

Abstract
Arginase deficiency is caused by deficiency of arginase 1 (ARG1), a urea cycle enzyme that converts arginine to ornithine. Clinical features of arginase deficiency include elevated plasma arginine levels, spastic diplegia, intellectual disability, seizures and growth deficiency. Unlike other urea cycle disorders, recurrent hyperammonemia is typically less severe in this disorder. Normalization of plasma arginine levels is the consensus treatment goal, because elevations of arginine and its metabolites are suspected to contribute to the neurologic features. Using data from patients enrolled in a natural history study conducted by the Urea Cycle Disorders Consortium, we found that 97% of plasma arginine levels in subjects with arginase deficiency were above the normal range despite conventional treatment. Recently, arginine-degrading enzymes have been used to deplete arginine as a therapeutic strategy in cancer. We tested whether one of these enzymes, a pegylated human recombinant arginase 1 (AEB1102), reduces plasma arginine in murine models of arginase deficiency. In neonatal and adult mice with arginase deficiency, AEB1102 reduced the plasma arginine after single and repeated doses. However, survival did not improve likely, because this pegylated enzyme does not enter hepatocytes and does not improve hyperammonemia that accounts for lethality. Although murine models required dosing every 48 h, studies in cynomolgus monkeys indicate that less frequent dosing may be possible in patients. Given that elevated plasma arginine rather than hyperammonemia is the major treatment challenge, we propose that AEB1102 may have therapeutic potential as an arginine-reducing agent in patients with arginase deficiency.
Introduction
Arginase deficiency (MIM#207800) is caused by a deficiency of arginase 1, a cytosolic urea cycle enzyme that converts arginine to ornithine. Characteristic phenotypic features of arginase deficiency include spastic diplegia, seizures, intellectual disability (ranging from mild to severe), self-protein restriction and growth deficiency (1,2). Less frequent complications include hepatic cirrhosis and fibrosis (3–6). Unlike other urea cycle disorders, hyperammonemia is less frequent and typically less severe. Elevated plasma arginine levels are diagnostic for arginase deficiency and are the basis of newborn screening for this disorder. Diagnosis can be confirmed with molecular studies or with red blood cell arginase enzyme activity.
Although seizures and intellectual disability may be observed in all urea cycle disorders, spastic diplegia is a unique feature of arginase deficiency that is not typical of any other urea cycle disorder. The etiology of the neurologic phenotype in arginase deficiency is unknown. However, increased levels of guanidino compounds, which are produced from arginine, have been detected in blood, urine and cerebrospinal fluid from patients with arginase deficiency, and these compounds have been hypothesized to cause the neurologic phenotype (7–11). Alternative hypotheses that have been proposed include dysregulation of other metabolites of arginine such as nitric oxide and agmatine (7).
Because arginine and its metabolites are suspected to cause the neurologic phenotype in arginase deficiency, the consensus goal of treatment has been reduction of plasma arginine with a low-protein diet, amino acid supplementation and administration of nitrogen-scavenging agents. Such treatment has been shown to decrease plasma arginine levels (2,12–14), and in some patients, it has resulted in stabilization or prevention of the neurologic phenotype (2,12–14). However, with this treatment strategy, normalization or near-normalization of plasma arginine in arginase deficiency is challenging.
The benefits of reducing the levels of plasma arginine and arginine metabolites have also been demonstrated in patients receiving liver transplantation. Long-term follow-up studies of two patients after orthologous liver transplantation demonstrated normalization of plasma arginine and plasma guanidino compounds (5,15). Furthermore, after transplantation, the stabilization or prevention of neurologic progression suggests that circulating arginine and its metabolites are the cause for the neurologic phenotype rather than intrinsic abnormalities in the central nervous system (5,15).
Because normalization of plasma arginine levels in arginase deficiency is challenging, the use of arginase enzyme therapy for lowering circulating plasma arginine levels has been suggested as an alternative treatment strategy. To test this approach, arginase has been introduced into hollow-fiber reactors and shown to reduce arginine levels in artificial buffers and in whole blood derived from rabbits (16). Likewise, arginase-loaded erythrocytes reduced plasma arginine levels in wild-type rats and in mice made hyperargininemic with arginine injections (17,18). As arginase is abundantly expressed in red blood cells, red blood cell transfusions have been used to provide an exogenous source of arginase in patients with mixed results (19–21).
Human recombinant arginase has recently emerged as a potential therapy for various forms of cancer (22). For example, arginase causes arginine depletion in tumors that do not express argininosuccinate synthase, an enzyme necessary for arginine synthesis, and hence, they are auxotrophic for arginine. In the setting of arginine depletion, cell proliferation is reduced. Recently, AEB1102, a human recombinant arginase enzyme produced in Escherichia coli was developed for this purpose (23,24). The enzyme is pegylated to improve stability and half-life. In addition, the replacement of two manganese (Mn2+) ions with cobalt (Co2+) resulted in increased catalytic activity and improved stability (23,24). In wild-type mice, twice weekly intraperitoneal delivery of AEB1102 resulted in depletion of plasma arginine (25). We hypothesized that this human recombinant arginase, at lower doses, could be used to reduce plasma arginine in patients with arginase deficiency.
In the present study, using data from the Urea Cycle Disorders Consortium (UCDC) of the Rare Disease Clinical Research Network (RDCRN), we confirmed that patients with arginase deficiency have elevated plasma arginine levels despite current treatment approaches. In a preclinical, translational study, we tested whether AEB1102 lowers plasma arginine levels in neonatal and inducible adult murine models of arginase deficiency. In addition, we evaluated the time course of plasma arginine recovery after administration of AEB1102 in cynomolgus monkeys. The objective of this study was to determine whether human recombinant arginase therapy could be a potential new treatment strategy for normalization of plasma arginine in patients with arginase deficiency.
Results
Elevated plasma arginine despite treatment in patients with arginase deficiency
A total of 22 subjects (11 males, 11 females) with arginase deficiency were enrolled in the Longitudinal Study of Urea Cycle Disorders conducted by the UCDC (ClinicalTrials.gov Identifier: NCT00237315). Of these 22 subjects, 3 were diagnosed because a sibling had the disorder, 2 were diagnosed by newborn screening and the remaining 17 subjects were diagnosed after developing symptoms of the disorder. The age of diagnosis ranged from 8 days to 12 years, and the age range at the most recent visit was 1 month to 50 years. Eleven patients had molecular diagnosis, and an additional five had red blood cell arginase enzyme activity that was between 1 and 2% of the control range. Confirmation of diagnosis for the remaining six patients was based on plasma arginine elevation. Growth parameters and neurologic parameters are presented in Table 1.
Table 1.
Characteristics of UCDC Longitudinal Study Cohort of subjects with arginase deficiency
| Diagnosis | Number assessed | Mean (range) or % |
|---|---|---|
| Age at first symptoms | 19 | 20.3 months (0–72 months) |
| Age at diagnosis | 22 | 39.1 months (0–144 months) |
| Age at most recent visit | 22 | 177 months (1–600 months) |
| Growth parameters (ages 2–20 years) | ||
| Mean weight Z-score | 13 | −0.36 (−2.36 to 1.22) |
| Mean height Z-score | 13 | −1.19 (−4.27 to 1.23) |
| FOC < 3rd %tile | 11 | 9% |
| Treatment | ||
| Presence of one or more hyperammonemia episodes | 22 | 55% |
| Nitrogen-scavenging agents | 22 | 86% |
| Liver transplantation | 22 | 5% |
| Neurologic phenotype | ||
| Abnormal tone | 21 | 62% |
| Abnormal reflexes | 21 | 52% |
| Seizures | 22 | 45% |
| Intellectual disability or developmental delaya | 19 | 89% |
| Non-ambulatoryb | 15 | 60% |
aData for patients 6 months old or greater were used.
bData for patients 3 years and older were used.
Despite dietary protein restriction, 97% of all plasma arginine levels collected at routine study visits were above the normal range (using an upper limit of the normal range = 150 µmol/l), and no subject had all plasma arginine levels within the normal range (Fig. 1A). Most subjects (86%) were on a nitrogen-scavenging agent at the most recent visit. However, only 55% of subjects reported a history of at least one hyperammonemia episode with most subjects reporting less than five total hyperammonemia episodes across their lifetime (Fig. 1B). One subject had received liver transplantation. These results clearly demonstrate that the present standard therapy is suboptimal in controlling the plasma arginine levels.
Figure 1.

Elevated plasma arginine levels in patients with arginase deficiency. (A) All plasma arginine levels for each subject are plotted (n = 22 subjects, range of 1–13 plasma arginine levels available for each subject). The black line indicates an estimated upper limit of normal (ULN). (B) Frequent hyperammonemia is not a complication observed in most patients with arginase deficiency. The total number of baseline and interim hyperammonemia episodes reported by each patient was calculated and plotted. The age range of the subjects at the most recent study visit is provided.
AEB1102 normalizes plasma arginine in neonatal arginase-deficient mice
The neonatal mouse model for arginase deficiency (Arg1−/−) demonstrates severe hyperargininemia and early mortality (10–14 days of age) from hyperammonemia (26). In proof-of-concept studies, we used this mouse model to test whether AEB1102 normalizes plasma arginine in this neonatal model of arginase deficiency. To this end, we performed pharmacokinetic studies to determine the optimal dose and frequency of AEB1102. We demonstrated that a single dose of AEB1102 (0.04 mg/kg) administered at postnatal day 4–5 normalized plasma arginine levels by 24 h in a dose-dependent manner (Fig. 2A). Results are not shown for doses of enzyme >0.05 mg/kg as they depleted plasma arginine levels. Time course studies of plasma arginine after a single dose (0.05 mg/kg) revealed that optimal dose frequency is 48 h (Fig. 2B). Repeated dose studies (0.04 mg/kg every 48 h) demonstrated persistence of reduced plasma arginine levels compared with mice treated with placebo (P < 0.0001) (Fig. 3A). However, repeated dosing did not result in improved survival in neonatal pups with arginase deficiency (Fig. 3C) likely because arginase enzyme therapy does not treat hyperammonemia, which is the cause of death in this model (26) as this pegylated enzyme is not expected to enter the liver. Neonatal pups treated with repeated dosing were sacrificed for compassionate reasons on postnatal day 10–12 which is consistent with the expected survival of 10–14 days in this mouse model (26). As expected, the levels of ornithine, the product of the arginase enzyme, rise during treatment with AEB1102 (P < 0.0001) (Fig. 3B).
Figure 2.

Dose and time-course studies of AEB1102 in neonatal arginase-deficient mice. (A) Plasma arginine in neonatal Arg1−/− mice decreases in a dose-dependent manner 24 h after intraperitoneal injection of AEB1102. (B) Plasma arginine levels in neonatal Arg1−/− increases to near baseline levels between 72 and 120 h after a single intraperitoneal injection (0.05 mg/kg) of AEB1102. −/− = arginase deficient, +/+ = wild type.
Figure 3.

Repeated dose studies of AEB1102 in neonatal arginase-deficient mice. (A) After repeat dosing every 48 h, plasma arginine levels show persistent reduction on postnatal day 11 in neonatal Arg1−/− mice. Treatment was started on Day 6 with a dose of 0.04 mg/kg/dose. (B) Plasma ornithine levels on Day 11 are higher in treated versus untreated neonatal Arg1−/− mice. (C) Treated and untreated neonatal Arg1−/− mice survive until Days 10–14 which is consistent with previously published reports (26). (D) Whole brain arginine levels are significantly higher in untreated versus treated neonatal Arg1−/− mice. (E) Liver arginine levels are not significantly different between untreated versus treated neonatal Arg1−/− mice. One-way ANOVA was used with Dunnett's post-test, and all groups were compared with the −/− enzyme-treated group. Tx = treatment, ***P < 0.0001, **P < 0.01, *P < 0.05.
AEB1102 reduces brain arginine levels in the neonatal model
Amino acid analysis performed in whole brain samples from Arg1−/−mice at postnatal day 11 after repeated dosing of arginase enzyme revealed a decrease in whole brain arginine level relative to whole brain from untreated mice (P = 0.005) (Fig. 3D). We suspect that this decrease in whole brain arginine level reflects circulating arginine in the blood that may still be present in the sample at the time of collection. In contrast, the level of liver arginine was unchanged with treatment, and this finding supports our hypothesis that this pegylated enzyme does not enter the liver and, thus, does not treat hyperammonemia (Fig. 3E).
AEB1102 reduces plasma arginine but does not improve survival in an adult arginase-deficient model
As described previously (27,28), we generated an adult inducible model of arginase deficiency. To confirm hyperargininemia in the model, we demonstrated that plasma arginine rises as expected after tamoxifen administration (Fig. 4A). As in the neonatal model, AEB1102 administered by intraperitoneal injection in the adult model reduced plasma arginine levels in a dose-dependent manner (Fig. 4B). A rise in plasma ornithine similar to the neonatal studies was also observed in the adult model (data not shown). Based on time-course studies (Fig. 4C), a dose frequency of every 48 h was selected for follow-up studies.
Figure 4.

Dose and time-course studies of AEB1102 in an adult arginase-deficient mouse model. (A) Plasma arginine begins to rise by Day 11 after tamoxifen treatment in the adult inducible arginase-deficient (Cre+) mice and continues to rise through Day 18. (B) Plasma arginine is reduced in a dose-dependent manner in the adult inducible arginase-deficient mice (Cre+) 24 h after treatment with AEB1102. For each dose, 1–2 outliers in each group did not respond to AEB1102. (C) Plasma arginine levels begin to rise by 72 h after AEB1102 delivery in the adult inducible arginase-deficient (Cre+) mice. Data for control mice are included for reference (Cre−). Doses are provided in mg/kg.
To assess whether repeated doses of AEB1102 resulted in sustained reduction in plasma arginine levels in the adult model, repeated doses of AEB1102 or PBS were given on Day 15 and Day 17. Plasma and tissues were collected on Day 18 (∼24 h after the last dose of enzyme or PBS). After two doses of AEB1102 (0.02 mg/kg/dose), plasma arginine levels were significantly lower than in mice treated with PBS (P < 0.0001) (Fig. 5A), although % weight loss was similar in the two groups (Fig. 5B). Plasma ornithine levels were significantly higher in the AEB1102-treated adult arginase-deficient mice (Cre+) compared with PBS-treated mice (Fig. 5C). Although the plasma ornithine rose slightly in the wild-type mice (Cre−) treated with AEB1102, the magnitude of the rise was not as high as that in AEB1102-treated arginase-deficient mice (Cre+) (Fig. 5C). Although plasma guanidinoacetate levels were higher in the adult arginase-deficient mice (Cre+), there were no significant differences in plasma guanidinoacetate levels between arginase-deficient mice (Cre+) that were treated with AEB1102 versus placebo possibly because the mice were exposed to decreased plasma arginine levels for only 4 days, and such a short time frame may not be sufficient to clear already elevated plasma guanidinoacetate levels (data not shown). In addition, although there was a trend toward a decrease in brain arginine levels after treatment (P = 0.0994, Fig. 5D) with AEB1102, liver arginine levels were similar in arginase-deficient mice (Cre+) treated with AEB1102 or PBS (Fig. 5E).
Figure 5.

Repeated dose studies of AEB1102 in an adult arginase-deficient mouse model. (A) Repeated doses of AEB1102 result in persistently decreased plasma arginine levels on Day 18 in adult inducible arginase-deficient mice (Cre+). Mice were treated with AEB1102 or PBS on Day 15 and on Day 17, and plasma was collected ∼24 h after the second dose. (B) Weight loss in adult inducible arginase-deficient mice (Cre+) treated with AEB1102 versus placebo is not significantly different at Day 18. Mice were monitored with weights daily and compassionately euthanized once weight loss was ≥20% (compared with Day 12) or on Day 18 (whichever was first). Five mice (n = 2 Cre+ and PBS-treated; n = 3 Cre+ and AEB1102-treated) were sacrificed on Day 17 because of weight loss ≥20% and were not included in the data analysis. (C) Repeated doses of AEB1102 result in persistent elevation in plasma ornithine on Day 18 in adult inducible arginase-deficient mice (Cre+). (D) Brain arginine levels in adult inducible arginase-deficient mice (Cre+) treated are not significantly different in mice treated with repeated doses of AEB1102 versus PBS. (E) Liver arginine levels are not significantly different in adult inducible arginase-deficient mice (Cre+) treated with repeated doses of AEB1102 versus PBS. All groups were compared with the Cre+ AEB1102-treated mice with a one-way ANOVA with Dunnett's post-test. ***P < 0.0001, *P < 0.01.
To test whether repeated doses of AEB1102 resulted in improved survival or hyperammonemia, the enzyme (0.02 mg/kg/dose) was dosed every 48 h. Survival until compassionate euthanasia (e.g. weight loss ≥20%, decreased movement in cage) was compared between the arginase-deficient (Cre+) mice treated with AEB1102 versus PBS, and no significant difference was observed (Fig. 6A). Elevated ammonia levels at the time of sacrifice were also not significantly different between the two groups (Fig. 6B), suggesting that AEB1102 does not improve survival, weight loss or hyperammonemia.
Figure 6.

Survival studies in adult arginase-deficient mice treated with AEB1102. (A) Survival in adult inducible arginase-deficient mice (Cre+) treated with AEB1102 versus PBS is not significantly different. (B) Plasma ammonia levels in adult inducible arginase-deficient mice (Cre+) treated with AEB1102 versus PBS are not significantly different. The dotted line represents the upper limit of the range in wild-type mice.
Studies of AEB1102 in cynomolgus monkeys support weekly dosing
To assess the translatability of such an approach in humans, dosing studies were conducted in non-human primates. The high concentrations of AEB1102 achieved subsequent to intravenous administration of all doses led to an immediate decrease in plasma arginine levels in cynomolgus monkeys without hyperargininemia (Fig. 7). Post-dose plasma arginine levels continued to be low until serum levels of AEB1102 dropped below 1–2 µg/ml. This observed threshold range for arginine reduction was consistent after dose administration on both Days 1 and 8 and in all three dose groups. At the highest dose level of 1 mg/kg on Days 1 and 8, there was minimal recovery of plasma arginine levels at 168 h post-dose on both Day 1 and Day 8, consistent with AEB1102 concentrations that did not decline below ∼1 µg/ml over most of this time frame. As the dose of AEB1102 decreased, there was progressively more recovery of plasma arginine levels. At the lowest dose (0.1 mg/kg), plasma arginine levels begin to rise by Day 4 and return to near-normal levels by Days 7–8 (Fig. 7).
Figure 7.
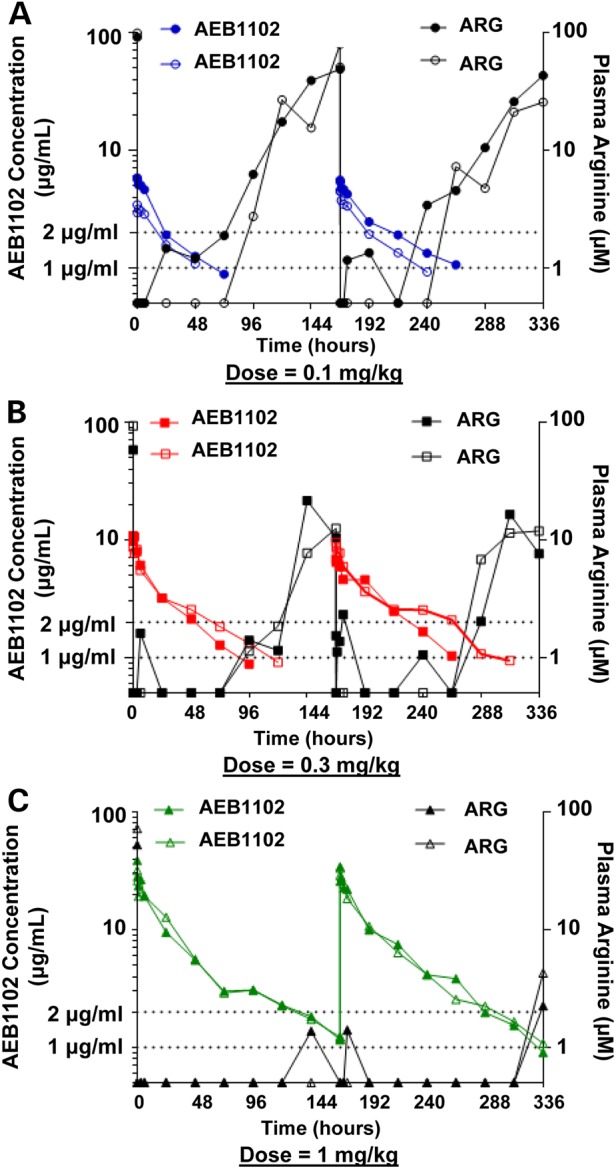
AEB1102 dose studies in cynomolgus monkeys. (A) The concentration of AEB1102 and plasma arginine levels are shown for monkeys after single and repeat intravenous dosing at 0.1 mg/kg (semi-log scale). Doses were administered at Days 1 and 8. The solid-filled circles represent Monkey #1 and unfilled circles represent Monkey #2. (B) The concentration of AEB1102 and plasma arginine levels are shown in monkeys after single and repeat intravenous dosing at 0.3 mg/kg (semi-log scale). Doses were administered at Days 1 and 8. The solid-filled squares represent Monkey #1 and unfilled squares represent Monkey #2. (C) The concentration of AEB1102 and plasma arginine levels in monkeys are shown after single and repeat intravenous dosing at 1 mg/kg (semi-log scale). Doses were administered at Days 1 and 8. The solid-filled triangles represent Monkey #1 and unfilled triangles represent Monkey #2.
Discussion
Using both neonatal and adult arginase-deficient mouse models, we have demonstrated that AEB1102 reduces plasma arginine levels in a dose-dependent manner and that this reduction is sustained with repeated dosing. The doses used were nearly 100-fold lower than doses used previously to deplete plasma arginine in wild-type adult mice, although we dosed more frequently (every 2 days versus twice weekly) (25). However, despite reduction in plasma arginine levels, human recombinant arginase 1 enzyme (AEB1102) did not improve survival in either model likely because of persistence of hyperammonemia. The data from the UCDC longitudinal study cohort of patients with arginase deficiency suggest that persistently elevated plasma arginine levels rather than severe, frequent, episodic hyperammonemia is the main treatment challenge in arginase deficiency (Fig. 1). This difference in the severity of hyperammonemia in mice compared with human patients has been attributed to the induction of arginase 2 enzyme activity in the kidney of humans with arginase 1 deficiency (29–32). Thus, given that circulating arginine and its metabolites have been proposed to be the cause for the neurologic phenotype in arginase deficiency, we suspect that AEB1102 may have therapeutic potential in patients with arginase deficiency.
Our results demonstrating that reduction in plasma arginine levels does not improve survival in the neonatal model confirms results obtained by Hu et al. (32) who demonstrated that myocyte-mediated expression of arginase in neonatal Arg1−/− mice resulted in improved plasma arginine levels without improvement in hyperammonemia or survival. Hu et al. (32) also detected a decrease in liver arginine levels in their model, but in our studies, no decrease in hepatic arginine levels was observed. One potential reason why hepatic arginine levels were not reduced in our models is that we suspect that this pegylated enzyme does not enter the liver. Furthermore, the skeletal muscle-specific gene therapy model by Hu et al. (32) was shown to exhibit some hepatic expression that was hypothesized to result from arginase 1 expression in hepatic myofibroblasts. Perhaps, expression in hepatic myofibroblasts contributed to the decrease in hepatic arginine in the gene therapy model. Alternatively, perhaps, the sustained decrease in plasma arginine over a longer time period with gene therapy contributed to the reduction in liver arginine levels in the gene therapy model.
Intraperitoneal injection was selected as the mode of AEB1102 delivery, because this mode of delivery had been used in prior studies in wild-type mice (25). However, with intraperitoneal delivery, we noted that a small subset of mice did not respond to AEB1102. We suspect that this lack of response was due to technical failures in delivery (e.g. leakage in the neonatal model or injection into the intestine). Perhaps, intravenous delivery via tail vein injection or retro-orbital injection would decrease some of the variability observed. Furthermore, we noted more variability in plasma arginine levels in the adult model which we attribute to the fact that the plasma arginine is rising in the adult model rather than stably elevated as in the neonatal model (Fig. 4A). The 48-h dose frequency used in mice would be challenging in human patients. However, although the studies were performed in monkeys with normal plasma arginine levels, the data from cynomolgus monkeys support a less frequent dosing regimen which may be more practical for human patients with arginase deficiency.
Plasma ornithine was noted to rise in both the neonatal and adult arginase-deficient mouse models. Elevated plasma ornithine is known to be associated with gyrate atrophy (33,34). Thus, strict monitoring of ornithine levels would be essential when considering administration in humans. However, a mild rise in plasma ornithine may also be beneficial in patients with arginase deficiency, because ornithine supplementation has been proposed as a treatment modality for arginase deficiency as ornithine reduces the activity of arginine-glycine amidinotransferase (AGAT), the enzyme that generates guanidinoacetate, an epileptogenic compound, from arginine (35). Because of differences in the metabolic rate between mice and humans, it is difficult to predict whether plasma ornithine will rise and if so, the magnitude of rise in humans.
Our murine studies clearly show a reduction in plasma arginine after administration of AEB102. We also detected a reduction in brain arginine in neonatal but not adult arginase-deficient mice. The reason for the discordance between the two models is unclear but may be due to the fact that the neonatal mice received an additional dose of enzyme compared with the adult mice. The survival in the treated neonatal and adult arginase-deficient mice was likely too short to allow for the study of the long-term impact on brain arginine and on arginine metabolites, such as guanidino compounds in both the plasma and brain. Hu et al. (32) demonstrated a reduction but not normalization of plasma guanidinoacetate levels in association with reduced plasma arginine levels in their myocyte-mediated arginase expression model, a result that supports our hypothesis that prolonged exposure to AEB1102 may lead to reduction in the circulating levels of arginine metabolites, such as guanidinoacetate. However, although data support that arginine and its metabolites cause the neurologic phenotype in this disorder, the exact etiology of the neurologic dysfunction remains unclear. Thus, if clinical trials of AEB1102 are pursued, long-term studies of neurologic outcome will be necessary to determine whether reduction of plasma arginine (and potentially its metabolites) is sufficient to improve neurologic outcome in this disorder.
The time course of exposure to AEB1102 was also likely too short to allow for evaluation of potential immunologic responses to the enzyme. It has been suggested that expression of ARG2 which has ∼92% homology with ARG1 in conserved regions of the protein and ∼42% homology in less conserved regions may provide some protection against an immune response to gene therapy, and the same may be true for an enzyme therapy (32,36). However, it is possible that antibodies formed to epitopes unique to ARG1 could still induce an immune response despite this homology. Data from the UCDC cohort together with published studies demonstrate that a variety of mutation types have been associated with arginase deficiency, and some mutations, but not all, are associated with the presence of immunoreactive material present on western blot (1,2). Thus, if an immune response is observed, it is possible that mutation type and the presence of cross-reactive immunologic material may predict which patients are at increased risk for an immune response.
Overall, we have demonstrated that human recombinant arginase 1 enzyme (AEB1102) lowers plasma arginine in a dose-dependent manner in both neonatal and adult inducible arginase-deficient mouse models. Although survival and hyperammonemia are not improved with AEB1102, we hypothesize that AEB1102 may have clinical utility in human patients with arginase deficiency since elevated plasma arginine rather than severe hyperammonemia is the greater challenge in treating patients with arginase deficiency.
Materials and Methods
Patient population and data collection
Data for patients with arginase deficiency were collected as part of the Longitudinal Study of Urea Cycle Disorders (ClinicalTrials.gov NCT00237315) (37), a natural history study conducted by the NIH-sponsored Rare Diseases Clinical Research Network's (RDCRN) Urea Cycle Disorders Consortium (UCDC). The UCDC includes 14 academic center sites in the USA, Canada and Europe (37). Data for all study visits at all centers were collected in a standardized format as detailed in the manual of operations, and the data were entered into the electronic database maintained by the Data Management and Coordinating Center of the RDCRN. For enrollment of patients with arginase deficiency, the following diagnostic criteria were used: ≥5-fold elevation of arginine in the blood and/or reduced arginase enzyme activity in red blood cells or other appropriate tissue and/or identification of pathogenic variants in ARG1. Data from all 22 subjects with arginase deficiency who were enrolled in the Longitudinal Study of Urea Cycle Disorders were included in this analysis. The data used in this analysis were obtained from the enrollment visit and subsequent follow-up study visits through May 2013. Plasma samples for amino acid analysis were collected before administration of nitrogen-scavenging agents and at least 3 h after the most recent meal. Plasma amino acid analysis and other laboratory analyses were performed at local CLIA-certified laboratories. EPI INFO software (http://wwwn.cdc.gov/epiinfo/) was used to calculate body weight and height z-scores based on the 2000 CDC growth charts at the most recent visit for all subjects between the ages of 2 and 20 years. For head circumference percentile, the appropriate reference data for age and gender were used (38,39). For all parameters studied, the number of subjects with data for that parameter is indicated in the text or table. For developmental delay and/or intellectual disability, data were obtained from parental report at baseline visit or from physical examination at either baseline or follow-up visits. For the single subject who had liver transplantation, data from the most recent visit prior to transplantation were used in the analysis.
Mouse colony management
All mouse studies were performed in the Transgenic Mouse Facility at Baylor College of Medicine. The mice were maintained in microisolator cages in ventilated racks with a 12:12 h, light:dark cycle. Mice were fed either Lab Diet 5V5R or Harlan 2020 chows, which have comparable nutritional content. All mouse studies were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee (IACUC).
Neonatal arginase 1 deficiency mouse studies
A neonatal mouse model for arginase deficiency (B6.129-Arg1tm1Rki/J) was obtained from Jackson Laboratories (Bar Harbor, ME, USA) (26). Because of initial poor litter survival in the mouse colony, the B6.129-Arg1tm1Rki/J female mice were crossed to 129/SvEv male mice, and the F1 offspring were intercrossed to generate F2 offspring with improved survival which were used for the study. Both male and female mice were used for all studies. All mating cages were provided with drinking water supplemented with sodium benzoate (Spectrum Chemical Manufacturing Company, Gardena, CA, USA), a nitrogen-scavenging agent, (1.5 mg/ml of drinking water, pH adjusted to 7–8, drug dosage: ∼250 mg/kg/day) which was introduced at least 48 h prior to delivery of the pups and continued through the course of the experiment. For initial pharmacokinetic studies, varied doses of AEB1102 (diluted in sterile phosphate-buffered saline, PBS, Mediatech, Manassas, VA, USA) were administered as a single dose by intraperitoneal injection using a 0.3 ml syringe at postnatal day 4–5. Placebo-treated mice were administered an equivalent dose of sterile PBS via intraperitoneal injection. The mice were sacrificed by decapitation under deep anesthesia with inhaled isoflurane (Butler Schein Animal Health, Dublin, OH, USA) 24 h after administration of AEB1102. Trunk blood was collected in a heparin-coated tube at sacrifice, and plasma was isolated for plasma amino acid analysis. Similar single-dose studies were performed with sacrifice 48 h or later after administration to collect plasma for time-course studies. For repeated dose studies, dosing was performed via intraperitoneal injection every 48 h starting at postnatal day 6, and mice were sacrificed by decapitation under deep anesthesia with inhaled isoflurane at postnatal day 11 (∼24 h after third dose). Trunk blood and tissues (liver and whole brain) were collected. For survival studies, mice were monitored daily and compassionately sacrificed when pups demonstrated decreased movement, constant tremor, seizure or ≥20% weight loss. All genotyping was performed using DNA isolated from toe tissue collected from neonatal pups using published primer sequences (26).
Adult arginase 1 deficiency mouse studies
The C57BL/6-Arg1tm1Pmu/J; Arg1f/f mouse was obtained from Jackson Laboratories (Bar Harbor, ME, USA) and crossed with a mouse that has a ubiquitously expressed tamoxifen-inducible Cre recombinase (B6.129-Gt(ROSA)26Sortm1(Cre/ERT2)Tyj/J; Cre+) to generate an inducible adult mouse model of arginase deficiency (27,28,40,41). Genotyping to identify Cre+ mice was performed using standard procedures with DNA isolated from tail tissue collected from pups at weaning. To generate the arginase-deficient model, tamoxifen (Sigma, St Louis, MO, USA) was administered (75 mg/kg or maximum dose of 100 µl of 20 mg/ml solution) via oral gavage for 5 consecutive days to 5- to 10-week-old male and female mice (referred to as Days 1–5). Tamoxifen was prepared as a solution of 20 mg/ml (200 mg in 0.5 ml of 100% ethanol and 9.5 ml of corn oil, mixed on rotator for 3–4 h at room temperature, stored at −20°C for no longer than 2–3 weeks). On Day 15 after first oral gavage dose of tamoxifen, male and female mice were administered AEB1102 (in sterile PBS) via intraperitoneal injection using a Hamilton syringe. Placebo-treated mice were administered an equivalent volume of sterile PBS via intraperitoneal injection. For repeat dose experiments, dosing of AEB1102 or PBS was repeated every 48 h. Mice were weighed on Day 12 and then daily starting on Day 15 until the end of the experiment. Mice that lost 20% or more of their body weight (based on weight on Day 12) or were in extremis were euthanized prior to study completion as per IACUC guidelines. All blood collections in adult animals were performed with retro-orbital collections into heparin-coated tubes after anesthesia with inhaled isoflurane in the fed state. Tissues were collected after cervical dislocation under anesthesia.
Plasma and tissue studies (mouse)
Plasma and tissue samples were stored at −80°C until time of analysis. Tissue samples for amino acid analysis were homogenized in Seraprep™ (10 µl/mg of tissue, Pickering Labs, Mountain View, CA, USA) and centrifuged at 2000 rpm (376 g) × 4 min in a microfuge tube. The supernatant was stored at −80°C until the time of analysis. Amino acid analysis was performed by Biochrom 30 Amino Acid Analyzer (Biochrom, UK) at the Baylor College of Medicine Medical Genetics Laboratories using standard methods. Ammonia levels were measured using the Ammonia Assay Kit (Sigma) immediately after sample collection. Guanidinoacetate concentrations in plasma were determined by UPLC-MS/MS (Waters, UK) with a previously published assay (42).
Cynomolgus monkey studies
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The contract testing facility selected to perform the animal studies (MPI Research, Inc., Mattawan, MI, USA) maintains an Animal Welfare Assurance statement with the National Institutes of Health, Office of Laboratory Welfare (A4192-01, A3181-01). To ensure compliance, protocols were approved by the MPI Research Institutional Animal Care and Use Committee before the initiation of treatment.
The repeat intravenous dose study was performed in naïve male and female cynomolgus monkeys that were 2–4 kg and 2–5 years of age at the time of the study. The human recombinant enzyme (AEB1102) was administered via a single intravenous injection into the saphenous or other suitable vein at Days 1 and 8. The dose volume for each animal was determined based on body weight. AEB1102 was formulated in 10% glycerol in PBS, pH 7.4, and provided to MPI at a nominal concentration of 3 mg/ml. This formulation was diluted at a 1:1 ratio with 0.45% sodium chloride on each day of administration. These formulations were stored at −60 to −90°C. Three doses (0.1 mg/kg, 0.3 mg/kg and 1 mg/kg) were used in two monkeys each in a repeated dose study.
Blood samples were collected prior to dose administration and at the following time points after dose administration: 5 min, 15 min, 3, 6, 24, 48, 72, 96, 120, 144 and 168 h. For analysis of serum AEB1102 concentration, blood samples were collected (0.5 ml/time point) from the femoral vein/artery (or other suitable vein) into tubes with no anticoagulant. Samples were allowed to clot at room temperature for at least 30 min and centrifuged to collect serum. Following centrifugation, the serum samples were split into two approximately equal aliquots and stored frozen (−70 ± 10°C) until analysis. For analysis of plasma arginine concentration, blood (1 ml/time point) was collected from the femoral vein/artery into K2EDTA tubes and centrifuged. After centrifugation, acetic acid (2%) was added to the plasma samples, and the aliquots were stored frozen (−70 ± 10°C) until analysis.
Analysis for serum AEB1102 concentrations was performed at Intertek Pharmaceutical Services (San Diego, CA, USA) using an enzyme activity assay in cynomolgus monkey serum. Analysis of plasma arginine concentrations also was performed at Intertek Pharmaceutical Services (Eldorado Hills, CA, USA) using an LC-MS/MS assay.
Statistical analysis
A one-way ANOVA with Dunnett's post-test with comparisons of all groups to the treated −/− (neonatal model) or treated Cre+ (adult model) group was performed for analyses of plasma and tissue metabolites. For two-sample comparisons, a Mann–Whitney test was used.
Acknowledgements
This work was supported by the Genzyme/ACMG Foundation for Genetic and Genomic Medicine Medical Genetics Training Award in Clinical Biochemical Genetics (L.C.B.), the National Urea Cycle Disorders Foundation Fellowship (L.C.B.), a fellowship from the Urea Cycle Disorders Consortium (UCDC; U54HD061221), which is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) (LCB), and the National Institutes of Health [T32 GM07526 to L.C.B., R01 DK102641 to B.L., and R01 CA154754 to G.G. and E.S.]. S.C.S.N. is a recipient of the DDCF Clinical Scientist Development Award (Grant #2013095); no part of this funding was used to support research with animal models or their primary tissues. This work was also supported by Baylor College of Medicine IDDRC Grant Number 1 U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development or the National Institutes of Health. This work was also funded by a series of cores supported by the National Institutes of Health [HD024064 to the BCM Intellectual and Developmental Disabilities Research Center, AI036211, CA125123, RR024574 to the Baylor College of Medicine Advanced Technology Cores, and DK56338 to the Texas Medical Center Digestive Disease Center]. The Urea Cycle Disorders Consortium (UCDC; U54HD061221) is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). Members of the Urea Cycle Disorders Consortium include Mark L. Batshaw, Mendel Tuchman, Marshall L. Summar, Nicholas Ah Mew, Matthias R. Baumgartner, Susan A. Berry, Stephen Cederbaum, Curtis Coughlin, III, George A. Diaz, Annette Feigenbaum, Renata C. Gallagher, Cary O. Harding, Georg Hoffmann, Douglas S. Kerr, Brendan Lee, Uta Lichter-Konecki, Shawn E. McCandless, J. Lawrence Merritt II, Sandesh CS Nagamani, Andreas Schulze, Margretta R. Seashore, Tamar Stricker, Susan Waisbren, James Weisfeld-Adams, Derek Wong, and Mark Yudkoff.
Conflict of Interest statement. G.G. serves on the board and has equity in Aeglea BioTherapeutics. S.E.A. and S.W.R. are employed by and have equity in Aeglea BioTherapeutics. E.S. owns stock in Aeglea BioTherapeutics. D.E.J. is a paid consultant of Aeglea BioTherapeutics. None of the other authors received research support or other financial support from Aeglea BioTherapeutics.
Contributor Information
Collaborators: Members of the Urea Cycle Disorders Consortium, Mark L. Batshaw, Mendel Tuchman, Marshall L. Summar, Nicholas Ah Mew, Matthias R. Baumgartner, Susan A. Berry, Stephen Cederbaum, Curtis Coughlin, III, George A. Diaz, Annette Feigenbaum, Renata C. Gallagher, Cary O. Harding, Georg Hoffmann, Douglas S. Kerr, Brendan Lee, Uta Lichter-Konecki, Shawn E. McCandless, J. Lawrence Merritt, II, Sandesh CS Nagamani, Andreas Schulze, Margretta R. Seashore, Tamar Stricker, Susan Waisbren, James Weisfeld-Adams, Derek Wong, and Mark Yudkoff
References
- 1.Carvalho D.R., Brum J.M., Speck-Martins C.E., Ventura F.D., Navarro M.M., Coelho K.E., Portugal D., Pratesi R. (2012) Clinical features and neurologic progression of hyperargininemia. Pediatr. Neurol., 46, 369–374. [DOI] [PubMed] [Google Scholar]
- 2.Uchino T., Snyderman S.E., Lambert M., Qureshi I.A., Shapira S.K., Sansaricq C., Smit L.M., Jakobs C., Matsuda I. (1995) Molecular basis of phenotypic variation in patients with argininemia. Hum. Genet., 96, 255–260. [DOI] [PubMed] [Google Scholar]
- 3.Cederbaum S.D., Shaw K.N., Spector E.B., Verity M.A., Snodgrass P.J., Sugarman G.I. (1979) Hyperargininemia with arginase deficiency. Pediatr. Res., 13, 827–833. [DOI] [PubMed] [Google Scholar]
- 4.Jorda A., Portoles M., Rubio V., Capdevila A., Vilas J., Garcia-Pino J. (1987) Liver fibrosis in arginase deficiency. Arch. Pathol. Lab. Med., 111, 691–692. [PubMed] [Google Scholar]
- 5.Santos Silva E., Martins E., Cardoso M.L., Barbot C., Vilarinho L., Medina M. (2001) Liver transplantation in a case of argininaemia. J. Inherit. Metab. Dis., 24, 885–887. [DOI] [PubMed] [Google Scholar]
- 6.Jorda A., Rubio V., Portoles M., Vilas J., Garcia-Pino J. (1986) A new case of arginase deficiency in a Spanish male. J. Inherit. Metab. Dis., 9, 393–397. [DOI] [PubMed] [Google Scholar]
- 7.Deignan J.L., De Deyn P.P., Cederbaum S.D., Fuchshuber A., Roth B., Gsell W., Marescau B. (2010) Guanidino compound levels in blood, cerebrospinal fluid, and post-mortem brain material of patients with argininemia. Mol. Genet. Metab., 100 (Suppl. 1), S31–S36. [DOI] [PubMed] [Google Scholar]
- 8.Marescau B., Qureshi I.A., De Deyn P., Letarte J., Ryba R., Lowenthal A. (1985) Guanidino compounds in plasma, urine and cerebrospinal fluid of hyperargininemic patients during therapy. Clin. Chim. Acta., 146, 21–27. [DOI] [PubMed] [Google Scholar]
- 9.Mizutani N., Hayakawa C., Ohya Y., Watanabe K., Watanabe Y., Mori A. (1987) Guanidino compounds in hyperargininemia. Tohoku J. Exp. Med., 153, 197–205. [DOI] [PubMed] [Google Scholar]
- 10.Wiechert P., Mortelmans J., Lavinha F., Clara R., Terheggen H.G., Lowenthal A. (1976) Excretion of guanidino-derivates in urine of hyperargininemic patients. J. Genet. Hum., 24, 61–72. [PubMed] [Google Scholar]
- 11.Deignan J.L., Marescau B., Livesay J.C., Iyer R.K., De Deyn P.P., Cederbaum S.D., Grody W.W. (2008) Increased plasma and tissue guanidino compounds in a mouse model of hyperargininemia. Mol. Genet. Metab., 93, 172–178. [DOI] [PubMed] [Google Scholar]
- 12.Snyderman S.E., Sansaricq C., Norton P.M., Goldstein F. (1979) Argininemia treated from birth. J. Pediatr., 95, 61–63. [DOI] [PubMed] [Google Scholar]
- 13.Snyderman S.E., Sansaricq C., Chen W.J., Norton P.M., Phansalkar S.V. (1977) Argininemia. J. Pediatr., 90, 563–568. [DOI] [PubMed] [Google Scholar]
- 14.Cederbaum S.D., Moedjono S.J., Shaw K.N., Carter M., Naylor E., Walzer M. (1982) Treatment of hyperargininaemia due to arginase deficiency with a chemically defined diet. J. Inherit. Metab. Dis., 5, 95–99. [DOI] [PubMed] [Google Scholar]
- 15.Silva E.S., Cardoso M.L., Vilarinho L., Medina M., Barbot C., Martins E. (2013) Liver transplantation prevents progressive neurological impairment in argininemia. JIMD Rep., 11, 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanalas J.J., Spector E.B., Cederbaum S.D. (1982) Hollow-fiber reactors containing mammalian arginase: an approach to enzyme replacement therapy. Biochem. Med., 27, 46–55. [DOI] [PubMed] [Google Scholar]
- 17.Adriaenssens K., Karcher D., Marescau B., Van Broeckhoven C., Lowenthal A., Terheggen H.C. (1984) Hyperargininemia: the rat as a model for the human disease and the comparative response to enzyme replacement therapy with free arginase and arginase-loaded erythrocytes in vivo. Int. J. Biochem., 16, 779–786. [DOI] [PubMed] [Google Scholar]
- 18.Kaminsky Y.G., Kosenko E.A. (2012) Argocytes containing enzyme nanoparticles reduce toxic concentrations of arginine in the blood. Bull. Exp. Biol. Med., 153, 406–408. [DOI] [PubMed] [Google Scholar]
- 19.Michels V.V., Beaudet A.L. (1978) Arginase deficiency in multiple tissues in argininemia. Clin. Genet., 13, 61–67. [DOI] [PubMed] [Google Scholar]
- 20.Sakiyama T., Nakabayashi H., Shimizu H., Kondo W., Kodama S., Kitagawa T. (1984) A successful trial of enzyme replacement therapy in a case of argininemia. Tohoku J. Exp. Med., 142, 239–248. [DOI] [PubMed] [Google Scholar]
- 21.Mizutani N., Hayakawa C., Maehara M., Watanabe K. (1987) Enzyme replacement therapy in a patient with hyperargininemia. Tohoku J. Exp. Med., 151, 301–307. [DOI] [PubMed] [Google Scholar]
- 22.Feun L.G., Kuo M.T., Savaraj N. (2015) Arginine deprivation in cancer therapy. Curr. Opin. Clin. Nutr. Metab. Care, 18, 78–82. [DOI] [PubMed] [Google Scholar]
- 23.Stone E., Chantranupong L., Gonzalez C., O'Neal J., Rani M., VanDenBerg C., Georgiou G. (2012) Strategies for optimizing the serum persistence of engineered human arginase I for cancer therapy. J. Contr. Release, 158, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stone E.M., Glazer E.S., Chantranupong L., Cherukuri P., Breece R.M., Tierney D.L., Curley S.A., Iverson B.L., Georgiou G. (2010) Replacing Mn(2+) with Co(2+) in human arginase i enhances cytotoxicity toward l-arginine auxotrophic cancer cell lines. ACS Chem. Biol., 5, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mauldin J.P., Zeinali I., Kleypas K., Woo J.H., Blackwood R.S., Jo C.H., Stone E.M., Georgiou G., Frankel A.E. (2012) Recombinant human arginase toxicity in mice is reduced by citrulline supplementation. Transl. Oncol., 5, 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iyer R.K., Yoo P.K., Kern R.M., Rozengurt N., Tsoa R., O'Brien W.E., Yu H., Grody W.W., Cederbaum S.D. (2002) Mouse model for human arginase deficiency. Mol. Cell Biol., 22, 4491–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasten J., Hu C., Bhargava R., Park H., Tai D., Byrne J.A., Marescau B., De Deyn P.P., Schlichting L., Grody W.W. et al. (2013) Lethal phenotype in conditional late-onset arginase 1 deficiency in the mouse. Mol. Genet. Metab., 110, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sin Y.Y., Ballantyne L.L., Mukherjee K., St Amand T., Kyriakopoulou L., Schulze A., Funk C.D. (2013) Inducible arginase 1 deficiency in mice leads to hyperargininemia and altered amino acid metabolism. PLoS One, 8, e80001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spector E.B., Rice S.C., Cederbaum S.D. (1983) Immunologic studies of arginase in tissues of normal human adult and arginase-deficient patients. Pediatr. Res., 17, 941–944. [DOI] [PubMed] [Google Scholar]
- 30.Grody W.W., Argyle C., Kern R.M., Dizikes G.J., Spector E.B., Strickland A.D., Klein D., Cederbaum S.D. (1989) Differential expression of the two human arginase genes in hyperargininemia. Enzymatic, pathologic, and molecular analysis. J. Clin. Invest., 83, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grody W.W., Kern R.M., Klein D., Dodson A.E., Wissman P.B., Barsky S.H., Cederbaum S.D. (1993) Arginase deficiency manifesting delayed clinical sequelae and induction of a kidney arginase isozyme. Hum. Genet., 91, 1–5. [DOI] [PubMed] [Google Scholar]
- 32.Hu C., Kasten J., Park H., Bhargava R., Tai D.S., Grody W.W., Nguyen Q.G., Hauschka S.D., Cederbaum S.D., Lipshutz G.S. (2014) Myocyte-mediated arginase expression controls hyperargininemia but not hyperammonemia in arginase-deficient mice. Mol. Ther., 22, 1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayasaka S., Kodama T., Ohira A. (2011) Retinal risks of high-dose ornithine supplements: a review. Br. J. Nutr., 106, 801–811. [DOI] [PubMed] [Google Scholar]
- 34.Simell O., Takki K. (1973) Raised plasma-ornithine and gyrate atrophy of the choroid and retina. Lancet, 1, 1031–1033. [DOI] [PubMed] [Google Scholar]
- 35.Amayreh W., Meyer U., Das A.M. (2014) Treatment of arginase deficiency revisited: guanidinoacetate as a therapeutic target and biomarker for therapeutic monitoring. Dev. Med. Child Neurol., 56, 1021–1024. [DOI] [PubMed] [Google Scholar]
- 36.Vockley J.G., Jenkinson C.P., Shukla H., Kern R.M., Grody W.W., Cederbaum S.D. (1996) Cloning and characterization of the human type II arginase gene. Genomics, 38, 118–123. [DOI] [PubMed] [Google Scholar]
- 37.Seminara J., Tuchman M., Krivitzky L., Krischer J., Lee H.S., Lemons C., Baumgartner M., Cederbaum S., Diaz G.A., Feigenbaum A. et al. (2010) Establishing a consortium for the study of rare diseases: The Urea Cycle Disorders Consortium. Mol. Genet. Metab., 100 (Suppl. 1), S97–S105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bushby K.M., Cole T., Matthews J.N., Goodship J.A. (1992) Centiles for adult head circumference. Arch. Dis. Child, 67, 1286–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nellhaus G. (1968) Head circumference from birth to eighteen years. Practical composite international and interracial graphs. Pediatrics, 41, 106–114. [PubMed] [Google Scholar]
- 40.El Kasmi K.C., Qualls J.E., Pesce J.T., Smith A.M., Thompson R.W., Henao-Tamayo M., Basaraba R.J., Konig T., Schleicher U., Koo M.S. et al. (2008) Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol., 9, 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ventura A., Kirsch D.G., McLaughlin M.E., Tuveson D.A., Grimm J., Lintault L., Newman J., Reczek E.E., Weissleder R., Jacks T. (2007) Restoration of p53 function leads to tumour regression in vivo. Nature, 445, 661–665. [DOI] [PubMed] [Google Scholar]
- 42.Sun Q., O'Brien W.E. (2010) Diagnosis of creatine metabolism disorders by determining creatine and guanidinoacetate in plasma and urine. Methods Mol. Biol., 603, 175–185. [DOI] [PubMed] [Google Scholar]