

The EphA2 receptor plays multiple roles in cancer through two distinct signaling mechanisms. In a novel cross-talk, the β2-adrenoceptor/cAMP/PKA axis can promote EphA2 pro-oncogenic, ligand-independent signaling, blocking cell repulsion induced by ligand-dependent signaling. PKA emerges as a third kinase, besides AKT and RSK, that can regulate EphA2.
Abstract
The EphA2 receptor tyrosine kinase plays key roles in tissue homeostasis and disease processes such as cancer, pathological angiogenesis, and inflammation through two distinct signaling mechanisms. EphA2 “canonical” signaling involves ephrin-A ligand binding, tyrosine autophosphorylation, and kinase activity; EphA2 “noncanonical” signaling involves phosphorylation of serine 897 (S897) by AKT and RSK kinases. To identify small molecules counteracting EphA2 canonical signaling, we developed a high-content screening platform measuring inhibition of ephrin-A1–induced PC3 prostate cancer cell retraction. Surprisingly, most hits from a screened collection of pharmacologically active compounds are agents that elevate intracellular cAMP by activating G protein–coupled receptors such as the β2-adrenoceptor. We found that cAMP promotes phosphorylation of S897 by protein kinase A (PKA) as well as increases the phosphorylation of several nearby serine/threonine residues, which constitute a phosphorylation hotspot. Whereas EphA2 canonical and noncanonical signaling have been viewed as mutually exclusive, we show that S897 phosphorylation by PKA can coexist with EphA2 tyrosine phosphorylation and block cell retraction induced by EphA2 kinase activity. Our findings reveal a novel paradigm in EphA2 function involving the interplay of canonical and noncanonical signaling and highlight the ability of the β2-adrenoceptor/cAMP/PKA axis to rewire EphA2 signaling in a subset of cancer cells.
INTRODUCTION
The Eph receptors are a large family of receptor tyrosine kinases with distinctive signaling abilities (Pasquale, 2005). Eph receptor canonical signaling, which is induced by ephrin ligands and depends on kinase activity, plays an important role in a variety of disease processes ranging from pathological forms of angiogenesis and inflammation to inhibition of tissue regeneration, exacerbation of neurodegenerative processes, and in some cases cancer progression (Boyd et al., 2014; Barquilla and Pasquale, 2015). Thus Eph receptor antagonists and kinase inhibitors can have useful therapeutic applications (Noberini et al., 2012a; Riedl and Pasquale, 2015).
Although several chemical compounds are known to inhibit Eph receptor signaling, the most promising compounds identified as antagonists of ephrin ligand binding have only moderate potency and selectivity (Barquilla and Pasquale, 2015; Tognolini and Lodola, 2015). Several kinase inhibitors targeting Eph receptors with high potency are also known but generally have poor selectivity (Chang et al., 2008; Karaman et al., 2008; Choi et al., 2009; Noberini et al., 2012a).
We therefore sought to develop a cell-based assay suitable for the high-throughput identification of Eph receptor inhibitors. This assay monitors “cell retraction”—a prototypical repulsive response induced by Eph receptor canonical signaling in response to ephrin stimulation (Miao et al., 2000; Huang et al., 2008; Astin et al., 2010). Cell-based high-content screens can circumvent drawbacks of high-throughput screens based on biochemical assays with purified proteins, which often identify artifactual pan-assay inhibitors and compounds difficult to develop for use in biological systems (Baell and Holloway, 2010; Baell and Walters, 2014). In addition, cell-based screens have the potential to identify compounds that inhibit Eph receptor function not only through direct mechanisms (such as inhibition of ephrin ligand binding or kinase activity) but also through indirect mechanisms (such as interference with downstream signaling pathways, including previously unknown ones).
Eph receptor kinase activity is induced by the binding of ephrins that are often located on the surface of neighboring cells and is thus initiated by cell–cell contact (Pasquale, 2005). Eph receptor–ephrin interaction and signaling usually lead to cell separation and retraction of the cell periphery. This repulsive cell retraction response plays a role in numerous physiological and disease processes, including axon guidance and tissue patterning during development and inhibition of tissue regeneration after injury (Pasquale, 2005, 2008). In cancer cells, Eph receptor repulsive responses can have different consequences on malignancy. In cases in which Eph receptors are expressed in cancer cells and ephrins in the surrounding normal tissues, cell–cell repulsion leads to contact inhibition of locomotion, thus limiting tumor expansion and metastatic dissemination (Clevers and Batlle, 2006; Astin et al., 2010; Pasquale, 2010). However, when both Eph receptors and ephrins are expressed by the tumor cells, their repulsive effects could facilitate the dissemination of cancer cells from the tumor mass (Sugiyama et al., 2013; Batson et al., 2014).
We chose EphA2 as the prototype Eph receptor to establish a high-content screening platform for the identification of inhibitors of canonical signaling. EphA2 is widely expressed in different cancer types and has been associated with a poor clinical prognosis (Landen et al., 2005; Pasquale, 2010; Biao-Xue et al., 2011; Tandon et al., 2011). EphA2 canonical signaling in cancer cells can have tumor-promoting or tumor-suppressing effects, depending on the cellular context (Pasquale, 2010). Among the tumor-promoting effects, EphA2 can mediate cancer cell dispersal from a tumor through cell–cell repulsion induced by ephrin-A ligands (Sugiyama et al., 2013; Batson et al., 2014), cell invasiveness through epithelial–mesenchymal transition and RhoA-dependent ameboid migration (Parri et al., 2009; Taddei et al., 2011), and increased cancer cell fitness through macropinocytosis (Choi et al., 2013; Commisso et al., 2013; Ha et al., 2014). In addition, EphA2 canonical signaling has been implicated in vasculogenic mimicry, tumor angiogenesis and other pathological forms of angiogenesis (Ogawa et al., 2000; Pasquale, 2010), inflammation, including neuroinflammation (Larson et al., 2008; Cercone et al., 2009; Thundyil et al., 2013; Hong et al., 2015), atherosclerosis (Funk et al., 2012; Jiang et al., 2014), and various types of infections (Chakraborty et al., 2012; Dutta et al., 2013; Kaushansky et al., 2015; Subbarayal et al., 2015).
We carried out an automated screen of a small library of drugs and other active compounds based on measuring inhibition of EphA2-dependent cell retraction, which uncovered a previously unsuspected cross-talk between EphA2 and the β2-adrenoceptor/cAMP/protein kinase A (PKA) signaling axis. In this cross-talk, EphA2 phosphorylation by PKA blocks ephrin-A1–induced cell retraction by potentiating EphA2 noncanonical signaling.
RESULTS
Screening of the Library of Pharmacologically Active Compounds identifies compounds that block EphA2-dependent cell retraction
To screen for inhibitors of EphA2 canonical signaling, we developed a cell-based, high-throughput assay to monitor cell retraction by measuring the “cytoplasmic area” in PC3 prostate cancer cells expressing farnesylated enhanced green fluorescent protein (EGFP-F; Supplemental Figure S1; see Materials and Methods for details on the assay). We chose PC3 cells for this assay because they have been extensively used to dissect EphA2 downstream signaling pathways involved in cell retraction (Miao et al., 2000; Parri et al., 2005, 2007; Huang et al., 2008; Taddei et al., 2009; Astin et al., 2010) and for the characterization of peptides and chemical compounds that activate or inhibit EphA2 canonical signaling (Noberini et al., 2008; Mitra et al., 2010; Petty et al., 2012; Castelli et al., 2015). Furthermore, EphA2 is the predominant Eph receptor that can be activated by ephrin-A1 Fc stimulation in PC3 cells (Fox et al., 2006; Noberini et al., 2012b; broadinstitute.org/ccle), causing dramatic retraction of their peripheral regions, cell rounding, and occupancy of a smaller area of the culture dish (Miao et al., 2000; Huang et al., 2008; Noberini et al., 2008). Therefore compounds that interfere with EphA2 canonical signaling should prevent PC3 cell retraction induced by ephrin-A1 Fc.
We screened the Library of Pharmacologically Active Compounds (LOPAC) collection, which comprises 1280 compounds with a known mechanism of action. Twenty-six compounds at concentrations of 2.5–5 μM restored average cell area to values ≥50% that of untreated cells in all three replicas of the screen and were thus considered hits (Supplemental Table S1). In addition, 5 borderline compounds gave cytoplasmic area values of ∼45% (D0676 and F6886) or <45% (S8442, C145, O100) in one of the three replicas. Examples of images analyzed for compounds considered and not considered hits are shown in Supplemental Figure S1A.
We verified the results with independent preparations of four hits representing different classes of compounds and of the five borderline compounds. Follow-up experiments included retraction assays carried out manually in 96-well plates using phalloidin staining to monitor the shape of parental PC3 cells or carried out in an automated manner with different compound concentrations using the same PC3–EGFP-F cells and imaging algorithm used for the screen. Both assays confirmed that the four hit compounds and two of the borderline compounds (D0676 and F6886) inhibit EphA2-mediated PC3 cell retraction (Figure 1 and Supplemental Table S1). These results demonstrate the robustness of the high-content assay we developed and its ability to effectively identify inhibitors of EphA2-mediated cell retraction.
FIGURE 1:

Inhibition of EphA2-dependent cell retraction by representative hit compounds. (A, B) “No compound” refers to cells treated with Fc control or ephrin-A1 Fc and DMSO. Other cells were pretreated for 40 min with the indicated compounds and then for 10 min with Fc or ephrin-A1 Fc in the presence of the compounds. Forskolin was used at 20 μM and dibutyryl cyclical AMP (dbcAMP) at 0.5 mM. The other compounds were used at 5 μM. NECA, 5′-(N-ethylcarboxamido) adenosine. Cells were stained for F-actin with fluorescent phalloidin (red), and nuclei were labeled with DAPI (blue). The inactive compound (O100, oxotremorine methiodide) did not inhibit retraction and rounding of the cells induced by ephrin-A1 Fc, similar to the control. All other compounds shown inhibited retraction, and the cells remained flat. (C) Dose–response curve for forskolin-mediated inhibition of cell retraction analyzed by automated image analysis in the 384-well format. Averages and SEs from quadruplicate measurements from two experiments carried out on different days.
Elevated cAMP blocks EphA2-mediated PC3 cell retraction
Most of the hits are agonists for β2-adrenergic receptors (adrenoceptors; Supplemental Table S1), which signal by activating adenylate cyclase, thus increasing intracellular cAMP levels. Some hits can also activate α2- and β1-adrenoceptors, and three of the hits activate the A2 adenosine receptor, all of which also signal by increasing cAMP levels. Hit compounds could in principle inhibit cell retraction induced by ephrin-A1 Fc through several mechanisms. For example, they could act as antagonists that prevent ephrin ligand binding to EphA2. Of interest, the α1-adrenoceptor antagonist doxazosin binds to the EphA2 ligand-binding domain, although it functions as an agonist (Petty et al., 2012). However, an enzyme-linked immunosorbent assay (ELISA) measuring interaction of EphA2 ligand-binding domain fused to alkaline phosphatase with immobilized ephrin-A1 Fc did not reveal significant antagonistic activity of several hit compounds of different classes even at concentrations of 20 μM (Supplemental Figure S2A).
An additional hit was forskolin, a compound that directly activates adenylate cyclase (Supplemental Table S1 and Figure 1, B and C). Therefore the nature of the hit compounds identified in the screen supports a role for cAMP in the inhibition of EphA2-mediated cell retraction. Indeed, treatment of PC3 cells with dibutyryl cAMP, a membrane-permeable cAMP analogue, also prevented cell retraction (Figure 1B). Thus the results of the screen reveal an unexpected and intriguing cross-talk between EphA2 and G protein–coupled receptors that elevate cAMP.
We used the hits forskolin and norepinephrine to further investigate the mechanism of EphA2 regulation by cAMP. The two compounds did not decrease EphA2 levels in unstimulated PC3 cells and did not substantially accelerate EphA2 degradation induced by the ephrin-A1 Fc ligand (Supplemental Figure S2, B and C). Forskolin also did not reduce EphA2 tyrosine phosphorylation (indicative of kinase activity) in cells treated with ephrin-A1 Fc (Supplemental Figure S2, B and C). In addition, forskolin did not detectably affect EphA2 endocytosis induced by ephrin-A1 Fc stimulation (Supplemental Figure S2D). Thus cAMP blocks PC3 cell retraction without down-regulating total or cell surface EphA2 and without inhibiting receptor canonical signaling, which depends on ephrin ligand-binding and kinase activity.
The β2-adrenoceptor/cAMP/PKA axis promotes EphA2 phosphorylation on S897 and several nearby residues
Given that EphA2 ligand binding, kinase activity, and protein stability were not affected by β2-adrenoceptor agonists or forskolin, we searched for other modifications in EphA2 that could explain the inhibition of cell retraction triggered by these compounds. Of interest, mass spectrometry experiments revealed that forskolin promotes phosphorylation of multiple serine/threonine residues in the linker segment connecting the kinase and SAM domains, markedly increasing the number of doubly phosphorylated peptides detected (Figure 2, A and B, and Supplemental Table S2). The EphA2 kinase–SAM linker segment comprises 20 amino acids (Seiradake et al., 2010), six of which (T883, S892, S897, T898, S899, and S901) could be phosphorylated. Analysis of the PhosphoSitePlus database (phosphosite.org) confirmed that five of the six residues form a cluster of phosphorylated residues (phosphorylation hotspot), whereas phosphorylation of T883 has not been documented (Figure 2C).
FIGURE 2:

Forskolin increases phosphorylation of S897 and other hotspot sites in the EphA2 kinase–SAM domain linker segment. (A) Number of peptides (spectral counts) from the EphA2 kinase–SAM linker segment identified by mass spectrometry as containing the indicated phosphosite in control and forskolin-treated PC3 cells. The sequence of the two peptides containing hotspot residues is shown at the top, with the assigned phosphorylated residues in bold. (B) Left, number of peptides from A with one or two phosphosites, illustrating the dramatic increase in doubly phosphorylated peptides. Right, total number of EphA2 peptides detected, which was similar in control and forskolin-treated cells, indicating that similar total EphA2 levels were analyzed. (C) Number of studies in which each hotspot phosphosite was identified, which provides an indication of the abundance of the phosphosite.
Phosphorylation of S897 is critical for the EphA2 noncanonical signaling mechanism that promotes cancer cell malignancy (Miao et al., 2009, 2015b; Paraiso et al., 2015). S897 is part of an RXXS motif (where R is arginine, X is any residue, and S is S897), which can be phosphorylated by the basophilic AGC kinases (Rust and Thompson, 2011; scansite3.mit.edu). AKT has indeed been reported to phosphorylate EphA2 S897 in many cancer cell lines, including PC3 cells (Miao et al., 2009). However, we did not detect substantially increased AKT phosphorylation on the T308 and S473 regulatory sites in PC3 cells treated with forskolin (Figure 3, A and B). In addition, forskolin increased EphA2 S897 phosphorylation even in the presence of the phosphoinositide 3 (PI3) kinase inhibitor wortmannin, which completely abolished AKT phosphorylation (Figure 3A). Taken together, these results imply that forskolin treatment does not increase S897 phosphorylation in PC3 cells by enhancing AKT activation. Ribosomal S6 kinase (RSK), another AGC kinase, has also been reported to phosphorylate EphA2 on S897 in various cancer cell lines downstream of the activated ERK pathway and in response to inflammatory cytokines (Moritz et al., 2010; Zhou et al., 2015). However, we detected only very low ERK1/2 and RSK1/2/3 phosphorylation (at sites indicative of activation) in PC3 cells, and RSK phosphorylation was not increased by forskolin (Figure 3B). Thus the two serine/threonine kinases known to phosphorylate S897 did not seem to be responsible for the effects of forskolin in PC3 cells.
FIGURE 3:

Forskolin and β2-adrenoceptor agonists increase EphA2 phosphorylation on S897 by PKA as well as phosphorylation on S901. (A) PC3 cells were treated with the PKA inhibitor H89 or the PI3 kinase inhibitor wortmannin (WTM) for 1 h and with forskolin (FSK) for the last 40 min of inhibitor treatment. Cell lysates were probed by immunoblotting with the indicated antibodies. (B) Phosphokinase array signals show the effects of 40-min forskolin treatment on the indicated phosphosites. Images of the duplicate spots on the arrays and exposure times for the autoradiographs are shown at the top. The histogram shows averages from quantification of the spots, with the error bars representing SDs. (C) In vitro kinase reactions with immunoprecipitated EphA2 WT (with and without the H89 PKA inhibitor) or S897A mutant. A control immunoprecipitate is also included. The immunoblot was probed with a phospho-S897–specific antibody and reprobed for EphA2. (D) PC3 cells transfected with empty vector or with constructs encoding PKA WT, the kinase-inactive PKA K72H mutant (PKA KD), or the PKA inhibitor peptide PKI were treated for 30 min with PBS as a control or 10 μM norepinephrine, and lysates were probed with the indicated antibodies. (E) PC3 cells were treated with vehicle DMSO as a control, 20 μM forskolin, or 10 μM indicated β-adrenoceptor agonists for 1 h. Where indicated, cells were treated with 1 μM β-adrenoceptor antagonist propanolol (prop.) for 1 h before the 1-h treatment with β-adrenoceptor agonists. EphA2 immunoprecipitates were probed by immunoblotting for phospho-S897 and reprobed for EphA2. (F) EphA2 WT immunoprecipitated from transiently transfected HEK293 cells was either left untreated or treated with calf-intestinal alkaline phosphatase (CIP) to dephosphorylate the receptor. The control immunoprecipitate was obtained with nonimmune rabbit immunoglobulin Gs. The immunoprecipitates and a cell lysate for comparison were probed with the phospho-S901–specific antibody, a phospho-S897–specific antibody, or an EphA2 antibody. (G) Lysates of HEK293 cells transiently transfected constructs encoding EphA2 WT and the indicated EphA2 mutants or empty vector as a control were probed with the phospho-S901–specific antibody, a phospho-S897–specific antibody, or an EphA2 antibody. (H) In vitro kinase reactions measuring the phosphorylation of immunoprecipitated EphA2 WT and the indicated phosphosite mutants incubated with recombinant CK1 or PKA. EphA2 immunoprecipitates incubated only with ATP without any recombinant kinase served as a control. The EphA2 immunoprecipitates were probed by immunoblotting for phospho-S901 and phospho-S897 and then reprobed for EphA2.
Another AGC kinase, protein kinase A (PKA), is directly regulated by cAMP and can also recognize the RXXS substrate motif containing S897 (Rust and Thompson, 2011). Indeed, previous proteomics studies showed that PKA (phosphorylated on T198 in the activation loop) colocalizes in protrusions at the leading edge of migrating cells together with EphA2 phosphorylated on S897 or both S897 and S901 (Wang et al., 2007). Supporting the hypothesis that forskolin-induced EphA2 S897 phosphorylation is mediated by PKA, the PKA kinase inhibitor N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H89) abolished the phosphorylation (Figure 3A). In addition, purified recombinant PKA phosphorylated S897 in an in vitro kinase assay, which was also inhibited by H89 (Figure 3C). Furthermore, overexpression of PKA wild type (WT) resulted in increased EphA2 phosphorylation on S897 in unstimulated as well as norepinephrine-stimulated cells, whereas expression of the PKA K72H kinase-dead mutant (Iyer et al., 2005) or the PKA inhibitor peptide PKI (Dalton and Dewey, 2006) inhibited EphA2 S897 phosphorylation induced by norepinephrine stimulation (Figure 3D). Additional experiments confirmed that different β2-adrenoceptor agonists that were identified as hits in the screen increase EphA2 S897 phosphorylation in PC3 cells and that the β-adrenoceptor antagonist propanolol greatly reduces this phosphorylation (Figure 3E). Taken together, these results implicate the β2-adrenoceptor/cAMP/PKA axis in the regulation of EphA2 S897 phosphorylation.
Because our mass spectrometry data and the PhosphoSitePlus database both suggest that S901 is an abundant hotspot phosphorylation site that is up-regulated by forskolin concomitant with the S897 phosphosite (Figures 2, A and C, and 3A), we generated phosphospecific antibodies to further investigate S901 phosphorylation (Figure 3, F and G). Of interest, immunoblotting revealed that phosphorylation on S901 is greatly reduced in the EphA2 S897A mutant expressed in HEK293 cells (Figure 3G), suggesting a mechanism by which S897 phosphorylation primes EphA2 for phosphorylation on S901. Thus S901 phosphorylation may depend on hierarchical phosphorylation by a kinase such as casein kinase 1 (CK1), which recognizes S/T residues located to the C-terminus of a phosphorylated S/T residue (Flotow et al., 1990; Flotow and Roach, 1991; Salazar and Hofer, 2009; scansite3.mit.edu). The EphA2 S897D mutant was only slightly more phosphorylated on S901 than the S897A mutant (Figure 3G), suggesting that a single aspartic acid poorly mimics the effect of phosphorylated S897, consistent with the reported specificity of CK1 (Flotow and Roach, 1991; Venerando et al., 2014) and with the inability of the EphA2 S897D mutant to stimulate cell migration as phosphoS897 EphA2 does (Miao et al., 2009). We confirmed that CK1 can phosphorylate EphA2 on S901 in an in vitro kinase reaction and found that the EphA2 S897A mutant displayed reduced phosphorylation on S901, suggesting that phosphorylation at S897 is a priming step required for S901 phosphorylation (Figure 3H). CK1 also appeared to weakly phosphorylate S897, despite the fact that this residue is not part of a typical CK1 substrate motif (Figure 3H). In summary, our data show that the β2-adrenoceptor/cAMP/PKA axis can increase phosphorylation of multiple S/T residues in the EphA2 kinase–SAM linker segment, which could play a role in counteracting cell retraction.
S897 phosphorylation by PKA is critical for inhibition of cell retraction induced by EphA2
We found that treatment of PC3 cells with the PKA-selective agonist 6-Benz-cAMP at a concentration of 200 μM partially inhibits ephrin-A1 Fc–induced cell retraction (Figure 4, A–C) and promotes S897 phosphorylation (albeit to a lesser extent than forskolin; Figure 4D), implicating PKA activation in inhibition of cell retraction.
FIGURE 4:

PKA activation inhibits EphA2-dependent cell retraction. (A) Representative images of phalloidin-labeled PC3 cells stimulated for 12 min with 0.5 μg/ml control Fc or ephrin-A1 Fc or pretreated for 40 min with 20 μM forskolin or 200 μM of the PKA agonist 6-Benz-cAMP before ephrin-A1 Fc stimulation. Scale bar, 50 μm. (B) Histogram showing average cell areas ± SE under the different conditions (693 cells/condition from three experiments in each of which 77 cells/well from three wells were counted). ****p < 0.0001 for the comparison with the ephrin-A1 Fc condition by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. (C) Cumulative distribution showing the relative frequencies of cells with areas smaller than indicated on the x-axis. (D) Immunoblot of PC3 cells treated with 6-Benz-cAMP or forskolin to assess the levels of EphA2 phosphorylation on S897.
To evaluate the importance of different EphA2 hotspot phosphosites in the inhibition of cell retraction by cAMP, we knocked down endogenous EphA2 in PC3 cell populations and replaced it with EphA2 mutants lacking the S892, S897, or S901 phosphosite. Remarkably, cells expressing the EphA2 S897A mutant were insensitive to the effects of forskolin and underwent extensive retraction after ephrin-A1 Fc stimulation even in the presence of forskolin, suggesting that S897 phosphorylation is critical for inhibition of cell retraction by cAMP/PKA (Figure 5). In contrast, ephrin-A1 Fc–induced retraction was completely inhibited by forskolin in PC3 cells expressing the S892A or S901A mutant, similar to the cells expressing EphA2 WT. Knockdown cells not reconstituted with EphA2 exhibited reduced retraction when stimulated with ephrin-A1 Fc, as expected, with some retraction still occurring both in the absence and in the presence of forskolin. This residual retraction is likely due to some remaining EphA2 and/or low expression of other EphA receptors responsive to ephrin-A1 Fc, such as possibly EphA4 (Astin et al., 2010; Seiradake et al., 2013). These results were confirmed using independently generated PC3 cell populations expressing WT or mutant EphA2 (Supplemental Figure S3). Thus phosphorylation of S897 by PKA, possibly in concert with phosphorylation of other hotspot sites primed by S897 phosphorylation, mediates the inhibitory effect of forskolin on PC3 cell retraction induced by ephrin-A1 stimulation.
FIGURE 5:

Inhibition of EphA2-dependent cell retraction by cAMP requires S897 phosphorylation. (A) Representative images of phalloidin-labeled, EphA2-knockdown PC3 cell populations infected with pLVX-IRES-Neo lentiviral vector (Vector) or lentivirus encoding WT EphA2 (WT) or the indicated EphA2 mutants. The cells, pretreated or not for 40 min with 20 μM forskolin, were stimulated for 12 min with 0.5 μg/ml control Fc or ephrin-A1 Fc. (B) Histogram showing average cell areas ± SE for the different conditions (160 cells/condition from an experiment in which 80 cells/well from two wells were measured). ****p < 0.0001 for the comparison of ephrin-A1 Fc–stimulated cells with the corresponding Fc-stimulated cells by one-way ANOVA followed by Sidak’s multiple comparisons test. (C) Immunoblot of PC3 cells transduced with empty lentiviral vector control and cells expressing the different EphA2 mutants to assess the levels of EphA2 expression and phosphorylation on S897 and S901.
EphA2 S897 phosphorylation by PKA is not mutually exclusive with ephrin-induced canonical signaling
Previous reports showed that ephrin stimulation of canonical signaling can rapidly decrease S897 phosphorylation, suggesting that EphA2 exists in two alternative signaling states with distinctive activities: tyrosine phosphorylated or phosphorylated on S897 (Miao et al., 2009, Miao et al., 2015b; Supplemental Figure S5A). Antibody phosphokinase array analysis of PC3 cells revealed a dramatic decrease in AKT phosphorylation on both T308 and S473 after 12-min ephrin-A1 Fc stimulation (Figure 6, A and B), in agreement with published data (Miao et al., 2009; Yang et al., 2011). Phosphorylation of a number of AKT substrates and downstream targets, including p70 S6 kinase, GSK3α/β, PRAS40, WNK1, and CREB (www.cellsignal.com/common/content/content.jsp?id=science-tables-akt-substrate), was also concomitantly reduced by ephrin-A1 Fc stimulation, as expected. AKT inhibition likely also contributes to the loss of EphA2 S897 phosphorylation induced by ephrin-A1 Fc stimulation in cancer cell lines with high AKT activation and low PKA activation and thus to the low EphA2 S897 phosphorylation when EphA2 is tyrosine phosphorylated in these cells (Miao et al., 2009; Yang et al., 2011).
FIGURE 6:

EphA2 canonical signaling rapidly inhibits AKT but not PKA. (A) PC3 cells stably expressing EphA2 WT or the S897A mutant were treated with 20 μM forskolin for 40 min and/or 0.5 μg/ml ephrin-A1 Fc for 12 min. Lysates were probed by immunoblotting with the indicated antibodies. (B) Quantification of pS897 and pS901 phosphorylation relative to total EphA2 levels and normalized to the value in forskolin-treated cells. Averages ± SE from four independent experiments. The pS897 and pS901 levels in cells stimulated with forskolin and ephrin-A1 Fc are not significantly different from those in cells stimulated only with forskolin by one-sample t test. (C) Normalized phosphokinase array signals show the effects of ephrin-A1 Fc stimulation, with or without forskolin treatment, on the indicated phosphosites. Images of the duplicate spots on the arrays and exposure times for the autoradiographs are shown at the top. The histogram shows averages from quantification of the spots, normalized to the control condition for each phosphosite, with the error bars representing SDs.
In contrast to AKT, we did not detect rapid loss of PKA activation after stimulation of EphA2 canonical signaling, based on the lack of effect of ephrin-A1 Fc on CREB S133 phosphorylation as well as EphA2 S897 phosphorylation in PC3 cells treated with forskolin (Figure 6). Thus EphA2 can be simultaneously phosphorylated on both S897 and tyrosine residues in forskolin-treated PC3 cells stimulated with ephrin-A1 Fc.
The cAMP/PKA signaling axis increases EphA2 S897 phosphorylation in a subset of cancer cell lines
Besides PC3 cells, cAMP/PKA signaling activated by forskolin can increase EphA2 S897 phosphorylation in other aggressive cancer cell lines examined, including the androgen-independent DU145 prostate cancer cell line and the pancreatic cancer cell lines PANC1 and MIA PaCa2 (Figure 7), consistent with the reported role of S897 phosphorylation in cancer malignancy (Miao et al., 2009, 2015b; Binda et al., 2012; Tawadros et al., 2012; Kawai et al., 2013; Paraiso et al., 2015; Zhou et al., 2015). Forskolin also modestly increased EphA2 S897 phosphorylation in the moderately aggressive BxPC3 pancreatic cancer cell line. In contrast, forskolin did not increase EphA2 S897 phosphorylation in less aggressive cell lines such as the androgen receptor–positive RWPE2 and LNCaP prostate cancer cell lines (the latter engineered to express EphA2), the Capan2 and PL45 pancreatic cancer cell lines, and immortalized nontumorigenic prostate epithelial cell lines (RWPE1 and BPH1; Figure 7B). Forskolin also did not increase the high EphA2 S897 phosphorylation observed in the aggressive Hs766T and AsPC1 pancreatic cancer cell lines. Control experiments in which AKT was inactivated by treatment with the PI3 kinase inhibitor wortmannin confirmed that the observed increase in EphA2 S897 phosphorylation induced by forskolin was independent of AKT (Supplemental Figure S4). In addition, forskolin promoted phosphorylation of the PKA substrate CREB, consistent with inducing PKA activation, even in cells in which EphA2 S897 phosphorylation was not increased. We also used epidermal growth factor (EGF), whose downstream signaling pathways are known to promote EphA2 S897 phosphorylation (Miao et al., 2009), to confirm that the S897 phosphosite can be regulated through PKA-independent mechanisms in cell lines that are insensitive to forskolin. Thus our findings indicate that PKA activation can promote EphA2 S897 phosphorylation in a variety of cancer cell lines, although this signaling connection might not be operational in immortalized prostate and pancreatic epithelial cells and in some of the cancer cell lines.
FIGURE 7:
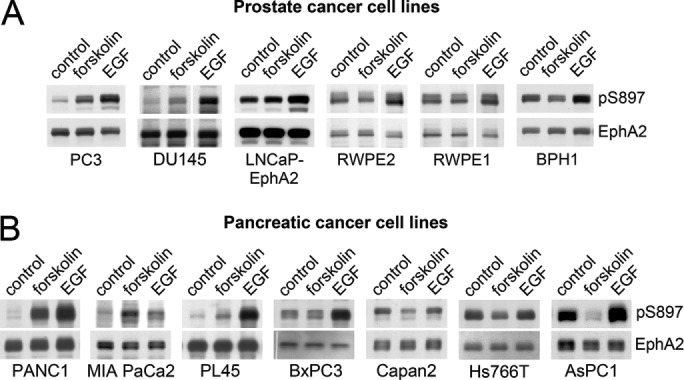
PKA phosphorylates EphA2 on S897 in a subset of cancer cells. (A) Prostate cancer and immortalized prostate epithelial cell lines and (B) pancreatic cancer cell lines were treated with vehicle DMSO as a control, 20 μM forskolin, or 10 ng/ml EGF for 20 min. Cell lysates were probed by immunoblotting with the indicated antibodies. pAKT indicates AKT phosphorylated on S473; pCREB indicates CREB phosphorylated on S133. A vertical gap indicates that irrelevant intervening lanes from the blot were removed. Additional control lanes and immunoblots with additional control antibodies are shown in Supplemental Figure S4.
DISCUSSION
We developed a cell-based, high-throughput screening platform that takes advantage of ephrin-A1–induced morphological changes in PC3 cells and allowed us to identify inhibitors of cell retraction induced by EphA2 canonical signaling. The compounds identified do not interfere with ephrin ligand binding, EphA2 kinase activity, or EphA2 endocytosis and do not promote EphA2 degradation. Instead, they function by elevating intracellular cAMP. Neutralization of the EphA2-repulsive effects by cAMP requires EphA2 phosphorylation on S897, since forskolin did not prevent the retraction of PC3 cells expressing the nonphosphorylatable EphA2 S897A mutant. This mutant induces normal cell retraction through canonical signaling but is incapable of mediating the opposing effects of noncanonical signaling, which depend on S897 phosphorylation. It will be interesting to investigate whether the effect of the S897A mutation on retraction also depends on concomitant impaired phosphorylation of S901 and possibly other hotspot sites or whether loss of only S897 phosphorylation is sufficient to restore cell retraction in the presence of forskolin.
The precise mechanism through which phosphorylation of S897, possibly in conjunction with phosphorylation of other hotspot sites, blocks cell retraction mediated by EphA2 canonical signaling remains to be elucidated. For example, EphA2 noncanonical signaling through S897 phosphorylation can increase RAC1 activity and cell protrusions through Ephexin4 and RhoG (Hiramoto-Yamaki et al., 2010), which could oppose RhoA-induced cell retraction (Miao et al., 2005; Astin et al., 2010). Regardless of its precise mechanism of action, EphA2 S897 phosphorylation might block cell retraction by acting in concert with other signaling events triggered by high cAMP levels in PC3 cells. cAMP can signal through two main pathways—PKA and the EPAC family of RAP1 exchange factors (Cheng et al., 2008; Gloerich and Bos, 2010). PKA-dependent regulation of a number of actin cytoskeleton regulatory proteins could contribute to cell spreading and inhibition of cell retraction (Howe, 2004, 2011; McKenzie et al., 2011). For example, PKA can phosphorylate and inactivate RhoA, which could both decrease actomyosin contractility and stabilize microtubules (Howe, 2004; Batson et al., 2014). PKA can also increase RAC1/CDC42 activation and cell protrusion as well as decrease myosin light chain phosphorylation and actomyosin contraction through several mechanisms (Howe, 2004; Howe et al., 2005). Although RAP1-induced integrin activation can inhibit EphA receptor–mediated cell retraction (Richter et al., 2007; Yang et al., 2011), a less critical role of RAP1 in forskolin-induced inhibition of PC3 cell retraction would be predicted. This is because PKA activated by cAMP can phosphorylate RAP1, decreasing its plasma membrane localization and activation (Takahashi et al., 2013), which would dampen possible effects of EPACs. In addition, EphA2 canonical signaling can down-regulate RAP1 activity in PC3 cells by inhibiting the C3G exchange factor and might also activate RAP1 GTPase-activating proteins (Richter et al., 2007; Huang et al., 2008).
Although AKT was originally reported as the predominant kinase responsible for EphA2 phosphorylation on S897, we report here that PKA can also robustly phosphorylate this residue. PKA-dependent phosphorylation of S897 is not surprising, given the similar substrate motifs recognized by PKA and AKT (Rust and Thompson, 2011). Indeed, recent evidence also shows that other AGC kinases, such as RSK, can phosphorylate EphA2 on S897, for example, downstream of transforming growth factor β or the activated ERK mitogen-activated protein kinase pathway (Moritz et al., 2010; Zhou et al., 2015). Thus multiple signaling pathways can converge on the regulation of EphA2 noncanonical signaling through S897 phosphorylation.
Previous reports showed that EphA2 S897 phosphorylation occurs independently of ephrin binding and that in some cells, tyrosine and S897 phosphorylation characterize two separate EphA2 signaling forms, presumably because EphA2 canonical signaling can rapidly inactivate AKT (Supplemental Figure S5A; Miao et al., 2009, 2015b; Yang et al., 2011). In contrast, our data show that when S897 is phosphorylated by a kinase that is not inactivated by EphA2, such as PKA, then noncanonical signaling can coexist with canonical signaling and block at least some of its functional effects (Supplemental Figure S5B). This represents a new paradigm in EphA2 signaling function.
We found that cAMP increases EphA2 phosphorylation on not only the previously characterized S897 but also three other nearby residues that are part of a phosphorylation hotspot in the EphA2 kinase–SAM linker segment. Of interest, phosphorylation of all five hotspot sites has been detected by mass spectrometry, often with concomitant phosphorylation of two or three sites in the same peptide (Figure 3C; www.phosphosite.org). Thus phosphorylation of the EphA2 kinase–SAM linker segment may serve to integrate coincident signals from different serine/threonine kinases. Because AGC kinases have similar substrate recognition motifs, additional kinases of this group might also phosphorylate S897 (scansite3.mit.edu). Furthermore, our data suggest that EphA2 S897 phosphorylation primes EphA2 for phosphorylation on S901 by CK1 and possibly other serine/threonine kinases with similar substrate specificity (Flotow and Roach, 1991; Venerando et al., 2014). Thus S897 phosphorylation appears to control at least one other hotspot phosphosite, and it will be interesting to investigate whether it can control others. This could explain how cAMP can increase phosphorylation of hotspot sites that do not conform to a PKA substrate motif. Of the five hotspot phosphosites, phospho-S897 appears to be the functionally most important one for regulation of PC3 cell retraction. However, other hotspot phosphosites may be required together with phospho-S897 to block cell retraction, whereas only a single phosphosite could be ineffective. Alternatively, other hotspot phosphosites may serve to reinforce/prolong the effects of S897 phosphorylation or confer additional, yet-unknown functional properties to EphA2.
Of interest, the results of our high-content screen reveal a previously unknown cross-talk between EphA2 and G protein–coupled receptors that stimulate adenylyl cyclase, thus elevating intracellular cAMP. Rewiring of EphA2 signaling by β2-adrenoceptors has potentially important translational implications since β2-adrenoceptors are present in many types of cancer cells and cells of the tumor microenvironment (Cole and Sood, 2012; Cole et al., 2015; Hanoun et al., 2015). β2-Adrenoceptors in tumors could be activated by stress hormones such as epinephrine (adrenaline) released in the circulation by the adrenal gland and norepinephrine (noradrenaline) released locally by sympathetic nerves innervating tumor tissue (Hassan et al., 2014; Cole et al., 2015; Hanoun et al., 2015). In fact, sympathetic nerves have been linked to malignancy in prostate and other cancers, and their density has been associated with poor clinical outcome (Magnon et al., 2013; Hanoun et al., 2015). Furthermore, many widely used drugs, such as bronchodilators, vasodilators, and muscle relaxants, function by activating β2-adrenoceptors, whereas other drugs, such as β-blockers, inhibit them. Thus increased EphA2 noncanonical signaling could contribute to the effects of stress and β2-adrenoceptor agonists on cancer malignancy, whereas its inhibition could contribute to the beneficial effects of β-blockers, as documented in mouse cancer models and human cancer epidemiologic studies (Chida et al., 2008; Powe and Entschladen, 2011; Cole and Sood, 2012; Cole et al., 2015; Tang et al., 2013).
EphA2 may be part of a PKA signaling program promoting malignancy through S897 phosphorylation (Miao et al., 2009, 2015b; Binda et al., 2012; Tawadros et al., 2012; Kawai et al., 2013; Paraiso et al., 2015; Zhou et al., 2015), since we detected PKA-dependent EphA2 phosphorylation on S897 in some aggressive prostate and pancreatic cancer cell lines. Besides the stress hormones/cAMP/PKA axis, a number of cytokines in the tumor microenvironment, including EGF, can promote S897 phosphorylation (Miao et al., 2009; Zhou et al., 2015). This is in addition to the genetic and epigenetic alterations that can lead to the constitutive activation of AKT and RSK in many tumor types. How pathways regulating the three serine/threonine kinases may converge to regulate EphA2 S897 phosphorylation to affect tumor progression likely depends on both the cellular context of the tumor cells and their environment. Indeed, PKA activation did not increase S897 phosphorylation in some of the cancer cell lines examined and the nontransformed prostate cell lines, consistent with differential effects of PKA, depending on the cellular context. Previous studies also documented dual effects of the cAMP/PKA axis in promoting or inhibiting cancer cell migration/invasiveness and malignancy, depending on an intricate combination of factors (Stork and Schmitt, 2002; Naviglio et al., 2009; Sadar, 2009; McKenzie et al., 2011; Braadland et al., 2014; Sapio et al., 2014). The signaling networks that enable PKA-dependent S897 phosphorylation could lead to a series of consequences ranging from increased invasiveness, metastatic ability, and stemness to drug resistance (Miao and Wang, 2009; Miao et al., 2015a,b; Zhuang et al., 2010; Binda et al., 2012; Sapio et al., 2014; Paraiso et al., 2015; Liu et al., 2016). Supporting this notion, PKA and the hotspot-phosphorylated form of EphA2 are both important for cell migration/invasiveness and enriched in protrusive structures at the leading edge of moving cells (Howe et al., 2005; Wang et al., 2007; McKenzie et al., 2011). In future studies, it will be interesting to investigate whether concomitant activation of EphA2 noncanonical signaling might subvert other effects of canonical signaling besides cell repulsion and whether the interplay of canonical and noncanonical signaling might lead to additional new activities of EphA2.
MATERIALS AND METHODS
Reagents
Chemical compounds.
The LOPAC 1280 library and individual chemical compounds for follow-up studies were purchased from Sigma-Aldrich (St. Louis, MO). They include forskolin (F6886), (–)-epinephrine (+)-bitartrate (E4375), (–)-3,4-dihydroxynorephedrine (D5290), ritodrine hydrochloride (R0758), (R)-(–)-phenylephrine hydrochloride (P6126), 5′-(N-ethylcarboxamido)adenosine (E2387), (–)-isoproterenol hydrochloride (I6504), terbutaline hemisulfate (T2528), (S)-(–)-propranolol hydrochloride (P8688), and l-(–)-norepinephrine (+)-bitartrate (A9512). N6,2′-O-Dibutyryladenosine 3′,5′-cyclical monophosphate (D0627) and wortmannin (W3144) were also from Sigma-Aldrich. N6-Benzoyladenosine-3′,5′-cyclical monophosphate (6-Benz; B009) was from BioLog (Hayward, CA), and H89 (H-5239) was from LC Laboratories (Woburn, MA).
Other reagents.
Lipofectamine 2000 (11668019) was from Thermo Fisher Scientific (Waltham, MA). 4′,6-Diamidino-2-phenylindole (DAPI; D9542) was from Sigma-Aldrich. GammaBind G Sepharose beads (17-0885-01) were from GE Healthcare (Little Chalfont, United Kingdom) and rhodamine-conjugated phalloidin was from Biotium (00027; Hayward, CA) or Thermo Fisher Scientific (R415). Oligonucleotides were from Integrated DNA Technologies (Coralville, IA). Bovine serum albumin (BSA; 3116964001) was from Roche Diagnostics (Indianapolis, IN).
Recombinant proteins.
Mouse ephrin-A1 Fc (602-A1-200) and recombinant human EGF (236-EG) were from R&D Systems (Minneapolis, MN); human Fc (ICN55911) was from MP Biomedicals (Santa Ana, CA); the PKA catalytic subunit used in Figure 3C was kindly provided by Susan Taylor (University of California, San Diego, La Jolla, CA); and that used in Figure 3H was purchased from New England Biolabs (P6000S; Ipswich, MA); casein kinase 1 (CK1, P6030S), calf intestinal alkaline phosphatase (M0290S), and restriction enzymes were also from New England Biolabs; Pfu Turbo DNA polymerase (600250) was from Agilent Technologies (Santa Clara, CA).
Antibodies.
EphA2 antibodies were from EMD Millipore (05-480 clone D7; Billerica, MA), Thermo Fisher Scientific (34-7400), Santa Cruz Biotechnology (SC-924; Dallas, TX), and R&D Systems (AF3035); antibodies to EphA2 phospho-S897 were from Cell Signaling Technology (6347; Danvers, MA) and Cell Applications (CY1108; San Diego, CA); antibodies to EphA2 phospho-Y588 (12677), CREB phospho-S133 (9196S), CREB (9197S), AKT phospho-S473 (4056S), and AKT (9272S) were from Cell Signaling Technology; the PY20 phosphotyrosine–horseradish peroxidase (HRP) antibody (610012) was from BD Biosciences (Franklin Lakes, NJ); the anti–β-tubulin antibody (T0198) was from Sigma-Aldrich; and the hemagglutinin (HA) antibody (MMS-101R, HA.11 clone 16B12) was from Covance (San Diego, CA). Secondary HRP-conjugated antibodies against rabbit (AP307PMI), mouse (AP124PMI), and goat (AP106P) were from EMD Millipore. The anti-FLAG M2 affinity gel (A2220) was from Sigma-Aldrich.
Cell lines and media.
The PC3 (CRL-1435), RWPE1 (CRL-11609), RWPE2 (CRL-11610), AsPC1 (CRL-1682), and Hs766T (HTB-134) cell lines were purchased from the American Type Culture Collection (Manassas, VA). The BPH1 cell line (originally generated at University of California, San Francisco, San Francisco, CA) was kindly provided by S. Hayward (Vanderbilt University, Nashville, TN), the DU145 cell line by E. Adamson (Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA), the LNCaP cell line by J. Reed (Sanford Burnham Prebys Medical Discovery Institute), the BxPC3 cell line by P. Itkin-Ansari (Sanford Burnham Prebys Medical Discovery Institute), the MIA PaCa2 cell line by C. Comisso (Sanford Burnham Prebys Medical Discovery Institute), the PANC1 cell line by F. Levine (Sanford Burnham Prebys Medical Discovery Institute), and the Capan2 and PL45 pancreatic cancer cell lines by G. Powis (Sanford Burnham Prebys Medical Discovery Institute). The Lenti-X HEK293T cell line (632180) was purchased from Takara Clontech (Mountain View, CA). RPMI-1640 medium (10-040-CV) and DMEM (10-013-CV) were purchased from Corning (Tewksbury, MA). Fetal bovine serum was from Hyclone (Logan, UT), Keratinocyte Serum Free Medium (17005-042), McCoy’s 5a Medium Modified (16600108), and Kaighn’s F12 medium (21127022) were purchased from Thermo Fisher Scientific. Puromycin (ant-pr-1) was purchased from Invivogen (San Diego, CA). Neomycin (04727894001) was purchased from Roche Diagnostics. Antibiotic antimycotic (45000-616) was purchased from Corning.
High-content screening assay to measure PC3 cell retraction
To highlight cell shape for automated imaging in high-throughput format, a PC3 cell population stably expressing the membrane-targeted EGFP-F was generated by infection of PC3 cells with a pLenti6.2/V5-DEST lentiviral vector encoding EGFP-F (from the Sanford Burnham Prebys Medical Discovery Institute’s Viral Vector Core Facility). To obtain a cell population with a more uniform fluorescence, cells with medium EGFP fluorescence intensity were selected by fluorescence-activated cell sorting (Supplemental Figure S1A).
The protocol to induce cell retraction was adapted for high-content screening in 384-well tissue culture plates by optimizing the cell density, ephrin-A1 Fc and dimethyl sulfoxide (DMSO) concentrations, and time of exposure to ephrin-A1 Fc. For automated measurement of cell retraction, it is desirable to capture a maximum number of cells per image field while keeping cells separate from each other to allow accurate detection of the outlines of individual cells. By seeding cells at different densities varying from 500 to 2500 cells/well in 384-well tissue culture plastic plates, an optimal cell density of ∼2200 cells/well was determined (Supplemental Figure S1A). Because chemical library compounds are usually dissolved in DMSO, DMSO sensitivity was also evaluated (Supplemental Figure S1C). This showed that the PC3-EGFP-F cells do not exhibit significant changes in cell area at DMSO concentrations of up to 0.5%. At higher DMSO concentrations, a reduction in cell area occurred, which could interfere with the cell retraction assay. To find the optimal ephrin-A1 Fc concentration inducing a consistent cell retraction, dose–response curves were obtained at varying incubation times (Supplemental Figure S1B and unpublished data). The optimal ephrin-A1 Fc concentration to induce cell retraction for the assay in the 384-well format was estimated to be slightly >1 μg/ml, which caused 80–100% maximal cell retraction at incubation times of 15–25 min. The time dependence of the ephrin-A1 Fc–induced EphA2-mediated cell retraction was evaluated over a 5- to 60-min time period. The optimal ephrin-A1 Fc incubation time, resulting in the highest amount of retraction with consistent repeatability across replicates, was determined to be 15–25 min (Supplemental Figure S1B and unpublished data). The retraction assay was carried out at 37°C because the assay did not yield satisfactory results at room temperature.
For high-content screening, PC3-EGFP-F cells cultured in Kaighn’s F12 medium with 10% fetal bovine serum (FBS) were trypsinized, resuspended in Kaighn’s F12 medium with 5% FBS at 2200 cells/well in 25 μl of culture medium, and grown overnight at 37°C in a 5% CO2 incubator. The next day, the cells were treated for 25 min with 5 μM chemical compounds in 25 μl of culture medium and then stimulated for 20 min with ephrin-A1 Fc added in 25 μl of cell culture medium to reach a final concentration of 1.25 μg/ml in the continued presence of the compounds (the final compound concentration after ephrin-A1 Fc addition was 2.5 μM, and the final DMSO concentration was 0.25%). The cells were then fixed with 4% formaldehyde in phosphate-buffered saline (PBS) for 30 min at room temperature, and nuclei were stained with DAPI (Supplemental Figure S1A).
Image acquisition was performed on an Opera QEHS (PerkinElmer, Waltham, MA) with 10×/0.45 numerical aperture air objective, since 10× magnification produced reproducible assay performance with a minimum amount of imaging time. Images were acquired sequentially for EGFP (using 488-nm laser excitation and a 540/70-nm emission filter) and DAPI (using 365-nm xenon lamp excitation and a 450/50-nm emission filter). An image analysis protocol was developed using Acapella 2.0 HCS software with the following analysis settings. Cell nuclei were detected from the DAPI images using Acapella’s <nuclei_detection_G> algorithm with threshold adjustment of 1.5, nuclear splitting adjustment of 7, minimum nuclear area of 50, and minimum nuclear contrast of 0.1. DAPI staining was used to help the automated identification of individual cells and assess the size and shape of the nuclei. In addition, the DAPI nuclear stain was used to monitor signs of cytotoxicity based on changes in nuclei (cell) count and nuclear morphometric features. Cell images were analyzed using Acapella’s <cytoplasm_detection_A> algorithm, with cytoplasm individual threshold adjustment set to 0.3 (Supplemental Figure S1D). A large number of parameters were extracted from the nuclei, cytoplasm, and cell masks, including cell count per well and well averages of cell-by-cell metrics such as nuclei area, total intensity of the nuclei, average intensity of the nuclei, nuclei elongation, nuclei roundness, cytoplasm area, cell area, equivalent radius, cell roundness, elliptical roundness, average cytoplasm intensity, total cytoplasm intensity, average cell intensity, and total cell intensity. Image analysis on a cell-by-cell basis was performed using cytoplasmic area (obtained by subtracting the nuclear area from the cell area) as the primary assay readout (Supplemental Figure S1D), which yielded a good dynamic range and an acceptable coefficient of variation Z′ of ∼0.5. Average cytoplasmic area in wells treated with ephrin-A1 Fc and 0.25% DMSO (corresponding to the final DMSO concentration in the assay) but without compounds (Supplemental Figure S1A, bottom left) was set as 0%, and average cytoplasmic area in wells treated only with 0.25% DMSO (Supplemental Figure S1A, top left) was set as 100%. Other parameters, such as cell count, nuclei area, nuclei intensity, and nuclei roundness, were also extracted and used for quality control and to evaluate compound cytotoxicity.
Nonautomated cell retraction assay in 96-well plates
For the retraction assays shown in Figure 1, PC3 cells were cultured in Kaighn’s F12 medium with 10% FBS, plated at 4000 cells/well in 96-well tissue culture plates (82050-748; Greiner Bio One, Frickenhausen, Germany), and grown overnight. The cells were then starved for 1 h in serum-free medium, incubated for 40 min with 5 μM compounds, 20 μM forskolin, 0.5 mM dibutyryl cyclical AMP, or DMSO as vehicle control, and stimulated for 10 min with 0.5 μg⁄ml ephrin-A1 Fc or Fc as a control. The cells were then fixed for 15 min in 4% formaldehyde in PBS, permeabilized for 3 min in 0.5% Triton X-100 in Tris-buffered saline, and stained with rhodamine-conjugated phalloidin to visualize F-actin. Nuclei were labeled with DAPI.
For the retraction assays shown in Figures 4 and 5 and Supplemental Figure S3, PC3 cells were plated overnight in RPMI-1640 medium with 10% FBS and penicillin/streptomycin in 96-well tissue culture plates at 4000 cells/well. The cells were then serum starved for 3 h and 20 min, followed by treatment with 20 μM forskolin, 200 μM 6-Benz-cAMP, or 0.2% DMSO for 40 min. Cell retraction was induced at 37°C by stimulation with 0.5 μg/ml ephrin-A1 Fc or Fc for 12 min in the continued presence of forskolin or 6-Benz-cAMP. The cells were then fixed in 4% paraformaldehyde in PBS for 15 min at 37°C, permeabilized in 0.1% Triton X-100 in PBS for 15 min, incubated with blocking solution (10% goat serum, 1% BSA in PBS) for 30 min, labeled for actin with rhodamine-conjugated phalloidin diluted in blocking buffer for 1 h, and stained with DAPI for 10 min. Cell images were captured using an Olympus Inverted IX81 fluorescence microscope with a Color CCD SPOT RT3 Camera and a 20× objective. Cell area was measured using ImageJ (National Institutes of Health, Bethesda, MD).
Generation of PC3 cell populations expressing EphA2 mutant constructs
For EphA2 knockdown, the GGATAAGTTTCTATTCTGT target sequence in the 3′ untranslated region of human EphA2 was identified using the Block-iT RNAi designer tool (Thermo Fisher Scientific) and the Whitehead siRNA Selection program (sirna.wi.mit.edu) and used to design the hairpin oligonucleotide ACCGGTGGATAAGTTTCTATTCTGTCTCGAGACAGAATAGAAACTTATCCTTTTTGAATTC, which was then cloned into the AgeI and EcoRI sites of the pLKO.1 puro lentivirus. The control hairpin oligonucleotide CACCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTTGAATTC was also in the pLKO.1 puro lentivirus. PC3 cells were transduced with the EphA2 short hairpin RNA (shRNA) lentivirus and selected for 7 d with 2 μg/ml puromycin. Down-regulation of EphA2 expression was confirmed by immunoblotting in comparison with cells transduced in parallel with a control shRNA lentivirus and the parental PC3 cells.
The pLVX lentivirus encoding FLAG-tagged EphA2 WT was obtained by replacing the NdeI-BamHI restriction fragment of the pLVX-IRES-Neo lentiviral vector (632181;Takara Clontech) with the corresponding fragment from the pFLAG-CMV3 plasmid (E6783; Sigma-Aldrich), which encodes a signal peptide followed by the FLAG tag sequence, and then cloning the human EphA2 mature coding sequence (nucleotides 175–3061; GenBank accession number BC037166) using the NotI site in the EphA2 sequence and a restriction site in the lentivirus multiple cloning site. The EphA2 mutants were generated by site-directed mutagenesis with Pfu Turbo DNA polymerase in pcDNA plasmids, verified by sequencing the entire EphA2 cDNA, and subcloned into the pLVX-FLAG-EphA2-IRES-Neo lentivirus by replacing the sequence between the NotI and BamHI sites within the EphA2 sequence. Nucleotide changes were TCT to GCT (S892A), AGC to GCC (S897A), AGC to GAC (S897D), and TCG to GCG (S901A).
All infectious lentiviruses were produced in the Lenti-X HEK293T cell line (632180; Takara Clontech) using the VSV-G envelope glycoprotein and pCMVΔR8.91 packaging constructs (Zufferey et al., 1997). The lentiviruses were then used to infect PC3 cells with a multiplicity of infection yielding ≤30% infected cells. Cell populations were then selected for 10 d with 150 μg/ml neomycin, and EphA2 expression levels were verified by immunoblotting.
ELISA to measure inhibition of EphA2–ephrin binding
Protein A–coated wells (PI15132; Pierce Biotechnology, Rockford, IL) were used to immobilize ephrin-A1 Fc incubated at 1 μg/ml in TBST (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.01% Tween 20). Culture supernatant from transfected HEK293 cells containing the EphA2 ligand-binding domain fused to alkaline phosphatase (EphA2 AP) was diluted in TBST and incubated for 3 h in the presence and in the absence of 20 μM compounds as previously described (Koolpe et al., 2002; Noberini et al., 2008). The amount of bound EphA2 AP was quantified using pNPP (PI34045; Thermo Fisher Scientific) as the substrate. Alkaline phosphatase activity from wells coated with Fc control was subtracted as background.
Immunoprecipitation and immunoblotting
Cells were collected when they reached ∼75% confluency in ice-cold lysis buffer (1% Triton X-100, 150 mM NaCl, 40 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, with 1 mM phenylmethylsulfonyl fluoride, 1 mM EDTA, 2 mM pyrophosphate, 2 mM glycerophosphate, 5 mM sodium fluoride, 1 mM sodium orthovanadate, 10 μg/ml leupeptin, 10 μg/ml apoprotinin, and 10 μg/ml pepstatin). Alternatively, the lysis buffer was supplemented with Halt Combined Protease and Phosphatase Inhibitor Cocktail (PI78442; Thermo Fisher Scientific). For immunoprecipitations, lysates were centrifuged at 13,000 × g for 10 min at 4°C to remove insoluble material. Protein concentration in the supernatant was determined using the Pierce BCA Protein Assay Kit (23227; Thermo Fisher Scientific). The supernatant was further precleared by incubation with Sepharose beads (4B200; Sigma-Aldrich) for 30 min at 4°C on a rotator. Each immunoprecipitation was performed using 1–2 μg of antibody coupled to 10 μl of GammaBind G Sepharose beads for 4 h at 4°C. Immunoprecipitates were washed four times with lysis buffer and eluted in SDS sample buffer by incubation at 55°C for 15 min. For immunoprecipitations with anti-FLAG M2-agarose affinity gel, the lysis buffer contained 50 mM Tris HCl instead of HEPES.
For immunoblotting, polyvinylidene fluoride membranes were incubated for 1 h in blocking buffer (0.1% Tween-20, 4% BSA in PBS) and then overnight at 4°C with primary antibodies diluted in blocking buffer. Membranes were then incubated for 1 h at room temperature with rabbit anti-goat, goat anti-rabbit, or goat anti-mouse secondary antibodies conjugated to HRP (AP106P, AP307P, and AP124PMI respectively; EMD Millipore), followed by ECL chemiluminescence detection (RNP2106; GE Healthcare) using x-ray film.
Generation and characterization of S901 phosphospecific antibody
The peptide KLPSTSGpSEGVPFR, corresponding to EphA2 residues 895–907 with S901 phosphorylated and an added N-terminal lysine, was coupled to BSA using glutaraldehyde and used to immunize rabbits. Antibodies were then affinity purified from immune serum using peptide coupled to an Affi-Gel 10 column (1536046; Bio-Rad Laboratories, Hercules, CA). Antibodies purified using the phosphorylated or the nonphosphorylated peptide preferentially recognize EphA2 phosphorylated on S901. They recognize EphA2 WT overexpressed by transient transfection in HEK293 cells but not EphA2 that has been treated with calf intestinal alkaline phosphatase, which indiscriminately dephosphorylates pS/pT/pY residues (Figure 3F). The antibodies also do not recognize the EphA2 S901A mutant, which lacks the S901 phosphorylation site (Figure 3G). These data confirm the desired specificity of the antibodies for the phosphorylated S901 motif.
In vitro kinase assays
PC3 cells stably expressing FLAG-tagged EphA2 were lysed in lysis buffer without phosphatase inhibitors and used to immunoprecipitate EphA2 with 40 μl of FLAG M2 affinity gel. Beads were washed twice with 1 ml of kinase buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 0.1 mM EDTA, 2 mM dithiothreitol [DTT], 0.01% Brij 35) and aliquoted in four tubes. Each aliquot was resuspended in 50 μl of kinase buffer with 1) no ATP or kinase (unpublished data), 2) 200 μM ATP (9804; Cell Signaling Technology) and no kinase, 3) ATP and CK1 kinase, and 4) ATP and PKA kinase. Immunoprecipitates were incubated for 30 min in an orbital shaker (300 rpm) at 30°C. Kinase reactions were stopped by the addition of SDS-containing sample buffer, followed by incubation at 65°C for 15 min.
Kinase phosphorylation antibody array
A membrane-based antibody array (Proteome Profiler, Human Phospho-Kinase Array Kit, ARY003B; R&D Systems)) was used to determine the relative levels of various human kinase phosphorylation sites according to the manufacturer’s instructions. PC3 cells expressing EphA2 WT or S897A were starved and treated with forskolin and/or ephrin-A1 Fc according to the same protocol used for the retraction assays shown in Figures 4 and 5, and 600 μg of protein lysate was used for each array set. Multiple exposures of the blotted membranes were captured using x-ray film, and the scanned spots were quantified using Photoshop (Adobe, San Jose, CA).
Cell surface biotinylation
PC3 cells reconstituted with EphA2 WT or the S897A mutant after knockdown of endogenous EphA2 (see earlier description) were starved for 3 h and 20 min in serum-free medium, treated for 40 min with 20 μM forskolin or DMSO as a control, and then stimulated for 10 or 20 min with 0.5 μg/ml ephrin-A1 Fc or human Fc control in the continued presence of forskolin. Cell surface proteins were then biotinylated on ice by two 20-min treatments with 1 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (21331; Thermo Fisher Scientific/Pierce) in PBS at 4°C with shaking, according to the manufacturer’s instructions. Cells were washed three times with 100 mM glycine in PBS to quench the biotinylation reaction, followed by PBS, and lysed in modified RIPA buffer (150 mM NaCl, 1 mM EDTA, 1 % Triton X-100, 1 % sodium deoxycholate, 0.1 % SDS, 20 mM Tris, pH 8.0) containing protease and phosphatase inhibitors. For pull downs, 50 μg of each lysate was incubated overnight at 4°C with 20 μl of streptavidin agarose beads (20347; Thermo Fisher Scientific). Beads were washed with modified RIPA buffer and eluted with 50 μl of SDS sample buffer containing β-mercaptoethanol as a reducing agent to cleave the biotin group from the cross-linked proteins.
Effects of modulating PKA activity on EphA2 S897 phosphorylation
PC3 cells were transfected with empty pcDNA3.1 vector or pcDNA3.1 vector encoding the HA-tagged WT PKA catalytic subunit, the HA-tagged kinase-inactive PKA K72H mutant catalytic subunit (PKA KD; Iyer et al., 2005), or the pRSV-PKI-v2 vector encoding the PKA inhibitor peptide PKI (plasmid 45066; Addgene [ Day et al., 1989]) together with a 1:5 ratio of pPUR DNA vector (631601; Takara Clontech) using Lipofectamine 2000. After 24 h, transfected cells were selected with 2 μg/ml puromycin for 4 d. The cells were then stimulated for 30 min with 10 μM norepinephrine or PBS as a control and collected in lysis buffer.
EphA2 degradation induced by ephrin-A1 Fc stimulation
PC3 cells were starved for 3 h and 20 min in serum-free medium, treated for 40 min with 20 μM forskolin or DMSO as a control, and then stimulated with 0.5 μg/ml ephrin-A1 Fc or human Fc control for different time periods in the continued presence of forskolin. Cells were collected in lysis buffer, and EphA2 was immunoprecipitated using a mouse monoclonal antibody (05-480). Immunoblots were probed with PY20 phosphotyrosine-HRP antibody and reprobed for total EphA2 (34-7400).
Liquid chromatography/mass spectrometry analysis of EphA2 phosphorylation sites
PC3 cells were starved for 3 h and 20 min and then stimulated for 40 min with 20 μM forskolin or DMSO vehicle control. Cells were collected in ice-cold lysis buffer. Lysates were centrifuged at 13,000 × g for 10 min at 4°C to remove insoluble material and further precleared by incubation with Sepharose beads for 30 min at 4°C on a rotator. EphA2 was pulled down using biotinylated-ephrinA1-Fc ligand (BT602; R&D Systems) coupled to streptavidin beads (PI20347; Thermo Fisher Scientific). Beads were washed, and EphA2 was eluted with 6 M urea. Eluted material was treated with 10 mM DTT for 30 min at 37°C, alkylated with 40 mM indoleacetamide for 45 min at 37°C, and digested with 500 ng of trypsin (V5280; Promega, Fitchburg, WI) overnight at 37°C in an orbital shaker. Peptides were desalted using a peptide microtrap (Michrom BioResources, Auburn, CA), and phosphopeptides were enriched using a batch TiO2-based enrichment method (Ma et al., 2013). Both nonenriched and enriched phosphopeptide fractions were individually separated using a MS2 HPLC connected to a 200 × 0.2–mm column, ionized using a Captive Spray Source (Michrom Bioresources), and analyzed using a decision-tree tandem mass spectrometry (MS/MS) method (Swaney et al., 2008) in an LTQ Orbitrap Velos mass spectrometer equipped with electron transfer dissociation (Thermo Fisher Scientific). MS/MS data were searched against a concatenated target-decoy ipi.HUMAN.v.3.73 protein database (89,652 entries) using semitryptic specificity, with Sorcerer-SEQUEST on Sorcerer Enterprise (Sage-N Research, Milpitas, CA). Precursor-ion mass tolerance was ≤5.00 ppm, and product-ion mass tolerance was 0.5 atomic mass units (amu). Static carbamidomethylation of Cys (+57.0214 amu), differential oxidation of Met (+15.9949 amu), and differential phosphorylation of S, T, and Y (+79.9663 amu) were specified. Only for ETD spectra, the Versasearch script (Sage-N) specified differential modification of peptide N-termini (b- to c-ions, +17.0265 amu) and C-termini (y- to z-radical ions, −16.0187 amu) because c- and z-ions are the prominent product ions in ETD MS/MS spectra (Syka et al., 2004). Filtering was with ProteinProphet (Trans-Proteomic Pipeline) at a false discovery rate of <0.01. Phosphopeptides were also filtered at the peptide level, and phosphopeptide spectra were manually inspected to verify that site-determining ions were present. Spectral quality allowed confident localization of phosphorylation sites.
Supplementary Material
Acknowledgments
We thank C. Fernandez for generating the EphA2 S897A mutant, R. Elward for help with experiments to characterize the pS901 antibody, S. Taylor for the purified PKA catalytic subunit and the PKA WT and PKA KD constructs, R. Maurer for the pRSV-PKI-v2 construct, C. Comisso for the MIA PaCa2 cell line, J. Reed for the LNCaP cell line, S. Hayward for the BPH1 cell line, P. Itkin-Ansari for the BxPC3 cell line, G. Powis for the Capan2 and PL45 cell lines, F. Levine for the PANC1 cell line, E. Adamson for the DU145 cell line, and A. D’Osualdo for the control pLK0.1 puro lentivirus, VSV-G envelope glycoprotein, and pCMVΔR8.91 packaging constructs. This work was supported by National Institutes of Health Grants P01 CA138390 and R21 NS066479 and Institutional Funding (E.B.P.), fellowships from the Spanish Ministry of Science and Education (A.B.P.), and National Cancer Institute Cancer Center Support Grant P30 CA030199 (supporting Sanford Burnham Prebys Medical Discovery Institute Core Facilities).
Abbreviations used:
- CK1
casein kinase 1
- EGF
epidermal growth factor
- EGFP-F
farnesylated enhanced green fluorescent protein
- LOPAC
Library of Pharmacologically Active Compounds
- PKA
protein kinase A
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0048) on July 6, 2016.
REFERENCES
- Astin JW, Batson J, Kadir S, Charlet J, Persad RA, Gillatt D, Oxley JD, Nobes CD. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat Cell Biol. 2010;12:1194–1204. doi: 10.1038/ncb2122. [DOI] [PubMed] [Google Scholar]
- Baell J, Walters MA. Chemistry: chemical con artists foil drug discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Barquilla A, Pasquale EB. Eph receptors and ephrins: therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2015;55:465–487. doi: 10.1146/annurev-pharmtox-011112-140226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batson J, Maccarthy-Morrogh L, Archer A, Tanton H, Nobes CD. EphA receptors regulate prostate cancer cell dissemination through Vav2-RhoA mediated cell-cell repulsion. Biol Open. 2014;3:453–462. doi: 10.1242/bio.20146601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biao-Xue R, Xi-Guang C, Shuan-Ying Y, Wei L, Zong-Juan M. EphA2-dependent molecular targeting therapy for malignant tumors. Curr Cancer Drug Targets. 2011;11:1082–1097. doi: 10.2174/156800911798073050. [DOI] [PubMed] [Google Scholar]
- Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, Huse JT, Cajola L, Zanetti N, Dimeco F, et al. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell. 2012;22:765–780. doi: 10.1016/j.ccr.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd AW, Bartlett PF, Lackmann M. Therapeutic targeting of EPH receptors and their ligands. Nat Rev Drug Discov. 2014;13:39–62. doi: 10.1038/nrd4175. [DOI] [PubMed] [Google Scholar]
- Braadland PR, Ramberg H, Grytli HH, Tasken KA. beta-Adrenergic receptor signaling in prostate cancer. Front Oncol. 2014;4:375. doi: 10.3389/fonc.2014.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli R, Tognolini M, Vacondio F, Incerti M, Pala D, Callegari D, Bertoni S, Giorgio C, Hassan-Mohamed I, Zanotti I, et al. delta-Cholenoyl-amino acids as selective and orally available antagonists of the eph-ephrin system. Eur J Med Chem. 2015;103:312–324. doi: 10.1016/j.ejmech.2015.08.048. [DOI] [PubMed] [Google Scholar]
- Cercone MA, Schroeder W, Schomberg S, Carpenter TC. EphA2 receptor mediates increased vascular permeability in lung injury due to viral infection and hypoxia. Am J Physiol Lung Cell Mol Physiol. 2009;297:L856–L863. doi: 10.1152/ajplung.00118.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Veettil MV, Bottero V, Chandran B. Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc Natl Acad Sci USA. 2012;109:E1163–1172. doi: 10.1073/pnas.1119592109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Jorgensen C, Pawson T, Hedley DW. Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br J Cancer. 2008;99:1074–1082. doi: 10.1038/sj.bjc.6604676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 2008;40:651–662. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chida Y, Hamer M, Wardle J, Steptoe A. Do stress-related psychosocial factors contribute to cancer incidence and survival. Nat Clin Pract Oncol. 2008;5:466–475. doi: 10.1038/ncponc1134. [DOI] [PubMed] [Google Scholar]
- Choi KM, Park GL, Hwang KY, Lee JW, Ahn HJ. Efficient siRNA delivery into tumor cells by p19-YSA fusion protein. Mol Pharm. 2013;10:763–773. doi: 10.1021/mp300344p. [DOI] [PubMed] [Google Scholar]
- Choi Y, Syeda F, Walker JR, Finerty PJ, Jr, Cuerrier D, Wojciechowski A, Liu Q, Dhe-Paganon S, Gray NS. Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorg Med Chem Lett. 2009;19:4467–4470. doi: 10.1016/j.bmcl.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, Batlle E. EphB/EphrinB receptors and Wnt signaling in colorectal cancer. Cancer Res. 2006;66:2–5. doi: 10.1158/0008-5472.CAN-05-3849. [DOI] [PubMed] [Google Scholar]
- Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer. 2015;15:563–572. doi: 10.1038/nrc3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SW, Sood AK. Molecular pathways: beta-adrenergic signaling in cancer. Clin Cancer Res. 2012;18:1201–1206. doi: 10.1158/1078-0432.CCR-11-0641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GD, Dewey WL. Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides. 2006;40:23–34. doi: 10.1016/j.npep.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Day RN, Walder JA, Maurer RA. A protein kinase inhibitor gene reduces both basal and multihormone-stimulated prolactin gene transcription. J Biol Chem. 1989;264:431–436. [PubMed] [Google Scholar]
- Dutta D, Chakraborty S, Bandyopadhyay C, Valiya Veettil M, Ansari MA, Singh VV, Chandran B. EphrinA2 regulates clathrin mediated KSHV endocytosis in fibroblast cells by coordinating integrin-associated signaling and c-Cbl directed polyubiquitination. PLoS Pathog. 2013;9:e1003510. doi: 10.1371/journal.ppat.1003510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem. 1990;265:14264–14269. [PubMed] [Google Scholar]
- Flotow H, Roach PJ. Role of acidic residues as substrate determinants for casein kinase I. J Biol Chem. 1991;266:3724–3727. [PubMed] [Google Scholar]
- Fox BP, Tabone CJ, Kandpal RP. Potential clinical relevance of Eph receptors and ephrin ligands expressed in prostate carcinoma cell lines. Biochem Biophys Res Commun. 2006;342:1263–1272. doi: 10.1016/j.bbrc.2006.02.099. [DOI] [PubMed] [Google Scholar]
- Funk SD, Yurdagul A, Jr, Albert P, Traylor JG, Jr, Jin L, Chen J, Orr AW. EphA2 activation promotes the endothelial cell inflammatory response: a potential role in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:686–695. doi: 10.1161/ATVBAHA.111.242792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- Ha KD, Bidlingmaier SM, Zhang Y, Su Y, Liu B. High-content analysis of antibody phage-display library selection outputs identifies tumor selective macropinocytosis-dependent rapidly internalizing antibodies. Mol Cell Proteomics. 2014;13:3320–3331. doi: 10.1074/mcp.M114.039768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoun M, Maryanovich M, Arnal-Estape A, Frenette PS. Neural regulation of hematopoiesis, inflammation, and cancer. Neuron. 2015;86:360–373. doi: 10.1016/j.neuron.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan S, Karpova Y, Flores A, D’Agostino R, Jr, Danhauer SC, Hemal A, Kulik G. A pilot study of blood epinephrine levels and CREB phosphorylation in men undergoing prostate biopsies. Int Urol Nephrol. 2014;46:505–510. doi: 10.1007/s11255-013-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramoto-Yamaki N, Takeuchi S, Ueda S, Harada K, Fujimoto S, Negishi M, Katoh H. Ephexin4 and EphA2 mediate cell migration through a RhoG-dependent mechanism. J Cell Biol. 2010;190:461–477. doi: 10.1083/jcb.201005141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JY, Shin MH, Chung KS, Kim EY, Jung JY, Kang YA, Kim YS, Kim SK, Chang J, Park MS. EphA2 receptor signaling mediates inflammatory responses in lipopolysaccharide-induced lung injury. Tuberc Respir Dis (Seoul) 2015;78:218–226. doi: 10.4046/trd.2015.78.3.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe AK. Regulation of actin-based cell migration by cAMP/PKA. Biochim Biophys Acta. 2004;1692:159–174. doi: 10.1016/j.bbamcr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Howe AK. Cross-talk between calcium and protein kinase A in the regulation of cell migration. Curr Opin Cell Biol. 2011;23:554–561. doi: 10.1016/j.ceb.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe AK, Baldor LC, Hogan BP. Spatial regulation of the cAMP-dependent protein kinase during chemotactic cell migration. Proc Natl Acad Sci USA. 2005;102:14320–14325. doi: 10.1073/pnas.0507072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wu D, Jin H, Stupack D, Wang JY. Induction of cell retraction by the combined actions of Abl-CrkII and Rho-ROCK1 signaling. J Cell Biol. 2008;183:711–723. doi: 10.1083/jcb.200801192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer GH, Garrod S, Woods VL, Jr, Taylor SS. Catalytic independent functions of a protein kinase as revealed by a kinase-dead mutant: study of the Lys72His mutant of cAMP-dependent kinase. J Mol Biol. 2005;351:1110–1122. doi: 10.1016/j.jmb.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Jiang H, Li X, Zhang X, Liu Y, Huang S, Wang X. EphA2 knockdown attenuates atherosclerotic lesion development in ApoE(-/-) mice. Cardiovasc Pathol. 2014;23:169–174. doi: 10.1016/j.carpath.2014.01.006. [DOI] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Kaushansky A, Douglass AN, Arang N, Vigdorovich V, Dambrauskas N, Kain HS, Austin LS, Sather DN, Kappe SH. Malaria parasites target the hepatocyte receptor EphA2 for successful host infection. Science. 2015;350:1089–1092. doi: 10.1126/science.aad3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai H, Kobayashi M, Hiramoto-Yamaki N, Harada K, Negishi M, Katoh H. Ephexin4-mediated promotion of cell migration and anoikis resistance is regulated by serine 897 phosphorylation of EphA2. FEBS Open Bio. 2013;3:78–82. doi: 10.1016/j.fob.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolpe M, Dail M, Pasquale EB. An ephrin mimetic peptide that selectively targets the EphA2 receptor. J Biol Chem. 2002;277:46974–46979. doi: 10.1074/jbc.M208495200. [DOI] [PubMed] [Google Scholar]
- Landen CN, Kinch MS, Sood AK. EphA2 as a target for ovarian cancer therapy. Expert Opin Ther Targets. 2005;9:1179–1187. doi: 10.1517/14728222.9.6.1179. [DOI] [PubMed] [Google Scholar]
- Larson J, Schomberg S, Schroeder W, Carpenter TC. Endothelial EphA receptor stimulation increases lung vascular permeability. Am J Physiol Lung Cell Mol Physiol. 2008;295:L431–L439. doi: 10.1152/ajplung.90256.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Yang Z, Wang T, Yang Z, Chen H, Hu Y, Hu C, Guo L, Deng Q, Liu Y, et al. beta2-AR signaling controls trastuzumab resistance-dependent pathway. Oncogene. 2016;35:47–58. doi: 10.1038/onc.2015.58. [DOI] [PubMed] [Google Scholar]
- Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, Castilla EA, Chen J, Yajima T, Porollo A, et al. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell. 2013;152:599–611. doi: 10.1016/j.cell.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, Frenette PS. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- McKenzie AJ, Campbell SL, Howe AK. Protein kinase A activity and anchoring are required for ovarian cancer cell migration and invasion. PLoS One. 2011;6:e26552. doi: 10.1371/journal.pone.0026552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- Miao H, Gale NW, Guo H, Qian J, Petty A, Kaspar J, Murphy AJ, Valenzuela DM, Yancopoulos G, Hambardzumyan D, et al. EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene. 2015b;34:558–567. doi: 10.1038/onc.2013.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao B, Ji Z, Tan L, Taylor M, Zhang J, Choi HG, Frederick DT, Kumar R, Wargo JA, Flaherty KT, et al. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 2015a;5:274–287. doi: 10.1158/2159-8290.CD-14-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Strebhardt K, Pasquale EB, Shen TL, Guan JL, Wang B. Inhibition of integrin-mediated cell adhesion but not directional cell migration requires catalytic activity of EphB3 receptor tyrosine kinase. J Biol Chem. 2005;280:923–932. doi: 10.1074/jbc.M411383200. [DOI] [PubMed] [Google Scholar]
- Miao H, Wang B. Eph/ephrin signaling in epithelial development and homeostasis. Int J Biochem Cell Biol. 2009;41:762–770. doi: 10.1016/j.biocel.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Duggineni S, Koolpe M, Zhu X, Huang Z, Pasquale EB. Structure-activity relationship analysis of peptides targeting the EphA2 receptor. Biochemistry. 2010;49:6687–6695. doi: 10.1021/bi1006223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3:ra64. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviglio S, Caraglia M, Abbruzzese A, Chiosi E, Di Gesto D, Marra M, Romano M, Sorrentino A, Sorvillo L, Spina A, et al. Protein kinase A as a biological target in cancer therapy. Expert Opin Ther Targets. 2009;13:83–92. doi: 10.1517/14728220802602349. [DOI] [PubMed] [Google Scholar]
- Noberini R, Koolpe M, Peddibhotla S, Dahl R, Su Y, Cosford ND, Roth GP, Pasquale EB. Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. J Biol Chem. 2008;283:29461–29472. doi: 10.1074/jbc.M804103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noberini R, Lamberto I, Pasquale EB. Targeting Eph receptors with peptides and small molecules: progress and challenges. Semin Cell Dev Biol. 2012a;23:51–57. doi: 10.1016/j.semcdb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noberini R, Rubio de la Torre E, Pasquale EB. Profiling Eph receptor expression in cells and tissues: a targeted mass spectrometry approach. Cell Adh Migr. 2012b;6:102–112. doi: 10.4161/cam.19620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Pasqualini R, Lindberg RA, Kain R, Freeman AL, Pasquale EB. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 2000;19:6043–6052. doi: 10.1038/sj.onc.1204004. [DOI] [PubMed] [Google Scholar]
- Paraiso KH, Das Thakur M, Fang B, Koomen JM, Fedorenko IV, John JK, Tsao H, Flaherty KT, Sondak VK, Messina JL, et al. Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discov. 2015;5:264–273. doi: 10.1158/2159-8290.CD-14-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri M, Buricchi F, Giannoni E, Grimaldi G, Mello T, Raugei G, Ramponi G, Chiarugi P. EphrinA1 activates a Src/focal adhesion kinase-mediated motility response leading to rho-dependent actino/myosin contractility. J Biol Chem. 2007;282:19619–19628. doi: 10.1074/jbc.M701319200. [DOI] [PubMed] [Google Scholar]
- Parri M, Buricchi F, Taddei ML, Giannoni E, Raugei G, Ramponi G, Chiarugi P. EphrinA1 repulsive response is regulated by an EphA2 tyrosine phosphatase. J Biol Chem. 2005;280:34008–34018. doi: 10.1074/jbc.M502879200. [DOI] [PubMed] [Google Scholar]
- Parri M, Taddei ML, Bianchini F, Calorini L, Chiarugi P. EphA2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid-like motility style. Cancer Res. 2009;69:2072–2081. doi: 10.1158/0008-5472.CAN-08-1845. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty A, Myshkin E, Qin H, Guo H, Miao H, Tochtrop GP, Hsieh JT, Page P, Liu L, Lindner DJ, et al. A small molecule agonist of EphA2 receptor tyrosine kinase inhibits tumor cell migration in vitro and prostate cancer metastasis in vivo. PLoS One. 2012;7:e42120. doi: 10.1371/journal.pone.0042120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powe DG, Entschladen F. Targeted therapies: using beta-blockers to inhibit breast cancer progression. Nat Rev Clin Oncol. 2011;8:511–512. doi: 10.1038/nrclinonc.2011.123. [DOI] [PubMed] [Google Scholar]
- Richter M, Murai KK, Bourgin C, Pak D, Pasquale EB. The EphA4 receptor regulates neuronal morphology through SPAR-mediated inactivation of Rap GTPases. J Neurosci. 2007;27:14215–14205. doi: 10.1523/JNEUROSCI.2746-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl SJ, Pasquale EB. Targeting the Eph system with peptides and peptide conjugates. Curr Drug Targets. 2015;16:1031–1047. doi: 10.2174/1389450116666150727115934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust HL, Thompson PR. Kinase consensus sequences: a breeding ground for crosstalk. ACS Chem Biol. 2011;6:881–892. doi: 10.1021/cb200171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadar MD. The role of cyclic AMP in regulating the androgen receptor. In: Tindall D, Mohler J, editors. Androgen Action in Prostate Cancer. New York: Springer; 2009. pp. 465–503. [Google Scholar]
- Salazar C, Hofer T. Multisite protein phosphorylation–from molecular mechanisms to kinetic models. FEBS J. 2009;276:3177–3198. doi: 10.1111/j.1742-4658.2009.07027.x. [DOI] [PubMed] [Google Scholar]
- Sapio L, Di Maiolo F, Illiano M, Esposito A, Chiosi E, Spina A, Naviglio S. Targeting protein kinase A in cancer therapy: an update. EXCLI J. 2014;13:843–855. [PMC free article] [PubMed] [Google Scholar]
- Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY. An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat Struct Mol Biol. 2010;17:398–402. doi: 10.1038/nsmb.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiradake E, Schaupp A, del Toro Ruiz D, Kaufmann R, Mitakidis N, Harlos K, Aricescu AR, Klein R, Jones EY. Structurally encoded intraclass differences in EphA clusters drive distinct cell responses. Nat Struct Mol Biol. 2013;20:958–964. doi: 10.1038/nsmb.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- Subbarayal P, Karunakaran K, Winkler AC, Rother M, Gonzalez E, Meyer TF, Rudel T. EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 2015;11:e1004846. doi: 10.1371/journal.ppat.1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama N, Gucciardo E, Tatti O, Varjosalo M, Hyytiainen M, Gstaiger M, Lehti K. EphA2 cleavage by MT1-MMP triggers single cancer cell invasion via homotypic cell repulsion. J Cell Biol. 2013;201:467–484. doi: 10.1083/jcb.201205176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat Methods. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei ML, Parri M, Angelucci A, Bianchini F, Marconi C, Giannoni E, Raugei G, Bologna M, Calorini L, Chiarugi P. EphA2 induces metastatic growth regulating amoeboid motility and clonogenic potential in prostate carcinoma cells. Mol Cancer Res. 2011;9:149–160. doi: 10.1158/1541-7786.MCR-10-0298. [DOI] [PubMed] [Google Scholar]
- Taddei ML, Parri M, Angelucci A, Onnis B, Bianchini F, Giannoni E, Raugei G, Calorini L, Rucci N, Teti A, et al. Kinase-dependent and -independent roles of EphA2 in the regulation of prostate cancer invasion and metastasis. Am J Pathol. 2009;174:1492–1503. doi: 10.2353/ajpath.2009.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Dillon TJ, Liu C, Kariya Y, Wang Z, Stork PJ. Protein kinase A-dependent phosphorylation of Rap1 regulates its membrane localization and cell migration. J Biol Chem. 2013;288:27712–27723. doi: 10.1074/jbc.M113.466904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets. 2011;15:31–51. doi: 10.1517/14728222.2011.538682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Li Z, Lu L, Cho CH. beta-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin Cancer Biol. 2013;23:533–542. doi: 10.1016/j.semcancer.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Tawadros T, Brown MD, Hart CA, Clarke NW. Ligand-independent activation of EphA2 by arachidonic acid induces metastasis-like behaviour in prostate cancer cells. Br J Cancer. 2012;107:1737–1744. doi: 10.1038/bjc.2012.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thundyil J, Manzanero S, Pavlovski D, Cully TR, Lok KZ, Widiapradja A, Chunduri P, Jo DG, Naruse C, Asano M, et al. Evidence that the EphA2 receptor exacerbates ischemic brain injury. PLoS One. 2013;8:e53528. doi: 10.1371/journal.pone.0053528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognolini M, Lodola A. Targeting the Eph-ephrin system with protein-protein interaction (PPI) inhibitors. Curr Drug Targets. 2015;16:1048–1056. doi: 10.2174/1389450116666150825144457. [DOI] [PubMed] [Google Scholar]
- Venerando A, Ruzzene M, Pinna LA. Casein kinase: the triple meaning of a misnomer. Biochem J. 2014;460:141–156. doi: 10.1042/BJ20140178. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ding SJ, Wang W, Jacobs JM, Qian WJ, Moore RJ, Yang F, Camp DG, 2nd, Smith RD, Klemke RL. Profiling signaling polarity in chemotactic cells. Proc Natl Acad Sci USA. 2007;104:8328–8333. doi: 10.1073/pnas.0701103104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang NY, Fernandez C, Richter M, Xiao Z, Valencia F, Tice DA, Pasquale EB. Crosstalk of the EphA2 receptor with a serine/threonine phosphatase suppresses the Akt-mTORC1 pathway in cancer cells. Cell Signal. 2011;23:201–212. doi: 10.1016/j.cellsig.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Yamada N, Tanaka T, Hori T, Yokoyama S, Hayakawa Y, Yano S, Fukuoka J, Koizumi K, Saiki I, et al. Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA2. Nat Commun. 2015;6:7679. doi: 10.1038/ncomms8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S, Jackson D, Muraoka-Cook R, Arteaga C, Chen J. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. 2010;70:299–308. doi: 10.1158/0008-5472.CAN-09-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.