

Abstract
Multicellular organisms encounter environmental conditions that adversely affect protein homeostasis (proteostasis), including extreme temperatures, toxins, and pathogens. It is unclear how they use sensory signaling to detect adverse conditions and then activate stress response pathways so as to offset potential damage. Here, we show that dopaminergic mechanosensory neurons in C. elegans release the neurohormone dopamine to promote proteostasis in epithelia. Signaling through the DA receptor DOP‐1 activates the expression of xenobiotic stress response genes involved in pathogenic resistance and toxin removal, and these genes are required for the removal of unstable proteins in epithelia. Exposure to a bacterial pathogen (Pseudomonas aeruginosa) results in elevated removal of unstable proteins in epithelia, and this enhancement requires DA signaling. In the absence of DA signaling, nematodes show increased sensitivity to pathogenic bacteria and heat‐shock stress. Our results suggest that dopaminergic sensory neurons, in addition to slowing down locomotion upon sensing a potential bacterial feeding source, also signal to frontline epithelia to activate the xenobiotic stress response so as to maintain proteostasis and prepare for possible infection.
Keywords: xenobiotic stress, ubiquitin, proteostasis, dopamine, C. elegans
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Neuroscience; Protein Biosynthesis & Quality Control
Introduction
Multicellular organisms develop and interact with an ever‐changing environment, which can present conditions that impair development as well as physiology at the cellular and organismal level. Extreme temperatures, toxins, and pathogens present major physiological challenges, including challenges to the cell for maintaining protein homeostasis (proteostasis)—the natural folding and function of the cellular proteome (Shore & Ruvkun, 2013; Warnatsch et al, 2013; Vilchez et al, 2014; Treweek et al, 2015). The failure to maintain proteostasis in the face of such environmental challenges results in physiological decline, as oxidized and unfolded proteins threaten cellular physiology.
Most organisms have evolved multiple stress response pathways that act at the cellular level to help maintain proteostasis. For example, elevated temperatures can result in the unfolding and misfolding of proteins, and cells respond by elevating the transcription of heat‐shock proteins (HSPs), some of which act as chaperones to help proteins fold properly even under higher temperature (Akerfelt et al, 2010; Murshid et al, 2013). Oxidative and xenobiotic stress can also damage proteins, DNA, and other macromolecules, and the oxidative stress response pathway, mediated by the transcription factor Nrf2, acts to offset the resulting reactive oxygen species (ROS) and protect macromolecules from oxidative and xenobiotic damage (Nguyen et al, 2009; Bock, 2014). Damaged and unfolded proteins are removed by the ubiquitin proteasome system (UPS), which covalently attaches chains of the small protein ubiquitin to lysine side chains of each protein substrate (Ciechanover & Stanhill, 2014). Such poly‐ubiquitinated substrates are removed by proteolysis via the 26S proteasome. Alternatively, poly‐ubiquitinated, aggregation‐prone proteins that are resistant to degradation by the proteasome can be isolated within cells and degraded in autophagosomes after recognition by the autophagy pathway (Kirkin et al, 2009; Ryno et al, 2013; Ciechanover & Kwon, 2015; Cortes & La Spada, 2015; Lim & Yue, 2015). Membrane proteins can also be refolded and/or removed in the ER by the unfolded protein response (UPR) pathway (Ryno et al, 2013). Whereas these different stress response pathways are effective at the level of individual cells, it remains unclear how multicellular organisms coordinate these pathways over multiple differentiated tissues.
Changes in proteostasis in one tissue can alter proteostasis in other tissues (Van Oosten‐Hawle & Morimoto, 2014). In C. elegans, the thermosensory neuron AFD senses acute heat stress and releases the neurotransmitter serotonin, which is then able to activate the expression of HSPs in distal tissues in response (Prahlad et al, 2008; Tatum et al, 2015). Similarly, the sensory neurons ASH and ASI can modulate the UPR in distal tissues like the intestine in a process that requires the octopamine receptor OCTR‐1 (although octopamine is unlikely to be the neuron‐to‐intestine signal itself) (Sun et al, 2011, 2012). Although neuronal signaling likely provides a primary mechanism for regulating stress response across tissues, direct signaling between non‐neuronal tissues has also been shown to regulate the heat‐shock response and longevity in a non‐autonomous fashion (Van Oosten‐Hawle et al, 2013; Van Oosten‐Hawle & Morimoto, 2014). Clearly, non‐autonomous signaling between tissues conveys information about changes in proteostasis internal to the organism. Yet it remains unclear how animals use sensory signaling to detect external (environmental) cues indicative of adversity and activate stress response pathways so as to offset potential damage (Aballay, 2013; Van Oosten‐Hawle & Morimoto, 2014; Volovik et al, 2014; Mardones et al, 2015).
Another proteostasis protection mechanism that can be modulated non‐autonomously is the ubiquitin proteasome system (UPS). For example, germ line ablation activates the expression of the proteasome subunit RPN‐6 in C. elegans via regulation by the DAF‐16 (FOXO) transcription factor (Vilchez et al, 2012). In addition, EGF signaling can augment UPS activity in epithelial cells as C. elegans enter their period of peak fecundity (Liu et al, 2011). Reduced UPS activity, particularly as an organism ages, leads to a decline in proteostasis. This is a bidirectional relationship, as a decline in proteostasis (e.g., due to the impairment of chaperones and other protein quality control mechanisms) leads to changes in UPS activity. Thus, the exact relationship between non‐autonomous sensory signaling, UPS activity, and other protein quality control systems remains uncertain.
To identify novel regulators of UPS activity and proteostasis, we used a transgenic approach in C. elegans by monitoring UPS activity using the ubiquitin fusion degradation (UFD) substrate UbG76V‐GFP, an unstable protein that is comprised of a non‐cleavable ubiquitin placed amino‐terminal to GFP (Liu et al, 2011; Segref et al, 2011). UbG76V‐GFP mimics a mono‐ubiquitinated protein, and additional ubiquitins are attached by the UFD complex to K48 residues located on the N‐terminal ubiquitin, eventually resulting in a poly‐ubiquitinated substrate that is degraded by the 26S proteasome (Butt et al, 1988; Johnson et al, 1995; Dantuma et al, 2000). UbG76V‐GFP degradation was previously monitored in C. elegans intestinal or hypodermal epithelia when this UFD substrate was expressed from either the sur‐5 or col‐19 promoter, respectively (Liu et al, 2011; Segref et al, 2011). Using this reporter, here we have screened for genes that play a role is regulating UbG76V‐GFP degradation in the epithelia. We found that the neurotransmitter dopamine (DA), which is made by mechanosensory neurons in C. elegans and used to transmit information about surrounding environmental viscosity to the motoneurons so as to modulate feeding behavior (Sawin et al, 2000; Chase et al, 2004; Kindt et al, 2007; Suo & Ishiura, 2013), also functions to modulate UbG76V‐GFP turnover in epithelia. In the absence of DA signaling or mechanosensation of the external environment, intestinal and hypodermal epithelia show increased stability of the otherwise unstable UbG76V‐GFP reporter protein. The regulation of UbG76V‐GFP degradation by DA signaling requires the cAMP response element binding protein transcription factor (CREB) and the ELT‐3 GATA transcription factor, and we find by expression profile analysis that DA signaling promotes the expression of the xenobiotic stress response genes. These genes, in turn, are required for DA to modulate UbG76V‐GFP degradation. Failure to signal through DA and its receptors results in animals with altered proteostasis, impaired survival at high temperature, increased sensitivity to infection by pathogenic bacteria, and increased sensitivity of unstable proteins to reactive oxygen species (ROS) exposure. Our results suggest that dopaminergic sensory neurons, in addition to slowing down locomotion upon sensing a potential bacterial feeding source, also signal to epithelial tissues to modulate proteostasis and prepare for infection by a potential bacterial pathogen.
Results
Dopaminergic signaling modulates protein turnover
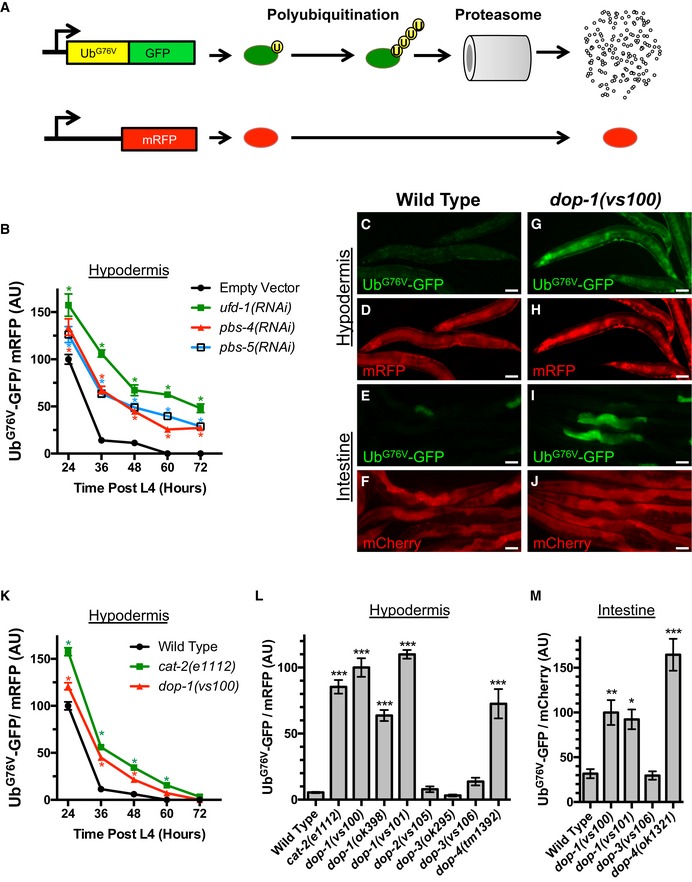
In order to examine changes in UPS activity, we employed previously described transgenic strains that express an UbG76V‐GFP reporter, allowing us to monitor UbG76V‐GFP degradation in C. elegans intestinal or hypodermal epithelia when the reporter was expressed from either the sur‐5 or col‐19 promoter, respectively (Liu et al, 2011; Segref et al, 2011). UbG76V‐GFP levels inversely reflect the ability of the UPS to remove damaged and unstable proteins. Moreover, UbG76V‐GFP levels can be compared to a relatively stable control protein (either mCherry or mRFP) expressed from the same promoter to rule out differences due to transgene expression (Fig 1A). In hypodermis, the expression of UbG76V‐GFP and mRFP from the col‐19 promoter beginning at the L4 stage of development results in the rapid accumulation of both proteins within 24 h (L4 + 24 h; Fig 1B). However, UbG76V‐GFP is rapidly degraded upon entering the period of peak fecundity (L4 + 48 h and onward, Fig 1B–D). We also examined UbG76V‐GFP in transgenic animals expressing UbG76V‐GFP from the sur‐5 promoter, focusing on intestinal expression of the reporter from this broadly expressed promoter. We found a similar turnover of the protein in intestine by L4 + 48 h as that observed in hypodermis (Fig 1E and F). UbG76V‐GFP degradation is mediated in part by poly‐ubiquitination by the UFD E3/E4 ligase complex and proteolysis by the 26S proteasome (Liu et al, 2011; Segref et al, 2011). We reconfirmed this finding by performing RNAi of subunits from these complexes, which resulted in increased peak accumulation of UbG76V‐GFP at L4 + 24 h, as well as a slower rate of UbG76V‐GFP turnover and a plateaued steady state level of UbG76V‐GFP at later time points (Fig 1B). The levels of the unstable UbG76V‐GFP protein therefore provide an index of UPS activity and proteostasis.
Figure 1. Dopaminergic signaling modulates protein turnover.
-
ASchematic representation of the UbG76V‐GFP chimeric reporter and its associated internal control (mRFP or mCherry, in red). The amino acid sequences for ubiquitin are in yellow, whereas the sequences for GFP are in green. The UbG76V‐GFP chimera, which contains a mutation in the terminal residue of ubiquitin, cannot be cleaved. The resulting protein is a substrate for poly‐ubiquitination (indicated by the circles labeled with “U”) and degradation by the proteasome. High levels of reporter poly‐ubiquitination and/or proteasome activity result in little or no GFP fluorescence.
-
BQuantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis from animals of the indicated time point (in hours) after the L4 stage. Animals have been exposed to the indicated knockdown RNAi bacterial strains or a strain that only contains the empty RNAi vector. *P < 0.001, Student's t‐test corrected for multiple comparisons at each time point using the Holm–Sidak method. N = 20 animals per genotype and time point. Error bars indicate SEM.
-
C, DCo‐expression via the col‐19 promoter of UbG76V‐GFP (green) and mRFP (red) in C. elegans hypodermis from a single integrated transgene at L4 + 48 h. Wild‐type animals are shown. Scale bar, 50 μm. UbG76V‐GFP fluorescence is barely detectable.
-
E, FCo‐expression via the sur‐5 promoter of UbG76V‐GFP (green) and mCherry (red) in C. elegans from two separate integrated transgenes (with focus on the intestine) at L4 + 48 h. Wild‐type animals are shown. Scale bar, 50 μm. UbG76V‐GFP fluorescence is barely detectable.
-
G, HCo‐expression of UbG76V‐GFP (green) and mRFP (red) in C. elegans hypodermis at L4 + 48 h in dop‐1(vs100) mutants. Scale bar, 50 μm. Abundant, stable UbG76V‐GFP fluorescence is observed.
-
I, JCo‐expression of UbG76V‐GFP (green) and mCherry (red) in C. elegans from the sur‐5 promoter at L4 + 48 h in dop‐1(vs100) mutants. Scale bar, 50 μm. Abundant, stable UbG76V‐GFP fluorescence is observed, particularly in the intestine.
-
KQuantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis from animals of the indicated time point (in hours) after the L4 stage and of the indicated genotype. *P < 0.001, Student's t‐test corrected for multiple comparisons at each time point using the Holm–Sidak method. N = 20 animals per genotype and time point. Error bars indicate SEM.
-
LQuantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis of animals at L4 + 48 h and of the indicated genotypes. ***P < 0.001, ANOVA with Dunnett's post hoc comparison to the wild‐type control. N = 20 animals per genotype and time point. Error bars indicate SEM.
-
MQuantified fluorescence of UbG76V‐GFP normalized to mCherry (both expressed by the sur‐5 promoter) in the intestine of animals at L4 + 48 h and of the indicated genotypes. ***P < 0.001, **P < 0.01, *P < 0.05, ANOVA with Dunnett's post hoc comparison to the wild‐type control. N = 20 animals per genotype and time point. Error bars indicate SEM.
We used the UbG76V‐GFP reporter in the hypodermis to perform an RNAi screen for regulators of UPS activity, screening an RNAi library enriched for signal transduction molecules, synaptic proteins cytoskeletal regulators, and membrane trafficking molecules (Sieburth et al, 2005). From 2,072 screened clones, we identified 31 RNAi candidates with altered UbG76V‐GFP/mRFP ratios at the L4 + 48 h time point. Particularly striking from this screen was the stabilization of UbG76V‐GFP in animals knocked down for a receptor for the neurotransmitter dopamine (DA). We hypothesized that dopamine signaling might regulate proteostasis and investigated this hypothesis by examining UbG76V‐GFP in true mutants for genes involved in DA signaling. For example, loss‐of‐function mutations in the DA receptor dop‐1 resulted in elevated UbG76V‐GFP levels in both the intestine and the hypodermis (Fig 1G–K). Animals deficient in DA synthesis itself showed a similar phenotype. CAT‐2 encodes tyrosine hydroxylase, which is essential for DA synthesis (Lints & Emmons, 1999), and we found that loss‐of‐function mutants for cat‐2 contained elevated ratios of UbG76V‐GFP to mRFP in the hypodermis at L4 + 48 h (Fig 1K and L). We found that three independently isolated loss‐of‐function alleles in the receptor dop‐1, a D1‐like DA receptor (DAR), resulted in elevated UbG76V‐GFP in both epithelial tissue types (Fig 1G–M). In addition, loss‐of‐function mutations in dop‐4, the other D1‐like DAR in C. elegans, also resulted in elevated UbG76V‐GFP (Fig 1L and M). By contrast, loss of dop‐2 or dop‐3, which encode D2‐like DARs, did not affect UbG76V‐GFP (Fig 1L and M). These results indicate that DA signaling promotes the degradation of unstable UPS substrates in epithelia via activation of D1‐like DARs.
Dopaminergic neurons secrete DA into the pseudocoelomic body cavity where it can diffuse to multiple tissues. For example, secreted DA acts through the receptors DOP‐2 and DOP‐3 on motoneurons to regulate body wall muscle contraction and locomotion (Chase et al, 2004). We reasoned that DA might regulate UbG76V‐GFP turnover in epithelia either directly (by acting through DA receptors like DOP‐1 on epithelial membranes) or indirectly (by acting through DA receptors on neurons, which in turn transmit a second signal to epithelia). Previous studies of DOP‐1 focused on its expression in neurons using GFP‐based expression reporters (Tsalik et al, 2003; Chase et al, 2004; Sanyal et al, 2004). We examined transgenic animals for one of these reporters, adEx1647, a translational reporter that contains about 4 kb of upstream promoter sequences and about 5 kb of the coding exons and introns (almost the entire coding region) (Tsalik et al, 2003). We found that DOP‐1, in addition to being expressed in several sets of neurons, is also expressed in adult epithelial cells, including the intestine and hypodermis (Fig EV1A and B). To address whether DOP‐1 function is required cell autonomously in epithelia to regulate UbG76V‐GFP turnover, we generated a transgene, P vha‐6 ::dop‐1(+), containing the vha‐6 promoter, which is specific for the intestine, and coding sequences for the wild‐type dop‐1 gene. As a positive control, we also generated a complete wild‐type rescuing dop‐1 transgene, called P dop‐1 ::dop‐1(+), containing the dop‐1 promoter and coding sequences. These transgenes were separately introduced into dop‐1 mutants containing the P sur‐5 ::Ub G76V ‐GFP. The resulting transgenic animals were examined for fluorescence at L4 + 48 h. In contrast to dop‐1 mutants lacking either rescuing transgene, dop‐1 mutants with either P dop‐1 ::dop‐1(+) or P vha‐6 ::dop‐1(+) showed wild‐type levels of UbG76V‐GFP turnover (Fig EV1C). These results demonstrate that DOP‐1 is required cell autonomously in epithelia to regulate UbG76V‐GFP turnover and support a model in which DA acts on receptors directly on epithelial cell membranes.
Figure EV1. DOP‐1 receptors act in epithelial cells.
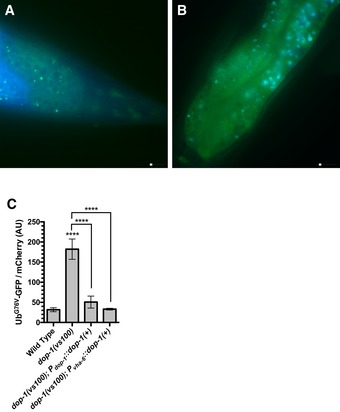
- Fluorescence image of the tail region of transgenic dop‐1::gfp animals, with GFP in green and endogenous autofluorescence (captured with DAPI filters) in blue. Scale bar, 10 μm.
- Fluorescence image of the intestine of transgenic dop‐1::gfp animals, with GFP in green and endogenous autofluorescence (captured with DAPI filters) in blue. Scale bar, 10 μm.
- Quantified fluorescence of UbG76V‐GFP normalized to mCherry in the intestine from L4 + 48 h animals of the indicated genotypes. ****P < 0.0001, ANOVA with Dunnett's post hoc comparison to the wild‐type control (for dop‐1 mutants) or with Sidak post hoc comparison between indicated genotypes (brackets). N = 20 animals per genotype and time point. Error bars indicate SEM.
Dopaminergic signaling promotes protein poly‐ubiquitination without perturbing proteasome function
The stabilization of UbG76V‐GFP protein in DA signaling mutants could be due to either a reduction in its poly‐ubiquitination or a reduction in its proteolysis by the proteasome in these mutants. To determine the molecular mechanism by which DA signaling mediates UbG76V‐GFP turnover, we examined UbG76V‐GFP protein by Western blot, detecting it with either anti‐ubiquitin antibodies or anti‐GFP antibodies (Fig 2A). We detected little UbG76V‐GFP protein when it was expressed in hypodermis in wild‐type animals at L4 + 48 h. In cat‐2, dop‐1, and dop‐4 mutants, however, we detected an increase in non‐ubiquitinated, mono‐ubiquitinated, and di‐ubiquitinated species, suggesting that these mutants either have a reduction in initial mono‐ubiquitination and subsequent poly‐ubiquitin chain extension, or have reduced ability to clear ubiquitinated substrates (Fig 2A). While we did not detect proteins larger than 50 kDa in DA signaling mutants using the anti‐GFP antibodies, we did detect additional proteins ranging from 50 to 80 kDa in these mutants using anti‐ubiquitin antibodies. We quantified the accumulation of these proteins by conducting Western blots using anti‐ubiquitin antibodies on multiple biological replicates of wild‐type animals and dop‐1 mutants (Fig EV2A). While we cannot determine whether such proteins are mono‐, di‐, or poly‐ubiquitinated, our results suggest that ubiquitinated forms of some endogenous proteins accumulate in DA signaling mutants. We also observed a similar increase in non‐ubiquitinated and partially ubiquitinated proteins in these mutants when the reporter was expressed from the sur‐5 promoter, which is more broadly expressed (Fig 2B). Together, these results suggest that DA signaling is required for normal levels of either protein ubiquitination, proteolysis by the proteasome, or both.
Figure 2. Dopaminergic signaling promotes protein poly‐ubiquitination.
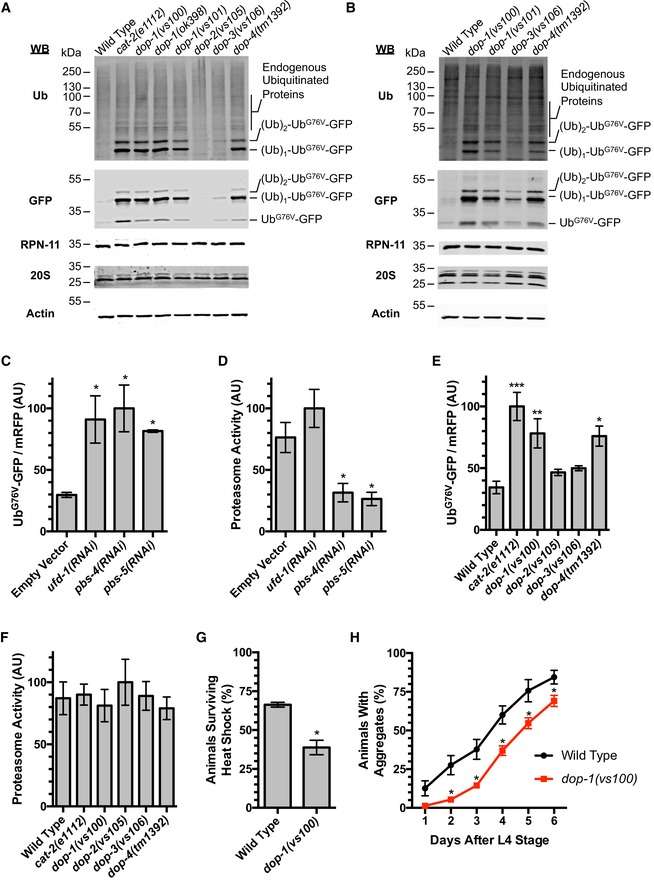
- Western blot of lysed nematodes at the L4 + 48 h stage, probed with antibodies recognizing ubiquitin, GFP, RPN‐11, multiple 20S proteasome subunits, or actin as a loading control. Animals express UbG76V‐GFP protein in their hypodermis. The position of molecular weight markers is shown on the left of each blot. The position of UbG76V‐GFP protein, as well as UbG76V‐GFP with the indicated number of additional ubiquitin moieties, based on molecular weight, is indicated on the right of each blot. Twenty animals were loaded per lane for each indicated genotype (WT indicates wild type).
- Western blot of lysed nematodes at the L4 + 48 h stage, probed and annotated as in (A). Animals express UbG76V‐GFP protein from the sur‐5 promoter. Thirty animals were loaded per lane for each indicated genotype.
- Quantified fluorescence of UbG76V‐GFP normalized to mRFP from lysates of transgenic nematodes at L4 + 48 h that express these reporters in the hypodermis. Animals were exposed to bacteria containing the indicated RNAi clone or an empty RNAi vector as a control. *P < 0.05, ANOVA with Dunnett's post hoc comparison to the empty RNAi vector control. N = 3 trials. Error bars indicate SEM.
- Quantified epoxomicin‐sensitive proteasome activity (as measured through the turnover of fluorescent chymotrypsin substrate) from the same lysates as in (C). *P < 0.05, ANOVA with Dunnett's post hoc comparison to the empty RNAi vector control. N = 3 trials. Error bars indicate SEM.
- Quantified fluorescence of UbG76V‐GFP normalized to mRFP from lysates of transgenic nematodes at L4 + 48 h that express these reporters in the hypodermis. Animals were of the indicated genotype. ***P < 0.001, **P < 0.01, *P < 0.05, ANOVA with Dunnett's post hoc comparison to wild type. N = 4 trials. Error bars indicate SEM.
- Quantified epoxomicin‐sensitive proteasome activity (as measured through the turnover of fluorescent chymotrypsin substrate) from the same lysates as in (E). No statistical difference (P < 0.05 cutoff) was detected by ANOVA. N = 4 trials. Error bars indicate SEM.
- Percentage of live animals of the indicated genotype assayed after 6‐h exposure to heat shock. 100% of control animals of the same genotype but not given heat shock survived. *P < 0.01, Student's t‐test. N = 3 trials. Error bars indicate SEM.
- Percent of animals of the indicated genotype containing aggregates of Poly(Q)44::YFP at the indicated time (in days) after L4 stage. *P < 0.01, Student's t‐test. N = 3 trials, 50–80 animals per trial per day. Error bars indicate SEM.
Source data are available online for this figure.
Figure EV2. Loss of proteasome activity and DOP‐1 results in an additive effect on protein turnover.
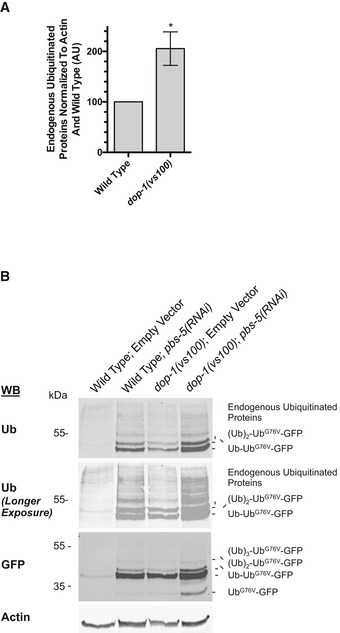
- Quantification of endogenous ubiquitinated proteins in the 50–80 kDa range detected from whole nematode lysates by Western blot using the anti‐ubiquitin antibody. Lysates are from animals of the indicated genotype, and quantification of each replicate blot (N = 4 biological replicates) was normalized to an anti‐actin probing of the same blot followed by normalization to the wild‐type ubiquitin/actin ratio for that blot. Averages are shown along with SEM for error bars. *P < 0.02 using a paired Student's t‐test.
- Western blot of lysed nematodes at the L4 + 48 h stage, probed with antibodies recognizing ubiquitin, GFP, or actin as a loading control. Animals express UbG76V‐GFP protein in their hypodermis. The position of molecular weight markers is shown on the left of each blot. The position of UbG76V‐GFP protein, as well as UbG76V‐GFP with the indicated number of additional ubiquitin moieties, based on molecular weight, is indicated on the right of each blot. Twenty animals were loaded per lane for each indicated genotype and RNAi treatment. For the anti‐ubiquitin by Western blot, a longer exposure is shown to make poly‐ubiquitinated species more visible.
We also directly examined the proteasome in DA signaling mutants. We did not observe changes in the levels of proteasome subunits in lysates for various DA signaling mutants (Fig 2A and B). To directly assay proteasome activity, we generated lysates of L4 + 48 h nematodes that express UbG76V‐GFP in the hypodermis. We first analyzed UbG76V‐GFP and mRFP levels in these lysates by fluorescence spectroscopy (Fig 2C). We observed low levels of UbG76V‐GFP at L4 + 48 h in wild‐type animals using this approach, similar to what we observed by epifluorescence microscopy and Western blot analysis. We also generated lysates from animals exposed to RNAi bacteria, including animals knocked down for the E4 poly‐ubiquitination chain extending enzyme ufd‐1, as well as for the proteasome subunits pbs‐4 and pbs‐5. Exposure to RNAi against any of these genes resulted in stabilized UbG76V‐GFP (Fig 2C). We incubated the same lysates with the fluorescent chymotryptic substrate Suc‐Leu‐Leu‐Val‐Tyr‐AMC in the presence or absence of the proteasome inhibitor epoxomicin to measure proteasome activity over time. As expected, RNAi knockdown of pbs‐4 and pbs‐5 resulted in diminished proteasome activity relative to an empty RNAi vector control (Fig 2D). By contrast, RNAi knockdown of ufd‐1, a gene that normally promotes poly‐ubiquitination of substrates but is not involved in proteasome catalytic activity, as expected did not reduce proteasome activity. These findings indicate that about a threefold decrease in proteasome activity results in about a threefold increase in UbG76V‐GFP levels in this assay.
We next generated lysates from multiple DA signaling mutants. Similar to what we observed by epifluorescence microscopy and Western blot analysis, we found that cat‐2, dop‐1, and dop‐4 mutants have elevated levels of UbG76V‐GFP protein (Fig 2E). We performed the same analysis of proteasome activity in lysates from these mutants; however, we did not observe a substantial change in proteasome activity in any of these mutants (Fig 2F). If the accumulation of UbG76V‐GFP in DA signaling mutants were due to a decrease in proteasome activity, then we should have (i) observed an increase in poly‐ubiquitinated (i.e., 4 or more ubiquitin moieties) UbG76V‐GFP relative to total UbG76V‐GFP in these mutants, and (ii) detected a healthy decrease in our direct measurement of proteasome activity. Instead, the observed decrease in poly‐ubiquitinated proteins and the unchanged activity in our proteasome assay strongly suggest that DA signaling does not regulate proteasome activity.
If DA signaling promotes the turnover of UbG76V‐GFP through increased poly‐ubiquitination instead of increased proteasome activity, then a knockdown of proteasome subunits should further enhance the stability of UbG76V‐GFP in dop‐1 mutants. We examined UbG76V‐GFP by Western blot in wild‐type and in dop‐1 mutants either exposed to empty RNAi vector or pbs‐5(RNAi). Wild‐type animals accumulate mono‐ubiquitinated, di‐ubiquitinated, and poly‐ubiquitinated UbG76V‐GFP when the proteasome is impaired by pbs‐5 RNAi‐mediated knockdown (Fig EV2B). Knockdown of pbs‐5 in dop‐1 mutants resulted in a dramatic additive increase in poly‐ubiquitinated UbG76V‐GFP, demonstrating that the proteasome remains active and degrades any residual poly‐ubiquitinated UbG76V‐GFP in dop‐1 mutants. Taken together, our results suggest that the proteasome is not the prime target of regulation by DA. Instead, DA signaling is required for the proper ubiquitination of unstable proteins like UbG76V‐GFP.
We performed two additional tests to detect changes in protein homeostasis in dop‐1 mutants. First, we examined the sensitivity of dop‐1 mutants to heat shock. After exposing adult nematodes to a 6‐h heat shock (34°C), we found that nearly 70% of wild‐type animals survived, whereas only about a third of dop‐1 mutant adults survived (Fig 2G), suggesting that dop‐1 mutants are less able to handle the unfolded protein burden generated by heat‐shock stress. Second, we examined a polyglutamine protein aggregation reporter expressed in the intestine from the transgene P vha‐6 ::Poly(Q)44::YFP (Mohri‐Shiomi & Garsin, 2008). Polyglutamine proteins form aggregates as C. elegans age, and this process, which is thought to be protective, is facilitated by poly‐ubiquitination (Morley et al, 2002; Hsu et al, 2003; Howard et al, 2007; Mohri‐Shiomi & Garsin, 2008; Kirkin et al, 2009). We found that dop‐1 mutants were less efficient at sequestering polyglutamine proteins into these aggregates during aging (Fig 2H). Taken together, the observed impairment of protein homeostasis in dop‐1 mutants is consistent with a role for DA signaling in promoting healthy proteostasis.
Dopamine signaling promotes protein turnover via the xenobiotic stress pathway
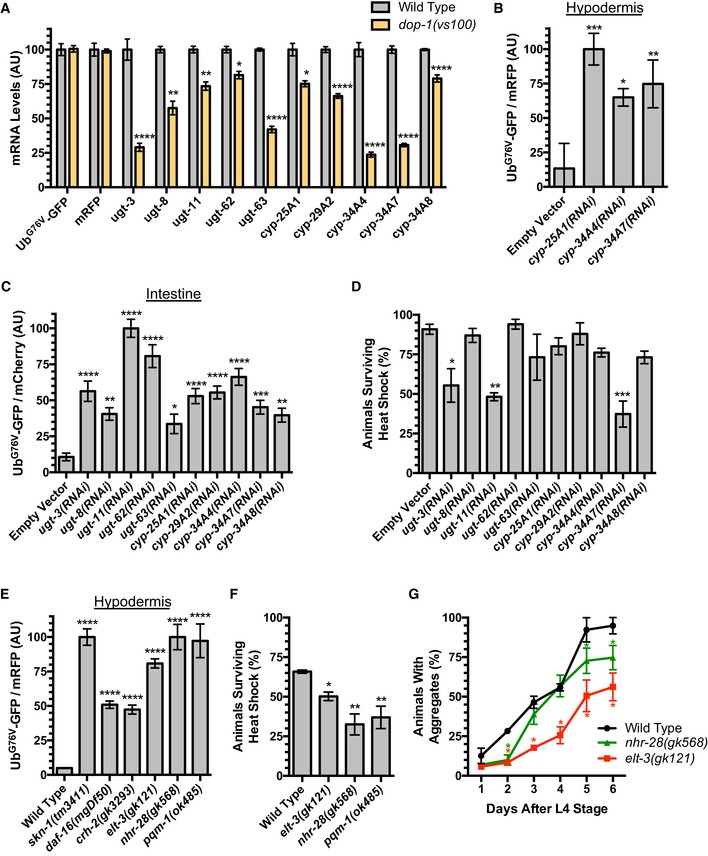
Dopamine signaling can regulate cellular physiology through a combination of transcriptional and posttranscriptional regulatory steps (Kebabian et al, 1972; Cooper et al, 1986; Cadet et al, 2010). To begin to understand the mechanism of DA action with regard to proteostasis, we compared the mRNA expression profile of dop‐1 mutants to wild‐type animals during adulthood (L4 + 48 h) using RNA‐seq. We isolated poly‐A(+) RNA from three replicates of each genotype and subjected each sample to RNA‐seq. We assessed differential gene expression between genotypes using EdgeR and an FDR‐adjusted q‐value (Anders & Huber, 2010), focusing only on genes with significant changes (q < 0.01). As expected, the levels of UbG76V‐GFP and mRFP mRNA did not vary between wild‐type and dop‐1 mutants (Fig 3A). We found that 334 genes were significantly downregulated at least 1.5‐fold in dop‐1 compared to wild type, whereas 436 genes were upregulated in dop‐1 relative to wild type (Table EV1). We used DAVID functional annotation clustering enrichment analysis (Da Huang et al, 2009a,b) on the genes regulated by DOP‐1 signaling (Table EV2). Particularly striking in the set of genes that were downregulated in dop‐1 mutants compared to wild type was the enrichment of annotation clusters for genes involved in xenobiotic and endobiotic detoxification and metabolism (Lindblom & Dodd, 2006), including oxidoreductase activity (multiple CYP or cytochrome P450 enzymes), lipid modification, and glycosyl transferases (multiple UGT or UDP‐glucuronosyltransferases) (Table 1). These results suggest that DA signaling through the DOP‐1 receptor promotes xenobiotic detoxification.
Figure 3. Dopamine signaling promotes protein turnover via the xenobiotic stress pathway.
-
ARelative abundance of mRNA for the indicated xenobiotic stress response gene (or UbG76V‐GFP or mRFP) detected in wild‐type versus dop‐1 mutants. Values are based on RNA‐seq/EdgeR analysis, with the relative levels for each gene normalized to the value observed in the wild‐type control. ****P < 0.0001, **P < 0.01, *P < 0.05, FDR from EdgeR analysis using a Benjamini and Hochberg correction. Error bars indicate SEM.
-
B, CQuantified fluorescence of UbG76V‐GFP normalized to (B) mRFP in the hypodermis or (C) mCherry in the intestine from L4 + 48 h animals exposed to feeding RNAi for either an empty vector control or the indicated xenobiotic stress gene. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ANOVA, Tukey's multiple comparison test compared to control. N = 20 animals per genotype and time point. Error bars indicate SEM.
-
DPercentage of live animals raised on the indicated feeding RNAi (or empty vector control) bacteria assayed after 6‐h exposure to heat shock. 100% of control animals of the same genotype but not given heat shock survived. ***P < 0.001, **P < 0.01, *P < 0.05, ANOVA, Tukey's multiple comparison test compared to wild type. N = 3 trials. Error bars indicate SEM.
-
EQuantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis from L4 + 48 h animals of the indicated genotype. ****P < 0.0001, ANOVA, Tukey's multiple comparison test compared to wild type. N = 20 animals per genotype and time point. Error bars indicate SEM.
-
FPercentage of live animals of the indicated genotype assayed after 6‐h exposure to heat shock. 100% of control animals of the same genotype but not given heat shock survived. **P < 0.01, *P < 0.05, ANOVA, Tukey's multiple comparison test compared to wild type. N = 3 trials. Error bars indicate SEM.
-
GPercent of animals of the indicated genotype containing aggregates of Poly(Q)44::YFP at the indicated time (in days) after L4 stage. *P < 0.001, Student's t‐test corrected for multiple comparisons at each time point using the Holm–Sidak method. N = 3 trials, 50–80 animals per trial per day. Error bars indicate SEM.
Table 1.
DAVID functional annotation clustering enrichment analysis for genes regulated by DOP‐1 signaling
| Annotation cluster | Cluster enrichment score | Cluster EASE score P‐value |
|---|---|---|
| Genes that are downregulated in dop‐1 mutants | ||
| Collagens and cuticle formation | 9.60 | 2.5 × 10−10 |
| Oxidoreductase activity and cytochrome P450s | 3.73 | 1.9 × 10−4 |
| Uncharacterized C. elegans proteins | 2.35 | 4.5 × 10−3 |
| UDP‐glucuronosyltransferases | 2.22 | 6.0 × 10−3 |
| Extracellular protease inhibitors | 2.11 | 7.8 × 10−3 |
| Genes that are upregulated in dop‐1 mutants | ||
| Hedgehog receptor signaling | 3.49 | 3.2 × 10−4 |
| C‐lectins | 2.73 | 1.9 × 10−3 |
| Heat‐shock proteins | 2.26 | 5.5 × 10−3 |
Individual annotation clusters generated by DAVID functional clustering are listed for genes that are downregulated (top half of table) and upregulated (bottom half of table) in dop‐1 mutants relative to wild type. The cluster enrichment score is the geometric mean (in –log scale) of the P‐values (from Fisher's exact test with DAVID EASE Score modification, also shown) of all of the individual annotations within that cluster.
We also noted that there were annotation clusters of genes that were upregulated in dop‐1 mutants compared to wild type and that these included C‐lectins and heat‐shock chaperones (Table 1). The elevated level of heat‐shock gene expression in dop‐1 mutants even at permissive temperatures (20°C) suggests that DA signaling is needed to prevent a heat‐shock response, which is consistent with impaired proteostasis in these mutants. The elevated levels of heat‐shock genes in dop‐1 mutants are not likely to be the cause of UbG76V‐GFP stabilization in those mutants because RNAi knockdown of those genes, including hsp‐4, hsp‐16.2, hsp‐16.11, hsp‐16.41, hsp‐16.48, hsp‐16.49, hsp‐17, and hsp‐43, did not suppress the stabilization of UbG76V‐GFP in dop‐1 mutants: All dop‐1 animals exposed to RNAi knockdown for these genes resembled dop‐1 mutants given empty vector RNAi, with nearly all animals showing UbG76V‐GFP fluorescence at L4 + 48 h. Indeed, loss‐of‐function mutations in the chaperones hsp‐16.2 and hsp‐43, as well as the heat‐shock transcriptional regulator hsf‐1, in an otherwise wild‐type background resulted in UbG76V‐GFP stabilization, suggesting that impaired heat‐shock proteostasis is sufficient to block UbG76V‐GFP turnover (Fig EV3A).
Figure EV3. Additional mutations that slow reporter protein turnover.
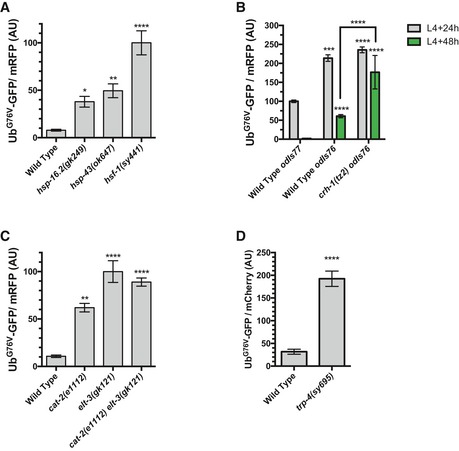
- Quantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis from L4 + 48 h animals of the indicated genotype. ****P < 0.0001, **P < 0.01, *P < 0.05, ANOVA with Dunnett's post hoc comparison to the wild‐type control for the respective time point. N = 20 animals per genotype and time point. Error bars indicate SEM.
- Whereas the odIs77[P col‐19 ::Ub G76V ‐GFP]‐integrated transgene is used for all other experiments, it is closely linked to the crh‐1 gene. Therefore, we examined UbG76V‐GFP levels in crh‐1 mutants using the odIs76[P col‐19 ::Ub G76V ‐GFP]‐integrated transgene, which expresses UbG76V‐GFP at higher levels than does the odIs77 transgene, but with the same pattern of increased turnover at L4 + 48 h (some residual UbG76V‐GFP remains at L4 + 48 h in odIs76 transgenics, but all of the protein is turned over by L4 + 72 h). Mutations in crh‐1 block UbG76V‐GFP turnover in odIs76 transgenics. Quantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis from animals of the indicated stage (gray for L4 + 24 h, green for L4 + 48 h) and the indicated mutant. ****P < 0.0001, ***P < 0.001, ANOVA with Dunnett's post hoc comparison to the wild‐type odIs77 transgenics at L4 + 24 h. Bracket indicates a Bonferroni post hoc comparison, ****P < 0.0001. N = 20 animals per genotype and time point. Error bars indicate SEM.
- Quantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis from L4 + 48 h animals of the indicated genotype. ****P < 0.0001, **P < 0.01, ANOVA with Dunnett's post hoc comparison to the wild‐type control for the respective time point. N = 20 animals per genotype and time point. Error bars indicate SEM.
- Quantified fluorescence of UbG76V‐GFP normalized to mCherry in the intestine from L4 + 48 h animals of the indicated genotypes. ****P < 0.0001, Student's t‐test. N = 20 animals per genotype and time point. Error bars indicate SEM.
Given the decreased expression of multiple xenobiotic detoxification genes in the absence of DA signaling (Fig 3A), we hypothesized that the changes in UbG76V‐GFP stability in DA signaling mutants might be due to impaired xenobiotic detoxification. We tested this idea by knocking down the expression of individual candidate genes from our RNA‐seq analysis using RNAi and examining the effect on UbG76V‐GFP levels. We found that the knockdown of multiple CYP and UGT genes resulted in the stabilization of UbG76V‐GFP in the hypodermis (Fig 3B) and the intestine (Fig 3C) relative to internal controls (mRFP and mCherry, respectively). We also examined the sensitivity to heat shock of animals knocked down for CYP and UGT genes. After a 6‐h heat shock (34°C), we found a significant reduction in the number of surviving animals knocked down by RNAi for ugt‐3, ugt‐11, and cyp‐34A7 compared to an empty vector control (Fig 3D), suggesting that animals lacking the activity of these xenobiotic stress genes are less able to handle heat‐shock stress. These results suggest that the turnover of unstable UbG76V‐GFP protein and cellular UPS activity is modulated by the levels of xenobiotic detoxification enzymes, and that DA signaling, at least in part, modulates proteostasis in epithelia by promoting xenobiotic metabolism.
Given the large number of CYP and UGT genes in the genome, we were concerned that there might be a fair amount of functional redundancy between the xenobiotic detoxification components that might preclude observing a full phenotype in any single RNAi experiment. We reasoned that mutants for known master regulators of these genes might represent a more comprehensive impairment of the xenobiotic response. Xenobiotic stress response genes contribute to innate immunity against bacterial pathogens (Papp et al, 2012; Runkel et al, 2013; Pellegrino et al, 2014; Pukkila‐Worley et al, 2014), and many of these genes are also regulated as part of the oxidative stress response (mediated by the SKN‐1/Nrf2 transcription factor) and the insulin signaling system (mediated by the DAF‐16/FOXO transcription factor) (An & Blackwell, 2003; Murphy et al, 2003; Oliveira et al, 2009; Park et al, 2009; Mair et al, 2011). Indeed, DAF‐16 ChIP‐seq sites are located at nearly all of the CYP and UGT genes in our RNA‐seq dataset, and cyp‐34A7 was identified as a positive transcriptional target of DAF‐16 through microarray analysis (Murphy et al, 2003). Many are also regulated by cAMP signaling and the CRH‐1/CREB transcription factor, which acts downstream of DA signaling for its role in behavior modulation (Cadet et al, 2010; Beaulieu & Gainetdinov, 2011; Mair et al, 2011; Suo & Ishiura, 2013). Xenobiotic genes are also regulated by the GATA transcription factor ELT‐3, with ChIP‐seq sites for ELT‐3 near ugt‐3, ugt‐8, ugt‐11, ugt‐63, cyp‐25A1, cyp‐29A2, cyp‐34A7, and cyp‐34A8 (Budovskaya et al, 2008). ChIP‐seq sites for the transcription factors NHR‐28 and PQM‐1 are also found at nearly every CYP and UGT gene identified in our RNA‐seq analysis (Miyabayashi et al, 1999; Gerstein et al, 2010; Tepper et al, 2013; Araya et al, 2014), suggesting that these might be additional regulators of the xenobiotic stress response genes.
We examined UbG76V‐GFP levels in loss‐of‐function mutants for skn‐1, daf‐16, the two C. elegans CREB genes (crh‐1 and crh‐2), elt‐3, nhr‐28, and pqm‐1. We found that UbG76V‐GFP was stabilized in each case (Figs 3E and EV3B), suggesting that broad regulators of the xenobiotic stress response and the detoxification pathway are required for UbG76V‐GFP turnover. The effects of cat‐2 and let‐3 mutations on UbG76V‐GFP turnover are not additive, as double mutants showed similar levels of stabilization relative to single mutants (Fig EV3C), consistent with these genes acting in the same genetic pathway. Consistent with UbG76V‐GFP stabilization in these mutants, daf‐16 mutants are sensitive to heat shock (Saul et al, 2008), and skn‐1 knockdown by RNAi results in impairment of UPS activity and elevated levels of heat‐shock proteins (Saul et al, 2008). We also examined elt‐3, nhr‐28, and pqm‐1 mutants for sensitivity to heat shock, finding a significant reduction in the number of surviving animals in these mutants compared to wild type after a 6‐h heat shock (34°C) (Fig 3F), suggesting that they are less able to handle heat‐shock stress. We also introduced the P vha‐6 ::Poly(Q)44::YFP into elt‐3 and nhr‐28 mutants (the reporter was too closely linked to pqm‐1 for analysis in pqm‐1 mutants) and found that these mutants were less efficient at sequestering polyglutamine proteins into aggregates (Fig 3G). Taken together, our results indicate that master regulators of the xenobiotic stress response are required for proper proteostasis.
Mechanosensory dopaminergic neurons are required to modulate protein turnover in epithelia
Mechanosensory neurons release DA in response to shear stress on the body wall as nematodes enter high‐viscosity bacterial lawns (Sawin et al, 2000; Kindt et al, 2007). Given that DA signaling promotes the expression of xenobiotic stress response genes, we reasoned that such signaling might be (i) activated by pathogenic bacteria and (ii) required for animals to survive pathogenic infection. To explore this possibility, we first tested whether mechanosensation is required to modulate UbG76V‐GFP turnover in epithelial tissues by examining mutants lacking mec‐5, a unique collagen required for all mechanosensation in the nematode, and trp‐4, a mechanosensory TRPN (NOMPC) channel expressed and required specifically in the mechanosensory dopaminergic neurons (Du et al, 1996; Emtage et al, 2004; Li et al, 2006; Kang et al, 2010). We found that UbG76V‐GFP turnover is reduced in mec‐5 and trp‐4 mutants (Figs 4A and EV3D), although not to the same extent as in DA signaling mutants, indicating that the activation of these neurons by environmental mechanical stimulation is required to fully promote UbG76V‐GFP turnover in epithelia, but that these dopaminergic neurons might also promote UbG76V‐GFP turnover in response to additional environmental cues besides mechanical stimulation.
Figure 4. Dopamine signaling modulates xenobiotic stress survival.
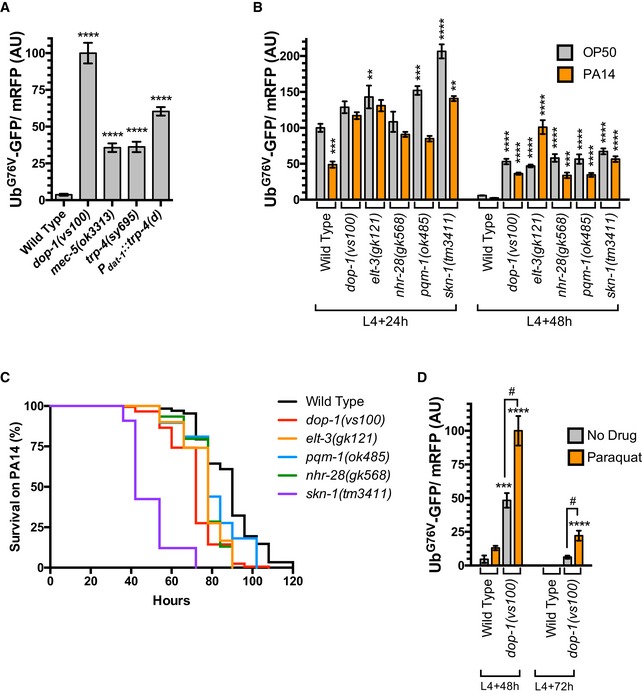
- Quantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis from L4 + 48 h animals of the indicated genotype. ****P < 0.0001, ANOVA, Tukey's multiple comparison test compared to wild type. N = 20 animals per genotype. Error bars indicate SEM.
- Quantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis from either L4 + 24 h animals (left half of graph) or L4 + 48 h animals (right half of graph) of the indicated genotype. Animals were either continually grown on regular OP50 food (gray bars) or switched to PA14 food at the L4 stage (orange bars). ****P < 0.0001, ***P < 0.001, **P < 0.01, ANOVA, Tukey's multiple comparison test compared to wild type. N = 20 animals per genotype. Error bars indicate SEM.
- Survival curves (percentage of live animals assayed each hour after PA14 exposure) for the indicated mutants at 20°C. P log‐rank < 0.0001. Median survival time was 90 h for wild type (N = 304), 72 h for dop‐1(vs100) (N = 180), 78 h for elt‐3(gk121) (N = 120), 78 h for pqm‐1(ok485) (N = 116), 78 h for nhr‐28(gk568) (N = 123), and 42 h for skn‐1(tm3411) (N = 33). Eight wild‐type animals and six dop‐1 mutants were censored (the animals crawled up the side of the plate and dessicated). The media contained 50 μg/ml of 5‐fluoro‐2′‐deoxyuridine (FUdR) to prevent the growth of egg offspring during the experiment.
- Quantified fluorescence of UbG76V‐GFP normalized to mRFP in the hypodermis from either L4 + 48 h animals (left half of graph) or L4 + 72 h animals (right half of graph) of the indicated genotype. Animals were either continually grown on regular OP50 food (gray bars) or switched to NGM plates containing 1 mM paraquat at the L4 stage (orange bars). ****P < 0.0001, ***P < 0.001, ANOVA, Tukey's multiple comparison test compared to wild type. # P < 0.001, ANOVA, Bonferroni multiple comparison as indicated by the bracketed lines. N = 20 animals per genotype. Error bars indicate SEM. No UbG76V‐GFP fluorescence was detected in wild type at L4 + 72 h on NGM plates containing either 1 mM paraquat or no drug at all.
TRP‐4 is expressed primarily in dopaminergic neurons (ADE, CEP, PDE), but it is also found in the neurons DVA and DVC. To determine whether the dopaminergic neurons specifically are required to promote UbG76V‐GFP turnover in epithelia, we took advantage of a transgene, P dat‐1 ::trp‐4(d), that expresses a toxic TRP‐4 mutant channel under the control of the dat‐1 promoter and is thus only active in dopaminergic neurons (Nagarajan et al, 2014). The transgene triggers a progressive cell death of the dopaminergic neurons but not DVA or DVC. We introduced this transgene into nematodes that express UbG76V‐GFP and found that UbG76V‐GFP turnover was reduced (Fig 4A). The magnitude of the effect caused by P dat‐1 ::trp‐4(d) was not to the same extent as that observed in DA signaling mutants; however, not all of dopaminergic neurons undergo neurodegeneration in many of the P dat‐1 ::trp‐4(d) transgenic animals (Nagarajan et al, 2014), so a partial phenotype relative to DA signaling mutants is expected. Taken together, our results show that the dopaminergic neurons are required non‐autonomously to regulate the turnover of unstable proteins like UbG76V‐GFP in epithelia.
Xenobiotic stress modulates protein turnover via dopamine‐dependent mechanism
We next tested whether exposure to a different type of bacteria—in this case the pathogenic PA14 strain of Pseudomonas aeruginosa—results in altered UbG76V‐GFP turnover compared to the standard, non‐pathogenic OP50 E. coli feeding strain. We found that UbG76V‐GFP levels relative to mRFP control were depressed (even at the earlier L4 + 24 h time point) in nematodes that were switched to PA14 lawns beginning at the L4 stage compared to those grown continually on OP50 (Fig 4B), suggesting that the ability of epithelial cells to remove unstable proteins like UbG76V‐GFP is enhanced in nematodes exposed to PA14 bacteria. Mutations in dop‐1 blocked the elevated turnover of UbG76V‐GFP at L4 + 24 h, as well as at later time points (Fig 4B). In addition, loss‐of‐function mutations in the xenobiotic stress regulators elt‐3, nhr‐28, and pqm‐1, as well as a mutation in skn‐1, which also depresses xenobiotic stress response gene expression and impairs innate immunity against bacterial pathogens (An & Blackwell, 2003; Oliveira et al, 2009; Park et al, 2009; Papp et al, 2012), all blocked UbG76V‐GFP turnover (Fig 4B). These results indicate that pathogenic bacteria like PA14 can promote the turnover of unstable proteins through a process that requires DA signaling and most likely the xenobiotic stress response. These results are consistent with previous observations that the specific strain of the bacterial food source can influence proteostasis and protein poly‐ubiquitination in feeding nematodes (Segref et al, 2011); however, unlike the variant non‐pathogenic E. coli strains used in the previous study, which reduced UbG76V‐GFP turnover, we find that pathogenic P. aeruginosa triggers accelerated turnover.
If DA signaling mediates a xenobiotic defense response, then one would expect that loss‐of‐function mutants for either DA signaling components or regulators of the xenobiotic defense response should show increased sensitivity to bacterial infection. We examined this idea by quantifying nematode survival on lawns of PA14 over time. Wild‐type nematodes begin to succumb to PA14 after about 70 h of exposure, with most animals dying after about 110 h (Fig 4C). We confirmed that mutants for skn‐1 die more rapidly (Fig 4C) when exposed to PA14 (Papp et al, 2012). Similarly, we found that mutants for dop‐1, elt‐3, pqm‐1, and nhr‐28 succumbed to PA14 more quickly than did wild type, although not quite at the rapid rate of skn‐1 mutants, indicating that DA signaling and regulators of the xenobiotic stress response are required for effective survival to pathogen exposure.
If the xenobiotic stress response is required for efficient UbG76V‐GFP turnover, then one can infer that xenobiotic toxins and ROS impair cellular proteostasis, particularly proteostasis maintained by the UPS. Indeed, UbG76V‐GFP turnover is sensitive to a variety of xenobiotic compounds and ROS, including ethanol, cadmium, and paraquat (Segref et al, 2011). We examined UbG76V‐GFP levels in the hypodermis of either untreated animals or animals treated with 1 mM paraquat (Fig 4D). Paraquat treatment only generated a minor increase in UbG76V‐GFP levels in the hypodermis of wild‐type animals. By contrast, paraquat treatment resulted in a twofold increase in UbG76V‐GFP in dop‐1 mutants at L4 + 48 h and persistent UbG76V‐GFP stabilization even out to L4 + 72 h. These results are consistent with the reduced levels of xenobiotic stress response genes in dop‐1 mutants and suggest that DA signaling is required to protect the epithelial proteome from ROS and xenobiotic toxins. Taken together, our findings suggest that DA signaling promotes healthy proteostasis through activation of the xenobiotic stress response in frontline barrier epithelia, and that this regulation is critical for innate immunity against invading pathogens.
Discussion
In previous studies, the GFP‐based UPS substrate UbG76V‐GFP was expressed from transgenes and employed as a reporter for UPS activity (Dantuma et al, 2000; Lindsten et al, 2003; Liu et al, 2011; Segref et al, 2011). In C. elegans, UbG76V‐GFP undergoes regular turnover when expressed in epithelial cells of either the intestine or the hypodermis, and this turnover is enhanced as animals enter peak fecundity. Here, we have used this reporter to screen for modulators of UPS activity (and by extension proteostasis) in epithelia. We found that the dopamine signaling system, including the D1‐like receptors DOP‐1 and DOP‐4, promotes healthy proteostasis and innate immunity by driving the expression of xenobiotic detoxification genes, the activity of which is required to remove xenobiotic compounds and ROS to prevent their impairment of the proteome. Mechanosensory neurons release dopamine in response to environmental viscosity, which is a hallmark of bacterial growth. Nematodes appear to use dopaminergic sensory neurons to sense the bacterial environment for potential pathogens, with the resulting neurohormone signaling in turn modulating proteostasis in epithelial barrier tissues in preparation for possible infection. These conclusions are supported by several pieces of data. First, in the absence of dopamine signaling, a UPS reporter shows depressed turnover in intestine and hypodermis. Second, in the absence of dopamine signaling, there are no detectable changes in proteasome activity, but there is a detectable increase in the fraction of non‐ubiquitinated proteins and lower mobility poly‐ubiquitinated proteins, suggesting that dopamine signaling mutants have impaired mono‐ and poly‐ubiquitination rather than impaired proteasome proteolysis activity. Third, in the absence of dopamine signaling, the levels of multiple xenobiotic detoxification enzymes are reduced. Fourth, RNAi knockdown of these individual xenobiotic detoxification enzymes results in reduced UPS reporter turnover, as does reduction of function in master regulators (crh‐1, crh‐2, skn‐1, daf‐16, elt‐3, nhr‐28, and pqm‐1) of the xenobiotic detoxification response. Fifth, mutations that either impair touch sensation alone in the dopaminergic sensory neurons or kill these neurons outright are sufficient to reduce the turnover of the UPS reporter in epithelia. Sixth, growth on pathogenic bacteria accelerates UPS reporter turnover, and this acceleration requires dopaminergic signaling. Seventh, exposure of nematodes to the ROS generating agent paraquat results in dramatic UPS reporter stabilization when dopaminergic signaling is impaired. Finally, mutations that impair dopaminergic signaling reduce the ability of nematodes to survive pathogenic infection and proteotoxic insult. Taken together, our findings demonstrate that mechanosensory dopaminergic neurons respond to environmental cues and use dopamine as a neurohormone to enhance the ability of frontline epithelial barrier cells to counter potential infection and protein damage.
Modulation of proteostasis by DA signaling likely complements the role of DA signaling in modulating behavior. Multiple published studies demonstrated that C. elegans employ their mechanosensory dopaminergic neurons to sense viscous bacterial lawns and release DA as a long‐distance neurohormone (Sawin et al, 2000; Chase et al, 2004; Kindt et al, 2007; Suo & Ishiura, 2013). DA acts through D2‐like receptors on the motoneurons to slow locomotion so as to facilitate feeding on the newly identified potential food source (Chase et al, 2004; Suo & Ishiura, 2013). We propose that DA simultaneously acts through D1‐like receptors to activate expression of xenobiotic detoxification genes and modulate protein homeostasis in epithelia (Fig EV4). Infection by pathogenic bacteria results in protein damage in barrier epithelia (Mohri‐Shiomi & Garsin, 2008). It is therefore reasonable that a multicellular organism like C. elegans might evolve the ability to sense bacteria and translate such sensation into a neurohormonal signal to activate mechanisms to counteract potential damage. Thus, this sensory‐based signaling mechanism, in addition to activating feeding behaviors in response to a potential bacterial food source, would also prepare the animal in case the food source turns out to be pathogenic. This touch‐based mechanism likely complements other sensory mechanisms that trigger an innate immunity response (Chang et al, 2011; Aballay, 2013; Meisel et al, 2014; Mardones et al, 2015).
Figure EV4. Dopamine regulates behavior and proteostasis through separate receptor pathways.
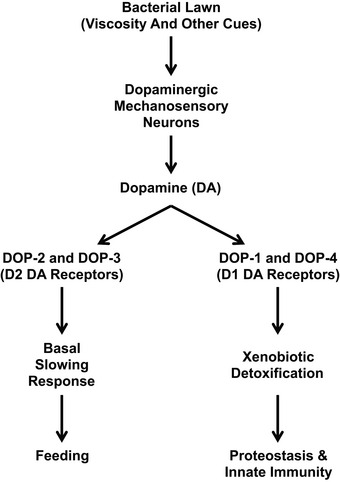
When nematodes encounter a bacterial lawn, the lawn's viscosity is sensed by dopaminergic mechanosensory neurons, which then release the neurohormone dopamine (DA) into the pseudocoelomic body cavity in response. DA binds to the D2‐like DA receptors DOP‐2 and DOP‐3 on motoneurons, resulting in reduced activity of the locomotion circuit and a basal slowing response behavior of the animal, allowing it to feed. DA also binds to the D1‐like DA receptors DOP‐1 and DOP‐4, and we propose that these receptors activate the expression of xenobiotic detoxification genes. The resulting detoxification enzymes maintain protein homeostasis in the barrier epithelia of the hypodermis and the intestine, as well as promote innate immunity.
Dopamine signaling is likely to act through CREB to promote the xenobiotic stress response. DA signaling regulates changes in gene expression through the transcription factor CREB, and the C. elegans CREB homolog CRH‐1 promotes the expression of xenobiotic detoxification genes (Mair et al, 2011; Suo & Ishiura, 2013). We found that mutations in the CREB homologs crh‐1 and crh‐2 result in reduced UPS activity, similar to that observed in DA signaling mutants. The Nrf2 homolog SKN‐1 also promotes the expression of xenobiotic detoxification genes and plays an important role in innate immunity (Inoue et al, 2005; Oliveira et al, 2009; Park et al, 2009; Choe et al, 2012; Papp et al, 2012). Mutations in skn‐1 also result in reduced UPS reporter turnover, but whether DA signaling acts through SKN‐1 remains unknown. The transcription factors DAF‐16, ELT‐3, NHR‐28, and PQM‐1 also regulate xenobiotic detoxification and also show stabilized UPS reporter protein, as does direct xenobiotic compound and ROS exposure, lending further support for the xenobiotic detoxification response as a proteostasis mechanism (Miyabayashi et al, 1999; Murphy et al, 2003; Budovskaya et al, 2008; Gerstein et al, 2010; Segref et al, 2011; Tepper et al, 2013; Araya et al, 2014). It will be interesting to determine whether DA signaling also acts through these transcription factors.
We previously observed that EGF signaling regulates the timing of UbG76V‐GFP turnover in the hypodermis, raising the possibility that DA signaling might act through EGF signaling (Liu et al, 2011). We think that this is unlikely, however, because mutants for EGF signaling and DA signaling have distinct gene expression profiles (EGF signaling does not regulate the same set of xenobiotic response genes as does DA signaling). Interestingly, we tried to generate double mutants between gain‐of‐function let‐23 EGF receptor mutations, which result in accelerated protein turnover, and dop‐1 loss‐of‐function mutations, which result in depressed protein turnover; however, we were not able to obtain viable double mutants, suggesting that these two pathways might act independently from one another and complement one another.
How the xenobiotic detoxification network in turn modulates UPS activity and proteostasis remains to be elucidated, but the simplest explanation might be that xenobiotic modification of cellular proteins increases their availability as poly‐ubiquitination substrates, most likely by damaging them. When confronted by xenobiotic toxin exposure, the increase in damaged and unfolded protein load might be so high that the UPS machinery required to remove them becomes saturated unless DA signaling is present to activate the xenobiotic stress response to prevent the toxins from reaching the proteome. We believe that in the absence of DA signaling, the multiple E3 and E4 ubiquitin ligases involved in removing damaged proteins become saturated rather than the proteasome because (i) non‐ubiquitinated, mono‐ubiquitinated, and di‐ubiquitinated UPS reporter proteins accumulated in DA signaling mutants rather than larger poly‐ubiquitinated reporter protein, which is more consistent with poly‐ubiquitination impairment than it is proteasome impairment, and (ii) additional UPS reporter stabilization could be observed in DA signaling mutants when the proteasome was impaired by RNAi knockdown, which indicates that the proteasome is still removing any poly‐ubiquitinated reporter protein that does form in these mutants. Our working model is that particularly unstable proteins like the UPS reporter can no longer be as efficiently poly‐ubiquitinated and removed because the poly‐ubiquitination machinery is otherwise occupied by trying to offset global proteome damage (Fig 5). Consistent with this model, we found that mutants with an impaired xenobiotic response are more sensitive to proteotoxic stress. Moreover, DA signaling mutants show an upregulation of proteins involved in unfolded protein stress response (e.g., heat‐shock protein expression) and proteins involved in the UPS (e.g., cul‐6 and multiple F‐box E3 ligases), suggesting that in the absence of DA signaling, epithelial cells are upregulating proteostasis mechanisms to compensate for the increased burden of damaged and misfolded proteins. In wild‐type animals, dopamine signaling and the resulting xenobiotic response would minimize the accumulation of damaged proteins to a low enough level that the various proteostasis mechanisms (i.e., the poly‐ubiquitination machinery, the proteasome, aggregation of ubiquitinated insoluble proteins into inclusion bodies, and autophagy) can handle them. It remains a formal possibility that the detoxification pathway is also required to remove one or more xenobiotic compounds that would otherwise impair the poly‐ubiquitination machinery directly, although this seems less likely given the diverse set of toxins (e.g., paraquat, heat shock, cadmium ions, ethanol) that can impair UPS reporter turnover (Segref et al, 2011). Regardless of the specific mechanism, the resulting maintenance of proteostasis, along with the expression of other innate immunity factors, helps barrier epithelia repel infection. This signaling system might counteract that of serotonin, which is released from the chemosensory neuron ADF to negatively regulate, through a separate mechanism, innate immune response, including the rectal swelling that occurs in nematodes exposed to the pathogenic bacteria Microbacterium nematophilum (Anderson et al, 2013). Our findings demonstrate a novel example of how a multicellular organism can use its nervous system to sense environmental challenges and modulate proteostasis in target tissues using a known neurohormonal signal.
Figure 5. A hypothesized model for dopamine signaling modulation of proteostasis.
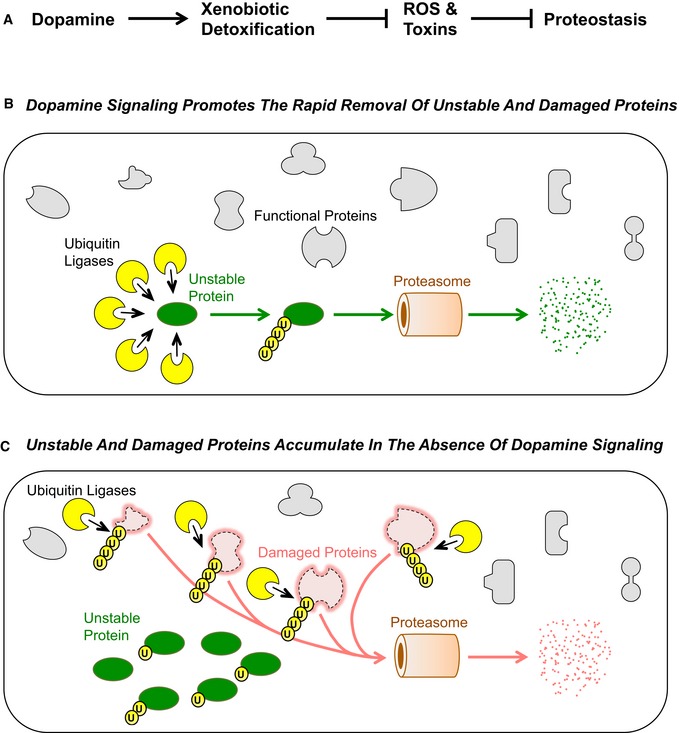
- A simple regulatory model for DA signaling and proteostasis. When nematodes encounter a bacterial lawn, the lawn's viscosity is sensed by dopaminergic mechanosensory neurons, which then release the neurohormone dopamine (DA) into the pseudocoelomic body cavity in response. DA signaling activates the expression of xenobiotic detoxification genes in epithelial tissues. The products of these genes remove toxins and ROS, which otherwise would damage proteins. Arrows indicate positive relationships, whereas T bars indicate inhibitory relationships.
- DA signaling, through activation of the xenobiotic response, protects most proteins (gray shapes) from proteotoxic stress due to toxins and ROS. Protein quality control mechanisms like the UPS (particularly, E3 and E4 ubiquitin ligases, indicated by yellow circles) are fully available to act on unstable proteins (green oval), thereby triggering the rapid ubiquitination and degradation of such proteins. Individual ubiquitin molecules are indicated by small circles with the letter “U”.
- In the absence of DA signaling, there is less activation of the xenobiotic response and thus many proteins within epithelial cells become damaged by toxins and ROS (red shapes comprising dotted lines). Protein quality control mechanisms like the UPS (yellow circles) become overwhelmed ubiquitinating and degrading the large load of stressed proteins, leaving few of them available to process unstable proteins (green oval). The result is an observed stabilization of such proteins, many of which are not ubiquitinated or only partially ubiquitinated. Individual ubiquitin molecules are indicated by small circles with the letter “U”.
A role for dopamine in proteostasis might transcend nematodes. In mammals, dopaminergic neurons are particularly susceptible to oxidative and metabolic stress, environmental toxins, and drugs of abuse, which in turn can increase the risk for Parkinson's disease (PD) (Ferrucci et al, 2008; Ares‐Santos et al, 2013; Taylor et al, 2013; Friend et al, 2014). The resulting changes in dopaminergic signaling in PD contribute to physiological decline in neuronal and non‐neuronal DA targets (Miller & O'Callaghan, 2014; Stayte & Vissel, 2014). Our findings suggest that DA regulates multiple factors that maintain proteostasis, and we speculate that loss of proteostasis due to impairments in DA signaling could be an important factor exacerbating physiological decline in PD‐ and DA‐associated drug abuse.
Materials and Methods
Strains and growth conditions
Standard methods were used to culture C. elegans (Brenner, 1974). Animals were grown at 20°C on standard NGM plates seeded with OP50 Escherichia coli. The following strains were provided by the Caenorhabditis Genetics Center and were backcrossed to the laboratory N2 wild‐type strain three to six times: cat‐2(e1112), dop‐1(vs100), dop‐1(vs101), dop‐1(ok398), dop‐2(vs105), dop‐3(ok295), dop‐3(vs106), dop‐4(tm1392), dop‐4(ok1321), dop‐5(ok568), mec‐5(ok3313), trp‐4(sy695), daf‐16(mgDf50), crh‐2(gk3293), skn‐1(tm3411). Transgenic strains odls77[P col−19 ∷Ub G76V ‐GFP, P col−19 ∷mRFP], norSci1[P dat‐1 ::trp‐4(d), unc‐119(+)]; unc‐119(ed3); vtIs1[P dat‐1 ::gfp, rol‐6dm] (a gift from M. Doitsidou, University of Edinburgh), dgEx80[P vha‐6 ::PolyQ44::YFP, rol‐6dm], vsIs28[P dop‐1 ::GFP], adEx1647[P dop‐1 ::GFP], and hhIs64[P sur‐5 ::Ub G76V ‐GFP, unc‐119(+)]III; hhIs73[P sur‐5 ::mCherry, unc‐119(+)] (a gift from T. Hoppe, University of Cologne) have been described previously (Tsalik et al, 2003; Chase et al, 2004; Mohri‐Shiomi & Garsin, 2008; Liu et al, 2011; Segref et al, 2011). When possible, researchers were blinded to the genotype of the observed sample.
Statistics
Twenty animals were chosen per genotype so as to observe an effect size at least as large as the coefficient of variation. All data with normal distributions were analyzed with GraphPad Prism 6 in most cases using ANOVA with Dunnett's post hoc test or a Student's t‐test (two‐tailed) with Holm–Sidak correction for multiple comparisons.
RNA‐seq analysis
Developmentally synchronized animals were obtained by hypochlorite treatment of gravid adults to release embryos. Synchronized embryos were hatched on NGM plates and grown at 20°C until 48 h after the L4 stage of development. Fluorodeoxyuridine was used to prevent the development of second‐generation embryos once animals reached fertile adulthood (Gandhi et al, 1980). For each RNA‐seq experiment, populations for odIs77[P col‐19 ::Ub G76V ‐GFP] and dop‐1(vs100); [P col‐19 ::Ub G76V ‐GFP] were grown simultaneously under the same conditions. Total RNA was isolated from animals using TRIzol (Invitrogen) combined with bead beater lysis in 3 biological replicates, and an mRNA library (single‐end, 50‐bp reads) was prepared for each sample/replicate using Illumina Truseq with PolyA selection (RUCDR). Libraries were sequenced on an Illumina HiSeq 2500 in Rapid Run Mode, resulting in high‐quality sequence reads (FAST QC Phred score average of 40 out of the full 50 bp length). Reads (45–49 million per genome and replicate) were analyzed using Geospiza Genesifter, with alignment to the C. elegans genome (WS220) via Illumina WTS BWA (GATKv3). From 89–90% of reads mapped to the genome (72–74% mapped to annotated ORFs). Normalization and statistical analysis was performed with EdgeR with a Benjamini and Hochberg correction. Lists of differentially expressed genes were analyzed by DAVID (v6.7, david.abcc.ncifcrf.gov/home.jsp) using functional annotation clustering and a low classification stringency as the method for assessing annotation enrichment. RNA‐seq datasets are available in NIH/NCBI GEO through accession number GSE80807.
RNAi feeding
RNAi feeding protocols were as described previously (Timmons et al, 2001). The RNAi screen was performed with 2,072 clones from the Ahringer RNAi library (Kamath et al, 2003; Sieburth et al, 2005) in animals with the odIs77 transgene in an otherwise wild‐type genetic background. E. coli (HT115) producing dsRNA for individual genes was seeded onto NGM plates containing 25 μg/ml carbenicillin and 0.2% lactose to induce the expression of the dsRNA for the gene of interest. The negative control was conducted by seeding the plates with HT115 containing empty vector pL4440. Synchronized embryos were hatched on each plate and grown at 20°C until 48 h after the L4 stage of development. Each clone in the screen was assessed in two independent RNAi experiments. Positives for one or both experiments were assessed a third time, and any clone that gave a positive phenotype for two or three of the experiments was included in the collection of 31 positives from the screen. It should be noted that UGT genes, CYP genes, and proteasome subunit genes were not included in the screened library. To assess knockdown phenotypes for these genes, we performed directed experiments with feeding RNAi bacteria using the procedure above. The respective clones contained sequences for ugt‐3, ugt‐8, ugt‐11, ugt‐13, ugt‐37, ugt‐57, ugt‐62, ugt‐63, ugt‐64, cyp‐25A1, cyp‐34A4, cyp‐34A7, cyp‐34A8, cyp‐29A2, rpn‐11, pbs‐4, pbs‐5, ufd‐1, hsp‐4, hsp‐16.2, hsp‐16.11, hsp‐16.41, hsp‐16.48, hsp‐16.49, hsp‐17, and hsp‐43 in RNAi vectors; these were obtained from Open Biosystems.
Fluorescence microscopy, imaging analysis, and intensity measurements
GFP‐, mRFP‐, and mCherry‐tagged fluorescent proteins were visualized in nematodes by mounting on 2% agarose pads with 10 mM tetramisole. Fluorescent images were observed using an Axioplan II (Carl Zeiss, Thornwood, NY). A 5× (numerical aperture 0.15) PlanApo objective was used to detect GFP and mRFP signal. Imaging was done with an ORCA charge‐coupled device camera (Hamamatsu, Bridgewater, NJ) by using iVision software (Biovision Technologies, Uwchlan, PA). Exposure times were chosen to capture at least 95% of the dynamic range of fluorescent intensity of all samples. GFP fluorescence was quantified by obtaining outlines of worms using images of the mRFP or mCherry control in case of hypodermis and intestine, respectively. The mean fluorescence intensity within each outline was calculated (after subtracting away background coverslip fluorescence) for UbG76V‐GFP, mRFP and mCherry signals.
Measurement of 26S proteasome activity, GFP levels, and RFP levels in lysates
Developmentally synchronized 48‐h staged animals were lysed in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM EDTA, 2 mM ATP, 1 mM dithiothreitol and protease inhibitor cocktail tablets; Roche) using a Mini Bead Beater 16 (Biospec). Lysates were transferred to microcentrifuge tubes and centrifuged at 18,000 g for 15 min at 4°C.
To measure proteasome activity, an equal amount of protein lysate (5 μg in 20 μl lysis buffer) was premixed with either 250 ng of proteasome inhibitor (epoxomicin, Boston Biochem, prepared in 50% dimethyl sulfoxide) or an equivalent volume of vehicle (50% dimethyl sulfoxide lacking the inhibitor), followed by the addition of proteasome assay buffer (200 μl; 25 mM HEPES, pH 7.5 and 0.5 mM ethylenediaminetetraacetic acid) containing a chymotryptic substrate (80 μM Suc‐Leu‐Leu‐Val‐Tyr‐AMC; Boston Biochem). Reactions were incubated at 25 °C for 1 h, and fluorescence (λex = 360 nm; λem = 465 nm) was detected after every 5 min using a Tecan Infinite F200 detector. Epoxomicin‐sensitive activity was generated with triplicate measurements from two independent experiments, quantified, and plotted.
To measure GFP and RFP in lysates, 5 μg of lysate in 100 μl of lysis buffer was placed in a 96‐well plate, in duplicate. GFP and RFP fluorescence was detected using a Tecan Infinite F200 detector using a filter set for either GFP (λex = 485 nm; λem = 535 nm) or RFP (λexe = 590 nm; λem = 635). Relative fluorescence values were calculated.
Slow‐killing assays and survival analysis
Using a slow‐killing (SK) assay, nematodes were challenged with an infective strain of Pseudomonas aeruginosa (PA14). More than 50 L4 stage nematodes were transferred from NGM‐OP50 to SK plates containing the pre‐grown bacterial lawn of P. aeruginosa and incubated at 20°C (Powell & Ausubel, 2008). SK plates were prepared with 50 μg/ml of 5‐fluoro‐2′‐deoxyuridine (FUdR), which prevents the growth of egg offspring during the experiment. E. coli OP50 was fed nematodes as a negative control strain with limited killing of C. elegans during experimental time course. Nematodes were monitored throughout a 150‐h period under a dissection microscope to detect unresponsive and dead worms. Survival analyses were performed using the Kaplan–Meier method, and the significance of differences between survival curves calculated using Prism (GraphPad Software). Heat‐shock survival was assessed by placing nematodes of the indicated stage at 34°C for 6 h, allowing them to recover for 1 h at 20°C, and then scoring live versus dead animals based on pharyngeal pumping and locomotion.
Western blotting and antibodies
Protein samples (30 μl) were prepared from 20 or 30 nematodes, depending upon hypodermis or intestine reporter, synchronized at L4, and lysed at 48 h post‐L4 for odIs77 and the various mutants. Equal amounts of protein samples were resolved by electrophoresis through 12% SDS–polyacrylamide gel (Bio‐Rad). Western blotting was performed using mouse anti‐Ub (1:2,000, Enzo), mouse anti‐GFP (1:2,000, Roche), rabbit anti‐PSMD14 (1:1,000, Novus), mouse anti‐20S alpha 1–7 (1:1,000, Abcam), and mouse anti‐actin (1:5,000, Millipore). Western blotted proteins were visualized and quantified using fluorescent‐conjugated secondary antibodies from Odyssey and Licor imaging system. Each experiment was repeated at least five times with lysates from separate nematode preparations.
Author contributions
KKJ designed the genetic, molecular, behavioral, and cell biological experiments under the direct supervision of CR. KKJ and TLM collected the data and analyzed the results under the direct supervision of CR. CR and KKJ wrote the manuscript, with significant editorial input and discussion of their results from TLM.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Source Data for Figure 2
Acknowledgements
We thank Gang Liu and Tejash Shah for sharing their initial studies using the UPS GFP reporter. We thank Stephanie Pyonteck for her guidance and help concerning the RNA‐seq analysis. We thank M. Doitsidou, A. Fire, the C. elegans Genetics Center, S. Mitani, D. Garsin, T. Hoppe, S. Xu, and the Japanese National Bioresource Project for reagents and strains. We thank Kiran Madura for advice on measuring the proteasome and the use of Tecan plate reader. We thank members of the Rongo laboratory for comments on the manuscript. The manuscript was funded by grants from the National Institutes of Health (R01 NS42023 and R01 GM101972, to C.R.), as well as the New Jersey Commission on Cancer Research and the Charles and Johanna Busch Fellowship (to K.K.J.); these agencies had no other role in the research or the manuscript.
The EMBO Journal (2016) 35: 1885–1901
See also: YL Chew & WR Schafer (September 2016)
References
- Aballay A (2013) Role of the nervous system in the control of proteostasis during innate immune activation: Insights from C. elegans . PLoS Pathog 9: e1003433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerfelt M, Morimoto RI, Sistonen L (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11: 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JH, Blackwell TK (2003) SKN‐1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 17: 1882–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson A, Laurenson‐Schafer H, Partridge FA, Hodgkin J, Mcmullan R (2013) Serotonergic chemosensory neurons modify the C. elegans immune response by regulating g‐protein signaling in epithelial cells. PLoS Pathog 9: e1003787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya CL, Kawli T, Kundaje A, Jiang L, Wu B, Vafeados D, Terrell R, Weissdepp P, Gevirtzman L, Mace D, Niu W, Boyle AP, Xie D, Ma L, Murray JI, Reinke V, Waterston RH, Snyder M (2014) Regulatory analysis of the C. elegans genome with spatiotemporal resolution. Nature 512: 400–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ares‐Santos S, Granado N, Moratalla R (2013) The role of dopamine receptors in the neurotoxicity of methamphetamine. J Intern Med 273: 437–453 [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63: 182–217 [DOI] [PubMed] [Google Scholar]
- Bock KW (2014) Homeostatic control of xeno‐ and endobiotics in the drug‐metabolizing enzyme system. Biochem Pharmacol 90: 1–6 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of C. elegans . Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budovskaya YV, Wu K, Southworth LK, Jiang M, Tedesco P, Johnson TE, Kim SK (2008) An elt‐3/elt‐5/elt‐6 gata transcription circuit guides aging in C. elegans . Cell 134: 291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt TR, Khan MI, Marsh J, Ecker DJ, Crooke ST (1988) Ubiquitin‐metallothionein fusion protein expression in yeast. a genetic approach for analysis of ubiquitin functions. J Biol Chem 263: 16364–16371 [PubMed] [Google Scholar]
- Cadet JL, Jayanthi S, Mccoy MT, Beauvais G, Cai NS (2010) Dopamine d1 receptors, regulation of gene expression in the brain, and neurodegeneration. CNS Neurol Disord Drug Targets 9: 526–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Paek J, Kim DH (2011) Natural polymorphisms in C. elegans hecw‐1 e3 ligase affect pathogen avoidance behaviour. Nature 480: 525–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase DL, Pepper JS, Koelle MR (2004) Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans . Nat Neurosci 7: 1096–1103 [DOI] [PubMed] [Google Scholar]
- Choe KP, Leung CK, Miyamoto MM (2012) Unique structure and regulation of the nematode detoxification gene regulator, skn‐1: implications to understanding and controlling drug resistance. Drug Metab Rev 44: 209–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47: e147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Stanhill A (2014) The complexity of recognition of ubiquitinated substrates by the 26s proteasome. Biochim Biophys Acta 1843: 86–96 [DOI] [PubMed] [Google Scholar]
- Cooper DM, Bier‐Laning CM, Halford MK, Ahlijanian MK, Zahniser NR (1986) Dopamine, acting through d‐2 receptors, inhibits rat striatal adenylate cyclase by a gtp‐dependent process. Mol Pharmacol 29: 113–119 [PubMed] [Google Scholar]
- Cortes CJ, La Spada AR (2015) Autophagy in polyglutamine disease: imposing order on disorder or contributing to the chaos? Mol Cell Neurosci 66: 53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Huang W, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Huang W, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG (2000) Short‐lived green fluorescent proteins for quantifying ubiquitin/proteasome‐dependent proteolysis in living cells. Nat Biotechnol 18: 538–543 [DOI] [PubMed] [Google Scholar]
- Du H, Gu G, William CM, Chalfie M (1996) Extracellular proteins needed for C. elegans mechanosensation. Neuron 16: 183–194 [DOI] [PubMed] [Google Scholar]
- Emtage L, Gu G, Hartwieg E, Chalfie M (2004) Extracellular proteins organize the mechanosensory channel complex in C. elegans touch receptor neurons. Neuron 44: 795–807 [DOI] [PubMed] [Google Scholar]
- Ferrucci M, Pasquali L, Paparelli A, Ruggieri S, Fornai F (2008) Pathways of methamphetamine toxicity. Ann N Y Acad Sci 1139: 177–185 [DOI] [PubMed] [Google Scholar]
- Friend DM, Fricks‐Gleason AN, Keefe KA (2014) Is there a role for nitric oxide in methamphetamine‐induced dopamine terminal degeneration? Neurotox Res 25: 153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Santelli J, Mitchell DH, Stiles JW, Sanadi DR (1980) A simple method for maintaining large, aging populations of Caenorhabditis elegans . Mech Ageing Dev 12: 137–150 [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, Alves P, Chateigner A, Perry M, Morris M, Auerbach RK, Feng X, Leng J, Vielle A, Niu W, Rhrissorrakrai K et al (2010) Integrative analysis of the Caenorhabditis elegans genome by the modencode project. Science (New York, NY) 330: 1775–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard RA, Sharma P, Hajjar C, Caldwell KA, Caldwell GA, Du Breuil R, Moore R, Boyd L (2007) Ubiquitin conjugating enzymes participate in polyglutamine protein aggregation. BMC Cell Biol 8: 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C (2003) Regulation of aging and age‐related disease by daf‐16 and heat‐shock factor. Science 300: 1142–1145 [DOI] [PubMed] [Google Scholar]
- Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, Blackwell TK, Matsumoto K (2005) The C. elegans p38 mapk pathway regulates nuclear localization of the transcription factor skn‐1 in oxidative stress response. Genes Dev 19: 2278–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ES, Ma PC, Ota IM, Varshavsky A (1995) A proteolytic pathway that recognizes ubiquitin as a degradation signal. J Biol Chem 270: 17442–17456 [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J (2003) Systematic functional analysis of the Caenorhabditis elegans genome using rnai. Nature 421: 231–237 [DOI] [PubMed] [Google Scholar]
- Kang L, Gao J, Schafer WR, Xie Z, Xu XZ (2010) C. elegans trp family protein trp‐4 is a pore‐forming subunit of a native mechanotransduction channel. Neuron 67: 381–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebabian JW, Petzold GL, Greengard P (1972) Dopamine‐sensitive adenylate cyclase in caudate nucleus of rat brain, and its similarity to the “dopamine receptor”. Proc Natl Acad Sci USA 69: 2145–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt KS, Quast KB, Giles AC, De S, Hendrey D, Nicastro I, Rankin CH, Schafer WR (2007) Dopamine mediates context‐dependent modulation of sensory plasticity in C. elegans . Neuron 55: 662–676 [DOI] [PubMed] [Google Scholar]
- Kirkin V, Mcewan DG, Novak I, Dikic I (2009) A role for ubiquitin in selective autophagy. Mol Cell 34: 259–269 [DOI] [PubMed] [Google Scholar]
- Li W, Feng Z, Sternberg PW, Xu XZ (2006) A C. elegans stretch receptor neuron revealed by a mechanosensitive trp channel homologue. Nature 440: 684–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Yue Z (2015) Neuronal aggregates: formation, clearance, and spreading. Dev Cell 32: 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblom TH, Dodd AK (2006) Xenobiotic detoxification in the nematode Caenorhabditis elegans . J Exp Zool A Comp Exp Biol 305: 720–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten K, Menendez‐Benito V, Masucci MG, Dantuma NP (2003) A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol 21: 897–902 [DOI] [PubMed] [Google Scholar]
- Lints R, Emmons SW (1999) Patterning of dopaminergic neurotransmitter identity among Caenorhabditis elegans ray sensory neurons by a tgfbeta family signaling pathway and a hox gene. Development 126: 5819–5831 [DOI] [PubMed] [Google Scholar]
- Liu G, Rogers J, Murphy CT, Rongo C (2011) EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J 30: 2990–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair W, Morantte I, Rodrigues AP, Manning G, Montminy M, Shaw RJ, Dillin A (2011) Lifespan extension induced by AMPK and calcineurin is mediated by crtc‐1 and CREB. Nature 470: 404–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardones P, Martinez G, Hetz C (2015) Control of systemic proteostasis by the nervous system. Trends Cell Biol 25: 1–10 [DOI] [PubMed] [Google Scholar]
- Meisel JD, Panda O, Mahanti P, Schroeder FC, Kim DH (2014) Chemosensation of bacterial secondary metabolites modulates neuroendocrine signaling and behavior of C. elegans . Cell 159: 267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DB, O'Callaghan JP (2014) Biomarkers of Parkinson's Disease (PD): present and future. Metabolism 64: S40–S46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi T, Palfreyman MT, Sluder AE, Slack F, Sengupta P (1999) Expression and function of members of a divergent nuclear receptor family in Caenorhabditis elegans . Dev Biol 215: 314–331 [DOI] [PubMed] [Google Scholar]
- Mohri‐Shiomi A, Garsin DA (2008) Insulin signaling and the heat shock response modulate protein homeostasis in the Caenorhabditis elegans intestine during infection. J Biol Chem 283: 194–201 [DOI] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI (2002) The threshold for polyglutamine‐expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans . Proc Natl Acad Sci USA 99: 10417–10422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, Mccarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C (2003) Genes that act downstream of daf‐16 to influence the lifespan of Caenorhabditis elegans . Nature 424: 277–283 [DOI] [PubMed] [Google Scholar]
- Murshid A, Eguchi T, Calderwood SK (2013) Stress proteins in aging and life span. Int J Hyperthermia 29: 442–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan A, Ning Y, Reisner K, Buraei Z, Larsen JP, Hobert O, Doitsidou M (2014) Progressive degeneration of dopaminergic neurons through trp channel‐induced cell death. J Neurosci 34: 5738–5746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Nioi P, Pickett CB (2009) The Nrf2‐antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284: 13291–13295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, Blackwell TK (2009) Condition‐adapted stress and longevity gene regulation by Caenorhabditis elegans SKN‐1/Nrf. Aging Cell 8: 524–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp D, Csermely P, Soti C (2012) A role for SKN‐1/Nrf in pathogen resistance and immunosenescence in Caenorhabditis elegans . PLoS Pathog 8: e1002673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Tedesco PM, Johnson TE (2009) Oxidative stress and longevity in Caenorhabditis elegans as mediated by skn‐1 . Aging Cell 8: 258–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM (2014) Mitochondrial UPR‐regulated innate immunity provides resistance to pathogen infection. Nature 516: 414–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR, Ausubel FM (2008) Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol Biol 415: 403–427 [DOI] [PubMed] [Google Scholar]
- Prahlad V, Cornelius T, Morimoto RI (2008) Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science 320: 811–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukkila‐Worley R, Feinbaum RL, Mcewan DL, Conery AL, Ausubel FM (2014) The evolutionarily conserved mediator subunit mdt‐15/med15 links protective innate immune responses and xenobiotic detoxification. PLoS Pathog 10: e1004143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runkel ED, Liu S, Baumeister R, Schulze E (2013) Surveillance‐activated defenses block the ros‐induced mitochondrial unfolded protein response. PLoS Genet 9: e1003346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryno LM, Wiseman RL, Kelly JW (2013) Targeting unfolded protein response signaling pathways to ameliorate protein misfolding diseases. Curr Opin Chem Biol 17: 346–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S, Wintle RF, Kindt KS, Nuttley WM, Arvan R, Fitzmaurice P, Bigras E, Merz DC, Hebert TE, Van Der Kooy D, Schafer WR, Culotti JG, Van Tol HH (2004) Dopamine modulates the plasticity of mechanosensory responses in Caenorhabditis elegans . EMBO J 23: 473–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul N, Pietsch K, Menzel R, Steinberg CE (2008) Quercetin‐mediated longevity in Caenorhabditis elegans: is daf‐16 involved? Mech Ageing Dev 129: 611–613 [DOI] [PubMed] [Google Scholar]
- Sawin ER, Ranganathan R, Horvitz HR (2000) C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26: 619–631 [DOI] [PubMed] [Google Scholar]
- Segref A, Torres S, Hoppe T (2011) A screenable in vivo assay to study proteostasis networks in Caenorhabditis elegans . Genetics 187: 1235–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore DE, Ruvkun G (2013) A cytoprotective perspective on longevity regulation. Trends Cell Biol 23: 409–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth D, Ch'ng Q, Dybbs M, Tavazoie M, Kennedy S, Wang D, Dupuy D, Rual JF, Hill DE, Vidal M, Ruvkun G, Kaplan JM (2005) Systematic analysis of genes required for synapse structure and function. Nature 436: 510–517 [DOI] [PubMed] [Google Scholar]
- Stayte S, Vissel B (2014) Advances in non‐dopaminergic treatments for Parkinson 's disease. Front Neurosci 8: 113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Liu Y, Aballay A (2012) Organismal regulation of xbp‐1‐mediated unfolded protein response during development and immune activation. EMBO Rep 13: 855–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Singh V, Kajino‐Sakamoto R, Aballay A (2011) Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332: 729–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo S, Ishiura S (2013) Dopamine modulates acetylcholine release via octopamine and creb signaling in Caenorhabditis elegans . PLoS ONE 8: e72578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatum MC, Ooi FK, Chikka MR, Chauve L, Martinez‐Velazquez LA, Steinbusch HW, Morimoto RI, Prahlad V (2015) Neuronal serotonin release triggers the heat shock response in C. elegans in the absence of temperature increase. Curr Biol 25: 163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JM, Main BS, Crack PJ (2013) Neuroinflammation and oxidative stress: co‐conspirators in the pathology of parkinson's disease. Neurochem Int 62: 803–819 [DOI] [PubMed] [Google Scholar]
- Tepper RG, Ashraf J, Kaletsky R, Kleemann G, Murphy CT, Bussemaker HJ (2013) PQM‐1 complements daf‐16 as a key transcriptional regulator of daf‐2‐mediated development and longevity. Cell 154: 676–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A (2001) Ingestion of bacterially expressed dsrnas can produce specific and potent genetic interference in Caenorhabditis elegans . Gene 263: 103–112 [DOI] [PubMed] [Google Scholar]
- Treweek TM, Meehan S, Ecroyd H, Carver JA (2015) Small heat‐shock proteins: important players in regulating cellular proteostasis. Cell Mol Life Sci 72: 429–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalik El, Niacaris T, Wenick AS, Pau K, Avery L, Hobert O (2003) LIM homeobox gene‐dependent expression of biogenic amine receptors in restricted regions of the C. elegans nervous system. Dev Biol 263: 81–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oosten‐Hawle P, Porter RS, Morimoto RI (2013) Regulation of organismal proteostasis by transcellular chaperone signaling. Cell 153: 1366–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oosten‐Hawle P, Morimoto RI (2014) Organismal proteostasis: role of cell‐nonautonomous regulation and transcellular chaperone signaling. Genes Dev 28: 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Morantte I, Liu Z, Douglas PM, Merkwirth C, Rodrigues AP, Manning G, Dillin A (2012) RPN‐6 determines C. elegans longevity under proteotoxic stress conditions. Nature 489: 263–268 [DOI] [PubMed] [Google Scholar]
- Vilchez D, Saez I, Dillin A (2014) The role of protein clearance mechanisms in organismal ageing and age‐related diseases. Nat Commun 5: 5659 [DOI] [PubMed] [Google Scholar]
- Volovik Y, Moll L, Marques FC, Maman M, Bejerano‐Sagie M, Cohen E (2014) Differential regulation of the heat shock factor 1 and daf‐16 by neuronal nhl‐1 in the nematode C. elegans . Cell Rep 9: 2192–2205 [DOI] [PubMed] [Google Scholar]
- Warnatsch A, Bergann T, Kruger E (2013) Oxidation matters: the ubiquitin proteasome system connects innate immune mechanisms with MHC class I antigen presentation. Mol Immunol 55: 106–109 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Source Data for Figure 2