

Extracellular adenosine–driven osteogenic differentiation of stem cells.
Keywords: Pluripotent stem cells, small molecule, adenosine signaling, adenosine receptors, osteogenic differentiation, bone repair
Abstract
The abilities of human pluripotent stem cells (hPSCs) to proliferate without phenotypic alteration and to differentiate into tissue-specific progeny make them a promising cell source for regenerative medicine and development of physiologically relevant in vitro platforms. Despite this potential, efficient conversion of hPSCs into tissue-specific cells still remains a challenge. Herein, we report direct conversion of hPSCs into functional osteoblasts through the use of adenosine, a naturally occurring nucleoside in the human body. The hPSCs treated with adenosine not only expressed the molecular signatures of osteoblasts but also produced calcified bone matrix. Our findings show that the adenosine-mediated osteogenesis of hPSCs involved the adenosine A2bR. When implanted in vivo, using macroporous synthetic matrices, the human induced pluripotent stem cell (hiPSC)–derived donor cells participated in the repair of critical-sized bone defects through the formation of neobone tissue without teratoma formation. The newly formed bone tissues exhibited various attributes of the native tissue, including vascularization and bone resorption. To our knowledge, this is the first demonstration of adenosine-induced differentiation of hPSCs into functional osteoblasts and their subsequent use to regenerate bone tissues in vivo. This approach that uses a physiologically relevant single small molecule to generate hPSC-derived progenitor cells is highly appealing because of its simplicity, cost-effectiveness, scalability, and impact in cell manufacturing, all of which are decisive factors for successful translational applications of hPSCs.
INTRODUCTION
Human pluripotent stem cells (hPSCs), which include both human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), are optimal cell sources for regenerative medicine, in vitro model systems to study embryogenesis and disease, and screening platforms for drug discovery (1–3). Although both cell sources have the ability to self-renew indefinitely and differentiate into three germ layers, hiPSCs offer additional clinical benefits because they can be derived from patients’ own cells. Despite the opportunities, cost-effective and efficient differentiation of hPSCs into tissue-specific cells with in vivo function is still a challenge, limiting their widespread applications (4, 5). A typical application of hPSCs often involves their in vitro differentiation into desired phenotypes before use, which will limit many unwanted outcomes associated with the potential of hPSCs to differentiate into all cell types in the human body.
Over the past decade, substantial strides have been made to control differentiation of stem cells into targeted phenotypes by a variety of approaches, such as genetic manipulation, biomaterials, growth factors, small molecules, or cocktails thereof (6–14). We, and others, have shown that biomaterials containing calcium phosphate (CaP) minerals direct osteogenic differentiation of stem cells (15–18). Extending these osteoinductive biomineralized matrices to understand the role of CaP minerals on osteogenesis has unearthed phosphate–adenosine triphosphate (ATP)–adenosine A2b receptor (A2bR) axis signaling in CaP-induced osteogenic differentiation of human bone marrow–derived mesenchymal stem cells (hMSCs) (19). Our study has also shown that the activation of A2bR inhibits adipogenic differentiation of hMSCs while promoting their osteogenesis (20). Leveraging these understandings, we propose that exogenous adenosine could be a powerful small molecule to derive osteoprogenitor cells and/or osteoblasts from hPSCs.
Approaches, such as using small molecules to direct differentiation of stem cells, are highly attractive and could significantly improve their therapeutic applications. Using small molecule(s) to direct stem cell fate has been an active area of research and has resulted in the identification of small molecules that promote self-renewal (21, 22), differentiation of stem cells (23–27), and dedifferentiation (28), transdifferentiation (29, 30), and reprogramming of somatic cells (31–33), as well as limit the aging of bone marrow–derived MSCs (34). It has been shown that small molecules can promote osteogenic differentiation of MSCs or osteoprogenitor cells (35, 36). For instance, purmorphamine and N6-benzoyladenosine-3′,5′-cyclic monophosphate have been reported to direct osteogenic differentiation of mesenchymal progenitor cells (35) and osteoblast-like MC3T3-E1 cells (36), respectively. Although these studies have shown the application of small molecules to direct osteogenic differentiation of progenitor cells, the efficacy of small molecules to direct osteogenic differentiation of naïve stem cells, like hPSCs, is yet to be established. In a recent study, Kanke et al. reported the sequential usage of four different small molecules to derive osteoblasts from PSCs (27). Specifically, the PSCs were cultured in medium supplemented with CHIR99021 and cyclopamine to achieve mesoderm induction. These mesoderm-committed cells were differentiated into osteoblasts by using conventional osteogenic induction medium supplemented with smoothened agonist and helioxanthin derivative 4-(4-methoxyphenyl)pyrido[40,30:4,5]thieno [2,3-b]pyridine-2-carboxamide.
Here, we determine the ability of exogenous adenosine to induce conversion of hPSCs into osteoblasts without the use of embryoid body formation. Adenosine is a naturally occurring small molecule, a purine nucleoside, and is a metabolite of ATP (37), which is known for its physiological and pharmacological functions via interactions with four subtypes of G protein (guanine nucleotide–binding protein)–coupled purinergic receptors (A1R, A2aR, A2bR, and A3R) and has been extensively used as a neuromodulator and vasodilator (37–40). Recent studies have implicated the importance of adenosine and its receptors in bone fracture repair and bone homeostasis (41).
RESULTS
Adenosine drives osteogenic differentiation of hPSCs
hiPSCs and hESCs were seeded onto Matrigel-coated coverslips and cultured in growth medium, growth medium supplemented with adenosine, or conventional osteogenic induction medium, and their osteogenic differentiation was determined. Previous studies have shown that hPSCs cultured in osteogenic induction medium containing β-glycerophosphate, ascorbic acid-2-phosphate, and dexamethasone undergo osteogenesis (42). The cells in all cultures grew to confluence within 10 days with no significant differences irrespective of the medium used (fig. S1, A and B). Analysis of gene expression profile of hiPSCs at 21 days of culture showed considerable up-regulation of various genes relevant to cells undergoing osteogenesis in growth medium supplemented with adenosine or osteogenic induction medium (Fig. 1A). Osteogenic differentiation of hiPSCs was further evaluated through time-resolved quantitative analyses for various osteogenic genes (RUNX2, OCN, and SPP1). The quantitative polymerase chain reaction (qPCR) analyses showed that hiPSCs cultured in the presence of exogenous adenosine, similar to osteogenic induction medium, consistently exhibited an up-regulation of RUNX2, OCN, and SPP1 expressions throughout 21 days of culture (Fig. 1B). The hiPSCs, cultured under all medium conditions, showed down-regulation of NANOG (a pluripotency marker), suggesting that the hiPSCs in all cultures lost pluripotency irrespective of whether the hiPSCs underwent osteogenic differentiation (Fig. 1B). Similar to hiPSCs, hESCs also showed up-regulation of osteogenic markers when cultured in growth medium supplemented with adenosine or osteogenic induction medium (fig. S2A). In addition to gene expression profiling, the conversion of hPSCs (hiPSCs and hESCs) into osteoblasts was evaluated by immunofluorescence staining for osteocalcin (an osteoblast-specific marker) (Fig. 1C and figs. S2B and S3). In accordance with the gene expression profile, the hPSCs cultured in the presence of adenosine or osteogenic induction medium stained positive for osteocalcin in most cells after 21 days of culture (Fig. 1C and fig. S2B). The intensity of osteocalcin staining for the hPSCs undergoing osteogenic differentiation was found to gradually increase with culture time (figs. S2B and S3). No such osteocalcin expression was found for the hPSCs cultured in growth medium despite their loss of pluripotency (Fig. 1C and figs. S2B and S3).
Fig. 1. Exogenous adenosine induced osteogenic differentiation of hiPSCs.
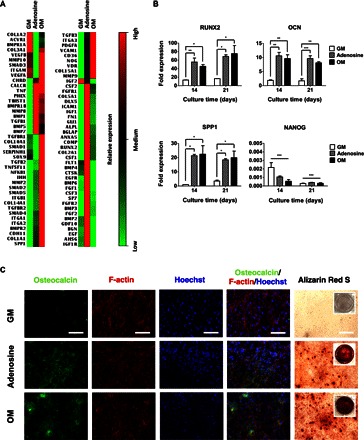
(A) Transcription profile of 84 genes relevant to osteogenesis for hiPSCs cultured for 21 days in growth medium (GM), adenosine-supplemented growth medium (Adenosine), and conventional osteogenic induction medium (OM). Relative expressions: red (high), black (medium), and green (low). (B) Time-dependent quantitative gene expressions of hiPSCs for osteogenic markers (RUNX2, OCN, and SPP1) and pluripotent marker (NANOG) cultured in GM, adenosine, and OM. (C) Immunofluorescence staining of osteocalcin (green), F-actin (red), and nuclei (blue), as well as Alizarin Red S staining of hiPSCs cultured for 21 days in GM, adenosine, and OM. Scale bars, 100 μm. Inset shows the stained image of the entire well. Data are presented as means ± SEs (n = 3). Data are shown as fold expression of target genes after normalization to undifferentiated, pluripotent hiPSCs. For RUNX2, OCN, and SPP1, the groups with various medium conditions at the same culture time were compared by using one-way analysis of variance (ANOVA) with Tukey-Kramer post hoc test. For NANOG, all the groups were compared to undifferentiated, pluripotent hiPSCs by two-way ANOVA with Bonferroni post hoc test. Asterisks were assigned to P values with statistical significances (*P < 0.05; **P < 0.01; ***P < 0.001).
To further confirm osteogenic differentiation of the hPSCs, we examined the calcification of cell cultures by staining for Alizarin Red S. The hPSCs cultured in both adenosine-supplemented growth medium and osteogenic-inducing medium stained positive for Alizarin Red S after 21 days of culture (Fig. 1C and fig. S2B). Similar to osteocalcin, the intensity for Alizarin Red S staining became progressively prevalent with culture time (figs. S2B and S3). On the contrary, no calcification was observed for the hPSCs cultured in growth medium.
Adenosine-driven osteogenesis of hPSCs involves A2bR
The hPSCs cultured in growth medium supplemented with adenosine showed an up-regulation of A2bR expression with no considerable differences in the expression of other adenosine (or P1) receptors, such as A1R, A2aR, or A3R (Fig. 2A and fig. S4A). No consistent up-regulation of any adenosine receptor subtypes was observed for hPSCs cultured under other medium (growth and osteogenic induction medium) conditions. To investigate the potential involvement of A2bR in adenosine-driven osteogenic differentiation of hPSCs, we used an A2bR antagonist, 8-[4-[4-(4-chlorophenzyl)piperazide-1-sulfonyl)phenyl]]-1-propylxanthine (PSB 603) (19). The presence of PSB 603 in cultures inhibited the exogenous adenosine–mediated up-regulation of osteogenic markers (Fig. 2B and fig. S4B). The hPSCs cultured in the presence of PSB 603 also showed down-regulation of NANOG when compared to undifferentiated hPSCs (Fig. 2B and fig. S4B). The PSB 603–mediated attenuation of osteogenic differentiation was further confirmed by immunofluorescence staining for osteocalcin and Alizarin Red S staining for calcification. Concurrent with the gene expression profile, the hPSCs cultured in the presence of PSB 603 showed a significant reduction in osteocalcin expression and calcification (Fig. 2C and fig. S4C).
Fig. 2. Adenosine-induced osteogenic differentiation of hiPSCs involves A2bR.
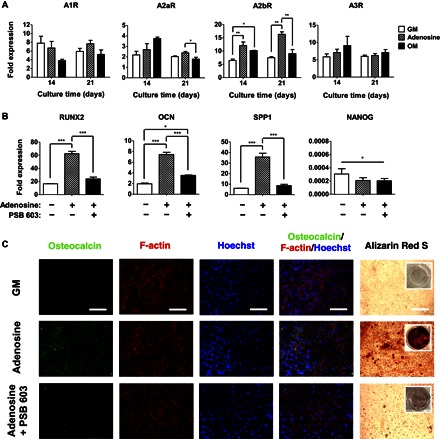
(A) Time-dependent quantitative gene expressions of hiPSCs for adenosine receptor subtypes (A1R, A2aR, A2bR, and A3R) cultured in growth medium (GM), adenosine-supplemented growth medium (Adenosine), and osteogenic induction medium (OM). (B) Quantitative gene expression analysis of osteogenic markers (RUNX2, OCN, and SPP1) and pluripotent marker (NANOG) for hiPSCs after 21 days of culture. The plus (+) and minus (−) symbols in gene expression denote growth medium in the presence and absence of adenosine or PSB 603 (an A2bR antagonist). (C) Immunofluorescence staining for osteocalcin (green), F-actin (red), and nuclei (blue), as well as Alizarin Red S staining of hiPSCs cultured for 21 days in the presence and absence of adenosine or PSB 603. Inset shows the stained image of the whole well. Various media include growth medium (GM), growth medium containing adenosine (Adenosine), and growth medium containing both adenosine and PSB 603 (Adenosine + PSB 603). Scale bars, 100 μm. Data are shown as means ± SEs (n = 3). Data are presented as fold expression of target genes after normalization to undifferentiated, pluripotent hiPSCs. For A1R, A2aR, A2bR, and A3R, as well as RUNX2, OCN, and SPP1, the groups with various medium conditions at the same culture time were compared by one-way ANOVA with Tukey-Kramer post hoc test. For NANOG, all the groups were compared to undifferentiated, pluripotent hiPSCs by two-way ANOVA with Bonferroni post hoc test. Asterisks indicate statistical significances according to P values (*P < 0.05; **P < 0.01; ***P < 0.001).
hiPSC-derived osteoblasts produce calcified bone tissue in vitro
Having established the ability of adenosine to direct osteogenic commitment of hPSCs, we next determined the bone tissue–forming ability of hiPSC-derived cells. The hiPSCs were cultured in the presence of exogenous adenosine (that is, growth medium supplemented with adenosine) for 21 days, and the differentiated cells (hereafter termed as Ad-hiPSCs) were loaded into poly(ethylene glycol) diacrylate-co-acryloyl 6-aminocaproic acid (PEGDA-co-A6ACA) macroporous hydrogels (figs. S5A and S6A). The hiPSCs cultured for 21 days in growth medium devoid of exogenous adenosine were used as a control, hereafter termed as d-hiPSCs. The schematic in Fig. 3A illustrates the experimental procedures.
Fig. 3. In vitro bone-forming ability of hiPSC-derived cells.
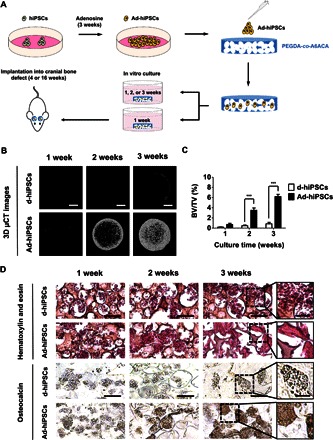
(A) Schematic of the experimental protocol used to examine the bone-forming ability of hiPSC-derived osteoblasts within the macroporous matrices. (B and C) Three-dimensional microcomputed tomography (μCT) images (B) and the corresponding bone mineral densities [bone volume per total volume (BV/TV)] (C) of cell-laden (Ad-hiPSCs and d-hiPSCs) matrices as a function of culture time. Scale bars, 2 mm. (D) H&E staining and immunohistochemical staining for osteocalcin of the macroporous matrices containing Ad-hiPSCs and d-hiPSCs cultured for 1, 2, and 3 weeks. Scale bars, 100 μm. High-magnification images are also provided. Scale bars, 50 μm. Data are displayed as means ± SEs (n = 4). Two groups at the same culture time were compared by two-tailed Student’s t test. Asterisks indicate statistical significances according to P values (***P < 0.01).
The cell-laden matrices were cultured in medium lacking osteogenic-inducing factors. After 3 days of cell seeding, live-dead staining showed that the cells were viable and homogeneously distributed within the macroporous matrices (figs. S5B and S6B). The gross appearance of the Ad-hiPSC–laden matrices as a function of culture time revealed the formation of calcified tissue (fig. S5C). No such intense calcification or hard tissue formation was observed for cell-laden matrices containing d-hiPSCs. Three-dimensional (3D) microcomputed tomography (μCT) images of the Ad-hiPSC–laden matrices showed progressive formation of calcified tissue throughout 3 weeks of culture (Fig. 3B). In stark contrast, the matrices containing d-hiPSCs showed minimal mineralization along the periphery of the matrices. Rotation views (360°) of the μCT models further showed dense and homogeneous calcified tissue formation for Ad-hiPSCs after 3 weeks of culture (movies S1 and S2). Quantification of hard tissue formation from the 3D μCT images corroborated the observations with significantly higher mineralization for Ad-hiPSCs as compared to d-hiPSCs (Fig. 3C). Two-dimensional cross-sectional images of the matrices containing Ad-hiPSCs, in three perpendicular planes, showed uniform hard tissue formation throughout the porous matrices after 3 weeks of culture (fig. S5D).
Histological examination of the calcified tissues was performed to determine the formation of bone tissue. Hematoxylin and eosin (H&E) staining showed the presence of dense extracellular matrix (ECM) resembling bone tissue for the cultures containing Ad-hiPSCs at 3 weeks of culture, whereas the cultures involving d-hiPSCs displayed minimal bone tissue formation (Fig. 3D). The bone tissue formation was also confirmed by immunohistochemical staining for osteocalcin. The cell-laden matrices containing Ad-hiPSCs showed the presence of osteocalcin-rich ECM, which was found to increase over the course of 3 weeks (Fig. 3D). On the contrary, minimal osteocalcin staining was observed in the cell-laden matrices containing d-hiPSCs.
hiPSC-derived osteoblasts directly participate in the healing of critical-sized cranial defects
The ability of hiPSC-derived osteoblasts (Ad-hiPSCs) to contribute to bone tissue repair was examined by using critical-sized cranial bone defects in mice. Ad-hiPSC–laden PEGDA-co-A6ACA macroporous matrices were cultured in vitro for 1 week before their implantation. μCT analyses of these cell-laden matrices before implantation showed minimal hard tissue formation (fig. S6C). The in vivo bone tissue formation of Ad-hiPSC–laden matrices was compared to those with acellular matrices and sham groups. Bone tissue formation at 4 and 16 weeks after implantation was evaluated through μCT analyses that showed formation of hard tissue in the defects treated with Ad-hiPSC–laden matrices (Fig. 4A and fig. S7A). The newly formed hard tissue was found to cover the entire defect. On the contrary, minimal to no hard tissue formation was observed in the sham control, and the defects treated with acellular matrices showed partial calcification. Quantification of μCT images showed significantly higher hard tissue formation in the defects treated with Ad-hiPSCs, compared to sham and acellular controls, with bone volume reaching similar to that of native bone by 16 weeks (Fig. 4B). The defects treated with Ad-hiPSCs showed homogeneous hard tissue formation throughout the cranial defect that was integrated with the adjacent native bone.
Fig. 4. hiPSC-derived osteoblasts (Ad-hiPSCs) contribute to the healing of critical-sized bone defects through the formation of vascularized neobone tissue.
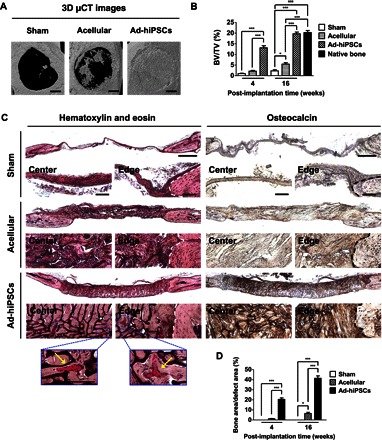
(A) Three-dimensional μCT images of cranial bone defects with no implantation (Sham), as well as defects treated with acellular and Ad-hiPSC–laden matrices [Ad-hiPSCs; hiPSC derivatives generated by culturing in growth medium supplemented with adenosine (30 μg/ml) for 21 days] after 16 weeks. Scale bars, 1 mm. (B) Bone mineral densities [bone volume per total volume (BV/TV)] of defects treated with acellular and Ad-hiPSC–laden matrices, as well as sham group following 4 and 16 weeks of implantation. Native mouse cranial bone was used as a control. (C) H&E staining and immunohistochemical staining for osteocalcin of cranial bone defects treated with acellular and Ad-hiPSCs–laden matrices, and sham group after 16 weeks. Scale bars, 500 μm. High-magnification images show the center and edge of cranial bone defects. White and black dashed lines mark the interface between newly formed tissue and the native bone tissue. Yellow arrows in H&E staining denote microvessels containing red blood cells. Scale bars, 100 μm. (D) Histomorphometric analysis for neobone density within cranial bone defects (Bone area/defect area) determined from H&E staining images for Sham, Acellular, and Ad-hiPSCs groups at 4 and 16 weeks after treatment. Data are shown as means ± SEs (n = 6). Multiple groups at the same post-implantation time were compared by one-way ANOVA with Tukey-Kramer post hoc test. Asterisks were assigned to P values with statistical significances (*P < 0.05; ***P < 0.001).
H&E and osteocalcin staining, at 4 and 16 weeks after treatment, confirmed the presence of neobone tissue in defects treated with Ad-hiPSCs (Fig. 4C and fig. S7B). H&E-stained images at 4 and 16 weeks showed formation of bone tissue resembling the morphology of native tissue. Histomorphometrical analysis corroborated the aforementioned observations that the defects treated with Ad-hiPSCs had significantly higher bone content compared to acellular and sham groups (Fig. 4D). The neobone tissue was also characterized by osteocalcin-rich ECM (Fig. 4C and fig. S7B). Compared to Ad-hiPSCs, sham and acellular control groups showed minimal bone formation, with hard tissue domains nonhomogeneously distributed within the defects. No evidence of teratoma formation or infection was observed in any of the animals treated with Ad-hiPSCs.
To determine the contribution of transplanted Ad-hiPSCs toward the formation of neobone tissue, we performed human-specific lamin A/C staining. Immunofluorescence staining of the newly formed bone tissue with Ad-hiPSCs at 4 and 16 weeks after implantation showed the colocalization of osteocalcin and human-specific lamin A/C, confirming the contribution of transplanted Ad-hiPSCs toward bone repair (Fig. 5A and fig. S8A). The new bone tissue was also filled with cells that were positive for osteocalcin but negative for lamin A/C, implicating the contribution of host cells, as well, toward bone formation. Osteocalcin-positive cells were also found in cohorts treated with acellular matrices and sham groups, indicating the infiltration of host cells into the implanted matrices and defect sites.
Fig. 5. hiPSC-derived osteoblasts contributed to the repair of critically sized cranial defects through the formation of vascularized neobone tissue.
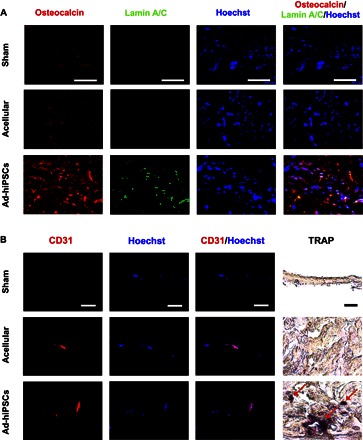
(A and B) Immunofluorescence staining for osteocalcin (red) and human-specific lamin A/C (green) (A) and CD31 (red) along with nuclei (blue; Hoechst), as well as TRAP staining (B) of cranial bone defects treated with Ad-hiPSC–laden (Ad-hiPSCs) and acellular matrices, and sham group after 16 weeks of implantation. Red arrows indicate TRAP-positive stains. Scale bars, 50 μm.
Not only did the transplanted cells survive and contribute to the bone tissue formation, but the newly formed bone tissue showed the presence of vascularization. Specifically, the cell-laden matrices were permeated with intraluminal red blood cells (Fig. 4C). Positive staining of the tissue sections with platelet endothelial cell adhesion molecule antibody (CD31, also known as PECAM-1) further confirmed the presence of vascularization (Fig. 5B and fig. S8B). The cell-laden matrices also stained positive for tartrate-resistant acid phosphatase (TRAP) at 4 and 16 weeks after treatment (Fig. 5B and fig. S8B). Positive TRAP staining indicates the presence of osteoclast-like cells within the regenerated bone tissue. No significant positive TRAP staining was observed for the defects treated with acellular matrices and sham groups.
DISCUSSION
Small molecule–directed differentiation of hPSCs could significantly contribute to their applications. Small molecules, which can be readily synthesized and remain relatively stable compared to growth factors, offer a simple, reproducible, and cost-effective strategy to generate large numbers of hPSC derivatives. Here, we describe the use of exogenous adenosine to derive osteoblasts from hPSCs and use these hiPSC-derived cells for the repair of critical-sized cranial defects through bone tissue formation.
Consistent with previous reports, hESCs and hiPSCs cultured in medium lacking basic fibroblast growth factors (bFGF) showed loss of pluripotency (43). Although cells cultured in all medium conditions showed down-regulation of the pluripotent marker NANOG, only the cells cultured in osteogenic-inducing medium and growth medium containing adenosine underwent osteogenic differentiation. The hPSCs exposed to exogenous adenosine showed an up-regulation of adenosine A2bR, with no significant differences in the expression levels of other adenosine subtype receptors (A1R, A2aR, and A3R). Concomitant with these findings, the blocking of A2bR with PSB 603, an A2bR antagonist, attenuated adenosine-induced osteogenic differentiation of the cells. Together, the results implicate the key role played by A2bR in exogenous adenosine–mediated osteogenic differentiation of hPSCs. These finding are in accordance with the emerging studies that show the pivotal role of adenosine and adenosine receptors in bone tissue function and homeostasis (41). Studies have also reported functional dominance of A2bR in human osteoprogenitor cells (44). In addition, there is evidence suggesting the secretion of extracellular adenosine by osteoprogenitor cells and MSCs and the influence of extracellular adenosine in promoting osteoblast functions (44, 45). MSCs undergoing osteogenic differentiation have been shown to exhibit transient up-regulation of A2bR (46). Similarly, overexpression of A2bR has been shown to induce osteogenic differentiation of MSCs (46), whereas blocking A2bR activity using pharmacological agents has been shown to inhibit their osteogenic commitment (47). Our recent studies show the involvement of extracellular adenosine (ATP-metabolized) and A2bR in mineralized matrix–mediated osteogenic differentiation of hMSCs (19) while inhibiting their adipogenesis (20). These in vitro findings are supported by the recent in vivo studies involving A2bR knockout mice, which exhibited lower bone development and delayed fracture repair compared to wild-type mice (48).
The osteogenic cells derived from hiPSCs by using exogenous adenosine resulted in 3D bone tissue formation in vitro, even in the absence of any osteoinductive molecules in culture medium. We choose macroporous PEGDA-co-A6ACA matrices as a 3D scaffold for both in vitro and in vivo bone tissue formation because our previous studies showed that they do not intrinsically have any osteoinductive function (16, 49). In addition to in vitro bone tissue formation, when implanted in vivo, the Ad-hiPSCs contributed to tissue repair through the regeneration of new bone tissue. The newly formed tissues resembled native bone in morphology and mineral density. Besides transplanted donor cells, the infiltrated host cells also contributed to bone tissue formation. However, as evident from the acellular control, the infiltration of host cells alone was not adequate to achieve homogeneous bone formation and complete healing of the defect.
Vascularization of the implant plays an important role in the survival of the transplanted cells and neobone tissue formation. The presence of a vascular network within the implant likely promoted the survival of the transplanted cells and also the recruitment of endogenous cells. The vascularization of the implant could be attributed to the macroporous structure of the PEGDA-co-A6ACA matrices that readily allowed the infiltration of host cells, including vascular endothelial cells (50, 51). Detection of TRAP-positive osteoclastic cells within the engineered bone could imply the initiation of bone resorption (52). It was previously reported that osteoblasts not only produce monocyte chemoattractant protein-1 (MCP-1) to recruit osteoclastic precursors but also express receptor activator of nuclear factor κB ligand (RANKL) to activate osteoclastic cells (53). The copresence of bone-synthesizing osteoblasts and bone-resorbing osteoclasts within the newly regenerated bone tissue suggests plausible physiological remodeling of the tissue, a key feature of native bone.
In summary, this study demonstrates that adenosine alone can direct osteogenic differentiation of hPSCs in growth medium lacking any other osteoinductive factors. The adenosine-mediated osteogenic commitment of hPSCs involved A2bR. To our knowledge, this is the first demonstration of direct conversion of hPSCs into osteoblasts solely by a single small molecule. Furthermore, hiPSC-derived osteoblasts contributed to the repair of critical-sized bone defects through formation of de novo bone, which was integrated with surrounding native tissue without teratoma formation. A simple, cost-effective, and efficient osteogenic induction protocol involving adenosine will allow obtaining a large number of clinically viable therapeutic cells to treat critical bone defects.
MATERIALS AND METHODS
Maintenance of hPSCs
hESCs (HUES9 cell line) and hiPSCs (IMR90p18-iPS; WiCell Research Institute) were maintained as described elsewhere (43). Briefly, hPSCs were cultured on feeder layers of mitotically inactivated mouse embryonic fibroblasts using a medium consisting of KnockOut DMEM (Life Technologies, catalog no. 10829-018), 10% (v/v) KnockOut Serum Replacement (Life Technologies, catalog no. 10828028), 10% (v/v) human plasmanate (Talecris Biotherapeutics), 1% (v/v) nonessential amino acids, 1% (v/v) GlutaMAX, 1% (v/v) penicillin/streptomycin, and 55 μM 2-mercaptoethanol. hPSCs were passaged using Accutase (Millipore) at approximately 80% confluence. The fresh medium was supplemented with bFGF (30 ng/ml) (Life Technologies) and replenished daily.
Differentiation of hPSCs
hPSCs were plated onto culture-grade coverslips (15 mm in diameter; Fisherbrand, catalog no. 1254582) coated with Matrigel (BD Biosciences, catalog no. 354277), following the manufacturer’s protocol (16). Matrigel coating was used to facilitate the adhesion of hPSCs. Briefly, Matrigel was diluted in chilled Dulbecco’s modified Eagle’s medium (DMEM) at a ratio of 1:86. The coverslips were incubated with the Matrigel solution at 4°C overnight and subsequently at 37°C for 1 hour. Pluripotent hPSCs were plated at a density of 10,000 cells/cm2 and cultured in maintenance medium at 37°C and 5% CO2 for 3 days to allow the attachment of the cells onto the coverslips. After 3 days of culture, the maintenance medium was gradually replaced with growth medium [high-glucose DMEM, 10% (v/v) fetal bovine serum (HyClone), 4 mM l-glutamine, and penicillin/streptomycin (50 U/ml)], growth medium supplemented with adenosine (30 μg/ml) (Sigma-Aldrich, catalog no. A4036), growth medium supplemented with adenosine (30 μg/ml) and 100 nM PSB 603 (Tocris Bioscience, catalog no. 3198), or osteogenic-inducing medium [growth medium containing 10 mM β-glycerophosphate (Calbiochem, catalog no. 35675), 50 μM ascorbic acid-2-phosphate (Sigma-Aldrich, catalog no. A8960), and 100 nM dexamethasone (Sigma-Aldrich, catalog no. D2915)]. Each medium was replenished every 2 days.
Reverse transcription polymerase chain reaction
hPSCs cultured under various medium conditions were examined for changes in the gene expression as a function of culture time. RNA was extracted from cell cultures (n = 3) using TRIzol according to the manufacturer’s instructions. For each sample, 1 μg of RNA was reverse-transcribed to complementary DNA (cDNA) using an iScript cDNA synthesis kit (Bio-Rad, catalog no. 170-8891). Real-time PCR reactions were run on ABI Prism 7700 Real-time PCR Cycler (Applied Biosystems). Human Osteogenesis PCR array (SABiosciences, catalog no. PAHS-026) was used to examine osteogenic differentiation of hiPSCs. In the case of PCR array, 84 genes were analyzed and their relative expressions were presented as a heat map. The colors of the heat map were scaled according to the relative expression of hiPSCs cultured under various medium conditions. Red color represents the highest expression, whereas green color represents the lowest expression. The color between red and green represents the intermediate expression level. For qPCR analysis of selective genes, SYBR Select Master Mix (Life Technologies, catalog no. 4472908) was mixed with various primers (GAPDH, RUNX2, OCN, SPP1, NANOG, A1R, A2aR, A2bR, and A3R). The primer sequences are listed in table S1. The expression of each target gene was normalized to that of corresponding GAPDH, a housekeeping gene. The expression levels of hPSCs cultured using various medium conditions were normalized to that of undifferentiated, pluripotent hPSCs and presented as fold expression.
Immunofluorescence staining of monolayer cultures
Monolayer cultures were fixed by using 4% (w/v) paraformaldehyde at 25°C for 10 min and washed with phosphate-buffered saline (PBS). The fixed cells were treated with blocking solution of 3% (w/v) bovine serum albumin and 0.1% (v/v) Triton-X in PBS at 25°C for 60 min. The treated cells were incubated with a primary antibody against osteocalcin (1:100, mouse; Santa Cruz Biotechnology, catalog no. sc-74495) in blocking solution at 4°C for 16 hours and washed with PBS. The cells were incubated with a secondary antibody (goat anti-mouse; 1:250, Life Technologies, Alexa Fluor 568) and phalloidin (1:100; Life Technologies, Alexa Fluor 488) in the blocking solution at 25°C for 60 min and washed with PBS. The nuclei were stained by using Hoechst 33342 (2 μg/ml) (Life Technologies, catalog no. H1399) in PBS at 25°C for 10 min and washed with PBS. The stained cells were mounted onto glass slides and visualized by using a fluorescence microscope (Carl Zeiss, Axio Observer.A1). The images were acquired in a linear mode using the same exposure time for all groups. The background of images was identically subtracted for all images by using ImageJ software by applying a rolling ball algorithm with a rolling ball radius of 750 pixels.
Alizarin Red S staining of monolayer cultures
The hPSC cultures were fixed with 4% (w/v) paraformaldehyde for 10 min and washed with PBS. The cells were stained with 2% (w/v) Alizarin Red S Solution (Sigma-Aldrich, catalog no. A5533; pH 4.2) for 10 min and washed with PBS. The stained cells were visualized using a microscope under H-filter in color mode.
Preparation of macroporous matrices
PEGDA-co-A6ACA macroporous hydrogels were prepared by either poly(methyl methacrylate) (PMMA) leaching method (16) or cryogelation (54). Synthesis of PEGDA [Mn (number-average molecular weight) = 3.4 kD] and A6ACA was carried out as previously reported (14, 55). Briefly, a cylindrical polypropylene mold (5 mm in diameter) was packed with 30 mg of PMMA microspheres (150 to 180 μm in diameter; Bangs Laboratories, catalog no. BB05N). Approximately, 18 μl of precursor solution containing 20% (w/v) PEGDA, 0.5 M A6ACA in 0.5 N NaOH, and 0.3% (w/v) Irgacure was added into the PMMA-packed mold and photopolymerized by using ultraviolet light (λ = 365 nm) for 10 min. PMMA beads were leached out by using acetone for 3 days and hydrated in deionized (DI) water to yield macroporous hydrogels measuring 7 mm in diameter and 2 mm in thickness.
In the case of cryogelation to create macroporous hydrogels, 75 μl of chilled precursor solution containing 20% (w/v) PEGDA, 0.5 M A6ACA in 0.5 N NaOH, 0.5% (w/v) ammonium persulfate, and 0.2% (v/v) N,N,N′-N′-tetramethylethylenediamine was dispensed into a chilled polystyrene dish. A chilled coverslip of 15 mm in diameter was placed onto the precursor solution and polymerized at −20°C for 24 hours. PBS was added into the dish to yield macroporous hydrogels by thawing ice crystals. The resultant macroporous hydrogels were reduced to a size of 4 mm in diameter and 0.7 mm in thickness before they were sterilized in 70% ethanol for 3 hours and washed with PBS for 5 days.
Scanning electron microscopy
Scanning electron microscopy (SEM) imaging was carried out to examine the morphology of the macroporous hydrogels. Briefly, the matrices were rinsed with DI water, cut into thin slices, and subjected to flash freezing and lyophilization for 24 hours. The sliced samples were iridium-coated for 7 s using a sputter coater (Emitech, K575X) and imaged by using a SEM machine (Philips XL30 ESEM).
Cell seeding and in vitro culture
To evaluate the bone-forming ability of hiPSC-derived cells, the hiPScs were cultured in growth medium supplemented with adenosine (30 μg/ml) for 21 days, and these cell derivatives termed as Ad-hiPSCs were seeded into the macroporous matrices. hiPSCs cultured under identical conditions in growth medium lacking exogenous adenosine (d-hiPSCs) were used as a control. Before cell (Ad-hiPSCs and d-hiPSCs) seeding, sterile macroporous matrices (7-mm diameter by 2-mm thickness) were incubated with 1.5 ml of growth medium at 37°C for 24 hours. Approximately 20 μl of the medium was removed, and 20 μl of the cell suspension containing 1.5 million cells was seeded into the matrices. The cell-laden matrices were incubated at 37°C for 2 hours to allow cell infiltration and cultured in 1.5 ml of the growth medium supplemented with 10 mM β-glycerophosphate and 50 μM ascorbic acid-2-phosphate but devoid of dexamethasone (56, 57). The medium was replenished every 2 days.
Live-dead assay
To examine the viability and distribution of the seeded cells within the macroporous matrices, live-dead staining was conducted. At 3 days after seeding, the samples were cut into thin slices and washed with PBS. The slices were incubated in a staining solution containing 0.05% (v/v) green fluorescent calcein-AM and 0.2% (v/v) red fluorescent ethidium homodimer-1 in DMEM at 37°C for 30 min according to the manufacturer’s instructions (Life Technologies, catalog no. L-3224). The stained slices were washed with PBS and imaged using a fluorescence microscope (Carl Zeiss, Axio Observer.A1).
Critical-sized cranial bone defects
For in vivo study, 12 immunodeficient mice (NOD.CB17-Prkdcscid/J, approximately 3 months old) were used with the approval of the Institutional Animal Care and Use Committee at the University of California, San Diego. Before surgery, the mice were administered with ketamine (Ketaset, 100 mg/kg) and xylazine (AnaSed, 10 mg/kg) through an intraperitoneal injection. The anesthetized mice were subjected to a 15-mm-long incision on their skin of parietal skull bones, and cranial surface was gently scrapped to remove the periosteum. The exposed cranial bone (4 mm in diameter) on the underlying dura mater was removed in full thickness to create critical-sized bone defects (58). Two cranial bone defects were created in the left and right parietal skull bones in each mouse. Acellular and Ad-hiPSC–laden macroporous matrices (4 mm in diameter and 0.7 mm in thickness) were cultured for 1 week in vitro and implanted into the defects. Sham controls that received no treatment were also included. The skin of parietal skull bones was then carefully sutured. After the surgery, the mice were housed in separate cages and sacrificed after 4 and 16 weeks of implantation.
Microcomputed tomography
μCT was used to examine the hard tissue formation both in vitro and in vivo. The in vitro cultured constructs and parietal skull bones including calvarial bone defects were fixed in 4% paraformaldehyde at 4°C for 5 days and washed with PBS. The fixed samples were tightly packed between styrofoam spacers in 50-ml centrifuge tubes containing wet tissue wipes to maintain the hydration of the samples. The samples were scanned by using a SkyScan 1076 μCT scanner (Bruker; pixel resolution, 9 μm; 50 kV, Al filter). The scanned images were reconstructed by using NRecon software (SkyScan, Bruker). The reconstructed images were assembled into 3D models by using CT Analyser software (SkyScan, Bruker) or converted into 2D cross-sectional images in coronal, transaxial, and sagittal planes of the matrices by using DataViewer software (SkyScan, Bruker). To selectively account for positive signals from the calcified tissue, background signals were uniformly subtracted from the reconstructed images of all groups by applying a thresholding range of 90 to 255. Using 3D models, mineral density within the matrices was quantified and presented as a percentage of bone volume per total volume. Rotational views (360°) of 3D models were generated into movies at approximately 30° rotation per second by using CTVol software (SkyScan, Bruker).
Histological analysis
To perform histochemical analyses, paraffin-embedded sections were prepared as previously reported (16). Briefly, the in vitro cultured matrices and parietal skull bones, including the neobone tissue within the calvarial bone defects, were fixed in 4% paraformaldehyde at 4°C for 5 days and washed with PBS for 6 hours. The fixed samples were decalcified in 10% EDTA (pH 7.3) at 4°C for 2 weeks and washed with PBS for 6 hours. The decalcified samples were dehydrated, equilibrated in CitriSolv, and incubated in a molten mixture of 95% (w/w) paraffin and 5% (w/w) poly(ethylene-co-vinyl acetate) (Sigma-Aldrich, catalog no. 437220) at 70°C under vacuum for 24 hours. The paraffin-embedded samples were sliced into 10-μm-thick sections using a rotary microtome (Leica, RM2255). Before staining, the sections were deparaffinized in CitriSolv for 15 min and rehydrated.
Histochemical staining and histomorphometry
For H&E staining, the rehydrated sections were incubated in hematoxylin solution (Ricca, catalog no. 3536-16) for 7 min and washed with DI water. The sections were then immersed in an Eosin-Y solution (Richard-Allan Scientific, catalog no. 7111) for 1 min and washed with DI water. The stained sections were dehydrated and imaged under H-filter in color mode. The stained images were stitched to acquire a continuous view of whole calvarial bone defects integrated with the surrounding native bone tissue.
For histomorphometrical analysis, six representative H&E-stained images were selected (n = 6), and the areal sum of the engineered bone resembling the morphology of native bone as well as the defect area were quantified by using ImageJ. The areal density of the newly formed bone was presented as the percentage of bone area per defect area.
For TRAP staining, a staining solution was prepared by following the manufacturer’s protocol (Acid Phosphatase kit, Sigma-Aldrich, catalog no. 387A). Briefly, 50 μl of Fast Garnet GBC base solution and 50 μl of sodium nitrite solution were mixed. After 2 min, the mixture was added into 4.5 ml of DI water prewarmed to 37°C. To this solution, 50 μl of Naphthol AS-Bl phosphate solution, 200 μl of acetate solution, and 100 μl of tartrate solution were sequentially added to yield the staining solution. The rehydrated sections were incubated in the staining solution at 37°C for 1 hour while protected from light. The stained sections were washed with DI water, dehydrated, and imaged under H-filter in color mode.
Immunohistochemical staining
The rehydrated sections were treated with proteinase K (20 μg/ml) (Invitrogen, catalog no. 100005393), dissolved in a mixture of 95% (v/v) TE buffer [50 mM tris-HCl, 1 mM EDTA, and 0.5% (v/v) Triton X-100; pH 8.0] and 5% (v/v) glycerol at 37°C for 15 min and washed with PBS. The treated sections were immersed in a blocking solution containing 3% (v/v) normal goat serum and 0.1% (v/v) Triton X-100 in PBS at 25°C for 1 hour and incubated with primary antibodies against osteocalcin (1:100, rabbit; Abcam, catalog no. ab93876) in the blocking solution at 4°C for 16 hours. The sections were washed with PBS, treated with 3% (v/v) hydrogen peroxide for 7 min, and washed with PBS. The treated sections were incubated with a horseradish peroxidase–conjugated secondary antibody (1:200, donkey anti-rabbit; Jackson ImmunoResearch, catalog no. 711-035-152) in the blocking solution at 25°C for 60 min and washed with PBS. The sections were developed in 3-3′ diaminobenzidine substrate solution (Vector Laboratories, catalog no. SK-4100) for 3 min. The stained sections were washed with PBS, dehydrated, and imaged under H-filter in color mode. The stained images were stitched to show the continuous view of whole calvarial bone defects integrated with the surrounding native bone.
Immunohistofluorescence staining
The rehydrated sections were treated with proteinase K (20 μg/ml) in TE buffer at 37°C for 15 min and washed with PBS. The treated sections were permeabilized in 0.1% (v/v) Triton-X in PBS at 25°C for 4 min and washed with PBS. The permeabilized sections were further treated in sodium borohydride (2.5 mg/ml) (Fisher Scientific, catalog no. 16940-66-2) in 50% EtOH at 25°C for 30 min and washed with PBS. The treated sections were immersed in a blocking solution of 10% (v/v) donkey serum in PBS at 25°C for 60 min. The sections were incubated in primary antibodies against lamin A/C (1:100, rabbit; Abcam, catalog no. ab108595), osteocalcin (1:100, mouse; Santa Cruz Biotechnology, catalog no. sc-74495), or CD31 (1:100, goat; Santa Cruz Biotechnology, catalog no. sc-1506) in the blocking solution at 4°C for 16 hours and washed with PBS. The sections were then incubated with secondary antibodies raised against rabbit (1:200, goat anti-rabbit; Life Technologies, Alexa Fluor 488, catalog no. A11008), mouse (1:200, goat anti-mouse; Life Technologies, Alexa Fluor 568, catalog no. A11004), or goat (1:200, donkey anti-goat; Jackson ImmunoResearch, allophycocyanin, catalog no. 705-136-147), as well as Hoechst 33342 (2 μg/ml) (Life Technologies, catalog no. H1399) in the blocking solution at 25°C for 60 min and washed with PBS. The stained sections were imaged using a fluorescence microscope (Olympus FluoView FV1000). All images were taken using the same exposure time. The background was uniformly subtracted from all images using ImageJ software by applying a rolling ball algorithm (rolling ball radius, 750 pixels).
Statistical analysis
All experiments were repeated independently at least twice in addition to the triplicates used in each experiment. Using GraphPad Prism 5, statistical significances were considered for P values less than 0.05. Two-tailed Student’s t test was used to compare two groups at the same time point. One-way ANOVA including Tukey-Kramer post hoc test was used to compare multiple groups at the same time point. Two-way ANOVA including Bonferroni post hoc test was used to compare multiple groups across different time points.
Supplementary Material
Acknowledgments
We thank P. Nayak for valuable discussions. Funding: This work was supported by NIH (grant 1 R01 AR063184-01A1). Author contributions: H. Kang, Y.-R.V.S., and S.V. designed the experiments; H. Kang, Y.-R.V.S., M.N., and H. Kabra performed the experiments; H. Kang, Y.-R.V.S., and S.V. analyzed the data; and H. Kang, Y.-R.V.S., and S.V. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/8/e1600691/DC1
fig. S1. Attachment and growth of hPSCs cultured under various medium conditions.
fig. S2. Exogenous adenosine induced osteogenic differentiation of hESCs.
fig. S3. Exogenous adenosine induced expressions of osteoblastic markers for hiPSCs.
fig. S4. Adenosine-induced osteogenic differentiation of hESCs uses A2bR.
fig. S5. In vitro hard tissue–forming ability of hiPSC-derived cells.
fig. S6. Minimal hard tissue formation for Ad-hiPSC–laden matrices before implantation.
fig. S7. hiPSC-derived osteoblasts (Ad-hiPSCs) facilitate the repair of critical-sized bone defects.
fig. S8. Donor hiPSC–derived cells (Ad-hiPSCs) contribute to the regeneration of vascularized neobone tissue.
table S1. List of primer sequences used for qPCR analysis.
movie S1. Minimal calcification from d-hiPSCs in vitro.
movie S2. Dense and homogeneous calcified tissue formation from Ad-hiPSCs in vitro.
REFERENCES AND NOTES
- 1.Pera M. F., Trounson A. O., Human embryonic stem cells: Prospects for development. Development 131, 5515–5525 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Wobus A. M., Boheler K. R., Embryonic stem cells: Prospects for developmental biology and cell therapy. Physiol. Rev. 85, 635–678 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Amabile G., Meissner A., Induced pluripotent stem cells: Current progress and potential for regenerative medicine. Trends Mol. Med. 15, 59–68 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Hwang N. S., Varghese S., Elisseeff J., Controlled differentiation of stem cells. Adv. Drug Deliv. Rev. 60, 199–214 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murry C. E., Keller G., Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell 132, 661–680 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Discher D. E., Mooney D. J., Zandstra P. W., Growth factors, matrices, and forces combine and control stem cells. Science 324, 1673–1677 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lebkowski J. S., Gold J., Xu C., Funk W., Chiu C.-P., Carpenter M. K., Human embryonic stem cells: Culture, differentiation, and genetic modification for regenerative medicine applications. Cancer J. 7 (Suppl. 2), S83–S93 (2001). [PubMed] [Google Scholar]
- 8.Lutolf M. P., Gilbert P. M., Blau H. M., Designing materials to direct stem-cell fate. Nature 462, 433–441 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schugar R. C., Robbins P. D., Deasy B. M., Small molecules in stem cell self-renewal and differentiation. Gene Ther. 15, 126–135 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Li W., Ding S., Small molecules that modulate embryonic stem cell fate and somatic cell reprogramming. Trends Pharmacol. Sci. 31, 36–45 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Brey D. M., Motlekar N. A., Diamond S. L., Mauck R. L., Garino J. P., Burdick J. A., High-throughput screening of a small molecule library for promoters and inhibitors of mesenchymal stem cell osteogenic differentiation. Biotechnol. Bioeng. 108, 163–174 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Levi B., Hyun J. S., Montoro D. T., Lo D. D., Chan C. K. F., Hu S., Sun N., Lee M., Grova M., Connolly A. J., Wu J. C., Gurtner G. C., Weissman I. L., Wan D. C., Longaker M. T., In vivo directed differentiation of pluripotent stem cells for skeletal regeneration. Proc. Natl. Acad. Sci. U.S.A. 109, 20379–20384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marolt D., Campos I. M., Bhumiratana S., Koren A., Petridis P., Zhang G., Spitalnik P. F., Grayson W. L., Vunjak-Novakovic G., Engineering bone tissue from human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 109, 8705–8709 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayala R., Zhang C., Yang D., Hwang Y., Aung A., Shroff S. S., Arce F. T., Lal R., Arya G., Varghese S., Engineering the cell–material interface for controlling stem cell adhesion, migration, and differentiation. Biomaterials 32, 3700–3711 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Phadke A., Shih Y.-R. V., Varghese S., Mineralized synthetic matrices as an instructive microenvironment for osteogenic differentiation of human mesenchymal stem cells. Macromol. Biosci. 12, 1022–1032 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Kang H., Wen C., Hwang Y., Shih Y.-R. V., Kar M., Seo S. W., Varghese S., Biomineralized matrix-assisted osteogenic differentiation of human embryonic stem cells. J. Mater. Chem. B Mater. Biol. Med. 2, 5676–5688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller P., Bulnheim U., Diener A., Lüthen F., Teller M., Klinkenberg E.-D., Neumann H.-G., Nebe B., Liebold A., Steinhoff G., Rychly J., Calcium phosphate surfaces promote osteogenic differentiation of mesenchymal stem cells. J. Cell. Mol. Med. 12, 281–291 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cameron K., Travers P., Chander C., Buckland T., Campion C., Noble B., Directed osteogenic differentiation of human mesenchymal stem/precursor cells on silicate substituted calcium phosphate. J. Biomed. Mater. Res. A 101A, 13–22 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Shih Y.-R. V., Hwang Y., Phadke A., Kang H., Hwang N. S., Caro E. J., Nguyen S., Siu M., Theodorakis E. A., Gianneschi N. C., Vecchio K. S., Chien S., Lee O. K., Varghese S., Calcium phosphate-bearing matrices induce osteogenic differentiation of stem cells through adenosine signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 990–995 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang H., Shih Y.-R. V., Varghese S., Biomineralized matrices dominate soluble cues to direct osteogenic differentiation of human mesenchymal stem cells through adenosine signaling. Biomacromolecules 16, 1050–1061 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsutsui H., Valamehr B., Hindoyan A., Qiao R., Ding X., Guo S., Witte O. N., Liu X., Ho C.-M., Wu H., An optimized small molecule inhibitor cocktail supports long-term maintenance of human embryonic stem cells. Nat. Commun. 2, 167 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen S., Do J. T., Zhang Q., Yao S., Yan F., Peters E. C., Schöler H. R., Schultz P. G., Ding S., Self-renewal of embryonic stem cells by a small molecule. Proc. Natl. Acad. Sci. U.S.A. 103, 17266–17271 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu S., Wurdak H., Wang J., Lyssiotis C. A., Peters E. C., Cho C. Y., Wu X., Schultz P. G., A small molecule primes embryonic stem cells for differentiation. Cell Stem Cell 4, 416–426 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Chen S., Borowiak M., Fox J. L., Maehr R., Osafune K., Davidow L., Lam K., Peng L. F., Schreiber S. L., Rubin L. L., Melton D., A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat. Chem. Biol. 5, 258–265 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Minami I., Yamada K., Otsuji T. G., Yamamoto T., Shen Y., Otsuka S., Kadota S., Morone N., Barve M., Asai Y., Tenkova-Heuser T., Heuser J. E., Uesugi M., Aiba K., Nakatsuji N., A small molecule that promotes cardiac differentiation of human pluripotent stem cells under defined, cytokine- and xeno-free conditions. Cell Rep. 2, 1448–1460 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Siller R., Greenhough S., Naumovska E., Sullivan G. J., Small-molecule-driven hepatocyte differentiation of human pluripotent stem cells. Stem Cell Reports 4, 939–952 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanke K., Masaki H., Saito T., Komiyama Y., Hojo H., Nakauchi H., Lichtler A. C., Takato T., Chung U.-i., Ohba S., Stepwise differentiation of pluripotent stem cells into osteoblasts using four small molecules under serum-free and feeder-free conditions. Stem Cell Reports 2, 751–760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen S., Zhang Q., Wu X., Schultz P. G., Ding S., Dedifferentiation of lineage-committed cells by a small molecule. J. Am. Chem. Soc. 126, 410–411 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Ladewig J., Mertens J., Kesavan J., Doerr J., Poppe D., Glaue F., Herms S., Wernet P., Kögler G., Müller F.-J., Koch P., Brüstle O., Small molecules enable highly efficient neuronal conversion of human fibroblasts. Nat. Methods 9, 575–578 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Wang H., Cao N., Spencer C. I., Nie B., Ma T., Xu T., Zhang Y., Wang X., Srivastava D., Ding S., Small molecules enable cardiac reprogramming of mouse fibroblasts with a single factor, Oct4. Cell Rep. 6, 951–960 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin T., Ambasudhan R., Yuan X., Li W., Hilcove S., Abujarour R., Lin X., Hahm H. S., Hao E., Hayek A., Ding S., A chemical platform for improved induction of human iPSCs. Nat. Methods 6, 805–808 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huangfu D., Maehr R., Guo W., Eijkelenboom A., Snitow M., Chen A. E., Melton D. A., Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 26, 795–797 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou P., Li Y., Zhang X., Liu C., Guan J., Li H., Zhao T., Ye J., Yang W., Liu K., Ge J., Xu J., Zhang Q., Zhao Y., Deng H., Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 341, 651–654 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Misra J., Mohanty S. T., Madan S., Fernandes J. A., Hal Ebetino F., Graham R., Russell G., Bellantuono I., Zoledronate attenuates accumulation of DNA damage in mesenchymal stem cells and protects their function. Stem Cells 34, 756–767 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X., Ding S., Ding Q., Gray N. S., Schultz P. G., A small molecule with osteogenesis-inducing activity in multipotent mesenchymal progenitor cells. J. Am. Chem. Soc. 124, 14520–14521 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Lo K. W.-H., Kan H. M., Ashe K. M., Laurencin C. T., The small molecule PKA-specific cyclic AMP analogue as an inducer of osteoblast-like cells differentiation and mineralization. J. Tissue Eng. Regen. Med. 6, 40–48 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Layland J., Carrick D., Lee M., Oldroyd K., Berry C., Adenosine: Physiology, pharmacology, and clinical applications. JACC Cardiovasc. Interv. 7, 581–591 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Daval J.-L., Nehlig A., Nicolas F., Physiological and pharmacological properties of adenosine: Therapeutic implications. Life Sci. 49, 1435–1453 (1991). [DOI] [PubMed] [Google Scholar]
- 39.Cunha R. A., Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: Different roles, different sources and different receptors. Neurochem. Int. 38, 107–125 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Collis M. G., The vasodilator role of adenosine. Pharmacol. Ther. 41, 143–162 (1989). [DOI] [PubMed] [Google Scholar]
- 41.Ham J., Evans B. A. J., An emerging role for adenosine and its receptors in bone homeostasis. Front. Endocrinol. 3, 113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karp J. M., Ferreira L. S., Khademhosseini A., Kwon A. H., Yeh J., Langer R. S., Cultivation of human embryonic stem cells without the embryoid body step enhances osteogenesis in vitro. Stem Cells 24, 835–843 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Chang C.-W., Hwang Y., Brafman D., Hagan T., Phung C., Varghese S., Engineering cell–material interfaces for long-term expansion of human pluripotent stem cells. Biomaterials 34, 912–921 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evans B. A. J., Elford C., Pexa A., Francis K., Hughes A. C., Deussen A., Ham J., Human osteoblast precursors produce extracellular adenosine, which modulates their secretion of IL-6 and osteoprotegerin. J. Bone Miner. Res. 21, 228–236 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Takedachi M., Oohara H., Smith B. J., Iyama M., Kobashi M., Maeda K., Long C. L., Humphrey M. B., Stoecker B. J., Toyosawa S., Thompson L. F., Murakami S., CD73-generated adenosine promotes osteoblast differentiation. J. Cell. Physiol. 227, 2622–2631 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gharibi B., Abraham A. A., Ham J., Evans B. A. J., Adenosine receptor subtype expression and activation influence the differentiation of mesenchymal stem cells to osteoblasts and adipocytes. J. Bone Miner. Res. 26, 2112–2124 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Costa M. A., Barbosa A., Neto E., Sá-e-Sousa A., Freitas R., Neves J. M., Magalhães-Cardoso T., Ferreirinha F., Correia-de-Sá P., On the role of subtype selective adenosine receptor agonists during proliferation and osteogenic differentiation of human primary bone marrow stromal cells. J. Cell. Physiol. 226, 1353–1366 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Carroll S. H., Wigner N. A., Kulkarni N., Johnston-Cox H., Gerstenfeld L. C., Ravid K., A2B adenosine receptor promotes mesenchymal stem cell differentiation to osteoblasts and bone formation in vivo. J. Biol. Chem. 287, 15718–15727 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phadke A., Hwang Y., Kim S. H., Kim S. H., Yamaguchi T., Masuda K., Varghese S., Effect of scaffold microarchitecture on osteogenic differentiation of human mesenchymal stem cells. Eur. Cell. Mater. 25, 114–129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wen C., Kang H., Shih Y.-R. V., Hwang Y., Varghese S., In vivo comparison of biomineralized scaffold-directed osteogenic differentiation of human embryonic and mesenchymal stem cells. Drug Deliv. Transl. Res. 6, 121–131 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Shih Y.-R., Phadke A., Yamaguchi T., Kang H., Inoue N., Masuda K., Varghese S., Synthetic bone mimetic matrix-mediated in situ bone tissue formation through host cell recruitment. Acta Biomater. 19, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teitelbaum S. L., Bone resorption by osteoclasts. Science 289, 1504–1508 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Matsuo K., Irie N., Osteoclast–osteoblast communication. Arch. Biochem. Biophys. 473, 201–209 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Hwang Y., Zhang C., Varghese S., Poly(ethylene glycol) cryogels as potential cell scaffolds: Effect of polymerization conditions on cryogel microstructure and properties. J. Mater. Chem. 20, 345–351 (2010). [Google Scholar]
- 55.Lin S., Sangaj N., Razafiarison T., Zhang C., Varghese S., Influence of physical properties of biomaterials on cellular behavior. Pharm. Res. 28, 1422–1430 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bellows C. G., Heersche J. N. M., Aubin J. E., Inorganic phosphate added exogenously or released from β-glycerophosphate initiates mineralization of osteoid nodules in vitro. Bone Miner. 17, 15–29 (1992). [DOI] [PubMed] [Google Scholar]
- 57.Cheng S. L., Yang J. W., Rifas L., Zhang S. F., Avioli L. V., Differentiation of human bone marrow osteogenic stromal cells in vitro: Induction of the osteoblast phenotype by dexamethasone. Endocrinology 134, 277–286 (1994). [DOI] [PubMed] [Google Scholar]
- 58.Cowan C. M., Shi Y.-Y., Aalami O. O., Chou Y.-F., Mari C., Thomas R., Quarto N., Contag C. H., Wu B., Longaker M. T., Adipose-derived adult stromal cells heal critical-size mouse calvarial defects. Nat. Biotechnol. 22, 560–567 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/8/e1600691/DC1
fig. S1. Attachment and growth of hPSCs cultured under various medium conditions.
fig. S2. Exogenous adenosine induced osteogenic differentiation of hESCs.
fig. S3. Exogenous adenosine induced expressions of osteoblastic markers for hiPSCs.
fig. S4. Adenosine-induced osteogenic differentiation of hESCs uses A2bR.
fig. S5. In vitro hard tissue–forming ability of hiPSC-derived cells.
fig. S6. Minimal hard tissue formation for Ad-hiPSC–laden matrices before implantation.
fig. S7. hiPSC-derived osteoblasts (Ad-hiPSCs) facilitate the repair of critical-sized bone defects.
fig. S8. Donor hiPSC–derived cells (Ad-hiPSCs) contribute to the regeneration of vascularized neobone tissue.
table S1. List of primer sequences used for qPCR analysis.
movie S1. Minimal calcification from d-hiPSCs in vitro.
movie S2. Dense and homogeneous calcified tissue formation from Ad-hiPSCs in vitro.