

Abstract
The outcome of drug therapy is often unpredictable, ranging from beneficial effects to lack of efficacy to serious adverse effects. Variations in single genes are 1 well-recognized cause of such unpredictability, defining the field of pharmacogenetics (see Glossary). Such variations may involve genes controlling drug metabolism, drug transport, disease susceptibility, or drug targets. The sequencing of the human genome and the cataloguing of variants across human genomes are the enabling resources for the nascent field of pharmacogenomics (see Glossary), which tests the idea that genomic variability underlies variability in drug responses. However, there are many challenges that must be overcome to apply rapidly accumulating genomic information to understand variable drug responses, including defining candidate genes and pathways; relating disease genes to drug response genes; precisely defining drug response phenotypes; and addressing analytic, ethical, and technological issues involved in generation and management of large drug response data sets. Overcoming these challenges holds the promise of improving new drug development and ultimately individualizing the selection of appropriate drugs and dosages for individual patients.
The concept that genetic variation contributes to variability in disease phenotypes and in drug responses is widely accepted and validated in many research settings. For some drugs, there are clear implications of genetic information for drug therapy to avoid toxicity and to optimize response (1, 2). In addition, understanding genetic contributors to variability in drug response provides a new tool in drug development that carries the hope of decreasing the risk for unexpected toxicities, identifying patients most likely to respond, and streamlining drug development (3).
The English physiologist Archibald Garrod (4) proposed that similar genetic factors might underlie inborn errors of metabolism and variable responses to drugs. The field of pharmacogenetics developed after variant drug responses due to large single gene effects were described (1, 5–8), as Garrod suggested. Genetic science is now increasingly turning its attention to how variation in large gene sets, in complex biological pathways (see Glossary), or in the whole genome, contributes to variable phenotypes, such as disease susceptibility (9). This evolution from genetics to genomics is paralleled by progress in understanding the genetic contribution to variable drug responses, from pharmacogenetics to pharmacogenomics (Table 1) (Figure 1). This review will outline progress in the field by describing mechanisms underlying variable drug responses, the potential role of genetic factors in their causes, and contemporary and evolving approaches to identifying these genetic factors. Examples are presented throughout, although it is not our goal to review these in a comprehensive fashion; rather it is our intent to identify the challenges that must be overcome and their potential solutions if genetic and genomic information is to be integrated into drug prescribing.
Table 1. Challenges in Pharmacogenomics.
| Challenge | Potential Approaches |
|---|---|
| Establishing that drug responses are heritable | Twin studies; family studies |
| Linkage between drug response and genomic loci in cell lines, or model organisms | |
|
| |
| Defining candidate genes | Pharmacokinetic |
| Pharmacodynamic | |
| Drug targets | |
| Biological milieu in which drugs act | |
| Disease genes and pathways | |
| Whole genome approaches | |
|
| |
| Defining drug responses | Biomarkers |
| Surrogates | |
| “Hard” end points | |
|
| |
| Data management, including uniform representation of phenotypic data | Improved informatics |
| Centralized, Web-accessible public database relating genetic variants and drug responses: www.PharmGKB.org | |
|
| |
| Reproducibility | Replication sets |
| Large study populations | |
|
| |
| Statistical analysis of associations | New statistical methods, including consideration of haplotypes |
|
| |
| Interrogating very large sets of polymorphisms in large numbers of patients | New platforms (e.g., chip- or bead-based) |
|
| |
| Moving to practice | Reproducible study results |
| Cost-effectiveness | |
| Health care provider education | |
Figure 1. The concept of pharmacogenetics.
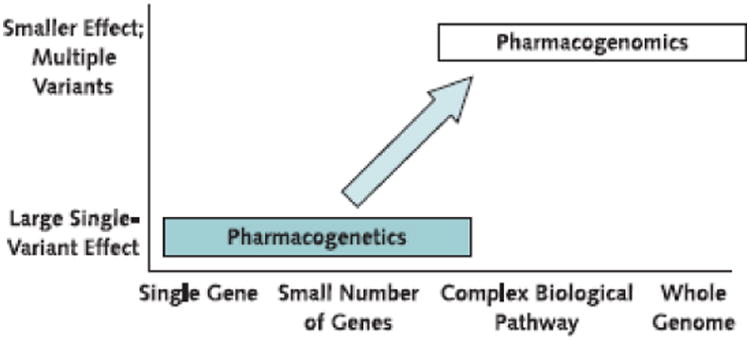
Pharmacogenetics focuses on large clinical effects of single gene variants in small numbers of patients. However, the concept of pharmacogenomics examines many genomic loci, including large biological pathways and the whole genome, to identify variants that together determine variability in response to drug therapy.
Mechanisms Underlying Variable Drug Responses
Clinicians and the lay public accept the notion that not all patients respond to drug therapy in the same fashion. An overarching challenge in contemporary therapeutics is to define the mechanisms underlying such variability. Occasionally, the distribution of drug responses across a population is clearly bimodal, suggesting a predominant role for a single variable that is often genetic (Figure 2, top). More commonly, drug responses in patient populations show a broad distribution (Figure 2, bottom). Studies with twins done as early as the 1960s support the idea that this pattern of responses may also include a prominent genetic component (12–14).
Figure 2. Two types of variability in drug action.
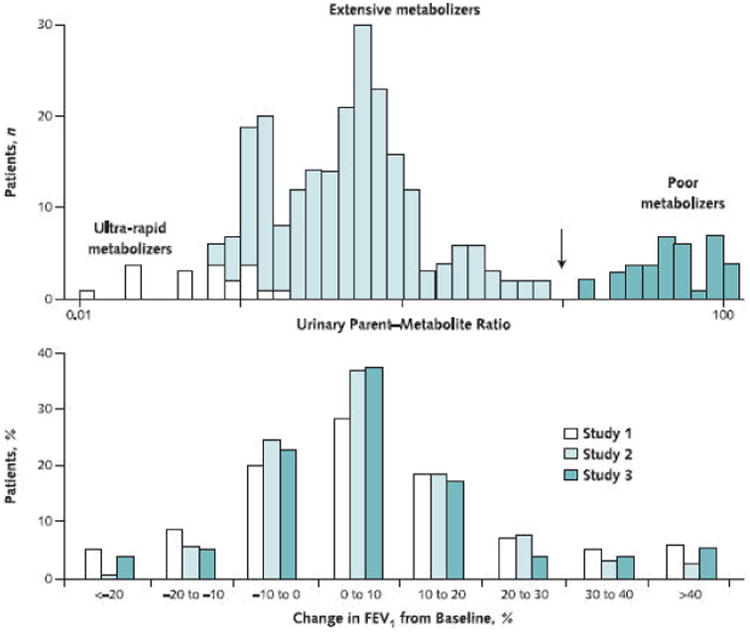
Top. Volunteers received 10 mg of the CYP2D6 substrate debrisoquine, and the ratio of urinary concentrations of the parent drug and its 4-hydroxy metabolite in urine were determined. This experiment identifies at least 2 distinct populations, extensive and poor metabolizers, separated at the antimode (arrow). Redrawn with permission from reference 10. Bottom. Change in FEV1 in 1117 participants in 3 different trials of antiasthmatic therapy (inhaled steroids). Although the responses vary markedly, from an apparently deleterious drug effect to a highly beneficial one, there is no antimode. Redrawn with permission from reference 11.
Two distinct processes, either of which can be influenced by genetic factors, underlie the generation of a clinical drug action: delivery to and removal from target sites in plasma on cell surfaces, or within cells (pharmacokinetics) (see Glossary) and interaction with the targets to generate a cellular effect that is translated to clinical effect (pharmacodynamics). Thus, the starting point for many contemporary pharmacogenetic studies is identification of variable drug responses in an individual patient or across a population. Then, an understanding of the underlying pharmacokinetics or pharmacodynamics can be used to identify individual candidate genes (see Glossary) in which variant function may “explain” the variable drug response. An alternate approach, to interrogate many candidate genes or even the whole genome (see Glossary), has emerged more recently and will be discussed further.
Genetically Determined Pharmacokinetics
Some of the earliest findings in pharmacogenetics involve variations in single genes encoding drug-metabolizing enzymes, which can underlie aberrant responses to substrate drugs (Table 2). The highest likelihood of aberrant drug responses occurs when genetically determined reduced function of 1 drug elimination pathway is coupled with the absence of alternate pathways that can readily subserve the same function (15). Individual cases were first identified in patients with clinically dramatic phenotypes (see Glossary) because these patients were homozygous (see Glossary) for loss of function in such alleles (see Glossary).
Table 2. Examples of Associations between Drug Response and Genetic Variants*.
| Drug | Variable Clinical Effect | Genes with Associated Variants | Possible Mechanism |
|---|---|---|---|
| Azathioprine and mercaptopurine | Bone marrow aplasia | TPMT | Hypofunctional alleles |
| Reduced therapeutic effect at standard doses | Wild-type alleles | ||
|
| |||
| Some antidepressants and β-blockers | Increased side effect risk | CYP2D6 | Hypofunctional alleles |
| Decreased efficacy | Gene duplication | ||
|
| |||
| Omeprazole | Helicobacter pylori cure rate | CYP2C19 | Hypofunctional alleles |
|
| |||
| Irinotecan | Neutropenia | UGT1A1 | Decreased expression due to regulatory polymorphism |
|
| |||
| HIV protease inhibitors | Central nervous system levels | MDR1 | Altered P-glycoprotein function |
|
| |||
| β-blockers | Blood pressure lowering and heart rate slowing | ADRB1 | Altered receptor function or number |
|
| |||
| Inhaled β2-agonists | Bronchodilation | ADRB2 | Altered receptor function or number |
|
| |||
| Diuretics | Blood pressure lowering | ADD1 | Altered cytoskeletal function by adducin variants |
|
| |||
| Warfarin | Anticoagulation | VKORC1 | Variant haplotypes in regulatory regions leading to variable expression |
| CYP2C9 | Coding region variants causing reduced S-warfarin clearance | ||
|
| |||
| Abacavir | Immunologic reactions | HLA variants | Altered immunologic responses |
|
| |||
| QT-prolonging antiarrhythmics | Drug-induced arrhythmia | Ion-channel genes | Exposure of subclinical reduction in repolarizing currents by drugs |
|
| |||
| General anesthetics | Malignant hyperthermia | RYR1 | Anesthetic-induced increased release of sarcoplasmic reticulum calcium by mutant channels |
|
| |||
| Inhaled steroids | Bronchodilation | CRHR1 | Unknown |
|
| |||
| HMG-CoA reductase inhibitors (statins) | Low-density lipoprotein cholesterol lowering | HMGCR | Altered HMG-CoA reductase activity |
ADD1 = the gene encoding α-adducin; ADRB1 = the gene encoding the β1-adrenergic receptor; ADRB2 = the gene encoding the β2-adrenergic receptor; CRHR1 = the gene encoding corticotrophin-releasing hormone receptor-1; CYP2C19 = the gene encoding the 2C19 cytochrome P450 isoform; CYP2C9 = the gene encoding the 2C9 cytochrome P450 isoform; CYP2D6 = the gene encoding the 2D6 cytochrome P450 isoform; HMG-CoA = 3-hydroxy-3-methylglutaryl coenzyme A; HMGCR = the gene encoding HMG-CoA reductase; MDR1 = the gene encoding P-glycoprotein; RYR1 = the gene encoding the skeletal muscle calcium-release channel; TPMT = the gene encoding thiopurine methyltransferase; UGT1A1 = the gene encoding uridine diphosphate glycosyltransferase 1 family, polypeptide A1; VKORC1 = the gene encoding vitamin K epoxide reductase complex, subunit 1.
Coding-Region Variants
Changes in DNA sequence that occur in regions that encode protein may lead to changes in the primary amino acid sequence and protein function. A well-studied example that is entering routine clinical practice is the thiopurine methyltransferase (TPMT) gene, whose protein product is responsible for bioinactivation of thiopurines, such as azathioprine or mercaptopurine (1, 16, 17). Rare individuals who are homozygous for loss of function variants are at high risk for bone marrow aplasia during therapy with standard doses, and this is stated in the package label. Ten percent of persons carry a single abnormal allele and are also at increased risk for bone marrow toxicity (18, 19). Conversely, “standard” doses of mercaptopurine that are used in the 90% of patients with functional alleles mutations (see Glossary) may in fact be inadequate for achieving an optimal antileukemic effect (20).
The most common mechanism for drug elimination is metabolism by members of the cytochrome P450 (CYP) superfamily. Common coding-region CYP variants that affect drug elimination and responses have now been described. The frequency of many variants varies by ethnicity, and this may be one factor determining ethnic-specific drug responses. Up to 10% of white and African-American persons are homozygous for loss of activity of a cytochrome P450 isoform, which is termed CYP2D6. Persons with this poor-metabolizer genotype have drug accumulation and increased side effects with some antidepressants (21). In addition, persons who are poor metabolizers do not metabolize codeine to its active metabolite morphine and thus have reduced analgesia (22). An important implication of the identification of highly variable CYP2D6 activity is that new drug candidates that are eliminated predominantly by this enzyme are often not further developed (23, 24). In contrast to CYP2D6, the poor metabolizer trait for a different CYP, CYP2C19, is more common in Asian persons, and persons with this genotype have higher drug concentrations and a greater cure rate of Helicobacter pylori infections during therapy with the CYP2C19 substrate omeprazole (25).
DNA Variants in Noncoding Regions
Only a small fraction of the human genome encodes proteins. One important role of noncoding DNA is to regulate the amount of messenger RNA (mRNA) transcribed, and thus protein generated, in the basal state or in response to many environmental stimuli. Sequence variants in regulatory regions that result in altered amounts of otherwise normally functioning protein can underlie abnormal drug responses. A good example is the repeat polymorphism (see Glossary) in the promoter of UGT1A1, which encodes the glucuronosyltransferase responsible for conjugation of bilirubin and many drugs. The most common hypofunctional allele, termed UGT1A1*28, is an insertion of 2 extra base pairs (TA) in a key regulatory region of the gene, resulting in decreased protein expression. Impaired elimination of bilirubin by this mechanism is the cause of the Gilbert syndrome. UGT1A1 is responsible for the metabolism of SN-38, the active metabolite of the anticancer drug irinotecan, and persons who are homozygous for UGT1A1*28 are at increased risk for serious adverse effects of the drug (26). This effect is of sufficient clinical importance that it is now described on the irinotecan product label (27, 28). Commercial tests for variants in TPMT, CYP2D6, CYP2C19, and UGT1A1 are now available (29).
Variable Drug Transport
Drug entry into and removal from cells are often active processes, accomplished by specific drug transport molecules (30, 31), and variants in the genes encoding these transporters have been implicated in variable drug responses. Thus, normal function of the drug efflux transporter P-glycoprotein is required for the biliary excretion of digoxin, and a common P-glycoprotein polymorphism has been associated with variable serum digoxin (32, 33). Similarly, polymorphisms in an organic anion (uptake) transporter have been implicated in the efficacy and some adverse effects of statins (34).
Genetically Determined Pharmacodynamics
Variability in the Genes Encoding Drug Targets
Drugs can produce highly variable effects, even in the absence of substantial variability in drug concentrations at target sites. This pharmacodynamic variability tends to be drug- or disease-specific, in contrast to pharmacokinetic variability that often extends across many drugs and disease processes. One obvious set of genes in which variants might account for such pharmacodynamic variability is those encoding drug targets. Thus, a plausible candidate gene for modulating variability in response to β2-agonists in asthma is the β2-receptor, and such variants in this gene have been reported (35).
Variants in multiple genes can contribute to variable drug actions, and recent studies with warfarin are an excellent example. Previous work had identified variable metabolism by CYP2C9 as a major contributor to variable responses to the drug (36). In 2004, coding-region mutations in VKORC1, encoding a subunit of the vitamin K epoxide reductase complex (the pharmacologic target for the drug), were found to cause a rare syndrome of warfarin resistance (37). Subsequently, common variants in VKORC1 have been found to account for a much greater fraction of variability in warfarin response (21%) than do variants in CYP2C9 (6%) (38). The CYP2C9 variants are in the coding region and alter enzyme activity, whereas the VKORC1 variants are noncoding and are thought to alter expression of the protein. The mechanisms underlying the remaining variability in warfarin effect have not been elucidated.
Variability in the Biological Context in Which Drugs Act
Variability in drug actions may reflect genetically determined changes not only in metabolism or in the drug target but also in the complex biological milieu in which drugs interact with their target molecules. An example that has received considerable attention is drug-induced prolongation of the QT interval and susceptibility to the ventricular tachycardia torsades de pointes, a common cause for drug withdrawal or relabeling (39). Although the length of the QT interval is determined by many ion-channel and other genes, only 1 (HERG [KCNH2]) is the target for QT-prolonging drugs; as a consequence, new drug candidates are now routinely screened for KCNH2-blocking activity (40). Of importance, surveys of patients with drug-induced torsades de pointes occasionally identify predisposing mutations not only in the drug target (the KCNH2 channel) but also in other genes that control the QT interval (41, 42); that is, variability in the physiologic control of the QT interval can contribute to variable effects of KCNH2 blockers.
Defining Drug Response
A key first step in any pharmacogenetic study, and indeed in all contemporary translational medicine, is precisely defining responders and nonresponders to therapy. Failure in this effort ensures failure for any subsequent genetic analysis. There is as much tension between well-defined clinical (often termed “hard”) end points, such as death, and biomarkers and surrogate end points, such as altered tumor size, change in blood pressure, or altered serum concentration of a disease-related protein, in pharmacogenetics as there is in all translational research. To the extent that biomarkers and surrogate end points can serve as relevant “intermediate phenotypes” that define important subsets of disease or drug responses, their definition is a critical component of minimizing confounding variables in any genetic-based analysis. A related challenge is the informatics problem of developing uniform methods to represent phenotypic data, such as drug responses, as is now the case with relatively standardized methods for representing genetic sequence and variation data.
Evolving Approaches to Identify Genes That Modulate Drug Responses
One approach in contemporary pharmacogenetics is to first use an understanding of pharmacokinetics and pharmacodynamics (see Glossary) to identify candidate genes that may determine the action of a drug. Then, the candidate genes are screened for variants, and the variants are screened for biological activity. This “genotype to phenotype” approach can lead not only to associations (see Glossary) between functional variants and drug responses but also to new insights into the basic biology of these proteins (43, 44). The converse strategy, “phenotype to genotype,” first defines variable drug response phenotypes and then searches in single genes, in pathways, or across the genome for sources of that variability.
Initial studies in pharmacogenetics often focused on small numbers of drug response outliers, studying candidate genes identified on the basis of an understanding of the underlying physiology and culminating in identification of variants associated with the outlier phenotype. This approach is intuitively appealing, especially if variants display abnormal function in vitro or in animal models that could plausibly explain the outlier drug response phenotype. CYP2D6, UGT1A1, and KCNH2 variants that mediate unusual responses to drug therapy are examples of this approach. Outlier drug responses that may lend themselves to such a strategy include the problems of rare and clinically severe adverse drug reactions, such as torsades de pointes, the lupus reaction, or hepatotoxicity (39, 45). Because these reactions often occur in an apparently unpredictable fashion, they have been termed “idiosyncratic,” and a genetic predisposition is often invoked. Accrual of patient sets in which to test this idea will require multi-center collaborations (46), and the initial focus of genetic analysis has been on a small number of genes chosen because they may be associated with disease susceptibility (41, 42, 47–50).
Disease Phenotypes
An obvious potential mechanism underlying variability in therapy for such common diseases as cancer, arrhythmia, or asthma is that these phenotypes represent an overlapping spectrum of mechanistically distinct diseases at the molecular or genetic level (51–53). In some cases, understanding the basis for a monogenic disease can inform and rationalize drug therapy; thus, mutations in KCNJ11, a component of a sulfonylurea-sensitive potassium channel, cause neonatal diabetes that is especially sensitive to sulfonylureas (54, 55). Disease-associated genetic variants (see Glossary) may be inherited (present in the germline) or arise later; such somatic cell mutations are often found in cancer, where they may arise as a consequence or a cause of disease and may contribute to variability in drug response. The epidermal growth factor receptor inhibitors gefitinib and erlotinib display highly variable responses in lung cancer, and dramatic tumor shrinkage now seems to be characteristic of a small group of patients whose tumors have somatic mutations in the tyrosine kinase domain of the receptor (56, 57). A screening test to identify such responders has now been commercialized (58).
Another example of the relationship between disease and drug response genetics is shown by a large study that implicated specific alleles in the 5-lipoxygenase gene as risk factors for myocardial infarction and stroke (59). On the basis of this finding, drugs to inhibit the 5-lipoxygenase–activating protein have been developed, and the first trial studied only patients selected for the at-risk genetic variant (60). This study paradigm shows how screening very large gene sets, up to the whole genome, can inform new biology and drug development, an advantage that the candidate gene approach generally lacks.
Genome-Wide Approaches
Although the approach of analyzing candidate genes has considerable intuitive appeal, it suffers from the criticism that it fails to consider a potential contribution of other genes, including those whose function is not yet well understood. Increasingly robust technologies are being developed to interrogate sets of single-nucleotide polymorphisms (SNPs) across the entire genome, and such whole genome association approaches have been applied to identify new disease susceptibility genes and pathways (61–63). This “unbiased” whole genome approach may also be applicable to the problem of identifying new loci-modulating variability in drug response.
Comparing the frequencies of hundreds of thousands of polymorphisms in patients with or without a specified genotype (see Glossary) will inevitably generate a very large number of false-positive associations. Thus, one way to interpret genome-wide association studies is that they may be able to identify genomic loci, which can then be studied in other patient subsets (that is, they are hypothesis-generating tools). Despite the very large number of polymorphisms in the human genome, it has also become apparent that these are not independent. Knowledge of the genotype at 1 locus informs genotype at other loci that are in linkage disequilibrium (64). The definition of such haplotype (see Glossary) structure across the genome and of “tag SNPs” that can thereby provide genotype information at multiple loci is a major goal of ongoing studies within industry and at the international HapMap consortium (65, 66). Such information will in turn be an enabling resource for large-scale genome-wide association studies by reducing the number of SNP sites for study.
The Concept of Drug Response “Pathways”
An appealing intermediate step between the single candidate gene approach and the nascent genome-wide approach is to consider that variable disease and drug response phenotypes reflect perturbations in function of increasingly well-characterized physiologic systems comprising dozens or hundreds of interacting gene products. Thus, studies of response to corticosteroids or β2-agonists in patients with asthma consider variations not only in corticosteroid or β2-adrenergic receptors but also in the downstream and interacting signaling pathways they modulate. In 1 such study, genotyping at 131 SNP sites in 14 genes in the steroid pathway identified and subsequently reproduced an association between response to inhaled steroids in asthma and polymorphisms in the type 1 corticotrophin-releasing hormone receptor (67). This identifies a new predictor of drug response and suggests that this receptor could be a target for new drug development. Similarly, screening 145 polymorphisms in 5 candidate ion-channel genes has identified a noncoding region in a potassium-channel gene as a potential contributor to risk for torsades de pointes (68). Screening 148 polymorphisms in 10 genes involved in cholesterol control identified non-coding variants in the 3-hydroxy-3-methylglutaryl coenzyme A reductase gene-modulating response to statin drugs that interact with this gene product (69). One advantage of this approach is that the number of rationally chosen candidate genes is markedly expanded but remains technologically and computationally tractable. In addition, increasingly sophisticated computational techniques are evolving to analyze how the behavior of a complex biological system changes in response to variable function of an individual component (70, 71).
Moving to Clinical Practice
An increasingly sophisticated understanding of the relationship between genetic variants and variability in drug response has identified and highlighted many important obstacles that must be overcome if this approach is to enter and enrich clinical practice.
Design and Interpretation of Pharmacogenetic and Pharmacogenomic Studies
Identification of an association between a clinical phenotype, such as drug response, and a genetic variant or a set of genetic variants is an increasing theme in the medical literature. However, many such associations were not reproduced in subsequent studies (72, 73); false-positive associations are one common reason. Another reason may be subtle differences in patient study groups, such as ethnicity or definition of end points. Thus, similar to other diagnostic approaches, pharmacogenetic testing will be implemented only when its predictive value is established.
Regulatory Issues in Genetic Testing
Questions regarding regulatory issues include mechanisms for regulating genotyping tests, the extent to which pharmacogenetic analyses should be incorporated into new drug development before or after large clinical trials, and whether and how pharmacogenetic information can be incorporated into the product labels that inform clinicians and patients. The U.S. Food and Drug Administration has launched an initiative to collect pharmacogenetic information during drug development that may help address some of these issues (74, 75); in addition, as already discussed, some drug labels have been changed to include pharmacogenetic information.
Development of New Genomic Technologies
Delivery of reliable genetic information to the clinician for decision making requires very rapid turn-around, or a setting in which drug therapy can be reasonably initiated after the results of a genetic test are available (for example, in chronic disorders, such as hypertension). Technology in this area is evolving very rapidly. A third scenario, in which an individual patient's whole genome is sequenced and available for decision making, seems likely within 5 to 10 years. This will require whole genome sequencing for less than several thousand dollars, appropriate information management systems, and new genomic statistical analysis approaches.
Ethical Issues
Identification of genetic variants associated with altered pathophysiology has raised questions of whether individuals or groups enriched for such genetic variants could be stigmatized (for example, by being denied insurance) (76, 77).
Education
The human genome is a new resource, and considerable education will be required to apply its promise in practice (78). Even for common and well-defined genetic variants with reproducible and important consequences for disease or drug therapy, wide acceptance by the medical community has been slow. This may reflect failure of education or lack of clear-cut studies showing added clinical value to understanding genetic information before prescribing drugs. Increasing educational efforts for the medical and lay communities will be required to understand the advantages and limitations of new genome-based clinical data.
Cost
One reason for slow adoption of pharmacogenetic testing is that data on cost-effectiveness are limited. This is obviously a complex issue that relates not only to the cost of genotyping itself but also to competing costs, such as those of caring for a patient with a catastrophic and predictable complication of drug therapy. Of note, however, a genotype needs to be established only once in a lifetime, and the costs of currently available tests are often less than those of the drugs themselves.
The Pharmacogenetics Research Network
Work toward overcoming the challenges described here has identified and highlighted specific obstacles in generating high-quality pharmacogenetic information and moving it from the research to the clinical setting and has, in some cases, pointed the way to solutions. Recognizing the challenges and the promise of pharmacogenetics, the National Institutes of Health initiated the Pharmacogenetics Research Network in 2000, with 3 overall goals: 1) to investigate the relationship of genetic variation to variable drug response; 2) to become an interactive network of investigators that impacts and elevates the field of pharmacogenetics with knowledge, tools, and resources; and 3) to create a publicly available knowledge base with reliable information that links phenotypes to genotypes. This knowledge base is available at www.pharmgkb.org and represents a central Web-enabled repository of variants in genes determining drug responses, their relationship to drug response phenotypes, and the strength of the evidence linking the two. This Web site is not only a repository but also a test bed that may enable advances in such areas as discovery of new associations or improved representations for drug response phenotypes.
Summary
Although the concept of pharmacogenetics, 1 allele at a time, was first proposed over a century ago, the more recent term pharmacogenomics captures the essence of contemporary work in this field. In addition to studying single allelic variants with large clinical effects, investigators are beginning to explore much larger sets of genes, including pathways up to the whole genome, and how variations in these pathways may affect drug response.
Pharmacogenomics is a young field that holds considerable promise of contributing to our understanding of the mechanisms underlying variability in human physiology and its response to drug therapy, with the potential to improve therapy. Indeed, studies in the field have discovered new biological mechanisms and have identified certain molecules, such as CYP2D6 or KCNH2, that are now routinely considered in drug development to enhance drug safety and are identifying new targets for drug action. Ultimately, identification of DNA variants before prescribing medication may become a routine feature of any patient–physician encounter. Although there are many challenges to be overcome in the implementation of pharmacogenetic vision in clinical practice, potential solutions are also evolving rapidly.
Acknowledgments
The authors thank Rochelle Long, Dina Paltoo, and Eileen Dolan for their comments on earlier versions of the manuscript.
Grant Support: Sites in the Pharmacogenetics Research Network are supported by the following U01 awards: GM61373, GM61390, GM74492, HL69757, GM61393, GM63340, HL65962, GM74518, GM61388, DA20830, and HL65899. The Pharmacogenetics and Pharmacogenomics Knowledge Base created by the network is housed at www.pharmgkb.org and is supported by U01 GM61374.
Glossary
- Alleles
Alternate sequences of the same gene, 1 inherited from each parent.
- Association
A statistical finding that the frequency of 1 or more genetic variants is significantly different in patients with a phenotype than in those without the phenotype. Often polymorphisms in candidate genes are studied. A more recent method, whole genome association, seeks to identify new genes involved in variable phenotypes; the technique uses new methods to compare genotypes at hundreds of thousands of polymorphic sites in large numbers of patients with and without a specific phenotype.
- Biological pathway
A set of proteins that interact to produce normal and abnormal physiology.
- Candidate gene
A gene in which variants could plausibly explain a given phenotype, such as severity of disease or variable response to drug. Methods to identify candidate genes include basic science studies, identifying DNA sequences conserved across species, human genetics, or genome-wide analyses. Candidate genes may be in pharmacokinetic or pharmacodynamic pathways.
- Genome
The collection of all DNA in an organism. Only a small proportion (probably <3%) of human genomes encodes proteins.
- Genetic variant
A difference in DNA sequence compared with a reference sequence.
- Genotype
The genetic makeup of an individual, which may refer to the whole genome or to specific genes or regions of genes.
- Haplotype
A set of genetic variants that are inherited together. Polymorphisms that are co-inherited more often than by chance alone are in linkage disequilibrium. Haplotype blocks may include many individual polymorphisms in high linkage disequilibrium; as a result, establishing genotype at any single polymorphic site with such a block may establish genotypes at linked sites within the block. Individual single-nucleotide polymorphisms (SNPs) that can be used to establish genotype within a haplotype block are termed tag SNPs.
- Homozygous
The same alleles in a specific region of DNA.
- Mutation
Rare variants, most often in coding regions, which are often associated with genetic diseases, such as cystic fibrosis or sickle cell anemia.
- Pharmacodynamics
The study of the relationship between drug concentrations and drug effects.
- Pharmacogenetics
The study of the relationship between individual gene variants and variable drug effects.
- Pharmacogenomics
The study of the relationship between variants in a large collection of genes, up to the whole genome, and variable drug effects.
- Pharmacokinetics
The study of the relationship between drug dose and drug concentrations (often as a function of time) in plasma or tissue.
- Phenotype
Measurable characteristics of an organism. These may derive from genotype, environment, or their combination. Organisms with the same phenotype can have different genotypes.
- Polymorphisms
DNA variants that are common, often defined as greater than 1% in a given population (although rare polymorphisms are increasingly being recognized). Polymorphisms can be in coding regions (where they may synonymous or nonsynonymous) or, more commonly, in noncoding regions, and often vary by ethnicity. The most common type of polymorphism is a change in 1 nucleotide (base pair) in a DNA sequence, referred to as an SNP. Other polymorphisms are insertion and deletion of multiple sequential nucleotides (“indels”); variable numbers of repeats, such as doublets or triplets; or large-scale duplications or deletions. Although some genetic variants are known to alter protein abundance or function, the functional consequences of most polymorphisms are unknown.
Footnotes
Potential Financial Conflicts of Interest: Consultancies: D.M. Roden (GlaxoSmithKline, Pfizer Inc., AstraZeneca, Abbott Laboratories, Novartis, 1st Genetic Trust), R.M. Krauss (Abbott Laboratories, AstraZeneca, Bristol-Myers Squibb, Merck & Co. Inc., Pfizer Inc., International Dairy Foods Association), M.J. Ratain (Prometheus, Genzyme Corp., Genentech), R.M. Weinshilboum (National Institutes of Health); S.T. Weiss (Glaxo-Wellcome, Roche Pharmaceuticals, Millennium Pharmaceuticals, Genetech, Schering-Plough, Variagenics, Genome Therapeutics, Merck Frost); Honoraria: D.M. Roden (GlaxoSmithKline, Pfizer, Inc., Astra-Zeneca, Abbott Laboratories, Novartis, 1st Genetic Trust), Dr. Krauss (Kos Pharmaceuticals, Pfizer); Stock ownership or options (other than mutual funds): M.J. Ratain (Variagenics, Nuvelo, Applera); Grants received: D.M. Roden (1st Genetic Trust), R.M. Krauss (King, Merck & Co. Inc., Schering-Plough, Pfizer Inc.), H.L.M. Leod (National Institutes of Health), M.J. Ratain (National Institutes of Health), R.M. Weinshilboum (Eli Lilly Inc.); S.T. Weiss (Glaxo Wellcome, AstraZeneca, Pfizer Inc.); Grants pending: D.M. Roden (GlaxoSmithKline), M.J. Ratain (National Institutes of Health); Patents received: M.J. Ratain (National Institutes of Health), M.V. Relling (National Institutes of Health); Patents pending: M.J. Ratain (National Institutes of Health); Royalties: D.M. Roden (Genaissance), M.J. Ratain (National Institutes of Health).
References
- 1.Weinshilboum R. Inheritance and drug response. N Engl J Med. 2003;348:529–37. doi: 10.1056/NEJMra020021. [DOI] [PubMed] [Google Scholar]
- 2.Evans WE, McLeod HL. Pharmacogenomics—drug disposition, drug targets, and side effects. N Engl J Med. 2003;348:538–49. doi: 10.1056/NEJMra020526. [DOI] [PubMed] [Google Scholar]
- 3.Roses AD. Pharmacogenetics and drug development: the path to safer and more effective drugs. Nat Rev Genet. 2004;5:645–56. doi: 10.1038/nrg1432. [DOI] [PubMed] [Google Scholar]
- 4.Garrod AE. Inborn errors of metabolism. 2nd. London: Henry Frowde and Hodder Stroughton; 1923. [Google Scholar]
- 5.Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–91. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 6.Kalow W. Pharmacogenetics—Heredity and responses to drugs. Philadelphia: WB Saunders; 1962. [Google Scholar]
- 7.Motulsky AG. Drug reactions enzymes, and biochemical genetics. J Am Med Assoc. 1957;165:835–7. doi: 10.1001/jama.1957.72980250010016. [DOI] [PubMed] [Google Scholar]
- 8.Price-Evans DA, Manley FA, McKusick VA. Genetic control of isoniazid metabolism in man. Br Med J. 1960;5197:485–91. doi: 10.1136/bmj.2.5197.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer UA. Pharmacogenetics—five decades of therapeutic lessons from genetic diversity. Nat Rev Genet. 2004;5:669–76. doi: 10.1038/nrg1428. [DOI] [PubMed] [Google Scholar]
- 10.Dahl ML, Johansson I, Bertilsson L, Ingelman-Sundberg M, Sjöqvist F. Ultrarapid hydroxylation of debrisoquine in a Swedish population. Analysis of the molecular genetic basis. J Pharmacol Exp Ther. 1995;274:516–20. [PubMed] [Google Scholar]
- 11.Tantisira KG, Lake S, Silverman ES, Palmer LJ, Lazarus R, Silverman EK, et al. Corticosteroid pharmacogenetics: association of sequence variants in CRHR1 with improved lung function in asthmatics treated with inhaled corticosteroids. Hum Mol Genet. 2004;13:1353–9. doi: 10.1093/hmg/ddh149. [DOI] [PubMed] [Google Scholar]
- 12.Vesell ES, Page JG. Genetic control of dicumarol levels in man. J Clin Invest. 1968;47:2657–63. doi: 10.1172/JCI105949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vesell ES, Page JG. Genetic control of drug levels in man: antipyrine. Science. 1968;161:72–3. doi: 10.1126/science.161.3836.72. [DOI] [PubMed] [Google Scholar]
- 14.Vesell ES, Page JG. Genetic control of drug levels in man: phenylbutazone. Science. 1968;159:1479–80. doi: 10.1126/science.159.3822.1479. [DOI] [PubMed] [Google Scholar]
- 15.Roden DM. Proarrhythmia as a pharmacogenomic entity: a critical review and formulation of a unifying hypothesis. Cardiovasc Res. 2005;67:419–25. doi: 10.1016/j.cardiores.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 16.Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr. 1991;119:985–9. doi: 10.1016/s0022-3476(05)83063-x. [DOI] [PubMed] [Google Scholar]
- 17.Schütz E, Gummert J, Mohr F, Oellerich M. Azathioprine-induced myelosuppression in thiopurine methyltransferase deficient heart transplant recipient [Letter] Lancet. 1993;341:436. doi: 10.1016/0140-6736(93)93028-y. [DOI] [PubMed] [Google Scholar]
- 18.Evans WE, Hon YY, Bomgaars L, Coutre S, Holdsworth M, Janco R, et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J Clin Oncol. 2001;19:2293–301. doi: 10.1200/JCO.2001.19.8.2293. [DOI] [PubMed] [Google Scholar]
- 19.Relling MV, Hancock ML, Rivera GK, Sandlund JT, Ribeiro RC, Krynetski EY, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–8. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 20.Stanulla M, Schaeffeler E, Flohr T, Cario G, Schrauder A, Zimmermann M, et al. Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA. 2005;293:1485–9. doi: 10.1001/jama.293.12.1485. [DOI] [PubMed] [Google Scholar]
- 21.Dahl ML, Bertilsson L. Genetically variable metabolism of antidepressants and neuroleptic drugs in man. Pharmacogenetics. 1993;3:61–70. doi: 10.1097/00008571-199304000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Caraco Y, Sheller J, Wood AJ. Impact of ethnic origin and quinidine coadministration on codeine's disposition and pharmacodynamic effects. J Pharmacol Exp Ther. 1999;290:413–22. [PubMed] [Google Scholar]
- 23.Walker DK. The use of pharmacokinetic and pharmacodynamic data in the assessment of drug safety in early drug development. Br J Clin Pharmacol. 2004;58:601–8. doi: 10.1111/j.1365-2125.2004.02194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee LS, Nafziger AN, Bertino JS., Jr Evaluation of inhibitory drug interactions during drug development: genetic polymorphisms must be considered. Clin Pharmacol Ther. 2005;78:1–6. doi: 10.1016/j.clpt.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Furuta T, Ohashi K, Kamata T, Takashima M, Kosuge K, Kawasaki T, et al. Effect of genetic differences in omeprazole metabolism on cure rates for Helicobacter pylori infection and peptic ulcer. Ann Intern Med. 1998;129:1027–30. doi: 10.7326/0003-4819-129-12-199812150-00006. [DOI] [PubMed] [Google Scholar]
- 26.Ratain MJ. From bedside to bench to bedside to clinical practice: an odyssey with irinotecan. Clin Cancer Res. 2006;12:1658–60. doi: 10.1158/1078-0432.CCR-06-0159. [DOI] [PubMed] [Google Scholar]
- 27.Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H, et al. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res. 2000;60:6921–6. [PubMed] [Google Scholar]
- 28.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–8. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 29.AmpliChip CYP450 test. Med Lett Drugs Ther. 2005;47:71–2. [PubMed] [Google Scholar]
- 30.Kim RB. Transporters and xenobiotic disposition. Toxicology. 2002:181–182. 291–7. doi: 10.1016/s0300-483x(02)00296-2. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Brett CM, Giacomini KM. Role of organic cation transporters in drug absorption and elimination. Annu Rev Pharmacol Toxicol. 1998;38:431–60. doi: 10.1146/annurev.pharmtox.38.1.431. [DOI] [PubMed] [Google Scholar]
- 32.Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmöller J, Johne A, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A. 2000;97:3473–8. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marzolini C, Paus E, Buclin T, Kim RB. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther. 2004;75:13–33. doi: 10.1016/j.clpt.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Kim RB. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) and genetic variability (single nucleotide polymorphisms) in a hepatic drug uptake transporter: what's it all about? Clin Pharmacol Ther. 2004;75:381–5. doi: 10.1016/j.clpt.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 35.Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, et al. Complex promoter and coding region beta 2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci U S A. 2000;97:10483–8. doi: 10.1073/pnas.97.19.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aithal GP, Day CP, Kesteven PJ, Daly AK. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet. 1999;353:717–9. doi: 10.1016/S0140-6736(98)04474-2. [DOI] [PubMed] [Google Scholar]
- 37.Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hörtnagel K, Pelz HJ, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427:537–41. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 38.Rieder MJ, Reiner AP, Gage BF, Nickerson DA, Eby CS, McLeod HL, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med. 2005;352:2285–93. doi: 10.1056/NEJMoa044503. [DOI] [PubMed] [Google Scholar]
- 39.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–22. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 40.Shah RR. Pharmacogenetic aspects of drug-induced torsades de pointes: potential tool for improving clinical drug development and prescribing. Drug Saf. 2004;27:145–72. doi: 10.2165/00002018-200427030-00001. [DOI] [PubMed] [Google Scholar]
- 41.Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci U S A. 2000;97:10613–8. doi: 10.1073/pnas.180223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–8. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 43.Leabman MK, Huang CC, DeYoung J, Carlson EJ, Taylor TR, de la Cruz M, et al. Natural variation in human membrane transporter genes reveals evolutionary and functional constraints. Proc Natl Acad Sci U S A. 2003;100:5896–901. doi: 10.1073/pnas.0730857100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drolet B, Simard C, Mizoue L, Roden DM. Human cardiac potassium channel DNA polymorphism modulates access to drug-binding site and causes drug resistance. J Clin Invest. 2005;115:2209–13. doi: 10.1172/JCI23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 2003;349:474–85. doi: 10.1056/NEJMra021844. [DOI] [PubMed] [Google Scholar]
- 46.James SP. This month at the NIH: NIDDK forms hepatotoxicity clinical research network. Gastroenterology. 2003;125:1008. [PubMed] [Google Scholar]
- 47.Woosley RL, Drayer DE, Reidenberg MM, Nies AS, Carr K, Oates JA. Effect of acetylator phenotype on the rate at which procainamide induces anti-nuclear antibodies and the lupus syndrome. N Engl J Med. 1978;298:1157–9. doi: 10.1056/NEJM197805252982101. [DOI] [PubMed] [Google Scholar]
- 48.Huang YS, Chern HD, Su WJ, Wu JC, Chang SC, Chiang CH, et al. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis. Hepatology. 2003;37:924–30. doi: 10.1053/jhep.2003.50144. [DOI] [PubMed] [Google Scholar]
- 49.Huang YS, Chern HD, Su WJ, Wu JC, Lai SL, Yang SY, et al. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitis. Hepatology. 2002;35:883–9. doi: 10.1053/jhep.2002.32102. [DOI] [PubMed] [Google Scholar]
- 50.Funk C, Pantze M, Jehle L, Ponelle C, Scheuermann G, Lazendic M, et al. Troglitazone-induced intrahepatic cholestasis by an interference with the hepatobiliary export of bile acids in male and female rats. Correlation with the gender difference in troglitazone sulfate formation and the inhibition of the canalicular bile salt export pump (Bsep) by troglitazone and troglitazone sulfate. Toxicology. 2001;167:83–98. doi: 10.1016/s0300-483x(01)00460-7. [DOI] [PubMed] [Google Scholar]
- 51.Fox CS, Parise H, D'Agostino RB, Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–5. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 52.Spooner PM, Albert C, Benjamin EJ, Boineau R, Elston RC, George AL, Jr, et al. Sudden cardiac death, genes, and arrhythmogenesis: consideration of new population and mechanistic approaches from a National Heart, Lung, and Blood Institute workshop, Part II. Circulation. 2001;103:2447–52. doi: 10.1161/01.cir.103.20.2447. [DOI] [PubMed] [Google Scholar]
- 53.Raby BA, Van Steen K, Celedón JC, Litonjua AA, Lange C, Weiss ST, et al. Paternal history of asthma and airway responsiveness in children with asthma. Am J Respir Crit Care Med. 2005;172:552–8. doi: 10.1164/rccm.200501-010OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sagen JV, Raeder H, Hathout E, Shehadeh N, Gudmundsson K, Baevre H, et al. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes. 2004;53:2713–8. doi: 10.2337/diabetes.53.10.2713. [DOI] [PubMed] [Google Scholar]
- 55.Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–49. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 56.Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 57.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 58.Couzin J. Pharmacogenomics. Cancer sharpshooters rely on DNA tests for a better aim. Science. 2004;305:1222–3. doi: 10.1126/science.305.5688.1222a. [DOI] [PubMed] [Google Scholar]
- 59.Helgadottir A, Manolescu A, Thorleifsson G, Gretarsdottir S, Jonsdottir H, Thorsteinsdottir U, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet. 2004;36:233–9. doi: 10.1038/ng1311. [DOI] [PubMed] [Google Scholar]
- 60.Hakonarson H, Thorvaldsson S, Helgadottir A, Gudbjartsson D, Zink F, Andresdottir M, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA. 2005;293:2245–56. doi: 10.1001/jama.293.18.2245. [DOI] [PubMed] [Google Scholar]
- 61.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 63.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 64.Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, et al. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science. 2001;294:1719–23. doi: 10.1126/science.1065573. [DOI] [PubMed] [Google Scholar]
- 65.A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, et al. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–9. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 67.Weiss ST, Lake SL, Silverman ES, Silverman EK, Richter B, Drazen JM, et al. Asthma steroid pharmacogenetics: a study strategy to identify replicated treatment responses. Proc Am Thorac Soc. 2004;1:364–7. doi: 10.1513/pats.200409-043MS. [DOI] [PubMed] [Google Scholar]
- 68.Kääb S, Pfeufer A, Hinterseer M, Näbauer M, Schulze-Bahr E. Long QT syndrome. Why does sex matter? Z Kardiol. 2004;93:641–5. doi: 10.1007/s00392-004-0129-6. [DOI] [PubMed] [Google Scholar]
- 69.Chasman DI, Posada D, Subrahmanyan L, Cook NR, Stanton VP, Jr, Ridker PM. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA. 2004;291:2821–7. doi: 10.1001/jama.291.23.2821. [DOI] [PubMed] [Google Scholar]
- 70.Rudy Y. From genetics to cellular function using computational biology. Ann N Y Acad Sci. 2004;1015:261–70. doi: 10.1196/annals.1302.022. [DOI] [PubMed] [Google Scholar]
- 71.Cohen JE. Mathematics is biology's next microscope, only better; biology is mathematics' next physics, only better. PLoS Biol. 2004;2:e439. doi: 10.1371/journal.pbio.0020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 73.Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29:306–9. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- 74.Lesko LJ, Woodcock J. Translation of pharmacogenomics and pharmacogenetics: a regulatory perspective. Nat Rev Drug Discov. 2004;3:763–9. doi: 10.1038/nrd1499. [DOI] [PubMed] [Google Scholar]
- 75.Salerno RA, Lesko LJ. Pharmacogenomic data: FDA voluntary and required submission guidance. Pharmacogenomics. 2004;5:503–5. doi: 10.1517/14622416.5.5.503. [DOI] [PubMed] [Google Scholar]
- 76.Breckenridge A, Lindpaintner K, Lipton P, McLeod H, Rothstein M, Wallace H. Pharmacogenetics: ethical problems and solutions. Nat Rev Genet. 2004;5:676–80. doi: 10.1038/nrg1431. [DOI] [PubMed] [Google Scholar]
- 77.Billings PR. Genetic nondiscrimination. Nat Genet. 2005;37:559–60. doi: 10.1038/ng0605-559. [DOI] [PubMed] [Google Scholar]
- 78.Frueh FW, Goodsaid F, Rudman A, Huang SM, Lesko LJ. The need for education in pharmacogenomics: a regulatory perspective. Pharmacogenomics J. 2005;5:218–20. doi: 10.1038/sj.tpj.6500316. [DOI] [PubMed] [Google Scholar]