml) were performed as previously reported (12).
Cell viability assays
Cell viability was assessed by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay in six replicates as previously reported (12).
Cell proliferation assay
The CellTiter-GLO® Luminescent Assay (Promega, Madison, WI), which quantifies intracellular ATP as an indicator of cell viability, was used to assess cell proliferation. MDA-MB-231 cells were seeded at a density of 2×104 cells/well in 24-well plates. Viable cell numbers were determined on the next day (designated day 0) and subsequently at days 1, 2 and 3, according to the manufacturer’s instructions. Luminescence intensities were measured with a Promega GloMax® 96 microplate luminometer.
RNA isolation and real-time qPCR
Cells were washed once with phosphate-buffered saline (PBS) and total RNA was isolated with TRIzol (Thermo Fisher Scientific, Waltham, MA) and reverse-transcribed into cDNA using the iScript™ cDNA Synthesis Kit (Bio-Rad; Hercules, CA). Real-time qPCR was performed on a CFX Connect™ Real-Time PCR Detection System using SsoAdvanced™ SYBR® Green Supermix (Bio-Rad). Primer sequences are provided in Supplementary Table 1, available at Carcinogenesis Online. All samples including the control without template were assayed in triplicate. The relative number of target transcripts was normalized to the number of human 18S transcripts found in the same sample. The relative quantification of target gene expression was performed with the comparative cycle threshold (CT) method.
Chromatin immunoprecipitation
After crosslinking with 1% formaldehyde for 10min at room temperature, cells were exposed to 125mM glycine followed by two washes with ice-cold PBS, and whole cell lysates were prepared with chromatin immunoprecipitation (ChIP) lysis buffer (50mM HEPES-KOH pH 7.5, 140mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS with protease inhibitor cocktail; 10min on ice). Cellular DNA fragments of 200–600bp in size were generated by sonication, followed by centrifugation at 12000rpm at 4°C for 15min. For immunoprecipitation, supernatants were incubated with anti-E2F1 or β-actin antibodies at 4°C overnight, followed by protein A/G agarose beads (Santa Cruz) at 4°C for 2h. The immunocomplexes were washed twice with the following sequence of buffers: ChIP lysis buffer, high-salt wash buffer (ChIP lysis buffer containing 500mM NaCl), LiCI wash buffer (10mM Tris, pH 8.0, containing 250mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate and 1mM EDTA) and then TE buffer (10mM Tris, pH 7.5, containing 1mM EDTA). The immunoprecipitates were eluted from the beads using 150 µl of elution buffer (50mM Tris pH 8.0, 1% SDS and 10mM EDTA), and incubated at 65°C overnight to reverse crosslinks. After incubation with RNase A (0.5mg/ml) at 65°C for 4h, DNA was extracted with a PCR purification kit (Qiagen, Valencia, CA), and analyzed by PCR using the e2TAK taq DNA polymerase (Clontech Laboratories, Mountain View, CA). The sequences of the primers used to amplify the proximal ILK promoter region are provided in Supplementary Table 1, available at Carcinogenesis Online.
Reporter assays
The NFκB and E2F transactivation activities were measured using Cignal Reporter Assay Kits (CCS-013L and CCS-003L, respectively; Qiagen) according to the manufacturer’s instructions. The reporter plasmids were transfected by Lipofectamine 2000 (Life Technologies), and the Dual-Luciferase Reporter Assay System (Promega) was used for detecting luciferase luminescence with a Promega GloMax® 96 microplate luminometer.
Electrophoretic mobility shift assay
Nuclear extracts were prepared by NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific), and double-stranded electrophoretic mobility shift assay (EMSA) probes were annealed from oligonucleotides with end-labeled biotin (Thermo Fisher Scientific). The oligonucleotide sequences of the ILK promoter region used as the probe are listed in Supplementary Table 1, available at Carcinogenesis Online. The LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific) was used to perform EMSA according to the manufacturer’s instruction. Briefly, 4 μg of nuclear extract in 20 μl binding buffer [5ng/μl Poly (dI-dC), 2.5% glycerol, 5 μg/μl bovine serum albumin (BSA), 1mM DTT, 100mM KCl, 0.2mM EDTA] was incubated with 2nM biotin-labeled probe at room temperature for 20min. For cold competition analysis, nuclear extracts were pre-incubated with 50-fold unlabeled ILK promoter probe, E2F1 consensus binding probe (sc-2507, Santa Cruz Biotechnology, Santa Cruz, CA) or E2F1 mutant binding probe (sc-2508, Santa Cruz, CA) for 15min on ice before the addition of biotin-labeled probe. DNA-protein complexes were resolved on 6% native polyacrylamide gels at 100V for 1.5h, and wet-transferred to Biodyne B Nylon Membrane (Thermo Fisher Scientific) at 250 mA overnight at 4°C. After cross-linking with the membrane by UV, the DNA-protein complex was detected by chemiluminescence according to the manufacturer’s instruction.
Enzyme-linked immunosorbent assay
IL-6 levels in culture medium were detected 24h after seeding equal numbers of cells on 12-well plates using the Human IL-6 DuoSet ELISA assay kit (R&D System; Minneapolis, MN) according to the manufacturer’s instruction.
Immunoblot analysis
Growing cells were harvested by scraping and lysed in the presence of RIPA lysis buffer containing protease inhibitor cocktail. An equal amount of protein with SDS sample buffer (60mM Tris–HCl, pH 6.8, 2% SDS, 10% glycerol, 0.01% bromophenol blue and 5% 2-mercaptoethanol) from each sample was loaded per lane, separated by SDS-PAGE, and transferred onto an NC membrane, and proteins were probed with specific antibodies. Secondary antibodies conjugated to horseradish peroxidase and Western Lighting Chemiluminescence Reagent Plus (Perkin-Elmer; Waltham, MA) were used to develop images.
Immunofluorescence
Cells were seeded on round-cover glasses in 24-well culture plates. After treatments, cells were fixed, permeabilized, blocked in 1% BSA in PBS buffer, and then incubated with primary antibodies to Stat3 (1:200 dilution) followed by Alexa 488-conjugated secondary antibodies (1:200 dilution), each for 2h in PBS at room temperature. Nuclei were stained with 4′6-diamidino-2-phenylindole (DAPI) contained in the Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Images were obtained with an Olympus FV1000 Filter Confocal Microscope (Olympus Corp., Japan) with optics field (40×).
Tissue microarrays and immunohistochemical analysis
Tissue microarray (TMA) slides from the Breast Cancer Progression TMA (Sets 4, 6, and 8) were provided by the NCI Cancer Diagnosis Program (CDP). Other investigators may have received slides from these same array blocks. Each TMA block contained 130–133 cores taken from paraffin-embedded specimens, including 112–115 breast cancer and normal breast specimens plus 18 control cores. Ductal carcinoma in situ and normal breast tissue cores were 1.5mm, and the remaining cores in the set were 0.6mm in diameter. Data provided for each case include tumor size, TNM stage, number of nodes positive, tumor grade, age at diagnosis and race. The TMA were deparaffinized in xylene and rehydrated in graded alcohol, and antigen retrieval was done by incubating slides for 20min in DAKO target retrieval solution (Dako, Carpinteria, CA) heated to 125°C. After cooling to 90°C for 10s, the slides were then cooled further in the same container for 10min, and incubated with 3% H2O2 for 10min. Non-specific binding was blocked by incubating slides in normal goat serum containing avidin for 30min. Anti-ILK antibody (1:100 dilution, #3856, Cell Signaling Technology, Beverly, MA) or anti-IL-6 antibody (1:150 dilution, ab6672, Abcam, Cambridge, UK) was incubated with slides in DAKO antibody diluent with background reducing component for 30min in a humidity chamber. Slides were then incubated with biotinylated goat anti-rabbit antibody (1:1000 dilution) for 30min, followed by VECTASTAIN Elite ABC Reagent (Vector Laboratories) for 30min, and DAB Peroxidase Substrate (Vector Laboratories) for 5min, and then counterstained with hematoxylin for 30s. TMA results were expressed in terms of staining intensities (0, 0.5, 1, 1.5, 2, 2.5 and 3) as scored by a board-certified pathologist, who was blinded to the corresponding clinical outcomes.
Mammosphere formation assays
A total of 500 single cells/well were plated on ultra-low attachment 24-well plates (Corning Inc.; Union City, CA) and maintained in MammoCult™ Human Medium (Stemcell Technologies; Vancouver, BC, Canada). Cells were treated with test agent or DMSO for 7 days, and the number of mammospheres with the diameters greater than 50 µm, was counted at 40× magnification using a microscope fitted with a ruler. All experiments were performed at least in triplicate.
ALDEFLUOR assays
Cell populations with high ALDH enzymatic activity were isolated using the ALDEFLUOR kit (StemCell Technologies) as described in the manufacturer’s instructions. Briefly, treated cells were trypsinized, harvested and suspended in buffer containing ALDH substrate (BAAA 1mM per 1×106 cells) and incubated at 37°C for 30min. The specific ALDH inhibitor, DEAB (50mM), was used as negative control. Using a FACSCalibur flow cytometer, ALDEFLUOR fluorescence was excited at 488nm and fluorescence emission was detected using a standard fluorescein isothiocyanate filter. The gating region of the ALDHbr population was established based on negative controls.
Flow cytometry
MDA-MB-468 cells were trypsinized, resuspended and incubated with APC conjugated mouse antihuman CD44 antibody (559942) and PE conjugated mouse antihuman CD24 antibody (555428) (BD Biosciences, Franklin Lakes, NJ) for 30min on ice in PBS as manufacturer’s suggested dilution. Double negative staining and single CD44, CD24 staining were used as control to define CD44+CD24− population. Side and forward scatter were used to exclude out debris and cell doublets. After washed of staining, CD44+CD24− and non-CD44+CD24− cells were sorted out by FACSAria III flow cytometer for further experiments.
Animal models
Female athymic nude mice (Athymic NCr-nu/nu; 5–6 weeks of age; NCI/Charles River Laboratories, Frederick, MD) were group-housed under conditions of constant photoperiod (12h light: 12h dark) with ad libitum access to sterilized food and water. All experimental procedures using mice were done in accordance with protocols approved by The Ohio State University Institutional Animal Care and Use Committee. To assess the mechanism by which ILK contributes to tumor growth in vivo, subcutaneous xenograft tumors were established in female athymic nude mice by injection of MCF-7IL-6/TRE-shILK, MDA-MB-231TRE-shILK, MDA-MB-231TRE-shILK/Akt-CA and MDA-MB-231TRE-shILK/RelA cells (1×106 cells/0.1ml in Matrigel). Beginning one day after tumor cell injection, mice were randomly assigned to receive doxycycline (2mg in 0.2ml) or sterile water once daily by oral gavage. Tumors were measured weekly, in an unblinded manner, with calipers and volumes were calculated using the formula, V = 0.52 × (width2 × length). Body weights were measured weekly. To assess the effect of T315 on IL-6-induced tumor growth, subcutaneous tumors were established by injection of MCF-7IL6 or parental MCF-7 cells (2×106 cells/0.1ml in Matrigel). Tumors and body weights were measured as described above. Mice with established MCF-7IL6 tumors (~50mm3) were randomized to two groups (n = 6) that received T315 (50mg/kg) or vehicle (10% DMSO/0.5% methylcellulose/0.1% Tween 80 in sterile water) twice daily by oral gavage.
Statistical analysis
Quantitative data are presented as means ± SD from three independent experiments, each performed in triplicate, unless indicated otherwise in the figure legends. The correlation between IL-6 and ILK expression in the breast cancer TMA was assessed by Pearson product moment correlation using R software package (version 3.0.1.; http://cran.r-project.org/). R was also used to perform survival analysis by the Kaplan-Meier method and log-rank test to compare sub-groups for significance. All other comparisons of group means were analyzed using two-tailed unpaired t-tests with assumptions of normality and equal variance (GraphPad Prism 5, GraphPad Software, Inc., La Jolla, CA). Differences were considered significant at P < 0.05.
Results
Evidence that IL-6 regulates ILK expression
The premise that ILK is an important mediator of the IL-6-NF-κB signaling loop was supported by the positive correlation between IL-6 production and ILK expression, at both protein and mRNA levels, in MCF-7 cells and three TNBC lines, including the PTEN-deficient MDA-MB-468, and the PTEN-positive SUM-159 and MDA-MB-231 (Figure 1A). These three TNBC cell lines were characterized by high secretion of IL-6 (MDA-MB-231, 120 pg/ml; SUM-159, 85 pg/ml; MDA-MB-468, 68 pg/ml) that paralleled expression levels of ILK, and the low levels of IL-6 in MCF-7 cells (0.1 pg/ml) correlated with low ILK expression. The activation status of Akt and Stat3 varied among these three cell lines due to differences in their PTEN functional status and endogenous IL-6 expression levels, respectively. While MCF-7, SUM-159 and MDA-MB-231 cells express functional PTEN, MDA-MB-468 cells are PTEN-deficient (15), which underlies their substantially upregulated phospho-Akt level relative to the other three cell lines (16). Several lines of evidence suggest that IL-6 regulates ILK expression in these cell lines. For example, activation of IL-6 signaling in MCF-7 cells by exogenous IL-6, as indicated by increased Stat3 phosphorylation, elevated ILK expression in a dose-dependent manner, accompanied by parallel increases in phospho-Akt (Figure 1B, left). This IL-6-induced upregulation of ILK was also evident in an IL-6-overexpressing stable clone of MCF-7 cells [MCF-7IL-6] (3) (Figure 1B, center), which secreted high levels of IL-6 into the medium (1ng/ml) relative to IL-6-deficient MCF-7 cells (right). Furthermore, we obtained two lines of evidence to verify that ILK was an IL-6-inducible kinase. First, exposure of MDA-MB-231 cells to IL-6 receptor (IL-6R) neutralizing antibodies led to decreased expression of ILK (Figure 1C). Second, we generated stable clones of MDA-MB-231 and SUM-159 cells that expressed doxycycline-inducible IL-6 shRNA. As shown, doxycycline-induced shRNA-mediated knockdown of IL-6 in MDA-MB-231 and SUM-159 cells suppressed ILK expression at both mRNA and protein levels (Figure 1D). The expression of non-specific control shRNA in stable MDA-MB-231 clones, in the presence and absence of doxycycline, had no effect on the levels of ILK protein or IL-6 mRNA, thereby ruling out off-target effects (Figure 1E). This correlation between IL-6 and ILK expression in cell lines was confirmed in clinical specimens included in the Breast Cancer Progression TMA Analysis Set (NCI Cancer Diagnosis Program, Rockville, MD). Immunohistochemical evaluation of IL-6 and ILK expression in 230 breast tumor samples showed a positive correlation between IL-6 and ILK expression (Pearson’s correlation coefficient (r) = 0.473; P < 0.0001) (Figure 1F). Subsequent subgroup analysis correlated high ILK expression (intensity, 2–3) with low relapse-free survival probability (P = 0.045) among patients with high IL-6 expression (intensity, 2–3) (Figure 1G). Together, these findings suggest a putative role of ILK as a downstream effector of IL-6 in breast cancer. The clinical information for all patient samples in the TMA, along with the corresponding scores for the IL-6 and ILK immunohistochemical staining intensities, are available in the format provided by the NCI CDP as Supplementary Tables 2A–C, available at Carcinogenesis Online.
Figure 1.

ILK expression correlates with IL-6 levels in breast cancer cell lines and clinical samples. (A) Left, Western blot analysis of the differential expression/activation status of ILK and various IL-6 downstream signaling markers, among four different breast cancer cell lines, MCF-7, MDA-MB-468 (468), SUM-159 (159) and MDA-MB-231 (231). Right, ELISA analysis of IL-6 levels in culture medium (top) and qPCR analysis of ILK mRNA levels in these four cell lines. Data are presented as mean ± SD (n = 3). (B) Western blot analyses of the expression/activation status of ILK and the aforementioned IL-6 downstream signaling markers in MCF-7 cells in response to 24-hour treatment with different concentrations of exogenous IL-6 (left), and in MCF-7 and MCF-7IL-6 cells (middle). Right, ELISA analysis of IL-6 levels in culture medium of MCF-7 and MCF-7IL-6 cells. (C) Western blot analysis of the effect of an IL-6 receptor (IL-6R) neutralizing antibody (10 μg/ml, 72-h treatment) or IgG control antibody on the expression of ILK in MDA-MB-231 cells. (D) qPCR and/or Western blot analyses of the effects of doxycycline (Dox)-inducible shRNA-mediated knockdown of IL-6 on the mRNA expression of IL-6 (left) and the mRNA and protein expression of ILK (center and right) in MDA-MB-231 (231) and SUM-159 (159) cells. Data are presented as mean ± SD (n = 3). (E) Effect of stable expression of non-specific control (Ctl) shRNA in MDA-MB-231 cells on ILK protein abundance (by western blot), IL-6 mRNA levels (by qPCR), and NF-κB reporter activity. Data are presented as mean ± SD (n = 3). (F) Immunohistochemical analysis of IL-6 and ILK in human breast tumors (n = 230) on a TMA. Left, Exemplar images showing cytoplasmic staining for IL-6 and ILK. Staining intensities were semi-quantitatively scored as follows: 0, negative; 1, weak; 2, moderate; 3, strong. Right, Pearson product-moment correlation analysis between IL-6 and ILK expression levels in the same TMA samples. (G) Subgroup analysis showing correlation between high (intensity, 2–3) (n = 29) or low (intensity, 0 < 2) (n = 89) ILK expression and survival time among patients with high IL-6 expression (intensity, 2–3). Data for the IL-6low/ILKlow subgroup is also shown (n = 93). The number of samples from the IL-6low/ILKhigh subgroup (n = 4) was inadequate for analysis.
IL-6 transcriptionally activates ILK gene expression through E2F1
To shed light onto how IL-6 activates ILK gene expression, we used the TRANSFAC database to predict transcription factor binding sites on the ILK promoter [http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3] (17,18). This analysis revealed 14 putative E2F1 binding motifs on the ILK promoter, located within a region approximately −506 to −89 nucleotides upstream from the transcription start site, and far exceeding the numbers of sites for the other transcription factors identified within the promoter region, including NF-κB (2 sites), Stat3 (1 site) and c-Jun (2 sites) (Supplementary Figure 1, available at Carcinogenesis Online). Functionally, the role of E2F1 in regulating ILK gene expression was supported by the ectopic expression of E2F1, which, unlike that of NF-κB or Stat3, significantly increased ILK promoter-luciferase reporter activity (Figure 2A, left) and mRNA levels (right) in MCF-7 cells. We also obtained several lines of evidence implicating E2F1 in IL-6-induced transcriptional activation of ILK gene expression. The siRNA-mediated silencing of E2F1 in MCF-7IL-6 cells resulted in decreased promoter-luciferase activity and expression of ILK at both mRNA and protein levels (Figure 2B). Similarly, siRNA-mediated silencing of E2F1 also reduced ILK expression in MDA-MB-231 cells (Figure 2C, upper), which exhibit high IL-6 expression, while overexpression of E2F1 in MCF-7 cells, which express low levels of IL-6, increased ILK expression (lower).
Figure 2.

IL-6 stimulates E2F1-dependent transcriptional activation of ILK gene expression. (A) Left, Effects of the ectopic expression of E2F1, NF-κB, and Stat3, relative to the pCMV control, on ILK promoter-driven luciferase activity in MCF-7 cells (upper panel; data are presented as mean ± SD, n = 3). Lower panel, Ectopic expression of protein was confirmed by western blot. Right, E2F1 and ILK mRNA levels in MCF-7 cells ectopically expressing E2F1 as determined by qPCR. Data are presented as mean ± SD (n = 3). (B) Left, Effects of siRNA-mediated knockdown of E2F1 on ILK promoter-driven luciferase activity (upper panel; data are presented as mean ± SD, n = 3) and ILK protein levels (lower) in MCF-7IL-6 cells. Right, E2F1 and ILK mRNA levels, as determined by qPCR, in MCF-7IL-6 cells with siRNA-mediated knockdown of E2F1. Data are presented as mean ± SD (n = 3). (C) Western blot analyses of the effects of ectopic expression of E2F1 on ILK expression in MCF-7 cells (upper panel), and siRNA-mediated knockdown of E2F1 on ILK expression in MDA-MB-231 cells (lower panel). (D) ChIP (left) and EMSA (right) analyses of the effect of exogenous IL-6 (50ng/ml) on E2F1 binding to the ILK promoter. Left, upper, Diagram of the ILK promoter showing the region containing E2F1 binding sites and the fragments amplified by the two primer pairs used for RT-PCR, of which pair #2 served as negative control. (E) EMSA analysis of the specificity of E2F1 binding to putative E2F1 binding sites on ILK promoter (-368 to -335) using MDA-MB-231 cell nuclear lysate. Lane 1 (probe-only control) shows non-specific binding and free probe. Lane 2 shows the shift after E2F1 binding. Lane 3 shows that the shift of protein-DNA complex includes ILK promoter probe by inclusion of a 50-fold molar excess of unlabeled probe. Lane 4 and 5 show that the shift of protein-DNA complex includes E2F1 by inclusion of 50-fold molar excess of E2F1 consensus probe or mutated E2F1 consensus probe, respectively.
Furthermore, ChIP analysis indicated that this IL-6-induced upregulation of ILK expression was associated with increased E2F1 binding to the ILK promoter in the E2F1 binding site-rich region identified above in response to IL-6 in MCF-7 cells (primer pair #1; Figure 2D, left). This finding was consistent with EMSA results showing increased E2F1 binding to DNA in IL-6-treated MCF-7 cells (Figure 2D, right). The specificity of this E2F1 binding was confirmed in two ways. First, use of a primer pair outside of this E2F1 binding region (pair #2) as negative control in the ChIP assay showed no appreciable E2F1 binding following IL-6 stimulation (Figure 2D, left). Second, competitions using 50-fold molar excess of an unlabeled competition probe or E2F1 consensus probe in the EMSA substantially reduced binding of E2F1 to the ILK promoter in MDA-MB-231 nuclear extracts, while a site-directed mutant of the E2F1 consensus probe provided no such protection (Figure 2E). Together, these findings suggest that IL-6 induced ILK expression via an E2F1-dependent mechanism.
IL-6 activates E2F1 expression through Stat3/cyclin D1/CDK2 signaling
It is noteworthy that, relative to MDA-MB-468, SUM-159, and MDA-MB-231 cells, MCF-7 cells, which express lower endogenous levels of IL-6 than these TNBC cell lines, exhibited substantially lower E2F1 expression at both protein and mRNA levels, in parallel with ILK and cyclin D1 protein expression (Figure 3A). Transient treatment of MCF-7 cells with exogenous IL-6 caused a dose-dependent increase in E2F1 and cyclin D1 expression, accompanied by parallel increases in the ILK promoter-luciferase activity and ILK protein expression (Figure 3B). Mechanistically, we attributed this IL-6-induced upregulation of E2F1 expression to the reported effect of IL-6 on Rb phosphorylation, leading to the release of E2F1 from the Rb-E2F1 complex and consequent E2F1-mediated transcription of its own and other E2F1-targeted genes (19,20). This premise was supported by the suppressive effect of the CDK2 inhibitor SU9516 (IC50, ~4 μM) (21) on IL-6-induced E2F1 transactivation activity, Rb phosphorylation, CDK2 activation, and ILK expression (Figure 3C). We obtained evidence that this CDK2 activation was, at least in part, due to the activation of CCND1 gene expression by Stat3 (22), as cyclin D1 overexpression is known to facilitate constitutive activation of CDK2 (23). As shown in Figure 3D, the Stat3 inhibitor S3I-201 (IC50, 100 μM) (24) suppressed cyclin D1 expression, Rb phosphorylation and E2F1 and ILK expression. These data suggest that the regulation of ILK expression by IL-6 is mediated by the Stat3-cyclinD1/CDK2-E2F1 signaling axis.
Figure 3.

IL-6 activates E2F1 expression through Stat3/CyclinD1/CDK2 signaling. (A) Western blot (upper) and qPCR (lower) analyses of the differential expression of E2F1, cyclin D1, and/or ILK in four breast cancer cell lines (MCF-7, MDA-MB-468 [468], SUM-159, MDA-MB-231 [231]). Data are presented as mean ± SD (n = 3). (B) Dose-dependent effect of 24-hour treatment with exogenous IL-6 on ILK promoter-driven luciferase activity (upper) and protein levels of E2F1, cyclin D1, and ILK (lower) in MCF-7 cells. Data are presented as mean ± SD (n = 3). (C) The effect of CDK2 inhibitor SU9516 (20 μM, 48-h treatment) on E2F reporter luciferase activity (upper) and expression levels of ILK and cyclin D1 protein and phosphorylation status of CDK2 and Rb (lower) in MCF-7IL-6 cells. Data are presented as mean ± SD (n = 3). (D) Western blot analysis of the effect of the Stat3 inhibitor S3I-201 (48-hour treatment) on ILK, E2F1 and cyclin D1 expression and phosphorylation status of Stat3 and Rb in MCF-7IL-6 cells. (E) The effect of shRNA-mediated knockdown of ILK in MCF-7IL-6, MDA-MB-231 and SUM-159 cells on the expression/activation status of ILK and the IL-6 downstream signaling markers, Akt and Stat3.
ILK is a downstream effector in the IL-6 signaling pathway
Based on the above findings, we hypothesized that ILK acted as a downstream effector of IL-6 signaling. To address this hypothesis, we examined the effects of shRNA-mediated knockdown of ILK on IL-6 downstream signaling markers in the IL-6-overexpressing cell lines, MCF-7IL-6, MDA-MB-231 and SUM-159, which revealed that silencing of ILK reduced the phosphorylation levels of Akt and Stat3 in all three of these cell lines (Figure 3E). In addition to ILK, mTORC2 and DNA-dependent kinase (DNA-PK) have been reported to function as PDK2 to facilitate Ser473-Akt phosphorylation in different cell types (25–27). Thus, to examine the possible role of these other PDK2 candidates as downstream effectors of IL-6 signaling, we assessed the effects of siRNA-mediated knockdown of LST8, an mTORC2 subunit (28), and the catalytic subunit of DNA-PK (DNA-PKCS) on Akt phosphorylation in MCF-7IL-6 cells. Silencing of neither protein caused appreciable changes in the p-Ser473-Akt level (Supplementary Figure 2, available at Carcinogenesis Online), suggesting that ILK is an inducible PDK2 that phosphorylates Ser473-Akt in response to IL-6.
Pursuant to the above findings, we employed a small-molecule inhibitor, T315 (structure, Figure 4A, left), to corroborate the role of ILK in mediating IL-6 signaling in breast cancer cells. T315 is a potent inhibitor of the in vitro kinase activity of immunoprecipitated ILK complex (IC50, 0.6 µM) (13) and the viability of breast cancer cells (IC50: SUM-159, 1.5 µM; MDA-MB-231, 1.8 µM; MCF-7, 2.7 µM; MCF-7IL-6, 3.5 µM; Supplementary Figure 3, available at Carcinogenesis Online). Because ILK lacks critical conserved residues in its kinase homology domain (29,30), the functional relevance of its putative kinase activity relative to ILK’s important role as a scaffold protein for integrin and growth factor signaling remains controversial. To further confirm the suppressive effect of T315 on ILK function, we exposed MCF-7 cells ectopically expressing constitutively active ILK (S343D) to T315 (2 µM). As shown, T315 was capable of inhibiting this constitutively active ILK-induced upregulation of Ser473-Akt phosphorylation (Figure 4A, middle), and, as determined by immunoprecipitation, blocked the interaction between ILK and Rictor, which is required for the ability of ILK to facilitate Ser473-Akt phosphorylation (Figure 4A, right). Together, these data suggest a kinase-independent mechanism of action by which T315 can inhibit ILK function. In MDA-MB-231, SUM-159, and MCF-7IL-6 cells, T315 dose-dependently decreased the phosphorylation/expression of Akt and Stat3 (Figure 4B), reminiscent of reductions seen with ILK shRNA. In contrast, T315O, an inactive T315 analog (structure, Figure 4A, left), showed no appreciable effect on any of these signaling markers in MDA-MB-231 cells, refuting the involvement of any nonspecific effect of T315. In addition to the inhibition of Stat3 phosphorylation, T315 suppressed Stat3 translocation into the nucleus in MDA-MB-231 cells (Supplementary Figure 4, available at Carcinogenesis Online).
Figure 4.
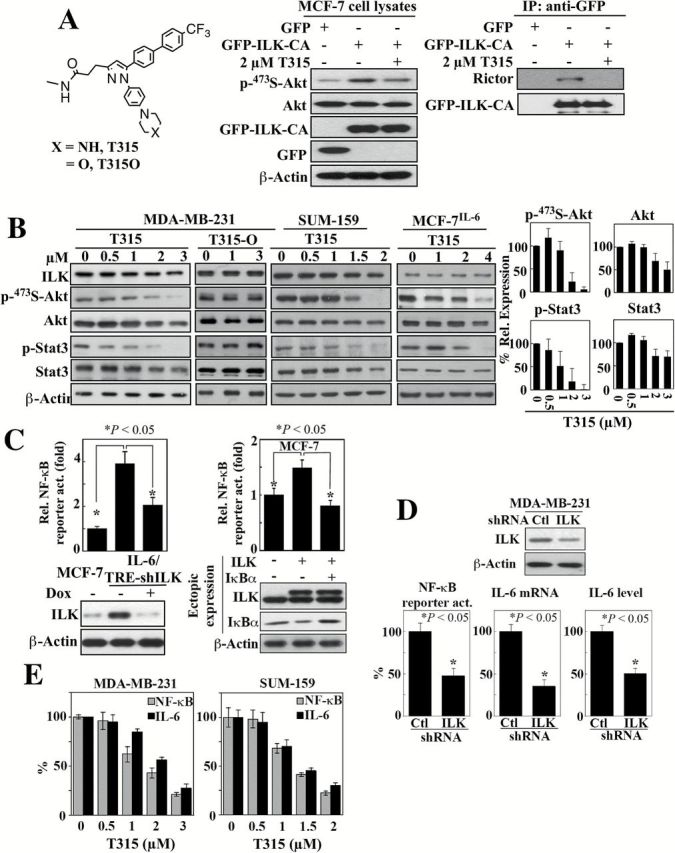
Evidence that ILK acts as a molecular switch for the IL-6-NF-κB feedback loop. (A) Left, structures of the ILK inhibitor T315 and its inactive analogue T315O. Middle and right, The effect of ILK inhibitor T315 (2 μM, 48-h treatment) on Akt phosphorylation (middle) and the interaction between ILK and Rictor (right) in MCF-7 cells ectopically expressiing GFP-tagged constitutively active (CA) ILK. IP, immunoprecipitation. (B) Western blot analysis of the dose-dependent effects of T315 and/or T315O (48-h treatment) on the expression/activation status of ILK and IL-6 downstream signaling markers, Akt and Stat3 in MDA-MB-231, SUM-159 and MCF-7IL-6 cells. Left, Representative blots. Right, Corresponding densitometric analysis of western blots showing relative abundance of p-473S-Akt and p-Stat3 after normalization to corresponding total protein, and Akt and Stat3 after normalization to β-actin in MDA-MB-231 cells. Data are presented as mean ± SD (n = 3). (C) Left, suppressive effect of shRNA-mediated knockdown of ILK on NF-κB luciferase reporter activity in MCF-7IL-6 cells. Data are presented as mean ± SD (n = 3). Right, effect of ectopic expression of ILK on NF-kB luciferase reporter activity in MCF-7 cells and its reversal by the enforced expression of the IκBα super-repressor. Data are presented as mean ± SD (n = 3). Changes in ILK and IκBα protein expression levels were confirmed by western blot (lower panels). (D) Suppressive effect of shRNA-mediated knockdown of ILK on NF-κB reporter activity, IL-6 mRNA expression, and secreted IL-6 abundance in culture medium in MDA-MB-231 cells. Data are presented as mean ± SD (n = 3). Change in ILK protein expression was confirmed by western blot (upper panel). (E) Dose-dependent suppressive effects of T315 (24-h treatment) on NF-κB reporter activity and IL-6 mRNA expression (by qPCR) in MDA-MB-231 and SUM-159 cells. Data are presented as mean ± SD (n = 3).
ILK as a molecular switch of the IL-6-NF-κB regulatory loop
Substantial evidence has demonstrated the existence of a positive feedback loop between the IL-6 and NF-κB pathways (31), which are known to concertedly expand the CSC population (32). In light of the inducible nature of ILK in response to IL-6 (Figures 1–3) and the reported activation of NF-κB by ILK in melanoma (33), we hypothesized that ILK acts as a novel molecular switch within this complex IL-6-NF-κB regulatory loop. To interrogate this link, we established MCF-7IL-6 stable clones with Tet/ON inducible ILK shRNA (MCF-7IL-6/TRE-shILK). The mechanistic link between ILK and NF-κB was corroborated by the ability of doxycycline-induced knockdown and ectopic expression of ILK in MCF-7IL-6/TRE-shILK and MCF-7 cells, respectively, to alter NF-κB-luciferase reporter activities (Figure 4C). Mechanistically, this ILK-induced NF-κB transactivation was associated with reduced IκBα expression, presumably the result of IKKα/β-facilitated IκBα degradation (31), which is consistent with the effect of ILK on Akt activation (Figure 4A). Accordingly, this NF-κB transactivation could be abrogated by the enforced expression of the IκBα super-repressor (S32A/S36A), which is resistant to phosphorylation-mediated degradation (Figure 4C, right).
As NF-κB is an important mediator for the transcriptional activation of IL-6 gene expression (34), the ability of ILK to induce NF-κB activity should be reflected in effects on IL-6 mRNA. The intricate role of ILK in regulating NF-κB-dependent IL-6 expression was demonstrated in two cell lines, MDA-MB-231 and MDA-MB-468. As shown in Figure 4D, shRNA-mediated knockdown of ILK in MDA-MB-231 cells resulted in parallel decreases in NF-κB transactivation activity and IL-6 expression at both mRNA and protein levels. Knockdown or overexpression ILK in MDA-MB-468 cells, in which ILK expression is intermediate between that in MCF7 and MDA-MB-231 cells, resulted in the down- and upregulation, respectively, of IL-6 mRNA expression (Supplementary Figure 5, available at Carcinogenesis Online). Similarly, this disruption of the IL-6-NF-κB regulatory circuitry was also noted after exposure to the ILK inhibitor T315, which resulted in dose-dependent reductions in NF-κB luciferase reporter activities and IL-6 mRNA levels in MDA-MB-231 and SUM-159 cells (Figure 4E).
Validation of the ILK-Akt-NF-κB-IL-6 signaling loop
To validate this ILK-Akt-NF-κB-IL-6 signaling loop, we established stable clones of MDA-MB-231 cells with Tet/ON inducible ILK shRNA (MDA-MB-231TRE-shILK), which were further stably transfected with a plasmid expressing constitutively active (CA)-Akt or the RelA/p65 subunit of NF-κB to generate MDA-MB-231TRE-shILK/CA-Akt and MDA-MB-231TRE-shILK/RelA cells, respectively. As shown, doxycycline-induced silencing of ILK in MDA-MB-231TRE-shILK cells (Figure 5A, left) led to parallel decreases in the NF-κB activity (49±2% of control activity) (Figure 5A, middle) and IL-6 mRNA levels (37±1% of control expression) (Figure 5A, right) (n = 3; P < 0.05), accompanied by a reduction in cell proliferation rates over a 3-day time course (Figure 5B; Supplementary Figure 6, , available at Carcinogenesis Online, MDA-MB-468 TRE-shILK). The roles of Akt and NF-κB as intermediary effectors coupling ILK to IL-6 expression was borne out by the abilities of the stably expressed CA-Akt and RelA to increase NF-κB activities (1.6±0.1-fold and 41.8±4.4-fold, respectively) and IL-6 mRNA levels (2.5±0.4-fold and 7.5±0.9-fold, respectively) in the face of doxycycline-induced ILK suppression. Moreover, ectopic expression of CA-Akt and RelA restored the proliferation rate of doxycycline-treated, ILK-knockdown MDA-MB-231TRE-shILK cells to that of their untreated counterparts (Figure 5B).
Figure 5.

Validation of the ILK-Akt-NF-κB-IL-6 signaling loop. (A) Left, confirmation of GFP, ILK, CA-Akt and RelA protein levels in MDA-MB-231TRE-shILK ectopically expressing CA-Akt, GFP-RelA or GFP, in the presence or absence of doxycycline (Dox) by Western blotting. Ectopic expression of CA-Akt and RelA reversed the suppressive effects of doxycycline-induced ILK knockdown on NF-κB luciferase reporter activity (middle) and IL-6 mRNA expression (right) in MDA-MB-231TRE-shILK cells. Data are presented as mean ± SD (n = 3). (B) Ectopic expression of CA-Akt and RelA restored the proliferation rate of ILK-knockdown MDA-MB-231TRE-shILK cells. Data are presented as mean ± SD (n = 3). +, doxycycline-treated. (C) Suppressive effects of doxycycline (Dox)-induced ILK knockdown on the growth of subcutaneous MDA-MB-231TRE-shILK xenograft tumors. Data are presented as mean ± SD (n = 6). (D) Ectopic expression of CA-Akt and RelA restored the growth of ILK-knockdown (+Dox) MDA-MB-231TRE-shILK tumors to a level comparable to that of ILK-intact controls (-Dox). Mean tumor volumes at 21 days after tumor cell injection are shown. Data are presented as mean ± SD (-Dox, n = 6; +Dox, n = 10; +Dox/+Akt, n = 10; +Dox/+RelA, n = 7). (E) Schematic diagram depicting the role of the Stat3-cyclin D1/CDK2-E2F1-ILK axis in mediating the IL-6-NF-κB feedback loop.
Based on these findings, we interrogated the effect of ILK knockdown on in vivo tumor growth by establishing xenograft tumors in athymic nude mice by subcutaneous injection of MDA-MB-231TRE-shILK cells. Mice that received daily oral doxycycline to suppress ILK expression exhibited significant retardation of tumor growth compared to untreated controls over a six-week period (n = 6; P < 0.05) (Figure 5C). In a follow-up experiment, xenograft tumors were established from MDA-MB-231TRE-shILK cells (n = 10) and the doubly transfected clones, MDA-MB-231TRE-shILK/CA-Akt (n = 10) and MDA-MB-231TRE-shILK/RelA (n = 7), to investigate the ability of CA-Akt and NF-κB to rescue tumor growth that was suppressed by doxycycline-induced ILK knockdown. At 21-days post-tumor cell injection, the tumors expressing CA-Akt and RelA were significantly larger than the MDA-MB-231TRE-shILK tumors, indicating that CA-Akt and RelA could restore the growth of ILK-deficient tumors to a level comparable to that of ILK-intact controls (Figure 5D).
ILK inhibition blocks IL-6-induced breast CSC properties and in vivo tumorigenesis
The findings above suggest that IL-6 and ILK form a positive feedback loop via NF-κB, which might provide a mechanistic basis to account for the constitutive activation of IL-6 signaling in TNBC cells (Figure 5E). As IL-6 plays an integral role in regulating the growth of TNBC cells (2), it is not surprising that, as shown above, shRNA-mediated knockdown of ILK suppressed proliferation of MDA-MB-231 (Figure 5B) and MDA-MB-468 (Supplementary Figure 6, available at Carcinogenesis Online) cells. In addition to cell proliferation, IL-6 has been reported to facilitate breast CSC expansion through the inflammatory feedback loop (35). From a mechanistic perspective, the role of ILK as an IL-6 downstream effector might provide a molecular basis to account for the unique ability of IL-6 to facilitate the conversion of non-stem cancer cells to CSCs (35). To verify this hypothesis, we assessed the effect of ILK inhibition on mammosphere formation in anchorage-independent, serum-free culture conditions, which represents a surrogate measure of CSC expansion (36,37). First, qPCR analysis of isolated MDA-MB-231 mammospheres exhibited significantly higher mRNA levels of IL-6, E2F1, ILK and the breast CSC marker c-Myc, relative to adherent parental cells, which is consistent with the proposed role of ILK in mediating IL-6-driven breast CSCs (Supplementary Figure 7, available at Carcinogenesis Online). Second, in line with the reported effect of IL-6 on CSCs (35), MCF-7IL-6 cells exhibited a significantly higher propensity, relative to MCF-7 cells, to form mammospheres (Figure 6A). Moreover, reminiscent of findings reported in TNBC cells (36,37), the mammosphere-forming capacity of MCF-7IL-6 cells was markedly enhanced by paclitaxel (10nM). In contrast, ILK inhibition in MCF-7IL-6 cells with T315 (1 µM) (Figure 6A) or doxycycline-induced ILK silencing in MCF-7IL-6/TRE-shILK cells (Figure 6B, left) suppressed mammosphere formation (P < 0.05) Also, flow cytometry-based analysis of aldehyde dehydrogenase (ALDH) activity, another breast CSC marker (38), showed a 2.8-fold increase in the ALDH-bright (ALDHbr) subpopulation in MCF-7IL-6 cells relative to the parental cells, which was suppressed by ILK knockdown (Figure 6B, right) or dose-dependently by T315 (Figure 6C, left), in a manner similar to that noted with mammosphere formation. These inhibitory effects of T315 on mammosphere formation and the ALDHbr population were confirmed in MDA-MB-231 cells (Supplementary Figure 8, available at Carcinogenesis Online). To examine whether T315 mediated this suppressive effect through the direct inhibition of ALDH activity, we compared the effects of T315 on the ALDHbr population in MCF-7 cells ectopically expressing ILK (MCF-7ILK) with that in cells overexpressing ALDH1A1 (MCF-7ALDH1). As shown, T315 (2 µM) inhibited the ALDHbr population in MCF-7ILK cells, but not in MCF-7ALDH1 cells (Figure 6C, right), thereby refuting the possibility that T315 might directly inhibit ALDH activity.
Figure 6.
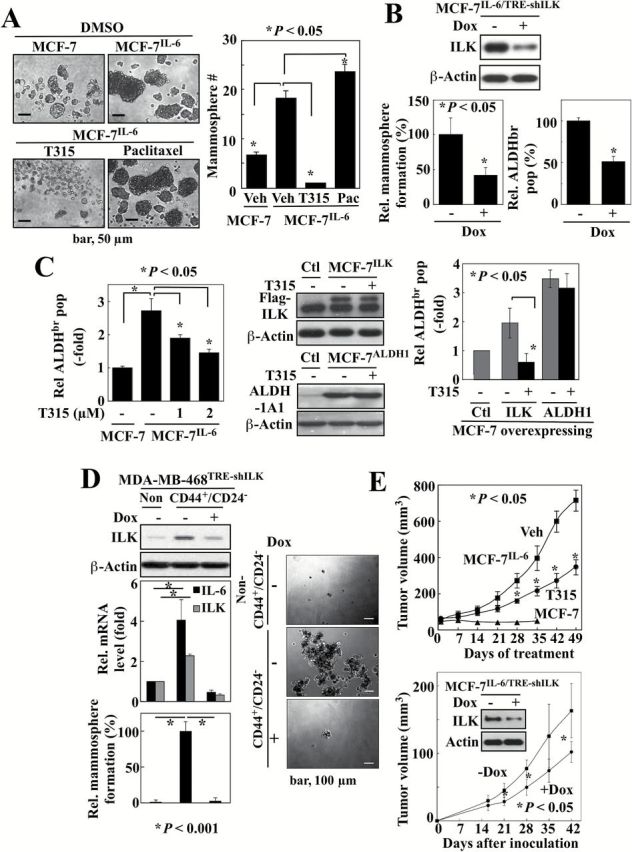
ILK inhibition blocks IL-6-induced breast CSC properties and in vivo tumorigenesis. (A) Effects of T315 (1 µM), and paclitaxel (10nM) versus DMSO (7-day treatment) on mammosphere formation in MCF-7IL-6 cells. Data are presented as mean ± SD (n = 3). (B) Suppressive effects of inducible ILK knockdown (Dox+) on mammo sphere formation (left) and the ALDHbr population (right) in MCF-7IL-6/TRE-shILK cells. Data are presented as mean ± SD (n = 3). (C) Left, Dose-dependent suppressive effect of T315 (48-hour treatment) on the ALDHbr population of MCF-7IL-6 cells by Aldefluor assays. Right, Effect of T315 (2 μM, 48-h treatment) on the ALDHbr population of MCF-7 cells ectopically expressing ILK or ALDH1A1 by Aldefluor assays. Middle, Ectopic expression of ILK and ALDH1A1 were confirmed by western blot. Data are presented as mean ± SD (n = 3). (D) Effect of doxycycline (Dox)-induced ILK knockdown (left, top) on IL-6 mRNA expression (left, middle) by qPCR, and on the mammosphere formation (left, bottom and right) in the CD44+CD24- versus the non-CD44+CD24- subpopulation of MDA-MB-468TRE shILK cells. ILK protein levels were confirmed by Western blotting. Data are presented as mean ± SD (n = 3). (E) Upper, Suppressive effect of oral T315 (50mg/kg; twice daily) on the growth of MCF-7IL-6 xenograft tumors in the absence of estrogen supplementation in nude mice. Lower, Suppressive effect of doxycycline (Dox)-induced ILK knockdown on the growth of MCF-7IL-6/TRE shILK xenograft tumors in the absence of estrogen supplementation in nude mice. Data are presented as mean ± SD (n = 5). Doxycycline (2mg in 0.2ml of solution) was given orally by gavage daily.
To verify the pivotal role of ILK in mediating IL-6-driven CSCs, we selected a stable clone of MDA-MB-468 cells with Tet/ON inducible ILK shRNA (MDA-MB-468TRE-shILK) to interrogate this mechanistic link in the CD44+/CD24− CSC subpopulation, of which the rationale was twofold. First, the parental MDA-MB-468 cells exhibited modest levels of IL-6 relative to MCF-7IL-6 and MDA-MB-231 cells. Second, the CD44+/CD24- subpopulation has been associated with the invasive/metastatic phenotype of breast cancer cells (39,40). In line with our hypothesis, Western blot and qPCR analyses revealed higher protein and mRNA expression levels of ILK and IL-6, respectively, in the CD44+/CD24- subpopulation relative to the non-CD44+/CD24- counterpart (Figure 6D, left top and center). Moreover, doxycycline-induced ILK knockdown in CD44+/CD24- cells led to reduced IL-6 expression, and equally important, abolished the ability of these cells to form mammospheres (left bottom and right), supporting an essential role of ILK in IL-6-induced breast CSC population.
The aggressive phenotype of MCF-7IL-6 cells was evidenced by rapid tumor growth in nude mice in the absence of estradiol supplementation, which, however, was suppressed by oral T315 at 50mg/kg twice daily (Figure 6E, upper). In contrast, the parental MCF-7 xenograft tumors failed to grow in the absence of exogenous estrogen. Additionally, we employed MCF-7IL-6/TRE-shILK cells to examine the effect of ILK knockdown on the IL-6-induced tumorigenicity of these cells in nude mice. As shown, doxycycline-induced a partial knockdown of ILK which suppressed xenograft tumor formation by MCF-7IL-6/TRE-shILK cells (1×106 cells) in nude mice (Figure 6E, lower) (P < 0.05, n = 5).
Discussion
Substantial evidence indicates that the IL-6 inflammatory loop is a major mechanism by which breast tumor cells acquire aggressive phenotype through interactions with the tumor microenvironment (3,6,7,35,41–43). In this study, we report a novel function of ILK as a key regulator of the IL-6-NF-kB regulatory loop in breast cancer cells. The upregulation of ILK expression by IL-6 is consistent with our previous finding that ILK is responsible for Ser-473-Akt phosphorylation and acquisition of mesenchymal phenotype in IL-6-producing aggressive cancer cell lines, including PC-3, MDA-MB-231 and MDA-MB-468 (12), and underlies the observed effect of exogenous or stably expressed IL-6 on Ser473-Akt phosphorylation in MCF-7 cells.
Conventional strategies to target this cytokine network include the use of humanized anti-IL-6 monoclonal antibody or inhibitors of IL-6 downstream effectors, such as Akt or Stat3 (32). However, cancer cells often acquire compensatory mechanisms through crosstalk with other signaling pathways to develop drug resistance, including those mediated by EGFR and transforming growth factor (TGF)-β (44). In addition to IL-6, evidence suggests that ILK might also be upregulated by other environmental stimuli/stresses, including hypoxia (45–47), TGFβ-1 (48), integrins (49) and Wnt (50). Thus, resistance to IL-6-targeted therapies might be overcome through the inhibition of its inducible downstream effector, ILK, which serves as a signaling node of the aforementioned signals from the tumor microenvironment to promote tumor progression, metastasis and chemoresistance. From a mechanistic perspective, our findings might have translational value in fostering new therapeutic strategies that target ILK for TNBC therapy.
It is noteworthy that ILK has been reported to regulate multiple oncogenic pathways (51), including those involving Akt/mTOR, glycogen synthase kinase 3β (52), Yes-associated protein/transcriptional co-activator with PDZ-binding motif (YAP/TAZ) (53), ZEB1 (54) and YB-1 (55). For example, through activation of YAP/TAZ, ILK is able to inhibit the Hippo tumor suppressor pathway (53), which is important to the maintenance of CSC phenotype (56). Of particular interest is YB-1, an oncogenic transcription/translation factor associated with poor prognosis, disease recurrence, and drug resistance of breast cancer (57), which has been reported to target the expression of a wide range of signaling effectors, including the growth factor receptors HER2 and EGFR, and the epithelial–mesenchymal transition (EMT) regulator Snail. Also noteworthy is ZEB1, an EMT activator that represses stemness-inhibiting microRNAs, including miR-200c, miR-203 and miR-183 (58). Consistent with our finding that IL-6 stimulates ILK expression, the expression of ZEB1 was increased and that of miR-200c was suppressed in IL-6-induced breast CSC (35). Equally important, we recently obtained evidence that ILK regulates IL-6-mediated Notch1 activation and CSC expansion in breast cancer cells by facilitating the membrane assembly of the γ-secretase complex in caveolae. Together, the interplay among these ILK downstream effectors in mediating IL-6-induced aggressive phenotypes in cancer cells warrants interrogation.
In this study, E2F1 was identified as the transcription factor responsible for IL-6-mediated up-regulation of ILK expression. The identification of abundant E2F1 binding sites within the ILK promoter suggests an interesting link between ILK and the cell cycle, as E2F1 is a well-known cell cycle regulatory protein, the activity of which is oppositely regulated by Rb and CDK (59). Thus, the abundance of ILK may oscillate in concert with E2F1 activity during the cell cycle progression. Furthermore, ILK regulates cyclinD1 expression (60) and localizes to the centrosome during mitosis (61). The relationship between E2F1 and ILK and its role in cell cycle regulation is worthy of further investigation.
In summary, as shown in Figure 5E, we obtained in vitro and in vivo evidence that ILK plays an important role, as an IL-6-inducible oncokinase, in mediating the effect of IL-6 on tumorigenesis and tumor progression via the IL-6-NF-κB feedback loop. Emerging evidence suggests that IL-6 induces the CSC population, drug resistance and metastasis in breast cancer, in which ILK might act to amplify the IL-6 signal to promote an aggressive tumor phenotype. From a mechanistic perspective, ILK represents a therapeutically advantageous target relative to NF-κB to suppress IL-6-driven breast cancer, of which the proof-of-concept was obtained by T315, a small-molecule inhibitor of ILK function. T315 showed in vitro efficacy in mimicking the effect of ILK silencing on the IL-6-NF-κB inflammatory loop and IL-6-induced Stat3 activation. Moreover, oral T315 was effective in vivo in suppressing estrogen-independent growth of high IL-6-expressing (MCF-7IL-6) xenograft tumors. Together, these findings establish ILK as an intermediary effector of the IL-6-NF-κB feedback loop and a promising therapeutic target in breast cancer cells.
Supplementary material
Supplementary Materials and Methods, Supplementary Figures 1–8, Supplementary Tables 1 and 2A-C can be found at http://carcin.oxfordjournals.org/
Funding
Stefanie Spielman Fund for Breast Cancer Research (to C.S.C.), the Lucius A. Wing Endowed Chair Fund (to C.S.C.), and a Pelotonia Graduate Student Fellowship (to E.C.H.) from The Ohio State University Wexner Medical Center and Comprehensive Cancer Center, a National Cancer Institute Cancer Center Support Grant (P30 CA016058), and predoctoral fellowships (to H.L.H. and M.W.C.) from the Graduate Student Study Abroad Program, National Science Council, Taiwan.
Supplementary Material
Acknowledgements
The authors express their appreciation for expert technical advice from Mr. Joseph P. Burnett (University of Michigan College of Pharmacy).
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- ALDH
aldehyde dehydrogenase
- BSA
bovine serum albumin
- ChIP
chromatin immunoprecipitation
- CSC
cancer stem cell
- EMSA
electrophoretic mobility shift assay
- ER
estrogen receptor
- ILK
integrin-linked kinase
- IL-6
interleukin-6
- PBS
phosphate-buffered saline
- PR
progesterone receptor
- TMA
tissue microarray
- TNBC
triple-negative breast cancer
References
- 1. Stratford A.L., et al. (2010) Targeting tumour-initiating cells to improve the cure rates for triple-negative breast cancer. Expert Rev. Mol. Med., 12, e22. [DOI] [PubMed] [Google Scholar]
- 2. Hartman Z.C., et al. (2013) Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res., 73, 3470–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sullivan N.J., et al. (2009) Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene, 28, 2940–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schafer Z.T., et al. (2007) IL-6 involvement in epithelial cancers. J. Clin. Invest., 117, 3660–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Conze D., et al. (2001) Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res., 61, 8851–8858. [PubMed] [Google Scholar]
- 6. Iliopoulos D., et al. (2009) An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell, 139, 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Korkaya H., et al. (2012) Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell, 47, 570–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sethi N., et al. (2011) Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell, 19, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bachelot T., et al. (2003) Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br. J. Cancer, 88, 1721–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salgado R., et al. (2003) Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer, 103, 642–646. [DOI] [PubMed] [Google Scholar]
- 11. Troussard A.A., et al. (2006) Preferential dependence of breast cancer cells versus normal cells on integrin-linked kinase for protein kinase B/Akt activation and cell survival. Cancer Res., 66, 393–403. [DOI] [PubMed] [Google Scholar]
- 12. Lee S.L., et al. (2013) Functional role of mTORC2 versus integrin-linked kinase in mediating Ser473-Akt phosphorylation in PTEN-negative prostate and breast cancer cell lines. PLoS One, 8, e67149. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13. Lee S.L., et al. (2011) Identification and characterization of a novel integrin-linked kinase inhibitor. J. Med. Chem., 54, 6364–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sasser A.K., et al. (2007) Interleukin-6 is a potent growth factor for ER-alpha-positive human breast cancer. FASEB J., 21, 3763–3770. [DOI] [PubMed] [Google Scholar]
- 15. Lacroix M., et al. (2004) Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res. Treat., 83, 249–289. [DOI] [PubMed] [Google Scholar]
- 16. Kenny P.A., et al. (2007) The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol., 1, 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Messeguer X., et al. (2002) PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics, 18, 333–334. [DOI] [PubMed] [Google Scholar]
- 18. Farre D., et al. (2003) Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res., 31, 3651–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen P.L., et al. (1989) Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell, 58, 1193–1198. [DOI] [PubMed] [Google Scholar]
- 20. DeCaprio J.A., et al. (1989) The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell, 58, 1085–1095. [DOI] [PubMed] [Google Scholar]
- 21. Lane M.E., et al. (2001) A novel cdk2-selective inhibitor, SU9516, induces apoptosis in colon carcinoma cells. Cancer Res., 61, 6170–6177. [PubMed] [Google Scholar]
- 22. Leslie K., et al. (2006) Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res., 66, 2544–2552. [DOI] [PubMed] [Google Scholar]
- 23. Junk D.J., et al. (2013) Constitutive CCND1/CDK2 activity substitutes for p53 loss, or MYC or oncogenic RAS expression in the transformation of human mammary epithelial cells. PLoS One, 8, e53776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin L., et al. (2009) The STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-beta signaling. Oncogene, 28, 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hresko R.C., et al. (2005) mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem., 280, 40406–40416. [DOI] [PubMed] [Google Scholar]
- 26. Sarbassov D.D., et al. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science, 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- 27. Bozulic L., et al. (2008) PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell, 30, 203–213. [DOI] [PubMed] [Google Scholar]
- 28. Guertin D.A., et al. (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell, 11, 859–871. [DOI] [PubMed] [Google Scholar]
- 29. Wickstrom S.A., et al. (2010) The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J., 29, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fukuda K., et al. (2009) The pseudoactive site of ILK is essential for its binding to alpha-Parvin and localization to focal adhesions. Mol. Cell, 36, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Makino K., et al. (2004) Upregulation of IKKalpha/IKKbeta by integrin-linked kinase is required for HER2/neu-induced NF-kappaB antiapoptotic pathway. Oncogene, 23, 3883–3887. [DOI] [PubMed] [Google Scholar]
- 32. Korkaya H., et al. (2011) Regulation of cancer stem cells by cytokine networks: attacking cancer’s inflammatory roots. Clin. Cancer Res., 17, 6125–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wani A.A., et al. (2011) Integrin-linked kinase regulates melanoma angiogenesis by activating NF-kappaB/interleukin-6 signaling pathway. Oncogene, 30, 2778–2788. [DOI] [PubMed] [Google Scholar]
- 34. Libermann T.A., et al. (1990) Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell Biol., 10, 2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iliopoulos D., et al. (2011) Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl Acad. Sci. USA, 108, 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liao M.J., et al. (2007) Enrichment of a population of mammary gland cells that form mammospheres and have in vivo repopulating activity. Cancer Res., 67, 8131–8138. [DOI] [PubMed] [Google Scholar]
- 37. Grimshaw M.J., et al. (2008) Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res., 10, R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ginestier C., et al. (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell, 1, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sheridan C., et al. (2006) CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res., 8, R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu S., et al. (2014) Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports, 2, 78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sansone P., et al. (2007) IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Invest., 117, 3988–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mani S.A., et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korkaya H., et al. (2011) Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Invest., 121, 3804–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yao Z., et al. (2010) TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl Acad. Sci. USA, 107, 15535–15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tan C., et al. (2004) Regulation of tumor angiogenesis by integrin-linked kinase (ILK). Cancer Cell, 5, 79–90. [DOI] [PubMed] [Google Scholar]
- 46. Abboud E.R., et al. (2007) Integrin-linked kinase: a hypoxia-induced anti-apoptotic factor exploited by cancer cells. Int. J. Oncol., 30, 113–122. [PubMed] [Google Scholar]
- 47. Tseng W.P., et al. (2010) Hypoxia induces BMP-2 expression via ILK, Akt, mTOR, and HIF-1 pathways in osteoblasts. J. Cell Physiol., 223, 810–818. [DOI] [PubMed] [Google Scholar]
- 48. Serrano I., et al. (2013) Role of the integrin-linked kinase (ILK)/Rictor complex in TGFbeta-1-induced epithelial-mesenchymal transition (EMT). Oncogene, 32, 50–60. [DOI] [PubMed] [Google Scholar]
- 49. Legate K.R., et al. (2006) ILK, PINCH and parvin: the tIPP of integrin signalling. Nat. Rev. Mol. Cell Biol., 7, 20–31. [DOI] [PubMed] [Google Scholar]
- 50. Oloumi A., et al. (2006) Modulation of Wnt3a-mediated nuclear beta-catenin accumulation and activation by integrin-linked kinase in mammalian cells. Oncogene, 25, 7747–7757. [DOI] [PubMed] [Google Scholar]
- 51. Hannigan G., et al. (2005) Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat. Rev. Cancer, 5, 51–63. [DOI] [PubMed] [Google Scholar]
- 52. Delcommenne M., et al. (1998) Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl Acad. Sci. USA, 95, 11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Serrano I., et al. (2013) Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat. Commun., 4, 2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matsui Y., et al. (2012) The importance of integrin-linked kinase in the regulation of bladder cancer invasion. Int. J. Cancer, 130, 521–531. [DOI] [PubMed] [Google Scholar]
- 55. Kalra J., et al. (2010) Suppression of Her2/neu expression through ILK inhibition is regulated by a pathway involving TWIST and YB-1. Oncogene, 29, 6343–6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hao J., et al. (2014) Role of Hippo signaling in cancer stem cells. J. Cell Physiol., 229, 266–270. [DOI] [PubMed] [Google Scholar]
- 57. Habibi G., et al. (2008) Redefining prognostic factors for breast cancer: YB-1 is a stronger predictor of relapse and disease-specific survival than estrogen receptor or HER-2 across all tumor subtypes. Breast Cancer Res., 10, R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wellner U., et al. (2009) The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell. Biol., 11, 1487–1495. [DOI] [PubMed] [Google Scholar]
- 59. Nevins J.R. (2001) The Rb/E2F pathway and cancer. Hum. Mol. Genet., 10, 699–703. [DOI] [PubMed] [Google Scholar]
- 60. D’Amico M., et al. (2000) The integrin-linked kinase regulates the cyclin D1 gene through glycogen synthase kinase 3beta and cAMP-responsive element-binding protein-dependent pathways. J. Biol. Chem., 275, 32649–32657. [DOI] [PubMed] [Google Scholar]
- 61. Fielding A.B., et al. (2008) Integrin-linked kinase localizes to the centrosome and regulates mitotic spindle organization. J. Cell Biol., 180, 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.