

Abstract
The characterization of evolutionary and biogeographical patterns is of fundamental importance to identify factors driving biodiversity. Due to their widespread but discontinuous distribution, deep‐sea hydrothermal vent barnacles represent an excellent model for testing biogeographical hypotheses regarding the origin, dispersal and diversity of modern vent fauna. Here, we characterize the global genetic diversity of vent barnacles to infer their time of radiation, place of origin, mode of dispersal and diversification. Our approach was to target a suite of multiple loci in samples representing seven of the eight described genera. We also performed restriction‐site associated DNA sequencing on individuals from each species. Phylogenetic inferences and topology hypothesis tests indicate that vent barnacles have colonized deep‐sea hydrothermal vents at least twice in history. Consistent with preliminary estimates, we find a likely radiation of barnacles in vent ecosystems during the Cenozoic. Our analyses suggest that the western Pacific was the place of origin of the major vent barnacle lineage, followed by circumglobal colonization eastwards through the Southern Hemisphere during the Neogene. The inferred time of radiation rejects the classic hypotheses of antiquity of vent taxa. The timing and the mode of origin, radiation and dispersal are consistent with recent inferences made for other deep‐sea taxa, including nonvent species, and are correlated with the occurrence of major geological events and mass extinctions. Thus, we suggest that the geological processes and dispersal mechanisms discussed here can explain the current distribution patterns of many other marine taxa and have played an important role shaping deep‐sea faunal diversity. These results also constitute the critical baseline data with which to assess potential effects of anthropogenic disturbances on deep‐sea ecosystems.
Keywords: cenozoic, dispersal, hydrothermal vents, polyphyly, Southern Hemisphere, species delimitation, RAD‐seq
Introduction
The characterization of evolutionary and biogeographical patterns is of fundamental importance for identifying the factors that shape the ranges of deep‐sea taxa and that ultimately drive biodiversity patterns in the ocean (McClain & Mincks 2010). This is particularly relevant in the light of the increasing interest in commercial resource extraction in the deep sea (Thurber et al. 2014). Mining of seafloor massive sulphide deposits at deep‐sea hydrothermal vent fields has become one of the main industrial targets for exploitation (Boschen et al. 2013). Understanding the biodiversity contained in these areas and its connection with the fauna found elsewhere is critical for assessing the potential impacts of exploiting these mineral resources (Van Dover 2010; Van Dover et al. 2012). Although organisms living at deep‐sea hydrothermal vents have adapted to cope with natural disturbances inherent to these ephemeral habitats, the intensity and frequency at which these occur can vary greatly depending on the particular geophysical nature of each system (Baker & German 2004). Thus, disturbance from mining could have additive or synergistic effects to natural disturbances at unprecedented scales, which could potentially lead to significant losses of biodiversity (Van Dover 2010). Due to their widespread distribution (Fig. 1), vent barnacles represent an excellent model for testing hypotheses regarding the historical biogeographical patterns of origin, dispersal and current diversity of modern deep‐sea chemosynthetic fauna; therefore, barnacles hold the promise of providing critical baseline data with which to assess potential effects of anthropogenic disturbances on deep‐sea ecosystems.
Figure 1.
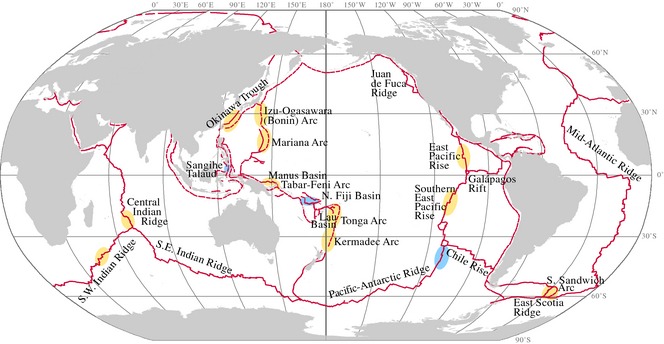
Global distribution map of hydrothermal vent barnacles. Ovals indicate regions where hydrothermal vent barnacles have been described (yellow: regions sampled in this study; blue: regions not sampled in this study). Red lines indicate active tectonic margins (solid lines: spreading centres; dotted lines: subduction zones).
Barnacles (Cirripedia Burmeister, 1834) are some of the most conspicuous organisms in deep‐sea hydrothermal vent ecosystems worldwide. These sessile crustaceans can be found in active vent fields in most of the major spreading ridge systems and volcanic arcs worldwide (Fig. 1), including the Central Indian Ridge (Van Dover et al. 2001; Nakamura et al. 2012), Southwest Indian Ridge (Tao et al. 2011), East Scotia Ridge (Rogers et al. 2012), northern and southern East Pacific Rise (Newman 1979; Jones 1993), Pacific–Antarctic Ridge (Southward 2005), Izu‐Ogasawara Arc (Ono et al. 1996), Okinawa Trough (Ohta 1990), Mariana Trough (Hessler & Lonsdale 1991), Sangihe Talaud (Herrera et al. 2010; Shank et al. 2010), Manus Basin (Tufar 1990), Edison Seamount (Tunnicliffe & Southward 2004), North Fiji Basin (Desbruyeres et al. 1994), Lau Basin (Southward & Newman 1998), Kermadec Arc (Buckeridge 2000), and are likely to be present in other unexplored areas. Hydrothermal vent barnacles inhabit areas of low‐temperature diffuse fluid flow. Populations can reach high densities and high biomass at over 1500 individuals per square metre (Tunnicliffe & Southward 2004; Marsh et al. 2012), playing key roles in vent communities as microhabitat engineers and funnelling the flow of energy through ecosystems from primary producers to higher trophic levels (Southward & Newman 1998; Van Dover 2002; Tunnicliffe & Southward 2004; Cubelio et al. 2007; Rogers et al. 2012; Reid et al. 2013).
Hydrothermal vent barnacles are presently grouped into four families belonging to the orders Pedunculata Lamarck, 1818 (suborder Scalpellomorpha, family Eolepadidae; commonly known as stalked or gooseneck barnacles) and Sessilia Lamarck, 1818 (suborder Verrucomorpha, family Neoverrucidae; suborder Brachylepadomorpha, family Neobrachylepadidae; and suborder Balanomorpha, family Chionelasmatidae; commonly known as acorn barnacles) (Newman et al. 2006). There are ca. 13 described vent barnacle species, with several new species awaiting description (Newman et al. 2006). A molecular phylogenetic study of the Cirripedia, employing nuclear ribosomal genes and the histone H3 gene, indicates that these morphologically based taxonomic groupings (particularly the orders) are polyphyletic and thus incongruent with evolutionary history (Pérez‐Losada et al. 2008). These results, together with those from Linse et al. (2013), also suggest that vent barnacles form a monophyletic clade that probably originated in the Cretaceous; however, the possibility of a single origin remains an open question due to the paucity of taxonomic sampling in that study. Furthermore, the relationships among morphospecies of vent barnacles also remain unresolved due to the low variability of markers examined to date.
Many putative species of vent barnacles appear to be restricted to particular ridge systems and neighbouring arc and back‐arc basins, and significant population structure has also been found at these scales (Watanabe et al. 2005). Together, these observations suggest a role of habitat discontinuity as an important mechanism of speciation. By far, the region of highest diversity of putative chemosynthetic barnacle species (measured as species richness) is the western Pacific, which is considered the centre of their distribution and possible place of origin (Newman et al. 2006). The western Pacific is also considered a biodiversity hotspot and potential place of origin of many modern groups of terrestrial and marine organisms, including deep‐sea taxa (Cairns 2007; Carpenter et al. 2011; Herrera et al. 2012). In a similar way, a recent biogeographical analyses using network theory hypothesizes a possible ancestral position for modern western Pacific fauna associated with hydrothermal vents, having exclusive edge connections (indicating faunal similarity and possible exchange paths) with the northeast Pacific, the East Pacific Rise and the Indian Ocean (Moalic et al. 2011).
In this study, we aim to characterize the global genetic diversity, and evolutionary and biogeographical history of barnacles from deep‐sea hydrothermal vents. Our approach was to build on previous phylogenetic studies by significantly expanding the taxonomic sampling and number of genetic markers. We targeted one mitochondrial gene region, the cytochrome c oxidase subunit I (coxI), and two nuclear gene regions, the large ribosomal subunit 28S and the histone H3 gene, obtaining complete sequences for 94 individuals, representing seven of the eight described genera, from 18 vent fields worldwide. We also performed restriction‐site‐associated DNA sequencing (RAD‐seq) on individuals from each identified species. Here, we (i) test the hypothesis of monophyly (i.e. a single evolutionary origin) of barnacles from deep‐sea hydrothermal vents; (ii) infer the place and time of origin and radiation of vent barnacles in geologic time; (iii) infer historical patterns of dispersal and colonization of vent barnacle taxa worldwide; and (iv) identify species boundaries and compare them to the current morphospecies hypotheses.
Materials and methods
Morphological identifications were performed on 94 barnacle specimens (Table S1, Supporting information) from deep‐sea hydrothermal vents using stereo‐microscopy and species descriptions as references. Individuals were collected from the Central Indian Ridge, East Pacific Rise, southern East Pacific Rise, Southwest Indian Ridge, East Scotia Ridge, Mariana Trough, Kermadec Arc, Lau Basin, Tonga Arc, Manus Basin, Izu‐Ogasawara (Bonin) Arc, and Okinawa Trough.
Partial DNA sequences of one mitochondrial (cytochrome c oxidase subunit I) and two nuclear markers (histone H3 gene and the ribosomal large subunit 28S) were generated for each individual. Additional sequences from the superorder Thoracica Darwin, 1854 were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/genbank/) and included in the analyses (Table S2, Supporting information).
Restriction‐site‐associated DNA sequencing (RAD‐seq) (Baird et al. 2008) was performed on selected individuals from each morphospecies (Table S1, Supporting information) to obtain a genome‐wide set of markers that could be used to infer a robust backbone of the vent barnacle phylogenetic tree, and to compare to topologies obtained from species‐tree analyses of traditional Sanger‐based markers.
Molecular laboratory methods
Total genomic DNA was extracted from tissue samples by the following methods: (i) digesting the tissue in 2 % CTAB buffer (Teknova) with proteinase K (Fermentas) and RNase A/T1 (Fermentas) for 1 h, (ii) separating nucleic acids with chloroform: isoamyl alcohol (24:1) (Fermentas) and phenol: chloroform: isoamyl alcohol (25:24:1, Tris‐buffered at pH 8.0) (Fermentas), (iii) precipitating nucleic acids with 100% ethanol (1:1) and (iv) washing the precipitate twice with 70% ethanol. Polymerase chain reactions of traditional Sanger‐based markers were prepared to a final volume of 25 μl (1 μl of template) resulting in the following final concentrations of reagents and enzymes: 1 X GoTaq Flexi Buffer (Promega), 2.5 X BSA, 1.0 mm dNTPs (0.25 mm each), 2.0 mm MgCl2, 1 U Taq polymerase (GoTaq, Promega) and 0.3 μm of each primer. Primer pairs used for amplifications were 28SF_330 5′‐ CGTGAAGCTGCCAVTATGG‐3′ (designed in this study) and 28S_B (Whiting 2002) for 28S, H3F and H3R (Colgan et al. 1998) for H3, and LCO1490 and HC02198 (Folmer et al. 1994) for coxI. Negative controls were included in every experiment to test for contamination. The reactions were carried out with an initial denaturation step of 5 min at 94 °C, 32 cycles (35 for coxI) of 60 s at 94 °C, 90 s at 48 °C and 90 s at 72 °C, and a final elongation step of 10 min at 72 °C. PCR products were cleaned using the MinElute PCR Purification Kit (Qiagen) following the manufacturer's protocols. Cycle‐sequencing reactions were performed using the ABI BigDye Terminator v3.1 kit (Life Technologies Corp., CarlsBad, CA, USA) following the manufacturer protocols. Subsequent purification was performed through isopropanol precipitation. Automated sequencing was completed using a 3730xl DNA analyzer (Life Technologies Corp., CarlsBad, CA, USA) at the Josephine Bay Paul Center of the Marine Biological Laboratory. Complementary chromatograms were assembled and edited using geneious v6.1.6 (Drummond et al. 2011).
Concentration‐normalized genomic DNA was submitted to Floragenex Inc., (Eugene, OR, USA) for library preparation and RAD sequencing. Individual libraries were produced from DNA digested with a high‐fidelity SbfI restriction enzyme, which is predicted to cut ca. 5000–15 000 times in the genome of a thoracican barnacle (Table S3, Supporting information). This predicted range was obtained using the observed frequency of the SbfI recognition sequence, and its probability using a trinucleotide composition model, in the genome of the crustacean Daphnia pulex (Herrera et al. 2014). Ranges of genome size for barnacles were obtained from the Animal Genome Size Database (http://www.genomesize.com). Barcode tags were 10 base pairs long. Libraries were sequenced by 96‐multiplex on a single lane of an Illumina Hi‐Seq 2000 sequencer.
Alignments, saturation analysis and model selection
Each set of sequences for Sanger‐based markers was aligned independently using mafft (Katoh et al. 2002), employing the G‐INS‐i and Q‐INS‐i algorithms (gap opening penalty = 1.53, offset value = 0.07) for protein‐coding and ribosomal regions, respectively. To correct possible mistakes, all alignments of protein‐coding sequences were visually inspected and translated to amino acids in geneious v6.1.6 (Drummond et al. 2011). No unusual stop codons, misplaced reading frames or suspicious substitutions were identified, indicating that amplification of nuclear pseudogenes was unlikely (Lopez et al. 1994; Bensasson et al. 2001). Possible substitution saturation in the DNA sequences was evaluated by implementing the Xia test (Xia et al. 2003), as implemented in dambe v5.3.48 (Xia 2013), and by plotting genetic distances (K80 model) against the number of transitions and transversions. Saturation in codon partitions was also evaluated for each coding region.
Phylogenetic inferences
Nonsaturated data sets from individual Sanger‐based markers were analysed in raxml‐hpc2 v8.0 (Stamatakis 2006), as implemented in the cipres Science Gateway v3.3 (http://www.phylo.org), for a first‐pass phylogenetic inference using the maximum‐likelihood optimality criterion. Branch support was assessed by 500 bootstrap replicates. A Thoracica‐wide concatenated data set was also analysed in this program. Only outgroups with data for at least two of the three markers were included in the concatenated data set. Phylogenetic estimation through Bayesian inference was performed with these data sets in mrbayes v3.2.2 (Ronquist et al. 2012), as implemented in the cipres Science Gateway v3.3. Models of nucleotide substitution were selected for each nonsaturated gene region using jmodeltest v2.0 (Darriba et al. 2012), following the Bayesian information criterion (Table S4, Supporting information). Four independent analyses of 200 million Markov chain Monte Carlo (MCMC) generations (4 chains) were run with a sampling frequency of 20 thousand generations (burn‐in = 25%). Combined analyses were performed with explicit character partitions for each concatenated region, along with their independently selected models of evolution. State frequencies were allowed to vary under a flat Dirichlet prior distribution to account for the rate variation among partitions. Nucleotide frequencies, substitution rates, gamma shape and invariant site proportion parameters were unlinked across partitions. Default prior distribution settings were assumed for all other parameters. MCMC runs were analysed with the programs tracer v1.5 (Rambaut & Drummond 2007) and awty (http://ceb.csit.fsu.edu/awty) (Nylander et al. 2008). Convergence among independent runs was supported by observed values of standard deviation of partition frequencies (<0.01), potential scale reduction factors (PSRF) (ca. 1.00) and effective sample sizes (EES) (>200), in addition to high correlations between runs and the flat shapes of the stationary posterior distribution traces of each parameter.
Topological hypothesis testing
To test the hypothesis that barnacles from deep‐sea hydrothermal vents form a monophyletic group, we performed a Bayes factor comparison (Kass & Raftery 1995) between this topological hypothesis and the alternative hypothesis of nonmonophyly of the group using the Thoracica‐wide concatenated data set. The marginal likelihood for each topology model was estimated through the stepping‐stone method (Fan et al. 2011; Xie et al. 2011) in mrbayes using 50 steps. The estimation was performed in two independent runs of 100 million generations, with a diagnostic frequency of 1 million generations, for each topology model. All other parameters were set to default. Convergence among runs was diagnosed by the standard deviation of partition frequencies (<0.01).
Divergence time estimations
Time calibration of the phylogenetic hypothesis was carried out through a Bayesian‐MCMC joint estimation of phylogeny and divergence times in beast v1.7.5 (Drummond et al. 2012), as implemented in the cipres Science Gateway v3.3, using the Thoracica‐wide concatenated Sanger‐based markers data set. Variation in mutation rates among branches was allowed by assuming an uncorrelated relaxed lognormal molecular clock model. The Yule constant speciation rate model and no extinction (Yule 1925), the birth–death constant speciation and extinction rates model (Gernhard 2008), and the birth–death constant speciation and extinction rates with incomplete taxonomic sampling model (Stadler 2009) were tested as tree priors. Unlinked character partitions were set for each concatenated region, along with their independently selected models of evolution. Three fossil calibration points (C1, C2 and C7) were selected from the studies by Pérez‐Losada et al. (2008) and Linse et al. (2013) based on well‐supported topological congruencies with our phylogenetic hypothesis. Fossil ages were used as lower boundary constraints assuming prior exponential distributions with mean values of 25 Myr. Default prior distribution settings were assumed for all other parameters. Three independent MCMC analyses were run for 200 million generations with a sampling frequency of 20 thousand. Convergence diagnostics were examined for the combined runs in tracer as mentioned above. Most probable trees, after 25% burn‐in, were summarized into a maximum clade credibility tree with median node heights using treeannotator v1.7.1 (Drummond et al. 2012).
Historical biogeography
To infer historical patterns of dispersal in deep‐sea hydrothermal vent barnacle lineages, we performed a Bayesian reconstruction of discrete character states of geographic location for ancestral nodes (Lemey et al. 2009) using beast v1.7.5 (Heled & Drummond 2010). In this framework, the geographical sampling locations were mapped to the timescaled phylogenetic tree. Parameters for tree inference were as described above.
Species delimitation
To identify species boundaries for vent barnacles in Clade A (see Results section), we employed generalized mixed Yule‐coalescent (GMYC) likelihood method (Pons et al. 2006; Monaghan et al. 2009; Fujisawa & Barraclough 2013), with a single threshold, as implemented in the splits r‐package (available from http://r-forge.r-project.org/projects/splits/). This method estimates species boundaries by identifying increases in branching rates that are characteristic of transition points between interspecific speciation–extinction processes and intraspecific coalescent processes.
Species‐tree estimation
Bayesian analyses of species‐tree estimation for vent barnacle species identified in Clade A (see Results section) were carried out using data from the Sanger‐based markers in the program *beast v1.7.5 (Heled & Drummond 2010). This approach was employed to take into account evolutionary coalescent processes and gene tree heterogeneity, and to evaluate the effects of gene concatenation on the phylogenetic inference (Brito & Edwards 2008; Edwards 2008). Species were defined after the species delimitation analyses. Unlinked character, clock and tree partitions were set for each marker, along with their independently selected models of evolution. We assumed a piecewise linear and constant root population size model. Other parameters for tree inference were as described above.
RAD‐seq data quality control and loci clustering
Sequence reads were demultiplexed and quality‐filtered with the process_radtags program from the package stacks v1.19 (Catchen et al. 2011, 2013). Barcodes and Illumina adapters were excluded from each read, and length was truncated to 90 bp (−t 90). Reads with ambiguous bases were discarded (−c). Reads with an average quality score below 10 (−s 10) within a sliding window of 15% of the read length (−w 0.15) were discarded (−r). The rescue barcodes and RAD‐tags algorithm was enabled (−r). Additional filtering, and the clustering within and between individuals to identify loci, was performed using the program py rad v2.01 (Eaton 2014). Reads with more than 33 bases with a quality score below 20 were also discarded. The minimum depth of coverage required to build a cluster was 5 (d 5). As in Hipp et al. (2014), three different clustering thresholds were explored (c 0.80, 0.85 and 0.90). Similarly, four different values for the minimum taxon coverage in a given locus were explored (m 4, 6, 8 and 10). The maximum number of shared polymorphic sites in a locus was set to 3 (p 3). Loci were concatenated into combined RAD‐seq matrices.
RAD phylogenetics
Phylogenetic inferences of RAD‐seq matrices, built with py rad under each combination of clustering threshold and minimum taxon coverage parameters (as outlined above), were carried out in raxml‐hpc2 v8.0. We assumed a generalized time‐reversible DNA substitution model with a gamma‐distributed rate variation across sites. Branch support was assessed by 500 bootstrap replicates.
Results
Complete Sanger‐based marker data sets were obtained for all 94 individuals, except for two specimens of Vulcanolepas osheai. Sequences are stored at the GenBank database of the U.S. National Center for Biotechnology Information (NCBI). Approximate sequence lengths for each marker were 700 bp for 28S, 657 bp for coxI and 327 bp for H3. Xia tests indicated substantial saturation at the Thoracica‐wide level at third codon positions of coxI (Table S5, Supporting information). Little saturation was found in all other partitions. Maximum‐likelihood and Bayesian phylogenetic inferences from each Sanger‐based marker produced mostly congruent trees that varied in the degree of resolution, yet all showed poorly supported branches (i.e. posterior probability <80, bootstrap support <80) (Supporting information). Analyses of the Thoracica‐wide concatenated data set generated a better‐supported and ‐resolved phylogeny overall (Fig. 2, Supporting information). The topologies of these trees were congruent with previously published phylogenetic hypotheses for the Thoracica (Pérez‐Losada et al. 2008; Linse et al. 2013).
Figure 2.
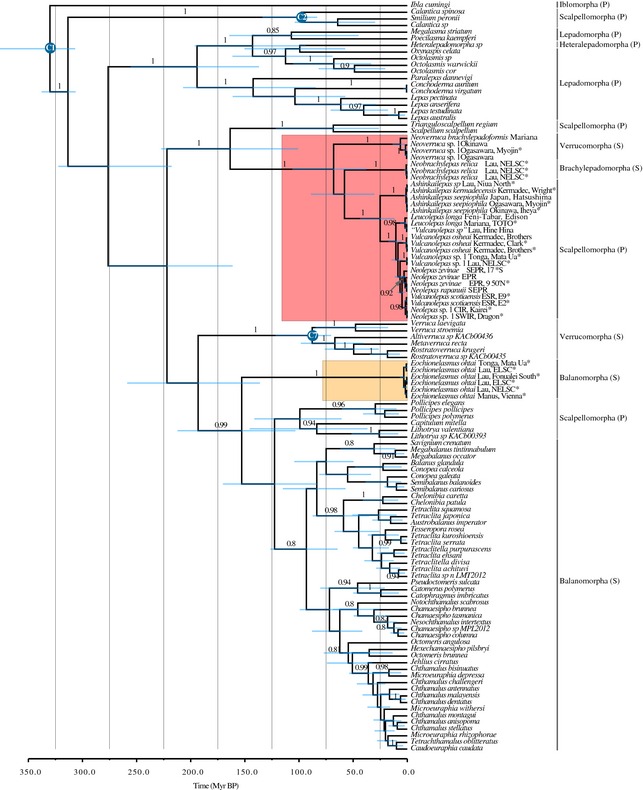
Maximum clade credibility ultrametric timescaled tree, generated under the birth–death model tree prior, for the Thoracica‐wide concatenated data set. Red square indicates hydrothermal vent Clade A. Yellow square indicates hydrothermal vent Clade B. Node bars represent the 95% highest posterior density intervals. Branch labels show posterior probabilities. Blue circles in nodes indicate fossil calibration points as in (Pérez‐Losada et al. 2008; Linse et al. 2013). Suborders belonging to the order Pedunculata (stalked or gooseneck barnacles) are indicated with (P). Suborders belonging to the order Sessilia (acorn barnacles) are indicated with (S). *Indicates data generated in this study.
RAD‐seq data sets were obtained from 13 individuals representing all currently described vent barnacle species (Table S1, Supporting information). An average of 843 541 reads (SD 589 377) were obtained per individual. Reads are stored at the Sequence Read Archive (SRA) of NCBI. The great variability in sequencing yield was largely a product of varying DNA integrity as some samples had notably degraded DNA (Table S6, Supporting information), as determined by agarose gel electrophoresis. An average of 712 306 reads per individual (SD 546 846), approximately 78% of all reads, were retained after quality‐filtering steps. In individuals with high‐integrity DNA, the number of RAD‐tag loci with depth of coverage >4 X was ca. 18 000, per individual. This number is congruent with the expected number of RAD‐tags, between 10 000 and 30 000, predicted for a barnacle, using the enzyme SbfI (Table S3, Supporting information). The average depth of coverage per locus was ca. 54 X (SD 13 X). As expected, the number of loci per individual was higher as the clustering threshold was larger (Table S7, Supporting information). Phylogenetic trees obtained from the RAD‐seq data sets were completely resolved, highly supported as indicated by bootstrap resampling, and were largely congruent with the trees produced with Sanger‐based data.
Phylogenetic inferences
Analyses of Sanger‐based markers revealed that barnacles from deep‐sea hydrothermal vents are divided into two well‐supported (posterior probability = 1, bootstrap support >99) main clades (Fig. 2): Clade A contains the genera Neobrachylepas Newman & Yamaguchi 1995 (order Sessilia, suborder Brachylepadomorpha), Neoverruca Newman, 1989, in Hessler & Newman, 1989 (order Sessilia, suborder Verrucomorpha), Ashinkailepas Yamaguchi et al. 2004 (order Sessilia, suborder Scalpellomorpha), Leucolepas Southward & Jones, 2003 (suborder Scalpellomorpha), Vulcanolepas Southward & Jones, 2003 (suborder Scalpellomorpha) and Neolepas Newman 1979 (suborder Scalpellomorpha), and Clade B was restricted to the genus Eochionelasmus Yamaguchi, 1990 (order Sessilia, suborder Balanomorpha). Clade A is well supported as the sister taxon to the predominantly deep‐sea clade of the Scalpellidae (Pérez‐Losada et al. 2008; Linse et al. 2013). Clade B Eochionelasmus is associated with the paraphyletic Balanomorpha group; however, the lack of support and resolution within the later group prevents an unambiguous phylogenetic placement.
Neobrachylepas and Neoverruca appear as the extant representatives of the earliest divergent lineages in Clade A; however, their order of divergence is unclear due to lack of strong branch support. The rest of the genera in Clade A belong to the family Eolepadidae. The genus Ashinkailepas belongs to the earliest divergent lineage in the family (Fig. 3) and contains two subclades, the first grouping individuals from the Izu‐Ogasawara (Bonin) Arc and the Okinawa Trough (identified as Ashinkailepas seepiophila), and the second grouping individuals from the Lau Basin and the Kermadec Arc. The latter subclade includes a paratype of A. kermadecensis. Neither genus Vulcanolepas nor Neolepas is monophyletic. The Vulcanolepas/Leucolepas from the Kermadec Arc, Lau Basin and Mariana Arc belong to lineages that appear to have diverged earlier in history with respect to a well‐supported and well‐resolved clade made up by N. zevinae/rapanuii from the East Pacific Rise and its sister subclade of V. scotiaensis from the East Scotia Ridge and Neolepas sp. 1 from the Southwest and Central Indian Ridge.
Figure 3.
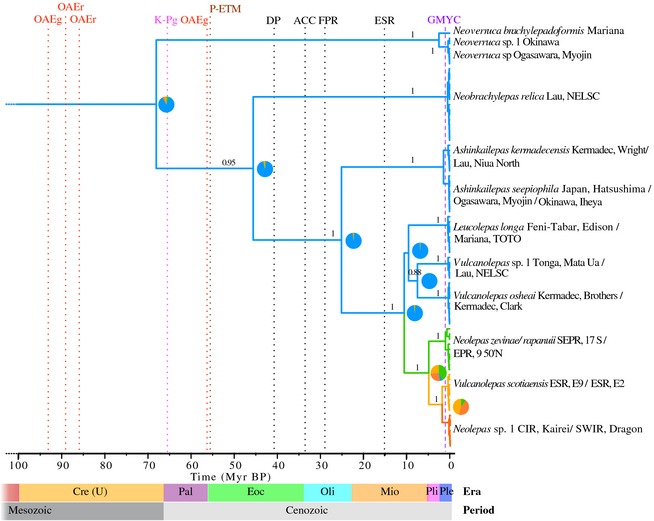
Clade A combined 28S, H3 and coxI maximum clade credibility ultrametric timescaled tree generated under the birth–death model. Branch colours show the most probable location states: western Pacific in blue, eastern Pacific in green, Southern Ocean south of the Atlantic in yellow and Indian Ocean in orange. Pie charts show the posterior probabilities of location states for each ancestral node (total pie area = 1). Branch labels show posterior probabilities. Purple vertical dashed line indicates the maximum‐likelihood‐inferred time for the speciation‐coalescent threshold for species delimitation (GYMC). Vertical dotted lines indicate important events in geologic time: Oceanic Anoxic Events (red, OAEg for global and OAEr for regional), Cretaceous–Paleogene mass extinction (fuchsia, K‐Pg), Palaeocene–Eocene Thermal Maximum (brown, P‐ETM), opening of the Drake Passage (black, DP), establishment of the Antarctic Circumpolar Current (black, ACC), disruption of the Farallon Pacific Ridge (black, FPR) and formation of the East Scotia Rise (black, ESR). Geologic periods and eras are indicated with horizontal bars: upper Cretaceous (Cre (U)), Palaeocene (Pal), Eocene (Eoc), Oligocene (Oli), Miocene (Mio), Pliocene (Pli) and Pleistocene (Ple). Species names are followed by the collection regions.
Topological hypothesis testing
None of the phylogenetic hypotheses inferred from the Thoracica‐wide concatenated Sanger‐based data set support the monophyly of barnacles from deep‐sea hydrothermal vents. The topological test showed that the hypothesis of monophyly was significantly less probable than the hypothesis of nonmonophyly (marginal log likelihoods −16928.21 and −16908.62, respectively). The large difference in log likelihoods (>5) (Kass & Raftery 1995) constitutes strong contradictory evidence against the monophyly of vent barnacles as originally suggested by Pérez‐Losada et al. (2008).
Divergence estimates and biogeographical history
Tree time calibrations of the combined Sanger‐based data set produced divergence estimates slightly older under the Yule tree prior of constant speciation, when compared with the nearly identical estimates obtained under the birth–death prior models (Fig. 2 and Supporting information). These divergence estimates are consistent with estimates from Linse et al. (2013). The tree obtained under the birth–death model had the best likelihood score; however, no significant differences were encountered among the models (log likelihood difference <1). The time to the most recent common ancestor (TMRCA) of Clade A was estimated at 68.0 million years before present (Myr BP) (95% highest posterior density interval [HPD]: 38.2–105.9) under the birth–death models (BD) and 79.3 Myr BP (95% HPD: 47.1–121.5) under the Yule model of constant speciation rate. The TMRCAs of the Eolepadidae and the Neolepas‐Vulcanolepas‐Leucolepas subclade were estimated at 25.1 Myr BP (95% HPD: 12.1–43.3) and 10.5 Myr BP (95% HPD: 5.4–17.3) under BD, and 31.2 Myr BP (95% HPD: 15.4–53.7) and 13.8 Myr BP (95% HPD: 7.5–23.1) under the Yule model, respectively. Divergence between Pacific and non‐Pacific Neolepas‐Vulcanolepas eolepadids was estimated to have occurred 4.8 Myr BP (95% HPD: 2.3–8.5) and 6.4 Myr BP (95% HPD: 3.0–11.2) under BD and Yule models, respectively. The split between the East Scotia Ridge and the Indian Ocean lineages occurred 1.7 Myr BP (95% HPD: 0.4–3.8) under BD and 2.3 (95% HPD: 0.5–4.9) under Yule. The TMRCA of Clade B Eochionelasmus was estimated at 3.2 Myr BP (95% HPD: 1.1–6.7) under the birth–death model and 4.2 Myr BP (95% HPD: 1.3–8.8) under Yule. The analysis of historical biogeography suggests with high probability that hydrothermal vent barnacles from Clade A originated in the western Pacific, and colonized the Eastern Pacific, the Atlantic sector of the Southern Ocean and the Indian Ocean during the late Miocene to early Pliocene.
Species delimitation
GMYC analyses of Clade A identified a transition point between interspecific speciation–extinction processes and intraspecific coalescent processes at 0.6 Myr BP for the timescaled combined Sanger‐based phylogeny estimated with the birth–death model tree prior (Fig. 3). The GMYC model showed a significant (α = 0.05) better fit to the data than the null model of uniform coalescent branching rates (likelihood ratio = 25.9, P < 0.0001). There were 12 distinct clusters identified, which largely corresponded to species already described or populations that were presumed to be new species. Genetic distances (coxI uncorrected distances) among individuals within clusters ranged between 0 and 0.9% (Table S8, Supporting information). Genetic distances among individuals from different clusters ranged between 2 and 17.8% (except for the two Neolepas zevinae/rapanuii clusters whose maximum distance was 0.9%). Similarly, in Clade B Eochionelasmus, the genetic distances among individuals ranged between 0 and 0.9%.
Species‐tree estimation
The topology of the inferred Sanger‐based species tree is fully congruent with the topology of the phylogenetic hypothesis obtained with the concatenated Sanger‐based markers data set, and the branch support values are mostly equal (Fig. 4). Poorly resolved regions of the tree include the relationships among lineages of Vulcanolepas/Leucolepas from the Kermadec Arc, Lau Basin and Mariana Trough, and basally the positions of Neoverruca and Neobrachylepas within Clade A.
Figure 4.
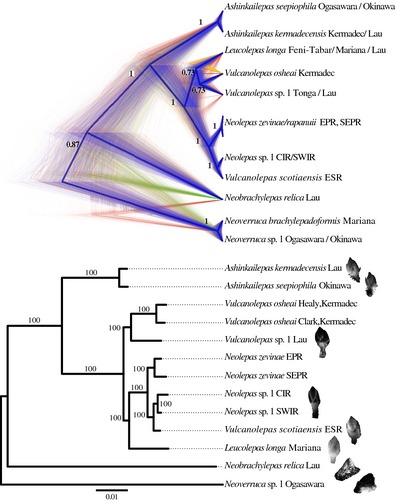
Top. Claudogram of the posterior distribution of species trees. High colour density is indicative of areas in the species trees with high topology agreement. Different colours represent different topologies. The maximum clade credibility species tree is shown with thicker branches. Branch labels show posterior probabilities. Trees with the same topology as the maximum clade credibility species tree are coloured in blue. Trees with different topologies are coloured yellow or red. Bottom. Maximum‐likelihood phylogenetic tree inferred with RAD‐seq data. The matrix used for this tree was obtained with a clustering threshold of 0.85 and minimum taxon coverage of 6. This matrix contains 828 960 nucleotide sites in 9766 loci. 76 353 of the sites are variable, and 26 955 are parsimony informative. This matrix contains 43.54% missing data. Branch labels show bootstrap support values. Scale bar indicates substitutions per site. Barnacle species images are from individuals included in this study. Species names are followed by the collection regions.
RAD phylogenetics
RAD‐seq matrices resulting from the three explored clustering thresholds (c 0.80, 0.85 and 0.90) contained similar numbers of loci and similar percentages of missing data per clustering parameter value used for the minimum taxon coverage in a given locus (approximately 15 500, 9600, 3800 and 600 loci, and 52%, 44%, 33% and 21% missing data, for m 4, 6, 8 and 10, respectively; see Table S9, Supporting information for details). The percentages of variable sites and parsimony informative sites across matrices ranged between 6.81–13.18% and 2.26–4.22%, respectively, being higher with smaller values of clustering thresholds and larger values of minimum taxon coverage. The tree topologies obtained from phylogenetic inferences of each matrix were identical to each other (Supporting information). These topologies from RAD‐seq matrices were also similar to the species tree obtained with Sanger‐based markers (Fig. 4), only differing in the position of Leucolepas, appearing in the RAD‐based trees as sister to the clade made up by N. zevinae/rapanuii from the East Pacific Rise, V. scotiaensis from the East Scotia Ridge and Neolepas sp. 1 from the Southwest and Central Indian Ridge. RAD‐based trees topologies were highly supported with bootstrap values of 100 for all branches, except for the ones from matrices generated with a minimum taxon coverage parameter of m10. In these cases, the branches supporting the clades of Vulcanolepas from the Lau Basin and the Kermadec Arc, and of Leucolepas–N. zevinae/rapanuii–V. scotiaensis–Neolepas sp. 1 have bootstrap support values >94 and 71, respectively.
Discussion
Are vent barnacles monophyletic?
The inferred evolutionary history of hydrothermal vent barnacles is not consistent with the hypothesis of monophyly (single ancestry) as proposed by the smaller taxon‐sampling studies of Pérez‐Losada et al. (2008) and Linse et al. (2013), which included only two of the four families of vent barnacles. Our analyses of a significantly expanded data set indicate that there are two main clades (Clade A and Clade B) (Fig. 2), thus suggesting that barnacles have colonized deep‐sea hydrothermal vents at least twice in history. The results from a concurrent study by Perez‐Losada et al. (2014) provide support to this inference by placing Clade B (Eochionelasmus ohtai) nested within the balanomorph barnacles, although the hypothesis of monophyly of vent barnacles was not explicitly tested in that study.
Deep‐sea hydrothermal vent barnacle Clade A is the more diverse of the two, containing six of the seven genera included in this study. This clade also contains a remarkable diversity of morphologies, including asymmetric (Neoverrucidae) and symmetric (Neobrachylepadidae), pedunculate (Eolepadidae) and sessile (Neoverrucidae and Neobrachylepadidae) forms (Fig. 4) (note that neoverrucid barnacles have a pedunculated stage during early ontogenesis (Newman & Hessler 1989)). The sister relationship between Clade A and the deep‐sea pedunculate Scalpellidae (Fig. 2) (Pérez‐Losada et al. 2008; Linse et al. 2013) suggests that the sessile state of the Neoverrucidae and Neobrachylepadidae is a derived state. This observation is consistent with the mounting evidence that the characters used to define higher taxonomic groups in Cirripedia need to be revised in the light of multilocus molecular phylogenetic hypotheses (Pérez‐Losada et al. 2008; Linse et al. 2013). A noteworthy example of this taxonomic and phylogenetic incongruence is the phylogenetic placement of N. relica nested in Clade A. N. relica is the sole living brachilepadoform species and until now was considered the most ‘primitive’ lineage of sessilian barnacles (Newman & Yamaguchi 1995). Clade B only contains the genus Eochionelasmus. Despite its morphological and phylogenetic affinities with the Balanomorpha, the phylogenetic position of Eochionelasmus in this study is unstable. Similarly, Perez‐Losada et al. (2014) found low support for the branches resolving the position Eochionelasmus ohtai within the balanomorphs. This instability is probably caused by the long branch supporting this clade, which may indicate a rapid evolutionary rate, old divergence or taxonomic undersampling (Fig. 2, Supporting information). Further taxonomic sampling of related genera and careful review of character use for systematics should help resolve its systematics.
Deep‐sea hydrothermal vent environments have been characterized as being patchy and ephemeral habitats with extreme spatial and temporal gradients of temperature, reduced chemicals, oxygen and food supply (Van Dover 2000). These conditions present significant physiological and ecological challenges to organisms and act as environmental filters that promote the evolution and distribution of species with specialized adaptations (Tunnicliffe et al. 2003; Fisher et al. 2007). The widespread persistence of vent chemosynthetic environments throughout earth's geologic history (Shock et al. 1995) has likely been an important factor enabling the independent colonization by multiple lineages of barnacles, as well as of other taxa, for example mussels (Lorion et al. 2013) and decapods (Yang et al. 2013). Clade A is nested within a predominantly deep‐sea clade Linse et al. (2013), suggesting a colonization of hydrothermal vents at depth. The nested position within Clade A of A. seepiophila – the only barnacle species known to live in both cold‐seep and hydrothermal vent environments – indicates a single colonization of seep environments by vent ancestors. This pattern contrasts with the stepwise colonization scenario of deep‐sea chemosynthetic environments, starting in organic substrates or cold‐seeps and then moving to hydrothermal vents, as suggested for other taxonomic groups (e.g. mussels (Lorion et al. 2013).
Historical biogeography
The most common recent ancestor of hydrothermal vent barnacles from Clade A probably lived in the late Mesozoic or early Cenozoic. The time to the most recent common ancestor inferred in this study is consistent with the timing inferred by Linse et al. (2013), but contrasts with the lower Cretaceous origin proposed by Pérez‐Losada et al. (2008) and with the classic hypotheses of antiquity of vent taxa, which proposed that hydrothermal vent barnacles were mid‐Mesozoic relict taxa (Newman 1979, 1985). The discrepancy with the results from Pérez‐Losada et al. (2008) is due to the exclusion of fossil calibration points given the uncertainty in the phylogenetic placement as described by Linse et al. (2013). The timing of radiation of Clade A during the Cenozoic is comparable to the estimates of origin and radiation in other chemosynthetic taxa, e.g. radiation of alvinocarid shrimp 6.7–11.7 Myr BP (Shank et al. 1999); origin of siboglinid tubeworms ca. 60 Myr BP (Chevaldonne et al. 2002); radiation of chemosynthetic mussels at ca. 45 Myr BP (Lorion et al. 2013); and radiation of kiwaid yeti crabs starting at ca. 30 Myr BP; also see reviews by Little & Vrijenhoek (2003) and Vrijenhoek (2013). A recent origin and radiation of most modern vent taxa and many other deep‐sea taxa (Little & Vrijenhoek 2003; Smith & Stockley 2005; Strugnell et al. 2008) is consistent with the inference of a major deep‐sea mass extinction event during the Cretaceous–Paleogene period boundary (Raup & Sepkoski 1982; Horne 1999; Harnik et al. 2012) (see Fig. 3). Several smaller‐scale extinction events linked to regional Oceanic Anoxic Events, ocean acidification and temperature changes also occurred during the Cretaceous period and at the Palaeocene–Eocene epoch boundary (Jacobs & Lindberg 1998; Rogers 2000; Harnik et al. 2012).
The most probable place of origin of the modern vent barnacle lineage from Clade A is the western Pacific, as indicated and highly supported by Bayesian ancestral state reconstruction. This is also the region where the oldest lineages and the highest diversity are found. The heterogeneity of depths in hydrothermal vent systems in the western Pacific and the close proximity to other chemosynthetic ecosystems such as cold seeps and organic enrichments, both shallow and deep, have been suggested as important factors driving the recolonization of vent environments and subsequent diversification (Moalic et al. 2011). Our analyses suggest that the most probable path of dispersal out of the western Pacific was a migration eastwards during the Miocene epoch, possibly following hydrothermal vent habitats along the Pacific–Antarctic Ridge, and colonization of the eastern Pacific. The neolepadids from the East Pacific Rise have a coalescence point that is posterior to the Oligocene disruption of the Pacific‐Farallon Ridge by subduction under the North American Plate, ca. 28.5 Myr BP (Fig. 3) (Atwater 1989), which can explain why barnacles are absent from the north eastern Pacific vents along the Juan de Fuca Ridge. A spreading through the Southern Hemisphere probably followed to the East Scotia Ridge and South Sandwich Arc during the late Miocene epoch, reaching the Southwest Indian Ridge and Central Indian Ridge during the Pliocene/Pleistocene epochs. No vent barnacle species have been found at Mid‐Atlantic Ridge hydrothermal vents, although the southern portion of this major mid‐ocean ridge remains largely unexplored.
The proposed history of dispersal is congruent with the timing of opening of the Drake Passage during the mid‐Eocene epoch, ca. 41 Myr BP (Scher 2006), the late Eocene establishment of the eastward‐flowing Antarctic Circumpolar Current (ACC), ca. 34 Myr BP (Scher 2006), and the mid‐Miocene formation of the East Scotia Rise, ca. 15 Myr BP (Livermore 2003) (see Fig. 3). Hydrothermal vent yeti crabs (Decapoda: Anomura: Kiwaidae) share an almost identical pattern of historical dispersal from the eastern Pacific to the East Scotia Ridge and the Southwest Indian Ridge (see Roterman et al. (2013) for a detailed hypothesis of vicariance in this group). A likely origin in the western or north‐western Pacific followed by migration and colonization eastwards throughout the Southern Hemisphere during the Miocene epoch has also been inferred for other nonvent deep‐sea taxa such as the octocoral Paragorgia arborea (Herrera et al. 2012) and other marine taxa such as the spiny dogfish Squalus acanthias (Verissimo et al. 2010) and the bryozoan Membranipora membranacea (Schwaninger 2008). These observations provide support for the biogeographical hypothesis proposed by Moalic et al. (2011) that the western Pacific was a centre of origin of modern vent fauna from which most taxa dispersed globally. However, our data do not support the idea of direct links between the western Pacific communities and the Indian Ocean, but rather a stepping‐stone mode of dispersal in the Southern Hemisphere following the direction of the dominant ACC. We suggest that the geological processes and dispersal mechanisms discussed here can explain the current distribution patterns of many other marine taxa and have played an important role shaping extant deep‐sea faunal diversity.
The history of Clade B is not well resolved. The phylogenetic hypothesis here presented suggests that the divergence of this lineage within the Balanomorpha occurred in the Mesozoic era (Fig. 2). However, this inferred antiquity is likely to be an artefact caused by taxonomic undersampling in this group. Additional data from other Echionelasmus populations, for example E. paquensis from the eastern Pacific, as well as from confamilial species and related groups would provide greater resolution of the evolution of Clade B.
Species delimitation and relationships
Inferences of species boundaries in Clade A, based on the generalized mixed Yule‐coalescent method, are largely congruent with descriptions of putative morphospecies. The identified species clusters are well constrained geographically by mid‐ocean spreading ridge system and neighbouring volcanic arc basins (Figs 3 and 4). Divergences among congeners in Ashinkailepas and Neoverruca are largely consistent with the biogeographical boundary between the north‐west and south‐west Pacific, inclusive of the Mariana Arc, proposed by Bachraty et al. (2009). Relationships among Vulcanolepas, Leucolepas and Neolepas species clusters remain contentious due to the nonmonophyly of all three genera as defined by Buckeridge et al. (2013) and thus require substantial revision.
There is a lack of overlap in genetic distances for the coxI barcode marker within and among inferred species clusters. The maximum genetic distance within species clusters of 0.9% and the minimum distance among species clusters of 2% are consistent with the proposed threshold value of ca. 2% to define species boundaries through DNA barcoding in Crustacea (Hebert et al. 2003; Lefebure et al. 2006). Similarly, the maximum genetic distance among individuals of Echionelasmus ohtai is 0.9%. The only exception to this pattern is found in the Neolepas zevinae/rapanuii species cluster pair, where the maximum distance between clusters is 0.9% (Table S8, Supporting information). There is no phylogenetic support for this split or geographic segregation between specimens from the East Pacific Rise and southern East Pacific Rise, thus suggesting that the division of Neolepas zevinae/rapanuii is not indicative of species‐level differentiation. A barcoding gap within and among species has been consistently found in other barnacle taxa (Tsang et al. 2008, 2009; Yoshida et al. 2011) and in crustaceans in general (Costa et al. 2007; Matzen da Silva et al. 2011); thus, our coxI genetic distance data provide further support to the species delimitations proposed for Clade A. The species delimitation framework developed in this study will enable rapid species assignments as specimens from newly‐ explored geographical regions become available.
RAD phylogenetics
Several sources of uncertainty have been associated with the use of the few traditional sequence markers available for nonmodel organisms (e.g. mitochondrial and ribosomal genes), including low variability, biased loci sampling, poor genomic representation, low statistical power and inclusion of pseudogenes, among others. The effects of these are often hard to identify due to the paucity of multilocus genomewide comparative data sets. Such problems have been recognized and accounted for in model organisms by comparing large numbers of genomic DNA sequences from various individuals and identifying thousands of variable regions across the genome (Rokas et al. 2003; Clark et al. 2007). Recent technological and methodological developments in next‐generation sequencing platforms and methodologies, such as RAD‐seq, have made genomic resources increasingly accessible and available for phylogenetics in nonmodel organisms (Wagner et al. 2012; Eaton & Ree 2013; Jones et al. 2013; Reitzel et al. 2013), thus offering a great opportunity to overcome the difficulties inherent to the use of traditional approaches in many taxa.
In this study, we demonstrated that RAD‐seq data provide strong support for the overall evolutionary history of vent barnacles inferred with traditional Sanger‐based markers, and allow the inference of a fully resolved and supported phylogenetic tree. The small difference in topology between the species tree inferred with Sanger‐based markers and the RAD‐seq trees does not alter any of the conclusions regarding the biogeographical history or species delimitation of vent barnacles, but does have taxonomic implications. Further sampling and a follow‐up morphological taxonomic revision would be needed to clarify the validity of the currently described genera. This study demonstrates the utility of comparative Sanger‐based and RAD sequencing as a means of comparative phylogenetic inference validation in poorly known taxa such as those thriving in the deep‐sea.
Conclusions
Phylogenetic inferences and topology tests indicate that hydrothermal vent barnacles are not a monophyletic group. The likely timing of barnacle radiation in hydrothermal vent ecosystems was during the late Cenozoic, consistent with the timing of other specific deep‐sea taxa, and correlated to the occurrence of major extinction events. Our analyses suggest that the western Pacific was the place of origin of the major hydrothermal vent barnacle lineage, followed by circumglobal colonization eastwards along the Southern Hemisphere during the Neogene period. Inferences of species boundaries based on generalized mixed Yule‐coalescent methods and DNA barcoding are largely congruent with morphological descriptions of putative species. RAD‐seq data provide strong support for the overall evolutionary history inferred from Sanger‐based markers and a fully resolved phylogenetic backbone for future studies of vent barnacle and hydrothermal faunal evolution. These results also constitute critical baseline data with which to assess potential effects of anthropogenic disturbances on deep‐sea ecosystems.
S.H. and T.M.S conceived and designed the research. S.H. performed the research. S.H. analysed the data. S.H., T.M.S. and H.W. collected and contributed the samples and reagents. S.H. wrote the manuscript with comments from T.M.S. and H.W.
Data accessibility
Supporting information: Nucleotide alignments, input files, are tree files available from the Dryad Digital Repository: http://doi.org/10.5061/dryad.7kn5k.
Raw data: Raw DNA sequences are available at the U.S. National Center for Biotechnology Information (NCBI) GenBank (Accession nos. KP294908‐KP295191) and Sequence Read Archive (SRA Accession nos. SRP051026).
Supporting information
Table S1 Collection and sequence information for the specimens used in this study.
Table S2 Accession numbers for sequences from the superorder Thoracica retrieved from GenBank.
Table S3 Predictions of number of RAD‐tags in thoracician barnacles using SbfI. Data for Daphnia pulex obtained from the U.S. National Center for Biotechnology Information (NCBI) WGS database. Observed frequency of recognition sequences and calculated probability based on a trinucleotide genome composition model were generated following the methodology described by Herrera et al. (2014). Data for known barnacle genome sizes obtained from the Animal Genome Size Database (http://www.genomesize.com). C‐value is defined as the amount of DNA in picograms in the nucleus, where the genome size in Mbp = 978 × C‐value.
Table S4 Nucleotide substitution models for each Sanger‐based genetic marker, as selected by the BIC criterion in jModeltest.
Table S5 Results from Xia saturation test for each Sanger‐based genetic marker.
Table S6 RAD sequencing results and filtering statistics.
Table S7 RAD clustering statistics.
Table S8 Uncorrected pairwise genetic coxI distances (%) among specimens from Clade A
Table S9 RAD‐seq matrices statistics.
Acknowledgements
This research was supported by the Office of Ocean Exploration and Research of the National Oceanic and Atmospheric Administration (NA09OAR4320129 to TMS); the Division of Ocean Sciences of the National Science Foundation (OCE‐1131620 to TMS); the Division of Polar Programs of the National Science Foundation (PLR‐0739675 to TMS); the Astrobiology Science and Technology for Exploring Planets program of the National Aeronautics and Space Administration (NNX09AB76G to TMS); and the Academic Programs Office (Ocean Ventures Fund to SH), the Ocean Exploration Institute (Fellowship support to TMS) and the Ocean Life Institute of the Woods Hole Oceanographic Institution (internal grant to TMS and SH). For enabling access to key specimens, we thank K. Iizasa (U. Tokyo), Y. Suzuki (U. Tokyo), S. Nakagawa (JAMSTEC), P. Tyler (NOCS), J. Copley (NOCS), A. Rogers (Oxford), N. Roterman (Oxford), K. Linse (BAS), M. Clark (NIWA), A. Rowden (NIWA), K. Schnabel (NIWA), S. Mills (NIWA), J. Resing (NOAA‐PMEL), R. Embley (NOAA‐PMEL), A. Reysenbach (PSU), M.K. Tivey (WHOI), P. Fryer (UH), C. Langmuir (Harvard), K. Von Damm (UNH), M. Lilley (UW), the masters, crew, scientific personnel and funding agencies of expeditions AT03‐28, AT07‐06, JC042, JC067, KM0417, KM0912, KOK0505, KOK0506, NT97‐10, NT97‐14, NT99‐09, RR1211, TAN1007, TAN1104, TAN1206, TN234, TN236, YK06‐13 and YK09‐13. Specimens provided by the National Institute of Water and Atmospheric Research (NIWA) were collected under the following research programs: Kermadec Arc Minerals, funded by the New Zealand Ministry of Business, Innovation & Employment (MBIE), Auckland University, Institute of Geological and Nuclear Science (GNS) and WHOI; Ocean Survey 20/20 funded by Land Information New Zealand; Impact of resource use on vulnerable deep‐sea communities (CO1X0906), funded by MBIE; and the Joint New Zealand‐USA 2005 NOAA Ring of Fire Expedition, part of NIWA's Seamount Program (FRST CO1X0508). We thank A.M. Tarrant, A.M. Reitzel, J.A.H. Benzie and three anonymous reviewers for providing helpful comments that improved this manuscript.
The copyright line for this article was changed on 25 November, 2015 after original online publication.
References
- Atwater T (1989) Plate tectonic history of the northeast Pacific and western North America In: The Eastern Pacific Ocean and Hawaii (eds Winterer EL, Hussong DM. & Decker RW.), pp. 21–72. Geological Society of America, Boulder, Colorado. [Google Scholar]
- Bachraty C, Legrende P, Desbruyeres D (2009) Biogeographic relationships among hydrothermal vent faunas on a global scale. Deep‐Sea Research Part I‐Oceanographic Research Papers, 56, 1371–1378. [Google Scholar]
- Baird N, Etter P, Atwood T, et al (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One, 3, e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker ET, German CR (2004) On the global distribution of hydrothermal vent fields In: Mid‐Ocean Ridges (eds German CR, Lin J, Parson LM.), pp. 245–266. American Geophysical Union, Washington, District of Columbia. [Google Scholar]
- Bensasson D, Zhang D, Hartl D, Hewitt G (2001) Mitochondrial pseudogenes: evolution's misplaced witnesses. Trends in Ecology & Evolution, 16, 314–321. [DOI] [PubMed] [Google Scholar]
- Boschen RE, Rowden AA, Clark MR, Gardner JPA (2013) Mining of deep‐sea seafloor massive sulfides: a review of the deposits, their benthic communities, impacts from mining, regulatory frameworks and management strategies. Ocean and Coastal Management, 84, 54–67. [Google Scholar]
- Brito PH, Edwards SV (2008) Multilocus phylogeography and phylogenetics using sequence‐based markers. Genetica, 135, 439–455. [DOI] [PubMed] [Google Scholar]
- Buckeridge J (2000) Neolepas osheai sp. nov., a new deep‐sea vent barnacle (Cirripedia: Pedunculata) from the Brothers Caldera, south‐west Pacific Ocean. New Zealand Journal of Marine and Freshwater Research, 34, 409–418. [Google Scholar]
- Buckeridge JS, Linse K, Jackson JA (2013) Vulcanolepas scotiaensis sp. nov., a new deep‐sea scalpelliform barnacle (Eolepadidae: Neolepadinae) from hydrothermal vents in the Scotia Sea, Antarctica. Zootaxa, 3745, 551–568. [DOI] [PubMed] [Google Scholar]
- Cairns S (2007) Deep‐water corals: an overview with special reference to diversity and distribution of deep‐water scleractinian corals. Bulletin of Marine Science, 81, 311–322. [Google Scholar]
- Carpenter KE, Barber PH, Crandall ED et al (2011) Comparative phylogeography of the Coral Triangle and implications for marine management. Journal of Marine Biology, 2011, 1–14. [Google Scholar]
- Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH (2011) Stacks: building and genotyping loci de novo from short‐read sequences. G3 1, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Molecular Ecology, 22, 3124–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaldonne P, Jollivet D, Desbruyeres D, Lutz RA, Vrijenhoek RC (2002) Sister‐species of eastern Pacific hydrothermal vent worms (Ampharetidae, Alvinellidae, Vestimentifera) provide new mitochondrial COI clock calibration. Cahiers de Biologie Marine, 43, 367–370. [Google Scholar]
- Clark AG, Eisen MB, Smith DR et al (2007) Evolution of genes and genomes on the Drosophila phylogeny. Nature, 450, 203–218. [DOI] [PubMed] [Google Scholar]
- Colgan DJ, McLauchlan A, Wilson GDF et al (1998) Histone H3 and U2 snRNA DNA sequences and arthropod molecular evolution. Australian Journal of Zoology, 46, 419–437. [Google Scholar]
- Costa FO, deWaard JR, Boutillier J et al (2007) Biological identifications through DNA barcodes: the case of the Crustacea. Canadian Journal of Fisheries and Aquatic Sciences, 64, 272–295. [Google Scholar]
- Cubelio SS, Tsuchida S, Watanabe S (2007) Vent associated Munidopsis (Decapoda: Anomura: Galatheidae) from Brothers Seamount, Kermadec Arc, Southwest Pacific, with description of one new species. Journal of Crustacean Biology, 27, 513–519. [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nature Methods, 9, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbruyeres D, Alayse‐Danet A‐M, Ohta S, the Scientific Parties of biolauands (1994) Deep‐sea hydrothermal communities in Southwestern Pacific back‐arc basins (the North Fiji and Lau Basins): composition, microdistribution and food web. Marine Geology 116, 227–242. [Google Scholar]
- Drummond AJ, Ashton B, Buxton S et al (2011) Geneious v6.1.6. Available from http://www.geneious.com/.
- Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton DA (2014) PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics, 30, 1844–1849.24603985 [Google Scholar]
- Eaton DAR, Ree RH (2013) Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Systematic Biology, 62, 689–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SV (2008) Is a new and general theory of molecular systematics emerging? Evolution, 63, 1–19. [DOI] [PubMed] [Google Scholar]
- Fan Y, Wu R, Chen MH, Kuo L, Lewis PO (2011) Choosing among partition models in Bayesian phylogenetics. Molecular Biology and Evolution, 28, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher CR, Takai K, Le Bris N (2007) Hydrothermal vent ecosystems. Oceanography, 20, 14–23. [Google Scholar]
- Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3, 294–299. [PubMed] [Google Scholar]
- Fujisawa T, Barraclough TG (2013) Delimiting species using single‐locus data and the Generalized Mixed Yule Coalescent approach: a revised method and evaluation on simulated data sets. Systematic Biology, 62, 707–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gernhard T (2008) The conditioned reconstructed process. Journal of Theoretical Biology, 253, 769–778. [DOI] [PubMed] [Google Scholar]
- Harnik PG, Lotze HK, Anderson SC et al (2012) Extinctions in ancient and modern seas. Trends in Ecology & Evolution, 27, 608–617. [DOI] [PubMed] [Google Scholar]
- Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Proceedings of the Royal Society B: Biological Sciences, 270, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J, Drummond AJ (2010) Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution, 27, 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera S, Munro C, Nganro N et al (2010) Biodiversity of the deep‐sea benthic fauna in the Sangihe‐Talaud region, Indonesia: observations from the INDEX‐SATAL 2010 expedition. AGU Fall Meeting Abstracts, 1, 1234. [Google Scholar]
- Herrera S, Shank TM, Sanchez JA (2012) Spatial and temporal patterns of genetic variation in the widespread antitropical deep‐sea coral Paragorgia arborea . Molecular Ecology, 21, 6053–6067. [DOI] [PubMed] [Google Scholar]
- Herrera S, Reyes‐Herrera PH, Shank TM (2014) Genome‐wide predictability of restriction sites across the eukaryotic tree of life. bioRxiv, doi: 10.1101/007781. [Google Scholar]
- Hessler RR, Lonsdale PF (1991) Biogeography of Mariana Trough hydrothermal vent communities. Deep‐Sea Research Part A‐Oceanographic Research Papers, 38, 185–199. [Google Scholar]
- Hipp AL, Eaton DAR, Cavender‐Bares J et al (2014) A framework phylogeny of the American Oak clade based on sequenced RAD data. PLoS One, 9, e93975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne DJ (1999) Ocean circulation modes of the phanerozoic: implications for the antiquity of deep‐sea benthonic invertebrates. Crustaceana, 72, 999–1018. [Google Scholar]
- Jacobs DK, Lindberg DR (1998) Oxygen and evolutionary patterns in the sea: onshore /offshore trends and recent recruitment of deep‐sea faunas. Proceedings of the National Academy of Sciences of the United States of America, 95, 9396–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DS (1993) A new Neolepas (Cirripedia: Thoracica: Scalpellidae) from an abyssal hydrothermal vent, southeast Pacific. Bulletin of Marine Science, 52, 937–948. [Google Scholar]
- Jones JC, Fan S, Franchini P, Schartl M, Meyer A (2013) The evolutionary history of Xiphophorus fish and their sexually selected sword: a genome‐wide approach using restriction site‐associated DNA sequencing. Molecular Ecology, 22, 2986–3001. [DOI] [PubMed] [Google Scholar]
- Kass RE, Raftery AE (1995) Bayes factors. Journal of the American Statistical Association, 90, 773–795. [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebure T, Douady CJ, Gouy M, Gilbert J (2006) Relationship between morphological taxonomy and molecular divergence within Crustacea: proposal of a molecular threshold to help species delimitation. Molecular Phylogenetics and Evolution, 40, 435–447. [DOI] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Drummond AJ, Suchard MA (2009) Bayesian phylogeography finds its roots. PLoS Computational Biology, 5, e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linse K, Jackson JA, Fitzcharles E, Sands CJ, Buckeridge JS (2013) Phylogenetic position of Antarctic Scalpelliformes (Crustacea: Cirripedia: Thoracica). Deep‐Sea Research Part I‐Oceanographic Research Papers, 73, 99–116. [Google Scholar]
- Little CTS, Vrijenhoek RC (2003) Are hydrothermal vent animals living fossils? Trends in Ecology & Evolution, 18, 582–588. [Google Scholar]
- Livermore R (2003) Back‐arc spreading and mantle flow in the East Scotia Sea. Geological Society, London, Special Publications, 219, 315–331. [Google Scholar]
- Lopez JV, Yuhki N, Masuda R, Modi W, O'Brien SJ (1994) Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. Journal of Molecular Evolution, 39, 174–190. [DOI] [PubMed] [Google Scholar]
- Lorion J, Kiel S, Faure B et al (2013) Adaptive radiation of chemosymbiotic deep‐sea mussels. Proceedings of the Royal Society B: Biological Sciences, 280, 20131243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh L, Copley JT, Huvenne VAI et al (2012) Microdistribution of faunal assemblages at deep‐sea hydrothermal vents in the Southern Ocean. PLoS One, 7, e48348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzen da Silva J, Creer S, dos Santos A et al (2011) Systematic and evolutionary insights derived from mtDNA COI barcode diversity in the Decapoda (Crustacea: Malacostraca). PLoS One, 6, e19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain C, Mincks SL (2010) The dynamics of biogeographic ranges in the deep sea. Proceedings of the Royal Society B: Biological Sciences, 277, 3533–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalic Y, Desbruyeres D, Duarte CM et al (2011) Biogeography revisited with network theory: retracing the history of hydrothermal vent communities. Systematic Biology, 61, 127–137. [DOI] [PubMed] [Google Scholar]
- Monaghan MT, Wild R, Elliot M et al (2009) Accelerated species inventory on madagascar using coalescent‐based models of species delineation. Systematic Biology, 58, 298–311. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Watanabe H, Miyazaki J et al (2012) Discovery of new hydrothermal activity and chemosynthetic fauna on the Central Indian Ridge at 18 degrees ‐20 degrees S. PLoS One, 7, e32965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman WA (1979) A new scalpellid (Cirripedia): a Mesozoic relic living near an abyssal hydrothermal spring. Transactions of the San Diego Society of Natural History, 19, 153–167. [Google Scholar]
- Newman WA (1985) The abyssal hydrothermal vent invertebrate fauna. A glimpse of antiquity? Bulletin of the Biological Society of Washington, 6, 231–242. [Google Scholar]
- Newman WA, Hessler RR (1989) A new abyssal hydrothermal verrucomorphan (Cirripedia; Sessilia): the most primitive living sessile barnacle. Transactions of the San Diego Society of Natural History, 21, 259–273. [Google Scholar]
- Newman WA, Yamaguchi T (1995) A new sessile barnacle (Cirripedia, Brachylepadomorpha) from the Lau Back‐Arc Basin, Tonga; first record of a living representative since the Miocene. Bulletin du Muséum National d'Histoire Naturelle. 4e Série. Section A, Zoologie, Biologie et Écologie Animales, 17, 221–243. [Google Scholar]
- Newman WA, Yamaguchi T, Southward AJ, Segonzac M (2006) Arthropoda, Crustacea, Cirripedia In: Handbook of Deep‐Sea Hydrothermal Vent Fauna (eds Desbruyeres D, Segonzac M, Bright M.), pp. 356–357. Denisia, Linz, Austria. [Google Scholar]
- Nylander JA, Wilgenbusch JC, Warren DL, Swofford DL (2008) AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinfomatics, 24, 581–583. [DOI] [PubMed] [Google Scholar]
- Ono T, Fujikura K, Hashimoto J, Fiujiwara Y, Segawa S (1996) The hydrothermal vent community at the Kaikata Seamount near Ogasawara (Bonin) Islands, South Japan. JAMSTEC Journal of Deep Sea Research, 12, 221–230. [Google Scholar]
- Ohta S (1990) Deep‐sea submersible survey of the hydrothermal vent community on the northeastern slope of the Iheya ridge, the Okinawa Trough. Proceedings of JAMSTEC Symposium on Deep Sea Research, 6, 145–156. [Google Scholar]
- Perez‐Losada M, Hoeg JT, Simon‐Blecher N et al (2014) Molecular phylogeny, systematics and morphological evolution of the acorn barnacles (Thoracica: Sessilia: Balanomorpha). Molecular Phylogenetics and Evolution, 81, 147–158. [DOI] [PubMed] [Google Scholar]
- Pérez‐Losada M, Harp M, Høeg JT et al (2008) The tempo and mode of barnacle evolution. Molecular Phylogenetics and Evolution, 46, 328–346. [DOI] [PubMed] [Google Scholar]
- Pons J, Barraclough TG, Gomez‐Zurita J et al (2006) Sequence‐based species delimitation for the DNA taxonomy of undescribed insects. Systematic Biology, 55, 595–609. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ (2007) Tracer v1.5, Available from http://beast.bio.ed.ac.uk/Tracer.
- Raup DM, Sepkoski JJJ (1982) Mass extinctions in the marine fossil record. Science, 215, 1501–1503. [DOI] [PubMed] [Google Scholar]
- Reid WDK, Sweeting CJ, Wigham BD et al (2013) Spatial differences in East Scotia Ridge hydrothermal vent food webs: influences of chemistry, microbiology and predation on trophodynamics. PLoS One, 8, e65553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzel AM, Herrera S, Layden MJ, Martindale MQ, Shank TM (2013) Going where traditional markers have not gone before: utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Molecular Ecology, 22, 2953–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers AD (2000) The role of the oceanic oxygen minima in generating biodiversity in the deep sea. Deep‐Sea Research Part II, 47, 119–148. [Google Scholar]
- Rogers AD, Tyler PA, Connelly DP et al (2012) The discovery of new deep‐sea hydrothermal vent communities in the southern ocean and implications for biogeography. PLoS Biology, 10, e1001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, Carroll SB (2003) Genome‐scale approaches to resolving incongruence in molecular phylogenies. Nature, 425, 798–804. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P et al (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roterman CN, Copley JT, Linse KT, Tyler PA, Rogers AD (2013) The biogeography of the yeti crabs (Kiwaidae) with notes on the phylogeny of the Chirostyloidea (Decapoda: Anomura). Proceedings of the Royal Society B: Biological Sciences, 280, 20130718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HD (2006) Timing and climatic consequences of the opening of Drake Passage. Science, 312, 428–430. [DOI] [PubMed] [Google Scholar]
- Schwaninger HR (2008) Global mitochondrial DNA phylogeography and biogeographic history of the antitropically and longitudinally disjunct marine bryozoan Membranipora membranacea L. (Cheilostomata): another cryptic marine sibling species complex? Molecular Phylogenetics and Evolution, 49, 893–908. [DOI] [PubMed] [Google Scholar]
- Shank TM, Black MB, Halanych KM, Lutz RA, Vrijenhoek RC (1999) Miocene radiation of deep‐sea hydrothermal vent shrimp (Caridea: Bresiliidae): evidence from mitochondrial cytochrome oxidase subunit I. Molecular Phylogenetics and Evolution, 13, 244–254. [DOI] [PubMed] [Google Scholar]
- Shank T, Herrera S, Bors E et al (2010) Hydrothermal vents and organic falls in the heart of the Coral Triangle: chemosynthetic communities discovered via relepresence in the Sangihe‐Talaud region, Northern Sulawesi, Indonesia. AGU Fall Meeting Abstracts, 1, 04. [Google Scholar]
- Shock EL, Mccollom T, Schulte MD (1995) Geochemical constraints on chemolithoautotrophic reactions in hydrothermal systems. Origins of Life and Evolution of the Biosphere, 25, 141–159. [DOI] [PubMed] [Google Scholar]
- Smith AB, Stockley B (2005) The geological history of deep‐sea colonization by echinoids: roles of surface productivity and deep‐water ventilation. Proceedings of the Royal Society B: Biological Sciences, 272, 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southward A (2005) Systematics and ecology of a new species of stalked barnacle (Cirripedia: Thoracica: Scalpellomorpha: Eolepadidae: Neolepadini) from the Pacific‐Antarctic Ridge at 38° S. Marine Biodiversity, 35, 147–156. [Google Scholar]
- Southward AJ, Newman WA (1998) Ectosymbiosis between filamentous sulphur bacteria and a stalked barnacle (Scalpellomorpha, Neolepadinae) from the Lau Back Arc Basin, Tonga. Cahiers de Biologie Marine, 39, 259–262. [Google Scholar]
- Stadler T (2009) On incomplete sampling under birth‐death models and connections to the sampling‐based coalescent. Journal of Theoretical Biology, 261, 58–66. [DOI] [PubMed] [Google Scholar]
- Stamatakis A (2006) RAxML‐VI‐HPC: maximum likelihood‐based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22, 2688–2690. [DOI] [PubMed] [Google Scholar]
- Strugnell JM, Rogers AD, Prodohl PA, Collins M, Allcock AL (2008) The thermohaline expressway: the Southern Ocean as a centre of origin for deep‐sea octopuses. Cladistics, 24, 853–860. [DOI] [PubMed] [Google Scholar]
- Tao C, Lin J, Guo S et al (2011) First active hydrothermal vents on an ultraslow‐spreading center: Southwest Indian Ridge. Geology, 40, 47–50. [Google Scholar]
- Thurber AR, Sweetman AK, Narayanaswamy BE et al (2014) Ecosystem function and services provided by the deep sea. Biogeosciences, 11, 3941–3963. [Google Scholar]
- Tsang L, Chan B, Wu T et al (2008) Population differentiation in the barnacle Chthamalus malayensis: postglacial colonization and recent connectivity across the Pacific and Indian Oceans. Marine Ecology Progress Series, 364, 107–118. [Google Scholar]
- Tsang LM, Chan BKK, Shih F‐L, Chu KH, Allen Chen C (2009) Host‐associated speciation in the coral barnacle Wanella milleporae (Cirripedia: Pyrgomatidae) inhabiting the Millepora coral . Molecular Ecology, 18, 1463–1475. [DOI] [PubMed] [Google Scholar]
- Tufar W (1990) Modern hydrothermal activity, formation of complex massive sulfide deposits and associated vent communities in the Manus back‐arc basin (Bismarck Sea, Papua New Guinea). Mitteilungen der Oesterreichischen Geologischen Gesellschaft, 82, 183–210. [Google Scholar]
- Tunnicliffe V, Southward AJ (2004) Growth and breeding of a primitive stalked barnacle Leucolepas longa (Cirripedia: Scalpellomorpha: Eolepadidae: Neolepadinae) inhabiting a volcanic seamount off Papua New Guinea. Journal of the Marine Biological Association of the UK, 84, 121–132. [Google Scholar]
- Tunnicliffe V, Juniper SK, Sibuet M (2003) Reducing environments of the deep‐sea floor In: Ecosystems of the World: The Deep Sea (ed. Tyler PA.), pp. 81–110. Elsevier Press, Amsterdam, Netherlands. [Google Scholar]
- Van Dover C (2000) The Ecology of Deep‐Sea Hydrothermal Vents. Princeton University Press, Princeton, New Jersey: [Google Scholar]
- Van Dover CL (2002) Trophic relationships among invertebrates at the Kairei hydrothermal vent field (Central Indian Ridge). Marine Biology, 141, 761–772. [Google Scholar]
- Van Dover CL (2010) Mining seafloor massive sulphides and biodiversity: what is at risk? ICES Journal of Marine Science, 68, 341–348. [Google Scholar]
- Van Dover CL, Humphris SE, Fornari D et al (2001) Biogeography and ecological setting of Indian Ocean hydrothermal vents. Science, 294, 818–823. [DOI] [PubMed] [Google Scholar]
- Van Dover CL, Smith CR, Ardron J et al (2012) Designating networks of chemosynthetic ecosystem reserves in the deep sea. Marine Policy, 36, 378–381. [Google Scholar]
- Verissimo A, McDowell JR, Graves JE (2010) Global population structure of the spiny dogfish Squalus acanthias, a temperate shark with an antitropical distribution. Molecular Ecology, 19, 1651–1662. [DOI] [PubMed] [Google Scholar]
- Vrijenhoek R (2013) On the instability and evolutionary age of deep‐sea chemosynthetic communities. Deep‐Sea Research Part II, 92, 189–200. [Google Scholar]
- Wagner CE, Keller I, Wittwer S et al (2012) Genome‐wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Molecular Ecology, 22, 787–798. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Tsuchida S, Fujikura K et al (2005) Population history associated with hydrothermal vent activity inferred from genetic structure of neoverrucid barnacles around Japan. Marine Ecology‐Progress Series, 288, 233–240. [Google Scholar]
- Whiting MF (2002) Mecoptera is paraphyletic: multiple genes and phylogeny of Mecoptera and Siphonaptera. Zoologica Scripta, 31, 93–104. [Google Scholar]
- Xia XH (2013) DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Molecular Biology and Evolution, 30, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Xie Z, Salemi M, Chen L, Wang Y (2003) An index of substitution saturation and its application. Molecular Phylogenetics and Evolution, 26, 1–7. [DOI] [PubMed] [Google Scholar]
- Xie WG, Lewis PO, Fan Y, Kuo L, Chen MH (2011) Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Systematic Biology, 60, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Lu B, Chen DF et al (2013) When did decapods invade hydrothermal vents? Clues from the western Pacific and Indian Oceans. Molecular Biology and Evolution, 30, 305–309. [DOI] [PubMed] [Google Scholar]
- Yoshida R, Osawa M, Hirose M, Hirose E (2011) A new genus and two new species of Peltogastridae (Crustacea: Cirripedia: Rhizocephala) parasitizing Hermit Crabs from Okinawa Island (Ryukyu Archipelago, Japan), and their DNA‐barcodes. Zoological Science, 28, 853–862. [DOI] [PubMed] [Google Scholar]
- Yule GA (1925) A mathematical theory of evolution, based on the conclusions of Dr. J.C. Willis. Philosophical Transactions of the Royal Society of London B Biological Sciences, 213, 21–87. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Collection and sequence information for the specimens used in this study.
Table S2 Accession numbers for sequences from the superorder Thoracica retrieved from GenBank.
Table S3 Predictions of number of RAD‐tags in thoracician barnacles using SbfI. Data for Daphnia pulex obtained from the U.S. National Center for Biotechnology Information (NCBI) WGS database. Observed frequency of recognition sequences and calculated probability based on a trinucleotide genome composition model were generated following the methodology described by Herrera et al. (2014). Data for known barnacle genome sizes obtained from the Animal Genome Size Database (http://www.genomesize.com). C‐value is defined as the amount of DNA in picograms in the nucleus, where the genome size in Mbp = 978 × C‐value.
Table S4 Nucleotide substitution models for each Sanger‐based genetic marker, as selected by the BIC criterion in jModeltest.
Table S5 Results from Xia saturation test for each Sanger‐based genetic marker.
Table S6 RAD sequencing results and filtering statistics.
Table S7 RAD clustering statistics.
Table S8 Uncorrected pairwise genetic coxI distances (%) among specimens from Clade A
Table S9 RAD‐seq matrices statistics.
Data Availability Statement
Supporting information: Nucleotide alignments, input files, are tree files available from the Dryad Digital Repository: http://doi.org/10.5061/dryad.7kn5k.
Raw data: Raw DNA sequences are available at the U.S. National Center for Biotechnology Information (NCBI) GenBank (Accession nos. KP294908‐KP295191) and Sequence Read Archive (SRA Accession nos. SRP051026).