

ABSTRACT
Purpose: To determine the effectiveness of adjuvant dendritic cell (DC) vaccination to induce tumor-specific immunological responses in stage III melanoma patients.
Experimental design: Retrospective analysis of stage III melanoma patients, vaccinated with autologous monocyte-derived DC loaded with tumor-associated antigens (TAA) gp100 and tyrosinase after radical lymph node dissection. Skin-test infiltrating lymphocytes (SKILs) obtained from delayed-type hypersensitivity skin-test biopsies were analyzed for the presence of TAA-specific CD8+ T cells by tetrameric MHC-peptide complexes and by functional TAA-specific T cell assays, defined by peptide-recognition (T2 cells) and/or tumor-recognition (BLM and/or MEL624) with specific production of Th1 cytokines and no Th2 cytokines.
Results: Ninety-seven patients were analyzed: 21 with stage IIIA, 34 with stage IIIB, and 42 had stage IIIC disease. Tetramer-positive CD8+ T cells were present in 68 patients (70%), and 24 of them showed a response against all 3 epitopes tested (gp100:154–162, gp100:280–288, and tyrosinase:369–377) at any point during vaccinations. A functional T cell response was found in 62 patients (64%). Rates of peptide-recognition of gp100:154–162, gp100:280–288, and tyrosinase:369–377 were 40%, 29%, and 45%, respectively. Median recurrence-free survival and distant metastasis-free survival of the whole study population were 23.0 mo and 36.8 mo, respectively.
Conclusions: DC vaccination induces a functional TAA-specific T cell response in the majority of stage III melanoma patients, indicating it is more effective in stage III than in stage IV melanoma patients. Furthermore, performing multiple cycles of vaccinations enhances the chance of a broader immune response.
KEYWORDS: Dendritic cell, DTH, immune response, skin-infiltrating lymphocytes, stage III melanoma, vaccination
Abbreviations
- CI
confidence interval
- DC
dendritic cell(s)
- DMFS
distant metastasis-free survival
- DTH
delayed-type hypersensitivity
- ELISPOT
enzyme-linked immunosorbent spot
- FDA
Food and Drug Administration
- HR
hazard ratio(s)
- HS
human serum
- IL
interleukin
- KLH
keyhole limpet hemocyanin
- MHC
major histocompatibility complex
- OS
overall survival
- PBMC
peripheral blood mononuclear cells
- PD-1
programmed death 1
- PGE2
prostaglandin E2
- RFS
recurrence-free survival
- SKIL(s)
skin-test infiltrating lymphocyte(s)
- TAA
tumor-associated antigen(s)
- Th
T-helper
- TNF-α
tumor necrosis factor-α.
Introduction
The incidence of stage III melanoma is rising and these patients are at high risk of relapse, even after a radical lymph node dissection with curative intention.1,2 Balch and colleagues showed that 5-y survival within substages of stage III were 78%, 59%, and 40% for patients with stage IIIA, IIIB, and IIIC melanoma, respectively.3 There is a great medical need for adjuvant treatments that will improve survival rates in this population.3,4 Until recently, interferon-α was the only approved adjuvant treatment, however it minimally improves overall survival (OS) and is not recommended in most European countries because of its side effect profile.5 For this reason, other adjuvant therapies are currently under investigation, among them ipilimumab,6 programmed death 1 (PD-1) inhibitors (NCT02388906, NCT02362594), and vaccination strategies (NCT00796445). Adjuvant ipilimumab 10 mg/kg showed a significant improvement of recurrence-free survival (RFS) compared to placebo, and it has been recently approved by the Food and Drug Administration (FDA). However, OS data have not yet been reported and treatment was accompanied by significant grade 3 and 4 toxicity.6
Dendritic cells (DC) are the most efficient antigen-presenting cells of the immune system due to their capacity to activate and prime naive T cells.7 They are essential in the induction of antitumor immunity, and may therefore play a vital role in anticancer immunotherapy.8,9 DC can be generated ex vivo, activated, and loaded with tumor antigens, and then injected into patients.7,10 We and others have treated stage IV melanoma patients with DC-based vaccines, and noted induction of tumor-specific immune responses, but long-lasting clinical responses are, thus far, limited.7,9,11,12 Since high tumor load causes immune suppression by secretion of immunosuppressive cytokines, and attraction of regulatory T cells and myeloid-derived suppressor cells in the tumor microenvironment, DC vaccination may be more effective as an adjuvant treatment in stage III patients.13,14
Because the immunotherapeutic options for melanoma are growing rapidly, immunomonitoring of patients is of great importance in these clinical trials. For instance, enzyme-linked immunosorbent spot (ELISPOT) assays and flow cytometric assays to detect antigen-specific T cells in peripheral blood samples, are commonly used in cancer immunotherapy trials. However, since tumor-associated antigen (TAA)-specific T cells are detected in only a minority of the peripheral blood samples, and to date no correlation has been found between immunological data in peripheral blood and clinical outcome, these peripheral blood assays might not be optimal for immunomonitoring.15
We previously described a bioassay evaluating skin-test infiltrating lymphocyte (SKIL) cultures from delayed-type hypersensitivity (DTH) skin-test biopsies after DC vaccination in stage IV melanoma patients.16 The mechanism of action of this bioassay is based on the fact that tumor-specific T cells should be able to invade peripheral tissues, recognize tumor antigens, proliferate, and deliver a functional response upon tumor-specific stimulation. Antigen-specific T cells which are also able to migrate to peripheral tissues are selected when a DTH with TAA-loaded DC is used. After short-time culturing of these SKILs from DTH skin biopsies, they are challenged by target cells loaded with defined tumor peptides and target cells that express naturally processed tumor antigens.10,16
The aim of this retrospective study was to explore the effectiveness of adjuvant DC vaccination to induce tumor-specific immunological responses in stage III melanoma patients.
Results
Patient and vaccine characteristics
The aim of this study was to evaluate immunological responses to DC vaccination. Since the DTH skin-test biopsies were taken after each cycle of three vaccinations, patients should have had completed at least one cycle to be immunologically evaluable. In total, 97 out of 99 (98%) HLA*02:01-positive stage III melanoma patients met this criterion. Baseline characteristics of the evaluable patients are listed in Table 1. Overall, 21 patients (22%) had stage IIIA, 34 (35%) had stage IIIB, and 42 (43%) had stage IIIC disease. The phenotype of the generated DC (expression of major histocompatibility complex (MHC) class I/II and co-stimulatory molecules) was determined by flow cytometry and the produced vaccines met the quality release criteria (Fig. S1A).11 Flow cytometry showed intracellular expression of both TAA (gp100 and tyrosinase) in DC electroporated with mRNA (Fig. S1B).
Table 1.
Baseline characteristics.
| Dendritic cell vaccinated stage III melanoma patients N = 97(%) | ||
|---|---|---|
| Sex | ||
| Male | 60 | (62) |
| Female | 37 | (38) |
| Age, years | ||
| Mean (range) | 50.6 | (22–79) |
| Age in categories, years | ||
| 21–30 | 4 | (4) |
| 31–40 | 19 | (20) |
| 41–50 | 23 | (24) |
| 51–60 | 31 | (32) |
| ≥61 | 20 | (21) |
| Breslow thickness primary melanoma, mm | ||
| ≤1.0 | 7 | (7) |
| 1.01–2.0 | 26 | (27) |
| 2.01–4.0 | 25 | (26) |
| >4.0 | 31 | (32) |
| No primary/unknown | 8 | (8) |
| Ulceration of primary melanoma | ||
| Absent | 68 | (70) |
| Present | 29 | (30) |
| Histological type melanoma | ||
| Superficial spreading | 54 | (56) |
| Nodular | 22 | (23) |
| Acral lentiginous | 1 | (1) |
| Lentigo maligna | 1 | (1) |
| Spitzoid | 2 | (2) |
| No primary/unknown | 17 | (18) |
| N stage at inclusion | ||
| N1a | 24 | (25) |
| N1b | 24 | (25) |
| N2a | 5 | (5) |
| N2b | 14 | (14) |
| N3 | 30 | (31) |
| AJCC pathological stage | ||
| IIIA | 21 | (22) |
| IIIB | 34 | (35) |
| IIIC | 42 | (43) |
| Adjuvant radiotherapya | ||
| No | 91 | (94) |
| Yes | 6 | (6) |
Adjuvant radiotherapy was given after the apheresis, but before the first vaccination.
The median follow-up time from apheresis to patient death or censoring was 47.4 mo (range 3.7–168.6 mo).
Immunological responses to KLH
Immunological responses to keyhole limpet hemocyanin (KLH) were used as a surrogate marker for immune competence. After vaccination, patients were assessed for the presence of KLH-specific T cells in their peripheral blood mononuclear cells (PBMC) and for antibody responses against KLH in their serum. After the first round of vaccinations, an increased T cell proliferation upon stimulation with KLH was found in 68 out of 80 patients (85%) tested (Fig. S2) and anti-KLH antibodies (IgG, IgA, and/or IgM) were detected in 62 out of 80 patients (78%) tested. Only four patients (4%) showed no increased KLH-specific T cell- or anti-KLH antibody-response. This was not associated with the incapacity to develop a TAA-specific CD8+ T cell response, since two of these patients had TAA-specific T cells in DTH skin-test biopsies. The above described results show that the majority of this patient population is well capable of inducing an immune response against the control antigen KLH.
Presence of TAA-specific CD8+ T cells
DTH skin-test biopsies were taken within 2 weeks after each cycle of vaccinations in all patients, and after short-time culture they were analyzed for the presence of TAA-specific CD8+ T cells by tetrameric MHC-peptide complexes (Fig. 1). The presence of TAA-specific CD8+ T cells recognizing any epitope (gp100:154–162, gp100:280–288, or tyrosinase:369–377) in SKIL cultures was shown in 68 patients (70%). A total of 24 patients (25%) had TAA-specific CD8+ T cells against all three epitopes, 27 patients (28%) showed tetramer-positive CD8+ T cells recognizing two epitopes, and there were 17 patients (17%) with T cells against only one epitope. Fifty-two patients (54%) had T cells against the epitope gp100:154–162, 51 patients (53%) T cells against gp100:280–288, and 40 patients (41%) against tyrosinase:369–377. In the majority of patients (72%), tetramer-positive T cells against any epitope were observed after the first cycle of vaccinations. Furthermore, 16 patients (24%) expanded their tetramer-positive T cell response after cycles 2 and 3. However, a notable portion of 72 patients with data available of consecutive rounds, lacked tetramer-positive T cells against an epitope after a positive result in a previous cycle; 29% for gp100:154–162, 35% for gp100:280–288, and 18% for tyrosinase:369–377 (Fig. 2A).
Figure 1.
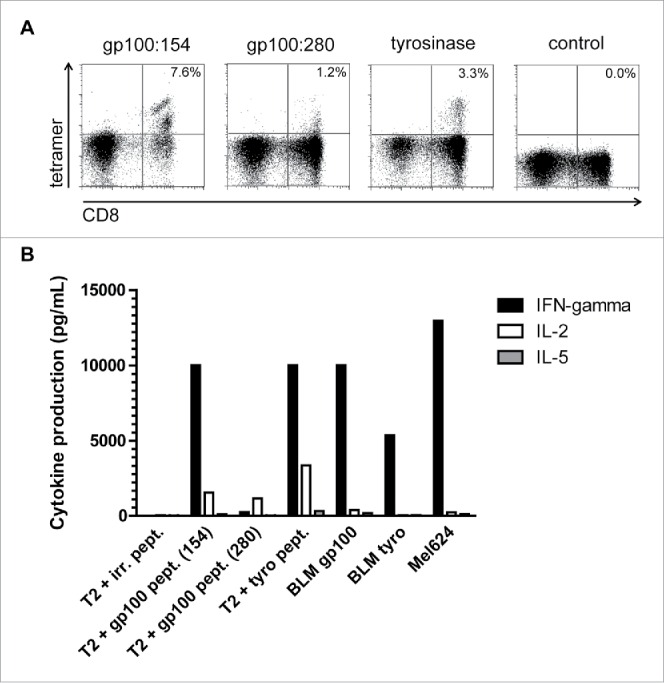
Tumor antigen-specific CD8+ (T)cell responses in SKIL cultures. An example is shown of a tetramer analysis of SKILs from patient II-D-12 (A). Cells were stained with tetramers encompassing the peptides gp100:154, gp100:280, tyrosinase or an irrelevant peptide (control) and with anti-CD8+ mAb. This analysis showes presence of TAA-specific CD8+ T cells against both gp100 epitopes and tyrosinase. Furthermore, a cytokine profile of the same T cells is shown after stimulation with T2 cells loaded with gp100 or tyrosinase peptide, BLM cells transfected with gp100 or tyrosinase, or MEL624 cells expressing both gp100 and tyrosinase (B). Functional TAA-specific T cells against gp100 and tyrosinase are present with a clear production of IFNγ and/or IL-2, but without IL-5 production.
Figure 2.

SKILs analysis in time. Skin-test infiltrating lymphocytes from DTH skin-test biopsies were analyzed for the presence of tetramer-positive CD8+ T cells (A) and a functional T cell response against individual epitopes (B). Three epitopes were tested, gp100:154, gp100:280, and tyrosinase. Unfilled/filled circles represent negative/positive tests. The black, red, and blue circles match to the first, second, and third cycle of vaccinations, respectively. Tumor-specific T cells are already seen after the first cycle in a large amount of patients. T cells against other epitopes were detected in a proportion of patients after cycle 2 and 3. However, in some patients T cells against an epitope were not detected after a positive result in the previous cycle. This is more often seen for tetramer-positive T cells than for a functional T cell response. The distant metastasis-free survival (DMFS) of each individual patient is shown with a cut-off at 5 y. Arrows indicate an ongoing DMFS.
Furthermore, the DTH skin-test biopsies were analyzed for the occurrence of a functional TAA-specific T cell response (Fig. 1), by measuring specific production of Th1/Th2 cytokines in response to T2 cells pulsed with gp100:154–162, gp100:280–288, or tyrosinase:369–377 (peptide-recognition), BLM transfected with gp100/tyrosinase and Mel624 (tumor-recognition). A functional TAA-specific T cell response (peptide-recognition and/or tumor-recognition) was found in 62 patients (64%), of whom 60 patients (97%) showed tetramer-positive T cells. Rates of peptide-recognition of gp100:154–162, gp100:280–288, and tyrosinase:369–377 were 40%, 29%, and 45%, respectively. Tumor-recognition was found in 47 patients (49%), of whom 25 patients showed a functional response against Mel624. As with the test using tetrameric MHC-peptide complexes, in some patients the functional assay for peptide-recognition showed no functional T cells after a positive result in a previous cycle; 6% for gp100:154–162, 8% for gp100:280–288, and 6% for tyrosinase:369–377 (Fig. 2B).
Besides the DTH skin-test biopsies, peripheral blood samples were analyzed for the occurrence of TAA-specific CD8+ T cells by tetrameric MHC-peptide complexes, and they were found in only 15 patients (15%) at any point during vaccinations. All these patients also showed tetramer-positive CD8+ T cells in the DTH skin-test biopsies.
Survival
Median RFS and distant metastasis-free survival (DMFS) of the whole study population were 23.0 mo (95% confidence interval (CI), 9.6–36.4 mo) and 36.8 mo (95% CI, 21.0–52.7 mo), respectively. Significant differences in DMFS were found between the absence or presence of ulceration and between the different AJCC pathological stages (Table S1). A trend toward an improved DMFS was seen in patients with tetramer-positive T cells at any point during vaccinations, compared to patients without tetramer-positive T cells; median DMFS 44.2 mo (95% CI, 2.2–18.0 mo) versus 10.1 mo (95% CI, 25.9–62.5 mo; p = 0.056). The same trend was found for a functional T cell response at any point during vaccinations; median DMFS 44.2 mo (95% CI, 26.5–61.9 mo) versus 14.0 mo (95% CI, 7.7–20.3 mo; p = 0.066). However, for tetramer-positive T cells, as well as for a functional T cell response, this trend was no longer present when a conditional landmark analysis was performed after each cycle of vaccinations in order to correct for potential guarantee-time bias, in which only patients were analyzed who completed that cycle (Fig. 3).
Figure 3.
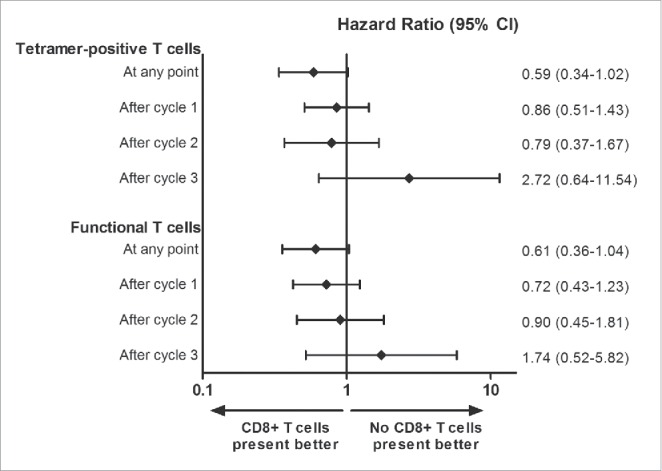
Analyses of distant metastasis-free survival. A graphical representation of the hazard ratios (HR) of distant metastasis-free survival (DMFS) comparing patients with or without tetramer-positive and a functional T cell response in DTH skin-test biopsies, using Cox proportional-hazard models. HR less than 1 defines a better DMFS for patients with tumor-specific T cells. Horizontal lines represent 95% confidence intervals. A trend for a lower risk of distant metastases is seen for patients with tetramer-positive T cells and a functional T cell response at any point during the vaccinations as compared with patients without a T cell response. However, conditional landmark analyses after cycle 1 (n = 97), cycle 2 (n = 72), and cycle 3 (n = 56) show no difference in risk of distant metastases between patients with or without tetramer-positive T cells and a functional T cell response.
Fifty-eight patients (60%) developed distant metastases, of whom 33 patients (57%) received some form of systemic treatment. Chemotherapy (DTIC) was given to 22 patients (67%), while 10 patients (17%) received ipilimumab and 5 patients (8%) targeted agents, because a considerable part of these patients was treated at a time when immune checkpoint inhibitors and targeted therapies were not yet available. At the time of data cut-off 48 deaths had occurred, and the median OS of the whole study population was 77.7 mo (95% CI, 48.1–107.3).
Discussion
Immunomonitoring is an important part of evaluating the effectiveness of immunotherapy. Besides peripheral blood samples, SKILs cultured from vaccine-challenged biopsies were used to detect TAA-specific T cells. The results of the here presented study show that a TAA-specific T cell response can be induced in the majority of stage III melanoma patients treated with adjuvant DC vaccination following a radical lymph node dissection.
Using SKIL cultures could have some advantages compared to commonly used tetramer analysis or ELISPOT assays of TAA-specific T cells in peripheral blood. First of all, SKIL cultures select TAA-specific T cells which are able to extravasate and migrate into peripheral tissues, while it is unknown if TAA-specific T cells in peripheral blood have these capabilities. Furthermore, peripheral blood samples show a lower prevalence of TAA-specific T cells, probably because most T cells are gathered in the lymphoid organs. Therefore, SKILs from DTH skin-test biopsies may be more suitable for immunomonitoring than PBMC in DC vaccinated patients, despite the fact that these biopsies are more time-consuming.
SKILs were evaluated according to the presence of TAA-specific CD8+ T cells by tetrameric MHC-peptide complexes. Tetramer-positive T cells recognizing one or more epitopes were found in 70% of patients, of whom a vast majority already showed T cells against an epitope after the first cycle of vaccinations. Furthermore, the chance of a broader immune response is enhanced by performing multiple cycles of vaccinations, because T cells against other epitopes were detected for the first time after the following cycles in many patients with tetramer-positive T cells during a previous cycle. Tumor-specific T cell responses might be more extensive than only against the three HLA-A*02:01-binding epitopes tested, especially in patients vaccinated with DC electroporated with mRNA, since these DC present more epitopes. Furthermore, because of epitope spreading the tumor-specific immune response might also be more widespread in patients vaccinated with peptide-pulsed DC.17 Besides using tetrameric MHC-peptide complexes, this SKIL analysis integrates the measurement of cytokine profiles, and therefore it is possible to differentiate between T-helper (Th)1 and Th2 immune responses. Th1-type cytokines are important in pro-inflammatory cellular responses, which are desirable for antitumor immunity, while Th2-type cytokines are more involved in anti-inflammatory responses.18 A functional T cell response, defined as specific production of Th1 cytokines and no Th2 cytokines in response to antigen-expressing target cells, was found in 64% of patients, which is clearly more frequent compared to patients with distant metastases receiving DC vaccinations (23%).19 A total of 40% of stage III patients already had a functional T cell response after the first cycle of vaccinations, indicating that the observed difference is not solely explained by more cycles of vaccinations. In addition, considerable more stage III than stage IV patients showed a broader immune response; tetramer-positive CD8+ T cells recognizing multiple epitopes were found in 52% and 14% of patients, respectively.19 Of course, false-positivity must always be considered in diagnostic tests, but the chance of false-positive results is assumed to be low since the different assays include negative controls as comparison. The occurrence of an adequate T cell response is negatively influenced by many factors, like immunosuppressive cytokines, immune checkpoints, regulatory T cells, and myeloid-derived suppressor cells. These factors are more frequently observed in patients with a higher tumor burden, and interact at different levels of the cancer immunity cycle, e.g., priming, activation, and proliferation of T cells, and infiltration of T cells in the tumor.14,20 In conclusion, DC vaccination is more effective in producing a tumor-specific T cell response in stage III than in stage IV melanoma patients.
Part of the patients with tetramer-positive T cells or a functional T cell response against a tumor-specific peptide did not show these T cells in the DTH skin test after a subsequent cycle of vaccinations. First, this might be caused by a low sensitivity of the test with false-negative results. Second, it might indicate that there is a lack of induction of memory in these patients. A final reason that TAA-specific T cells are no longer detected might be the absence of in vivo stimulation of these T cells by tumor cells, which might have been killed during the previous cycle.
A trend toward a better DMFS was found in patients with a tumor-specific T cell response at any point during vaccinations, when compared to patients without TAA-specific response. However, this difference might be explained by guarantee-time bias, since DMFS is compared across groups defined by an event occurring sometime during follow-up.21 Therefore, a conditional landmark analysis was performed after each cycle of vaccinations to correct for this type of bias, and this showed no difference in DMFS between the two groups. Therefore, it cannot be concluded that a tumor-specific T cell response in DTH skin-test biopsies can be used as a predictive marker for clinical outcome, despite the recognized disadvantages of a conditional landmark analysis, like the omission of events occurring earlier to the landmark.22
We already showed a favorable OS in stage III melanoma patients treated with adjuvant DC vaccination, compared to matched controls.23 Of course, no hard conclusions can be drawn from the retrospective data we present here, but in comparison to the EORTC phase 3 trial with adjuvant ipilimumab (10 mg/kg), both studies show comparable 3-y RFS rates (43% in our cohort versus 46.5% in the ipilimumab group), which is at least promising.6 A disadvantage of adjuvant ipilimumab is the toxicity profile, with grade 3 and 4 adverse events occurring in 54% of patients and 52% who stopped the treatment due to side-effects.6 This is in strong contrast to DC vaccination, where grade 3 and 4 toxicity is rarely observed.9 There is a risk of selection bias in the present study, since only patients were included in whom at least one DTH was performed. However, in only 2 out of 99 patients, DTH skin biopsies were not taken because of early relapse of disease after vaccinations were started. A prospective randomized clinical trial is necessary to determine the exact position of adjuvant DC vaccination in stage III melanoma patients.
The clinical effectiveness of DC vaccination might be improved by increasing the number of antigens. The melanoma differentiation antigens gp100 and tyrosinase were selected because they are commonly expressed on melanoma cells and have shown to be capable of inducing functional cytotoxic T cells.12,24,25 However, recent findings show that tumor-specific mutations, leading to neoantigens, may drive potent antitumor responses.26 Carreno and colleagues found that a DC vaccine with carefully selected patient-specific neoantigens, led to an increase in the breadth and diversity of melanoma neoantigen-specific T cells in peripheral blood samples of three stage IV melanoma patients.27 As most mutated proteins are essentially unique to a tumor, personalized antigen selection might be beneficial in vaccination strategies.26,27 A great challenge will be the identification of the right immunogenic neoantigens, especially in stage III melanoma patients, since only a minimal amount of tumor material might be available.28 For this reason, and in light of tumor heterogeneity, it might be preferable to combine commonly expressed melanoma differentiation antigens with patient-specific neoantigens in future DC vaccines.
In summary, DC vaccination is more effective in inducing functional TAA-specific CD8+ T cells in stage III than stage IV melanoma patients, and performing multiple cycles of vaccinations enhances the chance of a broader immune response. A prospective randomized clinical trial must determine the exact position of adjuvant DC vaccination in stage III melanoma patients.
Materials and methods
Patient characteristics
We retrospectively analyzed a cohort of 99 stage III melanoma patients, who were enrolled in our DC vaccination studies between December 1999 and July 2014. Patients received adjuvant DC vaccination within 2 mo after radical lymph node dissection. Inclusion criteria included melanoma with histologically proven regional metastatic disease without evidence of distant metastases; World Health organization (WHO) performance status of 0 or 1; HLA-A*02:01 phenotype; and histological proven melanoma expressing the melanoma-associated antigens gp100 (compulsory) and tyrosinase (non-compulsory). Patients with a second malignancy in the previous 5 y or serious concomitant disease were excluded. At least one DTH must have been performed to be included in this analysis, since the goal was to evaluate tumor-specific immunological responses. All studies were approved by the appropriate Medical Ethical Review Board and written informed consent was obtained from all patients.
Treatment schedule
All patients were vaccinated with autologous monocyte-derived DC loaded with TAA of gp100 and tyrosinase according to a schedule of three biweekly vaccinations, followed by a DTH skin test within 2 weeks after each vaccination cycle. Some patients received an extra vaccination before radical lymph node dissection for additional imaging studies. Patients received three cycles of vaccinations and a DTH skin-test at approximately 6-mo intervals when no signs of distant metastases were present. Differences in protocols included the route of administration, method of antigen loading, and combination treatment with interleukin (IL)-2 or cisplatin (Table 2). For the exact details of the vaccination protocols, we refer to these individual studies.12,29-32
Table 2.
Dendritic cell vaccination protocols.
| Protocol | Number of patients | Method of antigen loading | Route of administration | IL-2a | Cisplatin |
|---|---|---|---|---|---|
| 1 | 1 | Peptide: class I modb | i.n. | no | no |
| 2A | 10 | Peptide: class I wtc | i.d. | yes | no |
| 2B | 8 | Peptide: class I wtc | i.n. | yes | no |
| 2C | 10 | Peptide: class I wtc | i.n. | no | no |
| 2D | 10 | Peptide: class I wtc | i.d. | no | no |
| 2E | 10 | mRNAd | i.n. | no | no |
| 4A | 4 | Peptide: class I wtc + IIe | i.n. | no | no |
| 4B | 3 | Peptide: class I wtc | i.n. | no | no |
| 4C + D | 16 | mRNAd | i.n. | no | no |
| 5A | 5 | mRNAd | i.v./i.d. | no | no |
| 8A + B | 4 | mRNAd | i.n. | no | no |
| 9C | 9 | mRNAd | i.v./i.d. | no | yesf |
| 9D | 7 | mRNAd | i.v./i.d. | no | no |
| Total | 97 |
Low-dose IL-2 (9 MIU) was administered subcutaneously once daily for 1 week starting 3 d after each vaccination.
Class I mod; HLA class I-restricted modified gp100-derived peptides 154–162 Q→A and 280–288 A→V and HLA class I-restricted tyrosinase-derived peptide 369–377.
Class I wt; HLA class I-restricted wild-type gp100-derived peptides 154–162 and 280–288 and HLA class I-restricted tyrosinase-derived peptide 369–377.
mRNA; mRNA encoding full length gp100 and tyrosinase.
Class II; HLA class II-restricted gp100-derived peptide 44–59 and tyrosinase-derived peptide 448–462 analog.
Cisplatin (50 mg/m2) was administered intravenously before each vaccination.
Abbreviations: wt, wild type; mod, modified; i.d., intradermal; i.n., intranodal; i.v., intravenous; IL-2, Interleukin-2.
Vaccine preparation
Monocytes were enriched from leukapheresis products by plastic adherence of blood mononuclear cells or by counterflow centrifugation using Elutra-cell separator (Gambro BCT, Lakewood, CO) and single-use, functionally sealed disposable Elutra sets, as described before.33 Monocytes were cultured in the presence of IL-4 (500 U/mL), GM-CSF (800 U/mL) (both Cellgenix, Freiburg, Germany), and KLH (10 μg/mL, Calbiochem, Darmstadt, Germany) or Immucothel KLH (10 μg/mL, Biosyn, Carlsbad, CA). Most protocols used DC matured with autologous monocyte-conditioned medium (30%, v/v) supplemented with prostaglandin E2 (PGE2; 10 μg/mL, Pharmacia & Upjohn, Puurs, Belgium) and 10 ng/mL tumor necrosis factor-α (TNF-α; Cellgenix, Freiburg, Germany) for 48 h or a cocktail of 10 ng/mL TNF-α, 5 ng/mL IL-1β, 15 ng/mL, IL-6 (all Cellgenix), and 10 μg/mL PGE2.34,35 DC in protocols 5 and 8B were matured with a cocktail of prophylactic vaccines including BCG vaccine SSI (4% v/v, Nederlands Vaccin Instituut, Bilthoven, the Netherlands), Typhim Vi (4% v/v, Sanofi Pasteur MSD, Brussels, Belgium), and Act-HIB (4% v/v, Aventis Pasteur, Brussels, Belgium), supplemented with PGE2 (10 µg/mL) for 48 h (TLR-DC).36 In protocol 8A, DC were matured via electroporation with mRNA encoding CD40L, CD70, and constitutively active TLR4.32 DC were pulsed with two gp100-derived peptides (gp100:154–162 and gp100:280–288) and a tyrosinase-derived peptide (tyrosinase:369–377) or electroporated with mRNA encoding gp100 or tyrosinase.37 Quality control of DC was performed as described before.12 Cells were re-suspended and injected intradermally, or intranodally, or intradermally and intravenously. Patients received a maximum of 30 × 106 DC intradermally, 15 × 106 intranodally, or 10 × 106 DC intradermally plus 20 × 106 intravenously per vaccination.
KLH-specific proliferation
PBMC were isolated from heparinized blood by Ficoll–Paque density centrifugation, and stimulated with KLH (4 µg/2×105 PBMC) in ex vivo with 2% human serum (HS). After 3 d, cells were incubated with 3H-thymidine for 8 h and incorporation was measured with a β-counter. Experiments were performed in sextuplicate, non-specific proliferation upon stimulation with ovalbumin was used as control. Proliferative response to KLH is given as proliferation index (proliferation with KLH/proliferation without KLH).
KLH-specific antibody production
KLH-specific antibodies were measured in the sera of patients before and after vaccination by ELISA (www.klhanalysis.com). Briefly, microtiter plates (96 wells) were coated overnight at 4°C with KLH (25 μg/mL in PBS per well). After washing the plates, patient serum was added in duplicate for 1 h at room temperature. After extensive washing, patient KLH-specific antibodies were detected with mouse anti-human IgG, IgA, or IgM antibodies labeled with horseradish peroxidase (Invitrogen, Waltham, USA). 3,3′ 5,5-tetramethyl-benzidine (Dako, Glostrup, Denmark) was used as a substrate and plates were measured in a microtiter plate reader at 450 nm. For quantification an isotype-specific calibration curve for the KLH response was included in each microtiter plate.38
Skin-test infiltrating lymphocyte culture
Previously we observed that, in a series of patients who underwent pre-vaccination skin-test analysis, none of the patients had detectable levels of TAA-specific T cells prior to vaccination. Therefore, pre-vaccination skin-tests were not performed, and we focused on in-depth analysis on the post-vaccination samples. Skin tests were performed within 2 weeks after each vaccination cycle as described previously.10,16 Briefly, a maximum of 10 × 105 DC loaded with either gp100, tyrosinase or both antigens were injected intradermally in the skin of the back of patients at different sites, 4 cm apart from each other. After 48 h, punch biopsies (6 mm) were taken. Half of the biopsy was stored and half manually cut and cultured for 2–4 weeks in RPMI-1640 containing 7% HS and IL-2 (100 U/mL).
Tetramer staining of SKILs
SKIL cultures were stained with tetrameric-MHC complexes containing the HLA-A*02:01-binding epitopes gp100:154–162, gp100:280–288, or tyrosinase:369–377 as described previously.10 Tetrameric-MHC complexes recognizing HIV were used as correction for background binding. Tetramer positivity was defined as at least 2-fold increase in the double positive population compared to an irrelevant control antigen (HIV).
Functionality of SKILs
The production of Th1 (IFNγ and IL-2) and Th2 (IL-4 and IL-5) cytokines by SKILs was measured in supernatants after 16 h of co-culture with different target cells, to obtain a cytokine profile of post-vaccination SKILs. IL-4 production was not available for all patients. The target cells were T2 cells pulsed with gp100:154–162, gp100:280–288, and tyrosinase:369–377; BLM (a melanoma cell line expressing HLA-A*02:01 and no endogenous expression of gp100 and tyrosinase), transfected with control antigen G250, gp100 or tyrosinase; and an allogenic HLA-A*02:01-positive, gp100-positive, and tyrosinase-positive tumor cell line (Mel624). Cytokine analysis was done by cytometric bead array (Th1/Th2 Cytokine CBA 1; BD Pharmingen, San Diego, CA or human Th1/Th2 FlowCytomix multiplex kit; eBioscience, Vienna, Austria), according to the manufacturer instructions. A positive result was defined as a 2-fold increase compared to stimulation with the same cell lines pulsed with an irrelevant peptide.
SKIL culture evaluation
SKILs were evaluated according to two criteria. At first, the presence of TAA-specific CD8+ T cells, recognizing the pre-defined epitopes, by tetrameric MHC-peptide complexes.Secondly, the occurrence of a functional TAA-specific T cell response was measured, defined by peptide-recognition (T2 cells) and/or tumor-recognition (BLM and/or Mel624) with specific production of Th1 cytokines and no Th2 cytokines.
Statistical analysis
RFS, DMFS, and OS were calculated from the date of apheresis to the date of first recurrence, distant metastases and death, using the Kaplan–Meier method. Additionally, a conditional landmark analysis was performed to evaluate the effect of the occurrence of (functional) TAA-specific T cells on survival. Hazard ratios and associated 95% CI were assessed with the use of a Cox proportional-hazards model. A paired t-test was used to compare KLH proliferation in time. Ulceration was assumed absent if not reported in the pathology report. p values less than 0.05 were considered significant. SPSS version 20.0 software (SPSS Inc., Chicago, IL) and Graphpad Prism 5.03 (GraphPad Software inc, San Diego, CA) were used for statistical analysis.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the patients who participated in the studies and JFM Jacobs for analyzing antibody responses against KLH.
Funding
This work was supported by grants from the Dutch Cancer Society (KUN2009-4402), a Radboudumc PhD grant, and the stichting Afweer Tegen Kanker (ATK). C.F. received the NWO Spinoza award and an ERC Advanced grant. IJMdV received a NWO-VICI grant (918.14.655).
References
- 1.van Akkooi AC, Bouwhuis MG, van Geel AN, Hoedemaker R, Verhoef C, Grunhagen DJ, Schmitz PI, Eggermont AM, de Wilt JH. Morbidity and prognosis after therapeutic lymph node dissections for malignant melanoma. Eur J Surg Oncol 2007; 33:102-8; PMID:17161577; http://dx.doi.org/ 10.1016/j.ejso.2006.10.032 [DOI] [PubMed] [Google Scholar]
- 2.Erdmann F, Lortet-Tieulent J, Schuz J, Zeeb H, Greinert R, Breitbart EW, Bray F. International trends in the incidence of malignant melanoma 1953–2008–are recent generations at higher or lower risk? Int J Cancer 2013; 132:385-400; PMID:22532371; http://dx.doi.org/ 10.1002/ijc.27616 [DOI] [PubMed] [Google Scholar]
- 3.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S et al.. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009; 27:6199-206; PMID:19917835; http://dx.doi.org/ 10.1200/JCO.2009.23.4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eggermont AM, Testori A, Marsden J, Hersey P, Quirt I, Petrella T, Gogas H, MacKie RM, Hauschild A. Utility of adjuvant systemic therapy in melanoma. Ann Oncol 20 Suppl 6:vi30-4; PMID:19617295; http://dx.doi.org/12927565 10.1093/annonc/mdp250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wheatley K, Ives N, Hancock B, Gore M, Eggermont A, Suciu S. Does adjuvant interferon-α for high-risk melanoma provide a worthwhile benefit? A meta-analysis of the randomised trials. Cancer Treat Rev 2003; 29:241-52; PMID:12927565; http://dx.doi.org/ 10.1016/S0305-7372(03)00074-4 [DOI] [PubMed] [Google Scholar]
- 6.Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM et al.. Adjuvant ipilimumab vs. placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol 2015; 16:522-30; PMID:25840693; http://dx.doi.org/ 10.1016/S1470-2045(15)70122-1 [DOI] [PubMed] [Google Scholar]
- 7.Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013; 39:38-48; PMID:23890062; http://dx.doi.org/ 10.1016/j.immuni.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12:265-77; PMID:22437871; http://dx.doi.org/ 10.1038/nrc3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014; 15:e257-67; PMID:24872109; http://dx.doi.org/ 10.1016/S1470-2045(13)70585-0 [DOI] [PubMed] [Google Scholar]
- 10.de Vries IJ, Bernsen MR, Lesterhuis WJ, Scharenborg NM, Strijk SP, Gerritsen MJ, Ruiter DJ, Figdor CG, Punt CJ, Adema GJ. Immunomonitoring tumor-specific T cells in delayed-type hypersensitivity skin biopsies after dendritic cell vaccination correlates with clinical outcome. J Clin Oncol 2005; 23:5779-87; PMID:16110035; http://dx.doi.org/ 10.1200/JCO.2005.06.478 [DOI] [PubMed] [Google Scholar]
- 11.Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nature medicine 2004; 10:475-80; PMID:15122249; http://dx.doi.org/ 10.1038/nm1039 [DOI] [PubMed] [Google Scholar]
- 12.Aarntzen EH, Schreibelt G, Bol K, Lesterhuis WJ, Croockewit AJ, de Wilt JH, van Rossum MM, Blokx WA, Jacobs JF, Duiveman-de Boer T et al.. Vaccination with mRNA-electroporated dendritic cells induces robust tumor antigen-specific CD4+ and CD8+ T cells responses in stage III and IV melanoma patients. Clin Cancer Res 2012; 18:5460-70; PMID:22896657; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-3368 [DOI] [PubMed] [Google Scholar]
- 13.Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, Harlin H. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev 2006; 213:131-45; PMID:16972901; http://dx.doi.org/ 10.1111/j.1600-065X.2006.00442.x [DOI] [PubMed] [Google Scholar]
- 14.Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN et al.. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res 2012; 72:876-86; PMID:22174368; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer 2011; 11:805-12; PMID:22020206; http://dx.doi.org/ 10.1038/nrc3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aarntzen EH, Bol K, Schreibelt G, Jacobs JF, Lesterhuis WJ, van Rossum MM, Adema GJ, Figdor CG, Punt CJ, de Vries IJ. Skin-test infiltrating lymphocytes early predict clinical outcome of dendritic cell based vaccination in metastatic melanoma. Cancer Res 2012; 72:6102-10; PMID:23010076; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2479 [DOI] [PubMed] [Google Scholar]
- 17.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol 2002; 2:85-95; PMID:11910899; http://dx.doi.org/ 10.1038/nri724 [DOI] [PubMed] [Google Scholar]
- 18.Berger A. Th1 and Th2 responses: what are they? BMJ 2000; 321:424; PMID:10938051; http://dx.doi.org/ 10.1136/bmj.321.7258.424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aarntzen EH, Bol K, Schreibelt G, Jacobs JF, Lesterhuis WJ, Van Rossum MM, Adema GJ, Figdor CG, Punt CJ, De Vries IJ. Skin-test infiltrating lymphocytes early predict clinical outcome of dendritic cell-based vaccination in metastatic melanoma. Cancer Res 2012; 72:6102-10; PMID:23010076; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2479 [DOI] [PubMed] [Google Scholar]
- 20.Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013; 39:1-10; PMID:23890059; http://dx.doi.org/ 10.1016/j.immuni.2013.07.012 [DOI] [PubMed] [Google Scholar]
- 21.Giobbie-Hurder A, Gelber RD, Regan MM. Challenges of guarantee-time bias. J Clin Oncol 2013; 31:2963-9; PMID:23835712; http://dx.doi.org/ 10.1200/JCO.2013.49.5283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dafni U. Landmark analysis at the 25-year landmark point. Circ Cardiovasc Qual Outcomes 2011; 4:363-71; PMID:21586725; http://dx.doi.org/ 10.1161/CIRCOUTCOMES.110.957951 [DOI] [PubMed] [Google Scholar]
- 23.Bol KF, Aarntzen EHJG, in 't Hout FEM, Schreibelt G, Creemers JHA, Lesterhuis WJ, Gerritsen WR, Grunhagen DJ, Verhoef C, Punt CJA et al.. Favorable overall survival in stage III melanoma patients after adjuvant dendritic cell vaccination. Oncoimmunology 2016; 5:e1057673; PMID:26942068; http://dx.doi.org/ 10.1080/2162402X.2015.1057673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Topalian SL, Gonzales MI, Parkhurst M, Li YF, Southwood S, Sette A, Rosenberg SA, Robbins PF. Melanoma-specific CD4+ T cells recognize nonmutated HLA-DR-restricted tyrosinase epitopes. J Exp Med 1996; 183:1965-71; PMID:8642306; http://dx.doi.org/ 10.1084/jem.183.5.1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bakker AB, van der Burg SH, Huijbens RJ, Drijfhout JW, Melief CJ, Adema GJ, Figdor CG. Analogues of CTL epitopes with improved MHC class-I binding capacity elicit anti-melanoma CTL recognizing the wild-type epitope. Int J Cancer 1997; 70:302-9; PMID:9033632; http://dx.doi.org/ 10.1002/(SICI)1097-0215(19970127)70:3%3c302::AID-IJC10%3e3.0.CO;2-H [DOI] [PubMed] [Google Scholar]
- 26.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/ 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 27.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER et al.. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015; 348:803-8; PMID:25837513; http://dx.doi.org/ 10.1126/science.aaa3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delamarre L, Mellman I, Yadav M. Cancer immunotherapy. Neo approaches to cancer vaccines. Science 2015; 348:760-1; PMID:25977539; http://dx.doi.org/ 10.1126/science.aab3465 [DOI] [PubMed] [Google Scholar]
- 29.Lesterhuis WJ, Schreibelt G, Scharenborg NM, Brouwer HM, Gerritsen MJ, Croockewit S, Coulie PG, Torensma R, Adema GJ, Figdor CG et al.. Wild-type and modified gp100 peptide-pulsed dendritic cell vaccination of advanced melanoma patients can lead to long-term clinical responses independent of the peptide used. Cancer Immunol Immunother 2011; 60:249-60; PMID:21069321; http://dx.doi.org/ 10.1007/s00262-010-0942-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lesterhuis WJ, de Vries IJ, Schreibelt G, Lambeck AJ, Aarntzen EH, Jacobs JF, Scharenborg NM, van de Rakt MW, de Boer AJ, Croockewit S et al.. Route of administration modulates the induction of dendritic cell vaccine-induced antigen-specific T cells in advanced melanoma patients. Clin Cancer Res 2011; 17:5725-35; PMID:21771874; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-1261 [DOI] [PubMed] [Google Scholar]
- 31.Aarntzen EH, De Vries IJ, Lesterhuis WJ, Schuurhuis D, Jacobs JF, Bol K, Schreibelt G, Mus R, De Wilt JH, Haanen JB et al.. Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res 2013; 73:19-29; PMID:23087058; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-1127 [DOI] [PubMed] [Google Scholar]
- 32.Bol KF, Figdor CG, Aarntzen EH, Welzen ME, van Rossum MM, Blokx WA, van de Rakt MW, Scharenborg NM, de Boer AJ, Pots JM et al.. Intranodal vaccination with mRNA-optimized dendritic cells in metastatic melanoma patients. Oncoimmunology 2015; 4:e1019197; PMID:26405571; http://dx.doi.org/ 10.1080/2162402X.2015.1019197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger TG, Feuerstein B, Strasser E, Hirsch U, Schreiner D, Schuler G, Schuler-Thurner B. Large-scale generation of mature monocyte-derived dendritic cells for clinical application in cell factories. J Immunol Methods 2002; 268:131-40; PMID:12215381; http://dx.doi.org/ 10.1016/S0022-1759(02)00189-8 [DOI] [PubMed] [Google Scholar]
- 34.Reddy A, Sapp M, Feldman M, Subklewe M, Bhardwaj N. A monocyte conditioned medium is more effective than defined cytokines in mediating the terminal maturation of human dendritic cells. Blood 1997; 90:3640-6; PMID:9345048 [PubMed] [Google Scholar]
- 35.de Vries IJ, Adema GJ, Punt CJ, Figdor CG. Phenotypical and functional characterization of clinical-grade dendritic cells. Methods Mol Med 2005; 109:113-26; PMID:15585917; http://dx.doi.org/ 10.1385/1-59259-862-5:113 [DOI] [PubMed] [Google Scholar]
- 36.Bol KF, Aarntzen EH, Pots JM, Olde Nordkamp MA, van de Rakt MW, Scharenborg NM, de Boer AJ, van Oorschot TG, Croockewit SA, Blokx WA et al.. Prophylactic vaccines are potent activators of monocyte-derived dendritic cells and drive effective anti-tumor responses in melanoma patients at the cost of toxicity. Cancer Immunol Immunother 2016; 65:327-39; PMID:26861670; http://dx.doi.org/ 10.1007/s00262-016-1796-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuurhuis DH, Verdijk P, Schreibelt G, Aarntzen EH, Scharenborg N, de Boer A, van de Rakt MW, Kerkhoff M, Gerritsen MJ, Eijckeler F et al.. In situ expression of tumor antigens by messenger RNA-electroporated dendritic cells in lymph nodes of melanoma patients. Cancer Res 2009; 69:2927-34; PMID:19318559; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-3920 [DOI] [PubMed] [Google Scholar]
- 38.Aarntzen EH, de Vries IJ, Goertz JH, Beldhuis-Valkis M, Brouwers HM, van de Rakt MW, van der Molen RG, Punt CJ, Adema GJ, Tacken PJ et al.. Humoral anti-KLH responses in cancer patients treated with dendritic cell-based immunotherapy are dictated by different vaccination parameters. Cancer Immunol Immunother 2012; 61:2003-11; PMID:22527252; http://dx.doi.org/ 10.1007/s00262-012-1263-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.